

Abstract
T-cell prolymphocytic leukemia (T-PLL) is a poor-prognostic mature T-cell malignancy. It typically presents with exponentially rising lymphocyte counts, splenomegaly, and bone marrow infiltration. Effective treatment options are scarce and a better understanding of TPLL’s pathogenesis is desirable. Activation of the TCL1 proto-oncogene and loss-of-function perturbations of the tumor suppressor ATM are TPLL’s genomic hallmarks. The leukemic cell reveals a phenotype of active T-cell receptor (TCR) signaling and aberrant DNA damage responses. Regulatory networks based on the profile of microRNA (miR) have not been described for T-PLL. In a combined approach of small-RNA and transcriptome sequencing in 46 clinically and moleculary well-characterized T-PLL, we identified a global T-PLL-specific miR expression profile that involves 34 significantly deregulated miR species. This pattern strikingly resembled miR-ome signatures of TCR-activated T cells. By integrating these T-PLL miR profiles with transcriptome data, we uncovered regulatory networks associated with cell survival signaling and DNA damage response pathways. Despite a miR-ome that discerned leukemic from normal T cells, there were also robust subsets of T-PLL defined by a small set of specific miR. Most prominently, miR-141 and the miR- 200c-cluster separated cases into two major subgroups. Furthermore, increased expression of miR-223-3p as well as reduced expression of miR-21 and the miR-29 cluster were associated with more activated Tcell phenotypes and more aggressive disease presentations. Based on the implicated pathobiological role of these miR deregulations, targeting strategies around their effectors appear worth pursuing. We also established a combinatorial miR-based overall survival score for T-PLL (miROS-T-PLL), that might improve current clinical stratifications.
Introduction
T-cell prolymphocytic leukemia (T-PLL) is a neoplasm of post-thymic T cells.1 It represents the most frequent mature T-cell leukemia in Western countries, however, with an incidence of 2 per million per year, it is still classified as an orphan disease.2 T-PLL patients typically present at a median age of about 65 years, with exponentially rising lymphocytosis, marked bone marrow infiltration, and splenomegaly.3 Phenotypically, T-PLL cells resemble mature, antigenexperienced T cells.4,5 The aggressive growth of T-PLL cells is paralleled by a refractory behavior towards most conventional chemotherapies.6 The current treatment of choice, the CD52-antibody alemtuzumab, is efficient in inducing initial responses, but nearly all patients relapse within 12-24 months thereafter.7,8 Second-line options are even less efficient and the median overall survival (OS) of T-PLL patients is <2 years.1,3,9,10
Activating translocation of the T-cell leukemia/lymphoma 1 (TCL1) proto-oncogene is the most prevalent genetic aberration in T-PLL.11 In addition, loss-of-function perturbations of the tumor suppressor ataxia telangiectasia mutated (ATM) are reported for >80% of T-PLL patients.1 Both alterations contribute towards a phenotype of enhanced T-cell receptor signaling (TCR) and an aberrant DNA damage response, including resistance to p53-mediated cell death, deregulated cell cycle control, and deficient DNA repair mechanisms.1,11 Activating lesions in janus kinase (JAK) and signal transducer and activator of transcription (STAT) molecules as well as epigenetic aberrations have emerged as further hallmarks of T-PLL pathology, resulting in a sustained survival signaling and pro-oncogenic cell cycle deregulation.1,12–15 Despite these recent advances, a better understanding of T-PLL's pathobiology is of importance in order to identify novel treatment options.
MicroRNA (miR) have increasingly been recognized as relevant in the pathogenesis of hematopoetic and solid tumors. They are small non-coding RNA with an average length of 22 nucleotides. By targeting specific mRNA, miR function as posttranscriptional repressors.16 Importantly, most miR regulate a large set of genes, often resulting in a cooperative effect on a given cellular pathway, rather than a specific effect on a single gene. Both onco miR and tumor-suppressive miR have been causally implicated in mature B- and T-lymphoid malignancies.17– 19 As a prominent example, chronic lymphocytic leukemia (CLL) harbors an unique miR expression signature with miR-181b downregulation as the best investigated miR deregulation.20 When overexpressing miR- 181b in the Em-TCL1A CLL mouse model, leukemic expansion is decelerated.21 Moreover, miR-181b as well as the miR-29 and miR-34b/c were shown to target the proto-oncogene TCL1A, reflected by an association of their downregulation with oncogenic TCL1A overexpression in CLL.22 Likewise, specific miR have been identified to be involved in the pathogenesis of mature T-cell tumors such as cutaneous T-cell lymphoma (CTCL) (e.g., deregulation of miR-29 and miR-200)17,23 or NK/T-cell lymphoma (downregulated miR-150).24 In T-PLL, frequent genomic aberrations of argonaute RISC catalytic component 2 (AGO2), a master regulator of miR processing, 25 provide first hints for altered miR activity and miR expression signatures (miR-omes).1 However, global miR deregulations, likely involved in T-PLL’s pathophysiology, have not been reported.
In the presented study, we performed small-RNA sequencing to investigate the spectrum of differential miR expression in T-PLL. We identify global, T-PLL-specific miR alterations associated with gene signatures affiliated to functional categories of survival signaling and DNA damage response pathways. In addition, we show that the miR-omes of T-PLL cells and of activated T cells are remarkably similar. Finally, we identify associations of miR alterations with cellular activation, clinical tumor burden, and patient outcome, all underlining the impact of miR deregulations in T-PLL.
Methods
Patient cohort
Primary isolates of 48 well-annotated T-PLL patients and of T cells from six age-matched healthy donors were studied (banked 2009-2019; patient characteristics in Table 1). The diagnosis of TPLL was confirmed according to World Health Organization criteria26 and consensus guidelines.2 All patients (median age 68 years) provided informed consent according to the Declaration of Helsinki. Collection and use of the samples have been approved for research purposes by the ethics committee of the University Hospital of Cologne (#11–319). Most samples (82.6%) were collected prior to any first-line treatment (n=38 of 46).
Sequencing and data processing
RNA from peripheral blood mononuclear cells (PBMC) of T-PLL patients (median purity 95.4%) and CD3+ pan-T cells of six agematched healthy controls (median purity 90.2%) was subjected to library preparation and sequenced on the NovaSeq 6000 (n=48 TPLL) and the HiSeq4000 platform (n=46 T-PLL, Illumina, San Diego, USA) according to the manufacturer’s instructions for polyA-RNA and small-RNA sequencing, respectively. Details on cell isolation, stimulation, RNA isolation, library preparation, sequencing, and data processing are given in the Online Supplementary Methods.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) were performed on preranked lists using the GSEA-software (v3.0)27 and MSigDB (v7.0)28 HALLMARK gene sets. For each considered miR, Spearman correlation coefficients for this miR and all protein-coding genes were determined by comparing the respective gene's count per million (CPM) and fragments per kilobase of million (FPKM) mapped reads. Sorted lists of correlation coefficients were then used as input for the GSEA.
MiR target prediction
In order to obtain putative mRNA targets for each miR, predicted miR bindings were first determined using the R-package multiMiR (v1.6.0, Database Version 2.3.0),29 all of eight prediction databases (diana_microt, elmmo, microcosm, miranda, mirdb, pictar, pita, targetscan), and a 20% default prediction cutoff. All bindings predicted by <2 different databases were removed. From the remaining predicted genes, we chose those as putative miR targets that showed a negative Spearman correlation (rho<0; P<0.05; false discovery rate [FDR] <0.25) of their expression values with the expression of the respective miR.
Correlations with clinical data
In order to test for associations of miR-223-3p, miR-21, miR-29, and miR-200c/141 with clinical characteristics, cytogenetics, immunophenotypes, and outcome data, cases were divided into groups by the mean or tertiles as cutoffs according to the distribution of expression values within the patient cohort. Further details on statistics are provided in the Online Supplementary Methods.
Survival score
In order to develop a survival score, we (i) randomly divided our cohort into a training set (n=22) and a validation set (n=22). We then (ii) identified miR that were expressed in at least 80% of TPLL samples and (iii) that were highly associated with OS in the training set (upper tertile of patients with highest vs. tertile with the lowest expression) using log-rank tests. Several other parameters that had been described to be prognostically relevant in T-PLL (e.g., leukocyte counts, TCL1 mRNA expression) were added to the resulting miR with the strongest single OS associations (lowest P-values) towards a multi-parameter training model. Next, (iv) we subjected all parameters to a recursive partitioning algorithm using the rpart R package (v.4.1-15) to identify optimum individual cutoffs in our training set. Such deprioritizations finally retained the four factors miR-200a-3p, miR-223-3p, miR-424-5p, and TCL1A, for which optimum cutoffs best allowed discriminations of T-PLL patients with shorter versus longer OS. We then (v) built multivariate scores for all possible combinations of these four parameters, adding one point to the total score if the respective expression cutoff was passed, and calculated optimum thresholds for these scores. We (vi) selected the score which allowed best discrimination of OS in our training set and (vii) verified our score in the validation set as well as in the total cohort of 44 T-PLL patients.
Table 1.
Clinical, cytogenetic and immunophenotypic characteristics of analyzed T-cell prolymphocytic leukemia (n=46 cases analyzed by small- RNA sequencing).

Results
Global T-cell prolymphocytic leukemia-specific microRNA (miR) deregulations highlight differential expression of miR-200c and miR-141 clusters
In order to investigate the spectrum of cellular miR expressed in T-PLL, small-RNA sequencing was performed of peripheral blood (PB)-isolated tumor cells from 46 T-PLL patients and of pan-T cells of PB from six healthy donors. TPLL patient characteristics are presented in Table 1 and sample purities in the Online Supplementary Figure S1. As T-PLL cases show a spectrum of (often nonconventional) memory T-cell phenotypes and of small naïve subsets,5 we chose age-matched CD3+ pan-T cells as controls (reflecting a representative mix of populations) in these global profiling analyses.
In total, we identified 2,094 miR, of which 37 miR displayed a differential expression in T-PLL versus healthy donor T cells (q<0.05, Online Supplementary Table S1). Of these, 14 miR sequences were upregulated (0.7% of all identified miR) and 23 were downregulated in T-PLL (1.1%, Figure 1A). While miR-6724-5p (fold change [fc]=0.18, q<0.0001) and miR-206 (fc=0.04, q<0.0001) showed the strongest downregulation, miR-5699-3p (fc=122, q=0.02), miR-200c-3p (fc=38.2, q=0.005), and miR-141-3p (fc=43.2, q=0.005) were the most upregulated. Considering all T-PLL cases, miR-141-3p and miR-21-5p showed the highest absolute abundance while miR-206, miR-651-3p and miR- 6774-5p displayed the lowest absolute expression (Online Supplementary Figure S2). Among the 37 deregulated miR, miR-6724-5p was annotated four times due to its expression from four different genomic loci. The following analyses are, therefore, based on 34 miR, containing a sum expression for miR-6724-5p. In order to validate our results from small-RNA sequencing, we performed quantitative real-time polymerase chain reaction (qRT-PCR) analyses of three highly deregulated miR (miR-223-3p, miR-200c-3p, miR-141-3p, Online Supplementary Figure S3A to C) in eight T-PLL and four healthy donor T-cell controls. A strong correlation between the results from small-RNA sequencing and qRT-PCR (r2=0.84, P<0.0001, Pearson, Online Supplementary Figure S3D) confirmed differential (over)expression of these miR in T-PLL and underlines the robustness of the sequencing data. Using a previously published independent data set from single nucleotide polymorphism (SNP) arrays of 83 T-PLL1 (overlap of 23 cases with the cohort presented here), we identified only small fractions of cases to carry genomic losses of significantly downregulated miR: miR-140-3p, miR-196b-5p (both CN<1.5 in 4.82% of cases), miR-339-3p, and miR-589-5p (both CN<1.5 in 8.43%, Online Supplementary Table S2).
Figure 1.

Global microRNA-deregulations highlight differential expression of miR-141 and miR-200c clusters in T-cell prolymphocytic leukemia. (A) MicroRNA (miR) profiles were evaluated using small-RNA sequencing of primary human peripheral blood-derived T-cell prolymphocytic leukemia (T-PLL) cells (n=46 cases) and CD3+ healthy donor-derived pan-T cell controls (n=6 donors). Volcano plot showing fold changes (fc) and false discovery rates (FDR) of all identified miR (n=2,094). Differentially expressed miR (n=37, Online Supplementary Table S1) are highlighted in blue (downregulation) and red (upregulation). Relative proportions are based on all identified miR. (B) Principle component analysis (PCA) based on miR differentially expressed comparing T-PLL cases (n=46) to healthy donor controls (n=6). Separate clustering of cases and controls indicates global differences in miR expression profiles. (C) Heatmaps of differentially expressed miR (n=34; FDR<0.05) in T-PLL samples vs. CD3+ pan-T cells of healthy donors. Left two-column heatmap: mean counts per million (CPM) values compared between healthy donor controls (n=6) and T-PLL (n=46; red=higher expression; blue=lower expression). Right heatmap: colors represent z-scores of respective CPM values calculated for each miR (red=higher z-score; blue=lower z-score). Control samples with slightly lower purities (77% and 85%) and T-PLL cases presenting a rather unique transcriptome, which clusters closer to control T cells (Online Supplementary Figure S4C), are indicated by asterixis (brown: T-PLL cases; green: control cases).
Unsupervised clustering by principal component analysis (PCA) based on miR differentially expressed in T-PLL versus healthy donor T cells indicated a homogeneity across T-PLL samples and confirmed the global differences in the miR profiles between T-PLL and healthy controls (Figure 1B). Interestingly, unsupervised hierarchical clustering analysis of miR expression revealed two clusters of T-PLL cases, which were distinguished by expression of miR-200c and miR-141 family members (Figure 1C): 23 T-PLL cases showed low miR-141/-200c expression as compared to CD3+ pan-T cell controls whereas 23 T-PLL samples had higher than normal T-cell expression of miR-141 and -200c. Besides higher serum lactate dehydrogenase (LDH) levels (P=0.03, Mann-Whitney-Wilcoxon test [MWW]) and a lower incidence of TP53 deletions (by fluorescence in situ hybridisation [FISH], P=0.02, Fisher’s exact test) in the miR- 141/-200c high-expressing cohort, we did not find other differences between these two subsets (Online Supplementary Table S3). Comparing the transcriptomes of cases allocated to these two separate clusters, we identified 356 genes to be differentially expressed (Online Supplementary Table S4). In line with GSEA based on miR141/200c-correlated genes (see following analyses), we identified the HALLMARK pathways E2F TARGETS (normalized enrichment score [NES]=4.38, q<0.0001) and G2M CHECKPOINT (NES=2.68, q<0.0001) as significantly altered between the two clusters of T-PLL. Furthermore, global miR-ome profiles were not associated with distinct cellular immunophenotypes, e.g., neither with CD45RA/RO expression (i.e., “memory-like” vs. “naïve-like” T-PLL) nor with CD4/8 expression (Online Supplementary Figures S4A and B).
MicroRNA-profiles of T-cell prolymphocytic leukemia resemble those of T-cell receptor signaling-activated T cells and form regulatory networks around nodes of DNA damage response and prosurvival signaling
In order to align the miR-ome data with those of global transcriptome alterations, polyA-RNA sequencing was performed on PB-isolated tumor cells from 41 miR-characterized T-PLL patients and seven additional T-PLL patients as well as on CD3+ PB pan-T cells from six age-matched healthy donors. In total, we detected 948 protein-encoding mRNA to be differentially expressed (q<0.05, Online Supplementary Figure S5A; Online Supplementary Table S5). Using this set of deregulated genes, PCA corroborated homogeneity among T-PLL cases and a clear distinction to normal T-cell controls (Online Supplementary Figure S5B). In accordance with published data, TCL1A (fc=1843, q<0.0001) and CTLA4 (fc=0.06, q<0.0001) were among the most differentially expressed genes in T-PLL versus T-cell controls (Online Supplementary Figure S5C). In their transcriptome profiles two T-PLL clustered closer to control T cells than the bulk of cases. However, their miR expression signature (Figure 1C) and clinical presentation did not differ from the overall cohort.
GSEA (HALLMARK gene sets27) determined 34 gene sets as upregulated in T-PLL when compared to T-cell controls, of which 19 gene sets were significantly enriched at a FDR of <5%. Sixteen gene sets were downregulated in T-PLL (11 with FDR <0.05, Online Supplementary Table S6). The identified significantly altered pathways associated with cancer and/or immunology are presented in Figure 2A. These included several HALLMARK gene sets reflecting dysregulations in DNA damage response pathways (e.g., DNA REPAIR: NES=-2.18, q=0.005; E2F TAGETS: NES=2.05, q=0.01) and prosurvival signaling (e.g., INFLAMMATORY RESPONSE, NES=3.12, q<0.0001; TNFA SIGNALING VIA NFKB, NES=2.44, q=0.001), in line with previously published data.
As T-PLL cells generally display a mature, T-cell activated phenotype,1,11 we investigated whether T-PLL cell miRomes resemble those of TCR-activated healthy donorderived T cells. For that, PBMC (to avoid direct manipulation of T cells) of four healthy donors were cultured for 72 hours with and without stimulation by anti-CD3/CD28 crosslinking, followed by miR sequencing of CD3+-enriched cells. Sample purities and experimental controls are shown in the Online Supplementary Figure S6. We identified 56 miR which are differentially expressed in response to TCR activation (q<0.05, Online Supplementary Figure S7A; Online Supplementary Table S7). PCA indicated homogeneity within both groups (T-PLL and normal PBMC) as well as global differences between their TCR-induced miR profiles (Online Supplementary Figure S7B). We identified miR known to be affected by TCR activation (e.g., miR-150-5p)31 as well as previously unreported miR (e.g., miR-18a-5p; Online Supplementary Figure S7C). Integrative PCA based on differentially expressed miR in T-PLL versus healthy controls (Figure 1C) showed that the stimulated (over unstimulated) T cells clustered closer to T-PLL (Figure 2B). Fittingly, unsupervised clustering comparing TCR-stimulated T cells to unstimulated controls confirmed that the miR profiles of TPLL cells resemble the miR-ome of TCR-activated healthy donor-derived T cells (Figure 2C).
We next assessed implicated functional relationships by predicted mRNA targets for each deregulated miR in T-PLL. For that, we (i) ranked mRNA based on their degree of correlation with a specific miR and (ii) performed HALLMARK set GSEA on these ranked mRNA. Pathways reflecting dysregulations of DNA damage response and prosurvival signaling emerged as predominantly associated with the alterations of miR expression. Exemplary HALLMARK pathways are shown in Figure 3A, a full list of gene sets is displayed in Online Supplementary Figure S8. For example, we obtained highly significant NES for the E2F TARGET and the IL2 STAT5 SIGNALING HALLMARK gene sets for (i) the transcriptome of T-PLL as compared to the one of healthy controls (E2F TARGETS: NES=1.92, q=0.02; IL2 STAT5 SIGNALING: NES=-2.86, q<0.0001, Online Supplementary Table S6) and (ii) for most of the miR differentially expressed in T-PLL (Figure 3A).
Overall, there was a striking similarity of the miR profiles of T-PLL cells with those of TCR-activated T cells. By integrating T-PLL’s miR profiles with transciptome data via GSEA (based on differentially expressed mRNA and on mRNA ordered by their correlation to the respective miR), we uncovered prominent regulatory networks around DNA damage response and prosurvival pathways in T-PLL.
Figure 2.

Global alterations of T-cell prolymphocytic leukemia microRNA expression signatures/ transcriptome networks resemble those of T-cell receptor-activated T cells. (A) Gene set enrichment analysis (GSEA) of differentially expressed mRNA in T-cell prolymphocytic leukemia (T-PLL) using HALLMARK gene sets (n=948 genes, Online Supplementary Table S6; Online Supplementary Figure S5; total n=48 gene sets). Gene expression profiles were assessed using global mRNA sequencing of primary TPLL cells (n=48 cases) and healthy donor-derived CD3+ pan-T cells (n=6 donors). Color code represents assignment to dysregulation to either altered DNA damage response pathways (black) or prosurvival signaling (grey). Exemplary GSEA plots are presented (DNA REPAIR: normalized enrichment score [NES]=-2.18, q=0.008, E2F TARGETS: NES=2.05, q=0.01, INFLAMMATORY RESPONSE: NES=3.12, q<0.0001). (B and C) T-PLL miR-omes resembled those of activated healthy donor T cells: age-matched healthy donor-derived peripheral blood mononuclear cells (PBMC) were isolated via density gradient centrifugation. T-cell activation was achieved via antibody-mediated CD3/CD28 crosslinking. After 72 hours (hrs), CD3+ primary T cells were isolated by magnetic-activated cell sorting (MACS) (negative selection; see Method section for details; cell purities are given in Online Supplementary Figure S6A, see Online Supplementary Figure S6B and C for control experiments on stimulation). MiR profiles were generated using small-RNA sequencing. Unstimulated cultured controls clustered together with directly isolated control samples, indicating that there was a negligible cell culture effect on miR expression profiles. Color code: T-PLL in brown, controls in green colors (light green: CD3+ T cells cultured in vitro for 72 hrs. without stimulation; green: CD3+ T cells submitted to miR-ome sequencing directly after enrichment; dark-green: CD3+ T-cell cultures at 72 hrs. subsequent to T-cell receptor [TCR] activation). (B) PCA based on differentially expressed miR comparing T-PLL to healthy donor-derived T cells (n=37 miR, Online Supplementary Table S1). TCR-activated healthy donor-derived T cells clustered with T-PLL cases. (C) Heatmap and clustering based on differentially expressed miR comparing healthy donor-derived T cells: unstimulated controls versus TCR-activated condition (n=56 miR, FDR<0.05). Colors represent z-scores of CPM values calculated for each miR (blue=lower z-score; red=higher z-score).
Figure 3.

Functional affiliations of predicted microRNA-targets reflect associations with processes of altered DNA damage response and prosurvival signaling. (A) Gene set enrichment analysis (GSEA) heatmap based on microRNA (miR) associated mRNA. GSEA were conducted for all significantly deregulated miR (n=34 miR; Online Supplementary Table S1) using ranked correlation indices between mRNA and miR expression in 41 T-PLL and six healthy donor-derived T-cell samples. Exemplary HALLMARK pathways are displayed. Full figure is displayed in Online Supplementary Figure S8. Color code summarizes normalized enrichment scores (NES, blue=negative NES; red=positive NES). Statistical significance is summarized via asterisks (*P<0.05; **P<0.01 ; ***P<0.001 ; Kolmogorov-Smirnov-test). (B) Differential expression of miR-223-3p as analyzed by small-RNA sequencing shows significant upregulation in T-PLL (n=46) over normal T-cell controls (n=6); (fc=9.85; P=0.0002). (C) Exemplary GSEA plots of miR-223-3p correlated mRNA: P53 PATHWAY: NES=2.59, q<0.0001, G2M CHECKPOINT: NES=-2.34, q=0.003). (D) Predicted targets (by seed sequences, see Methods section for details) that correlated negatively with miR-223-3p expression in all analyzed cases and controls represent regulatory networks involved in DNA damage response and prosurvival signaling. Font color represents differential expression of mRNA comparing T-PLL cells (n=48 cases) and healthy donor-derived CD3+ pan-T cells (n=6 donors; for description of global mRNA sequencing results refer to Online Supplementary Figure S5 and Online Supplementary Table S5, blue= lower expression; red= higher expression). Color of highlighted boxes represents assignment of genes to functional groups of DNA damage response pathways (black) and prosurvival signaling (grey). (E) Groups of low and high miR-233-3p expression were assigned by results of small-RNA sequencing via comparison of the lower versus upper tertile of cases. Primary T-PLL cases were evaluated for CD38 and CD69 surface expression using flow cytometry (see Online Supplementary Table S8 for the comprehensive dataset). T-PLL with high miR-223-3p expression levels presented with a more activated T-cell phenotype (median CD38 expression: 65.5% vs. 3.6%, P=0.006; median CD69 expression: 8.5% vs. 1.0%, P=0.1; Mann–Whitney–Wilcoxon [MWW] test).
Increased miR-223-3p expression is linked to activated T-cell phenotypes and associates with signatures of altered DNA damage response and cell cycle deregulation
We next focused on phenotypic and clinical associations of miR (i) differentially expressed in our cohort and (ii) already linked to B- and / or T-cell leukemogenesis. In our sequencing analysis of small-RNA, miR-223-3p was significantly upregulated in T-PLL over CD3+ pan-T cells from age-matched healthy donors (fc=9.85, P=0.0002, Figure 3B). GSEA based on mRNA ranked by their correlation to miR- 223-3p expression revealed an association of miR-223-3p with signatures of altered DNA damage responses (e.g., P53 PATHWAY: NES=2.59, q<0.0001) and deregulated cell-cycle mediators (e.g., G2M CHECKPOINT: NES=-2.34, q=0.003, Figure 3C). We further evaluated potential miR-223-3p target mRNA, by combining target prediction databases and miR-ome – transcriptome correlations. Defining criteria of mRNA as putative miR targets in T-PLL are outlined in the Methods section. In total, we identified eight putative targets of miR-223-3p in T-PLL (Figure 3D). Notably, FOXO1 (rho=-0.41, P=0.005) was identified as one of them and was significantly downregulated in T-PLL (n=32 of 48 cases with fc<0.5 compared to normal T-cell controls). Tumor suppressive FOXO1 is a prominent regulator of redox balances and DNA insult-mediated cell death.32
When T-PLL cases were divided into three subgroups based on miR-223-3p expression (high, medium, low), we found that miR-223-3p expression correlated significantly with surface expression levels of the TCR activation markers CD38 (mean surface expression: 65.5% vs. 3.6%, P=0.006, MWW) and CD69 (mean 8.5% vs. 1.0%, P=0.1, MWW, Figure 3E, Online Supplementary Table S8 with summary of clinical data).
Increased levels of miR-200c and miR-141 species are associated with deregulated cell cycle molecules, activated phenotypes, and more aggressive presentations
Another miR family, miR-200c/-141, was significantly upregulated in a subset of 23 T-PLL cases (Figures 4A and 1C). Upregulation for miR-141-3p was 43.2-fold (P<0.0001), for miR-141-5p 29.0-fold (P=0.0001), for miR- 200c-3p 38.2-fold (P<0.0001), and for miR-200c-5p 56.6- fold (P=0.003) over all cases. MiR-141-3p showed the highest absolute CPM values among all deregulated miR in the entire cohort of T-PLL (mean CPMmiR-141-3p =26,561; mean CPM of all significantly deregulated miR in T-PLL =1,440, fc=18.4; Online Supplementary Figure S2). GSEA based on mRNA ranked by their correlation to miR-200c/-141 family members revealed significant enrichments of the HALLMARK E2F TARGET (NES=3.64, q<0.0001) and HALLMARK G2M CHECKPOINT gene sets (NES=3.05, q<0.0001, both based on miR-141-3p correlated mRNA, Figure 4B). Additionally, we identified 93 mRNA as potential targets of miR-200c/-141 (Figure 4C) in T-PLL. While nine mRNA showed an overlap between miR-141-3p and miR-200c-3p target mRNA, we found only a small set of putative targets which were shared between either miR- 141-3p or miR-200c-3p and miR-141-5p. Exemplarily, KAT2B, a known tumor suppressor affecting DNA damage and cell cycle regulation,33 emerged as a potential target of miR-200c-3p (rho=-0.41, P=0.005, Spearman). Surface expression of the T-cell activation marker CD40L was elevated in cases with high miR-141-3p (mean 16.5% vs. 0.05%, P=0.03, MWW), high miR-200c-3p (mean 16.5% vs. 0.05%, P=0.03, MWW), and high miR-200c-5p expression (mean 20.0% vs. 0.0%, P=0.009, MWW, Figure 4D). Serum LDH levels at the time of sample correlated with increased miR-141-3p (mean 898 U/L vs. 509 U/l, P=0.03, MWW, Figure 4E) and elevated miR-200c-3p expression (mean 917 U/L vs. 509 U/L, P=0.02, MWW, Online Supplementary Table S9 with summary of clinical data).
Reduced miR-21 expression is linked to features of advanced or aggressive disease
The small-RNA sequencing analysis also revealed a 3.7- fold reduction of miR-21-3p expression (fc=0.27, P<0.0001) and a 3.2-fold reduction of miR-21-5p expression (fc=0.31, P<0.0001) in T-PLL (Online Supplementary Figure S9A). Interestingly, absolute expression (CPM) values of miR-21-5p were the second most altered among all deregulated miR in T-PLL (mean CPM of all significantly deregulated miR in T-PLL of 1440 vs. mean CPMmiR-21-5p of 15,526, fc=10.8, Online Supplementary Figure S2), suggesting a highly T-PLL-specific loss of expression. GSEA of miR-21-associated mRNA implicated relevance of this miR in apoptosis and cell cycle regulation, as gene sets like HALLMARK P53 PATHWAY and G2M CHECKPOINT were significantly deregulated (NES of APOPTOSIS PATHWAY considering miR-21-5p-associated mRNA=3.89, q<0.0001; NES of G2M CHECKPOINT considering miR-21-3p-associated mRNA=2.39, q=0.001, Online Supplementary Figure S9B). Furthermore, we assessed potential target mRNA of miR-21-3p and miR-21-5p as described above (n=42, e.g., MAP3K1 as a putative target of both miR-21-3p and miR-21-5p, Online Supplementary Figure S9C). In contrast to the current concept of miR-21 being a potent suppressor of cell cycle arrest and apoptosis induction, we did not find negative correlations with mRNA mediating these published effects (e.g., PDCD4: rho=0.04, P=0.79; BTG2: rho=0.28, P=0.05; correlations based on miR-21-5p expression, Spearman).34 Dichotomized by mean miR-21-5p expression, T-PLL with low miR-21-5p levels revealed higher white blood cell (WBC) counts (mean 154 G/L vs. 90.0 G/l, P=0.02, MWW) and lower platelet counts (mean 110 G/L vs. 153 G/L, P=0.03, MWW, Online Supplementary Figure S9D) at the time of sampling, indicating a more active growth behavior of T-PLL with low miR- 21 expression. Fittingly, serum levels of LDH (mean 933 U/L vs. 522 U/L, P=0.02, MWW, Online Supplementary Figure S9E) were elevated in patients with low cellular miR-21-5p expression (Online Supplementary Table S10 with summary of clinical data).
Reduced expression of miR-29 clusters is associated with alterations of survival signaling and cell cycle regulators reflected in features of a more active disease
As analyzed by small-RNA sequencing, the miR-29 family members miR-29a-3p (fc=0.29, P<0.0001), miR-29b-1-5p (fc=0.47, P=0.001), and miR-29c-3p (fc=0.29, P<0.0001) showed a homogenous downregulation in T-PLL over healthy donor T cells (Online Supplementary Figure S10A). GSEA of associated mRNA showed an enrichment of genes of the HALLMARK E2F TARGET (NES=-3.52, q<0.0001) and of the TNFA SIGNALING VIA NFKB gene sets (NES=- 2.08, q=0.01, both miR-29a-3p-correlated mRNA) in low miR-29 expressing T-PLL (Online Supplementary Figure S10B). Furthermore, we identified 79 putative-target mRNA of the miR-29a-3p / miR-29b-1-5p / miR-29c-3p cluster in T-PLL by the above described strategy (Online Supplementary Figure S10C). Indicating an association of reduced miR-29 species with aberrant survival signaling, surface TCR activation markers were elevated in T-PLL with low miR-29a-3p (Online Supplementary Figure S10D), namely CD38 (mean expression 43.1% vs. 12.9%, P=0.02, MWW) and CD69 (mean 27.9% vs. 0.89%, P=0.02, MWW). A more active disease state as indicated by lower platelet counts (mean 110 G/L vs. 186 G/L, P=0.15, MWW) and higher LDH serum levels (mean 1,850 U/L vs. 708 U/l, P=0.06, MWW) at sampling was linked to low miR-29b-1-5p expression (Online Supplementary Figure S10E and F). Lower miR-29c expression tended to be associated with a higher incidence of effusions (six of 12 over one of ten with high miR-29c-3p-expression; P=0.07, Fisher’s exact test, Online Supplementary Figure S11A). In addition, genomic ATM deletions were more frequent in cases with low miR- 29b-1-5p expression (nine of 11 vs. one of 11 with high miR-29b-1-5p; P=0.002, Fisher’s exact test, Online Supplementary Figure S11B, Online Supplementary Table S11 with summary of clinical data).
Figure 4.

Increased expression of miR-200c and miR-141 clusters is associated with deregulation of cell cycle regulators reflected in more activated phenotypes and more aggressive disease course. (A) Differential expression of miR-141 and miR-200 family members as analyzed by small-RNA sequencing showed significant upregulation of miR-141-3p (fold change [fc]=43.2; P<0.0001), miR-141-5p (fc=29.0; P=0.0001), miR-200c-3p (fc=38.2; P<0.0001) and miR-200c-5p (fc=56.6; P=0.003, n=46 T-cell prolymphocytic leukemia [T-PLL], n=6 controls). (B) Exemplary gene set enrichment analysis (GSEA) plots of miR-200/-miR-141 correlated mRNA: E2F TARGETS: NES=3.64, q<0.0001, G2M CHECKPOINT: NES=3.05, q<0.0001 (both based on miR-141-3p correlated mRNA). (C) Predicted targets (by seed sequences, see Methods section for details) correlating negatively with miR-141/-miR-200 expression in all analyzed cases and controls showed regulatory networks involved in DNA damage response and prosurvival signaling. Font color represents differential expression of mRNA comparing T-PLL cells (n=48 cases) and healthy donor-derived CD3+ pan-T cells (n=6 donors; for description of global mRNA sequencing results refer to Online Supplementary Figure S5 and Online Supplementary Table S5, blue= lower expression; red= higher expression). Color of highlighting boxes represents assignment of genes to functional groups of DNA damage response pathways (black) and prosurvival signaling (grey). (D) Groups of low and high miR-141/-200 expression were assigned by results of small-RNA sequencing: after division into three tertiles, cases of the lower were compared to those of the upper tertile. Cases were evaluated for CD40L surface expression using flow cytometry. TPLL with higher miR-141-5p, miR-200c-3p, and miR-200c-5p expression presented with a more activated T-cell phenotype (median CD40L expression: 16.5% vs. 0.05%, P=0.03; 16.5% vs. 0.05%, P=0.03, 20.0% vs. 0.0%, P=0.009; MWW). (E) Higher serum LDH levels (see Online Supplementary Table S9 for a summary of clinical data) were associated with high expression of miR-141-3p and miR-200c-3p (median 898 U/L vs. 509 U/L; P=0.03; median 917 G/L vs. 509 G/L; P=0.02; MWW). Groups were divided into three tertiles and the lower was compared against the upper tertile.
A combinatorial miR-based overall survival score for T-cell prolymphocytic leukemia
Based on the observed correlation of miR expression with clinical parameters, we aimed to establish a prognostic score that stratifies T-PLL patients according to miR expression levels. In order to identify best candidates in an unbiased fashion, we first associated miR expression levels with OS for all miR detected in at least 80% of T-PLL samples (n>36 cases) and compared T-PLL patients with highest expression levels (upper tertile) to those with lowest expression of the respective miR (lower tertile; Online Supplementary Table S12 with summary of clinical data). In this analysis, miR-98-3p (median OS in high vs. low expression: 16.3 months vs. 29.4 months, P=0.0008, log-rank, Figure 5A), miR-200a-3p (52.7 months vs. 19.1 months, P=0.001, log-rank, Figure 5B), miR- 223-3p (14.9 months vs. 26.0 months, P=0.001, log-rank, Figure 5C), and miR-424-5p (14.4 months vs. 26.0 months, P=0.0007, log-rank, Figure 5D) were most significantly correlated with OS. We subjected a training model composed of these four miR (miR-98-3p, miR-200a-3p, miR-223-3p, miR-424-5p) and of selected factors that had shown to be of prognostic relevance in T-PLL (e.g., WBC counts, TCL1A mRNA level)1,11,30 to parameter shaving by recursive partitioning. This algorithm identified optimum individual thresholds stratifying OS in the randomly created 22-case training set. Using these cutoffs, three miR (200a-3p, miR- 223-3p, miR-424-5p) and TCL1A expression remained as most significant discriminators for OS. Multiple combinatorial scores of these four parameters were built and for these scores optimum thresholds for discrimination of OS were calculated (recursive partitioning, see Methods). Best separation was obtained using a miR-exclusive 3-tier score: miR- 200a-3p fc<2.21, miR-223-3p fc≥9.8, miR-424-5p fc≥0.91; relative to healthy donor T cells) with a cutoff of ≥2 sum points (Table 2). Finally, we verified the miROS-T-PLL score in the 22-case validation set (P=0.0004, log-rank) and in the total cohort of 44 T-PLL (median OS high vs. low miROS-TPLL: 14.4 months vs. 29.4 months, P<0.0001, log-rank, Figure 5E).
In order to identify variables underlying (as potential confounders) the miR-based prognostic separation, we associated the expression of miR, which we used for the score, as well as the miROS-T-PLL score itself, with genomic, mRNA expression, immunophenotypic, and clinical data (Online Supplementary Tables S13 and S14). We did not detect significant differences in the distribution of these parameters between the groups determined by expression of the three miR or by the miROS-T-PLL score, further validating the newly established score.
Discussion
Here, we report a pilot analysis of cellular miR expression in a cohort of 46 T-PLL patients. We identified 34 miR to be significantly deregulated in comparison to PB-derived T cells from age-matched healthy donors. These miR included those which had already been reported as altered in Tcell acute lymphoblastic leukemia (T-ALL, e.g., miR-223-3p)35 and in CTCL (e.g., miR-29 and miR-200).17,23 They also contained miR that had not been described in the neoplastic context (e.g., miR-10395-5p). The global profiles of deregulated miR in T-PLL showed a rather uniform pattern across the analyzed cases. Together with the integrated information from transcriptome sequencing, this set of data allows for the first time insights into miR-based regulatory networks in T-PLL.
It is important to mention, that four of the 34 differentially expressed miR presented with low CPM values (<1), either being unspecific background in the sequencing technology or representing biological relevant miR expressed at low levels. In addition, for two of the small-RNA identified to be differentially expressed in T-PLL (miR-6724-5p, miR- 5699-3p), miR-base36 assigned questionable confidence in their annotation, although their expression was previously reported in other entities (e.g., bladder cancer).37
In line with T-PLL’s phenotype of augmented TCR activation, 1,5,11 our comparative profiling revealed a resemblance of T-PLL’s miR-ome to the one of TCR-activated healthy donor-derived T cells. A specific remodeling of the miR repertoire upon TCR activation had been shown,38 however, analysis of full spectrum miR expression by small RNA sequencing upon TCR activation in healthy donor-derived pan-T cells had not been reported before. We identified here previously unknown miR (e.g., upregulation of miR- 18a-5p) to be altered upon TCR activation in addition to those that had been described (miR-17-5p or miR-150-5p).31,39 We conclude that constitutive TCR activation shapes the characteristic miR-ome of T-PLL cells.
As hallmarks of T-PLL’s miR-ome, we identified miR-223-3p, miR-21, the miR-29 family, and the miR-200c/-141 cluster as significantly deregulated. These miR have previously emerged as either onco miR or tumor-suppressive miR in other T- or B-cell malignancies.23,40–43 We further identified putative target signatures, potentially mediating the postulated effects of prosurvival signaling and aberrant DNA damage responses (e.g., downregulation of FOXO1 upon miR-223 upregulation). Limiting, the postulated target genes are, although predicted through multiple robust algorithms, based on associations without proven biological effects, which has to be addressed in future experiments. Deregulation of these hallmark miR in T-PLL was further associated with either a pronounced cellular activation phenotype or more aggressive clinical presentations.
MiR-21 stood out as one of the most abundant miR in TPLL. In contrast to the current concept of miR-21 being an onco miR,44 we detected significantly downregulated miR- 21 levels in T-PLL samples as compared to healthy donor T cells. Our integrative correlations further revealed an association of low miR-21 expression with more aggressive disease presentations. We identified SKP2 and MAP3K1 as predicted targets of miR-21 in T-PLL, potentially mediating these features. Notably, we did not find significant negative correlations of miR-21 with those mRNA that were previously described to mediate the effect of this miR as a potent suppressor of cell cycle inhibition and apoptosis.34 This indicates a T-cell specific and context-dependent function of miR-21.
Figure 5.

miR-based overall survival score for T-cell prolymphocytic leukemia (miROS-T-PLL) stratifies patients based on miR-200a-3p, miR-223-3p, and miR-424-5p expression. (A to D) Associations of microRNA (miR) expression with overall survival (OS) from diagnosis (see Online Supplementary Table S12 for the comprehensive dataset). (A) miR-98-3p (median OS high vs. low expression: 16.3 months vs. 29.4 months, P=0.0008, log-rank test); (B) miR-200a-3p (median OS high vs. low expression: 52.7 months vs. 19.1 months, P=0.001, log-rank test); (C) miR-223-3p (median OS high vs. low expression: 14.9 months vs. 26.0 months, P=0.001, log-rank test), and (D) miR-424-5p (median OS high vs. low expression: 14.4 months vs. 26 months, P=0.0007, log-rank test) were most significantly associated with OS, when comparing the tertile of T-PLL patients with the highest expression to the tertile with the lowest expression of the respective miR. (E) Analysis via training (n=22) and validation data sets (n=22): optimum thresholds were calculated by recursive partitioning for all possible combinations of miR-200a-3p, miR-223-3p, and miR-424-5p expression levels, adding one point to the total score if the respective expression cutoff was passed. Best results were obtained using the following thresholds: miR-200a-3b: fold change (fc)<2.21, miR-223-3p: fc ≥9.8, miR-424-5p: fc≥0.91 (fc relative to CD3+ pan-T cells derived from healthy donors) and a cutoff of ≥2 points. A significant OS association was observed in the validation set (P=0.0004, log-rank test) as well as (E) in the total cohort of 44 T-PLL patients (median OS high vs. low expression: 14.4 months vs. 29.4 months, P<0.0001, log-rank test).
Our computational analyses of mRNA targeted by deregulated miR in T-PLL suggest a strong impact of altered miR clusters on activation, death resistance, and aberrant DNA damage responses. Abnormal activity of these pathways, triggered by TCL1A overexpression and damaging ATM aberrations, has emerged as a hallmark of T-PLL pathobiolo gy.1,11 We propose that protumorigenic miR networks in their function as posttranscriptional regulators may further enhance the effects of these key genomic lesions, contributing substantially to the pathogenesis of TPLL. Similar cooperative miR-mRNA networks were postulated for T-ALL and CLL.35,45 The causes of the miR deregulations we observed here, remain unknown and are, besides TCR activation, likely multifactorial. As T-PLL is characterized by a strong genomic instability and high burdens of reactive oxygen species,1,46 mutations or copy number alterations of miR-encoding genes provide possible explanations, in addition to epigenetic mechanisms. Notably, incidences of genomic losses of the downregulated miR-140-3p, miR-196b-5p, miR-339-3p, and miR-589-5p were in the order of 4-9% of T-PLL cases in our cohort.
Table 2.
Prognostic score (miROS-T-PLL) including miR-200a-3p, miR- 223-3p, and miR-424-5p expression levels.
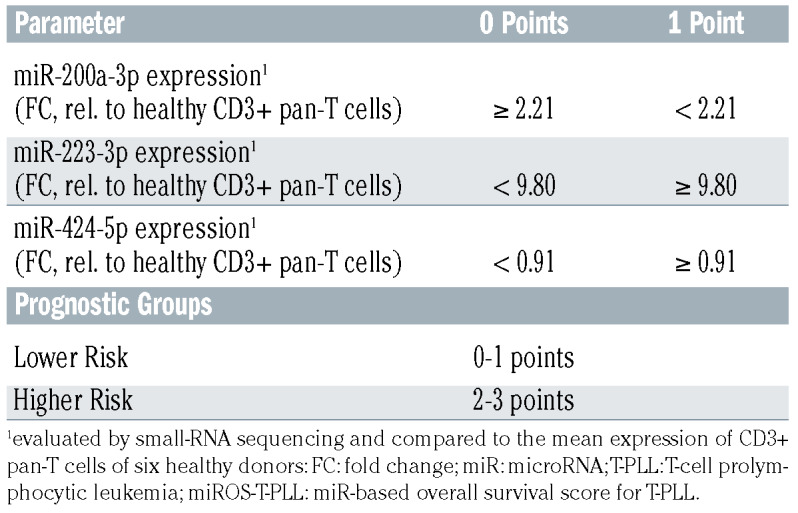
In summary, we identified a T-PLL-specific miR-ome, with 34 differentially deregulated miR, that appears instructed by TCR activation. By integrating altered miR expression with the information derived from transcriptome analyses, we postulate that the miR-ome of T-PLL shapes (dys)regulated networks towards apoptotic resistance, cell cycle abrogation, and defective DNA damage repair (Figure 6). Highlighting the pathobiological impact of the discovered miR deregulations, we developed the first clinical survival score that predicts T-PLL patients’ outcomes, based on expression levels of miR-200a-3p, miR- 223-3p, and miR-424-5p.
Figure 6.

Graphical summary of postulated microRNA/mRNA -based deregulated networks in T-cell prolymphocytic leukemia. T-cell activity shaped microRNA expression signatures (miR-omes)/ transcriptome networks are displayed as identified by our combinatorial approach of small-RNA and transcriptome sequencing analyses in 41 clinically well characterized T-cell prolymphocytic leukemia (T-PLL) cases. MiR differentially expressed in T-PLL and mRNA associated with these miR (P<0.05) are presented. The background color of miR and mRNA (dots) indicates the fold changes of differential expression as compared to age-matched healthy donor-derived CD3+ pan-T cells (blue=downregulation, red=upregulation). MiR-223-3p, the miR-21 family, the miR-29 family, and the miR-200c/141 family emerged as hallmarks of the T-PLL miR-ome, as they were (i) significantly deregulated among T-PLL cases, (ii) presented putative targets involved in oncogenic pathways of TPLL’s pathobiology, (iii) showed associations with prognostic parameters, and (iv) were already described in the leukemogenesis of other B- and T-cell malignancies. Deregulations of these four miR-families were associated with cooperative effects on DNA damage response pathways as well as on pro-proliferative and cell survival signaling (as revealed by gene set enrichment analysis based on correlated mRNA). Genes previously described as hallmarks of T-PLL (e.g., TCL1A, CTLA4, and MYC) were found within the network of the identified miR-associated mRNA.
Clinical management of T-PLL remains challenging and current studies mainly focus on the development of targeted treatments directed against anti-apoptotic factors like BLC247 or sustained prosurvival signaling mediated via JAK/STAT pathways.48,49 Our study suggests that developing a miR-directed targeting strategy could allow tackling a combination of those pathways and might therefore be a promising approach to successfully eradicate T-PLL cells. However, these concepts have to be tested in subsequent preclincal studies. Furthermore, the presented miROS-TPLL survival score might help in better discriminating rather indolent versus aggressive T-PLL disease phases at the time of diagnosis.
Supplementary Material
Funding Statement
Funding: This research was funded by the DFG Research Unit FOR1961 (Control-T; HE3553/4-2), the Köln Fortune program, and the Fritz Thyssen Foundation (10.15.2.034MN). This work was also funded by the EU Transcan-2 consortium ‘ERANET-PLL’ and by the ERAPerMed consortium ‘JAKSTAT-TARGET’. A.S. was supported by a scholarship of the German José Carreras Leukemia Foundation (DJCLS 03 F/2016). We gratefully acknowledge patients with their families for their invaluable contributions.
References
- 1.Schrader A, Crispatzu G, Oberbeck S, et al. Actionable perturbations of damage responses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat Commun. 2018;9(1):697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Staber PB, Herling M, Bellido M, et al. Consensus criteria for diagnosis, staging, and treatment response assessment of Tcell prolymphocytic leukemia. Blood. 2019;134(14):1132-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braun T, von Jan J, Wahnschaffe L, Herling M. Advances and perspectives in the treatment of T-PLL. Curr Hematol Malig Rep. 2020;15(2):113-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matutes E, Brito-Babapulle V, Swansbury J, et al. Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood. 1991;78(12):3269-3274. [PubMed] [Google Scholar]
- 5.Oberbeck S, Schrader A, Warner K, et al. Non-canonical effector functions of the Tmemory- like T-PLL cell are shaped by cooperative TCL1A and TCR signaling. Blood. 2020. Dec 10;136(24):2786-2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hopfinger G, Busch R, Pflug N, et al. Sequential chemoimmunotherapy of fludarabine, mitoxantrone, and cyclophosphamide induction followed by alemtuzumab consolidation is effective in T-cell prolymphocytic leukemia. Cancer. 2013;119(12):2258-2267. [DOI] [PubMed] [Google Scholar]
- 7.Cross M, Dearden C. B and T cell prolymphocytic leukaemia. Best Pract Res Clin Haematol. 2019;32(3):217-228. [DOI] [PubMed] [Google Scholar]
- 8.Dearden CE, Khot A, Else M, et al. Alemtuzumab therapy in T-cell prolymphocytic leukemia: comparing efficacy in a series treated intravenously and a study piloting the subcutaneous route. Blood. 2011;118(22):5799-802. [DOI] [PubMed] [Google Scholar]
- 9.Dearden C. How I treat prolymphocytic leukemia? Blood. 2012;120(3):538-551. [DOI] [PubMed] [Google Scholar]
- 10.Pflug N, Cramer P, Robrecht S, et al. New lessons learned in T-PLL: results from a prospective phase-II trial with fludarabine– mitoxantrone–cyclophosphamide–alemtuzumab induction followed by alemtuzumab maintenance. Leuk Lymphoma. 2019;60(3):649-657. [DOI] [PubMed] [Google Scholar]
- 11.Herling M, Patel KA, Teitell MA, et al. High TCL1 expression and intact T-cell receptor signaling define a hyperproliferative subset of T-cell prolymphocytic leukemia. Blood. 2008;111(1):328-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wahnschaffe L, Braun T, Timonen S, et al. JAK/STAT-activating genomic alterations are a hallmark of T-PLL. Cancers (Basel). 2019;11(12):1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellanger D, Jacquemin V, Chopin M, et al. Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia. 2014;28(2):417-419. [DOI] [PubMed] [Google Scholar]
- 14.Kiel MJ, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124(9):1460-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82-94. [DOI] [PubMed] [Google Scholar]
- 16.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16(7): 421-433. [DOI] [PubMed] [Google Scholar]
- 17.Kohnken R, Wen J, Mundy-Bosse B, et al. Diminished microRNA-29b level is associated with BRD4-mediated activation of oncogenes in cutaneous T-cell lymphoma. Blood. 2018;131(7):771-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zenz T, Mohr J, Eldering E, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113(16):3801-3808. [DOI] [PubMed] [Google Scholar]
- 19.Wallaert A, Van Loocke W, Hernandez L, Taghon T, Speleman F, Van Vlierberghe P. Comprehensive miRNA expression profiling in human T-cell acute lymphoblastic leukemia by small RNA-sequencing. Sci Rep. 2017;7(1):7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calin GA, Ferracin M, Cimmino A, et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353(17): 1793-1801. [DOI] [PubMed] [Google Scholar]
- 21.Bresin A, Callegari E, D’Abundo L, et al. miR-181b as a therapeutic agent for chronic lymphocytic leukemia in the Eμ-TCL1 mouse model. Oncotarget. 2015;6(23): 19807-19818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pekarsky Y, Santanam U, Cimmino A, et al. Tcl1 Expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66(24):11590-11593. [DOI] [PubMed] [Google Scholar]
- 23.Shen X, Wang B, Li K, et al. microRNA signatures in diagnosis and prognosis of cutaneous T cell lymphoma. J Invest Dermatol. 2018;138(9):202. [DOI] [PubMed] [Google Scholar]
- 24.Wu SJ, Chen J, Wu B, Wang YJ, Guo KY. MicroRNA-150 enhances radiosensitivity by inhibiting the AKT pathway in NK/T cell lymphoma. J Exp Clin Cancer Res. 2018;37(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye Z, Jin H, Qian Q. Argonaute 2: a novel rising star in cancer research. J Cancer. 2015;6(9):877-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. A systematic approach to diagnosis of mature T-cell leukemias reveals heterogeneity among WHO categories. Blood. 2004;104(2):328-335. [DOI] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge- based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545-15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database hallmark gene set collection. Cell Syst. 2015;1(6): 417-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ru Y, Kechris KJ, Tabakoff B, et al. The multiMiR R package and database: integration of microRNA–target interactions along with their disease and drug associations. Nucleic Acids Res. 2014;42(17):e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jain P, Aoki E, Keating M, et al. Characteristics, outcomes, prognostic factors and treatment of patients with T-cell prolymphocytic leukemia (T-PLL). Ann Oncol. 2017;28(7):1554-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodríguez-Galán A, Fernández-Messina L, Sánchez-Madrid F. Control of immunoregulatory molecules by miRNAs in T cell activation. Front Immunol. 2018;9:2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dansen TB, Burgering BMT. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008;18(9):421-429. [DOI] [PubMed] [Google Scholar]
- 33.Zhu C, Qin Y-R, Xie D, et al. Characterization of tumor suppressive function of P300/CBP-associated factor at frequently deleted region 3p24 in esophageal squamous cell carcinoma. Oncogene. 2009;28(31):2821-2828. [DOI] [PubMed] [Google Scholar]
- 34.Buscaglia LEB, Li Y. Apoptosis and the target genes of microRNA-21. Chin J Cancer. 2011;30(6):371-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mavrakis KJ, Van Der Meulen J, Wolfe AL, et al. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat Genet. 2011;43(7):673-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013;42(D1):D68-D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Usuba W, Urabe F, Yamamoto Y, et al. Circulating miRNA panels for specific and early detection in bladder cancer. Cancer Sci. 2019;110(1):408-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bronevetsky Y, Villarino AV, Eisley CJ, et al. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. J Exp Med. 2013;210(2):417-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sousa IG, do Almo MM, Simi KCR, et al. MicroRNA expression profiles in human CD3(+) T cells following stimulation with anti-human CD3 antibodies. BMC Res Notes. 2017;10(1):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pomari E, Lovisa F, Carraro E, et al. Clinical impact of miR-223 expression in pediatric T-Cell lymphoblastic lymphoma. Oncotarget. 2017;8(64):107886-107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwon JJ, Factora TD, Dey S, Kota J. A systematic review of miR-29 in cancer. Mol Ther Oncolytics. 2018;12:173-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humphries B, Yang C. The microRNA-200 family: small molecules with novel roles in cancer development, progression and therapy. Oncotarget. 2015;6(9):6472-6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garzon R, Heaphy CEA, Havelange V, et al. MicroRNA 29b functions in acute myeloid leukemia. Blood. 2009;114(26):5331-5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng YH, Tsao CJ. Emerging role of microRNA-21 in cancer (Review). Biomed Rep. 2016;5(4):395-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Qin Y, Zhang H. Identification of key miRNA-gene pairs in chronic lymphocytic leukemia through integrated analysis of mRNA and miRNA microarray. Oncol Lett. 2018;15(1):361-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prinz C, Vasyutina E, Lohmann G, et al. Organometallic nucleosides induce nonclassical leukemic cell death that is mitochondrial- ROS dependent and facilitated by TCL1-oncogene burden. Mol Cancer. 2015;14:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boidol B, Kornauth C, van der Kouwe E, et al. First-in-human response of BCL-2 inhibitor venetoclax in T-cell prolymphocytic leukemia. Blood. 2017;130(23):2499-2503. [DOI] [PubMed] [Google Scholar]
- 48.Gomez-Arteaga A, Margolskee E, Wei MT, van Besien K, Inghirami G, Horwitz S. Combined use of tofacitinib (pan-JAK inhibitor) and ruxolitinib (a JAK1/2 inhibitor) for refractory T-cell prolymphocytic leukemia (T-PLL) with a JAK3 mutation. Leuk Lymphoma. 2019;60(7):1626-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bailey NG, Elenitoba-Johnson KSJ. What is new in mature T-cell leukemias? Semin Diagn Pathol. 2020;37(1):72-78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.