

Summary
It is unknown whether antibody-mediated enhancement (ADE) contributes to the pathogenesis of COVID-19, and the conditions for ADE needs to be elucidated. We demonstrated that without inducing an ACE2-independent ADE on Raji cells, the neutralizing antibody CB6, a mouse anti-S1 serum and convalescent plasma, induced ADE on cells expressing FcγRIIA/CD32A and low levels of endogenous ACE2. ADE occurred at sub-neutralizing antibody concentrations, indicating that unneutralized S protein was required for ADE. The enhanced infectivity of 614G variant was higher than that of 614D wildtype in the presence of antibodies, further suggesting that ADE may be influenced by virus strains with different ACE2-binding affinity. Finally, knockdown of ACE2 or treatment with a fusion-inhibition peptide EK1C4 significantly reduced ADE. In conclusion, we identified an ADE mechanism mediated by neutralizing antibodies against SARS-CoV-2. ACE2 may act as a secondary receptor required for the antibody- and FcγR-mediated enhanced entry of SARS-CoV-2.
Subject areas: Immunology, Virology
Graphical abstract
Highlights
-
•
NAbs induced ADE of SARS-CoV-2 on cells expressing FcγRIIA and ACE2
-
•
Unneutralized S protein was required for ADE
-
•
ADE may be influenced by virus strains with different ACE2-binding affinity
-
•
ACE2 acts as a secondary receptor required for the Ab- and FcγR-mediated ADE
Immunology; Virology
Introduction
Antibody-dependent enhancement (ADE) is considered to be a potential challenge in the development of COVID-19 vaccine and the application of therapeutic antibodies (Lee et al., 2020; Ricke, 2021). Although introducing mutations in the Fc region of a therapeutic antibody may fully abolish its interaction with Fc receptors and its potential ADE effect (Chu et al., 2020; Shi et al., 2020), an intervention method is not available for use with the antibodies persistently present in the convalescent patients as well as in vaccinated individuals. Therefore, it is critically important to study the ADE potential of SARS-CoV-2 antibodies generated in human bodies, and to elucidate the underlying mechanism of ADE in SARS-CoV-2 infection.
Evidence acquired in vitro shows that antibodies targeting specific epitopes may have the ADE potential in SARS-CoV-2 infection (Chu et al., 2020; Li et al., 2021; Liu et al., 2021; Wu et al., 2020; Zhou et al., 2021), although the pathogenic consequences of ADE in humans is controversial (Chan et al., 2020; Garcia-Nicolas et al., 2021). However, some evidences showed that afucosylated antiviral IgG1 may be the trigger of the excessive inflammation observed in patients with severe COVID-19 (Larsen et al., 2021) and increased lung inflammation occurred in rare SARS-CoV-2 antibody-infused macaques (Li et al., 2021). Although the in vivo ADE may be milder than observed in the in vitro systems, the reason for this inconsistency is not clear, and the in vivo conditions for ADE to occur is critically important to understand.
The effect of ADE was first detected in dengue virus infections (Dejnirattisai et al., 2010) and subsequently in patients infected with HIV (Robinson et al., 1988), influenza virus (Ochiai et al., 1992), FIPV (Olsen et al., 1992), and Ebola virus (Kuzmina et al., 2018). Most of these viruses can infect immune cells, particularly macrophages. However, it is unknown whether a virus receptor-mediated basal level of infection is required for ADE. The possibility of simultaneous involvement of the virus receptor and Fcγ receptor (FcγR) in ADE is called the “dual-receptor mechanism” (Arvin et al., 2020). However, this hypothesis has long been debated, for example, the role of CD4 in the ADE of HIV (Homsy et al., 1989; Perno et al., 1990; Takeda et al., 1990). The inability of ACE2 antibodies to inhibit SARS-CoV ADE (Kam et al., 2007) and the lack of or low levels of ACE2 expression on the target cells for SARS-CoV or SARS-CoV-2 ADE assays also challenges this hypothesis (Chu et al., 2020; Jaume et al., 2011; Zhou et al., 2021). In contrast, other reports support this hypothesis, such as antibodies could mediate ADE of SARS-CoV on a cell line HL-CZ coexpressing ACE2 and FcγRII (Wang et al., 2014); and some immune cell lines with FcγRII expression do not support ADE of SARS-CoV or SARS-CoV-2 infection (Chu et al., 2020; Jaume et al., 2011).
Importantly, ADE always occurs at sub-neutralizing antibody concentrations (Jaume et al., 2011; Wan et al., 2020), which means that when the ADE effect reaches the peak, it usually decreases with the further increase of antibody concentration. In case of dengue virus, at the concentration of sub-neutralizing antibody, the virus binds to FcγR through the antibody, and the viral E protein directly binds to LILR-B1, which results in the prevention of the expression of interferon-stimulated genes and lysosomal degradation of the virus. At high antibody concentration, the binding of the E protein to LILR-B1 was completely blocked by the antibody, resulting in FcγR internalized virus neutralization (Kulkarni, 2020). It is also possible that antibody at high concentration will block the interaction of the virus with other cellular factors, such as the virus receptor. Based on this, we hypothesized that FcγR alone is insufficient for the ADE process, and the ADE process may also require other cellular factors.
Virus, antibodies, and target cells with essential receptor expression are major components of the ADE system. However, their involvement in SARS-CoV-2 ADE is insufficiently understood. For the involvement of FcγRs on target cells, it has been demonstrated that while some antibodies targeting N-terminal domain (NTD) domain of S protein mediates ADE effect in an FcγR-independent manner (Li et al., 2021; Liu et al., 2021), a group of neutralizing monoclonal antibody against receptor-binding domain (RBD) can enhance the infectivity of SARS-CoV-2 pseudovirus, which is mediated by FcγRIIB or FcγRI in vitro (Chu et al., 2020; Li et al., 2021; Zhou et al., 2021). However, it remains controversial whether antibody or convalescent plasma could mediate ADE on cells without ACE2 expression, which is the same virus receptor as used by SARS-CoV (Zhou et al., 2020). A preprint report showed that part of plasma from convalescent patient showed ADE effect on Raji cells and even peripheral blood mononuclear cells (PBMCs), with a much higher percentage in patients with severe vs. mild infections (Wu et al., 2020), while convalescent plasma could not mediate ADE on Raji cells in another study (Zhou et al., 2021). On the aspect of antibodies, available evidence does not clarify whether the ADE effect correlated with concentrations of neutralizing antibodies (Zhou et al., 2021). On the aspect of virus, no study, to our knowledge, approached yet whether the epidemic strains with different binding affinities to ACE2 affect the level of ADE.
To answer these critically important questions and to further illuminate the role of the ACE2-mediated viral entry pathway in the SARS-CoV-2 ADE process, we used the monoclonal antibody CB6, a mouse anti-S1 serum and convalescent plasma, to study the effect of ADE on immune cells or on exogenic FcγR-expressing cells. We show here that these antibodies can elicit ADE on cells with FcγRIIA/CD32A expression and low level of endogenous ACE2. We conclude that FcγR, ACE2 and membrane fusion are essential for this ADE process, which is different from known mechanisms of ADE.
Results
ADE of 614G variant can be detected on Daudi immune cells
To investigate the ADE ability of SARS-CoV-2, we prepared HIV/SARS-CoV-2 pseudovirus bearing the S protein with 614D or 614G mutation as previously reported (Wang et al., 2020) (Figure S1A). The infectivity of 614G virus on difference cells was correlated with cellular ACE2 expression (Figures 1A and 1B). We also prepared neutralizing monoclonal antibodies CB6 (Shi et al., 2020) and XG005 (Figure 1C), the non-neutralizing anti-RBD N1G12 and anti-S2 monoclonal antibodies in human IgG1 isotype (Figures S2A–S2D), and mouse antisera by immunizing Balb/c mice with S1 protein.
Figure 1.

Daudi immune cells support weak ADE of SARS-CoV-2
(A) Infectivity of HIV/SARS-CoV-2 614G on a panel of cell lines and monocytes and non-monocytes from PBMCs.
(B) ACE2 mRNA levels detected by real-time RT PCR in a panel of cell lines and monocytes and non-monocytes from PBMCs.
(C) Neutralization ability of monoclonal antibodies CB6 and XG005 IgG1 on 614D pseudoviruses.
(D) Infectivity of HIV/SARS-CoV-2 614G in the presence of different concentrations of XG005 on immune cell lines.
(E) Infectivity of HIV/SARS-CoV-2 614G (solid line) or 614D (dashed line) in the presence of different concentrations of CB6 IgG1 on immune cells.
(F) Infectivity of SARS-CoV-2 pseudovirus bearing 614G mutation in the presence of different concentrations of mouse anti-S1 serum or convalescent plasma from patients (No. 66, 109, 144) on immune cells. Data were derived from three independent experiments, and are presented as mean ± SD. One-way ANOVA was used to compare the difference between HeLa cell and other cell groups. Repeated measurement of one-way ANOVA was used to compare the difference between the enhanced infectivity and the infectivity at basal level.∗p<0.05; ∗∗p<0.01; ∗∗∗p<0.001; ∗∗∗∗p<0.0001.
We first evaluated the ADE ability of XG005 on Raji, THP-1, and Daudi cells. As previously reported, XG005 mediated enhanced infectivity of 614G pseudovirus on Raji cells (Zhou et al., 2021), as well as Daudi cells, but not THP-1 cells (Figure 1D). Next, we tested CB6 and convalescent plasma from three patients on these cell lines, as well as PBMCs. All these immune cells expressed FcγRs on their surfaces (Figures S3A and S3B). THP-1 has been used in ADE assays for SARS-CoV, MERS-CoV, and Ebola virus. However, CB6 antibody did not mediate ADE of SARS-CoV-2 614G on THP-1 cells (Figure 1E). In contrast, Daudi cells supported a weak ADE of the 614G but not 614D pseudovirus, with the relative luminescence unit (RLU) increased from 51.3 ± 5.1 to 93 ± 24.3 (Figure 1E). Because the SARS-CoV-2 pseudovirus bearing 614G mutation achieved 2-fold higher infectivity (Korber et al., 2020) than the 614D pseudovirus on Cos7 cells overexpressing human ACE2 (Cos7-hACE2) (Figure S1A), we suspected that the infectivity of 614D was undetectable even in the presence of ADE on Daudi cells, because the immune cells were generally not susceptible to SARS-CoV-2 infection (Figure 1A). Based on its higher infectivity and detection sensitivity, 614G was used in most of our subsequent ADE assays. Consistent with these findings, Daudi cells also supported a weak ADE mediated by mouse anti-S1 serum on 614G variant, with the RLU increased from 51.3 ± 5.1 to 111.7 ± 21.2 (Figure 1F). Although the convalescent plasma did not generate detectable ADE on these cells (Figure 1F), we reasoned that it was partially caused by the insufficient sensitivity of ADE assay based on the use of these cells. As controls, the ADE of MERS-CoV and MARV pseudoviruses on THP-1 cells was tested parallelly, whose ADE were obviously detected in the presence of their respective specific neutralizing antibodies (Figures S4A–S4D).
CD32A serves as the functional FcγR that mediates ADE
Because the detection window was quite narrow using Daudi cells to evaluate SARS-CoV-2 ADE, we sought to establish a more sensitive ADE assay system. Based on the observation that THP-1 cells showed a certain degree of susceptibility to MERS-CoV and MARV, and could supportADE of both viruses, we suspected that THP-1 cells could not support ADE of SARS-CoV-2 because of complete lack of susceptibility, which was affected by ACE2 expression (Figures 1A and 1B). We chose to overexpress FcγRs in Huh7 cells, which showed a degree of ACE2 expression and were partially susceptible to SARS-CoV-2 614G pseudovirus (Figures 1A and 1B).
Evidence indicates that FcγRs mediate ADE of several species of viruses including flavivirus, filovirus, and coronavirus. However, there is no consensus yet which FcγR plays a vital role in SARS-CoV-2 ADE effect. We overexpressed FcγRIA (CD64A), FcγRIIA (CD32A), FcγRIIB (CD32B), or FcγRIIIA (CD16A) on Huh7 (Figures S5A and S5B). Because FCER1G could facilitate the surface expression of CD16A (Hibbs et al., 1989), we also co-expressed CD16A and FCER1G, which generated an enhanced surface expression of CD16A (Figure S5E). In the ADE assay mediated by CB6 IgG1, Huh7 cells overexpressing CD32A (Huh7-CD32A) supported an obvious ADE for 614G variant. CB6 inhibited the infection at high concentrations and enhanced the infection at a lower concentration, with the maximum induction at 100 ng/mL. With a further decrease in antibody concentration, the effect of ADE gradually disappeared. CD16A-, CD32B-, and CD64A-overexpressing Huh7 cells supported the neutralizing effect of CB6 at high concentrations. No significant enhancement was observed at lower concentrations tested (Figure 2A and S5F). These results indicate that CD32A may function in the IgG1-mediated infection enhancement of SARS-CoV-2. We therefore employed Huh7-CD32A in the subsequent ADE assays.
Figure 2.

Establishment of a sensitive ADE assay system based on Huh7-CD32A cells
(A) Relative infectivity of SARS-CoV-2 pseudovirus (614G) on Huh7 cells overexpressing different FcγRs in the presence of different concentrations of CB6 IgG1.
(B and C) Relative infectivity of SARS-CoV-2 pseudovirus (614G) on HeLa cells overexpressing different FcγRs (B) or HeLa-ACE2-CD32A cells (C) in the presence of different concentrations of CB6 IgG1.
(D) Relative infectivity of SARS-CoV-2 pseudovirus (614G) on Huh7-CD32A cells in the presence of different concentrations of anti-S2, N1G12, or CB6 antibodies.
(E and F) Neutralization (E) or ADE assay (F) of SARS-CoV-2 pseudovirus bearing different mutations in S performed in the presence of CB6 IgG1. Data were derived from three independent experiments, and are presented as mean ± SD. Repeated measurement of one-way ANOVA was used to compare the difference between the enhanced infectivity and the infectivity at basal level. ∗p<0.05.
To further investigate the requirement of cell susceptibility for ADE induction, we also overexpressed FcγRs in HeLa cells (Figures S5C and S5D). In the ADE assay of 614G variant mediated by CB6 IgG1, none of the FcγR-overexpressing HeLa cells supported ADE (Figure 2B). When CD32A was overexpressed in HeLa-hACE2 cells to generate HeLa-hACE2-CD32A (Figure S5G), only neutralizing ability was detected for CB6 at high concentrations and ADE was undetectable at lower concentrations (Figure 2C). These results indicate that an appropriate level of susceptibility, e.g., not completely unsusceptible or highly susceptible, may be suitable for ADE induction.
We found that the non-neutralizing antibodies N1G12 and anti-S2 (Figures S2A–S2C) showed no ADE effect for SARS-CoV-2 614G pseudovirus (Figure 2D). When we evaluated the ADE potential of different isotypes of CB6 antibodies, we found that only CB6 IgG1 mediated the ADE effect on Huh7-CD32A cells. The IgM and IgA isotypes of CB6 possessed neutralizing ability similar to that of CB6 IgG1 (Figures S2A and S2B), but did not show ADE effect on Huh7-CD32A cells (Figure 2D). Because IgM and IgA bind to Fc α receptors (FcαRs) instead of FcγRs, this result implies that the interaction of Fc domain with its specific receptor was required for the ADE effect.
The maximum ADE induction concentration correlates with IC50
Recently, numerous SARS-CoV-2 variants of concern show different infectivity and different susceptibility to neutralizing mAbs. We therefore prepared pseudotyped viruses bearing S with N501Y or E484K-N501Y mutations based on the 614G variant, as well as all mutations in B.1.351 strain. CB6 showed similar neutralizing ability to the 614D and 614G variants, but impaired neutralizing ability to the N501Y variant, and further impaired neutralization of the E484K-N501Y variant, while no neutralization was observed on B.1.351 strain (Figure 2E). In ADE assays, CB6 mediated enhanced infectivity of the variants it could neutralize, but not that of B.1.351 (Figure 2F). Interestingly, the maximum ADE induction concentration of CB6 for different variants showed a trend of correlating with its IC50 (r = 0.8693, p = 0.0677) (Figure S5H); ADE occurred at the sub-neutralizing concentrations of CB6 for all the four variants.
We raised mouse antisera immunized with SARS-CoV-2 S1 protein (614D) to simulate vaccinated antisera. Antisera obtained from three mice showed strong neutralization of SARS-CoV-2, with similar IC50 values for 614D and 614G (Figures 3A and 3B). The three antisera showed an ADE effect on Huh7-CD32A cells for both 614D and 614G viruses (Figures 3C and 3D). It was noted that IC50 of serum from mouse 1 was lower than that of mouse 2 and 3 (Figures 3A and 3B), and the maximum ADE induction concentration of mouse 1 serum was also lower than that of mouse 2 and 3 (Figures 3C and 3D), suggesting that the ADE induction concentration of the antibody may be related to its neutralization activity.
Figure 3.

Mouse antisera against S1 show neutralization ability and ADE potential
(A and B) Neutralization ability of mouse antisera against SARS-CoV-2 pseudovirus bearing 614D (A) or 614G (B) on Cos7-hACE2 cells.
(C and D) Relative infectivity of SARS-CoV-2 pseudovirus bearing 614D (C) or 614G (D) on Huh7-CD32A cells in the presence of different concentrations of mouse antisera against S1 protein. Data were derived from assays in duplicates with mean values presented.
To further investigate the relationship of the neutralizing ability of the antibody and the maximum induction concentration of ADE, we collected plasma samples from 150 convalescent patients six months after discharge. The disease severity scores of all patients ranged from 3 (hospitalized, not requiring supplemental oxygen) to 6 (hospitalized, requiring ECMO, invasive mechanical ventilation, or both) according to the seven-category ordinal scale (Cao et al., 2020). We performed neutralization and ADE assays using these plasma samples. We tested the neutralization ability against 614G variant as well as the ADE potential of plasma from 93 convalescent patients, including all patients with severity scales of 3 (14 patients), 5 (8 patients), and 6 (1 patient), and equal numbers (35 patients) of male and female patients with a severity scale of 4. All 93 plasma samples showed neutralization ability against 614G variant with IC50 values ranging from 0.06% to 0.76% (Figure 4A).
Figure 4.

Maximum ADE induction concentration of convalescent plasma correlates with IC50
(A and B) Representative results of neutralization (A) and ADE (B) assays for SARS-CoV-2 pseudovirus (614G) using 7 convalescent plasma and 1 healthy control plasma. Data were derived from assays in duplicates with mean values presented.
(C) Correlation of maximum ADE induction concentration with IC50 for 614G variant. Data were derived from 90 convalescent plasma which showed canonical bell-shaped ADE curves. A linear regression model was used for correlation analysis.
Among 93 plasma samples tested, 90 plasma exhibited canonical bell-shaped ADE curves, suggesting that most had the potential to cause ADE. Seven representative ADE curves generated by plasma taken from patients with different disease severities are shown (Figure 4B). To determine the maximum ADE induction concentration for each convalescent plasma, a non-linear regression curve was fitted according to the relative infectivity values, and the plasma concentration corresponding to the maximum infection value was calculated (Figure S6A). Based on the 90 plasma samples with an ADE potential, a medium correlation was found between the IC50 of 614G and the maximum induction concentration of ADE (Pearson r = 0.7207, p < 0.0001). The maximum induction concentration of ADE was generally lower than the IC50 (Figure 4C).
Convalescent plasma from the severe groups mediate a stronger ADE effect than those from the mild group
We analyzed the relationship of the neutralization and ADE characteristics of the convalescent plasma with clinical features. A weak negative correlation between IC50 of 614D wildtype virus and age was observed (Pearson r = −0.2639, p = 0.0012) (Figure S6B), with the decrease in IC50 paralleling the increasing age. In particular, the prediction of a high IC50 was significant among patients aged ≥60 years (Figure S6C). In contrast, from IC50, the peak level of ADE of 614G virus showed no correlation with age (Figure S6D).
Among 93 convalescent plasma samples, although the IC50 of the 614G virus did not differ among severity groups, there was a trend showing that the IC50 was lower in the scale 5 + 6 group compared with those of the scale 3 (p = 0.12) and scale 4 (p = 0.06) groups (Figure 5A). The maximum infectivities in the presence of ADE differed significantly between scale 3 and scale 5 + 6 (p = 0.004), with convalescent plasma from scale 5 + 6 showing a stronger ADE effect compared with that of scale 3. There was no significant difference between scales 3 and 4, scale 4, and scale 5 + 6 (Figure 5B).
Figure 5.

Convalescent plasma from severe groups mediate a stronger ADE effect
(A) Comparison of IC50 for 614G variant in different disease severity scales.
(B) Comparison of the maximum infectivity (614G) in the presence of plasma from convalescent patients with different disease severity. Data are plotted as box and whiskers (minimum to maximum). One-way ANOVA was used to compare the difference between groups. ∗p<0.05; ns, not significant.
ACE2 can act as the secondary receptor in the FcγR-dependent ADE of SARS-CoV-2
Because binding to ACE2 causes a conformational change of the virus S protein, which leads to the protease cleavage and membrane fusion, we suspected that in the absence of the conformational changes caused by S-ACE2 binding, the virus may not be able to complete the subsequent proteolysis, membrane fusion, and invasion, which are important for conventional infection as well as being indispensable for the ADE process.
To directly study the role of ACE2 in the ADE process, we first performed blocking experiments using the antibody against ACE2 or soluble ACE2 (sACE2). As a control, antibody against CD32 showed strong inhibition effect on 614G ADE (Figure 6A). Anti-ACE2 antibody could also inhibit pseudovirus infection in the ADE assay (Figure 6A). However, the control IgG antibody also caused detectable inhibition (Figure 6A), suggesting nonspecific binding through the IgG-CD32A interaction. Simultaneously, sACE2 significantly inhibited virus infection in the presence of antibodies (Figure 6A). This may be resulted from two possibilities: the sACE2 inhibited the binding of the antibodies to the virus; or the sACE2 blocked the binding of the virus to the membrane-localized ACE2 receptors.
Figure 6.

ACE2-mediated entry pathway is involved in SARS-CoV-2 ADE
(A) Blockade assay performed on Huh7-CD32A cells. HIV/SARS-CoV-2 was incubated with 100 ng/mL CB6 IgG1 antibody as indicated, and applied to cells preincubated with anti-ACE2, anti-CD32, or IgG control as indicated. Or sACE2 was co-incubated with HIV/SARS-CoV-2 614G and 100 ng/mL CB6 IgG1 antibody, and then applied to the cells.
(B) Knockdown of ACE2 expression in 293T-CD32A cells by siRNA transfection. ACE2 protein level was detected by western blot. ACE2 mRNA level was detected by real-time RT PCR.
(C) Effect of ACE2 knockdown on SARS-CoV-2 614G ADE. 293T-CD32A cells transfected with or without siACE2 were infected with HIV/SARS-CoV-2 in the presence of different concentrations of CB6.
(D) Inhibition efficiency of fusion inhibitory peptide EK1C4 on HIV/SARS-CoV-2 614G on Cos7-hACE2 cells.
(E and F)Effect of EK1C4 on CB6 mediated SARS-CoV-2 614G ADE on 293T-CD32A (E) and Huh7-CD32A (F) cells. Data were derived from three independent experiments, and are presented as mean ± SD. One-way ANOVA was used to compare the difference between CB6 group and other groups. ∗∗p<0.01; ns, not significant.
To further elucidate the role of ACE2 in the ADE process, we established another CD32A-expressing cell line based on 293T cells, in order to utilize its high transfection efficiency. 293T cells had a similar ACE2 expression level as Huh7 cells and were partially susceptible to SARS-CoV-2 infection (Figures 1A and 1B). As expected, 293T-CD32A cells could support 614G ADE mediated by CB6 antibody. We then knocked down the ACE2 expression by siRNA transfection (Figure 6B) and measured the virus infectivity in the presence of CB6. Compared to the cells transfected with a non-targeting siRNA, the ADE level was significantly reduced in the siACE2 transfected cells (Figure 6C), proving that ACE2 was involved in the ADE process.
Further, to investigate whether the downstream event of S-ACE2 binding, such as membrane fusion, was involved in the ADE process, we applied the fusion inhibitory peptide EK1C4 to the ADE assay (Xia et al., 2020). We first verified that EK1C4 could inhibit the conventional 614G virus entry into Cos7-hACE2 cells (Figure 6D). Then, we showed that compared with the control peptide, EK1C4 completely inhibited the 614G virus infectivity in the presence of CB6 antibody on both 293T-CD32A cells and Huh7-CD32A cells (Figures 6E and 6F), indicating that the membrane fusion is also required for ADE.
Next, we asked whether the membrane fusion was involved in XG005-mediated enhanced infection, which could occur on Raji cells without ACE2 expression (Figure 1D) (Zhou et al., 2021). EK1C4 could completely inhibit the XG005-enhanced 614G infection on Raji cells (Figure S7), indicating that the membrane fusion is also important for the ACE2-independent ADE.
Discussion
In this study, we utilized HIV-based lentiviral vectors to study the characteristics of the ADE effect of SARS-CoV-2. First, we found that immune cells were not susceptible to SARS-CoV-2 pseudovirus infection, and among the immune cells tested, including THP-1, Raji, Daudi, and PBMC, only Daudi cells supported a weak ADE effect mediated by CB6 monoclonal antibody or mouse anti-S1 serum. THP-1 and Raji cells are often used in the ADE research (Jaume et al., 2011; Kuzmina et al., 2018; Wan et al., 2020). We verified that THP-1 could supportADE for MARV and MERS-CoV; and Raji cells could support ADE mediated by another SARS-CoV-2 RBD-targeting monoclonal antibody XG005 (Zhou et al., 2021). These results indicate that different antibodies may elicit ADE by different mechanisms, and FcγR may not be the only receptor required for ADE.
In our investigations of CB6-mediated ADE of SARS-CoV-2, we first identified CD32A as the functional FcγR molecule. We found that the exogenic CD32A-expressing cells are more sensitive and reliable than immune cell lines for performing ADE assays of CB6 as well as mouse antisera and convalescent plasma. For dengue virus, both CD32A and CD64A can mediate ADE, while CD32A functions more efficiently (Rodrigo et al., 2006). In MERS-CoV and SARS-CoV infection, CD32A has been proposed as the functional Fc receptor that mediates ADE (Jaume et al., 2011; Wan et al., 2020). While in filovirus infection, the binding affinity of Fc receptors to the IgG isoforms determines the extent of ADE. For antibodies in IgG1 isoform, blocking of CD64A significantly inhibits the ADE effect, which is consistent with the highest binding affinity of IgG1 to CD64A (Kuzmina et al., 2018). However, it is unclear which FcγR contributes to the SARS-CoV-2 ADE effect. CD32 (Wu et al., 2020), CD32B (Chu et al., 2020; Li et al., 2021), or CD64A (Li et al., 2021) have been shown to mediate ADE, which is different from our present findings. Based on the overexpression of individual FcγR molecules in Huh7 cells, we identified CD32A as the most efficient Fc receptor isoform that supported CB6 IgG1-mediated ADE effect.
The Fc-Fc receptor interaction plays a central role in the ADE process. In the previous reports, cells preincubated with antibodies against FcγRs could reduce the ADE effect. Removal of the Fc region (Wu et al., 2020), or introducing L234A and L235A (LALA) mutations into the Fc region of the antibody (Chu et al., 2020) compromises the binding of the Fc fragment to the Fc receptor (Reusch and Tejada, 2015) and abolished the ADE effect of SARS-CoV-2, further supporting the conclusion that ADE is dependent on Fc-Fc receptor interaction. Our present result shows that while CB6 IgG1 could mediate ADE effect on Huh7-CD32A cells, CB6 in IgM and IgA isotypes could not. Because IgM and IgA naturally bind to FcαRs instead of FcγRs, they could not interact with overexpressed CD32A on the surface of Huh7 cells, therefore could not initiate the ADE process. This result provides new evidence showing that the binding of an antibody to its specific Fc receptor is indispensable for the ADE effect. It will be interesting to investigate whether FcαRs could mediate ADE in the presence of IgM or IgA.
Here, we reveal the involvement of the ACE2-mediated virus entry pathway in the CB6 and antisera/plasma-mediated SARS-CoV-2 ADE process. This argument relies on the evidence as follows. First, the antibody-enhanced infectivity of the 614G variant was much higher than that of the 614D wild-type virus, which is consistent with their ACE2-binding affinities (Korber et al., 2020). This led us to doubt whether SARS-CoV-2 ADE was based on the ACE2-mediated entry pathway.
Further, we found that the maximum ADE induction plasma concentration (plasma dilution) correlated with IC50 (plasma dilution) and ADE occurred only at sub-neutralizing concentrations, suggesting the possibility that the unneutralized S protein could bind to ACE2 to initiate the entry process.
Moreover, we found that besides CD32A, a small amount of ACE2 expression was necessary for the ADE effect. For example, Huh7-CD32A or 293T-CD32A cells supported ADE. In contrast, HeLa-CD32A cells with insufficient ACE2 expression did not support SARS-CoV-2 ADE and HeLa-ACE2-CD32A cells overexpressing ACE2 could not either. In 293T-CD32A cells, knockdown of ACE2 expression significantly inhibited ADE. Further, application of the virus-cell fusion peptide inhibitor EK1C4 blocked ADE on both 293T-CD32A and Huh7-CD32A cells, further providing compelling evidence for the involvement of ACE2-mediated entry pathway in the SARS-CoV-2 ADE. Interestingly, EK1C4 not only inhibits CB6-mediated ACE2-dependent ADE but also XG005-mediated ACE2-independent ADE, revealing the involvement of membrane fusion in different types of ADE. This implies that this fusion inhibitor may be combined with therapeutic antibodies for clinical applications to prevent potential ADE.
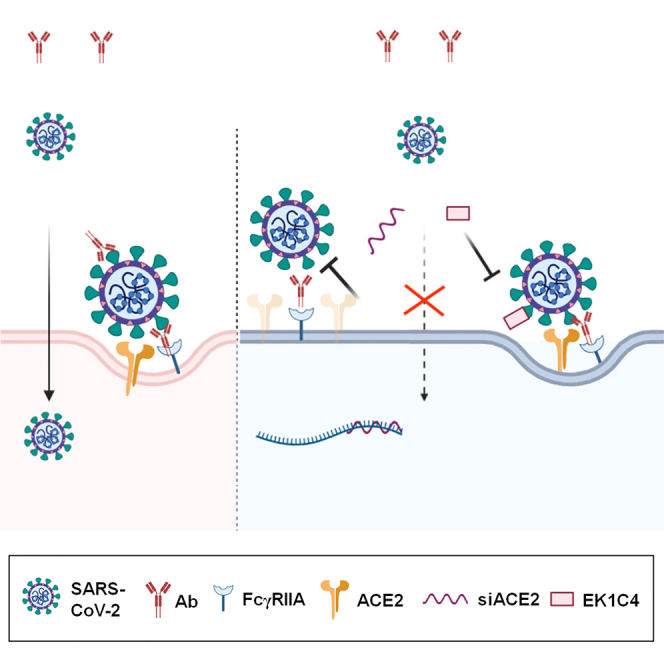
Together, our present findings support the conclusion that different antibodies may mediate ADE through different mechanisms. Under certain circumstance, ACE2 may act as the secondary receptor in the antibody- and FcγR-mediated enhanced entry of SARS-CoV-2. In cells expressing FcγR but not ACE2, or in cells coexpressing ACE2 and FcγR, but in the presence of a high concentration of neutralizing antibodies, the virus can be bridged to FcγRs by the antibodies, but is unable to bind to ACE2. Owing to the lack of ACE2-induced conformational changes of the S protein, the virus cannot complete the entry process required for the subsequent transport to the cytoplasm, and will be ultimately digested in the lysosomes. In cells coexpressing ACE2 and FcγR, sub-neutralizing antibody concentrations promote the attachment of the virus to the cells through Fc-FcγR interaction, as well as enable the unneutralized S protein to bind to ACE2 and initiate the entry procedure. Therefore, FcγR acts as an attachment factor to enhance the virus receptor-mediated invasion in the presence of sub-neutralizing antibodies (Figure 7).
Figure 7.

ADE of ACE2-mediated entry pathway in SARS-CoV-2 infection
FcγR acts as an attachment factor to enhance the virus receptor-mediated invasion in the presence of sub-neutralizing antibodies. This figure was created with BioRender.com.
We emphasize that although we found that most convalescent plasma exhibited ADE potential on Huh7-CD32A cells, they did not enhance virus infectivity of immune cell lines or PBMCs. Our results indicate that ADE might not be a common event in infected individuals. However, considering the diversity of antibodies and the variable status of immune cells in the human bodies, we cannot exclude the possibility that ADE may occur in special cases upon SARS-CoV-2 infection.
ACE2 is mainly expressed in type 2 alveolar epithelial cells, intestinal epithelial cells, bile duct cells, myocardial cells, and renal proximal tubular cells, but rarely expressed in immune cells (Qi et al., 2020), implying that immune cells are not commonly susceptible to SARS-CoV-2 (Zhou et al., 2020). However, the expression of ACE2 may be regulated by the pathophysiological state. For instance, IFNα and IFNγ increase the level of ACE2 in epithelial cells and enhance the susceptibility to SARS-CoV-2 (Busnadiego et al., 2020). Therefore, the expression of ACE2 may be upregulated because the inflammatory pathway is activated in patients with COVID-19 (Huang et al., 2020). Therefore, the mechanisms of the expression regulation of ACE2 in immune cells under pathological conditions and its effect on potential ADE are critically important to explore in the future.
Limitations of the study
Owing to the lack of live virus test, we cannot know whether the ADE process simulated by the pseudovirus is consistent with that of live virus. Especially, whether there is enhanced viral replication followed by the enhanced entry is unclear. In addition, the coexpression pattern of ACE2 and CD32A is not analyzed in the cells from individuals infected with SARS-CoV-2, which makes the possibility of ADE occurring in COVID-19 unclear.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SARS-CoV-2 S2, human IgG1 | Sino Biological Inc | CAT#40590-D001; RRID:AB_2857932 |
| MERS-CoV S R723, rabbit | Sino Biological Inc | CAT#40069-R723; RRID:AB_2860455 |
| MERS-CoV S T62, rabbit | Sino Biological Inc | CAT#40069-T62; RRID:AB_2904183 |
| MERS-CoV S MM23, mouse | Sino Biological Inc | CAT#40069-MM23; RRID:AB_2860454 |
| ACE2, rabbit | Abcam | CAT#ab15348; RRID:AB_301861 |
| β-actin, mouse | Sigma | CAT#A5316; RRID:AB_476743 |
| p24, rabbit | Sino Biological Inc | CAT#11695-RP02; RRID:AB_2904185 |
| anti-CD16 conjugated with BV421, mouse IgG1 | BD Biosciences | CAT#562878; RRID:AB_2737861 |
| anti-CD32 conjugated with FITC, mouse IgG1 | BD Biosciences | CAT#552883; RRID:AB_394512 |
| anti-CD64 conjugated with APC, mouse IgG1 | BD Biosciences | CAT#561189; RRID:AB_10611566 |
| control mouse IgG1 conjugated with BV421 | BD Biosciences | CAT#562438; RRID:AB_11207319 |
| control mouse IgG1 conjugated with FITC | BD Biosciences | CAT#555748; RRID:AB_396090 |
| control mouse IgG1 conjugated with APC | BD Biosciences | CAT#554681; RRID:AB_398576 |
| CD16, mouse | BD Biosciences | CAT#555404; RRID:AB_395804 |
| CD32, mouse | BD Biosciences | CAT#555447; RRID:AB_395840 |
| CD64, mouse | BD Biosciences | CAT#555525; RRID:AB_395911 |
| anti-CD16 conjugated with Pacific blue | Beijing 4A Biotech | CAT#FHW016-100 |
| HRP-conjugated goat anti-human IgG | Solarbio | CAT#SE101; RRID:AB_2904186 |
| goat anti-human IgG conjugated with PE | Abcam | CAT#ab98596; RRID:AB_10673825 |
| IRDye® 800CW Goat anti-Mouse IgG | LI-COR Biosciences | CAT#925-32210; RRID:AB_2687825 |
| IRDye® 680RD Goat anti-Rabbit IgG | LI-COR Biosciences | CAT#925-68071; RRID:AB_2721181 |
| anti-SARS-CoV-2 CB6, human IgG1 | this paper | N/A |
| anti-SARS-CoV-2 CB6, human IgM | this paper | N/A |
| anti-SARS-CoV-2 CB6, human IgA | this paper | N/A |
| anti-SARS-CoV-2 N1G12, human IgG1 | this paper | N/A |
| mouse serum anti-SARS-CoV-2 S1 | this paper | N/A |
| anti-SARS-CoV-2 XG005 | Dr. Qiao Wang | N/A |
| anti-EBOV ADI15878 | Dr. Linqi Zhang | N/A |
| anti-EBOV 206 | Dr. Linqi Zhang | N/A |
| anti-EBOV 314 | Dr. Linqi Zhang | N/A |
| anti-MARV MR191 | Dr. Linqi Zhang | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| EK1C4 | Dr. Shibo Jiang & Dr. Lu Lu | N/A |
| control peptide: YDHTKNYPFDVDQ | this paper | N/A |
| sACE2 | this paper | N/A |
| Critical commercial assays | ||
| Vironostika HIV-1 Antigen MicroELISAKit | Biomerieux bv, Boxtel | CAT#VPK-107 |
| Experimental models: Cell lines | ||
| 293T | ATCC | CAT#CRL-3216 |
| THP-1 | ATCC | CAT#TIB-202 |
| Raji | ATCC | CAT#CCL-86 |
| Daudi | Nanjing Cobioer Gene Technology | CAT#CBP60262 |
| Cos7-hACE2 | Beijing Vitalstar Biotechnology | CAT#hACE2-COS7 |
| Huh7 | Dr. Linqi Zhang | N/A |
| HeLa | Dr. Linqi Zhang | N/A |
| Oligonucleotides | ||
| siNT: UUCUCCGAACGUGUCACGUTT | this paper | N/A |
| siACE2-1: CCAUCUACAGUACUGGAAATT | this paper | N/A |
| siACE2-2: GGCCAUUAUAUGAAGAGUATT | this paper | N/A |
| CD16A forward: GACAGCGGCTCCTACTTCTG | this paper | N/A |
| CD16A reverse: AGTCCTGTGTCCACTGCAAA | this paper | N/A |
| CD32A forward: TCCCACAAGCAAACCACAGT | this paper | N/A |
| CD32A reverse: TGCTACAGCAGTCGCAATGA | this paper | N/A |
| CD32B forward: AGCGGATTTCAGCCAATCCC | this paper | N/A |
| CD32B reverse: ATACGGTTCTGGTCATCAGGC | this paper | N/A |
| CD64A forward: AAGTCACAATGGCACCTACC | this paper | N/A |
| CD64A reverse: GCTCAGGGTGACCAGATTCC | this paper | N/A |
| ACE2 forward: CATTGGTCTTCTGTCACCCGA | this paper | N/A |
| ACE2 reverse: ATGCGGGGTCACAGTATGTT | this paper | N/A |
| GAPDH forward: CTGCACCACCAACTGCTTAG | this paper | N/A |
| GAPDH reverse: GAGCTTCCCGTTCAGCTCAG | this paper | N/A |
| Recombinant DNA | ||
| pSARS-CoV-2-S, Wuhan-Hu-1 strain | this paper | N/A |
| pSARS-CoV-2-S, D614G | this paper | N/A |
| pSARS-CoV-2-S, N501Y-D614G | this paper | N/A |
| pSARS-CoV-2-S, E484K-N501Y-D614G | this paper | N/A |
| pSARS-CoV-2-S, B.1.351 strain | this paper | N/A |
| pMERS-CoV-S | Wang et al. (2020) | N/A |
| pMARV-GP | Dr. Linqi Zhang | N/A |
| pVSVG | Addgene | CAT#35616 |
| pLP1 | YouBio | CAT#VT1489 |
| pLP2 | YouBio | CAT#VT1490 |
| pNL Luc E- R- | Wang et al. (2020) | N/A |
| pLenti-Cas9-Blast | Addgene | CAT#52962 |
| pLenti-CD16A-Blast | this paper | N/A |
| pLenti-CD32A-Blast | this paper | N/A |
| pLenti-CD32B-Blast | this paper | N/A |
| pLenti-CD64A-Blast | this paper | N/A |
| pcDH-EF1-MCS-T2A-puro | SBI System Biosciences | CAT#CD527A-1 |
| pcDH-FCER1G-puro | this paper | N/A |
| Software and algorithms | ||
| ImageStudio software | LI-COR Biosciences | https://www.licor.com/bio/image-studio/ |
| GraphPad Prism software version 8.01 | GraphPad Software, Inc. | https://www.graphpad.com/scientificsoftware/prism/ |
| FlowJo V10 | TreeStar | https://www.flowjo.com/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Bin Cao (caobin_ben@163.com)
Materials availability
Constructs generated as part of this study are available upon request via an appropriate material transfer agreement. Sharing of antibodies with academic researchers may require a payment to cover the cost of generation and a completed Material Transfer Agreement.
Experimental models and subject details
Antibodies, antisera/plasma and soluble ACE2
Humanized N1G12 and CB6 antibodies were generated by Shenzhen HeavyBio Technology.
The amino acid sequences of variable regions of heavy chain (CB6VH) and light chain (CB6VL) were obtained from GenBank (MT470197 and MT470196) (Shi et al., 2020).
CB6VH
EVQLVESGGGLVQPGGSLRLSCAASGFTVSSNYMSWVRQAPGKGLEWVSVIYSGGSTFYADSVKGRFTISRDNSMNTLFLQMNSLRAEDTAVYYCARVLPMYGDYLDYWGQGTLVTVSS.
CB6VL
DIVMTQSPSSLSASVGDRVTITCRASQSISRYLNWYQQKPGKAPKLLIYAASSLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQSYSTPPEYTFGQGTKLEIKCB6.
To generate the CB6 expression vectors, the signal peptide from CD33 (MPLLLLLPLLWAGALA) was added to the N-terminal of the peptides. The constant region of human Kappa was added to the C-terminal of CB6VL. The constant region of human IgM (Mu), IgA (Alpha) or IgG (Gamma) was added to the C-terminal of CB6VH. The CB6 antibodies were then expressed by HEK293S cells (originating from a female fetus) and purified with Protein L columns for IgM and IgA antibodies (Smart-Lifesciences, Changzhou, China) and Protein A column for IgG antibody (Bestchrom, Shanghai, China).
Kappa
TVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC.
Mu
GSASAPTLFPLVSCENSPSDTSSVAVGCLAQDFLPDSITFSWKYKNNSDISSTRGFPSVLRGGKYAATSQVLLPSKDVMQGTDEHVVCKVQHPNGNKEKNVPLPVIAELPPKVSVFVPPRDGFFGNPRKSKLICQATGFSPRQIQVSWLREGKQVGSGVTTDQVQAEAKESGPTTYKVTSTLTIKESDWLGQSMFTCRVDHRGLTFQQNASSMCVPDQDTAIRVFAIPPSFASIFLTKSTKLTCLVTDLTTYDSVTISWTRQNGEAVKTHTNISESHPNATFSAVGEASICEDDWNSGERFTCTVTHTDLPSPLKQTISRPKGVALHRPDVYLLPPAREQLNLRESATITCLVTGFSPADVFVQWMQRGQPLSPEKYVTSAPMPEPQAPGRYFAHSILTVSEEEWNTGETYTCVVAHEALPNRVTERTVDKSTGKPTLYNVSLVMSDTAGTCY.
Alpha
ASPTSPKVFPLSLCSTQPDGNVVIACLVQGFFPQEPLSVTWSESGQGVTARNFPPSQDASGDLYTTSSQLTLPATQCLAGKSVTCHVKHYTNPSQDVTVPCPVPSTPPTPSPSTPPTPSPSCCHPRLSLHRPALEDLLLGSEANLTCTLTGLRDASGVTFTWTPSSGKSAVQGPPERDLCGCYSVSSVLPGCAEPWNHGKTFTCTAAYPESKTPLTATLSKSGNTFRPEVHLLPPPSEELALNELVTLTCLARGFSPKDVLVRWLQGSQELPREKYLTWASRQEPSQGTTTFAVTSILRVAAEDWKKGDTFSCMVGHEALPLAFTQKTIDRLAGKPTHVNVSVVMAEVDGTCY.
Gamma
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK.
To obtain the mouse monoclonal antibodies against SARS-CoV-2, the RBD domain with His tag was expressed from HEK293S cells and immunized 6-8-week female Balb/c mice. Monoclonal antibodies were purified from the supernatant of hybridoma cells subcloned with spleen cells from immunized mice and SP2/0 myeloma cells (Shenzhen HeavyBio Technology).
N1G12 antibody sequence was retrieved from one of the above bybridoma clones. The amino acid sequences of variable regions of heavy chain (N1GVH) and light chain (N1GVL) are as follows:
N1GVH
QIQLVQSGPELKKPGETVKISCKASGYIFRNYGMNWVKQSPGKTLKWMGWINTYTGEPTYADDFKGRIALSLETSANTAYLQINNLKNEDMATYFCARSTGTEWFGYWGQGTLVTVSA.
N1GVL
DIVMSQSPSSLAVSAGEKVTMSCKSSQSLLNSRNRKNYLAWYQQKPGQSPKLLIYWASTRESGVPDRFRGSGSGTDFTLTISSVQAEDLAVYYCKQSYNLITFGAGTKLELK.
To generate the N1G12 expression vector, the signal peptide from CD33 was added to the N-terminal of the peptides. The constant region of human Kappa was added to the C-terminal of N1GVL. The constant region of human IgG (Gamma) was added to the C-terminal of N1GVH. The expression and purification of the antibody were similar to that of CB6 IgG antibody.
Anti-S2 of SARS-CoV-2, MERS-CoV S antibodies R723, T62, MM23 were from Sino Biological Inc. EBOV and MARV Antibodies ADI15878, 206, 314 and MR191 were gifts from Dr. Linqi Zhang (Tsinghua University). Monoclonal antibody GX005 (Zhou et al., 2021) targeting SARS-CoV-2 RBD was provided by Dr. Qiao Wang (Fudan University).
To obtain anti-SARS-CoV-2 mouse serum, 100 μg SARS-CoV-2 S1 protein (Shenzhen HeavyBio Technology) was mixed with Freund's adjuvant, immunized 6-8-week female Balb/c mice in the hindlimbs, boosted 3 weeks later, and the serum was collected 3 weeks later (Ruibiotech).
The recruitment criteria for COVID-19 patients and the sampling strategies have been reported previously (Cao et al., 2020). All patients were hospitalized with COVID-19 of grade 3 or more on the seven-category ordinal scale (Cao et al., 2020). The plasma from convalescent patients were collected 6 months after their discharge, with informed consent signed by every patient. The procedures were approved by the Ethics Committee of China-Japan Friendship Hospital (Beijing, China; KY-2020–78.01), and complied with all relevant ethical regulations regarding human research.
The extracellular domain of ACE2 (17–740 aa) with mutation in the enzyme activity site (H374N & H378N) and with a His tag (GGGSASHHHHHHHHHH) at C-terminus was expressed from 293T cells (originating from a female fetus) (Shenzhen HeavyBio Technology).
Constructions
The spike gene of SARS-CoV-2 (Wuhan-Hu-1 strain and B.1.351 strain) was codon-optimized and cloned into BamHI/EcoRI in pcDNA3.1(+) with C-terminal 19-aa deletion 3 to generate pcDNA-SARS2-S (Wang et al., 2020). The spike genes with D614G, N501Y-614G, E484K-N501Y-D614G mutants, were generated by site-directed point mutation (TransGen Biotech). The MERS-CoV S gene was codon-optimized and cloned into pcDNA3.1(+) (Wang et al., 2020). The MARV glycoprotein expression vector was a gift from Dr. Linqi Zhang (Tsinghua University).
Human FcγR genes CD16A (GenBank: NM_000569.8), CD32A (GenBank: NM_001136219.3), CD32B (GenBank: NM_001002273.3) and CD64A (GenBank: NM_000566.4) were synthesized and cloned into XbaI/BamHI sites in pLenti-Cas9-Blast (Addgene) to substitute the Cas9 gene, and pLenti-CD16A-Blast, pLenti-CD32A-Blast, pLenti-CD32B-Blast and pLenti-CD64A-Blast were obtained. Human FCER1G gene was synthesized and cloned into EcoRI/BamHI sites in pCDH-EF1-MCS-IRES-puro to generate pcDH-FCER1G-puro. The vector pCDH-EF1-MCS-IRES-puro was constructed based on pcDH-EF1-MCS-T2A-puro (SBI System Biosciences) by synthesizing IRES sequence according to pIRES2-EGFP (Clontech) followed by puromycin coding sequence and cloning into NotI/SalI sites.
Cells
293T, THP-1 (male) and Raji (male) cells were from ATCC. Daudi (male) cells were from Nanjing Cobioer Gene Technology. Cos7-hACE2 cells (male) were from Beijing Vitalstar Biotechnology. Huh7 (male) and HeLa (female) cells were gifts from Dr. Linqi Zhang (Tsinghua University). PBMCs were separated from the whole blood donated by the authors (male) by a density gradient centrifugation method using histopaque 1077 (Sigma). Monocytes were purified using anti-CD14 beads (Miltenyi Biotec). 293T, Cos7-hACE2, Huh7 and its derivative cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Immune cells and cell lines were maintained in 1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were cultured at 37°C with 5% CO2.
To generate FcγR-expressing cells, 1.5 μg pVSVG (Addgene), 2 μg pLP1, 2 μg pLP2 (YouBio) and 5 μg pLenti-CD16A/CD32A/CD32B/CD64A-Blast or pcDH-FCER1G-puro were cotransfected into 2.5 x 106 293T cells. The medium was changed 12 hours post transfection. The supernatants were collected 48 hours post transfection, and passed through a 0.45 μm filter (milipore). The supernatants were added to Huh7 or HeLa culture media with 8 μg/mL polybrene. 48 hours post infection, cells were selected by 10 μg/mL blasticidin or 5 μg/mL puromycin to generate Huh7-or HeLa- CD16A/CD32A/CD32B/CD64A cells or FCER1G expressing cells. 293T-CD32A cells were also generated according to the above method.
Method details
siRNA transfection
293T cells were seeded at 2 x 105 per 3.5 cm dishes 24 hours before transfection. 4 μg siRNA was transfected with 6 μL lipofectamine 3000 (Life Technologies) according to the protocol. siRNAs were synthesized by GenePharma (Shanghai, China): siNT: UUCUCCGAACGUGUCACGUTT; siACE2-1: CCAUCUACAGUACUGGAAATT; siACE2-2: GGCCAUUAUAUGAAGAGUATT. For detection of mRNA and protein levels, cells were harvested 48 hours post transfection, and were subjected to the real-time RT PCR or Western blot. For ADE assay, cells were trypsinized and seeded in 96-well plates at the density of 7,500 cells/well 24 hours post transfection. ADE assay was performed another 24 hours later.
ELISA
0.1 μg His-tagged SARS-CoV-2 S1 protein expressed by HEK293S cells (Shenzhen HeavyBio Technology) in CBS buffer (1.6 g Na2CO3, 2.92 g NaHCO3/L) were coated on a 96-well microtiter plate (Solarbio) overnight at 4°C. Wells were blocked with 5% non-fat milk in CBS buffer for 1 hour at 37°C, followed by incubation with serially diluted antibodies in 1% BSA/PBS for 1 hour at 37°C. Horseradish peroxidase (HRP)-conjugated goat anti-human IgG antibody (Solarbio) was added at 1:5000 dilution for 1 hour at 37°C. Colors were developed by adding TMB substrate for 10 minutes at 37°C and then stop buffer (Thermo Fisher Scientific) was added. Plate was read at 450 nm on a Multiskan FC plate reader (Thermo Scientific, USA).
Real-time RTPCR
5 x 105 cells were seeded in 6 cm dish, lyzed by Trizol, and the total RNA was isolated. After reverse transcription, real-time PCR was performed using the following primers: CD16A: forward: GACAGCGGCTCCTACTTCTG; reverse: AGTCCTGTGTCCACTGCAAA; CD32A: forward: TCCCACAAGCAAACCACAGT; reverse: TGCTACAGCAGTCGCAATGA; CD32B: forward: AGCGGATTTCAGCCAATCCC; reverse: ATACGGTTCTGGTCATCAGGC; CD64A: forward: AAGTCACAATGGCACCTACC; reverse: GCTCAGGGTGACCAGATTCC; ACE2: forward: CATTGGTCTTCTGTCACCCGA; reverse: ATGCGGGGTCACAGTATGTT; GAPDH: forward: CTGCACCACCAACTGCTTAG; reverse: GAGCTTCCCGTTCAGCTCAG.
Western blot
To detect ACE2 expression in 293T-CD32A cells, cells in 3.5 cm dishes were lysed in 100 μL SDS loading buffer. To detect S proteins in pseudotyped viruses, 5 mL 293T supernatant containing virus particles was passed through 0.22 μm filter, ultracentrifuged at 110,000 g × 90 min, resuspended in 50 μL 1 X SDS loading buffer, The lysates were subjected to 4–20% PAGE gel (Yeasen), transferred onto PVDF Immobilon®-Psq transfer membrane (Millipore). Blots were blocked with Odyssey® Blocking Buffer (TBS) (LI-COR Biosciences). First antibodies used were rabbit anti-ACE2 (Abcam), mouse anti–β-actin (Thermo Fisher Scientific), anti-S1 mouse serum and rabbit anti-p24 (Sino Biological Inc) as indicated. Secondary antibodies used were IRDye® 800CW Goat anti-Mouse IgG and IRDye® 680RD Goat anti-Rabbit IgG (LI-COR Biosciences). The blots were imaged on the Odyssey® CLx imaging system using 700 nm and 800 nm channels and analyzed using ImageStudio software (LI-COR Biosciences).
Flow cytometry
To detect CD16A, CD32A, CD32B and CD64A expression, 5 x 105 cells were incubated with anti-CD16 conjugated with BV421, anti-CD32 conjugated with FITC and anti-CD64 conjugated with APC (BD Biosciences) in 100 μL PBS containing 1% BSA. To detect CD16A expression in FCER1G co-expressed cell, anti-CD16 conjugated with Pacific blue (Beijing 4A Biotech) was used. After washing for three times with PBS, cells were resuspended in 500 μL PBS containing 1% BSA, passed through 40 μm cell strainer (BD Falcon) and then subjected to flow cytometry analysis. Non-targeting antibodies in IgG1 isotype conjugated with the same fluorophore were used as negative controls for each immune cell type. Huh7 or HeLa cells without FcγR overexpression stained with the same antibodies were used as the negative controls for their derivatives.
To test the binding ability of anti-S antibodies with S protein, pcDNA-SARS2-S was transfected into 293T cells. 48 hours later, the cells were trypsinized. 5 x 105 cells were resuspended in 100 μL binding buffer (Biolegend) and incubated with 2.5 or 10 ng/mL antibodies as indicated for 45 minutes on ice. After washing for three times, cells were resuspended in 100 μL binding buffer containing goat anti-human IgG conjugated with PE (Abcam) and incubated for 30 minutes on ice. After washing for three times, the cells were resuspended in 500 μL binding buffer, passed through 40 μm cell strainer (BD Falcon) and then subjected to flow cytometry analysis. Flow cytometry data were analyzed by FlowJo V10 (TreeStar).
Pseudotyped virus production
To produce HIV-based pseudoviruses, 10 μg pNL Luc E− R- and 10 μg virus envelope protein expression vectors were co-transfected into 4.5 x 106 293T cells. The medium was replaced with fresh medium 12 hours later, and the supernatants of transfected cells were harvested 48 hours post transfection and passed through a 0.45 μm filter. The pseudotyped virus was normalized by p24 ELISA using a Vironostika HIV-1 Antigen MicroELISA Kit (Biomerieux bv, Boxtel). The supernatant containing 5 ng pseudotyped virus (p24) was used to infect cells in 96-well plates.
Neutralization, blockade and ADE assays
Antibodies or antisera/plasma with a series of dilution was incubated with pseudovirus for 1 hour at 37°C and then added to 7.5 x 103 Cos7-hACE2 cells for SARS-CoV-2 neutralization assay, to Huh7 cells for MERS-CoV and MARV neutralization assays; or added to 7.5 x 103 Huh7-derived cells, 293T-CD32A cells or 5 x 104 immune cells for ADE assays as indicated in the text.
For blockade assays by antibodies, 10 μg/mL antibodies were incubated with Huh7-CD32A cells at 4°C for one hour. At the same time, 5 ng pseudotyped virus was incubated with 100 ng/mL CB6 antibody at 37°C for one hour, and then the mixture was applied to the antibody-treated cells.
EK1C4 peptide (Xia et al., 2020) was kindly provided by Dr. Shibo Jiang and Dr. Lu Lu (Fudan University). Control peptide (YDHTKNYPFDVDQ) was derived from royal jelly and had no inhibition effect on SARS-CoV-2 entry as previously tested. For peptide blocking assay on Cos7-hACE2 cells, 5 ng pseudotyped virus was incubated with different concentrations of peptides at 37°C for 30 minutes, and then the mixture was applied to the cells for 12 hours before the medium was changes. For peptide blocking assay for ADE, 5 ng pseudotyped virus was incubated with different concentrations of CB6 antibody as well as 3.2 μM peptides at 37°C for 30 minutes, and then the mixture was applied to the cells for 12 hours before the medium was changes.
In all the above assays, the cells were lysed at 48 hours post infection, and luciferase activity was measured by the addition of equal volumn of luciferase substrate (Promega) and measured in a Spark® multimode microplate reader (Tecan). The titers of neutralizing antibodies were calculated as 50% inhibitory concentration (IC50) in neutralization assays.
Quantification and statistical analysis
Student's t-test was used to compare the differences of continuous variables that met a normal distribution between two groups, and if the distribution deviates from a normal distribution, the Mann-Whitney U-test was used. In case of more than two-group comparisons, one-way analysis of variance followed by the Tukey's post hoc test was used. To calculate IC50, a non-linear regression curve was fitted based on the relative inhibition values. To derive the concentration of maximum induction of ADE, a non-linear regression curve was fitted based on the relative infectivity values. Repeated measurement of one-way ANOVA was used to compare the difference between the antibody-mediated enhanced infectivity and the infectivity at basal level. A linear regression model was used for correlation analysis. Cubic spline curve was plotted to illustrate the prediction for binarized IC50 of 614D variant with increasing age, and the magnitude of prediction is expressed as odds ratio (OR) and 95% confidence interval (CI). All statistical analyses were performed using the GraphPad Prism software version 8.01 (GraphPad Software, Inc.). P < 0.05 was considered statistically significant. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, not statistically significant.
Acknowledgments
We thank Dr. Linqi Zhang and Dr. Xuanling Shi (Tsinghua University), Dr. Shibo Jiang and Dr. Lu Lu, and Dr. Qiao Wang (Fudan University) for sharing experimental materials, Dr. Fei Guo (Chinese Academy of Medical Sciences and Peking Union Medical College), Dr. Aihua Zheng (Chinese Academy of Sciences), Dr. Peigang Wang (Capital Medical University), and Dr. Kehu Yuan (Shenzhen HeavyBio Technology) for discussion of the manuscript. This work is supported by National Key R&D Program of China (2020YFA0707600), Medical and Health Science Technology Innovation Project of Chinese Academy of Medical Sciences (2020-I2M-2-014), Project of BRC-BC (Biomedical Translational Engineering Research Center of BUCT-CJFH) (XK2020-09), Natural Science Foundation of China (82170015, 82041011 & 81800002); Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS 2018-I2M-1-003& 2020-I2M-CoV19-005).
Author contributions
ZW, HBL, and BC conceived and designed the project. ZW, TTD, and YLZ carried out the main part of experiments. QQN, SNY, and PPL contributed to the preparation of the pseudotyped virus. PFP and LC contributed to the fusion peptide inhibition assays. WQN contributes to the statistical analysis. ZW and YLZ wrote the manuscript. HBL and BC revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103720.
Contributor Information
Haibo Li, Email: shrineswe@vip.qq.com.
Bin Cao, Email: caobin_ben@163.com.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Arvin A.M., Fink K., Schmid M.A., Cathcart A., Spreafico R., Havenar-Daughton C., Lanzavecchia A., Corti D., Virgin H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature. 2020;584:353–363. doi: 10.1038/s41586-020-2538-8. [DOI] [PubMed] [Google Scholar]
- Busnadiego I., Fernbach S., Pohl M.O., Karakus U., Huber M., Trkola A., Stertz S., Hale B.G. Antiviral activity of type I, II, and III interferons counterbalances ACE2 inducibility and restricts SARS-CoV-2. mBio. 2020;11:e01928-20. doi: 10.1128/mBio.01928-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., et al. A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020;382:1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F., Zhang A.J., Yuan S., Poon V.K., Chan C.C., Lee A.C., Chan W.M., Fan Z., Tsoi H.W., Wen L., et al. Simulation of the clinical and pathological manifestations of coronavirus disease 2019 (COVID-19) in a golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin.Infect.Dis. 2020;71:2428–2446. doi: 10.1093/cid/ciaa325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H., Chan J.F., Wang Y., Yuen T.T., Chai Y., Hou Y., Shuai H., Yang D., Hu B., Huang X., et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin.Infect. Dis. 2020;71:1400–1409. doi: 10.1093/cid/ciaa410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W., Jumnainsong A., Onsirisakul N., Fitton P., Vasanawathana S., Limpitikul W., Puttikhunt C., Edwards C., Duangchinda T., Supasa S., et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science. 2010;328:745–748. doi: 10.1126/science.1185181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nicolas O., V'Kovski P., Zettl F., Zimmer G., Thiel V., Summerfield A. No evidence for human monocyte-derived macrophage infection and antibody-mediated enhancement of SARS-CoV-2 infection. Front Cell Infect Microbiol. 2021;11:644574. doi: 10.3389/fcimb.2021.644574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs M.L., Selvaraj P., Carpen O., Springer T.A., Kuster H., Jouvin M.H., Kinet J.P. Mechanisms for regulating expression of membrane isoforms of Fc gamma RIII (CD16) Science. 1989;246:1608–1611. doi: 10.1126/science.2531918. [DOI] [PubMed] [Google Scholar]
- Homsy J., Meyer M., Tateno M., Clarkson S., Levy J.A. The Fc and not CD4 receptor mediates antibody enhancement of HIV infection in human cells. Science. 1989;244:1357–1360. doi: 10.1126/science.2786647. [DOI] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaume M., Yip M.S., Cheung C.Y., Leung H.L., Li P.H., Kien F., Dutry I., Callendret B., Escriou N., Altmeyer R., et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcgammaR pathway. J. Virol. 2011;85:10582–10597. doi: 10.1128/JVI.00671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam Y.W., Kien F., Roberts A., Cheung Y.C., Lamirande E.W., Vogel L., Chu S.L., Tse J., Guarner J., Zaki S.R., et al. Antibodies against trimeric S glycoprotein protect hamsters against SARS-CoV challenge despite their capacity to mediate FcgammaRII-dependent entry into B cells in vitro. Vaccine. 2007;25:729–740. doi: 10.1016/j.vaccine.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e819. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni R. In: Dynamics of Immune Activation in Viral Diseases. Bramhachari P.V., editor. Springer Singapore; Singapore: 2020. Antibody-dependent enhancement of viral infections; pp. 9–41. [Google Scholar]
- Kuzmina N.A., Younan P., Gilchuk P., Santos R.I., Flyak A.I., Ilinykh P.A., Huang K., Lubaki N.M., Ramanathan P., Crowe J.E., Jr., et al. Antibody-dependent enhancement of Ebola virus infection by human antibodies isolated from survivors. Cell Rep. 2018;24:1802–1815.e1805. doi: 10.1016/j.celrep.2018.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M.D., de Graaf E.L., Sonneveld M.E., Plomp H.R., Nouta J., Hoepel W., Chen H.J., Linty F., Visser R., Brinkhaus M., et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science. 2021;371:eabc8378. doi: 10.1126/science.abc8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.S., Wheatley A.K., Kent S.J., DeKosky B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020;5:1185–1191. doi: 10.1038/s41564-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Edwards R.J., Manne K., Martinez D.R., Schafer A., Alam S.M., Wiehe K., Lu X., Parks R., Sutherland L.L., et al. In vitro and in vivo functions of SARS-CoV-2 infection-enhancing and neutralizing antibodies. Cell. 2021;184:4203–4219.e32. doi: 10.1016/j.cell.2021.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Soh W.T., Kishikawa J.I., Hirose M., Nakayama E.E., Li S., Sasai M., Suzuki T., Tada A., Arakawa A., et al. An infectivity-enhancing site on the SARS-CoV-2 spike protein targeted by antibodies. Cell. 2021;184:3452–3466.e3418. doi: 10.1016/j.cell.2021.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai H., Kurokawa M., Matsui S., Yamamoto T., Kuroki Y., Kishimoto C., Shiraki K. Infection enhancement of influenza A NWS virus in primary murine macrophages by anti-hemagglutinin monoclonal antibody. J. Med. Virol. 1992;36:217–221. doi: 10.1002/jmv.1890360312. [DOI] [PubMed] [Google Scholar]
- Olsen C.W., Corapi W.V., Ngichabe C.K., Baines J.D., Scott F.W. Monoclonal antibodies to the spike protein of feline infectious peritonitis virus mediate antibody-dependent enhancement of infection of feline macrophages. J. Virol. 1992;66:956–965. doi: 10.1128/jvi.66.2.956-965.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perno C.F., Baseler M.W., Broder S., Yarchoan R. Infection of monocytes by human immunodeficiency virus type 1 blocked by inhibitors of CD4-gp120 binding, even in the presence of enhancing antibodies. J. Exp. Med. 1990;171:1043–1056. doi: 10.1084/jem.171.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F., Qian S., Zhang S., Zhang Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem.Biophys. Res. Commun. 2020;526:135–140. doi: 10.1016/j.bbrc.2020.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch D., Tejada M.L. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25:1325–1334. doi: 10.1093/glycob/cwv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke D.O. Two different antibody-dependent enhancement (ADE) risks for SARS-CoV-2 antibodies. Front Immunol. 2021;12:640093. doi: 10.3389/fimmu.2021.640093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson W.E., Jr., Montefiori D.C., Mitchell W.M. Antibody-dependent enhancement of human immunodeficiency virus type 1 infection. Lancet. 1988;1:790–794. doi: 10.1016/s0140-6736(88)91657-1. [DOI] [PubMed] [Google Scholar]
- Rodrigo W.W., Jin X., Blackley S.D., Rose R.C., Schlesinger J.J. Differential enhancement of dengue virus immune complex infectivity mediated by signaling-competent and signaling-incompetent human Fcgamma RIA (CD64) or FcgammaRIIA (CD32) J. Virol. 2006;80:10128–10138. doi: 10.1128/JVI.00792-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Shan C., Duan X., Chen Z., Liu P., Song J., Song T., Bi X., Han C., Wu L., et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584:120–124. doi: 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- Takeda A., Sweet R.W., Ennis F.A. Two receptors are required for antibody-dependent enhancement of human immunodeficiency virus type 1 infection: CD4 and Fc gamma R. J. Virol. 1990;64:5605–5610. doi: 10.1128/jvi.64.11.5605-5610.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Shang J., Sun S., Tai W., Chen J., Geng Q., He L., Chen Y., Wu J., Shi Z., et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J. Virol. 2020;94:e02015–e02019. doi: 10.1128/JVI.02015-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.G., Tang D.J., Hua Z., Wang Z., An J. Sunitinib reduces the infection of SARS-CoV, MERS-CoV and SARS-CoV-2 partially by inhibiting AP2M1 phosphorylation. Cell Discov. 2020;6:71. doi: 10.1038/s41421-020-00217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.F., Tseng S.P., Yen C.H., Yang J.Y., Tsao C.H., Shen C.W., Chen K.H., Liu F.T., Liu W.T., Chen Y.M., et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem.Biophys. Res. Commun. 2014;451:208–214. doi: 10.1016/j.bbrc.2014.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Yan R., Liu M., Liu Z., Wang Y., Luan D., Wu K., Song Z., Sun T., Ma Y., et al. Antibody-dependent enhancement (ADE) of SARS-CoV-2 infection in recovered COVID-19 patients: studies based on cellular and structural biology analysis. medRxiv. 2020 doi: 10.1101/2020.10.08.20209114. [DOI] [Google Scholar]
- Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liu Z., Li S., Xu W., Zhang Q., Silva I.T., Li C., Wu Y., Jiang Q., Liu Z., et al. Enhancement versus neutralization by SARS-CoV-2 antibodies from a convalescent donor associates with distinct epitopes on the RBD. Cell Rep. 2021;34:108699. doi: 10.1016/j.celrep.2021.108699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.