

Abstract
Recombinant viral vectors have become integral tools for basic in vivo research applications. Helper-dependent adenoviral (HdAd) vectors have a large packaging capacity of ∼36 kb of DNA that mediate long-term transgene expression in vitro and in vivo. The large carrying capacity of HdAd enables basic research or clinical applications requiring the delivery of large genes or multiple transgenes, which cannot be packaged into other widely used viral vectors. Currently, common HdAd production systems use an Ad helper virus (HV) with a packaging signal (Ψ) that is flanked by either loxP or FRT sites, which is excised in producer cells expressing Cre or Flp recombinases to prevent HV packaging. However, these production systems prevent the use of HdAd vectors for genetic strategies that rely on Cre or Flp recombination for cell-type-specific expression. To overcome these limitations, we developed the VikAD production system, which is based on producer cells expressing the Vika recombinase and an HV that contains a Ψ flanked by vox sites. The availability of this production system will greatly expand the utility and flexibility of HdAd vectors for use in research applications to monitor and manipulate cellular activity with increased specificity.
Keywords: adenovirus, viral vectors, production, helper-dependent adenovirus, genetic intersectional strategies, site-specific recombinases, cell-type-specific expression
Graphical abstract
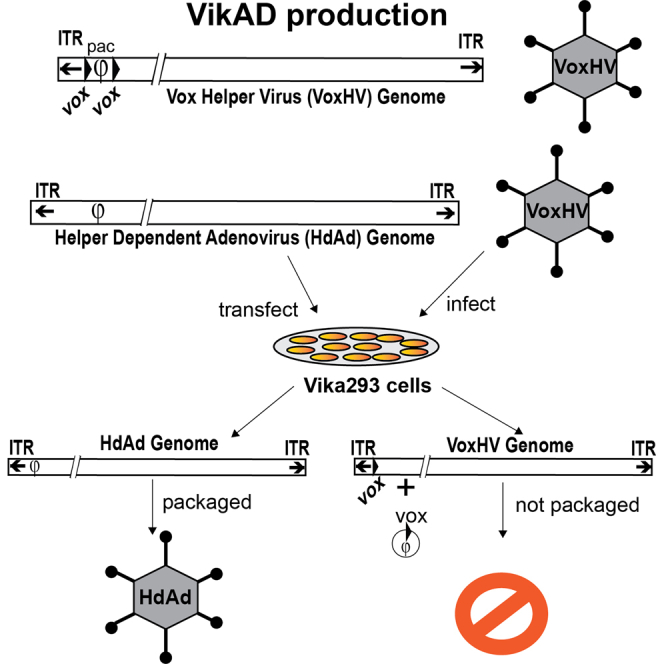
VikAD is a helper-dependent adenovirus (HdAd) production platform that utilizes a cell line expressing the Vika site-specific recombinase and an Ad HV that contains the packaging sequence flanked by vox sites. VikAD enables use of HdAd with genetic intersectional strategies using Cre and Flp or other site-specific recombinases.
Introduction
Recombinant viral vectors are key research tools for elucidating the cell-type-specific mechanisms that control cellular activity in vivo.1,2 Although adeno-associated virus (AAV) vectors (rAAV) and lentiviral viral vectors (LVV) are the most widely used viral vectors in both the basic research and clinical setting, they have a relatively small packaging capacity of ∼5 and ∼9 kb, respectively.3,4 Their small packaging capacity results in severe limitations in addressing research questions centered on proteins with large coding sequences or that require the delivery of transgene cassettes. Although not as widely used as rAAV and LVV, HdAd vectors have had great success in basic research and clinical applications.5 HdAd vectors lack all viral genes, have a packaging capacity of ∼36 kb,6 and provide long-term transgene expression.7, 8, 9, 10, 11, 12, 13, 14, 15 More importantly, they enable the ability to address long-standing research questions not possible with the limited packaging capacity of rAAV or LVV.7,10,14
The only viral sequences in the HdAd genome are the inverted terminal repeats (ITRs) and packaging signal sequence (Ψ), which are required for genome replication and packaging into vector particles.16 Therefore, HdAd production requires that the Ad viral proteins are supplied in trans with correct temporal regulation.16 The most successful HdAd production systems utilize a first-generation E1-deleted Ad vector as a helper virus (HV) to mediate HdAd genome replication and high titer HdAd vector production in producer cells expressing the Ad E1 proteins.17,18 Since HV is required to produce high titers of HdAd, strategies that utilize either Cre/loxP or Flp/FRT site-specific recombination have been designed to prevent HV genome packaging, while allowing HdAd genome packaging into progeny vector particles.17, 18, 19 These production systems use a combination of HEK293 (293) producer cells expressing the Ad E1 proteins and high levels of either Cre or Flp recombinases and an HV Ψ that is flanked by either loxP or FRT sites. These systems and their purification strategies allow for the generation of HdAd vectors with similar titers to first-generation Ad vectors with minor amounts of HV contamination (∼0.5%–0.01%).17, 18, 19
Genetic strategies relying on cell-type-specific Cre or Flp recombination to induce expression from rAAV or LVV vectors are important tools in elucidating the cellular and molecular mechanisms controlling cellular activity in vivo and organismal behavior.20, 21, 22 However, the limited packaging capacity of these vectors results in the inability to probe many molecules and their signaling cascades in their native context. Although HdAd is well positioned to fill this void, the dependency of Cre or Flp recombination prevents the production of HdAd vectors containing loxP or FRT sites. Therefore, there is a critical need for a system to produce HdAd in a Cre- or Flp-independent manner. Vika recombinase (Vika) from Vibrio coralliilyticus recognizes a 34-bp palindromic target site (vox) and exhibits efficient site-specific recombination in both bacterial and mammalian cells.23 Vika has no activity on pseudo vox sites present in the mammalian genome and has significantly lower genotoxicity and cytotoxicity than Cre.23
Since a Vika-based HdAd production system would overcome the current limitations of using HdAd in research applications, we developed a l HdAd production method using Vika/vox recombination. To do so, we created a producer cell line that expresses Vika and an HV containing the Ad ITRs with a Ψ flanked by vox sites. We show that this system, called VikAD, supports efficient replication of HdAd vectors to high titers with low levels of contaminating HV. VikAD was used to produce an HdAd FLEX EGFP vector and demonstrate Cre-dependent expression of EGFP in 1) a cell line, 2) a mouse Cre driver line, and 3) using dual delivery of both HdAd Cre and HdAd FLEX EGFP in vivo. Our results demonstrate that VikAD enables the production of HdAd vectors containing loxP sites. Since Vika does not cross-recombine on Frt, Cre and Cre-derivative, or noxP and pox sites,23,24 VikAD will enable the production of HdAd vectors that utilize Cre, Flp, or Cre-derivative sites for transgene expression.21,22,25 Therefore, VikAD has the ability to greatly expand the utility of HdAd for research applications that rely on genetic intersectional strategies using Cre and Flp or other site-specific recombinases in vitro and in vivo that are not possible with rAAV or LVV.
Results
Development of Vika293 cell line
To develop a Vika/vox-based HdAd production system, our first step was to generate a producer cell line that stably expresses high levels of Vika and Ad E1 proteins. We chose HEK293 (293) cells as the platform, since they stably express the Ad E1 proteins, are readily available, and support high levels of Ad replication.26 The Vika cDNA was codon optimized for human expression and an N-terminal HA epitope tag and nuclear localization signal (NLS) were added to create HA-NLS-Vika. We then created an LVV expressing HA-Vika-NLS to generate a pooled population of stably transduced HEK293 cells. To confirm Vika-mediated site-specific excision of DNA in our pooled population, we transfected a reporter plasmid containing a CMV-vox-dsRed-vox-EGFP cassette (Figure 1A). In the absence of Vika, dsRed is expressed from the CMV promoter, while in the presence of Vika, the dsRed ORF is excised, leading to expression of EGFP. Our initial characterization of the pooled population of Vika cells showed that the majority of cells expressed EGFP, indicating that Vika site-specific excision was successful. However, we found a small but significant fraction of dsRed expressing cells, which indicated heterogeneity in Vika excision activity (Figure 1B). Therefore, to exclude cells with little or no Vika activity, we established clonal populations of Vika cells to identify individual clones with high levels of excision activity. Parental Vika cells were transfected with pCMV-vox-dsRed-vox-EGFP followed by live single-cell fluorescence-activated cell sorting (FACS) to select for individual cells that expressed EGFP but not dsRed (Figure 1B). Clones with high levels of EGFP and undetectable dsRed expression were then amplified. Clones in which the reporter plasmid was found to have integrated into the chromosomal DNA were excluded from further analysis. Based on our screening, we identified a clone, Vika 24, which showed almost no dsRed expression compared with the pooled population (Figure 1B). Western blot analysis for Vika expression demonstrated that this clone had higher levels of Vika expression compared with the pooled population (Figure 1C). This clone was chosen for subsequent HdAd production and is renamed as Vika293 cells.
Figure 1.

Characterization of Vika expression and activity in HdAd producer cells
(A) Schematic of the reporter cassette used to indicate the presence of Vika activity. (B) Comparison of Vika activity before and after clonal sorting and expansion. 293 pooled Vika cells and Vika Clone 24 were transfected with reporter plasmid and images were captured 24 h post-transfection. (C) Western blot analysis of Vika expression. Vika protein was detected in total cell lysates using an antibody against the HA epitope tag on the N-terminus of Vika. Actin is the loading control.
Creation and characterization of voxHV
To create an HV genome that is unpackageable after Vika excision, the Ad Ψ was flanked by vox sites to create a vox HV plasmid, pVoxHV, which was transfected into 293 HEK producer cells. VoxHV was amplified and purified according to standard protocols.16 Restriction digest analysis was performed to confirm the integrity of the voxHV genome (Figure 2A). Viral DNA from purified viral particles was digested with MfeI to demonstrate that addition of vox sites did not result in genomic rearrangement of the HV genome. MfeI digestion of VoxHV genomic DNA showed the expected banding pattern based on the predicted MfeI fragment sizes (Figure 2B). We found that thirty 15-cm plates yielded 3.5 × 1012 VoxHV particles with an infectious titer of 2.1 × 1011 infectious units (IU)/ml in a final volume of 1 mL. These values are similar to titers obtained using the loxP HV, as reported (NG163-R2).17,27, 28, 29, 30 Taken together, these data demonstrate that the vox sites have no effect on viral DNA replication, assembly, or infectivity of the HV.
Figure 2.

VikAD enables excision of VoxHV packaging signal and amplification of HdAd
(A) Map of VoxHV genome with MfeI restriction sites. Vox sites and packaging signal are indicated. (B) MfeI digestion of purified VoxHV DNA showing banding pattern that corresponds to expected fragment sizes of MfeI digested VoxHV genome). (C) Total DNA was purified from infected cell lysates at each passage P0–P4 during HdAd amplification using Vika293 and VoxHV. PCR amplification was performed using primers that flank the HV Ψ. 116 cells expressing Cre recombinase infected with a loxP HV serve as a positive control for excision, resulting in a 345-bp amplicon. 293 parental cells infected with HV serve as a negative control, resulting in a 605-bp amplicon. (D) Equal amounts of cell lysate from passages 0–3 during HdAd production were used to transduce 293 cells. Images were captured at 24 h post-transduction, showing expression of the fluorescent reporter mClover3 from the HdAd genome. Images for passages 0 and 1 were captured using an exposure time of 400 ms. Images for passages 2 and 3 were captured using an exposure time of 25 ms.
VikAD production of HdAd
An effective HdAd production system must support production of HdAd to high titers that contain low levels of contaminating HV. To test the efficiency of our VikAD production system, we amplified an HdAd CMV mClover3 vector. Crude lysates were examined at each passage of HdAd production for both excision of VoxHV Ψ and HdAd amplification. To evaluate excision of the Ψ from VoxHV in Vika293 cells, standard non-quantitative PCR using 30 cycles of amplification and primers that flank the Ψ from VoxHV was performed (Figure 2C). As a positive control for excision, 116 (Cre-expressing) cells were infected with the loxP HV.17 As a negative control for excision, 293 cells were infected with the voxHV. Based on our PCR excision assay we were unable to detect VoxHV genome with the Ψ intact at any passage of HdAd amplification.
To assess the efficiency of HdAd amplification, a portion of the crude lysate from an equal number of Vika293 cells at each passage was used to transduce 293 cells. Images were captured 24 h post-transduction to monitor expression of the mClover3 reporter from the HdAd vector (Figure 2D). We observed a dramatic increase in mClover3 signal at each HdAd amplification passage as evidenced by the relative number of cells expressing mClover3 and the decrease in the exposure times necessary to visualize transduced cells. Analysis of HdAd titer revealed a physical titer of 2.0 × 1012 viral particles (vp)/ml and an infectious titer of 1.0 × 1011 infectious units (IU)/ml. These values are similar to the titers of an identical HdAd CMV mClover3 vector that was produced using the loxP HV and 116 cells using an identical vector amplification protocol, which grew to 2.3 × 1012 viral (vp)/mL with an infectious titer of 5.0 × 1010 (IU)/mL).
Production of HdAd containing loxP sites
Genetic intersectional strategies relying on Cre or Flp have become critical tools in understanding cellular and molecular mechanisms of cellular network activity.20,21 Since VikAD produced high titer HdAd, we wanted to determine if we could create an HdAD virus that was dependent on Cre-mediated excision for transgene expression. To do so, we cloned FLEX EGFP (Addgene#59,331, gift of Dr. Ian Wickersham) under control of the hsyn promoter into an HdAd genomic plasmid that also contained the hsyn mCherry transgene expression cassette (Figure 3A).27 HdAd FLEX EGFP virus was produced and purified to high titers using standard methods.31
Figure 3.

VikAD production of HdAd FLEX EGFP enables Cre dependent expression in vitro
(A) Schematic of HdAd genome containing a flexed EGFP reporter. In the absence of Cre, only mCherry is expressed from the vector. In the presence of Cre, both mCherry and EGFP are expressed. (B) 293 and 116 cells were transduced with 200 viral particles per cell. Fluorescent images were captured 72 h post-transduction.
To determine the excision efficiency of the VikaAD system during amplification, we carried out more extensive characterization of HdAd FLEX EGFP to determine HV contamination prior to density purification. Although excision of the Ψ in the VoxHV was comparable with the commonly used loxP HV when measured by non-quantitative PCR, it is well known from the Cre and Flp production systems that excision efficiency is not 100% and that there are differences in excision efficiencies between the Cre and Flp producer cell lines.17,19,32 Therefore, to determine Vika excision efficiency on the Ψ in VoxHV, we carried out ddPCR on crude lysates of HdAd FLEX EGFP prior to final purification. Crude lysate from the final passage contained ∼0.4% HV contamination, which is similar to previous reports of HV contamination with Cre and Flp production systems prior to density gradient purification.17, 18, 19 We found that HV contamination after CsCl purification of HdAd FLEX EGFP was ∼0.05%. Based on these results, our VikAD system mediates efficient production of HdAd with HV contamination levels comparable with the Cre and Flp systems before and after density gradient purification.
VikaAd enables the production of HdAd vectors with Cre excision-dependent transgene expression
To determine if Cre-mediated excision led to EGFP expression from HdAd FLEX EGFP in an in vitro context, we transduced 116 cells17 (Cre+) or 293 cells (Cre-). Although the hSyn promoter is a neurospecific promoter,33 it has low levels of activity in 293 cells and their derivatives, as 293 are related to neuronal cells.34 Therefore, we used a high MOI to see sufficient expression at detectable levels. Analysis of the 116 and 293 cells 72 h after transduction revealed that 293 cells only expressed mCherry (Figure 3B). However, 116 cells that were mCherry positive also expressed EGFP, demonstrating efficient Cre-mediated inversion of the EGFP sequence. To test functionality in vivo with a mouse Cre driver line in a native neuronal context, we injected HdAd FLEX EGFP into the P1 mouse cochlear nucleus (CN)35 of the Math5 mouse Cre driver line or C57BL/6J (wild-type, wt) mouse line (Figure 4A). Math5 (Atoh7) is a transcription factor that leads to selective labeling of the globular bushy cells (GBCs) in the CN.36,37 Analysis of the P9 auditory brainstem revealed EGFP expression in the CN, the GBC axons, and the calyces of Held, large glutamatergic presynaptic terminals, which arise from the GBCs axon in the Math5 Cre mouse line but not wt (Figures 4B–4D). Finally, to test whether co-delivery of HdAd FLEX EGFP with an HdAd Cre vector leads to efficient EGFP expression, we co-injected an HdAd Cre, which contains an hsyn mCherry reporter,27 with HdAd FLEX EGFP into the P1 mouse CN (Figure 5A). Analysis of the P9 auditory brainstem revealed EGFP expression that co-localized with EGFP expression in the CN and axons that projected from the CN (Figure 5B). In particular, the calyces of Held were co-labeled with EGFP and mCherry in the contra-lateral medial nucleus trapezoid body (MNTB) from the injection site (Figures 5C and 5D). Based on these results from cell lines, mouse Cre driver line, and dual viral injections, we demonstrate that VikAD leads to effective production of an HdAd vector for cell-type-specific labeling using Cre-dependent expression for in vitro and in vivo applications.
Figure 4.

VikAD production of HdAd FLEX EGFP enables Cre-dependent expression in a mouse Cre driver line
(A) Graphical representation of injection site (CN) and the presynaptic terminals (Calyx of Held at MNTB region) in the contralateral MNTB in the auditory brainstem. HdAd FLEX EGFP was injected at P1 in the CN of either Math5 Cre or wt mice (C57BL/6J). (B) Tile-scanned confocal images of (wt) (top) and Math5 Cre (bottom) mouse auditory brain stem. (C and D) Magnified images of CN and MNTB. (C) (top) CN injected with HdAd FLEX EGFP shows that only mCherry is expressed in the absence of Cre. (bottom) Math5 Cre CN demonstrates EGFP expression in the presence of Cre. (D) (top) Calyces of Held in the contralateral MNTB of wt mice. (bottom) Calyces of Held in the contralateral MNTB of Math5 Cre mice.
Figure 5.

Co-injection of HdAd Cre and HdAd FLEX EGFP results in Cre-dependent EGFP expression in a native neuronal circuit
(A) Cartoon of the auditory brainstem where HdAd Cre and HdAd FLEX EGFP were co-injected into the P1 CN of C57BL6/J mice. (B) Representative images of mCherry and EGFP expression in the P9 auditory brainstem. (C and D) Magnified images of mCherry and EGFP expression from the (C) CN and (D) contralateral MNTB.
Discussion
By generating a cell line that expresses Vika and an Ad HV that contains the Ψ flanked by vox sites, we created an HdAd production platform, VikAD, that will enable the use of HdAd with genetic intersectional strategies using Cre and Flp or other site-specific recombinases in vitro and in vivo. Therefore, VikAD fills an important need for intersectional genetic strategies for cell-type-specific expression of large genes and transgene expression cassettes not possible with rAAV or LVV.
VikAD and HdAd production
Our PCR excision assay revealed similar excision efficacy of the Ψ between 116 and Vika293 cells, and analysis of HV contamination of crude lysate during VikAD production showed that Vika293 produced HdAd with a similar level of HV contamination as the 116 and Flp producer cell lines.17, 18, 19 This demonstrates that VikAD is as efficient as these producer systems in excising the Ψ. However, it is possible that VikAD can be improved with regard to HV contamination prior to purification. A previous study showed that the increased levels of Cre recombinase in 116 cells compared with 293NS4 cells lead to significant increases in excision efficiency with a significant reduction in HV contamination prior to density gradient purification.17 Therefore, the excision efficacy in Vika293 cells could be improved by creating additional clonal cell lines with higher levels of Vika expression or by delivery of an additional Vika coding sequence to the current cell line. Although Vika has significantly lower toxicity compared with Cre,23 Vika expression may be at maximal levels in Vika293 cells, since Vika is codon optimized for expression in human cells. Another possibility to determine if we could increase Vika excision activity would be to use molecular evolution techniques.38 This strategy has been successfully used to create Flp recombinases with high thermostability and more efficient Flp excision.39 Finally, HV contamination might be further reduced by altering the spacing of the Ψ between the vox sites or the distance between the ITR and vox Ψ vox site.40 Future studies are needed to test these hypotheses.
VikAD for use with genetic intersectional strategies
VikAD enabled the production an HdAd FLEX EGFP vector that enables cell-type-specific expression in the context of Cre in vitro and in vivo. Vika is extremely specific and does not cross-recombine on Frt, Cre and Cre-derivative, or noxP and pox sites.23,24 Therefore, VikAD will enable the production of HdAd vectors that utilize Cre, Flp, or Cre-derivative sites for transgene expression21,22,25 to permit cell-type-specific and sparse labeling of cells in animals. In addition, since HdAd can package up to 36 kb of foreign DNA, VikAD can be used to create HdAd vectors that utilize these recombinase-dependent expression cassettes to express cDNAs, sensors, or other constructs that are too large to be packaged into AAV and LVV. We demonstrate that co-delivery of HdAd Cre with the HdAd FLEX EGFP vector leads to Cre-dependent reporter expression, showing that VikAD enables the ability to exploit the advantages of HdAd in combination with cell-type-specific rAAV and LVV expression vectors to probe neuronal circuit function across multiple animal species. While Ad Type 5 vectors have broad tropism, there are cell types that are refractory to Ad Type 5 but not rAAV or LVV; therefore, it should first be determined that the viral vectors in a dual-injection approach have overlapping tropism. Taken together, VikAD is an important addition to the viral vector field that increases the number of tools for elucidating the cellular and molecular mechanisms controlling cellular activity in vivo and regulation of organismal behavior.
Materials and methods
Ethical approval
All mice were used in accordance with animal welfare laws approved by the Institutional Committee for Care and Use of Animals at the University of Iowa (0,021,952). Animals were housed with 12-h light/dark cycle and ab libitum food/water supply. Mice of both sexes were used for all experiments. C57BL/6J mice wt were obtained from Jax labs and bred inhouse. Atoh7 (Math5) Cre lines were generated using by the University of Iowa Genome Editing Facility.
Generation of Math5 Cre mice
C57BL/6J mice were purchased from Jackson Labs (000,664; Bar Harbor, ME). Male mice older than 8 weeks were used to breed with 3- to 5-week-old super-ovulated females to produce zygotes for pronuclear injection. Female ICR (Envigo; Hsc:ICR(CD-1)) mice were used as recipients for embryo transfer. All animals were maintained in a climate-controlled environment at 25°C and a 12/12 light/dark cycle. Animal care and procedures conformed to the standards of the Institutional Animal Care and Use Committee of the Office of Animal Resources at the University of Iowa.
Preparation of Cas9 RNPs and the injection mix
Chemically modified CRISPR-Cas9 crRNAs and CRISPR-Cas9 tracrRNA were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA, Alt-R CRISPR-Cas9 crRNA; Alt-R CRISPR-Cas9 tracrRNA (Cat# 1,072,532)). The crRNAs and tracrRNA were suspended in T10 × 100.1 and combined with 1 μg/ul (∼29.5 uM) final concentration in a 1:2 (μg:μg) ratio. The RNAs were heated at 98°C for 2 min, then allowed to cool slowly to 20°C in a thermal cycler. The annealed cr:tracrRNAs were aliquoted to single-use tubes and stored at −80°C. Cas9 nuclease was also purchased from IDT (Alt-R S.p. HiFi Cas9 Nuclease). Individual cr:tracr:Cas9 ribonucleoprotein (RNP) complexes were made by combining Cas9 protein and cr:tracrRNA in T10 × 100.1 (final concentrations: 300 ng/μL (∼1.9 μM) Cas9 protein and 200 ng/μL (∼5.9 μM) cr:tracrRNA). The Cas9 protein and annealed RNAs were incubated at 37°C for 10 min. The RNP complexes were combined with single-stranded repair template and incubated an additional 5 min at 37°C. The concentrations in the injection mix were 60 ng/μL (∼0.37 μM) Cas9 protein, 20 ng/μL (∼0.59 μM) each cr:tracrRNA and 20 ng/μL single-stranded repair template.
Collection of embryos and injection
Pronuclear-stage embryos were collected using standard methods.41 Embryos were collected in KSOM media (Millipore; MR101D) and washed 3 times to remove cumulous cells. Cas9 RNPs and single-stranded repair template were injected into the pronuclei of the collected zygotes and incubated in KSOM with amino acids at 37°C under 5% CO2 until all zygotes were injected.42 Fifteen to 25 embryos were immediately implanted into the oviducts of pseudo-pregnant ICR females.
Cell lines
293 human embryonic kidney (293) cells and HeLa cells were maintained in Dulbecco's Modified Eagle Medium (DMEM), containing 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, and 100 U/mL penicillin and streptomycin. 293 cells contain the left hand 4,344 bp of the adenovirus serotype 5 genome, which expresses the viral E1A and E1B genes necessary for the replication of E1-deleted adenoviral vectors40 . Vika293 cells were maintained in the same medium containing 1 mg/mL G418 (IBI Scientific, Dubuque, IA, USA). 116 cells (kind gift from Philip Ng, Baylor College of Medicine 17) were maintained in Modified Eagle Medium (MEM), containing 10% FBS, 100 U/ml penicillin and streptomycin, and 100 μg/mL hygromycin (Life Technologies, Carlsbad, CA, USA).
Vika293 creation
Codon optimized Vika recombinase (GenBank: NZ_ACZN01000014.1 ZP_05884863) was synthesized with an N-terminal hemagglutinin (HA) epitope tag (YPYDVPDYA) and NLS (KKKRKV) (Thermo Fisher Scientific, Waltham, MA, USA). The synthesized fragment was cloned into the pFIV3.2CMVmcwtIRESneomycin lentiviral vector (The University of Iowa Viral Vector Core Facility, Iowa City, IA, USA), which uses the CMV promoter to drive expression of Vika. The HA epitope tag, NLS, and the Vika cDNA were synthesized directly adjacent without intervening linker sequences. Lentivirus was produced by The University of Iowa Viral Vector Core. 293 cells were transduced with Vika lentivirus, and transduced cells were selected by growth in medium containing 1 mg/mL G418. To establish clonal populations exhibiting high levels of Vika activity, Vika cells were transfected with a plasmid containing a CMV-vox-dsRed-vox-EGFP reporter cassette. Twenty-four hours post-transfection, cells were subjected to live cell FACS, gating on cells that were EGFP positive and dsRed negative (UIOWA flow Cytometry Core). Single cells were sorted into poly-L-lysine-coated 96-well plates (Corning Life Sciences, Tewksbury, MA, USA). Clones were expanded and assessed for excision activity and expression by reporter plasmid transfection and Western blot, respectively.
Vox HV
pVoxHV plasmid was created by removing the loxP sites from the pNG163-R2 (kind gift from Philip Ng, Baylor College of Medicine,17) HV plasmid and replacing them with the 34-bp vox sites in a parallel orientation (5′-AATAGGTCTGAGAACGCCCATTCTCAGACGTATT-3′). To do so, we used CRISPR/Cas9-mediated cleavage of the regions containing the loxP sites in pNG163-R2 using the following sgRNA sequences: upstream of loxP 5′-GTGTGATGTTGCAAGTGTGG-3′, downstream of loxP 5′-AATGCTTCCATCAAACGAGT-3′. sgRNA synthesis was carried out using the EnGen sgRNA Synthesis Kit, and sgRNAs were purified using the Monarch RNA Clean Up Kit (New England Biolabs (NEB), Ipswich, MA, USA). Plasmid DNA was digested in vitro with Cas9 nuclease from S. Pyogenes (NEB, Ipswich, MA, USA). Reactions were treated with proteinase K and heat inactivated. Two DNA fragments containing the vox sites and terminal 20-bp homology arms were synthesized (Integrated DNA Technologies, Coralville, IA, USA) and inserted into the digested pNG163-R2 using NEBuilder HiFi DNA Assembly, according to the manufacturer's protocol (NEB, Ipswich, MA, USA). Proper insertion of the vox sites was confirmed by Sanger sequencing. Ten micrograms of the pVoxHV was digested with PacI and transfected into a 6 cm dish of 293 HEK cells, using the ProFection Mammalian Transfection System (Promega, Madison, WI, USA). VoxHV production was performed according to standard protocols.43 Viral DNA from purified stocks was prepared using the PureLink Viral RNA/DNA Mini Kit (Thermo Fisher, Waltham, MA, USA).
HdAd mClover3 and HdAd FLEX EGFP production
The green fluorescent reporter mClover344 was cloned into a shuttle plasmid with CMV promoter and SV40 poly(A) signal and subsequently were transferred to the HdAd plasmid C4HSU27 to create pHdAd CMV mClover3. To create pHdAd FLEX EGFP, the FLEX EGFP sequence from Addgene plasmid 59,331 (pAAV-CAG-FLEX-EGFP, a gift from Ian Wickersham; http://n2t.net/addegen:59931 RRID:Addgene 5,933) was cloned into an HdAd shuttle plasmid with the hsyn promoter and BGH poly(A) signal. The syn FLEX EGFP bgh poly(A) cassette was then cloned into the HdAd plasmid C4HSU syn mCherry27,29 to create pHdAd syn FLEX EGFP syn mCherry, which contains two separate transgene cassettes for reporter expression.
All HdAd vectors were created using standard protocols.16,31 Ten micrograms of either PmeI digested pHdAd mClover3 or pHdAd FLEX EGFP syn mCherry was transfected into Vika293 cells using the ProFection Mammalian Transfection System (Promega, Madison, WI, USA) to create HdAd CMV mClover3 or HdAd FLEX EGFP viral vectors. Twenty-four hours later, the cells were infected with voxHV, and cells were harvested 48 h post-infection when the cells exhibited full cytopathic effect. HdAd vector was released by three cycles of freeze/thaw, and a portion of the cleared cell lysate was used to infect a new dish of Vika293 cells along with voxHV. A total of four serial amplifications was performed with the final passage consisting of thirty 15-cm diameter dishes. Vector was purified using two discontinuous cesium chloride gradients (1.25 and 1.35 g/mL) and one continuous cesium chloride gradient (1.35 g/mL), and the final prep was dialyzed into 250 mM sucrose, 40 mM HEPES pH 7.4, and 1 mM MgCl2.
Determination of viral titer
Physical titers were determined by spectrophotometry to measure absorbance at 260 nm. Physical titer (vp/mL) was calculated using the following equation: vp/mL = (A260 × dilution factor × 1.1 × 1012 × 36)/vector genome size in kb.16 Determination of HdAd was determined by FACS of infected HeLa cells. VoxHV infectious titers were determined by AdenoX Rapid Titer Kit (Takara Bio, Mountain View, CA, USA). HV contamination was measured by digital drop PCR (ddPCR) to quantify viral genomes from infected cells. Briefly, HeLa cells in 6-well dishes were infected with 5 μL purified virus stock for four hours. Cells were washed with PBS and lysed in buffer containing 0.65% NP-40, 150 mM NaCl, and 10 mM Tris HCl pH 8.0. Nuclei were pelleted by centrifugation at 3,000 × g for 2 min and washed with 1 mL fresh lysis buffer. Nuclei were pelleted again, and the supernatant discarded. Pelleted nuclei were resuspended in 200 μL PBS, and DNA was purified using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, DE). DNA was diluted 1:500 and used as a template for ddPCR, using the following primer/probe sets: HdAd forward 5′-CCCCGCTACCCCAATCC-3′, HdAd reverse 5′-TTAGCTTTTTTGGGTGATTTTTCC-3′, HdAd probe VIC-5′- AGCCTCTCTCATCTCACAGT -3′-MGBNFQ, HV forward 5′-TGGGCGTGGTGCCTAAAA-3′, HdAd reverse 5′-GCCTGCCCCTGGCAAT-3′, HV probe FAM-5′-TGTCTTTCAGTAGCAAGCT-3′-TAMRA. Template, primers, and probes were mixed with ddPCR™ Supermix for Probes (Bio-Rad Laboratories, Hercules, CA, USA). Droplets were formed using ddPCR™ Droplet Reader Oil (Bio-Rad Laboratories, Hercules, CA, USA). Cycling parameters were as follows: initial denaturation at 95°C for 10 min followed by 40 cycles of 94°C for 30 s and 58°C for 1 min. A final incubation at 98°C for 10 min was performed prior to droplet count. Droplets were generated and read using the Bio-Rad QX200 Droplet Digital PCR System.
PCR excision assay
Total DNA from crude cell lysates was purified using the PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific, Waltham, MA, USA). PCR was carried out using EconoTaq Master Mix (Lucigen, Middleton, WI, USA), 10 ng DNA template, and the following primers: psi forward 5′-GGAAGTGTGATGTTGCAAGT-3′ and psi reverse 5′-CAATGCTGGAGCCCATC-3′. PCR products were run on a 1.5% agarose gel stained with ethidium bromide.
Western blot
Cells were lysed in buffer containing 10 mM Tris HCl pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, and complete protease inhibitor cocktail (Roche, Basel, CH). Lysates were cleared by centrifugation at 20K × g for 30 min at 4°C, and protein was quantified using the Pierce BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). Twenty micrograms of protein was boiled in Laemmli sample buffer (32.9 mM Tris HCl pH 6.8, 13.2% glycerol, 1% SDS, 0.005% bromophenol blue, and 355 mM 2-mercaptoethanol). Proteins were separated using 4%–20% tris-glycine PAGE and transferred to polyvinylidene fluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA, USA). Blots were blocked in 5% non-fat dry milk in TBS-T and incubated at 4°C overnight with 0.5 μg/mL mouse monoclonal anti-HA antibody clone 5B1D10 (Thermo Fisher, Waltham, MA, USA). A duplicate set of samples on the same blot was incubated with 0.4 μg/mL mouse monoclonal anti-actin antibody clone AC-15 (Sigma-Aldrich, St. Louis, MO, USA). Blots were then incubated with 0.2 μg/mL goat anti-mouse-HRP, cat. #32,230 (Thermo Fisher Scientific, Waltham, MA, USA) and developed using Clarity Western ECL (Bio-Rad Laboratories, Hercules, CA, USA).
Infection of 293 and 116 cells with HdAd FLEX EGFP
Six-centimeter dishes were seeded with 7.4 × 105 293 and 116 cells. Dishes were infected 24 h later at 70% confluency (2 × 106 cells) with 200 vp/cell of HdAd FLEX EGFP. Images were taken 72 h post-infection in the EGFP, mCherry, and bright field channels. An overlay image of the EGFP and mCherry channels was created using Fiji software.
Stereotactic surgery
Stereotactic injection of HdAds into the CN was performed at postnatal day 1 (P1) as previously described.35 Briefly, mice were placed in a rubber glove and cooled for 5 min in an ice bath and secured on a chilled aluminum block. Depth of anesthesia was checked throughout the surgery by the absence of tail pinch response. Surgery took place under aseptic conditions. After the animals were anesthetized, a glass needle with tip opening ∼20 μm that was filled with viral vector was injected into the CN. For dual HdAd Cre27 and HdAd FLEX EGFP injection, vectors were mixed at a 1:1 particle ratio for a final concentration of 1 × 109 particles/ul. A total of approximately ∼1–1.5 μL HdAd Cre and HdAd FLEX EGFP (∼1 × 107 transducing units/μL) was injected into C57Bl/6J mice. For experiments with Math5 Cre mice, approximately ∼1–1.5 μL HdAd FLEX EGFP (∼1 × 107 transducing units/μL) was injected into the CN. In both cases, the viral vector solution was injected at a flow rate of approximately 0.5 μL/min. After injection, the glass needle was left in place for 1 min to dissipate potential back pressure issues and then slowly removed. Animals were then placed under a warm lamp. Upon full recovery, visual inspection, and tail pinch responsiveness, animals were returned to their respective cage with the dam. If animals did not recover from the surgery, they were immediately euthanized. In the case of runting or if animals were found in distress at later time point (days after surgery), the veterinary staff were consulted and their recommendation for further action was followed.
Imaging cell lines
All microscopic images of cell lines were captured at 10× magnification using an Olympus CKX41 microscope with an EGFP filter set and mCherry filter set equipped with DP21 camera and X-Cite Series 120Q Fluorescence Lamp.
Imaging auditory brainstem
To analyze transduction in the auditory brainstem, brains were dissected out from the injected animals at age postnatal day 9 (P9). Meningeal layers were carefully removed from brain samples, followed by a brief wash in 0.1 M PBS and stored in 4% PFA (in 1X PBS) for 16–24 h. Brain samples were washed 3 times for 10 min with 0.1 M PBS. Coronal auditory brainstem slices of 50-μm thickness containing CN and MNTB from the fixed brain samples were prepared using a Leica VT 1200 vibratome. Free-floating slices were incubated with DAPI (1 μg/mL) for ∼5 min and briefly washed with 0.1 M PBS. Sections then were mounted with Vectashield hardset antifade mounting medium with DAPI (H-1500, Vector laboratories, Burlingame, CA, USA). To analyze transduction in the auditory brainstem. Confocal images were acquired with a Zeiss LSM 880. Auditory brainstem slices were imaged using z stack and tilescan mode (bounding grid) with Plan Neofluar 10X/0.3 and 20X/0.8 and online stitched with overlap 10%–25%. Emission signal intensity for each channel was adjusted to below the saturation level. Images were further processed in Fiji (ImageJ 1.53c) and Adobe Photoshop.
Acknowledgments
This work was supported by the National Institute on Deafness and Other Communication Disorders (R01DC014093, R21DC018242), the National Institute of Neurological Disorders and Stroke (R01NS110742), and the University of Iowa (to S.M.Y., Jr.). We thank The University of Iowa Viral Vector Core (http://www.medicine.uiowa.edu/vectorcore) for providing lentivirus and assisting with vector quantification. We thank the University of Iowa Flow Cytometry Core for help and assistance with cell sorting. Transgenic mice were generated at the University of Iowa Genome Editing Core Facility directed by William Paradee, PhD and supported in part by grants from the NIH and from the Roy J. and Lucille A. Carver College of Medicine. We wish to thank Norma Sinclair, Patricia Yarolem, Joanne Schwarting, and Rongbin Guan for their technical expertise in generating transgenic mice.
Author contributions
Conceptualization, S.M.Y.; methodology, S.P. and S.M.Y.; validation, S.P. and S.M.Y.; investigation, S.P., P.V.R., and P.V.; resources, S.P. and S.M.Y.; writing—original draft, S.P. and S.M.Y.; writing—review and editing, S.P., P.V.R., P.V., and S.M.Y.; visualization, S.P., P.V.R., P.V., and S.M.Y.; supervision, S.P. and S.M.Y.; project administration, S.M.Y.; funding acquisition, S.M.Y.
Declaration of interests
The authors declare no conflicts of interest.
References
- 1.Nectow A.R., Nestler E.J. Viral tools for neuroscience. Nat. Rev. Neurosci. 2020;21:669–681. doi: 10.1038/s41583-020-00382-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y.H., Keiser M.S., Davidson B.L. Viral vectors for gene transfer. Curr. Protoc. Mouse Biol. 2018;8:e58. doi: 10.1002/cpmo.58. [DOI] [PubMed] [Google Scholar]
- 3.Li C., Samulski R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020;21:255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- 4.Anguela X.M., High K.A. Entering the modern era of gene therapy. Annu. Rev. Med. 2019;70:273–288. doi: 10.1146/annurev-med-012017-043332. [DOI] [PubMed] [Google Scholar]
- 5.Brunetti-Pierri N., Ng P. Gene therapy with helper-dependent adenoviral vectors: lessons from studies in large animal models. Virus Genes. 2017;53:684–691. doi: 10.1007/s11262-017-1471-x. [DOI] [PubMed] [Google Scholar]
- 6.Palmer D.J., Ng P. Characterization of helper-dependent adenoviral vectors. Cold Spring Harb. Protoc. 2011;2011:867–870. doi: 10.1101/pdb.prot5628. [DOI] [PubMed] [Google Scholar]
- 7.Kim I.H., Jozkowicz A., Piedra P.A., Oka K., Chan L. Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. U S A. 2001;98:13282–13287. doi: 10.1073/pnas.241506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toietta G., Mane V.P., Norona W.S., Finegold M.J., Ng P., McDonagh A.F., Beaudet A.L., Lee B. Lifelong elimination of hyperbilirubinemia in the Gunn rat with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. U S A. 2005;102:3930–3935. doi: 10.1073/pnas.0500930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmitt F., Pastore N., Abarrategui-Pontes C., Flageul M., Myara A., Laplanche S., Labrune P., Podevin G., Nguyen T.H., Brunetti-Pierri N. Correction of hyperbilirubinemia in gunn rats by surgical delivery of low doses of helper-dependent adenoviral vectors. Hum. Gene Ther. Methods. 2014;25:181–186. doi: 10.1089/hgtb.2013.236. [DOI] [PubMed] [Google Scholar]
- 10.Rastall D.P., Seregin S.S., Aldhamen Y.A., Kaiser L.M., Mullins C., Liou A., Ing F., Pereria-Hicks C., Godbehere-Roosa S., Palmer D., et al. Long-term, high-level hepatic secretion of acid alpha-glucosidase for Pompe disease achieved in non-human primates using helper-dependent adenovirus. Gene Ther. 2016;23:743–752. doi: 10.1038/gt.2016.53. [DOI] [PubMed] [Google Scholar]
- 11.Mian A., McCormack W.M., Jr., Mane V., Kleppe S., Ng P., Finegold M., O'Brien W.E., Rodgers J.R., Beaudet A.L., Lee B. Long-term correction of ornithine transcarbamylase deficiency by WPRE-mediated overexpression using a helper-dependent adenovirus. Mol. Ther. 2004;10:492–499. doi: 10.1016/j.ymthe.2004.05.036. [DOI] [PubMed] [Google Scholar]
- 12.Oka K., Belalcazar L.M., Dieker C., Nour E.A., Nuno-Gonzalez P., Paul A., Cormier S., Shin J.K., Finegold M., Chan L. Sustained phenotypic correction in a mouse model of hypoalphalipoproteinemia with a helper-dependent adenovirus vector. Gene Ther. 2007;14:191–202. doi: 10.1038/sj.gt.3302819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilbert R., Dudley R.W., Liu A.B., Petrof B.J., Nalbantoglu J., Karpati G. Prolonged dystrophin expression and functional correction of mdx mouse muscle following gene transfer with a helper-dependent (gutted) adenovirus-encoding murine dystrophin. Hum. Mol. Genet. 2003;12:1287–1299. doi: 10.1093/hmg/ddg141. [DOI] [PubMed] [Google Scholar]
- 14.Kiang A., Hartman Z.C., Liao S., Xu F., Serra D., Palmer D.J., Ng P., Amalfitano A. Fully deleted adenovirus persistently expressing GAA accomplishes long-term skeletal muscle glycogen correction in tolerant and nontolerant GSD-II mice. Mol. Ther. 2006;13:127–134. doi: 10.1016/j.ymthe.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Ghulam Muhammad A.K., Xiong W., Puntel M., Farrokhi C., Kroeger K.M., Salem A., Lacayo L., Pechnick R.N., Kelson K.R., Palmer D., et al. Safety profile of gutless adenovirus vectors delivered into the normal brain parenchyma: implications for a glioma phase 1 clinical trial. Hum. Gene Ther. Methods. 2012;23:271–284. doi: 10.1089/hgtb.2012.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmer D.J., Ng P. Rescue, amplification, and large-scale production of helper-dependent adenoviral vectors. Cold Spring Harb. Protoc. 2011;2011:857–866. doi: 10.1101/pdb.prot5627. [DOI] [PubMed] [Google Scholar]
- 17.Palmer D., Ng P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 2003;8:846–852. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 18.Umana P., Gerdes C.A., Stone D., Davis J.R., Ward D., Castro M.G., Lowenstein P.R. Efficient FLPe recombinase enables scalable production of helper-dependent adenoviral vectors with negligible helper-virus contamination. Nat. Biotechnol. 2001;19:582–585. doi: 10.1038/89349. [DOI] [PubMed] [Google Scholar]
- 19.Ng P., Beauchamp C., Evelegh C., Parks R., Graham F.L. Development of a FLP/frt system for generating helper-dependent adenoviral vectors. Mol. Ther. 2001;3:809–815. doi: 10.1006/mthe.2001.0323. [DOI] [PubMed] [Google Scholar]
- 20.Weissman T.A., Pan Y.A. Brainbow: new resources and emerging biological applications for multicolor genetic labeling and analysis. Genetics. 2015;199:293–306. doi: 10.1534/genetics.114.172510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atasoy D., Aponte Y., Su H.H., Sternson S.M. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J. Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fenno L.E., Mattis J., Ramakrishnan C., Deisseroth K. A guide to creating and testing new INTRSECT constructs. Curr. Protoc. Neurosci. 2017;80 doi: 10.1002/cpns.30. 4.39.31-4.39.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karimova M., Abi-Ghanem J., Berger N., Surendranath V., Pisabarro M.T., Buchholz F. Vika/vox, a novel efficient and specific Cre/loxP-like site-specific recombination system. Nucleic Acids Res. 2013;41:e37. doi: 10.1093/nar/gks1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karimova M., Splith V., Karpinski J., Pisabarro M.T., Buchholz F. Discovery of Nigri/nox and Panto/pox site-specific recombinase systems facilitates advanced genome engineering. Sci. Rep. 2016;6:30130. doi: 10.1038/srep30130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fenno L.E., Ramakrishnan C., Kim Y.S., Evans K.E., Lo M., Vesuna S., Inoue M., Cheung K.Y.M., Yuen E., Pichamoorthy N., et al. Comprehensive dual- and triple-feature intersectional single-vector delivery of diverse functional payloads to cells of behaving mammals. Neuron. 2020;107:836–853.e11. doi: 10.1016/j.neuron.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kovesdi I., Hedley S.J. Adenoviral producer cells. Viruses. 2010;2:1681–1703. doi: 10.3390/v2081681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lubbert M., Goral R.O., Satterfield R., Putzke T., van den Maagdenberg A.M., Kamasawa N., Young S.M., Jr. A novel region in the CaV2.1 alpha1 subunit C-terminus regulates fast synaptic vesicle fusion and vesicle docking at the mammalian presynaptic active zone. eLife. 2017;6:e28412. doi: 10.7554/eLife.28412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong W., Radulovic T., Goral R.O., Thomas C., Suarez Montesinos M., Guerrero-Given D., Hagiwara A., Putzke T., Hida Y., Abe M., et al. CAST/ELKS proteins control Voltage-gated Ca(2+) channel density and synaptic release probability at a mammalian central synapse. Cell Rep. 2018;24:284–293.e6. doi: 10.1016/j.celrep.2018.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lubbert M., Goral R.O., Keine C., Thomas C., Guerrero-Given D., Putzke T., Satterfield R., Kamasawa N., Young S.M., Jr. CaV2.1 alpha1 subunit expression regulates presynaptic CaV2.1 abundance and synaptic strength at a central synapse. Neuron. 2019;101:260–273.e6. doi: 10.1016/j.neuron.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radulovic T., Dong W., Goral R.O., Thomas C.I., Veeraraghavan P., Montesinos M.S., Guerrero-Given D., Goff K., Lubbert M., Kamasawa N., et al. Presynaptic development is controlled by the core active zone proteins CAST/ELKS. J. Physiol. 2020;598:2431–2452. doi: 10.1113/JP279736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montesinos M.S., Satterfield R., Young S.M., Jr. Helper-dependent adenoviral vectors and their use for neuroscience applications. Methods Mol. Biol. 2016;1474:73–90. doi: 10.1007/978-1-4939-6352-2_5. [DOI] [PubMed] [Google Scholar]
- 32.Parks R.J., Chen L., Anton M., Sankar U., Rudnicki M.A., Graham F.L. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. U S A. 1996;93:13565–13570. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kugler S., Meyn L., Holzmuller H., Gerhardt E., Isenmann S., Schulz J.B., Bahr M. Neuron-specific expression of therapeutic proteins: evaluation of different cellular promoters in recombinant adenoviral vectors. Mol. Cell Neurosci. 2001;17:78–96. doi: 10.1006/mcne.2000.0929. [DOI] [PubMed] [Google Scholar]
- 34.Shaw G., Morse S., Ararat M., Graham F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z., Cooper B., Kalla S., Varoqueaux F., Young S.M., Jr. The Munc13 proteins differentially regulate readily releasable pool dynamics and calcium-dependent recovery at a central synapse. J. Neurosci. 2013;33:8336–8351. doi: 10.1523/JNEUROSCI.5128-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saul S.M., Brzezinski J.A.t., Altschuler R.A., Shore S.E., Rudolph D.D., Kabara L.L., Halsey K.E., Hufnagel R.B., Zhou J., Dolan D.F., Glaser T. Math5 expression and function in the central auditory system. Mol. Cell Neurosci. 2008;37:153–169. doi: 10.1016/j.mcn.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J., Wu X.S., Sheng J., Zhang Z., Yue H.Y., Sun L., Sgobio C., Lin X., Peng S., Jin Y., et al. Alpha-synuclein mutation Inhibits endocytosis at mammalian central nerve terminals. J. Neurosci. 2016;36:4408–4414. doi: 10.1523/JNEUROSCI.3627-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang K.K., Wu Z., Arnold F.H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods. 2019;16:687–694. doi: 10.1038/s41592-019-0496-6. [DOI] [PubMed] [Google Scholar]
- 39.Buchholz F., Angrand P.O., Stewart A.F. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat. Biotechnol. 1998;16:657–662. doi: 10.1038/nbt0798-657. [DOI] [PubMed] [Google Scholar]
- 40.Hearing P., Samulski R.J., Wishart W.L., Shenk T. Identification of a repeated sequence element required for efficient encapsidation of the adenovirus type 5 chromosome. J. Virol. 1987;61:2555–2558. doi: 10.1128/JVI.61.8.2555-2558.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.PInkert C. Second edition. Elsevier Press; 2002. Transgenic Animal Technology. [Google Scholar]
- 42.Miura H., Quadros R.M., Gurumurthy C.B., Ohtsuka M. Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat. Protoc. 2018;13:195–215. doi: 10.1038/nprot.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ng P., Graham F.L. Construction of first-generation adenoviral vectors. Methods Mol. Med. 2002;69:389–414. doi: 10.1385/1-59259-141-8:389. [DOI] [PubMed] [Google Scholar]
- 44.Bajar B.T., Wang E.S., Lam A.J., Kim B.B., Jacobs C.L., Howe E.S., Davidson M.W., Lin M.Z., Chu J. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 2016;6:20889. doi: 10.1038/srep20889. [DOI] [PMC free article] [PubMed] [Google Scholar]