

Abstract
Introduction
Analyses of off‐label use of acetylcholinesterase inhibitors (AChEIs) in mild cognitive impairment (MCI) has produced mixed results. Post hoc analyses of observational cohorts, such as the Alzheimer's Disease Neuroimaging Initiative (ADNI), have reported deleterious effects in AChEI‐treated subjects (AChEI+). Here, we used neuroimaging biomarkers to determine whether AChEI+ subjects had a greater rate of neurodegeneration than untreated (AChEI–) subjects while accounting for baseline differences.
Methods
We selected 121 ADNI MCI AChEI+ subjects and 151 AChEI– subjects with a magnetic resonance imaging (MRI) scan; 82 AChEI+ and 110 AChEI– also had a fluorodeoxyglucose (FDG) scan. A subset (83 AChEI+ and 98 AChEI–) had cerebrospinal fluid (CSF) or amyloid positron emission tomography (PET) assessment for amyloid positivity. Linear regression models were used to compare the effect of treatment on changes in Mini‐Mental State Examination and Clinical Dementia Rating‐Sum of Boxes scores. We used standard regression in SPM (for baseline) and the SPM toolbox sandwich estimator, SwE (for longitudinal) for comparisons of AChEI+ and AChEI– FDG PET and MRI data.
Results
At baseline, the AChEI+ group had significantly reduced cortical gray matter density (GMD) and more hypometabolism than AChEI– subjects. The greater rate of atrophy and hypometabolic changes over time in AChEI+ compared to AChEI– subjects did not survive correction for baseline differences. AChEI+ participants were more likely to be amyloid‐positive and have lower GMD and FDG standardized uptake value ratio than AChEI– at baseline. AChEI+ subjects showed greater atrophy over time, which remained significant after controlling for amyloid status.
Discussion
Our data suggest that the observed differences in rates of cognitive decline, atrophy, and hypometabolism are likely the result of significant baseline differences between the groups. Furthermore, the data indicate no treatment effect of AChEI (positive of negative), rather that physicians prescribe AChEI to subjects who present with more severe clinical impairment. This alone may account for the negative effect seen previously in the ADNI population of AChEI use among MCI subjects.
Keywords: acetylcholinesterase inhibitor, Alzheimer's Disease Neuroimaging Initiative, fluorodeoxyglucose, longitudinal, magnetic resonance imaging, mild cognitive impairment
1. INTRODUCTION
Alzheimer's disease (AD) affects as many as 10% of all Americans over the age of 65 and is the fifth‐leading cause of death for that age group. 1 Currently there are no cures or viable treatments that can reverse, halt, or even slow cognitive decline for AD patients, making drug discovery at the forefront of AD research.
Central to the research effort is understanding the exact cause of AD, which has entertained a variety of hypotheses through the years, the oldest of which is the “cholinergic hypothesis.” 2 Cholinergic deficits are commonly noted in AD along projections from the basal forebrain to neocortex and associated limbic structures. 2 , 3 , 4 , 5 , 6 While this deficit is now considered to be a consequence of AD rather than a cause, it was the first target for drug intervention. Increasing acetylcholine concentration at the synapses of cholinergic neurons is still the most commonly prescribed symptomatic treatment used by physicians through a class of drugs known as anticholinergics or acetylcholinesterase inhibitors (AChEIs).
Currently there are three Food and Drug Administration (FDA)‐approved commonly prescribed reversible AChEIs: donepezil, rivastigmine, and galantamine. 7 , 8 , 9 Although these inhibitors are only FDA approved for use in mild to moderate AD, physicians frequently prescribe AChEIs off‐label to subjects in the pre‐dementia stages, such as those with mild cognitive impairment (MCI), in hopes that they might relieve the cognitive symptoms or even slow progression. 10 , 11 , 12 To date, six randomized clinical trials to test the efficacy of AChEI use in MCI patients have been completed. 13 , 14 , 15 , 16 , 17 , 18 Two of these studies, Petersen et al. and Winblad et al., used conversion to dementia as the primary outcome measure and failed to find a significant treatment effect. 15 , 16 However, Petersen et al. found that donepezil slowed conversion rate through the first year of treatment. 15 Secondary cognitive outcome measures reflected this finding, with little to no decline through the first 18 months, but rates comparable to placebo from 18 to 36 months. 15
The use of cognitive outcomes as primary and secondary efficacy measures has been met with moderate success. Koontz et al. found improvements on two of six Cambridge Automated Neuropsychological Test Assessment Battery (CANTAB) measures, as well as on two secondary outcomes—California Verbal Learning Test (CVLT) and Functional Activities Questionnaire (FAQ)—in response to treatment with galantamine. 14 Salloway et al. failed to find a significant treatment effect of donepezil on their primary outcomes–New York University (NYU) Delayed Paragraph Recall Test and the Alzheimer's Disease Cooperative Study–Clinical Global Impression of Change for MCI (ADCS CGIC‐MCI) measure—but reported improvement on secondary measures of Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐Cog) and Patient Global Assessment (PGA). 13 Doody et al., using both ADAS‐Cog and Clinical Dementia Rating sum of boxes (CDR‐SB) as primary efficacy measures found that only ADAS‐Cog had a significant treatment effect over 48 weeks, while finding little to no change in their secondary measures of cognition, behavior and function. 17 In 2015, Dubois et al. 18 used a neuroimaging primary outcome measure and found that the annual percent change (APC) of hippocampal volume was significantly lower (i.e., less atrophy was observed) in patients treated with donepezil after 1 year. Their secondary measures of APC in left/right hippocampal volume, global cerebral volume, and ventricular volume showed a similar effect. 18
Post hoc and meta‐analyses have been used to further test efficacy and assess risk of adverse effects. From these, there was little to no evidence of an AChEI slowing of disease progression, 19 , 20 , 21 while only ADAS‐Cog showed the desired treatment effect on cognition. 21 Imaging, however, has shown more promising results. AChEI‐treated MCI patients showed a reduction in cortical thinning rates, 22 basal forebrain atrophy, 23 whole brain APC, 24 whole brain atrophy, 25 and even increased frontal cortex activation in functional magnetic resonance imaging (fMRI). 26 However, no treatment effect was seen by Jack et al. on APC of any MRI measure in a post hoc examination of the donepezil and vitamin E trial. 27
In contrast to the results from randomized controlled studies listed above, analyses of observational studies such as the Alzheimer's Disease Neuroimaging Initiative (ADNI), have actually reported a worsening of cognitive symptoms measured by Mini‐Mental State Examination (MMSE), CDR, and ADAS‐Cog. 28 , 29 Notably, in both studies, the treatment population was significantly more impaired at baseline in CDR and ADAS‐Cog measures, respectively. 28 , 29 This baseline discrepancy was not added as a covariate in either analysis, potentially confounding the interpretation of the results.
To clarify the impact of baseline factors on AChEI treatment in MCI, we analyzed baseline and longitudinal changes in MMSE, CDR‐SB, and neurodegeneration—brain atrophy and hypometabolism—in AChEI‐treated and ‐untreated MCI subjects from the ADNI.
2. METHODS
2.1. Subjects
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). ADNI was launched in 2003 as a public–private partnership, led by principal investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial MRI, positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessments can be combined to accurately measure and predict the progression of MCI and early AD. ADNI has undergone three complete funding cycles to date: ADNI 1, ADNI GO, and ADNI 2. ADNI 3 is ongoing. ADNI GO and ADNI 2 included 18F‐Florbetapir amyloid PET imaging.
The clinical and biomarker characteristics of the ADNI cohort have been previously published. 30 ADNI has enrolled clinically diagnosed cognitively normal (CN), amnestic MCI, and dementia (DEM) subjects with an amnestic presentation thought to be due to AD (probable AD). All diagnostic criteria can be found on ADNI's website under the clinical protocols documents (http://adni.loni.usc.edu/methods/documents/). Briefly, DEM diagnosis is based on the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) criteria. 31 ADNI probable AD DEM subjects were 56 to 90 years old at enrollment, and scored between 20 and 26 on the MMSE 32 and 0.5 and 1 on the CDR global score. 33 Subjects diagnosed as amnestic MCI ranged from 55 to 91 years old at enrollment, had no significant functional impairment, scored between 24 and 30 on the MMSE, had a global CDR of 0.5 (memory score ≥ 0.5), and impairment on Wechsler Memory Scale—Logical Memory II test. 34 CN subjects had MMSE between 24 and 30, a global CDR of 0, and did not meet criteria for MCI or DEM. Subjects were excluded due to inability to undergo MRI; if they had other neurological disorders, active depression, or history of other psychiatric diagnosis, alcohol, or substance dependence within the past 2 years; <6 years of education; or were not fluent in English or Spanish. The full list of inclusion/exclusion criteria may be accessed in the ADNI procedures documents (http://adni.loni.usc.edu/methods/documents/). Written informed consent was obtained from all participants.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed relevant published primary research and reviews using both Google Scholar and PubMed. Randomized clinical trials, as well as post hoc and meta‐analyses of trials have been used to measure efficacy of acetylcholinesterase inhibitor (AChEI) treatment in mild cognitive impairment (MCI) patients. The relevant citations for these publications have been included.
Interpretation: Our findings suggest that previous reports citing AChEI use in MCI patients as deleterious or harmful may be reflective of the significant differences in impairment between treatment groups at baseline.
Future directions: The results from this research do not support previous claims of harmful effect by AChEI in MCI.
Subjects were selected according to the schematic seen in Figure 1. Briefly, our sample included both subjects taking AChEI (AChEI+) and not taking AChEI (AChEI–) who met criteria for MCI (ADNI 1) or late MCI (ADNI 2/GO) at the initial timepoint and had longitudinal MMSE and/or CDR‐SB. Medications were found in the “Key Background Medications” sheet in ADNI's study data. AChEI+ subjects were selected who were on any one of three AChEIs: donepezil, rivastigmine, or galantamine. All subjects had longitudinal MRI; 192 subjects (82 AChEI+ and 110 AChEI‐) also had longitudinal fluorodeoxyglucose (FDG) scans. One hundred eighty‐one subjects from the MRI and 137 subjects from the FDG cohort received either cerebrospinal fluid (CSF) collection or amyloid PET scans (AD biomarker‐validated cohort), which allowed us to interpret our results in light of biomarker evidence of amyloid pathology and to control for amyloid status in our subanalysis in the AD biomarker‐validated cohort. Amyloid positivity was defined as CSF amyloid beta (Aβ)1‐42 < 192 pg/mL using the multiplex xMAP Luminex platform (Luminex Corp) with Innogenetics (INNO‐BIA AlzBio3) immunoassay kit–based reagents or florbetapir PET whole brain standardized uptake value ratio (SUVR) ≥1.17. 35 , 36 In cases in which both CSF and amyloid PET were available, PET SUVR was preferred for determining amyloid positivity. In addition to florbetapir imaging, ADNI has a small subset of subjects with [11C]‐Pittsburgh compound B (PiB) imaging. To introduce the smallest amount of noise possible, the decision was made to use a single tracer for these analyses. 37
FIGURE 1.
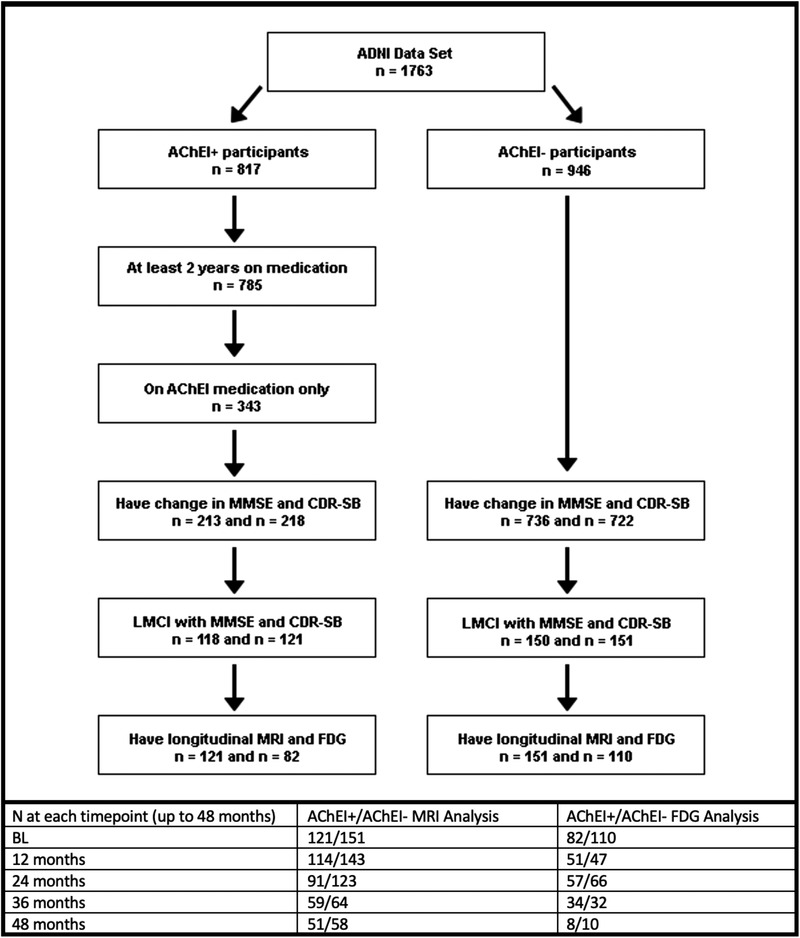
Schematic describing the selection of subjects for analysis, as well as number of subjects with data at each timepoint. AChEI, acetylcholinesterase inhibitor; ADNI, Alzheimer's Disease Neuroimaging Initiative; CDR‐SB, Clinical Dementia Rating–Sum of Boxes; FDG, fluorodeoxyglucose; MMSE, Mini‐Mental State Examination; MRI, magnetic resonance imaging
2.2. MRI and PET acquisition and analyses
ADNI MRI and PET acquisition and preprocessing protocols can be found at www.adni‐info.org. The MRI data acquisition and preprocessing have been previously described elsewhere and can be found in Table S1 in supporting information. 38 Preprocessed MRIs were downloaded from LONI IDA (https://ida.loni.usc.edu ) and analyzed using voxel‐based morphometry (VBM) in SPM12 as described previously. 39 , 40 Briefly, baseline scans were co‐registered to Montreal Neurological Institute (MNI) space, while follow‐up or longitudinal scans were co‐registered to their corresponding baseline scan. MRIs were then segmented into gray matter (GM), white matter (WM), and CSF components, then bias corrected and spatially normalized. 41 After this step, we used a nonlinear registration procedure, DARTEL, on each subject's GM and WM tissue maps, which iteratively matches to a subject‐specific mean template. 42 GM and WM maps were then normalized to MNI space as 1 × 1 × 1 mm voxels and smoothed using 10 mm full‐width half maximum (FWHM) Gaussian kernel. Additional processing using FreeSurfer version 5.1 was done to extract intracranial volume (ICV or eTIV) and baseline bilateral hippocampal volume. 43 , 44
As with MRI, PET acquisition and ADNI preprocessing normalization standards are well documented. 33 We downloaded preprocessed (averaged, aligned to standard space, re‐sampled to a standard image and voxel size of 2 × 2 × 2 mm and smoothed to a uniform resolution) amyloid and FDG PET data from LONI IDA (https://ida.loni.usc.edu). 45 PET images were co‐registered to MRI scans and warped into MNI space using transformation parameters obtained from the nonlinear registration of MRIs in SPM12. FDG PET scans were intensity normalized to mean pons uptake, while amyloid PET scans were intensity normalized using the whole cerebellum as a reference region. Whole brain SUVR was extracted as the region of interest for amyloid PET. 46 Baseline posterior cingulate cortex (PCC) SUVR was extracted as the region of interest in FDG PET scans, where hypometabolism serves as a sensitive marker for early AD and predicts the conversion from MCI to AD. 47 , 48
2.3. Statistical analyses
2.3.1. Demographic analyses
The statistical distribution of clinical and demographic characteristics—age, education, baseline CDR‐SB, baseline MMSE, baseline everyday cognition patient and informant scores, florbetapir PET mean cortical SUVR, and bilateral hippocampal volume were analyzed in SPSS version 24 using one‐way analysis of variance. Sex and apolipoprotein E (APOE) ε4 carrier (percentage of subjects carrying 0, 1 or 2 copies of the ε4 allele) comparisons were done using a chi‐square test with two‐sided P‐values. The alpha for all comparisons was P < .05.
Longitudinal change in CDR‐SB and MMSE was modeled using autoregressive linear mixed effects models in SAS 9.4, controlling for age, sex, and education. All visits up to 48 months were included (see Figure 1 for number of subjects at each timepoint).
2.3.2. Baseline parametric mapping
We generated MRI and FDG PET voxel‐wise regression maps on the baseline scans that directly compared AChEI+ and AChEI‐ groups using SPM12, with age, sex, and education as covariates, as well as field strength (1.5T vs. 3T) and ICV for the MRI analysis. Results are displayed at family‐wise error (FWE) cluster‐level correction of P < .05 to correct for multiple comparisons. The primary or cluster‐defining threshold was selected at an uncorrected P < .01.
The analyses were repeated in the AD biomarker–validated cohort while also controlling for baseline amyloid positivity. This allowed us to determine whether differences in cognitive or neurodegenerative outcomes are fully attributable to AD pathology.
2.3.3. Longitudinal parametric mapping
To visualize the longitudinal neurodegenerative effects in 3D, we relied on the Sandwich Estimator (SwE) SPM toolbox. 49 SwE not only has the flexibility to handle an unbalanced dataset, but the ability to estimate a covariance matrix, overcoming traditional pitfalls seen by spatially homogenous correlation assumptions. 49 Scans up to 48 months were selected to analyze the group effect (AChEI+ vs. AChEI–) while controlling for age, sex, and education, as well as ICV and field strength for MRI. To control for the likelihood that baseline cognitive and neurodegenerative differences (potentially suggestive of greater disease severity in one group vs. another) were contributing to longitudinal group differences, we repeated this analysis, while controlling for baseline PCC SUVR (FDG) and bilateral hippocampal volume (MRI). Additional models were run with an APOE ε4 covariate (Figures S1 and S2 in supporting information), an APOE ε4 x AChEI group covariate (Figures S3 and S4 in supporting information) and an APOE ε4 x amyloid covariate (Figure S5 in supporting information).
The analyses were repeated in our AD biomarker–validated cohort, with and without additionally controlling for amyloid status. All longitudinal results are displayed at a false discovery rate (FDR) of P < .05.
3. RESULTS
3.1. Cognitive outcomes
3.1.1. Full cohort
Full cohort analysis of demographic, clinical, and imaging variables for subjects with MRI and FDG PET scans can be seen in Table 1. We found no significant differences in age, sex, or education between AChEI+ and AChEI– subjects. There were significantly more APOE ε4 carriers in the AChEI+ than AChEI– group for both MRI and FDG analysis (P = .005 and .025, respectively). The AChEI+ group had a significantly worse baseline CDR‐SB (P = .009 and P = .041 for the MRI and FDG cohorts, respectively) and MMSE score (P = .008 and P = .011 for MRI and FDG cohorts, respectively) than AChEI– subjects. In the MRI analyses, AChEI+ voiced significantly greater subjective complaints compared to AChEI– (Everyday Cognition questionnaire participant score, P = .025). Informants relayed significantly greater concerns regarding the cognition of AChEI+ versus AChEI– in both the FDG and MRI samples (Everyday Cognition questionnaire informant score, P < .001, both). Longitudinally, AChEI+ subjects exhibited a significantly greater rate of decline in both CDR‐SB (P < .001 for both cohorts) and MMSE (P < .001 and P = .005 for the MRI and FDG cohorts, respectively).
TABLE 1.
Demographic and clinical comparisons between AChEI groups—Full cohort. Bolded P‐values are significant at P<.05
| AChEI Group (N = 272)—MRI analysis | AChEI+ (121) | AChEI– (151) | P‐value |
|---|---|---|---|
| Baseline age, years mean (SD) | 73.0 (7.1) | 73.5 (8.2) | 0.612 |
| Sex, male % | 66.9 | 57.0 | 0.093 |
| Education, years mean (SD) | 16.0 (2.9) | 16.1 (2.8) | 0.747 |
| APOE ε4 alleles, % 0/1/2 | 40/45/16 | 58/34/7 | 0.005 |
| Baseline CDR‐SB, mean (SD) | 1.67 (0.88) | 1.39 (0.88) | 0.009 |
| Change in CDR‐SB [95% CI] | 2.66 [2.05, 3.27] | –0.06 [–0.61, 0.49] | <0.001 |
| Baseline MMSE, mean (SD) | 27.2 (1.73) | 27.8 (1.72) | 0.008 |
| Change in MMSE [95% CI] | –3.33 [–4.17, –2.48] | –0.20 [–0.97, 0.56] | <0.001 |
| Baseline Everyday Cognition Total Patient Score, mean (SD) | 1.61 (0.57) | 1.44 (0.39) | 0.025 |
| Baseline Everyday Cognition Total Informant Score, mean (SD) | 2.42 (0.83) | 1.65 (0.70) | <0.001 |
| Amyloid positive at baseline, positive % | 75.9 | 48.0 | <0.001 |
| Baseline bilateral hippocampal volume, mean mm3 (SD) | 6237.9 (1163.7) | 6915.0 (1071.9) | <0.001 |
| AChEI group (N = 192)—FDG analysis | AChEI+ (82) | AChEI– (110) | P‐value |
| Age, years mean (SD) | 73.1 (7.0) | 73.5 (8.1) | 0.700 |
| Sex, male % | 69.5 | 57.3 | 0.080 |
| Education, years mean (SD) | 16.1 (2.7) | 16.3 (2.5) | 0.600 |
| APOE ε4 alleles, % 0/1/2 | 37/48/16 | 56/36/8 | 0.025 |
| Baseline CDR‐SB, mean (SD) | 1.69 (0.90) | 1.43 (0.83) | 0.041 |
| Change in CDR‐SB [95% CI] | 1.97 [1.31, 2.63] | 0.33 [–0.31, 0.97] | <0.001 |
| Baseline MMSE, mean (SD) | 27.4 (1.8) | 28.0 (1.6) | 0.011 |
| Change in MMSE [95% CI] | –2.78 [–3.74, –1.81] | –0.88 [–1.80, 0.04] | 0.005 |
| Baseline Everyday Cognition Total Patient Score, mean (SD) | 1.61 (0.61) | 1.49 (0.42) | 0.181 |
| Baseline Everyday Cognition Total Informant Score, mean (SD) | 2.28 (0.79) | 1.65 (0.70) | <0.001 |
| Amyloid positive, positive % | 80.7 | 51.3 | <0.001 |
| Baseline PCC SUVR, mean (SD) | 1.53 (0.13) | 1.58 (0.15) | 0.011 |
Abbreviations: AChEI, acetylcholinesterase inhibitor; APOE, apolipoprotein E; CDR‐SB, Clinical Dementia Rating–Sum of Boxes; CI, confidence interval; FDG, fluorodeoxyglucose; MMSE, Mini‐Mental State Examination; MRI, magnetic resonance imaging; PCC, posterior cingulate cortex; SD, standard deviation; SUVR, standardized uptake value ratio.
3.1.2. AD biomarker‐validated cohort
The AD biomarker‐validated cohort analysis of demographic, clinical, and imaging variables for subjects with MRI (AChEI+ N = 83 and AChEI– N = 98) and FDG PET scans (AChEI+ N = 57 and AChEI– N = 80) can be seen in Table 2. Every subject included in this group had either CSF Aβ1‐42 or whole brain florbetapir PET SUVR data, allowing us to determine their amyloid status and to additionally control for that in our analyses. We thought this was critical to do as a greater proportion of the AChEI+ subjects were amyloid‐positive compared to AChEI– (75.9 vs. 48.0% for MRI and 80.7 vs. 51.3% for FDG, P < .001 for both), which could imply that the rate of decline seen in one group versus another could be attributed to the underlying pathology. There were no significant differences between AChEI+ and AChEI– in age, sex, and education in the MRI cohort, or age and education in the FDG cohort. There were significantly more males in the FDG AChEI+ AD biomarker–validated group (P = .023). There were significantly more APOE ε4 AChEI+ carriers in the AChEI MRI analysis (P = .036). AChEI+ subjects were significantly more impaired in baseline MMSE (P = .013 and P = .034, MRI and FDG cohorts, respectively) compared to AChEI– subjects. As with the main, in both the MRI and FDG analyses AChEI+ subjects performed significantly worse on informant‐answered baseline Everyday Cognition (P < .001, both), while the AChEI+ performed worse in the patient‐answered Everyday Cognition in only the MRI analysis (P = .046). Additionally, AChEI+ subjects declined more rapidly in both CDR‐SB (P < .001 P = .028 for the MRI and FDG cohort, respectively) as well as MMSE (MRI cohort only, P = .005).
TABLE 2.
Demographic and clinical comparisons between AChEI groups–Biomarker validated cohort. Bolded P‐values are significant at P<.05
| AChEI Group (N = 181)—MRI analysis | AChEI+ (83) | AChEI– (98) | P‐value |
|---|---|---|---|
| Baseline age, years mean (SD) | 72.6 (7.1) | 72.8 (8.5) | 0.859 |
| Sex, male % | 67.5 | 57.1 | 0.102 |
| Education, years mean (SD) | 16.0 (2.9) | 16.4 (2.4) | 0.272 |
| APOE ε4 alleles, % 0/1/2 | 41/45/15 | 58/36/6 | 0.036 |
| Baseline CDR‐SB, mean (SD) | 1.63 (0.85) | 1.38 (0.88) | 0.062 |
| Change in CDR‐SB [95% CI] | 2.58 [1.84, 3.33] | –0.39 [–1.09, 0.32] | <0.001 |
| Baseline MMSE, mean (SD) | 27.2 (1.8) | 27.9 (1.7) | 0.013 |
| Change in MMSE [95% CI] | –3.06 [–3.25, –1.86] | –0.70 [–1.84, 0.44] | 0.005 |
| Baseline Everyday Cognition Total Patient Score, mean (SD) | 1.62 (0.59) | 1.44 (0.40) | 0.046 |
| Baseline Everyday Cognition Total Informant Score, mean (SD) | 2.28 (0.78) | 1.62 (0.67) | <0.001 |
| Amyloid positive at baseline, positive % | 75.9 | 48.0 | <0.001 |
| Baseline Bilateral Hippocampal Volume, mean mm3 (SD) | 6384.9 (1132.0) | 7013.9 (1063.1) | <0.001 |
| AChEI Group (N = 137)—FDG Analysis | AChEI+ (57) | AChEI– (80) | P‐value |
| Age, years mean (SD) | 73.0 (6.8) | 73.1 (8.3) | 0.967 |
| Sex, male % | 71.9 | 53.8 | 0.023 |
| Education, years mean (SD) | 16.1 (2.6) | 16.5 (2.3) | 0.369 |
| APOE ε4 alleles, % 0/1/2 | 39/47/14 | 55/38/8 | 0.133 |
| Baseline CDR‐SB, mean (SD) | 1.67 (0.91) | 1.46 (0.88) | 0.176 |
| Change in CDR‐SB [95% CI] | 1.89 [1.34, 2.64] | 0.71 [–0.02, 1.44] | 0.028 |
| Baseline MMSE, mean (SD) | 27.4 (1.8) | 28.0 (1.6) | 0.034 |
| Change in MMSE [95% CI] | –3.15 [–4.51, –1.79] | –1.91 [–3.23, –0.58] | 0.197 |
| Baseline Everyday Cognition Total Patient Score, mean (SD) | 1.62 (0.64) | 1.48 (0.43) | 0.191 |
| Baseline Everyday Cognition Total Informant Score, mean (SD) | 2.20 (0.76) | 1.63 (0.67) | <0.001 |
| Amyloid positive, positive % | 80.7 | 51.3 | <0.001 |
| Baseline PCC SUVR, mean (SD) | 1.54 (0.13) | 1.57 (0.13) | 0.109 |
Abbreviations: AChEI, acetylcholinesterase inhibitor; APOE, apolipoprotein E; CDR‐SB, Clinical Dementia Rating–Sum of Boxes; CI, confidence interval; FDG, fluorodeoxyglucose; MMSE, Mini‐Mental State Examination; MRI, magnetic resonance imaging; PCC, posterior cingulate cortex; SD, standard deviation; SUVR, standardized uptake value ratio.
3.2. Imaging outcomes
3.2.1. Full cohort
AChEI+ subjects displayed significantly smaller baseline hippocampal volume and lower FDG PCC SUVR than AChEI– (P < .001 and P = .011, MRI and FDG PET cohorts, respectively). Baseline 3D comparisons of AChEI+ to AChEI– within the MRI cohort while controlling for age, sex, and education differences resulted in voxelwise maps seen in Figure 2 (top left panel). AChEI+ subjects had significantly less (cluster‐level p FWE < 0.05) gray matter density (GMD) in the temporal and left parietal cortices. Longitudinally, additional atrophy was also observed in the right parietal and bilateral prefrontal cortex (p FDR < 0.05). When baseline hippocampal volume was added as a covariate, no significant voxels of atrophy remained (Figure 2, top middle and right panels, respectively).
FIGURE 2.
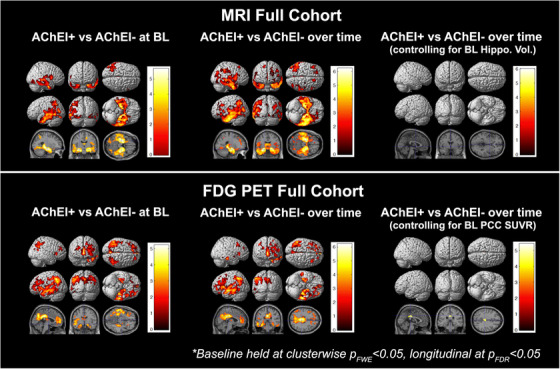
Significance maps for full cohort. AChEI, acetylcholinesterase inhibitor; BL, baseline; FDG, fluorodeoxyglucose; MRI, magnetic resonance imaging; PCC, posterior cingulate cortex; PET, positron emission tomography; SUVR, standardized uptake value ratio
Baseline comparison of AChEI+ to AChEI– within the FDG PET cohort yielded significant hypometabolism in left greater than right temporal, parietal, and frontal cortices. In addition, there was a large cluster of hypometabolism in posterior and anterior cingulate cortex (Figure 2, bottom left panel). Longitudinal comparisons of metabolic changes between AChEI+ to AChEI– showed a similar pattern of hypometabolism as the one seen at baseline—left greater than right temporal and frontal and left parietal hypometabolism. When baseline PCC SUVR was added as a covariate, only a small cluster of hypometabolism survived localized to the PCC (Figure 2, bottom middle and right panels, respectively).
3.2.2. AD biomarker–validated cohort
AChEI+ subjects had significantly smaller hippocampal volume (P < .001) but did not differ in PCC metabolism. At baseline, direct 3D comparison of AChEI+ to AChEI– subjects in our biomarker‐validated cohort revealed less GMD in bilateral temporal, parietal, and frontal cortices. When controlling for amyloid positivity, the statistical maps did not change, outside of a decrease in overall number of significant voxels (k of 111,031 voxels vs. 75,612, Figure 3, top left panels).
FIGURE 3.

Significance maps for biomarker validated cohort. AChEI, acetylcholinesterase inhibitor; BL, baseline; FDG, fluorodeoxyglucose; MRI, magnetic resonance imaging; PCC, posterior cingulate cortex; PET, positron emission tomography; SUVR, standardized uptake value ratio
Comparison of AChEI+ to AChEI– over 48 months revealed a pattern of atrophy similar to what was seen at baseline with significant voxels in bilateral temporal, parietal, and frontal cortices. After controlling for amyloid status the signal became largely restricted to bilateral medial and lateral temporal lobes (Figure 3, top right panels).
Baseline FDG PET comparison in our AD biomarker–validated cohort showed significantly greater hypometabolism of the right lateral temporal, bilateral parietal, and occipital cortices in AChEI+. Adding amyloid positivity as a covariate resulted in reduction of the effect to significant hypometabolism restricted to left parietal cortex (Figure 3, bottom left panels).
Comparison of longitudinal metabolic changes between AChEI+ and AChEI– subjects (with or without amyloid status in the model) failed to show significant differences between the groups (Figure 3, bottom right panels).
4. DISCUSSION
The effectiveness of off‐label AChEI use as a treatment of MCI has produced mixed results. To date, studies in the ADNI population have focused solely on cognitive outcomes that have suggested a negative or harmful effect of AChEIs on cognition, which authors have speculated may be due to the criteria of MCI in ADNI at the time and its overlap with early AD. 28 , 29 To further investigate the relative worsening of AChEI users in ADNI, we studied the longitudinal effect of AChEIs on imaging outcomes in addition to cognitive outcomes while accounting for amyloid status and baseline neurodegeneration differences.
We found that AChEI+ subjects were significantly more likely to be amyloid positive than AChEI– subjects. This indicates that physicians are more inclined to prescribe AChEIs to MCI patients due to AD perhaps basing their decision on the greater neurodegeneration or the greater patient and informant complaints observed in these subjects. This then brings up the point that any differences in cognitive and imaging outcomes might be due to baseline cognitive and neurodegenerative differences between the groups rather than medication effects. To this end, when adding baseline hippocampal volume and PCC SUVR as covariates in our regression models the vast majority of voxels indicative of faster atrophy and hypometabolic changes in those on AChEI did not survive. Hence, not accounting for baseline amyloid status and degree of neurodegeneration might be why cognitively harmful effects of AChEI in MCI were reported. 28 , 29 However, controlling for amyloid status did not fully explain the greater neurodegeneration in AChEI+ versus AChEI– subjects. Thus, we conclude that the faster progression observed in the AChEI+ group cannot be fully explained by the presence of amyloid but can be fully attributed to baseline between‐group differences.
The study presented here has several strengths and limitations. A major strength of this study is the broad range of longitudinal clinical and imaging measures available in ADNI. Additionally, ADNI uses stringent standardization procedures for clinical and imaging data collection across all sites. However, the rigorous inclusion/exclusion criteria used in ADNI means that this cohort, while representative of clinical trial participants, is not necessarily representative of patients with MCI in the general population. A further limitation is that we are comparing groups with some baseline differences in cognitive impairment, which is reflected in the imaging analysis. In an ideal study of longitudinal AChEI effects on MCI subjects, both the AChEI+ and AChEI– groups would be relatively equally impaired at baseline, as it may not be possible to fully control for baseline differences.
In conclusion, our study of the longitudinal effect of AChEIs on ADNI MCI subjects revealed no treatment effect. There was, however, a significant difference in the baseline cognitive deficit and neurodegeneration seen between AChEI+ and AChEI– subjects, suggesting that physicians’ AChEI‐prescribing behaviors favor subjects with more severe cognitive impairment (resulting in greater neurodegeneration), which may account for the results seen previously in the ADNI population.
CONFLICTS OF INTEREST
Eddie Stage, Diana Svaldi, Sophie Sokolow, Shannon L. Risacher, Krisztina Marosi, and Jerome I. Rotter have nothing to disclose.
Andrew J. Saykin has received research support from Eli Lilly and PET tracer precursor from AVID Radiopharmaceuticals. He also served on an advisory board for Bayer Oncology and received support from Springer‐Nature as Editor‐in‐Chief of Brain Imaging and Behavior. Liana G. Apostolova has served on an advisory board for Eli Lilly and Biogen and on the Speakers Bureau for Piramal and Eli Lilly. Dr. Apostolova has received research support from GE Healthcare; AVID Radiopharmaceuticals, Inc.; and Piramal Imaging. Liana G. Apostolova, MD, MS, has served on Advisory Boards for Eli Lilly, Biogen and Two Labs. Dr. Apostolova has received research support from GE Healthcare, AVID Radiopharmaceuticals, Inc., Life Molecular Imaging and Roche Diagnostics. Dr. Apostolova serves on a DSMB for IQVIA.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wpcontent/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. The analyses reported in this manuscript were funded by the NIA U01 AG057195, NIA R01 AG040770, NIA K02 AG048240, NIA P30 AG010133, NIA K01 AG049050, NIA R56 AG057195, and the Easton Consortium for Alzheimer's Drug Discovery and Biomarker Development.
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol‐Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Stage E, Svaldi D, Sokolow S, et al., Prescribing cholinesterase inhibitors in mild cognitive impairment—Observations from the Alzheimer's Disease Neuroimaging Initiative. Alzheimer's Dement. 2021;7:e12168. 10.1002/trc2.12168
REFERENCES
- 1. Alzheimers Association. 2019 Alzheimer's Disease Facts and Figures. 2019.
- 2. Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bowen DM, Smith CB, White P, Davison AN. Neurotransmitter‐related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain. 1976;99:459‐496. [DOI] [PubMed] [Google Scholar]
- 4. Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet. 1976;2:1403. [DOI] [PubMed] [Google Scholar]
- 5. Perry EK, Gibson PH, Blessed G, Perry RH, Tomlinson BE. Neurotransmitter enzyme abnormalities in senile dementia. Choline acetyltransferase and glutamic acid decarboxylase activities in necropsy brain tissue. J Neurol Sci. 1977;34:247‐265. [DOI] [PubMed] [Google Scholar]
- 6. Whitehouse P, Price D, Struble R, Clark A, Coyle J, Delon M. (1982). Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science, 215, (4537), 1237–1239. 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 7. Stahl SM. The new cholinesterase inhibitors for Alzheimer's disease, Part 1: their similarities are different. J Clin Psychiatry. 2000;61:710‐711. [DOI] [PubMed] [Google Scholar]
- 8. Galimberti D, Scarpini E. Old and new acetylcholinesterase inhibitors for Alzheimer's disease. Expert Opin Investig Drugs. 2016;25:1181‐1187. [DOI] [PubMed] [Google Scholar]
- 9. Colovic MB, Krstic DZ, Lazarevic‐Pasti TD, Bondzic AM, Vasic VM. Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr Neuropharmacol. 2013;11:315‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosler M, Retz W, Retz‐Junginger P, Dennler HJ. Effects of two‐year treatment with the cholinesterase inhibitor rivastigmine on behavioural symptoms in Alzheimer's disease. Behav Neurol. 1998;11:211‐216. [DOI] [PubMed] [Google Scholar]
- 11. Lee JH, Jeong SK, Kim BC, Park KW, Dash A. Donepezil across the spectrum of Alzheimer's disease: dose optimization and clinical relevance. Acta Neurol Scand. 2015;131:259‐267. [DOI] [PubMed] [Google Scholar]
- 12. Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev. 2006:CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Salloway S, Ferris S, Kluger A, et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo‐controlled trial. Neurology. 2004;63:651‐657. [DOI] [PubMed] [Google Scholar]
- 14. Koontz J, Baskys A. Effects of galantamine on working memory and global functioning in patients with mild cognitive impairment: a double‐blind placebo‐controlled study. Am J Alzheimers Dis Other Demen. 2005;20:295‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379‐2388. [DOI] [PubMed] [Google Scholar]
- 16. Winblad B, Gauthier S, Scinto L, et al. Safety and efficacy of galantamine in subjects with mild cognitive impairment. Neurology. 2008;70:2024‐2035. [DOI] [PubMed] [Google Scholar]
- 17. Doody RS, Ferris SH, Salloway S, et al. Donepezil treatment of patients with MCI: a 48‐week randomized, placebo‐controlled trial. Neurology. 2009;72:1555‐1561. [DOI] [PubMed] [Google Scholar]
- 18. Dubois B, Chupin M, Hampel H, et al. Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer's disease. Alzheimers Dement. 2015;11:1041‐1049. [DOI] [PubMed] [Google Scholar]
- 19. Raschetti R, Albanese E, Vanacore N, Maggini M. Cholinesterase inhibitors in mild cognitive impairment: a systematic review of randomised trials. PLoS Med. 2007;4:e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Russ TC, Morling JR. Cholinesterase inhibitors for mild cognitive impairment. Cochrane Database Syst Rev. 2012:CD009132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Birks J, Flicker L. Donepezil for mild cognitive impairment. Cochrane Database Syst Rev. 2006:CD006104. [DOI] [PubMed] [Google Scholar]
- 22. Cavedo E, Dubois B, Colliot O, et al. Reduced regional cortical thickness rate of change in donepezil‐treated subjects with suspected prodromal Alzheimer's disease. J Clin Psychiatry. 2016;77:e1631. e8. [DOI] [PubMed] [Google Scholar]
- 23. Cavedo E, Grothe MJ, Colliot O, et al. Reduced basal forebrain atrophy progression in a randomized Donepezil trial in prodromal Alzheimer's disease. Sci Rep. 2017;7:11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schuff N, Suhy J, Goldman R, et al. An MRI substudy of a donepezil clinical trial in mild cognitive impairment. Neurobiol Aging. 2011;32:2318. e31‐41. [DOI] [PubMed] [Google Scholar]
- 25. Prins ND, van der Flier WA, Knol DL, et al. The effect of galantamine on brain atrophy rate in subjects with mild cognitive impairment is modified by apolipoprotein E genotype: post‐hoc analysis of data from a randomized controlled trial. Alzheimers Res Ther. 2014;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saykin AJ, Wishart HA, Rabin LA, et al. Cholinergic enhancement of frontal lobe activity in mild cognitive impairment. Brain. 2004;127:1574‐1583. [DOI] [PubMed] [Google Scholar]
- 27. Jack CR Jr, Petersen RC, Grundman M, et al. Longitudinal MRI findings from the vitamin E and donepezil treatment study for MCI. Neurobiol Aging. 2008;29:1285‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schneider LS, Insel PS, Weiner MW. Alzheimer's disease neuroimaging I. Treatment with cholinesterase inhibitors and memantine of patients in the Alzheimer's Disease neuroimaging initiative. Arch Neurol. 2011;68:58‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kennedy RE, Cutter GR, Fowler ME, Schneider LS. Association of concomitant use of cholinesterase inhibitors or memantine with cognitive decline in Alzheimer's clinical trials: a meta‐analysis. JAMA Netw Open. 2018;1:e184080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer's disease neuroimaging initiative (ADNI): clinical characterization. Neurology. 2010;74:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA work group under the auspices of department of health and human services task force on Alzheimer's disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 32. Folstein MF, Folstein SE, McHugh PR. Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 33. Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology. 1993;43:2412‐2414. [DOI] [PubMed] [Google Scholar]
- 34. D W Wechsler Memory Scale – Revised. Psychological Corporation; San, Antonio, TX: 1987. 1987. [Google Scholar]
- 35. Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer's disease. Arch Neurol. 2011;68:1404‐1411. [DOI] [PubMed] [Google Scholar]
- 36. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Su Y, Flores S, Wang G, et al. Comparison of Pittsburgh compound B and florbetapir in cross‐sectional and longitudinal studies. Alzheimers Dement (Amst). 2019;11:180‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jack CR Jr, Bernstein MA, Fox NC, et al. The Alzheimer's disease neuroimaging initiative (ADNI): mRI methods. J Magn Reson Imaging. 2008;27:685‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Risacher SL, Kim S, Shen L, et al. The role of apolipoprotein E (APOE) genotype in early mild cognitive impairment (E‐MCI). Front Aging Neurosci. 2013;5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Whitwell JL. Voxel‐based morphometry: an automated technique for assessing structural changes in the brain. J Neurosci. 2009;29:9661‐9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. D'Agostino E, Maes F, Vandermeulen D, Suetens P. Atlas‐to‐image non‐rigid registration by minimization of conditional local entropy. Inf Process Med Imaging. 2007;20:320‐332. [DOI] [PubMed] [Google Scholar]
- 42. Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38:95‐113. [DOI] [PubMed] [Google Scholar]
- 43. Risacher SL, Shen L, West JD, et al. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging. 2010;31:1401‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buckner RL, Head D, Parker J, et al. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas‐based head size normalization: reliability and validation against manual measurement of total intracranial volume. Neuroimage. 2004;23:724‐738. [DOI] [PubMed] [Google Scholar]
- 45. Jagust WJ, Bandy D, Chen K, et al. The Alzheimer's disease neuroimaging initiative positron emission tomography core. Alzheimer's Dement. 2010;6:221‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid‐beta plaques: a prospective cohort study. Lancet Neurol. 2012;11:669‐678. [DOI] [PubMed] [Google Scholar]
- 47. Jagust WJ, Landau SM, Shaw LM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Landau SM, Harvey D, Madison CMet al. Associations between cognitive, functional, and FDG‐PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32:1207‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guillaume B, Hua X, Thompson PM, Waldorp L, Nichols TE. Alzheimer's disease neuroimaging I. Fast and accurate modelling of longitudinal and repeated measures neuroimaging data. Neuroimage. 2014;94:287‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information