

Abstract
Nonsense-mediated mRNA decay (NMD) degrades transcripts with premature stop codons. Given the prevalence of nonsense single nucleotide polymorphisms (SNPs) in the general population, it is urgent to catalog the effects of clinically approved drugs on NMD activity: any interference could alter the expression of nonsense SNPs, inadvertently inducing adverse effects. This risk is higher for patients with disease-causing nonsense mutations or an illness linked to dysregulated nonsense transcripts. On the other hand, hundreds of disorders are affected by cellular NMD efficiency and may benefit from NMD-modulatory drugs. Here, we profiled individual FDA-approved drugs for their impact on cellular NMD efficiency using a sensitive method that directly probes multiple endogenous NMD targets for a robust readout of NMD modulation. We found most FDA-approved drugs cause unremarkable effects on NMD, while many elicit clear transcriptional responses. Besides several potential mild NMD modulators, the anticancer drug homoharringtonine (HHT or omacetaxine mepesuccinate) consistently upregulates various endogenous NMD substrates in a dose-dependent manner in multiple cell types. We further showed translation inhibition mediates HHT's NMD effect. In summary, many FDA drugs induce transcriptional changes, and a few impact global NMD, and direct measurement of endogenous NMD substrate expression is robust to monitor cellular NMD.
Keywords: Ptbp2, Hnrnpl, Tra2b, mitoxantrone, topotecan hydrochloride, montelukast sodium, artesunate, albendazole, gabexate mesilate, trazodone hydrochloride
Graphical abstract
Zheng and colleagues determined the effects of individual FDA-approved drugs on NMD activity by quantitatively assessing multiple endogenous NMD target isoforms. While many drugs induce transcriptional responses, some show the potentials of modulating NMD to varying degrees, including HHT, identified as a robust NMD inhibitor.
Introduction
Nonsense-mediated mRNA decay (NMD) selectively degrades mutant transcripts with a premature termination codon (PTC) to prevent translation of C-terminal, truncated proteins.1, 2, 3 As a result, nonsense mutations often lead to the loss of functional proteins, accounting for molecular pathogenesis of over 20% monogenic diseases.4, 5, 6, 7, 8 For example, nonsense mutation is the leading cause of cystic fibrosis (CF) and Duchenne muscular dystrophy (DMD).9, 10, 11, 12 Since most nonsense mutation-associated disorders are rare diseases, therapeutic development is strategized toward targeting their commonalities (e.g., the underlying nonsense mutations and associated pathways) rather than individual symptoms.
Nonsense transcripts can also be generated by erroneous mRNA processing. NMD therefore acts as a post-transcriptional quality control mechanism, ensuring the fidelity of gene expression.1,3,13,14 In diseases associated with aberrant mRNA processing, excessive NMD-sensitive transcripts are produced. For example, in amyotrophic lateral sclerosis (ALS), cancers, and some multisystem disorders, defective splicing regulators cause dysregulated splicing, which shifts open reading frames and creates PTCs in many transcripts.15, 16, 17, 18, 19 These cryptic transcript variants reduce gene expression output and may gain toxic functions, leading to disturbance of cellular homeostasis and contributing to pathogenesis.20,21
Targeting cellular NMD therefore presents an opportunity to modify disease outcomes. In monogenic diseases, if the PTC-containing transcript encodes a (partially) functional protein, lower NMD efficiency is desirable. NMD inhibition can also enhance the effectiveness of nonsense suppression therapy, which allows a translational readthrough of the PTC to produce full-length functional proteins.22 Indeed, lower cellular NMD efficiency is associated with milder symptoms caused by the same nonsense mutation and better response to nonsense suppression therapy.10 On the other hand, if the PTC-containing transcript encodes a dominant negative or a toxic truncated protein, higher NMD efficiency is preferred to eliminate the production of the deleterious protein.23 NMD potentiation thus suppresses syndromes caused by nonsense mutations and/or cryptic splicing. Theoretically, NMD efficiency can modulate hundreds of different disorders caused by nonsense mutations or aberrant mRNA processing, with the caution that the effect depends on whether the nonsense transcript encodes a functional or deleterious protein.24
On the flip side, perturbing NMD efficiency by otherwise “safe” approved medicine could be a hidden mechanism causing unforeseen adverse effects, because even a healthy individual contains a dozen nonsense single nucleotide polymorphisms (SNPs). Early studies estimated that the human genome contained a thousand nonsense SNPs, half of which induced NMD of transcripts.25 By profiling 805 randomly selected nonsense SNPs in populations, Yngvadottir et al. found that the average individual carried ∼46 nonsense SNPs, including 14 PTC/PTC homozygous SNPs and 18 PTC/normal heterozygous SNPs.26 Because Yngvadottir et al. analyzed only a fraction of reported nonsense SNPs, the average individual likely contains many more nonsense variants. The biological impact of these genetic variants on individuals is generally unknown. These nonsense SNPs appear tolerated in normal circumstances, probably because their expression is silenced by NMD, but they may no longer be “benign” if NMD is attenuated in unfavorable conditions. Therefore, nonsense SNPs present an overlooked risk factor that may compound aging and health status to induce complications.
One way for nonsense SNPs to reveal themselves is due to treatment: a drug that modulates NMD efficiency alters the expression of nonsense SNPs and amplifies their associated risks, possibly leading to unanticipated adverse events. Theoretically, these adverse effects vary by individual for carrying different nonsense SNPs and likely only manifest in a subpopulation. Because of this inconsistency and low sampling probability, any adverse or side effects cannot be fully determined during clinical trials. However, this hidden risk could be substantial once drugs are approved for use in a larger population.
Given the prevalence of nonsense SNPs in the general population, it is urgent to evaluate FDA-approved drugs for their possible impact on the NMD pathway. Patients experience adverse effects from medicine, usually because we lack knowledge of the drug's full mechanism. In one potentially debilitating scenario, a patient with disease-causing nonsense mutations is administered a drug for a second illness unrelated to the nonsense mutations but the drug turns out to modulate NMD.
Several small molecules were identified as NMD inhibitors.27 For example, Martin et al. screened 31 selected compounds that fit into the SMG7 pocket to prevent SMG7 binding to UPF1, and identified NMDI14.28 Bhuvanagiri et al. used a PTC-containing luciferase reporter to screen the Prestwick library and showed 5-azacytidine induced ∼2-fold upregulation of a luciferase-based NMD reporter.29 While the mechanism of 5-azacytidine inhibiting NMD is unclear, its effect requires Myc upregulation.29 Gonzalez-Hilarion et al. also used a luciferase-based reporter to screen drugs and identified amlexanox, which promotes readthrough of PTC-containing mRNAs and modestly upregulates nonsense mutation-containing mRNAs, but somehow does not affect natural NMD substrate expression.30,31 Dang et al. showed that pateamine A interacts with eIF4AIII to repress NMD.32 Durand et al. reported that NMDI1 disrupted the UPF1-SMG5 interaction and inhibited NMD.33 Exogenous reporter-based systems have also been applied in genetic screens to identify NMD-modulating genes. Alexandrov et al. developed an NMD-sensitive fluorescence reporter system in a CRISPR screen and identified 11 candidate NMD regulators in addition to five known NMD pathway components.34 Casadio et al. conducted an RNAi screen in C. elegans expressing a PTC-containing GFP reporter and identified five novel NMD regulators.35 These studies demonstrate the power of NMD reporters to identify NMD modulators.36
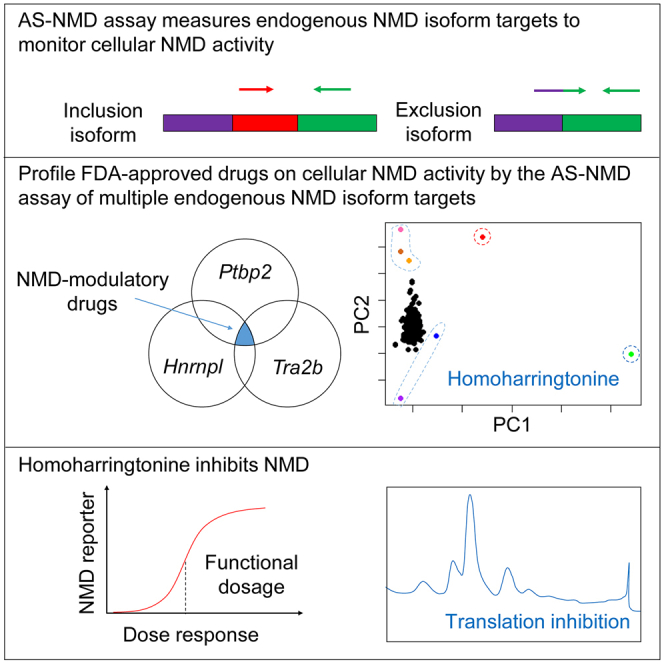
Because false positives and negatives are common in any drug screen and because previous screens relied on exogenous reporters, we decided to individually assess and rigorously confirm the influence of each FDA-approved drug on cellular NMD activity. Individually profiling each drug met the urgent need to evaluate these approved drugs for their possible adverse effects on nonsense SNPs. To do so unbiasedly, we directly analyzed the endogenous NMD targets upon each drug treatment (Figure 1A). Simultaneous measurements of multiple NMD targets for a consistent NMD effect enhances the rigor of calling NMD modulators.
Figure 1.

Schematic of the workflow for profiling FDA-approved drugs and preliminary analysis
(A) N2a cells were cultured in vitro the day before treatment. After a uniform 5 μM treatment for 5 h, cells were then directly collected in TRIzol reagents for total RNA extraction. Next, standard reverse transcription assay was performed. We applied our RT-qPCR-based AS-NMD assay to identify drugs with uniform reporter genes response. We then selected NMD modulator candidates for further analyses including dose test and mechanism study. (B) Isoform specific primers, including exon-exon junction primers, were used to detect inclusion or exclusion isoform. (C) PCA analysis of reporter gene isoform expression after drug treatment (the six RQ values). Each dot represents an FDA-approved drug. TG is included as a positive control.
Specifically, we applied an RT-qPCR-based quantitative method, termed AS-NMD assay, to examine expression of a panel of endogenous NMD-targeted isoforms, including Ptbp2, Hnrnpl, and Tra2b. Alternative mRNA splicing of these genes shifts the reading frame and produces both NMD and non-NMD isoforms.1,13,37, 38, 39 These NMD isoforms contain a PTC >50 nt upstream of the last exon-exon junction. In mammals, this sequence feature (termed the 50-nt rule) has the strongest potential in triggering NMD among the known NMD-inducing characteristics.40 Therefore, these endogenous NMD reporters have a robust predictive power to identify NMD modulators.
We separately measured the expression of their non-NMD isoforms to gauge whether the expression change in the NMD isoform was due to NMD modulation, transcriptional regulation, or splicing changes. NMD modulation should only affect the NMD transcripts without influencing the non-NMD transcripts. By contrast, transcriptional regulation is expected to change both NMD and non-NMD isoforms in the same direction. Alternative splicing (AS) alters expression levels of NMD and non-NMD transcripts in the opposite direction: upregulating one isoform and downregulating the other. Because the expression of NMD and non-NMD isoforms are individually measured and quantified by sensitive RT-qPCR assay, the method has low incidence of false positives and negatives.
Results
The workflow of profiling each drug effect on NMD
We profiled 704 FDA-approved drugs curated by the National Center for Advancing Translational Sciences (NCATS, NIH) (Table S1). The overall workflow is described in Figure 1A. We plated 650,000 cells in each well of a 6-well plate. The next morning, we applied individual drugs at the final concentration of 5 μM. Each treatment batch contained a negative control DMSO treatment and a positive control 0.2 μM thapsigargin (TG) treatment, previously shown to inhibit NMD.39 Because all the drugs were divided into many treatment group batches, these controls served as both a benchmark and a quality control for the assay. After 5 h of treatment cells were collected and total RNA extracted. We performed RT-qPCR analysis of NMD and non-NMD isoforms to determine their individual fold changes relative to the DMSO treatment (Table S2).
Specifically, we assayed three different endogenous NMD targets, Ptbp2, Hnrnpl, and Tra2b, to increase the robustness of determining NMD-modulatory drugs. As a result, our profiling method is expected to have a low false-positive rate. Normal full-length Ptbp2 transcript (herein F_Ptbp2) contains exon 10. Skipping exon 10 produces an NMD-sensitive isoform (herein N_Ptbp2).41,42 For Hnrnpl, the NMD-sensitive isoform (N_Hnrnpl) includes exon 6, and the non-NMD isoform (herein F_Hnrnpl) excludes exon 6.43 The Tra2b NMD isoform includes exon 2 (N_Tra2b), and its non-NMD isoform excludes exon 2 (herein F_Tra2b).44 We designed exon-exon junction primers specific to the inclusion or exclusion isoform (Figure 1B and Table S3) for quantitative measurement of each isoform expression using RT-qPCR. If a drug alters only NMD efficiency, the NMD isoforms are expected to change expression levels, whereas the non-NMD isoforms do not change. When all three NMD target genes behave similarly in response to a treatment, the drug is a high-confidence NMD modulator.
AS-NMD assay revealed cellular responses to FDA-approved drugs
We first examined the global pattern of drug responses regarding the endogenous reporter isoform expression. We conducted unbiased hierarchical clustering of the normalized expression levels (log2 transformation of RQ (relative quantification)). The heatmap showed varying effects of the FDA-approved drugs (Figure S1). The positive control TG, situated at the bottom of the heatmap, is clustered with homoharringtonine (HHT) and stands out with strong induction of the three NMD isoforms. We also performed principal component analysis (PCA) to determine whether the drug responses could be categorized by lower dimensions. Interestingly, most drugs were not clearly separated by any distinguishing component, except TG, HHT, and montelukast sodium (Figure 1C).
We show the responses of the NMD and non-NMD isoforms to each drug treatment in a scatterplot (Figures 2A–2C). Positive control TG treatments on average increased the expression of N_Ptbp2, N_Hnrnpl, and N_Tra2b by 7.5 ± 0.7-, 12.7 ± 1.0-, and 15.6 ± 1.0-fold, respectively (their averaged fold changes are shown as the red dots in Figures 2A–2C), but slightly affected their corresponding non-NMD isoforms (1.5 ± 0.1-, 1.1 ± 0.1-, 1.4 ± 0.1-fold, respectively), confirming the assay's effectiveness in identifying NMD modulators.
Figure 2.

AS-NMD assay showed reporters' responses to each FDA drug treatment
(A–C) Expression level changes in non-NMD isoform and NMD isoform of Ptbp2 (A), Hnrnpl (B), and Tra2b (C) were shown as scatterplots. Upregulation and downregulation of reporter gene expression were indicated by the 2-fold-change lines. Each dot represented the average of three replicates of one drug treatment, and the red dot represented the averaged expression level change in TG. (D–F) Identification of drugs that altered NMD isoforms >2-fold or <0.5-fold and altered the non-NMD isoforms between 0.5- and 2-fold of Ptbp2 (D), Hnrnpl (E), and Tra2b (F). (G and H) Venn diagrams were used to identify drugs exhibiting similar responses in all three reporter genes. (G) One drug changed NMD isoforms <0.5-fold and non-NMD isoforms between 0.5- and 2-fold. (H) Two drugs changed NMD isoforms >2-fold and non-NMD isoforms between 0.5- and 2-fold. Positive control, TG, is not included in (D–H).
We were most interested in drugs that alter the expression of NMD isoforms without substantially changing the non-NMD isoforms. Because each drug was individually applied to cells and analyzed by RT-qPCR, each drug treatment could be analyzed independently. Our initial criteria for categorizing NMD modulator candidates were the following: drugs inducing >2-fold change in NMD isoforms, while minimally (<2-fold) affecting their non-NMD counterparts. For the Ptbp2 reporter, we found nine drugs (enhancers) that decreased N_Ptbp2 expression by more than 2-fold, and 24 drugs (repressors) that increased N_Ptbp2 expression by more than 2-fold, while the fold change in F_Ptbp2 was less than 2-fold (Figure 2D). Similarly, for the Hnrnpl and Tra2b reporters, we identified one and eight enhancers and four and 31 repressors, respectively (Figures 2E and 2F).
Drugs inducing a uniform response in all three reporter genes were classified as probable NMD modulators. Overlapping these results yielded one NMD enhancer candidate (mitoxantrone hydrochloride) and two NMD repressor candidates (HHT and secoisolariciresinol) that uniformly affected the three reporters in the same direction (Figures 2G and 2H). Interestingly, all three drugs and their derivatives are anticancer drugs that work by suppressing cancer cell proliferation and viability.45, 46, 47, 48, 49 Mitoxantrone has been implicated in RNA regulation, as it binds to tau pre-mRNA, splicing regulatory elements to stabilize the RNA secondary structure and prevent erroneous splicing caused by gene mutations.50,51
FDA-approved drugs induce substantial transcriptional response
Many drugs elicited strong transcriptional responses. They altered the NMD and non-NMD isoform expression in the same direction with a similar magnitude, as most drugs were situated close to the diagonal lines, indicative of a transcriptional change following drug treatment (Figures 2A–2C). We found no drug changed the two isoforms (>2-fold) in the opposite direction. The wide range of fold changes in either Ptbp2, Hnrnpl, or Tra2b also demonstrated the effectiveness of the drug dose in causing cellular response.
In total, 235 of 704 drugs induced a 2-fold change in either the NMD or non-NMD isoforms of at least one reporter gene (Figure 3A). Among them, 119 drugs induced a 2-fold change in both NMD and non-NMD isoforms of at least one reporter gene. These numbers increased to 476 and 285, respectively, when we set the threshold at 1.5-fold change (Figure 3A). These results show that transcriptional response, either direct or indirect, constitutes a major molecular effect of FDA-approved drugs.
Figure 3.

Normalizing transcriptional responses using the NMD/non-NMD ratio of reporter genes to assess drug's impact on cellular NMD regulation
(A) Many FDA-approved drugs induced transcriptional responses of the reporter genes. (B) PCA analysis of NMD/non-NMD ratios (RQ ratios) of three reporters. (C–E) Ratio of NMD/non-NMD isoforms were calculated for Ptbp2 (C), Hnrnpl (D), and Tra2b (E) reporter genes. Red lines indicated 2-fold upregulation or downregulation of the ratios, and the red dot represented the averaged NMD/non-NMD ratio of TG.
Normalizing transcriptional response to define drug effect on NMD activity
If a drug affects both transcription and NMD, the large effect size on transcription can obscure its effects on NMD and hinder the discovery of NMD-modulatory drugs using our preset criteria. Theoretically, when a drug inhibits transcription of a reporter gene and represses NMD, the non-NMD isoform decreases and the NMD isoform may or may not change (Figure S2, upper left). When a drug induces transcription and simultaneously represses NMD, the non-NMD isoform is upregulated, though not as strongly as the NMD isoform (Figure S2, upper right). When a drug inhibits transcription and enhances NMD, the non-NMD isoform is downregulated and the NMD isoform is further reduced (Figure S2, lower left). Finally, when a drug induces transcription of a reporter gene and enhances NMD, the non-NMD isoform is upregulated and the NMD isoform could either change or not (Figure S2, lower right). In all cases, a drug's effect on NMD cannot be determined based on the stringent criteria that the non-NMD isoform should not change (Figure S2). Furthermore, a weak NMD effect could be canceled by a stronger transcription effect, rendering the change in NMD isoforms unnoticeable or in an opposite direction.
Given the widespread transcriptional responses and the possibility that a drug's effect on NMD was masked, we applied a second analysis metric. To remove the confounding effect from transcription changes, we normalized the expression of the NMD isoform by its non-NMD counterpart (the NMD/non-NMD ratio). We found the mean NMD/non-NMD ratio was 1.04, 1.04, and 1.06 for Ptbp2, Hnrnpl, and Tra2b, respectively, consistent with the notion that this normalization method effectively removes the impact of transcription. Interestingly, PCA analysis of the NMD/non-NMD ratios revealed additional drugs distinguishable from the larger population (Figure 3B).
The NMD/non-NMD ratio means (close to 1) suggested most drugs did not obviously influence cellular NMD activity. Indeed, with a 2-fold change (in the NMD/non-NMD ratio) as the threshold, only 12 drugs substantially altered the NMD/non-NMD ratio for the Ptbp2 reporter, nine drugs for the Hnrnpl reporter, and 14 drugs for the Tra2b reporter (Figures 3C–3E). Note that a ratio change can be induced by NMD or splicing regulation, so true NMD modulators are rarer.
The top 15 ranked drugs that increased the NMD/non-NMD ratio for the Ptbp2, Hnrnpl, and Tra2b reporters are listed in Figures 4A–4C, respectively. Among them, naloxone hydrochloride, brimonidine, amlodipine, and CGS 15943 are cellular receptor antagonists.52, 53, 54, 55 Tocainide, procaine hydrochloride, and mepivacaine hydrochloride are voltage-gated sodium channel modulators.56, 57, 58 Vindesine sulfate, podofilox, albendazole, and vinorelbine tartrate are microtubule modulators.59, 60, 61 We also listed the top 15 ranked drugs that decreased the N_Ptbp2/F_Ptbp2, N_Hnrnpl/F_Hnrnpl, and N_Tra2b/F_Tra2b ratio (Figures 4D–4F). Annotations of these drugs from PubChem indicate a wide range of actions from cellular enzyme inhibitors (bestatin, naproxen sodium, pyridostigmine bromide, and pravastatin sodium),62, 63, 64, 65 inflammatory response regulators (triamcinolone acetonide, naproxen sodium, cortisone acetate, and diflunisal),66, 67, 68, 69 to antibiotic reagents (sulfacetamide, cefixime trihydrate, mafenide acetate, and ampicillin sodium).70, 71, 72, 73
Figure 4.

Top 15 ranked drugs based on the NMD/non-NMD ratios of each reporter gene
(A–C) Top 15 ranked drugs with possible NMD inhibition activity. TG is included as a positive control. Original values of the NMD/non-NMD ratio are showed next to drugs. Base-2 log scale is used for the Y axis. (D–F) Top 15 ranked candidates with enhanced NMD activity. Original values of the NMD/non-NMD ratio are showed next to drugs. All three Y axes used the linear scale. Error bar represented RQ±SEM for each reporter.
Several drugs have been implicated in RNA processing. Topotecan decreases splicing efficiency by interfering with the interaction between NHP2L1 and U4 snRNA.74 Its effect on Ptbp2 and Hnrnpl may therefore relate to its splicing activity. Idarubicin, increasing the N_Tra2b/F_Tra2b ratio, competes with HNRNPA1 for binding internal ribosomal entry sites (IRES) to inhibit IRES-dependent translation.75 Epirubicin promotes miR-503 export by disrupting the interaction between hnRNPA2B1 and miR-503.76
Most drugs exhibit differential ranking among the reporter genes, arguing for integrating the responses of all reporter genes to rigorously identifying NMD modulators. Note that the ratio does not distinguish NMD regulation from AS regulation. Drugs affecting the ratio of one reporter gene but not the others likely influence AS rather than NMD. By contrast, if all three reporter genes universally change their ratios in response to one drug, it is more likely a result of NMD regulation.
To rigorously determine NMD modulators, we first calculated the standard deviation (SD) of NMD/non-NMD ratios across all drugs. We then used 3xSD from the mean as a threshold to call a drug's effect on NMD activity. We found five possible NMD repressors from the Ptbp2 reporter assay, one from Hnrnpl and one from Tra2b (Figure 5A). Overlapping these results identified HHT (besides positive control TG) as the sole repressor candidate. Satisfactorily, HHT showed the largest NMD/non-NMD ratios consistently across all three reporter genes (RatioPtbp2 = 5.0, RatioHnrnpl = 25.0, and RatioTra2b = 11.7). At 3xSD, no NMD enhancer was identified for any reporter assay.
Figure 5.

Combining the responses of endogenous reporters to identify potential NMD modulators
(A) Venn diagrams using a threshold of the NMD/non-NMD ratio above (the average + 3xSD). (B) Venn diagrams using a threshold of the NMD/non-NMD ratio above (the average + 2xSD). (C) Venn diagrams using a threshold of the NMD/non-NMD ratio above (the average +1.5xSD). (D) Venn diagrams using a threshold of the NMD/non-NMD ratio below (the average −1.5xSD). (E) Fisher tests obtained 9 inhibitors and 1 enhancer with a threshold of FDR_Fisher ≤0.01. (F) Randomization tests obtained 3 inhibitors and 3 enhancers with FDR_ permutation ≤0.01.
When we set a 2xSD threshold, we found 14, two, and eight possible NMD repressors from the Ptbp2, Hnrnpl, and Tra2b reporter assays. Again, HHT stood out as the only overlapping hit (Figure 5B). With a lower 1.5xSD threshold, despite more candidate repressors being identified (35, two, and 16 for the Ptbp2, Hnrnpl, and Tra2b reporters), HHT was still the only drug affecting all reporters (Figure 5C). As for possible NMD enhancers, none were identified with the 2xSD threshold. A lower 1.5xSD threshold led to 10, 0, and one candidates from the Ptbp2, Hnrnpl, and Tra2b reporter assays, respectively, yielding no overlap (Figure 5D).
Although most drugs were not as robust as HHT in altering the expression of these three NMD substrates, some mild NMD-regulatory drugs may not have been captured by our stringent criteria. We applied two independent methods to statistically determine the possibility of weak NMD regulation. First, we used Fisher's method to measure the significance of a drug effect combining all three reporters. Specifically, a Z test was developed for the RQ ratio (the NMD transcript versus the non-NMD-transcript) at the log2 scale. The three p values corresponding to the three reporter genes were then combined through the Fisher's method to assess the overall significance of each drug's effect (P_Fisher). In the second method, we performed randomization tests to assess the statistical significance for two out of the three reporter genes responding to a drug (P_permutation). Specifically, we randomly permuted the drug response data for each reporter gene and counted the occurrences, where two of the three random values were more extreme than the observed expression changes (i.e., random chance). P_Fisher and P_permutation were each subject to multiple testing correction through the FDR control. These statistics provide tentative evidence for a drug to regulate NMD. The detailed results are listed in Table S4.
Using Fisher's method and with FDR≤0.01, we identified nine drugs inhibiting NMD, including HHT, TG, artesunate, albendazole, montelukast sodium, diphenylcyclopropenone, cladribine, hexachlorophene, and topotecan hydrochloride (Figure 5E). Relaxing the FDR threshold to 0.05 did not identify additional candidates. Using the permutation test, only HHT, TG, and topotecan hydrochloride were identified with FDR ≤0.01 (Figure 5F). When the FDR cutoff was increased to 0.05, a total of 22 drugs were tentative NMD inhibitors. As for NMD enhancers, the Fisher's method identified mitoxantrone hydrochloride with an FDR threshold at either 0.01 or 0.05 (Figure 5E). The permutation test identified mitoxantrone hydrochloride, trazodone hydrochloride, and gabexate mesilate with an FDR cutoff of 0.01 (Figure 5F) and 32 more drugs with an FDR cutoff of 0.05. These drugs are potential NMD modulators.
HHT inhibits NMD in a dosage-dependent manner in various cell types
We decided to follow up with HHT, since it was ranked No.1 by multiple independent methods of calling NMD modulators and for all three reporter genes. HHT is a natural cytotoxic alkaloid extracted from Cephalotaxus Harringtonia and used to treat acute myeloid leukemia, chronic myeloid leukemia, and other types of cancer.77,78 It functions as a cell cycle blocker that stops the progression from G1 to S phase and from G2 to M phase and, as a result, efficiently induces apoptosis and necroptosis in cancer cells.79, 80, 81
We first examined the minimal required dose of HHT to inhibit NMD and its dose-response curves. We applied a range of HHT dosages from 0.0001 to 25 μM for 5 h and quantified the expression of NMD and non-NMD isoforms (Figures 6A–6C). Compared with DMSO treatment, the non-NMD isoform levels of three reporter genes changed minimally; therefore, HHT does not significantly affect transcription of these genes. The NMD isoform levels were unchanged up to 0.002 μM. At 0.01 μM, the NMD isoforms of Ptbp2 and Tra2b were not changed, but the Hnrnpl NMD isoform was modestly and significantly upregulated. At 0.05 μM, the NMD isoforms of all three reporter genes were upregulated by 7–10-fold. They were further increased in concordance with higher concentration of HHT. In summary, HHT inhibits NMD at concentrations as low as 0.05 μM and in a dosage-dependent manner.
Figure 6.

HHT inhibits NMD in a dosage-dependent manner
(A–C) NMD isoform expression level changes in Ptbp2 (A), Hnrnpl (B), and Tra2b (C) showed a dosage-dependent manner compared with non-NMD isoform in N2a cells. HHT started to inhibit NMD as low as 0.05 μM.
(D–F) NMD isoform expression level changes in PTBP2 (D), HNRNPL (E), and TRA2B (F) showed the same dosage-dependent manner in HEK 293T cells. Data were shown as mean ± SEM of three biological replicates. ∗, p < 0.05, ∗∗, p < 0.01, Student's t test.
To test whether HHT inhibits NMD in other cell types, we treated the human embryonic kidney-originated (HEK) 293T cells with different dosages of HHT. Again, HHT upregulated the three NMD isoforms as low as 0.05 μM and in a dosage-dependent manner (Figures 6D–6F). Non-NMD isoforms exhibited no changes between DMSO- and HHT-treated samples. We further tested whether HHT was effective in primary cells besides transformed cell lines. We isolated mouse neural progenitor cells (NPCs) from embryonic mouse neocortices and treated them with the same range of HHT for 5 h. A similar expression profile of the three reporter genes was observed under HHT treatment (Figure S3A–S3C). All three NMD isoforms began to show significant upregulation (∼3–7-fold) after treatment of 0.05 μM HTT. As the dosage increased, the fold changes went up to a 9- to 28-fold increase at 25 μM HTT. These results show that the HHT effect on NMD is conserved between human and mouse and in multiple cell types of different tissue origins.
HHT inhibits other NMD substrates
Among the known NMD-inducing sequence features, the presence of a terminal codon >50 nt upstream of the last exon-exon junction (in short, the 50-nt rule) has the strongest predictive value for NMD susceptibility.40 This is the basis of our AS-NMD assay. However, NMD can also target genes and transcripts without this feature.82, 83, 84 We therefore explored whether HHT could affect NMD substrates without the 50-nt rule. We selected some well-established NMD targets in literature, such as Gadd45b, Ddit3, Atf3, and Atf4,2,85, 86, 87 and tested them in HHT-treated N2a, 293T, and primary NPCs.
These four genes all were upregulated in multiple cell types upon HHT treatment (Figure S4). In N2a cells, we observed clear upregulation of Gadd45b (10.1–16.9-fold), Ddit3 (3.2–3.9-fold), and Atf3 (5.8–6.7-fold) with 0.05 μM HHT and above (Figures S4A–S4C). Atf4 showed a modest but significant increase (∼1.7-fold, Figure S4D). In 293T cells, ATF3 abundance increased after as low as 0.01 μM HHT treatment and reached up to 10.8-fold induction at high HHT concentration (Figure S4G). GADD45B exhibited a clear dosage-dependent upregulation (2.6–5.6-fold) upon treatment of 0.05 μM–25 μM HHT (Figure S4E). DDIT3 and ATF4 had a relatively weaker response but still displayed about 2- to 3-fold upregulation (Figures S4F and S4H). In NPCs, the 0.01 μM HHT treatment induced significant upregulation in Gadd45b (2.8 ± 0.3 fold), Atf3 (8.4 ± 1.2 fold), and Atf4 (3.9 ± 0.5 fold). With higher dosages, their mRNA levels increased by 5.5–26.4-fold (Figures S4I, S4K, and S4L). Ddit3 was unchanged with 0.01 μM HHT but showed a 3.0-fold increase at 0.05 μM and increased to 5.7-fold at 25 μM (Figure S4J). In summary, although these four NMD targets exhibited differential upregulation in various cell types, they all respond to HHT treatment in agreement with NMD inhibition.
Translational inhibition mediates HHT's NMD effect
Since HHT is a potent cell cycle blocker and cell death inducer, these effects may confound studies of its mechanism. To examine this, we first tested whether the NMD-inhibiting doses of HHT (0.05–5 μM) caused a reduction in cell numbers or cell death. Cell counts showed no difference among different treatment groups (Figure S5A). Using Annexin V and 7-AAD to label dying and dead cells, we conducted fluorescence-activated cell sorting (FACS) to test whether HHT activated apoptosis when inhibiting NMD. We found that all HHT-treated groups had a similar level of apoptotic and dead cells as the DMSO-treated group (Figures S5B–S5F and S5H). By contrast, 0.3 μM staurosporine (STS) clearly induced apoptosis as a positive control (Figures S5G–S5H).88 Therefore, neither cell proliferation nor cell survival are significantly affected during the treatment of 0.05–5 μM HHT, which inhibits NMD.
To understand the mechanism by which HHT inhibits NMD, we examined whether translation could be involved, since NMD is a translation-dependent process. 35S methionine, methionine analog L-azidohomoalanine (L-AHA), and puromycin are commonly used to assay global translational activity, as they incorporate into newly synthesized proteins.89, 90, 91 Puromycin is a tyrosyl-tRNA mimic that binds in the acceptor site of elongating ribosomes and subsequently attaches to nascent peptide chains. A short pulse (e.g., 10 min) of puromycin to label newly synthesized peptides, followed by Western blot with anti-puromycin antibody, is a common and effective method to measure the global level of active translation in cells.92, 93, 94, 95 We treated N2a cells with a range of HHT dosages from 0.0001 to 5 μM, DMSO or TG control. After 5 h, puromycin was added to the media for 10 min to label protein translation. We found that 0.0001–0.002 μM HTT did not induce noticeable changes in translation compared with DMSO treatment. These doses have no effect on NMD reporters either (Figures 6, S3, and S4). By contrast, inhibition of protein synthesis, demonstrated by the loss of puromycin-labelled proteins, was evident after treatment with 0.2 μM TG and as low as 0.01 μM HHT. With a 0.05 μM or higher dose of HTT treatment, little new protein synthesis was observed, despite loading the same amount of protein lysates (Figures 7A and S6).
Figure 7.

HHT treatment inhibits protein synthesis and prevents polysome formation
(A) N2a cells pulse-labeled with puromycin were lysed and subject to Western blot analysis with anti-Puromycin antibody and anti-GAPDH antibody staining (as the loading control). Cells treated with HHT concentration lower than 0.01 μM showed no difference compared with DMSO. Starting at 0.01 μM, there was clear protein synthesis inhibition. From 0.05 to 5 μM, the protein synthesis was almost completely inhibited. Three biological replicates show similar results. Polysome graphs of N2a samples treated with DMSO (B), 0.05 μM HHT (C), 1 μM HHT (D), and 0.2 μM TG (E). DMSO control showed normal polysome integrity compared with significant lower polysome portions in HHT- and TG-treated samples.
We also performed polysome fractionation using sucrose density gradient centrifugation of drug-treated cells and examined polysome integrity as another indicator of translational activity. DMSO-treated control cells showed a normal polysome profile consisting of individual ribosomal subunit peaks (40S and 60S), the monosomes (80S), and staggering peaks of polysomes (Figure 7B). TG, which inhibits translation, was used as a control (Figure 7E). Polysome was visibly dissociated in cells treated with 0.2 μM TG, a concentration that inhibits NMD. The reduced optical density of heavier polysomes was accompanied by an increase in the monosome peak, another indication of translation inhibition.96 Similar to TG, both 0.05 and 1 μM HHT reduced the heights of the polysomes to no visible peaks (Figures 7C and 7D). Concurrently, the monosome peak increased substantially as the polysomes became free monosomes. Since active global translation is indicated by discrete peaks of “lighter” to “heavier” (corresponding to 2× to 8×) polysomes, their absence or reduction in magnitudes indicates global translation inhibition. Therefore, HHT inhibits translation at the minimum concentration (0.05 μM) that represses NMD, showing that translational inhibition is the underlying mechanism of NMD inhibition.
Discussion
Approximately 3 million individuals in the US alone are afflicted with genetic diseases caused by nonsense mutations that convert a protein-coding codon into a PTC.4 PTC-harboring transcripts are degraded by NMD. These patients can benefit from NMD-modulatory drugs acting in a preferable direction, which depends on whether the NMD activity is beneficial or detrimental in the disease condition. If the C-terminal truncated proteins are deleterious or dominant negatives, NMD is beneficial, and enhancing NMD may be favorable.23 On the other hand, if the C-terminal truncated proteins are (partially) functional, NMD ablates functional proteins, and NMD repression is more desirable.22
In addition to disease-causing nonsense mutations, an increasing number of illnesses are associated with NMD. For example, mutations in RNA splicing factors leading to the appearance of PTC-containing transcripts have been shown as the most common mechanism in myelodysplastic syndromes.97,98 In other cases, NMD activity eliminates potentially functional products and leads to a more severe phenotype.30,99 Therefore, systematic profiling for chemical modulators among existing drugs is an immediate path to identify and distinguish drugs that can ameliorate versus worsen NMD-associated diseases. Furthermore, while FDA-approved drugs have not been rigorously analyzed for their influence on NMD, patients with underlying nonsense mutations can be affected unknowingly by approved drugs. It is therefore urgent to profile FDA-approved drugs for their effect on cellular NMD activity.
In this study, we applied a robust assay to profile each of 704 FDA-approved compounds from the NCATS/NIH clinical collection on NMD. Both NMD and non-NMD isoforms of Ptbp2, Hnrnpl, and Tra2b were measured simultaneously after DMSO or drug treatments. This AS-NMD assay is based on one NMD-inducing feature, namely, an exon-exon junction >50 nt downstream of a stop codon. Although the 50-nt rule is the most predictive feature of NMD targeting in mammals,40 our study may not identify compounds that act on NMD substrates of different features.
We found a wide range of drug-induced transcriptional responses. For example, the fold change in Ptbp2 isoform ranged from 0.08 to 7.7, and its NMD isoform from 0.13 to 10.1, showing an approximate 100-fold difference. Transcriptional regulation also appeared in a large proportion of the drugs we profiled (Figure 3A). However, we did not directly measure the transcription rates. As RT-qPCR can only measure the level of steady-state mRNA, these expression level changes reflect the combined effect of transcription, splicing regulation, NMD modulation, and other RNA decay regulation. Nevertheless, the transcriptional responses to FDA-approved drugs are worthy of further investigation, as they may constitute a major mechanism of drug effect, either positively or negatively.
The transcriptional response could mask the drug effect on NMD. We therefore calculated NMD/non-NMD ratios to remove the transcription effect with the precaution that the ratio can be influenced by splicing regulation. The average fold change in NMD/non-NMD ratio for three reporter genes fell within 1.04–1.06. Using the stringent criteria requiring similar responses of the three reporters, most drugs insignificantly influenced cellular NMD. We then took advantage of a large amount of expression data from all 704 drugs on 6 isoform expression and developed a randomization method and a Fisher method to determine weak NMD regulation. As such, we assigned statistics and FDR controls for each drug (Table S4). Only a small group of FDA drugs show tentative evidence of affecting cellular NMD (Figures 5E and 5F).
The knowledge that most FDA drugs act minimally on NMD is valuable for patients comprising 20% monogenic diseases caused by nonsense mutations as well as for an increasing number of illnesses linked to overloaded nonsense transcripts.4,6 One implication is that these drugs do not interfere with NMD activity to induce adverse effects, excluding NMD as a source of safety concern. This knowledge is also valuable to the general population who bear thousands of nonsense SNPs.
HHT is the most outstanding NMD modulator from this profiling. It is best known for its anti-cancer activity, and various cellular mechanisms have been proposed, including blocking cell cycle, inducing cell apoptosis, and inhibiting tumor growth.100,101 At the molecular level, HHT inhibits SP1, TET1, 5hmC, and FLT3 signaling pathways in acute myeloid leukemia by directly binding to SP1 at concentrations between 0.04 and 2 μM.102 HHT was also shown to target transcription factor NF-кB to reduce MYC gene expression.103 We found HHT is an effective NMD inhibitor while inhibiting translation, although to ultimately prove this causality would require de-repressing translation after HHT treatment. Further supporting HHT's translation inhibition effect is that HHT can bind to the A-side cleft of the ribosome, a common mechanism by which antibiotics inhibit translation.104 Studies of HHT have shown its effective dosages ranging from 0.04 to 2 μM. Since HHT inhibits NMD as low as 0.05 μM, our finding implied that NMD inhibition might be a contributing mechanism in prior studies.
Many previously reported NMD drugs (e.g., NMDI-1, NMDI-14, and pateamine A) are not included in the NCATS library. Amlexanox and 5-azacytidine are included, and we found that they did not robustly affect the endogenous reporter genes at 5 μM. In the original report of amlexanox as an NMD inhibitor,30 amlexanox was effective in upregulating nonsense mutation-containing mRNAs at 0.2, 1, 5, and 25 μM concentrations. However, the authors also showed, and concluded, that the same dosages did not affect natural NMD substrate expression. This is in line with our observation that amlexanox induced a 1.03- to 1.21-fold change in the expression of three endogenous reporter genes.
As for 5-azacytidine, the difference between ours and Bhuvanagiri et al. could be the duration of the treatment. Bhuvanagiri et al. treated cells for 18 h at 1.56–20 μM,29 whereas we treated cells for 5 h. Since 5-azacytidine requires Myc upregulation to inhibit NMD,29 MYC protein may not be sufficiently induced at 5 h after 5-azacytidine treatment. Since previous studies did not release the full data of screened drugs other than reported positive hits, it is unclear whether HHT was tested before.
Our study can be seen as testing each FDA drug with a validation assay without a primary screen. Primary screens using exogenous NMD reporters require more direct and precise validation assays to confirm a positive hit, while non-hits, including false negatives, are simply discarded. Protein-based exogenous reporters intrinsically contain noise from regulatory processes other than NMD. Our RNA-based AS-NMD assay directly examines endogenous NMD targets and distinguishes NMD regulation from transcription and AS regulation. Furthermore, multiple endogenous NMD targets (in addition to those in this study) can be assayed at the same time to increase the confidence of calling an NMD modulation, whereas exogenous NMD reporters are usually tested one at a time. Although our assay is costly and time consuming, we provide the highest-quality data possible to date on NMD regulation for FDA-approved drugs by using an assay that normally would be deployed only during the validation phase.
Here, we have profiled 704 FDA-approved drugs. The chemical space is vaster than our assays, and genuine NMD modulators still await discovery. The AS-NMD assay is robust and flexible and can be adopted for a larger library and/or platforms. The assay directly probes endogenous NMD targets as a readout, which can be easily applied to some transfection-resistant cell lines or tissues. The assay is both sensitive and highly specific and can be expanded to include additional NMD reporters to further enhance the robustness of the discovery. We will improve the assay and throughput to facilitate profiling of larger chemical libraries with the goal of identifying more specific NMD modulators and evaluating new drugs for their effects on NMD.
Materials and methods
Cell culture and FDA drug treatments
The 704 FDA-approved drug candidates are part of the NCATS/NIH Clinical Collection (Table S1). Six hundred and fifty thousand Neuro-2a (N2a) cells per well were plated on 6-well BioLite TC plates (Thermo Fisher) and incubated with 2 mL of N2a complete media. The N2a complete media consists of L-glutamine-free Dulbecco's modified Eagle's medium (DMEM) (Thermo Fisher), 10% FBS (VWR), and 1× GlutaMAX (Thermo Fisher). Cells were incubated overnight before treatment with the drugs at 37°C, 5% CO2. Separate TG (VWR)-treated cells at indicated concentrations were harvested along with other drug treatments. Due to the numbers of treatments, the experiments were divided into treatment groups of ∼12 samples per group. All groups were collected at 5 h post-treatment.
RNA extraction and cDNA synthesis
After aspirate media, 1 mL of TRIzol (Thermo Fisher) was directly added to the cells to extract total RNA following the manufacture protocol TRIzol reagent. DNase treatment was performed at 37°C for 35 min to degrade residue genomic DNA after TRIzol RNA isolation with 4 units of Turbo DNase (Thermo Fisher). Following the DNase treatment, RNA was purified again using phenol-chloroform pH 4.5 (VWR). RNA concentrations were measured using a Nanodrop 2000c (Thermo Fisher). cDNA was synthesized using 1 μg of DNA-free total RNA. The reverse transcription used 1 μL of 30 μM random hexamers (IDT) and 200 units of Promega M-MLV reverse transcriptase (Promega) in a 20 μL reaction following the Promega protocol. The completed 20 μL cDNA reactions were diluted with 180 μL of nuclease free H2O (1:9) as the working concentration.
AS-NMD drug profiling using RT-qPCR
RT-qPCR primers were designed using Primer 3 software and purchased from IDT. Information of primers location was listed with the sequence information (Table S3). For RT-qPCR primer details and quality control information, refer to prior publications.4, 5, 6, 7 RT-qPCR experiments were performed using 2× Power SYBR Green PCR master mix (Thermo Fisher), following the manufacturer protocol. A 10-μL reaction consisted of 5 μL 2× Power SYBR Green PCR master mix, 0.3 μL of 10 μM forward primer, 0.3 μL of 10 μM reverse primer, and 3 μL of cDNA. A QuantStudio 6 Real-Time PCR instrument (Thermo Fisher) was used for the RT-qPCR assay. The run program was as follows: 50°C for 2 min, 95°C for 15 s, and 60°C for 1 min, with the 95°C and 60°C steps repeated for 40 cycles. A melting curve test from 60°C to 95°C at a 0.05°C/s measuring rate was performed after each official run for quantity control. All reactions were conducted with three replicates with a non-cDNA control (NTC, not amplified).
Apoptosis analysis
Apoptosis analysis was performed with FITC Annexin V Apoptosis Detection Kit I (BD science). Six hundred and fifty thousand N2a cells per well were plated on 6-well BioLite TC plates with 2 mL of N2a complete media. Cells were cultured overnight before treatment. Cells were treated with different dosages of HHT from 0.01 to 5 μM, along with a negative control DMSO and a positive control 0.3 μM STS for 5 h. All groups were dissociated with 0.2 mL TrypLE and collected with 2 mL N2a complete media. Cells were washed twice with cold 1× PBS and then resuspended in 1× binding buffer at a concentration of 1 × 10ˆ6 cells/mL. One hundred microliters was taken out for staining with 1 μL FITC Annexin V and 1 μL 7-AAD. The mixture was gently tapped and incubated for 15 min at room temperature (RT) in the dark. Two hundred microliters 1× binding buffer per tube was added to the mixture before FACS with flow cytometer (NovoCyte).
Data analysis
All RT-qPCR data were analyzed using the QuantStudio 6 software. The software used the ΔΔCt method to calculate RQ (i.e., fold changes). Two housing-keeping genes, Gapdh and Sdha, were included for normalization using their geometric averaging.105 Outliers were omitted when the coefficient of variation of Ct among the three technical replicates was larger than 0.3. Statistical analysis and figure generation were performed in Microsoft Excel.
To consider combined effects of three reporters, we applied a Z test to measure the statistical significance of the drug effect. Specifically, the RQ ratio between the NMD and the non-NMD transcripts at the log2 scale was Z score normalized and tested whether the drug had an upregulation effect (μ>0) or a downregulation effect (μ<0) for a reporter gene. For each drug, the three p values corresponding to the three reporter genes were combined through the Fisher's method to assess the overall significance of its effect (P_Fisher). Secondly, we performed a randomization test to assess the significance of extreme values from two out of the three reporter genes. We randomly permuted the drug response data for each reporter gene for 10,000 times and counted the occurrences that the two random ratios were more extreme than the observed ones (P_permutation). Both p values were further subject to multiple testing correction through the FDR control. PCA was performed for the six RQ values or the three RQ ratios. The first two principal components were visualized to show the overall clusters of drugs based on their effect similarity. Hierarchical clustering and heat maps were plotted for the RQ values at the log2 scale using R software.
Puromycin labeling and Western blot
Six hundred and fifty thousand N2a cells per well were plated on 6-well BioLite TC plates with 2 mL of N2a complete media. Cells were cultured overnight before being treated with different dosages of HHT from 0.01 to 5 μM, along with one DMSO (negative control) and one 0.2 μM TG (positive control) for 5 h. An amount of 10 μg/mL puromycin was added to the media for a 10 min pulse labeling, then all media were removed, and cells were washed twice with cold 1× PBS. One hundred microliters lysis buffer (RIPA buffer with 1× Phosstop, 1× Protease inhibitor and 1 U/mL BenzonaseRTM Nuclease) was added to each well, and cells were collected into the lysis buffer using cell scrapers. Lysate from each well was transferred to one 1.5 mL tube, and all tubes were rotated at 4°C for 30 min to increase lysis efficiency. Samples were centrifuged at 14,800 rpm for 15 min, and supernatants were transferred to new tubes for further analysis. For Western blot analysis, 50 μg total protein per sample was loaded to SDS-PAGE gel. After proteins transferred to the PVDF membrane, the membrane was first stained with Ponceau S. Membrane was blocked by 1× TBST with 5% BSA before being incubated with primary antibodies. Anti-puromycin (Sigma-Aldrich, MABE343) and anti-GAPDH (Cell signaling, 2118S) primary antibodies were diluted at 1:1,000 ratio in 1× TBST with 5% BSA and incubated with PVDF membrane at 4°C overnight. The next day, after washing, the membrane was incubated with the corresponding fluorescence-conjugated secondary antibodies at RT for 30 min. Finally, the fluorescent signals were detected with the Typhoon FLA9000.
Polysome fractionation
For polysome fractionation experiments, 1.0 × 107 N2a cells were plated in 150 mm petri dishes overnight with 20 mL N2a complete media. Cells were treated with the indicated concentrations of HHT and TG on the next day before sample processing. Cycloheximide (Thermo Fisher) was added at a final concentration of 100 μg/mL followed by an incubation of 10 min at 37°C before lysate collection. The cells were washed twice with 10 mL cold 1× PBS containing 100 μg/mL cycloheximide and collected in 4 mL of the same cold solution with cell scrapers. The cells were lysed with 0.5 mL lysis buffer (20 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2, 2 mM DTT, 100 μg/mL cycloheximide, 1% Triton X-100, 50 μg/mL RNaseout, and 1× EDTA-free protease inhibitor cocktail). Roughly, 450 μL (6,000 optical units) of lysate was loaded onto premade sucrose gradients (60%–15%) and balanced (within 0.5 mg) before ultracentrifugation at 4°C at 41,000 rpm for 1.5 h using an SW41 Ti rotor. The centrifugation tubes were carefully removed from the ultracentrifuge and loaded onto the fractionation system consisting of the gradient fractionator (Brandel), the ISCO absorbance detector (ISCO), and fraction collector (R1 Fraction Collector, Brandel) at 2.0 sensitivity and 150 cm/h chart speed to record absorbance data and collect fractionations.
Acknowledgments
We want to thank the Zheng lab members (University of California, Riverside, USA) for their helpful discussion. We thank Dr. Yongsheng Shi and Kristianna Sophie Kera Sarkan for help with the puromycin labeling experiment.
Funding: This research was supported by a pilot grant from the MolMed Center at the University of California, Riverside, United States, and by the National Institute of Health, United States [grant numbers: R01NS104041 and R01MH116220 to S.Z. and R01GM137428 to L.C.].
Author contributions
Conceptualization, S.Z.; methodology, S.Z., Z.L., J.Z., and L.C.; formal analysis, Z.L., J.Z., and L.C.; investigation, J.L., Z.L., R.P., K.L., I.N., and L.C.; writing—original draft, Z.L. and J.Z.; writing—review and editing, S.Z., L.C., and J.Z.; supervision, S.Z.; funding acquisition, L.C. and S.Z.
Declaration of interests
The authors declare no conflict of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.12.003.
Supplemental information
References
- 1.Lykke-Andersen S., Jensen T.H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- 2.Kurosaki T., Popp M.W., Maquat L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019;20:406–420. doi: 10.1038/s41580-019-0126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang Y.-F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 4.Mort M., Ivanov D., Cooper D.N., Chuzhanova N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008;29:1037–1047. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 5.Bidou L., Allamand V., Rousset J.-P., Namy O. Sense from nonsense: therapies for premature stop codon diseases. Trends Mol. Med. 2012;18:679–688. doi: 10.1016/j.molmed.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Holbrook J.A., Neu-Yilik G., Hentze M.W., Kulozik A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 7.Kurosaki T., Maquat L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016;129:461–467. doi: 10.1242/jcs.181008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X., Gregory-Evans K., Wasan K.M., Sivak O., Shan X., Gregory-Evans C.Y. Efficacy of postnatal in vivo nonsense suppression therapy in a Pax6 mouse model of aniridia. Mol. Ther. Nucleic Acids. 2017;7:417–428. doi: 10.1016/j.omtn.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilschanski M., Yahav Y., Yaacov Y., Blau H., Bentur L., Rivlin J., Aviram M., Bdolah-Abram T., Bebok Z., Shushi L., et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med. 2003;349:1433–1441. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 10.Linde L., Boelz S., Nissim-Rafinia M., Oren Y.S., Wilschanski M., Yaacov Y., Virgilis D., Neu-Yilik G., Kulozik A.E., Kerem E., et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J. Clin. Invest. 2007;117:683–692. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linde L., Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552–563. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Finkel R.S., Flanigan K.M., Wong B., Bönnemann C., Sampson J., Sweeney H.L., Reha A., Northcutt V.J., Elfring G., Barth J., et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One. 2013;8:e81302. doi: 10.1371/journal.pone.0081302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popp M.W.-L., Maquat L.E. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rebbapragada I., Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr. Opin. Cell Biol. 2009;21:394–402. doi: 10.1016/j.ceb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Jaffrey S.R., Wilkinson M.F. Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat. Rev. Neurosci. 2018;19:715–728. doi: 10.1038/s41583-018-0079-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.da Costa P.J., Menezes J., Romão L. The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease. Int. J. Biochem. Cell Biol. 2017;91:168–175. doi: 10.1016/j.biocel.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Gardner L.B. Nonsense-mediated RNA decay regulation by cellular stress: implications for tumorigenesis. Mol. Cancer Res. 2010;8:295–308. doi: 10.1158/1541-7786.MCR-09-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen L.S., Wilkinson M.F., Gecz J. Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci. Biobehav. Rev. 2014;46(Pt 2):175–186. doi: 10.1016/j.neubiorev.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daar I.O., Maquat L.E. Premature translation termination mediates triosephosphate isomerase mRNA degradation. Mol. Cell. Biol. 1988;8:802–813. doi: 10.1128/mcb.8.2.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S.C.-W., Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016;22:976–986. doi: 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dvinge H., Kim E., Abdel-Wahab O., Bradley R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer. 2016;16:413–430. doi: 10.1038/nrc.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai B., Li Z., Ma M., Zhang J., Kong S., Abdalla B.A., Xu H., Jebessa E., Zhang X., Lawal R.A., et al. Long noncoding RNA SMUL suppresses SMURF2 production-mediated muscle atrophy via nonsense-mediated mRNA decay. Mol. Ther. Nucleic Acids. 2020;23:512–526. doi: 10.1016/j.omtn.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khajavi M., Inoue K., Lupski J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006;14:1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- 24.Miller J.N., Pearce D.A. Nonsense-mediated decay in genetic disease: friend or foe? Mutat. Res. Rev. Mut. Res. 2014;762:52–64. doi: 10.1016/j.mrrev.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamaguchi-Kabata Y., Shimada M.K., Hayakawa Y., Minoshima S., Chakraborty R., Gojobori T., Imanishi T. Distribution and effects of nonsense polymorphisms in human genes. PLoS One. 2008;3:e3393. doi: 10.1371/journal.pone.0003393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yngvadottir B., Xue Y., Searle S., Hunt S., Delgado M., Morrison J., Whittaker P., Deloukas P., Tyler-Smith C. A genome-wide survey of the prevalence and evolutionary forces acting on human nonsense SNPs. Am. J. Hum. Genet. 2009;84:224–234. doi: 10.1016/j.ajhg.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pawlicka K., Kalathiya U., Alfaro J. Nonsense-mediated mRNA decay: pathologies and the potential for novel therapeutics. Cancers. 2020;12:765. doi: 10.3390/cancers12030765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin L., Grigoryan A., Wang D., Wang J., Breda L., Rivella S., Cardozo T., Gardner L.B. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014;74:3104–3113. doi: 10.1158/0008-5472.CAN-13-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhuvanagiri M., Lewis J., Putzker K., Becker J.P., Leicht S., Krijgsveld J., Batra R., Turnwald B., Jovanovic B., Hauer C., et al. 5-azacytidine inhibits nonsense-mediated decay in a MYC-dependent fashion. EMBO Mol. Med. 2014;6:1593–1609. doi: 10.15252/emmm.201404461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Hilarion S., Beghyn T., Jia J., Debreuck N., Berte G., Mamchaoui K., Mouly V., Gruenert D.C., Déprez B., Lejeune F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012;7:58. doi: 10.1186/1750-1172-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atanasova V.S., Jiang Q., Prisco M., Gruber C., Piñón Hofbauer J., Chen M., Has C., Bruckner-Tuderman L., McGrath J.A., Uitto J., et al. Amlexanox enhances premature termination codon read-through in COL7A1 and expression of full length type VII collagen: potential therapy for recessive dystrophic epidermolysis Bullosa. J. Invest. Dermatol. 2017;137:1842–1849. doi: 10.1016/j.jid.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dang Y., Low W.-K., Xu J., Gehring N.H., Dietz H.C., Romo D., Liu J.O. Inhibition of nonsense-mediated mRNA decay by the natural product pateamine A through eukaryotic initiation factor 4AIII. J. Biol. Chem. 2009;284:23613–23621. doi: 10.1074/jbc.M109.009985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durand S., Cougot N., Mahuteau-Betzer F., Nguyen C.-H., Grierson D.S., Bertrand E., Tazi J., Lejeune F. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J. Cell Biol. 2007;178:1145–1160. doi: 10.1083/jcb.200611086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexandrov A., Shu M.-D., Steitz J.A. Fluorescence amplification method for forward genetic discovery of factors in human mRNA degradation. Mol. Cell. 2017;65:191–201. doi: 10.1016/j.molcel.2016.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casadio A., Longman D., Hug N., Delavaine L., Vallejos Baier R., Alonso C.R., Cáceres J.F. Identification and characterization of novel factors that act in the nonsense-mediated mRNA decay pathway in nematodes, flies and mammals. EMBO Rep. 2015;16:71–78. doi: 10.15252/embr.201439183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hug N., Longman D., Cáceres J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016;44:1483–1495. doi: 10.1093/nar/gkw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan Q., Saltzman A.L., Kim Y.K., Misquitta C., Shai O., Maquat L.E., Frey B.J., Blencowe B.J. Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev. 2006;20:153–158. doi: 10.1101/gad.1382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lareau L.F., Brooks A.N., Soergel D.A.W., Meng Q., Brenner S.E. The coupling of alternative splicing and nonsense-mediated mRNA decay. Adv. Exp. Med. Biol. 2007;623:190–211. doi: 10.1007/978-0-387-77374-2_12. [DOI] [PubMed] [Google Scholar]
- 39.Li Z., Vuong J.K., Zhang M., Stork C., Zheng S. Inhibition of nonsense-mediated RNA decay by ER stress. RNA. 2017;23:378–394. doi: 10.1261/rna.058040.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colombo M., Karousis E.D., Bourquin J., Bruggmann R., Mühlemann O. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA. 2017;23:189–201. doi: 10.1261/rna.059055.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spellman R., Llorian M., Smith C.W.J. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol. Cell. 2007;27:420–434. doi: 10.1016/j.molcel.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boutz P.L., Stoilov P., Li Q., Lin C.-H., Chawla G., Ostrow K., Shiue L., Ares M., Black D.L. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saltzman A.L., Kim Y.K., Pan Q., Fagnani M.M., Maquat L.E., Blencowe B.J. Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol. Cell. Biol. 2008;28:4320–4330. doi: 10.1128/MCB.00361-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stoilov P., Daoud R., Nayler O., Stamm S. Human tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum. Mol. Genet. 2004;13:509–524. doi: 10.1093/hmg/ddh051. [DOI] [PubMed] [Google Scholar]
- 45.Collins R., Trowman R., Norman G., Light K., Birtle A., Fenwick E., Palmer S., Riemsma R. A systematic review of the effectiveness of docetaxel and mitoxantrone for the treatment of metastatic hormone-refractory prostate cancer. Br. J. Cancer. 2006;95:457–462. doi: 10.1038/sj.bjc.6603287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imran M., Ahmad N., Anjum F.M., Khan M.K., Mushtaq Z., Nadeem M., Hussain S. Potential protective properties of flax lignan secoisolariciresinol diglucoside. Nutr. J. 2015;14:71. doi: 10.1186/s12937-015-0059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kantarjian H.M., O’Brien S., Cortes J. Homoharringtonine/omacetaxine mepesuccinate: the long and winding road to food and drug administration approval. Clin. Lymphoma Myeloma Leuk. 2013;13:530–533. doi: 10.1016/j.clml.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fabian C.J., Kimler B.F., Zalles C.M., Klemp J.R., Petroff B.K., Khan Q.J., Sharma P., Setchell K.D.R., Zhao X., Phillips T.A., et al. Reduction in Ki-67 in benign breast tissue of high-risk women with the lignan secoisolariciresinol diglycoside. Cancer Prev. Res. 2010;3:1342–1350. doi: 10.1158/1940-6207.CAPR-10-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ayella A., Lim S., Jiang Y., Iwamoto T., Lin D., Tomich J., Wang W. Cytostatic inhibition of cancer cell growth by lignan secoisolariciresinol diglucoside. Nutr. Res. 2010;30:762–769. doi: 10.1016/j.nutres.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 50.Zheng S., Chen Y., Donahue C.P., Wolfe M.S., Varani G. Structural basis for stabilization of the tau pre-mRNA splicing regulatory element by novantrone (mitoxantrone) Chem. Biol. 2009;16:557–566. doi: 10.1016/j.chembiol.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Agostino V.G., Adami V., Provenzani A. A novel high throughput biochemical assay to evaluate the HuR protein-RNA complex formation. PLoS One. 2013;8:e72426. doi: 10.1371/journal.pone.0072426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodrich P.M. Naloxone hydrochloride: a review. AANA J. 1990;58:14–16. [PubMed] [Google Scholar]
- 53.Doozandeh A., Yazdani S. Neuroprotection in glaucoma. J. Ophthalmic Vis. Res. 2016;11:209–220. doi: 10.4103/2008-322X.183923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meredith P.A., Elliott H.L. Clinical pharmacokinetics of amlodipine. Clin. Pharmacokinet. 1992;22:22–31. doi: 10.2165/00003088-199222010-00003. [DOI] [PubMed] [Google Scholar]
- 55.Williams M., Jarvis M.F. Adenosine antagonists as potential therapeutic agents. Pharmacol. Biochem. Behav. 1988;29:433–441. doi: 10.1016/0091-3057(88)90182-7. [DOI] [PubMed] [Google Scholar]
- 56.Vaughan Williams E.M. The relevance of cellular to clinical electrophysiology in classifying antiarrhythmic actions. J. Cardiovasc. Pharmacol. 1992;20:S1–S7. [PubMed] [Google Scholar]
- 57.Sheeba Sherlin Y., Vijayakumar T., Binoy J., Roy S.D.D., Jayakumar V.S. Büchi’s model based analysis of local anesthetic action in procaine hydrochloride: vibrational spectroscopic approach. Spectrochim. Acta A. Mol. Biomol. Spectrosc. 2018;205:55–65. doi: 10.1016/j.saa.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 58.Mosqueira M., Aykut G., Fink R.H.A. Mepivacaine reduces calcium transients in isolated murine ventricular cardiomyocytes. BMC Anesthesiol. 2020;20:10. doi: 10.1186/s12871-019-0926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hardin C., Shum E., Singh A.P., Perez-Soler R., Cheng H. Emerging treatment using tubulin inhibitors in advanced non-small cell lung cancer. Expert Opin. Pharmacother. 2017;18:701–716. doi: 10.1080/14656566.2017.1316374. [DOI] [PubMed] [Google Scholar]
- 60.Cohen T., Schwarz T.M., Vigant F., Gardner T.J., Hernandez R.E., Lee B., Tortorella D. The microtubule inhibitor podofilox inhibits an early entry step of human cytomegalovirus. Viruses. 2016;8:295. doi: 10.3390/v8100295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turan Ç., Metin N. Albendazole-induced anagen effluvium: a brief literature review and our own experience. Acta Dermatovenerol. Alp. Pannonica Adriat. 2020;29:161–163. [PubMed] [Google Scholar]
- 62.Mathé G. Bestatin, an aminopeptidase inhibitor with a multi-pharmacological function. Biomed. Pharmacother. 1991;45:49–54. doi: 10.1016/0753-3322(91)90122-a. [DOI] [PubMed] [Google Scholar]
- 63.Dawood M.Y. Primary dysmenorrhea: advances in pathogenesis and management. Obstet. Gynecol. 2006;108:428–441. doi: 10.1097/01.AOG.0000230214.26638.0c. [DOI] [PubMed] [Google Scholar]
- 64.Henderson J.D., Glucksman G., Leong B., Tigyi A., Ankirskaia A., Siddique I., Lam H., DePeters E., Wilson B.W. Pyridostigmine bromide protection against acetylcholinesterase inhibition by pesticides. J. Biochem. Mol. Toxicol. 2012;26:31–34. doi: 10.1002/jbt.20410. [DOI] [PubMed] [Google Scholar]
- 65.Raasch R.H. Pravastatin sodium, a new HMG-CoA reductase inhibitor. DICP. 1991;25:388–394. doi: 10.1177/106002809102500410. [DOI] [PubMed] [Google Scholar]
- 66.Lindholm J.-M., Taipale C., Ylinen P., Tuuminen R. Perioperative subconjunctival triamcinolone acetonide injection for prevention of inflammation and macular oedema after cataract surgery. Acta Ophthalmol. 2020;98:36–42. doi: 10.1111/aos.14175. [DOI] [PubMed] [Google Scholar]
- 67.Hsueh M.-F., Bolognesi M.P., Wellman S.S., Kraus V.B. Anti-inflammatory effects of naproxen sodium on human osteoarthritis synovial fluid immune cells. Osteoarthritis Cartilage. 2020;28:639–645. doi: 10.1016/j.joca.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chapman K.E., Coutinho A.E., Gray M., Gilmour J.S., Savill J.S., Seckl J.R. The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol. Cell. Endocrinol. 2009;301:123–131. doi: 10.1016/j.mce.2008.09.031. [DOI] [PubMed] [Google Scholar]
- 69.De Leo F., Quilici G., Tirone M., De Marchis F., Mannella V., Zucchelli C., Preti A., Gori A., Casalgrandi M., Mezzapelle R., et al. Diflunisal targets the HMGB1/CXCL12 heterocomplex and blocks immune cell recruitment. EMBO Rep. 2019;20:e47788. doi: 10.15252/embr.201947788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hincapié-Mejía G., Granda-Ramírez F., Ferraro F., Serna-Galvis E.A., Torres-Palma R.A. Dataset on application of electrochemical and photochemical processes for sulfacetamide antibiotic elimination in water. Data Brief. 2020;29:105158. doi: 10.1016/j.dib.2020.105158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Al-Tamimi M., Abu-Raideh J., Albalawi H., Shalabi M., Saleh S. Effective oral combination treatment for extended-spectrum beta-lactamase-producing Escherichia coli. Microb. Drug Resist. 2019;25:1132–1141. doi: 10.1089/mdr.2019.0065. [DOI] [PubMed] [Google Scholar]
- 72.Afshari A., Nguyen L., Kahn S.A., Montgomery A.C., Shinha T., Stratton C., Summitt B. The effective duration of antimicrobial activity of mafenide acetate after reconstitution. J. Burn Care Res. 2018;39:736–738. doi: 10.1093/jbcr/irx029. [DOI] [PubMed] [Google Scholar]
- 73.Chopra S.L., Dale D.G., Blackwood A.G. Effect of ampicillin on E. Coli of swine origin. Can. J. Comp. Med. Vet. Sci. 1963;27:223–227. [PMC free article] [PubMed] [Google Scholar]
- 74.Diouf B., Lin W., Goktug A., Grace C.R.R., Waddell M.B., Bao J., Shao Y., Heath R.J., Zheng J.J., Shelat A.A., et al. Alteration of RNA splicing by small-molecule inhibitors of the interaction between NHP2L1 and U4. SLAS Discov. 2018;23:164–173. doi: 10.1177/2472555217735035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hou H.-Y., Lu W.-W., Wu K.-Y., Lin C.-W., Kung S.-H. Idarubicin is a broad-spectrum enterovirus replication inhibitor that selectively targets the virus internal ribosomal entry site. J. Gen. Virol. 2016;97:1122–1133. doi: 10.1099/jgv.0.000431. [DOI] [PubMed] [Google Scholar]
- 76.Pérez-Boza J., Boeckx A., Lion M., Dequiedt F., Struman I. hnRNPA2B1 inhibits the exosomal export of miR-503 in endothelial cells. Cell. Mol. Life Sci. 2020;77:4413–4428. doi: 10.1007/s00018-019-03425-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quintás-Cardama A., Cortes J. Homoharringtonine for the treatment of chronic myelogenous leukemia. Expert Opin. Pharmacother. 2008;9:1029–1037. doi: 10.1517/14656566.9.6.1029. [DOI] [PubMed] [Google Scholar]
- 78.Yakhni M., Briat A., El Guerrab A., Furtado L., Kwiatkowski F., Miot-Noirault E., Cachin F., Penault-Llorca F., Radosevic-Robin N. Homoharringtonine, an approved anti-leukemia drug, suppresses triple negative breast cancer growth through a rapid reduction of anti-apoptotic protein abundance. Am. J. Cancer Res. 2019;9:1043–1060. [PMC free article] [PubMed] [Google Scholar]
- 79.Feldman E., Arlin Z., Ahmed T., Mittelman A., Puccio C., Chun H., Cook P., Baskind P. Homoharringtonine in combination with cytarabine for patients with acute myelogenous leukemia. Leukemia. 1992;6:1189–1191. [PubMed] [Google Scholar]
- 80.Ma F.S., Gao R.L., Jin J.M. Leukemic clonogenic assay and drug sensitivity test of homoharringtonine and cytarabine in acute myeloid leukemia. Zhonghua Nei Ke Za Zhi. 1989;28:737–740. 769. [PubMed] [Google Scholar]
- 81.Zhou D.C., Zittoun R., Marie J.P. Homoharringtonine: an effective new natural product in cancer chemotherapy. Bull. Cancer. 1995;82:987–995. [PubMed] [Google Scholar]
- 82.Brogna S., McLeod T., Petric M. The meaning of NMD: translate or perish. Trends Genet. 2016;32:395–407. doi: 10.1016/j.tig.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 83.MacDonald C.C., Grozdanov P.N. Nonsense in the testis: multiple roles for nonsense-mediated decay revealed in male reproduction. Biol. Reprod. 2017;96:939–947. doi: 10.1093/biolre/iox033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Somers J., Pöyry T., Willis A.E. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013;45:1690–1700. doi: 10.1016/j.biocel.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mendell J.T., Sharifi N.A., Meyers J.L., Martinez-Murillo F., Dietz H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004;36:1073–1078. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- 86.Lou C.-H., Dumdie J., Goetz A., Shum E.Y., Brafman D., Liao X., Mora-Castilla S., Ramaiah M., Cook-Andersen H., Laurent L., et al. Nonsense-mediated RNA decay influences human embryonic stem cell fate. Stem Cell Rep. 2016;6:844–857. doi: 10.1016/j.stemcr.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gardner L.B. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol. Cell. Biol. 2008;28:3729–3741. doi: 10.1128/MCB.02284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Belmokhtar C.A., Hillion J., Ségal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354–3362. doi: 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- 89.Wang D., Zavadil J., Martin L., Parisi F., Friedman E., Levy D., Harding H., Ron D., Gardner L.B. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol. Cell. Biol. 2011;31:3670–3680. doi: 10.1128/MCB.05704-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kobayashi M., Aida M., Nagaoka H., Begum N.A., Kitawaki Y., Nakata M., Stanlie A., Doi T., Kato L., Okazaki I.-M., et al. AID-induced decrease in topoisomerase 1 induces DNA structural alteration and DNA cleavage for class switch recombination. Proc. Natl. Acad. Sci. U S A. 2009;106:22375–22380. doi: 10.1073/pnas.0911879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Starck S.R., Green H.M., Alberola-Ila J., Roberts R.W. A general approach to detect protein expression in vivo using fluorescent puromycin conjugates. Chem. Biol. 2004;11:999–1008. doi: 10.1016/j.chembiol.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 92.Liu J., Xu Y., Stoleru D., Salic A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc. Natl. Acad. Sci. U S A. 2012;109:413–418. doi: 10.1073/pnas.1111561108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmidt E.K., Clavarino G., Ceppi M., Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 94.Aviner R., Geiger T., Elroy-Stein O. Genome-wide identification and quantification of protein synthesis in cultured cells and whole tissues by puromycin-associated nascent chain proteomics (PUNCH-P) Nat. Protoc. 2014;9:751–760. doi: 10.1038/nprot.2014.051. [DOI] [PubMed] [Google Scholar]
- 95.Brar G.A., Weissman J.S. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol. 2015;16:651–664. doi: 10.1038/nrm4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arribere J.A., Doudna J.A., Gilbert W.V. Reconsidering movement of eukaryotic mRNAs between polysomes and P bodies. Mol. Cell. 2011;44:745–758. doi: 10.1016/j.molcel.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shiozawa Y., Malcovati L., Gallì A., Sato-Otsubo A., Kataoka K., Sato Y., Watatani Y., Suzuki H., Yoshizato T., Yoshida K., et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018;9:3649. doi: 10.1038/s41467-018-06063-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rahman M.A., Lin K.-T., Bradley R.K., Abdel-Wahab O., Krainer A.R. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev. 2020;34:413–427. doi: 10.1101/gad.332270.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Du M., Liu X., Welch E.M., Hirawat S., Peltz S.W., Bedwell D.M. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc. Natl. Acad. Sci. U S A. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kuroda J., Kamitsuji Y., Kimura S., Ashihara E., Kawata E., Nakagawa Y., Takeuichi M., Murotani Y., Yokota A., Tanaka R., et al. Anti-myeloma effect of homoharringtonine with concomitant targeting of the myeloma-promoting molecules, Mcl-1, XIAP, and β-catenin. Int. J. Hematol. 2008;87:507–515. doi: 10.1007/s12185-008-0081-8. [DOI] [PubMed] [Google Scholar]
- 101.Chen R., Guo L., Chen Y., Jiang Y., Wierda W.G., Plunkett W. Homoharringtonine reduced Mcl-1 expression and induced apoptosis in chronic lymphocytic leukemia. Blood. 2011;117:156–164. doi: 10.1182/blood-2010-01-262808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li C., Dong L., Su R., Bi Y., Qing Y., Deng X., Zhou Y., Hu C., Yu M., Huang H., et al. Homoharringtonine exhibits potent anti-tumor effect and modulates DNA epigenome in acute myeloid leukemia by targeting SP1/TET1/5hmC. Haematologica. 2020;105:148. doi: 10.3324/haematol.2018.208835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen X.-J., Zhang W.-N., Chen B., Xi W.-D., Lu Y., Huang J.-Y., Wang Y.-Y., Long J., Wu S.-F., Zhang Y.-X., et al. Homoharringtonine deregulates MYC transcriptional expression by directly binding NF-κB repressing factor. Proc. Natl. Acad. Sci. U S A. 2019;116:2220–2225. doi: 10.1073/pnas.1818539116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gürel G., Blaha G., Moore P.B., Steitz T.A. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 2009;389:146–156. doi: 10.1016/j.jmb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.