

Abstract
Introduction
This work investigated the relationship between cerebrovascular disease (CVD) markers and Alzheimer's disease (AD) biomarkers of amyloid beta deposition, and neurofibrillary tau tangles in subjects spanning the AD clinical spectrum.
Methods
A total of 136 subjects participated in this study. Four groups were established based on AD biomarker positivity from positron emission tomography (amyloid [A] and tau [T]) and clinical diagnosis (cognitively normal [CN] and impaired [IM]). CVD markers were derived from structural and quantitative magnetic resonance imaging data.
Results
Transcapillary pulse wave delay was significantly longer in controls compared to AT biomarker–confirmed groups (A+/T–/CN P < .001, A+/T+/CN P < .001, A+/T+/IM P = .003). Intracranial low‐frequency oscillations were diminished in AT biomarker–confirmed groups both CN and impaired (A+/T–/CN P = .039, A+/T+/CN P = .007, A+/T+/IM P = .011). A significantly higher presence of microhemorrhages was measured in A+/T+/CN compared to controls (P = .006).
Discussion
Cerebrovascular markers indicate increased vessel stiffness and reduced vasomotion in AT biomarker–positive subjects during preclinical AD.
Keywords: Alzheimer's disease, amyloid imaging, low‐frequency oscillations, vascular imaging, vessel stiffness
1. INTRODUCTION
Alzheimer's disease (AD) is biologically defined by the National Institute on Aging amyloid, tau, and neurodegeneration (NIA ATN) biomarker research framework 1 consisting of amyloid beta (Aβ) accumulation (A) and the subsequent development of tau neurofibrillary tangles (T) and neurodegeneration (N). Emerging evidence suggests that once initiated Aβ accumulation follows a remarkably predictable time course, 2 , 3 and may be present 20 or more years prior to the dementia stage of the disease. 4 , 5 However, the mechanisms initiating accumulation remain uncertain and the time course of tau pathology 6 and eventual symptom expression is heterogeneous and moderately predicted by current Aβ markers. 7 Cerebrovascular disease (CVD) is considered a major risk factor for dementia. 8 , 9 , 10 Recent evidence indicates that vascular disease can interact with Aβ and tau pathology, 8 , 10 , 11 including a mechanistic overlap. 12 To this point, the addition of vascular disease to the ATN framework has been proposed; 13 however, the exact vascular mechanisms that might influence AD pathology have yet to be fully characterized in vivo and few non‐invasive quantitative markers provide specificity to CVD.
Magnetic resonance imaging (MRI) provides a unique platform to probe various aspects of brain pathology and function, including vascular disease. Past and ongoing studies in AD have largely inferred CVD based on the quantification of white matter hyperintensities (WMH) seen on T2 weighted fluid‐attenuated inversion recovery (T2‐FLAIR); 14 from measures of cerebral blood flow (CBF) using, for example, arterial spin labeling (ASL); and from microhemorrhages seen on T2* MRI. Many studies using these techniques have found correlations with AD biomarkers and cognition and hypo‐perfusion, 15 WMH burden, 16 and decreased brain blood flow. 17 Recently, white matter abnormalities have been used to characterize vascular contributions to cognitive impairment and dementia. 18 While these studies have demonstrated potential correlations between CVD and AD, these measures provide information that is non‐specific, hindering the potential to identify CVD and AD interacting pathways. Both hypoperfusion and WMH are not specific to CVD pathology, occurring in other diseases and normal aging. 19 Moreover, perfusion is intrinsically coupled to metabolic demand and thus correlated with neurodegeneration. 20 , 21
To provide vascular markers that are more specific to CVD, quantitative 4D flow MRI can be used. This technique allows for dynamic flow measurements in major arteries and veins. Unlike alternative measurements from transcranial Doppler, 4D flow MRI provides the ability to quantify velocities and flow rates in any of the intracranial arteries and veins. 4D flow MRI has been used to study clinically diagnosed AD subjects. 22 , 23 These studies found an association of reduced blood flow, increased flow pulsatility, and faster flow waveform transmission across the capillary bed. More recent studies have identified cerebrovascular stiffening 24 and decreased low‐frequency oscillations (LFOs) 25 in clinically diagnosed AD subjects, indicating vascular and/or tissue stiffening occurs concomitant to AD dementia clinical syndrome. However, these studies were limited to clinically diagnosed AD subjects and did not characterize cerebrovascular health during the preclinical phase of AD. Preclinical AD begins with the accumulation of amyloid pathology in the brain and it can last decades. 1 During this phase, pathophysiological changes are occurring and it is likely when therapeutic interventions would be most effective. Therefore, to truly understand the relationship and interaction pathways between CVD and AD, studies in preclinical AD biomarker–confirmed populations (e.g., Aβ, tau) are necessary. In this study we investigated the relationship between MRI‐based CVD markers including WMHs from T2‐FLAIR, microhemorrhages from susceptibility weighted imaging (SWI), perivascular spaces from structural images (T1‐weighted), transcapillary pulse wave delay (a measure of vascular compliance) and LFOs from 4D flow with AD biomarkers of Aβ deposition and neurofibrillary tau tangles. Positron emission tomography (PET) imaging was used to quantify AD biomarkers in subjects spanning the AD clinical spectrum.
HIGHLIGHTS
Cerebrovascular blood flow dynamics were characterized in Alzheimer's disease (AD) biomarker–confirmed subjects using 4D flow magnetic resonance imaging.
A significantly shorter transcapillary pulse wave delay was measured across the brain circulation in cognitively normal (CN) amyloid and tau biomarker–positive subjects.
A significant reduction in internal carotid artery (ICA) low‐frequency flow oscillations (0.01–0.10 Hz) was observed in CN amyloid and tau biomarker–positive subjects.
Dynamic vascular flow markers indicate that cerebrovascular modifications are occurring parallel to AD pathophysiological changes during preclinical AD.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts and presentations. Studies using dynamic vascular imaging markers to characterize cerebrovascular health in Alzheimer's disease (AD) biomarker–confirmed subjects are currently lacking; however, relevant work including studies on white matter alterations and cerebral blood flow deficits in AD are appropriately cited.
Interpretation: Our findings indicate a high degree of intracranial vessel stiffness and reduced low‐frequency flow oscillations are present in amyloid and tau biomarker positive cognitively normal individuals. These results suggest vascular alterations are occurring during the preclinical phase of AD.
Future directions: This article proposes the use of novel vascular imaging methodologies to characterize the type of interactions between cerebrovascular disease (CVD) and AD. Future studies will be focused on acquisition of longitudinal data from magnetic resonance imaging and positron emission tomography, fluid‐based vascular and AD biomarkers (e.g., cerebrospinal fluid, blood), and cognitive testing to study moderation effects of CVD in the neuropathologic trajectory of AD.
2. METHODS
2.1. Study participants and clinical classification
A total of 136 study participants (age = 71 ± 5 years) were recruited from the Wisconsin Alzheimer's Disease Research Center and Wisconsin Registry for Alzheimer's Prevention. 26 Demographic data are summarized in Table 1. Four groups were established based on combinations of PET biomarker status and clinical diagnosis. Three cognitively normal (CN) groups were defined: (1) amyloid and tau biomarker negative (A–/T–; n = 68), (2) amyloid positive and tau negative (A+/T–; n = 33), and (3) amyloid and tau positive (A+/T+; n = 21). A group of tau positive and amyloid negative (T+/A–) subjects was not included due to lack of statistical power (n = 2). A fourth group included cognitively impaired (IM) A+/T+ subjects (n = 14; n = 9 mild cognitively impaired [MCI], n = 5 dementia). The control group subjects were age and sex matched to the AT biomarker–positive groups using propensity scores in R statistical package v4.0.3. 27 All participants were diagnostically characterized in multidisciplinary consensus conferences using applicable criteria 1 , 26 , 28 , 29 , 30 , 31 and blind to AD biomarker status. The University of Wisconsin Institutional Review Board approved all study procedures and protocols following the policies and guidance established by the campus Human Research Protection Program. All study procedures were performed according to the Declaration of Helsinki, including obtaining written informed consent from each participant. Exclusion criteria included significant medical conditions such as major systemic illness. Cognitively impaired subjects in the fourth group were excluded if given a consensus diagnosis of a non‐Alzheimer's variant of dementia. MRI scans were reviewed by two neuroradiologists (H.A.R., L.E.) for incidental findings.
TABLE 1.
Participant demographics and summary of cardiovascular and Alzheimer's disease biomarkers
| Demographics | Total (n = 136) | 1. A–/T–/CN (n = 68) | 2. A+/T–/CN (n = 33) | 3. A+/T+/CN (n = 21) | 4. A+/T+/IM (n = 14) | Group test P‐value | Pairwise differences | Cohen's d |
|---|---|---|---|---|---|---|---|---|
| Age at MRI (years) | 71 ± 5 | 71 ± 5 | 70 ± 5 | 73 ± 5 | 72 ± 5 | 0.281 | – | – |
| Female (n, %) | 81, (60%) | 40, (59%) | 19, (58%) | 12, (57%) | 10, (71%) | 0.821 | – | – |
| Parental dementia history positive (n, %) | 87, (64%) | 39, (57%) | 22, (67%) | 17, (81%) | 9, (64%) | 0.157 | – | – |
| APOE ɛ4 carrier (n, %)* | 59, (45%) | 17, (25%) | 20, (61%) | 12, (60%) | 10, (91%) | <0.001 | 1 vs. 2, 3, 4 | 0.78, 0.77, 1.6 |
| Time between MRI and PiB PET (months) | 1.0 ± 9.2 | 0.1 ± 12 | 1.6 ± 5.5 | 1.2 ± 7.0 | 4.6 ± 6.3 | 0.376 | – | – |
| Time between MRI and MK‐6240 PET (months) | 1.6 ± 8.2 | 0.8 ± 9.2 | 1.4 ± 6.0 | 0.8 ± 7.1 | 6.0 ± 7.3 | 0.179 | ‐ | – |
| Body mass index (kg/m2) | 27.7 ± 5.2 | 28.6 ± 5.3 | 26.0 ± 5.2 | 28.5 ± 5.2 | 25.7 ± 4.3 | 0.036 | – | – |
| Hypertension and/or cholesterol medication (n, %) | 85, (63%) | 44, (65%) | 15, (45%) | 16, (80%) | 10, (71%) | 0.061 | – | – |
| Cardio‐ and cerebrovascular biomarkers | ||||||||
| SBP (mmHg) | 133 ± 17 | 135 ± 16 | 129 ± 18 | 133 ± 18 | 135 ± 18 | 0.406 | – | – |
| DBP (mmHg) | 79 ± 10 | 81 ± 9 | 77 ± 10 | 78 ± 9 | 77 ± 9 | 0.259 | – | – |
| HR (bpm) | 67 ± 11 | 68 ± 12 | 67 ± 11 | 67 ± 9 | 65 ± 9 | 0.853 | – | – |
| CHS scores | 2.3 ± 1.5 | 2.4 ± 1.6 | 2.2 ± 1.4 | 2.0 ± 1.1 | 3.0 ± 1.4 | 0.244 | – | – |
| Fazekas score (deep WMHs) | 1.3 ± 0.8 | 1.2 ± 0.8 | 1.3 ± 0.8 | 1.4 ± 0.5 | 1.7 ± 0.5 | 0.192 | – | – |
| Fazekas score (periventricular WMHs) | 1.4 ± 0.8 | 1.3 ± 0.8 | 1.5 ± 0.8 | 1.4 ± 0.5 | 1.8 ± 0.8 | 0.197 | – | – |
| Presence of Microhemorrhages (n, %) | 23, (17%) | 6, (9%) | 5, (15%) | 8, (40%) | 4, (29%) | 0.006 | 1 vs. 3 | 0.88 |
| Number of Microhemorrhages | 2.1 ± 2.9 | 1.5 ± 0.6 | 1.4 ± 0.6 | 3.0 ± 4.9 | 2.0 ± 1.4 | 0.752 | – | – |
| Presence of Perivascular spaces (n, %) | 28, (21%) | 10, (15%) | 10, (30%) | 6, (30%) | 2, (14%) | 0.237 | – | – |
| WMH lesion volume, %ICV** | 0.17 ± 0.29 | 0.17 ± 0.28 | 0.16 ± 0.27 | 0.11 ± 0.12 | 0.31 ± 0.48 | 0.213 | – | – |
| Total cerebral blood flow (mL/s/L) | 8.0 ± 1.6 | 7.9 ± 1.5 | 8.3 ± 1.4 | 7.9 ± 1.3 | 8.0 ± 2.6 | 0.503 | – | – |
| ICA‐SSS transcapillary pulse wave delay (ms) | 74 ± 39 | 93 ± 35 | 48 ± 40 | 53 ± 29 | 59 ± 27 | <0.001 | 1 vs. 2, 3, 4 | 0.74, 0.87, 0.81 |
| ICA low frequency flow range (mL/s/L) | 0.60 ± 0.17 | 0.66 ± 0.19 | 0.52 ± 0.14 | 0.55 ± 0.12 | 0.53 ± 0.14 | <0.001 | 1 vs. 2, 3, 4 | 0.59, 0.67, 0.64 |
| ICA low frequency flow σ (mL/s/L) | 0.13 ± 0.04 | 0.14 ± 0.04 | 0.12 ± 0.03 | 0.12 ± 0.03 | 0.11 ± 0.03 | <0.001 | 1 vs. 2, 3, 4 | 0.54, 0.69, 0.62 |
| ICA LFOs (1/Hz) [0.003‐0.100 Hz] | 7.8 ± 6.5 | 9.3 ± 8.0 | 7.1 ± 5.2 | 7.2 ± 3.3 | 6.3 ± 3.5 | <0.001 | 1 vs. 2, 3, 4 | 0.35, 0.51, 0.56 |
| Alzheimer's disease biomarkers | ||||||||
| Hippocampal volume, %ICV | 0.50 ± 0.07 | 0.52 ± 0.06 | 0.53 ± 0.05 | 0.48 ± 0.08 | 0.43 ± 0.06 | <0.001 | 1, 2 vs. 3, 4 | 0.65, 1.5; 0.78, 1.8 |
| PiB global DVR | 1.27 ± 0.30 | 1.03 ± 0.03 | 1.39 ± 0.20 | 1.58 ± 0.25 | 1.71 ± 0.16 | <0.001 | 1 vs. 2, 3, 4; 2 vs. 3, 4 | 3.1, 4.4, 9.3; 0.85, 1.7 |
| MK‐6240 SUVR (70–90 min) entorhinal cortex | 1.25 ± 0.47 | 1.00 ± 0.11 | 1.04 ± 0.14 | 1.77 ± 0.36 | 2.13 ± 0.46 | <0.001 | 1, 2 vs. 3, 4 | 3.9, 5.3; 2.9, 4.0 |
Abbreviations: σ, standard deviation; APOE, apolipoprotein E; CHS, Cardiovascular Health Study; CN, cognitively normal; DBP, diastolic blood pressure; DVR, distribution volume ratio; HR, heart rate; ICA, internal carotid artery; ICV, intracranial volume; IM, cognitively impaired; LFO, low‐frequency oscillations; MRI, magnetic resonance imaging; PET, positron emission tomography; PiB, Pittsburgh compound B; SBP, systolic blood pressure; SSS, superior sagittal sinus; WMH, white matter hyperintensities.
*Four subjects were not genotyped (three A+/T+/IM, one A+/T+/CN); carrier refers to presence of at least one APOE ɛ4 allele.
**One subject (A+/T+/CN) WMH lesion volume was not quantified because the segmentation did not pass neuroradiologist (LE) review.
Notes: A± and T± represent amyloid beta and tau tangle positivity groups determined by 11C‐PiB and 18F‐MK‐6240 PET, respectively. Pairwise differences were assessed using analysis of variance followed by post hoc analysis using the Tukey‐Kramer method (P < .05 significance).
2.2. Neuroimaging protocols
2.2.1. 4D flow MRI
Whole brain, time‐resolved 4D flow MRI data were acquired using 3.0T systems (SIGNA Premier and MR750, GE Healthcare) and a 3D radially undersampled sequence. 32 Data were acquired with the following imaging parameters: Venc = 80 cm/s, imaging volume = 22 × 22 × 10 cm 3 , TR/TE = 7.4/2.7 ms, scan time ≈7 min, acquired spatial resolution = 0.7 mm isotropic, flip angle = 8°, bandwidth = 250 kHz, number of projections ≈11,000. 25 , 32 Cardiac triggers were collected for each subject from a photoplethysmogram on a pulse oximeter (GE Healthcare) worn on the subject's finger during the MRI exam. Additional physiologic monitoring included cuff‐based blood pressures (Veris Vital Signs, Medrad) measured once at the start of the MRI exam.
Flow images were reconstructed via an offline reconstruction for all subjects. 22 , 25 Magnitude and velocity data were generated using GPU accelerated (SigPy) iterative SENSE with JSENSE and a local low rank temporal constraint. 24 , 33 , 34 , 35 Two imaging series were generated: (1) a time‐resolved series with a temporal resolution of ≈3.8s (0.26 Hz) (spatial res ≈1.72 mm 3 ) for assessment of LFOs and (2) a cardiac‐resolved series with a temporal resolution of ≈49 ms (20 Hz; spatial res ≈0.7 mm 3 ) for assessment of transcapillary pulse wave delay (e.g., high‐frequency oscillations). Time series were motion corrected in MATLAB (Mathworks) using a rigid body registration. The rigid body registration was performed by setting the magnitude image in the middle of the time series as the reference image, subsequently each image on the time series was translationally and rotationally registered to the fixed image. The registration transformation matrix from each timeframe was applied to the velocity images. Velocity images were corrected for background phase errors by fitting a second order polynomial to the background tissue, and phase aliasing was corrected using a Laplacian‐based phase unwrapping approach. 36 Intracranial arteries (internal carotid arteries [ICAs] and basilar artery [BA]) and veins (posterior inferior portion of the superior sagittal sinus [SSS]) were segmented automatically in MATLAB using a centerline process with local cross‐sectional cut‐planes automatically placed in every centerline point perpendicular to the axial direction of the vessel. 37 Regions of interest (ROIs) were automatically contoured using a k‐means clustering approach under the assumption that any cross section will contain a low‐signal background and a vessel region. 37 Blood flow rates were estimated from the product of cross‐sectional areas and velocities from the automatic segmentation. Total CBF was defined as the sum of blood flow rates in the ICAs and BA and was normalized to total brain volume (intracranial volume [ICV] – cerebrospinal fluid [CSF] volume). Transcapillary pulse wave delay (capillary stiffness) was defined from analysis of the wave propagation using a time maximal upslope algorithm for determination of the time shift between arteries (ICAs, BA) and veins (SSS). 22 Measurement landmarks included distal cervical ICAs, BA origin, and the posteroinferior portion of the SSS.
Low‐frequency flow range (max–min) and flow standard deviations (σ) were estimated from the time‐resolved velocity series as a marker for LFOs and were normalized to total brain volume. The rationale for normalizing flow measures to brain volume was to account for brain volume changes (e.g., metabolism driven perfusion), and search for other factors that can lead to blood flow changes. 20 , 21 Finally, LFOs were calculated as follows: time‐resolved velocity series were Fourier transformed, and LFOs were defined as the power spectral density (PSD) content in the frequency range of 0.003–0.10 Hz. LFOs were quantified by the summation of PSD in this frequency range. 38 To compare LFOs’ hemodynamic response between groups, mean PSD were also estimated. LFOs were assessed in the ICAs and SSS (BA LFOs’ analyses was limited by the lower spatial resolution of the time‐resolved series [spatial res ≈1.72 mm3], a necessary trade‐off for the high temporal resolution ≈3.8s).
2.2.2. Structural imaging
T1‐weighted, T2‐FLAIR images, and SWI data were collected to derive traditional markers of CVD and brain health. 26 T1‐weighted and T2‐FLAIR data were N4 bias field corrected. ICV and CSF were segmented from T1‐weighted data using SPM12 (www.fil.ion.ucl.ac.uk/spm). 39 Hippocampal volume was estimated using FSL FIRST and normalized by total ICV (sum of gray matter, white matter, and CSF volumes from SPM segmentation). WMH lesion volumes were segmented from 3D T2‐FLAIR images using the Lesion Segmentation Toolbox in SPM12. 40 This approach estimates the lesion probability at each voxel, outputting a lesion probability map. The lesion volume was the sum of voxels in which the probability was > = 0.5. The output underwent visual quality assessment by trained reviewers. In addition to WMH lesion volumes, Cardiovascular Health Study (CHS) and Fazekas scores were also derived from T2‐FLAIR images by an experienced neuroradiologist (L.E.) for a robust evaluation of WMH lesions. Overall, CHS scores take into account subject's age and all white matter involvement, while Fazekas scores do not take into account subject's age and can be used for regional assessment of white matter involvement. In the CHS scale a “0” represents no evidence of WMH lesion and a “9” reflects involvement of all white matter. 41 , 42 , 43 Fazekas scores were used to independently score periventricular and deep WMHs. 44 In the periventricular WMHs Fazekas scale a “0” represents a region absent of lesions and a “3” corresponds to irregular periventricular signal extending into the deep white matter. In deep WMHs Fazekas scale a “0” represents absence of lesions and “3” large confluent areas. In addition, the presence of microhemorrhages and perivascular spaces were characterized from SWI and T1‐weighted images, respectively, and a score of 1 was assigned in the presence of a lesion, otherwise a rating of 0 was assigned. The total number of microhemorrhages per subject was also estimated.
2.2.3. PET imaging
Amyloid and tau burden were assessed using 11C‐PiB and 18F‐MK‐6240 PET imaging, respectively. Details on PET imaging (Siemens EXACT HR+) methodology including radiopharmaceutical production, acquisition protocols, and image reconstruction; processing and quantification of PiB and MK‐6240 PET data are extensively described elsewhere. 45 , 46 , 47 In short, PiB images were transformed into voxel‐wise parametric images representing PiB binding using the cerebellar cortex as a reference region of negligible binding. 48 The cerebellar time activity curve was extracted from the PET data using a cerebellar gray matter mask image derived from the co‐registered T1‐weighted MRI. PiB distribution volume ratio (DVR) was estimated from a 70‐minute dynamic acquisition using reference Logan graphical analysis (t* = 35 min, k2 = 0.149 min–1, cerebellum gray matter reference region). 48 A global cortical DVR average was calculated and used to classify individuals as amyloid positive or negative using a global DVR threshold of > 1.19 derived from a composite measurement from eight bilateral ROIs (SPM12; angular gyrus, anterior cingulate gyrus, posterior cingulate gyrus, frontal medial orbital gyrus, precuneus, supramarginal gyrus, middle temporal gyrus, and superior temporal gyrus). 49 The reconstructed MK‐6240 PET time series was co‐registered to T1‐weighted MRI (SPM12). PET time series MK‐6240 standardized uptake value ratios (SUVRs) were calculated from a 20‐minute dynamic acquisition (4 × 5‐minute frames) beginning 70 minutes after bolus injection also using the inferior cerebellum gray matter as a reference region. Tau positivity was established from a previous analysis as entorhinal cortex MK‐6240 SUVR two standard deviations above the mean of the Aβ negative group (entorhinal MK‐6240 SUVR > 1.27). 45
2.3. Statistical analysis
This study included various biomarkers: (1) imaging‐based AD biomarkers (amyloid and tau burden, hippocampal volume), (2) non–imaging‐based CVD markers (heart rate [HR], systolic blood pressure [SBP], diastolic blood pressure [DBP]), (3) imaging‐based CVD markers (WMHs, microhemorrhages, perivascular spaces, transcapillary pulse wave delay, LFOs), and (4) other markers of overall health (body mass index [BMI]). Both imaging‐ and non–imaging‐based CVD markers were compared between AT biomarker groups. Linear regression modeling was used to study the correlation between transcapillary pulse wave delay, LFOs, and AT biomarkers including PiB DVR and MK‐6240 entorhinal cortex SUVR. Significance of group differences was assessed using analysis of variance followed by post hoc analysis using the Tukey‐Kramer method. Statistical analysis was performed in MATLAB. P < .05 was set as the threshold for statistical significance. Cohen's d effect size coefficients were estimated for groups with significant differences. 50
3. RESULTS
Traditional cardiovascular, CVD, and AT biomarkers are summarized in Table 1. No significant AT biomarker group differences were observed across traditional systemic cardiovascular metrics including HR, SBP, and DBP. Structural CVD imaging markers including WMH lesion volumes, CHS and Fazekas scores, and perivascular spaces were not significantly different between groups. However, in a supplementary analysis in which A+/T+/IM subjects were separated into subgroups (e.g., A+/T+/MCI [n = 9], A+/T+/dementia [n = 5]; Table S1 in supporting information), significantly higher WMH lesion volumes were measured in the A+ /T+/dementia group compared to the other groups (P = .005), albeit the sample size of the A+/T+/dementia group was small. A significantly larger presence of microhemorrhages was observed in A+/T+/CN (P = .006) compared to controls. In those A+/T+/CN subjects with detectable microhemorrhages (n = 8), blood pressure and HR were within normal levels (SBP = 125 ± 15 mmHg, DBP = 76 ± 10 mmHg, HR = 67 ± 14 bpm, age = 72 ± 6 years, 2 subjects with self‐report hypertension). The A+/T+/IM group also had a larger presence of microhemorrhages compared to controls, but not statistically different (P = .280). The number of microhemorrhages in subjects with at least one microhemorrhage were not significantly different among groups. Hippocampal volumes were significantly reduced in tau‐positive groups (A+/T+/CN, P = .031; A+/T+/IM, P < .001). Average times between MRI and PET acquisitions were 1.0 ± 9.2 months (PiB) and 1.6 ± 8.2 months (MK‐6240; see Table 1).
All study participants had a usable 4D flow dataset for LFO characterization; however, four participants (1f A–/T–/CN, 1f A+/T+/CN, 2 m A+/T+/IM) did not have usable cardiac gating data for transcapillary pulse wave delay assessment because of gating system failure during the MRI acquisition. Brain volume normalized total CBF was similar between groups (Figure 1A). The rationale for normalizing total blood flow to brain volume was to account for brain volume changes (e.g., metabolism), and search for other factors that can lead to blood‐flow reduction. 21 However, ICA and BA transcapillary pulse wave delay (Figure 1B) were significantly different and longer in controls (A–/T–/CN) compared to AT biomarker–confirmed groups including A+/T–/CN (ICA P < .001; BA P = .001), A+/T+/CN (ICA P < .001; BA P = .050) and A+/T+/IM (ICA P = .003; BA P = .026). The effect sizes of group differences are summarized in Table 1.
FIGURE 1.
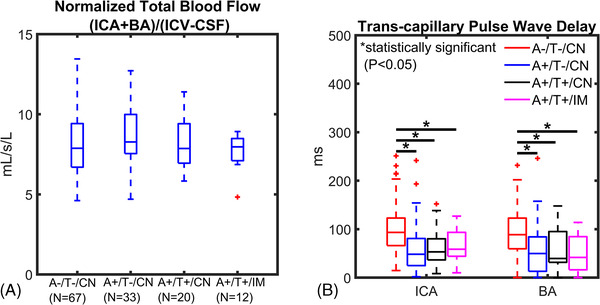
Blood flow and transcapillary pulse wave delay from cardiac‐resolved 4D flow magnetic resonance imaging. Data are summarized for controls and amyloid/tau (AT) biomarker–confirmed groups (amyloid positive [A+], tau positive [T+], cognitively normal [CN], cognitively impaired [IM]). A, Cerebral blood flow (basilar [BA] + internal carotid artery [ICA]) was normalized to brain volume and was similar between groups. B, ICA and BA transcapillary pulse wave delay time was significantly longer in the control group (A–/T–/CN) compared to A+/T–/CN (ICA P < .001; BA P = .001), A+/T+/CN (ICA P < .001; BA P = .050) and (A+/T+/IM; ICA P = .003, BA P = .026) indicating a significantly faster transmission of the cardiac pulse wave in the intracranial vascular network of AT biomarker–confirmed groups
Low‐frequency vascular markers derived from time‐resolved 4D flow data were negatively correlated with AT biomarkers (Figure 2). Brain volume normalized arterial (ICA) low‐frequency flow range (Figure 2A) was significantly decreased in the AT biomarker–confirmed groups A+/T–/CN (P < .001), A+/T+/CN (P < .001), and A+/T+/IM (P = .004) relative to controls (A–/T–/CN). Flow standard deviations (σ) (Figure 2B) were also significantly lower in the AT biomarker–confirmed groups A+/T–/CN (P < .001), A+/T+/CN (P < .001), and A+/T+/IM (P = .005) compared to controls (A–/T–/CN). Venous (SSS) measurements were diminished in AT biomarker groups; however, differences were not statistically different.
FIGURE 2.
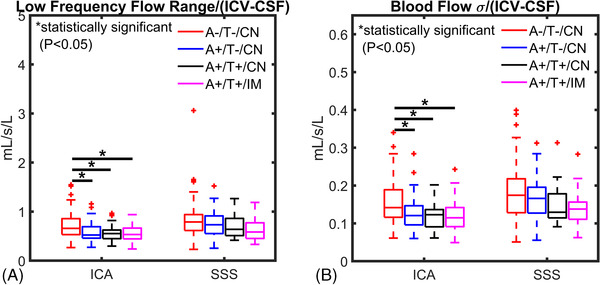
Low‐frequency flow range and standard deviations from time‐resolved 4D flow magnetic resonance imaging (MRI). A, Low‐frequency flow range and (B) flow standard deviation (σ) measured from time‐resolved 4D flow MRI data are summarized for controls and amyloid/tau (AT) biomarker–confirmed groups (amyloid positive [A+], tau positive (T+), cognitively normal [CN], cognitively impaired [IM)]). Brain volume normalized low frequency flow range and flow σ were significantly lower in the arterial circulation (internal carotid artery [ICA]) in AT‐positive groups (A+/T–/CN, A+/T+/CN, A+/T+/IM) compared to controls (A–/T–/CN; flow range: P < .001, P < .001, P = .004; flow σ: P < .001, P < .001, P = .005) respectively. These results suggest diminished autoregulation‐related vasomotion in Alzheimer's disease biomarker‐confirmed groups. A similar trend of lower flow range and σ in AT biomarker–positive groups was observed in the venous circulation
Power spectrum density analysis was used to characterize LFOs across groups (Figure 3). Overall, higher LFOs were measured in both arteries (ICA) and veins (SSS) of controls (A–/T–/CN; Figure 3A, B). A significantly higher amount of LFOs in the range of [0.003, 0.100] Hz (e.g., sum of power from PSD) was measured in the ICAs of the control group (A–/T–/CN) compared to AT biomarker–positive groups including A+/T–/CN (P = .039), A+/T+/CN (P = .007), A+/T+/IM (P = .011). In a more limited frequency range, [0.01, 0.10] Hz, still a significantly higher amount of LFOs was observed in the arteries (ICAs) of the control group (A–/T–/CN) than in AT biomarker–positive groups including A+/T–/CN (P = .027), A+//T+CN (P = .046), and A+/T+/IM (P = .046).
FIGURE 3.
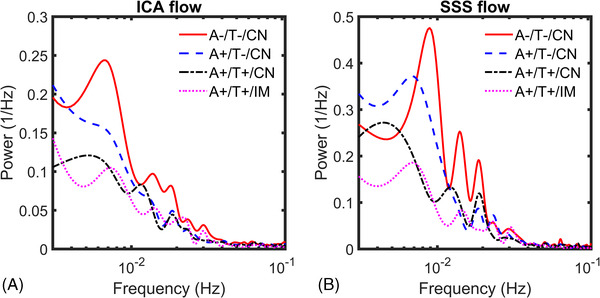
Low‐frequency flow oscillation power spectrum density analysis. Group average power spectrum density low frequency oscillations (LFOs) derived from time‐resolved 4D flow data in the internal carotid artery (ICA) (A) and superior sagittal sinus (SSS) (B) for tau negative (A–/T–/CN, A+/T–/CN) and tau positive groups (A+/T+/CN, A+/T+/IM; cognitively normal [CN], cognitively impaired [IM]). Diminished frequency content in ICAs and SSS was measured in biomarker‐positive groups compared to controls. ICAs’ autoregulation‐related LFO content [0.003, 0.100 Hz] was significantly reduced in these groups compared to controls (P = .039 A+/T–/CN; P = .007 A+/T+/CN; P = .011 A+/T+/IM). The threshold for statistical significance was P < .05
Linear regression modeling (Figure 4) showed a relatively weak correlation between ICA transcapillary pulse wave delay and global PiB DVR and MK‐6240 entorhinal cortex SUVR (R2 = 0.12, R2 = 0.04; Figure 4A, B). LFOs ([0.003, 0.100] Hz) were also weakly correlated with PiB and MK‐6240 (R2 = 0.11, R2 = 0.07; Figure 4C, D).
FIGURE 4.
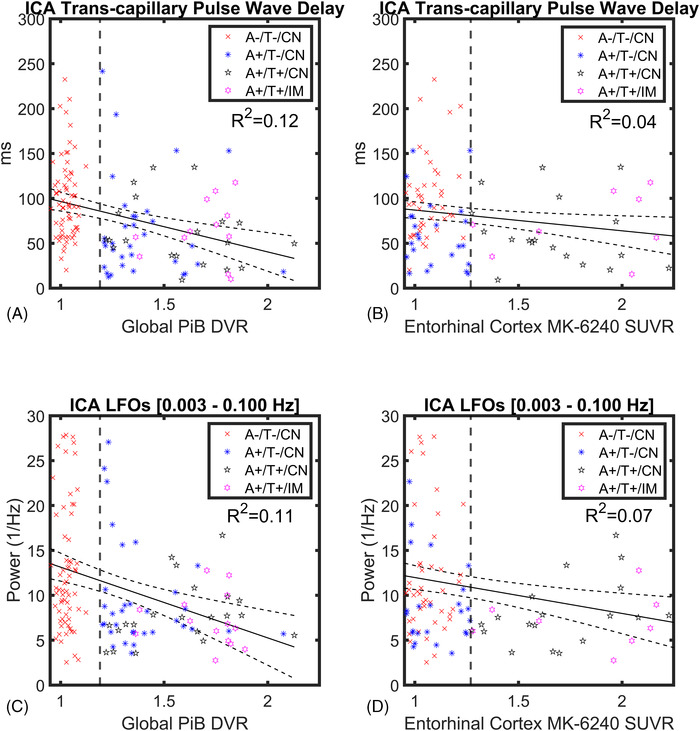
Linear regression modeling of dynamic vascular markers and amyloid/tau (AT) biomarkers. Linear regressions with 95% confidence intervals showing internal carotid artery (ICA) transcapillary pulse wave delay (A, B) and low‐frequency oscillations (LFOs; C, D) as a function of global Pittsburg compound B (PiB) distribution volume ratio (DVR) and enthorinal cortex MK‐6240 standardized uptake value ratio (SUVR) in all participants (n = 136). Linear correlations were relatively weak with coefficients of R2 = 0.12 (transcapillary pulse wave delay vs. PiB DVR), R2 = 0.04 (MK‐6240 SUVR) and R2 = 0.11 (LFOs vs. PiB DVR), R2 = 0.07 (MK‐6240 SUVR). Vertical lines mark global PiB DVR threshold > 1.19 for A+ (A, C) and entorhinal cortex MK‐6240 SUVR threshold > 1.27 for T+ (B, D)
4. DISCUSSION
This study aimed to improve our understanding of the interaction between CVD and AD pathologies by cross‐sectionally comparing CVD imaging markers to ATN markers in preclinical and cognitively impaired AD subjects. We specifically characterized the association between traditional systemic markers of vascular health and dynamic CVD imaging markers from MRI with AD biomarkers including Aβ and neurofibrillary tau from PET imaging in AD risk‐enriched cohorts. This includes unique and quantitative markers from 4D flow MRI that provide metrics of cerebrovascular dysfunction dynamics and vessel stiffness in the large vessels and capillaries. Non‐invasive methods and biomarkers for evaluating intracranial vessel health are of great interest for diagnosis, treatment monitoring, and study of disease interactions. For example, 4D flow allows assessment of the pulse wave velocity across the vasculature, which is indicative of vessel stiffness. 24 , 51 , 52 In our cohort, commonly used traditional CVD markers (both non‐imaging and imaging based) were unable to detect AT biomarker group differences, with the exception of microhemorrhages assessment. However, quantitative 4D flow measures of transcapillary pulse wave delay and LFOs were significantly different between AT biomarker–confirmed and age‐matched control groups. Specifically, a significantly shorter transcapillary pulse wave delay related to a faster pulse wave transmission across the brain circulation was measured in AT biomarker–positive subjects. In addition, lower arterial LFOs were measured in AT biomarker–positive subjects. These results suggest stiffer vessels and/or capillaries and decreased autoregulation–related vasomotion in the intracranial vasculature of amyloid‐ and tau‐positive subjects. 24 , 25 , 53 , 54 Importantly, these vascular alterations were observed in CN AT biomarker–positive subjects. Taken together, evidence suggests cerebrovascular modifications are occurring parallel to AD pathophysiological changes during preclinical AD.
During the last decade numerous experiments have posited mechanisms in which CVD and AD interact. This includes models suggesting vascular contributions to altered brain waste clearance, and more direct interactions through ischemic and inflammatory events. In both interaction mechanisms, altered vascular dynamics, vascular stiffness, and vessel wall dysfunction have been indicated as pathologic conditions. High‐frequency cardiac pulsations and low‐frequency oscillatory vasomotion are hypothesized to drive brain metabolite clearance pathways including the glymphatic system and intramural periarterial drainage (IPAD). 55 , 56 These clearance systems are hypothesized to remove toxic waste products including Aβ from the neuropil environment and are altered in the presence of vascular dysfunction. For example, researchers have demonstrated a reduction in glymphatic flow of CSF through perivascular spaces in hypertensive animal models with decreased cardiac pulsations. 57 Loss of vascular smooth muscle cells and LFO‐reduced vasomotion have been correlated with impaired clearance of amyloid in cerebral amyloid angiopathy (CAA) models. 58 These studies suggest a synergistic interaction between CVD and AD leading to exacerbation of AD pathology. Our human study findings align well with these hypotheses tested on animal models, indicating both increased intracranial vessel stiffness and LFO‐reduced vasomotion in AT biomarker–positive CN subjects.
By combining AT biomarker data and dynamic CVD imaging markers, a significant reduction of transcapillary pulse wave delay and LFOs was measured in the cerebrovasculature of AT biomarker–positive subjects both CN and impaired. Transcapillary pulse wave delay describes high‐frequency cardiac wave transmission along arteries, capillaries, and veins and is a proposed marker for vessel and capillary stiffness. 22 Arterial and venous LFOs in a frequency range < 0.10 Hz can be related to smooth muscle cell–driven vasomotion. 38 , 59 Cerebral autoregulation helps maintain relatively constant levels of CBF within large variations of arterial blood pressure. 59 Moreover, dynamic autoregulation is characterized by both spontaneous physiological fluctuations in arterial blood pressure and CBF. Frequencies in the range of 0.01to 0.10 Hz (LFOs/Mayer waves), measured from cerebral blood circulation using near‐infrared spectroscopy, have been associated with fluctuations in mean arterial pressure, and dynamic autoregulation mechanisms. 38 LFOs in this range have been found to be of similar amplitude and synchronized to dynamic autoregulation processes. 59 In addition, functional MRI (fMRI) experiments have found that a proportion of LFOs are due to localized control of CBF independent of local neural activity. 53 Therefore, our study results indicate vessel and capillary stiffness together with reduced autoregulation related vasomotion are concomitant with amyloid and tau accumulation. Data trends indicated vascular burden worsens along the biomarker trajectory of AD with further reduced arterial LFOs in presence of tau biomarkers. Although decreased LFOs were observed in both arteries and veins of AT biomarker–positive subjects, a significant drop in LFO amplitude was only observed in the arteries. These findings might reflect differences in venous and arterial walls, including the thinner venous wall compared to the thicker arterial wall with more smooth muscle and connective tissue. While other studies have previously characterized the relationship between CVD and AT biomarkers (e.g., amyloid and tau burden), to our knowledge, this is the first study that characterizes 4D flow–based cerebrovascular markers in CN AT biomarker–positive subjects and observed flow‐related vascular alterations on such cohorts. 60 These results agree well with another study that characterized cerebrovascular resistance from perfusion measures in non‐demented (e.g., MCI) amyloid‐positive populations. 11 In that study, the researchers found a synergistic relationship between vascular alterations and amyloidosis that produced cognitive decline. The presence of vascular dysfunction in preclinical AD might help explain the heterogeneity that is often observed in the cognitive trajectories of AD patients. Furthermore, therapies targeting improved cerebrovascular health during preclinical AD might hold potential to delay the onset of clinical symptoms in subjects that have begun Aβ accumulation. To date pharmacology and exercise therapies have found mixed results reducing risk for dementia; however, there are some indications suggesting benefits to brain health. 61 , 62 , 63
Traditional markers of cardiovascular disease including blood pressure and heart rate were similar between groups. Self‐reported use of hypertension‐ and cholesterol‐reducing medications was comparable between groups (P > .05). Moreover, total CBF as measured in the arteries was also similar between groups after adjusting for brain volume, demonstrating that after accounting for neurovascular coupled metabolic demand by brain volume normalization, no global cerebral perfusion deficits could be detected. 21 Markers of CVD such as WMH burden and perivascular spaces, which are derived from structural imaging and are typically used to characterize CVD, were also not significantly different between AT biomarker groups. However, a significantly higher number of microhemorrhages was detected in A+/T+/CN subjects compared to controls. Microhemorrhages are markers of CAA and hypertensive small vessel disease and can characterize post CVD and AD changes. Our results indicate CAA as a primary cause of the microhemorrhages observed in the A+/T+/CN group. The observation of a significantly higher presence of microhemorrhages in the CN/A+/T+ group, but not in the cognitively impaired A+/T+ group, supports the notion that CAA presence increases with AD pathology burden (A+/T+), followed by increased neurodegeneration and cognitive decline. However, by the onset of cognitive impairment development of new CAA‐induced microhemorrhages might be diminished, marking advanced neurodegeneration. In this study, the lowest hippocampal volumes were measured in cognitively impaired A+/T+ subjects. Perivascular spaces were not significantly different among groups. Others have found perivascular spaces strongly correlate with clinically diagnosed subcortical vascular cognitive impairment, but not with amyloid burden from PiB‐PET. 64 This suggests perivascular markers are sensitive to advanced vascular dysfunction in the brain not yet present in our cohort.
Flow‐based CVD imaging markers including transcapillary pulse wave delay and LFOs were significantly different between biomarker‐negative and AT biomarker‐positive subjects; however, measures such as mean blood flow and WMHs were not. The lack of group differences in WMHs might reflect the limitations of structural measures to detect subtle CVD modifications. Others have also detected vascular alterations in the cerebral macro‐ and micro‐circulation including changes in the hippocampus using 4D flow and resting‐state fMRI in healthy older adults, but no significant differences in WMH burden. 65 These results demonstrate dynamic markers of cererbrovascular health can provide access to unique information otherwise not accessible with WMHs or perfusion. Nonetheless, markers of white matter alterations are still useful to quantify CVD burden contributions to cognitive decline. 18
This study has a number of limitations. Capillary stiffness was probed globally using transcapillary pulse wave delay transit times; however, markers that can regionally probe the capillary bed are desirable. All groups including controls were risk enriched for AD including high incidence of parental family history of dementia due to AD; however, apolipoprotein E ɛ4, a known gene that increases the risk of AD, was more present in AT biomarker–positive groups.
5. CONCLUSION
In this work noninvasive quantitative MRI revealed cerebrovascular dysfunction was correlated with AT biomarker levels from PET imaging during preclinical AD. Traditional CVD markers (both non‐imaging and imaging based) were insensitive to vascular alterations with the exception of cerebral microhemorrhages. Significantly shorter transcapillary pulse wave delay transit times and weaker LFOs were measured in the intracranial arteries of AT biomarker–confirmed groups. These markers indicate presence of vascular and capillary stiffness and reduced autoregulation‐related vasomotion. Longitudinal studies are necessary to elicit CVD and amyloid chronicity interactions.
CONFLICTS OF INTEREST
K.A. Cody, T. Reher, and R.V. Cadman have nothing to disclose. L.A. Rivera‐Rivera's effort on this work was supported by the Alzheimer's Association AARFD‐20‐678095 paid to the institution. L. Eisenmenger's effort on this work was supported by the Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR002373 and KL2TR002374, as well as the Wisconsin Alzheimer's Disease Research Center grant P30‐AG062715 paid to the institution. T. Betthauser has also received grant support outside the submitted work from Alzheimer's Association AARF‐19‐614533 paid to the institution. H.A. Rowley has received consulting fees from GE HealthCare, iSchemaView and for accredited lectures and podcasts for Northwest Imaging Forums and the Data Safety Monitoring Board of HL Gore. H.A. Rowley has also received grant support outside the submitted work from the Radiological Society of North America, Fellow Grant paid to the institution. C.M. Carlsson has received payments for her participation in an NIH study at UCLA (D‐CARE) and study drugs from Amarin Corporation (Vascepa and placebo) for VA Merit Study. C.M. Carlsson has also received grant support outside the submitted work from NIH/Lilly, NIH/Esai, VA Merit, and NIH to the institution. N.A. Chin has received payments for being a member of the Med‐Sci committee for the WI Alzheimer's Association and member of the Med‐Sci committee for the Alzheimer's Foundation of America. S.C. Johnson serves on an advisory board for Roche Diagnostics for which he receives an honorarium and is principal investigator of an equipment grant from Roche. He receives research funding from Cerveau Technologies. K.M. Johnson has received royalties for patents licensed by the institution (UW‐Madison) unrelated to this project. K.M. Johnson has also received grant support outside the submitted work from NIH and GE Healthcare as research support to the institution.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We gratefully acknowledge research support from GE Healthcare. Additionally we would like to acknowledge Cerveau Technologies for providing MK‐6240 standard and precursor. This work was supported by the Alzheimer's Association (AARFD‐20‐678095); and the National Institutes of Health (R01NS066982, RF1AG027161, P30AG062715, R01AG021155, S10OD025245 and R01EB027087). This work was also supported by the Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR002373 and KL2TR002374. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Rivera‐Rivera LA, Eisenmenger L, Cody KA, et al. Cerebrovascular stiffness and flow dynamics in the presence of amyloid and tau biomarkers. Alzheimer's Dement. 2021;13:e12253. 10.1002/dad2.12253
REFERENCES
- 1. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018;14(4):535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bilgel M, An Y, Zhou Y, et al. Individual estimates of age at detectable amyloid onset for risk factor assessment. Alzheimer's Dement. 2016;12(4):373‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koscik RL, Betthauser TJ, Jonaitis EM, et al. Amyloid duration is associated with preclinical cognitive decline and tau PET. Alzheimer's Dement: Diagnos, Assessment Dis Monitor. 2020;12(1):e12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jagust WJ, Landau SM. Temporal Dynamics of β‐Amyloid accumulation in aging and Alzheimer's disease. Neurology. 2021;96(9):e1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. The Lancet Neurol. 2013;12(4):357‐367. [DOI] [PubMed] [Google Scholar]
- 6. Doré V, Krishnadas N, Bourgeat P, et al. Relationship between amyloid and tau levels and its impact on tau spreading. Euro J Nuclear Med Molecular Imaging. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jack CR, Wiste HJ, Weigand SD, et al. Predicting future rates of tau accumulation on PET. Brain. 2020;143(10):3136‐3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12(12):723‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer's disease dementia and cognitive function in elderly people: a cross‐sectional study. Lancet Neurol. 2016;15(9):934‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sweeney MD, Sagare AP, Zlokovic BV. Blood–brain barrier breakdown in Alzheimer's disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yew B, Nation DA. for the Alzheimer's disease neuroimaging initiative. Cerebrovascular resistance: effects on cognitive decline, cortical atrophy, and progression to dementia. Brain. 2017;140(7):1987‐2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hendrickx JO, Martinet W, Van Dam D, De Meyer GRY. Inflammation, nitro‐oxidative stress, impaired autophagy, and insulin resistance as a mechanistic convergence between arterial stiffness and Alzheimer's disease. Front Mol Biosci. 2021;8:651215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sweeney MD, Montagne A, Sagare AP, et al. Vascular dysfunction—The disregarded partner of Alzheimer's disease. Alzheimer's Dement. 2019;15(1):158‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12(8):822‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clark LR, Berman SE, Rivera‐Rivera LA, et al. Macrovascular and microvascular cerebral blood flow in adults at risk for Alzheimer's disease. Alzheimer's Dement : Diagnos, Assessment Dis Monitor. 2017;7:48‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Birdsill AC, Koscik RL, Jonaitis EM, et al. Regional white matter hyperintensities: aging, Alzheimer's disease risk, and cognitive function. Neurobiol Aging. 2014;35(4):769‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berman SE, Clark LR, Rivera‐Rivera LA, et al. Intracranial arterial 4D flow in individuals with mild cognitive impairment is associated with cognitive performance and amyloid positivity. J Alzheimers Dis. 2017;60(1):243‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vemuri P, Graff‐Radford J, Lesnick TG, et al. White matter abnormalities are key components of cerebrovascular disease impacting cognitive decline. Brain Communications. 2021;3(2):fcab076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alber J, Alladi S, Bae H‐J, et al. White matter hyperintensities in vascular contributions to cognitive impairment and dementia (VCID): knowledge gaps and opportunities. Alzheimer's Dement (N Y). 2019;5:107‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Watts ME, Pocock R, Claudianos C. Brain energy and oxygen metabolism: emerging role in normal function and disease. Front Mole Neurosci. 2018;11:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zarrinkoob L, Ambarki K, W∖a ahlin A, Birgander R, Eklund A, Malm J. Blood flow distribution in cerebral arteries. J Cerebral Blood Flow Metabolism. 2015;35(4):648‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rivera‐Rivera LA, Schubert T, Turski P, et al. Changes in intracranial venous blood flow and pulsatility in Alzheimer's disease: a 4D flow MRI study. J Cerebral Blood Flow Metabolism. 2017;37(6):2149‐2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rivera‐Rivera LA, Turski P, Johnson KM, et al. 4D flow MRI for intracranial hemodynamics assessment in Alzheimer's disease. J Cerebral Blood Flow Metabolism. 2016;36(10):1718‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rivera‐Rivera LA, Cody KA, Eisenmenger L, Cary P, Rowley HA, Carlsson CM, Johnson SC, Johnson KM. Assessment of vascular stiffness in the internal carotid artery proximal to the carotid canal in Alzheimer’s disease using pulse wave velocity from low rank reconstructed 4D flow MRI. Journal of Cerebral Blood Flow & Metabolism. 2021;41(2):298–311. 10.1177/0271678x20910302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rivera‐Rivera LA, Cody KA, Rutkowski D, et al. Intracranial vascular flow oscillations in Alzheimer's disease from 4D flow MRI. NeuroImage: Clin. 2020;28:102379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson SC, Koscik RL, Jonaitis EM, et al. The Wisconsin registry for Alzheimer's prevention: a review of findings and current directions. Alzheimer's Dement: Diagnosis, Assess Dis Monitor. 2018;10:130‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ho DE, Imai K, King G, Stuart EA. Matching as nonparametric preprocessing for reducing model dependence in parametric causal inference. Polit Analy. 2007;15(3):199‐236. [Google Scholar]
- 28. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 2011;7(3):270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 2011;7(3):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 2011;7(3):280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koscik R, Hermann BP, Allison S, et al. Validity evidence for the research category, “cognitively unimpaired – declining,” as a risk marker for mild cognitive impairment and Alzheimer's disease. Front Aging Neurosci. 2021;13:688478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson KM, Lum DP, Turski PA, Block WF, Mistretta CA, Wieben O. Improved 3D phase contrast MRI with off‐resonance corrected dual echo VIPR. Magn Reson Med. 2008:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ong F, Lustig M, SigPy: a python package for high performance iterative reconstruction. 27th Annual ISMRM Meeting, Montreal, Canada, p. 4819. [Google Scholar]
- 34. Pruessmann KP, Weiger M, Scheidegger MB, Boesiger P. others. SENSE: sensitivity encoding for fast MRI. Magn Reson Med. 1999;42(5):952‐962. [PubMed] [Google Scholar]
- 35. Ying L, Sheng J. Joint image reconstruction and sensitivity estimation in SENSE (JSENSE). Magn Reson Med. 2007;57(6):1196‐1202. [DOI] [PubMed] [Google Scholar]
- 36. Loecher M, Schrauben E, Johnson KM, Wieben O. Phase unwrapping in 4D MR flow with a 4D single‐step laplacian algorithm. J Magn Reson Imaging. 2016;43(4):833‐842. [DOI] [PubMed] [Google Scholar]
- 37. Schrauben E, Wåhlin A, Ambarki K, et al. Fast 4D flow MRI intracranial segmentation and quantification in tortuous arteries. J Magn Reson Imag : JMRI. 2015;42(5):1458‐1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tong Y, Hocke LM, Frederick BB. Low frequency systemic hemodynamic “Noise” in resting state BOLD fMRI: characteristics, causes, implications, mitigation strategies, and applications. Front Neurosci. 2019;13:787‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Allison SL, Koscik RL, Cary RP, et al. Comparison of different MRI‐based morphometric estimates for defining neurodegeneration across the Alzheimer's disease continuum. NeuroImage: Clin. 2019;23:101895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmidt P, Gaser C, Arsic M, et al. An automated tool for detection of FLAIR‐hyperintense white‐matter lesions in multiple sclerosis. NeuroImage. 2012;59(4):3774‐3783. [DOI] [PubMed] [Google Scholar]
- 41. Manolio TA, Kronmal RA, Burke GL, et al. Magnetic resonance abnormalities and cardiovascular disease in older adults. The cardiovascular health study. Stroke. 1994;25(2):318‐327. [DOI] [PubMed] [Google Scholar]
- 42. Moghekar A, Kraut M, Elkins W, et al. Cerebral white matter disease is associated with Alzheimer's pathology in a prospective cohort. Alzheimer's Dement. 2012;8(5):S71‐S77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kraut MA, Beason‐Held LL, Elkins WD, Resnick SM. The impact of magnetic resonance imaging‐detected white matter hyperintensities on longitudinal changes in regional cerebral blood flow. J Cereb Blood Flow Metab. 2008;28(1):190‐197. [DOI] [PubMed] [Google Scholar]
- 44. Fazekas F, Chawluk J, Alavi A, Hurtig H, Zimmerman R. MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. Am J Roentgenol. 1987;149(2):351‐356. [DOI] [PubMed] [Google Scholar]
- 45. Betthauser TJ, Koscik RL, Jonaitis EM, et al. Amyloid and tau imaging biomarkers explain cognitive decline from late middle‐age. Brain. 2020;143(1):320‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Betthauser TJ, Cody KA, Zammit MD, et al. In vivo characterization and quantification of neurofibrillary Tau PET Radioligand 18F‐MK‐6240 in humans from Alzheimer disease dementia to young controls. J Nuclear Med. 2019;60(1):93‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnson SC, Christian BT, Okonkwo OC, et al. Amyloid burden and neural function in people at risk for Alzheimer's Disease. Neurobiol Aging. 2014;35(3):576‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Price JC, Klunk WE, Lopresti BJ, et al. Kinetic modeling of amyloid binding in humans using PET Imaging and Pittsburgh Compound‐B. J Cereb Blood Flow Metab. 2005;25(11):1528‐1547. [DOI] [PubMed] [Google Scholar]
- 49. Racine AM, Clark LR, Berman SE, et al. Associations between performance on an abbreviated cogstate battery, other measures of cognitive function, and biomarkers in people at risk for Alzheimer's disease. J Alzheimer's Dis. 2016;54(4):1395‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lakens D. Calculating and reporting effect sizes to facilitate cumulative science: a practical primer for t‐tests and ANOVAs. Front Psychol. 2013;4:863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Björnfot C, Garpebring A, Qvarlander S, Malm J, Eklund A, Wåhlin A. Assessing cerebral arterial pulse wave velocity using 4D flow MRI. J Cereb Blood Flow Metab. 2021:0271678X211008744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ruesink T, Medero R, Rutkowski D, Roldán‐Alzate A. In vitro validation of 4D Flow MRI for local pulse wave velocity estimation. Cardiovasc Eng Technol. 2018;9(4):674‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Whittaker JR, Driver ID, Venzi M, Bright MG, Murphy K. Cerebral autoregulation evidenced by synchronized low frequency oscillations in blood pressure and resting‐state fMRI. Front Neurosci. 2019;13:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim H‐L, Kim S‐H. Pulse wave velocity in atherosclerosis. Front Cardiovasc Med. 2019;6:41‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front Aging Neurosci. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mestre H, Mori Y, Nedergaard M. The Brain's glymphatic system: current controversies. Trends Neurosci. 2020;43(7):458‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mestre H, Tithof J, Du T, et al. Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Communicat. 2018;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Veluw SJ, Hou SS, Calvo‐Rodriguez M, et al. Vasomotion as a driving force for paravascular clearance in the awake mouse brain. Neuron. 2020;105(3):549‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andersen AV, Simonsen SA, Schytz HW, Iversen HK. Assessing low‐frequency oscillations in cerebrovascular diseases and related conditions with near‐infrared spectroscopy: a plausible method for evaluating cerebral autoregulation?. Neurophotonics. 2018;5(3):030901‐030901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Graff‐Radford J, Arenaza‐Urquijo EM, Knopman DS, et al. White matter hyperintensities: relationship to amyloid and tau burden. Brain. 2019;142(8):2483‐2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. The SPRINT MIND Investigators for the SPRINT Research Group, The SPRINT MIND Investigators for the SPRINT Research Group. Effect of intensive vs standard blood pressure control on probable dementia: a randomized clinical trial. JAMA. 2019;321(6):553‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Torrandell‐Haro G, Branigan GL, Vitali F, Geifman N, Zissimopoulos JM, Brinton RD. Statin therapy and risk of Alzheimer's and age‐related neurodegenerative diseases. Alzheimer's Dement: Translat Res Clin Intervent. 2020;6(1):e12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alfini AJ, Weiss LR, Nielson KA, Verber MD, Smith JC. Resting cerebral blood flow after exercise training in mild cognitive impairment. JAD. 2019;67(2):671‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Banerjee G, Kim HJ, Fox Z, et al. MRI‐visible perivascular space location is associated with Alzheimer's disease independently of amyloid burden. Brain. 2017;140(4):1107‐1116. [DOI] [PubMed] [Google Scholar]
- 65. Vikner T, Eklund A, Karalija N, et al. Cerebral arterial pulsatility is linked to hippocampal microvascular function and episodic memory in healthy older adults. J Cereb Blood Flow Metab. 2021:0271678X20980652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information