

Abstract
Inhibitors, activators, and substrates of cyclin-dependent kinases (cdks) utilize a cyclin-binding sequence, known as a Cy or RXL motif, to bind directly to the cyclin subunit. Alanine scanning mutagenesis of the Cy motif of the cdk inhibitor p21 revealed that the conserved arginine or leucine (constituting the conserved RXL sequence) was important for p21's ability to inhibit cyclin E-cdk2 activity. Further analysis of mutant Cy motifs showed, however, that RXL was neither necessary nor sufficient for a functional cyclin-binding motif. Replacement of either of these two residues with small hydrophobic residues such as valine preserved p21's inhibitory activity on cyclin E-cdk2, while mutations in either polar or charged residues dramatically impaired p21's inhibitory activity. Expressing p21N with non-RXL Cy sequences inhibited growth of mammalian cells, providing in vivo confirmation that RXL was not necessary for a functional Cy motif. We also show that the variant Cy motifs identified in this study can effectively target substrates to cyclin-cdk complexes for phosphorylation, providing additional evidence that these non-RXL motifs are functional. Finally, binding studies using p21 Cy mutants demonstrated that the Cy motif was essential for the association of p21 with cyclin E-cdk2 but not with cyclin A-cdk2. Taking advantage of this differential specificity toward cyclin E versus cyclin A, we demonstrate that cell growth inhibition was absolutely dependent on the ability of a p21 derivative to inhibit cyclin E-cdk2.
Progression through the eukaryotic cell cycle requires the activity of a family of kinases known as cyclin-dependent kinases (cdks). cdks are inactive as monomers but become active upon heterodimerization with regulatory subunits known as cyclins. The assembly of these cyclin-cdk complexes is further regulated by the temporal expression of different cyclins so that only certain cyclin-cdk complexes are present during a given phase of the cell cycle. In early G1 phase of the cell cycle, cyclin D is complexed with Cdk4 or Cdk6 and phosphorylates the retinoblastoma protein (pRb), an early event in the G1-to-S transition. This is followed by the activation of cyclin E-cdk2 (late G1) and then cyclin A-cdk2 (S phase), which are responsible for the initiation of DNA replication and progression through S phase. In G2 phase of the cell cycle, cyclin B-cdc2 begins to accumulate and drives cells through mitosis, at which time the cell cycle is allowed to begin again. These topics have been extensively reviewed (16, 17, 20).
In addition to the temporal control of cyclin-cdk complexes, which restricts their activity to distinct periods in the cell cycle, cells have devised mechanisms for targeting cdks to specific proteins during their window of activity. We first noted this targeting mechanism in structure-function studies of p21; two independent motifs were identified, either of which could target p21 to cyclins and at least one of which was essential for optimal inhibition of kinase activity by p21 (5). This targeting sequence, also known as a Cy or RXL motif, is not limited to inhibitors but also forms the basis of the association of an activator, the cdc25a phosphatase, with cyclin-cdk complexes (23). Other groups identified a similar targeting sequence on many substrates that associate directly with the cdks (e.g., p107, p130, and Rb) and have inferred that this sequence targets the substrate to the cyclin part of the cyclin-cdk complex (1, 2, 24, 26). Similar Cy motifs have surfaced in the targeting of other cellular proteins to cyclin-cdk complexes, e.g., Myt1 to cyclin B-cdc2 (14), SSeCKS to cyclin D (13), CDC6 to cyclin A-cdk2 (11, 19, 22), human papillomavirus E1 protein to cyclin E-cdk2 (15), and β3-endonexin to cyclin A (18).
Although the use of Cy motifs as a key mechanism for targeting cellular factors to cdks is now well established, relatively little is known about the exact nature of this motif. Based on sequence conservation among known Cy motifs the core of the Cy motif appears to consist of an arginine and a leucine separated by a single amino acid, giving rise to the moniker RXL motif (Fig. 1). High-resolution structures of the cdk inhibitor p27 complexed with cyclin A-cdk2 and a Cy motif-containing peptide from p107 also complexed with cyclin A-cdk2 suggest that the Cy motif interacts with a hydrophobic groove on the surface of cyclin A (3, 7, 21). It is still not known, however, what amino acids, if any, can be tolerated at different positions of the Cy motif and whether different Cy motifs associate differentially with particular cyclin-cdk complexes. We have recently shown that the presence of a Cy motif on a cdk substrate results in a 100-fold decrease in Km (25). Such a large contribution to the efficiency of phosphorylation of a substrate makes it likely that most physiological cdk substrates will have a functional Cy motif. Yet a conserved RXL sequence has not been noted in the vicinity of the phosphoacceptor serine in many of the cdk substrates studied, suggesting that an RXL sequence is not necessary to form a cyclin-binding Cy motif.
FIG. 1.
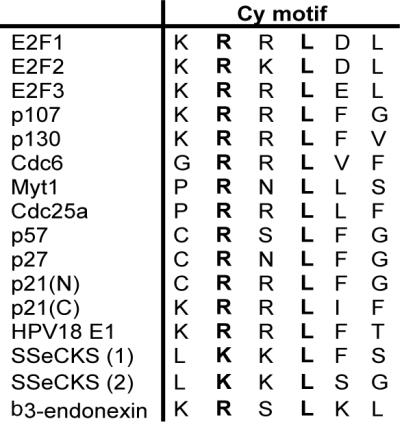
Sequence alignment of known Cy motifs. HPV, human papillomavirus. Conserved residues are in boldface.
Here we report the results of an extensive mutational analysis of the Cy motif from cdk inhibitor p21. The data indicate that the inhibitory activity of p21 is tolerant of a number of different amino acids in key positions of the Cy motif. We show that this high degree of tolerance is also true for Cy motifs present in cdk substrates. Finally, our results suggest that the mode of binding of p21 with cyclin E-cdk2 is different from its mode of binding with cyclin A-cdk2, and we take advantage of this observation to demonstrate that inhibition of cyclin E-cdk2 is absolutely essential for the growth suppression activity of p21, while the inhibition of cyclin A-cdk2 activity alone is insufficient for this effect.
MATERIALS AND METHODS
Plasmid construction and expression and purification of p21N Cy mutants.
Bacterial expression plasmid pGEX-p21N (residues 1 to 90) was constructed as described previously (4). The Stratagene Quikchange system was used to introduce a HindIII site upstream of the region of the gene coding for the Cy motif. Oligonucleotide cassettes flanked by HindIII and BlpI sites and containing either the desired mutation or a randomized codon were inserted into pGEX-p21N. The mutations were identified using standard DNA sequencing. The p21N-ΔCy mutant had a deletion that removed amino acids 17 to 24 from p21N. Plasmids encoding the mutants were then transformed into BL21(DE3). The glutathione S-transferase (GST) fusion proteins were expressed and purified as before (4, 5). Protein concentrations were determined using the Bio-Rad protein assay.
Expression and purification of cyclin-cdk complexes.
All cyclin-cdk complexes used in the in vitro kinase assays were expressed and purified by coinfecting Sf9 cells with baculoviruses encoding the appropriate cyclin-cdk pair. Cells were harvested 48 to 72 h later and purified as described previously (8). Baculoviruses expressing GST-cyclin E, GST-cyclin A, cdk2, GST-cdk4, and cyclin D1 were all kind gifts from Helen Piwnica-Worms.
The cyclin and cyclin-cdk complexes used in the in vitro binding assay were expressed in Escherichia coli. The genes for cyclin A and cyclin E were cloned using PCR into pHisTrx (a kind gift from Christophe Briand) and expressed as six-His–thioredoxin (His-Trx) fusion proteins (12). BL21(DE3) cells carrying this plasmid were grown at 25°C, induced with the addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 1 mM, and harvested 6 to 8 h later. The genes encoding Cdk2 and Civ1 (gift from Mark Solomon) were also cloned by PCR in the pMM vector (gift from Steve Blacklow) and expressed from a single promoter as a bicistronic message. BL21(DE3) cells carrying this plasmid were grown at 30°C, induced with IPTG, and harvested 24 h later. Recombinant bacterial cyclin-cdk complexes were assembled by mixing bacterial lysates containing either His-Trx–cyclin A or His-Trx–cyclin E and cdk2 and purified using Ni-nitrilotriacetic acid beads as described by the manufacturer (Qiagen). Additional information regarding plasmid construction and protein purification is available upon request.
Construction and expression of substrate peptides.
A plasmid encoding the substrate CDC6(wt) peptide has been described previously (25). Oligonucleotide cassettes were used to replace the Cy motif-encoding region of the CDC6 (wt) peptide expression plasmid with desired sequences allowing expression of CDC6-derived peptides containing either the arginine-to-valine or arginine-to-tryptophan mutations. Peptides were expressed and purified according to previously published methods (S. C. Backlow and P. S. Kim, Letter, Nat. Struct. Biol. 3:758–762, 1996).
Kinase assays.
All in vitro kinase assays were performed as described previously (25). For cyclin A-cdk2 and cyclin E-cdk2 in vitro kinase assays, the CDC6(wt) peptide was used as the phosphoacceptor substrate at a final concentration of 16 μM. For cyclin D1-cdk4 kinase assays, a substrate peptide derived from the CDC6(wt) peptide in which the SPPK phosphorylation site was changed to SPKK was used at a final concentration of 133 μM. The concentration of ATP was 65 μM in these assays. Substrate peptides with the mutant Cy motif were tested for their ability to be phosphorylated by cyclin A-cdk2 or cyclin E-cdk2 using 13 μM peptide and 85 μM ATP.
In vitro binding assays.
Purified GST-p21N mutants were mixed with bacterial lysates containing His-Trx–cyclin A or His-Trx–cyclin E or with purified His-Trx–cyclin A–cdk2 or purified His-Trx–cyclin E–cdk2. The GST-p21N-associated proteins were purified on agarose beads coupled to glutathione; washed extensively with a buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, and 0.01% NP-40; and analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Immunoblotting with anti-His antibodies (Santa Cruz Biotechnology) revealed the presence or absence of cyclins in the GST pull-down.
Cell cycle arrest by p21N mutants.
Coding sequences for the genes encoding wild-type p21N, p21N-ΔCy, p21N-R19V, and p21N-R19W were cloned into mammalian GST expression plasmid pEBG (23). U2OS cells were cotransfected with 5 μg of the expression plasmid for each p21N mutant and 1 μg of a plasmid encoding farnesylated green fluorescent protein (GFP; pEGFP-F; Clontech) using Lipofectamine-mediated transfection (Gibco BRL). Cells were harvested 48 h after transfection and prepared for fluorescence-activated cell sorting (FACS) as described by Jiang and Hunter (10). A Coulter Epics XL flow cytometer was used to determine the DNA profile of GFP-positive cells, which should also express the p21N plasmid. The cell cycle distribution for a given population was determined using the WinCycle DNA analysis software. All experiments were done in duplicate.
RESULTS
Alanine scanning mutagenesis of Cy motif.
To address the role of individual amino acids in the Cy motif-cyclin binding interface, we performed a mutational analysis of cdk inhibitor p21. All our experiments were done with a derivative of p21 containing only the N-terminal 90 amino acids, called p21N. This strategy eliminates a second Cy motif present in the C-terminal half of p21 that is redundant to the first Cy motif in the N-terminal half and so interferes with our functional analysis of the latter (5). Our earlier studies have shown that p21N is sufficient for interacting with and inhibiting cdks and for suppressing cell growth (4). Further, the cdk inhibitors related to p21, p27, and p57 are homologous to p21 only over this N-terminal half, and the crystal structure of the N-terminal half of p27 complexed with cyclin A-cdk2 demonstrates that this region contains all the sequences necessary for interacting with the cyclin-cdk complex.
pGEX-p21N is a bacterial GST expression vector that encodes p21N (residues 1 to 90), which includes the N-terminal Cy motif (residues 19 to 23) and the cdk-interacting domain (residues 53 to 58). Each residue in the core Cy motif from p21 (RRLFG, corresponding to residues 19 to 23) was mutated to alanine by inserting an oligonucleotide cassette into pGEX-p21N. These mutants were then tested for their ability to inhibit the kinase activity of cyclin D10-cdk4, cyclin E-cdk2, and cyclin A-cdk2. We also tested a p21N derivative lacking a functional Cy motif, p21N-ΔCy, in which residues 17 to 24, a region overlapping the Cy motif, were deleted. The 50% inhibitory concentrations (IC50) for each of these mutants with different cyclin-cdk complexes are shown in Fig. 2. Wild-type p21N potently inhibited cyclin E-cdk2, whereas deletion of the Cy motif abrogated this inhibitory activity. Similarly, mutations of the conserved arginine and leucine (R19 and L21) to alanine abolished the inhibitory activity of p21N, while mutations in the other positions had little effect, suggesting that R19 and L21 are the critical mediators of cyclin binding. In contrast to what was found for cyclin E-cdk2, the Cy mutants appeared to have little to no effect on p21N′s ability to inhibit cyclin A-cdk2, an observation that we address later (see Fig. 7). None of the p21N mutants inhibited cyclin D1-cdk4 at the concentrations tested (up to 1 μM).
FIG. 2.

Alanine scanning mutagenesis of the Cy motif from p21N reveals the importance of R19 and L21 in cdk inhibition. All five residues of the core Cy motif of p21N (RRLFG) were mutated to alanine and tested for their ability to inhibit cyclin E-cdk2 (gray bars), cyclin D1-cdk4 (black bars), and cyclin A-cdk2 (white bars). IC50 are plotted for each mutant.
FIG. 7.

In vitro binding assays demonstrate differential Cy motif requirements for the binding of p21N to cyclin A-cdk2 and cyclin E-cdk2. Different GST-p21N Cy mutants were tested for their ability to bind cyclin A or cyclin E either alone or in complex with cdk2. Binding reactions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by immunoblotting with an anti-His antibody.
Arginine 19 and leucine 21 mutations.
Having shown that R19 and L21 are critical binding determinants for the Cy motif-cyclin interaction, we created additional mutations at these two sites and monitored their effect on cyclin-cdk inhibition. By inserting mutant oligonucleotide cassettes, in which the codon for either R19 or L21 was randomized, into pGEX-p21N, we created a series of different mutations at each of these two positions. The IC50 for each mutant are shown in Fig 3.
FIG. 3.

Mutational analysis of residues R19 and L21 shows limited sequence degeneracy. Residues R19 (A) and L21 (B) were mutated to a number of other amino acids and then tested for their ability to inhibit cyclin E-cdk2 (gray bars), cyclin D1-cdk4 (black bars), and cyclin A-cdk2 (white bars). IC50 are plotted for each mutant.
For both the R19 and L21 positions, we found that only a narrow range of substitutions would allow the p21N mutant to retain its ability to inhibit cyclin E-cdk2. Replacement of R19 with either valine or leucine, both small hydrophobic residues, resulted in functional p21N derivatives with inhibitory activity. On the other hand, insertion of charged or polar residues or large aromatic residues at this position dramatically impaired the ability of p21N to inhibit cyclin E-cdk2 kinase. As for the L21 position, replacement of the leucine with other small hydrophobic residues such as valine or isoleucine preserved p21N′s inhibitory activity on cyclin E-cdk2, whereas less-conservative mutations to polar and charged residues abolished it. Although there were strong amino acid preferences for both of these positions, it is important to note that the requirements for arginine and leucine are not absolute, as might be suggested by sequence alignments of known Cy motifs (Fig. 1). In both cases, mutants that maintained the inhibitory activity of p21N were found. As was found with the alanine mutants, none of the R19 or L21 mutants showed a significantly impaired ability to inhibit cyclin A-cdk2 or increased inhibitory activity for cyclin D1-cdk4.
RXL is not sufficient for a Cy motif.
In addition to the single-amino-acid substitutions, we also examined a limited set of double mutants. The results from the alanine mutations suggested that residues R20, F22, and G23 were dispensable for the Cy motif-cyclin association. It is possible, however, that each of these residues makes a small but additive contribution to the binding interface that was undetectable using single-amino-acid substitutions but that would become noticeable if we combined two point mutations that had no effect alone. The IC50 of several of these mutants are shown in Fig. 4. For the majority of these mutants, we clearly see that the combination of two mutants which had little effect on their own was able to impair p21N′s inhibitory activity. For example, mutants RRLAA and RALAG both lost their inhibitory activity on cyclin E-cdk2 even though R20A, F22A, and G23A had no significant effect on their own. From these results, we conclude that the amino acid context in which the RXL residues exist is critical for its high-affinity interaction with the cyclin and that the other positions of the Cy motif play an important role in cyclin binding.
FIG. 4.

Analysis of double mutants. A number of double mutations were made in the core Cy motif of p21N and were tested for their ability to inhibit cyclin-cdk complexes. IC50 for each mutant with cyclin E-cdk2 (gray bars), cyclin D1-cdk4 (black bars), and cyclin A-cdk2 (white bars) are plotted for each mutant.
Cell cycle arrest by p21N Cy mutants.
To prove that the non-RXL Cy mutants are functional in vivo, we tested a subset of the p21N Cy mutants for their ability to block cell cycle progression in cell culture. Wild-type p21N, p21N-ΔCy, p21N-R19V, and p21N-R19W were expressed from a strong EF1α promoter in EBG plasmids, which were cotransfected with a plasmid encoding farnesylated GFP into U2OS cells. Cells were harvested 40 h later, stained with propidium iodide, and then analyzed by FACS to determine the DNA content of the transfected, GFP-positive cells. The data are shown in Fig. 5. Cells transfected with empty vector showed a normal cell cycle distribution (percentage of cells in G1 [percent G1] = 59.7% ± 8.2%), while cells transfected with wild-type p21N were completely blocked in G1 phase of the cell cycle (percent G1 = 95.1% ± 1.2%). p21N-R19V, which contains a VXL type Cy motif, which inhibited both cyclin E-cdk2 and cyclin A-cdk2 in vitro, halted cell cycle progression (percent G1 = 90.2% ± 5.8%), suggesting that VXL was a functional Cy motif in vivo. p21N-ΔCy, which did not inhibit cyclin E-cdk2 but which did inhibit cyclin A-cdk2 in vitro, did not significantly affect cell cycle distribution (percent G1 = 58.9% ± 7.9%), suggesting that inhibition of cyclin E-cdk2 was essential for p21N to block cell cycle progression.
FIG. 5.

p21N Cy mutants are able to cause G1 cell cycle arrest in mammalian cells. Different Cy mutants were overexpressed in U2OS cells and then analyzed by FACS to determine their cell cycle distributions.
Effects of Cy mutants on CDC6-derived peptide substrates.
Although Cy motifs have been discovered independently on cdk inhibitors and on substrates, we do not know whether the same attributes of a Cy motif are necessary for docking to the cyclin in the two cases. Further, as pointed out in the introduction, no sequence other than the SPXK sequence has been conserved near the phosphoacceptor serine in traditional cdk substrates, suggesting that Cy motifs on substrates are degenerate in sequence. Because we had variant Cy motifs that were functional in a cdk inhibitor, we could address these issues by testing a few of the variant Cy mutations for their ability to target a substrate for phosphorylation by cyclin-cdk complexes.
We have previously shown that a peptide containing residues 70 to 102 from replication factor CDC6 was a high-affinity substrate for cyclin-cdk complexes and that the efficient phosphorylation of this substrate required an intact Cy motif (25). Using this peptide as a template, we replaced the wild-type Cy motif (RRLVF) with the Cy mutant from p21N (RRLFG) as well as mutants R1V (VRLFG) and R1W (WRLFG). For the negative control, we used the previously characterized CDC6(null) peptide, which contained Cy motif RAARA (25). The rates of phosphorylation of each of these peptides with cyclin A-cdk2 and cyclin E-cdk2 were measured, and the results are shown in Fig. 6. As expected, the CDC6 peptide containing the Cy motif from p21N was phosphorylated very well, 39- and 24-fold better than the null peptide for cyclin E-cdk2 and cyclin A-cdk2, respectively. This shows that although a Cy motif appears nonessential in the interaction of p21N with cyclin A-cdk2, it is required for the efficient interaction of a substrate with this kinase. Likewise, the R19V substrate peptide is phosphorylated 14-fold better than the null peptide by cyclin E-cdk2 and 25-fold better than the null peptide by cyclin A-cdk2. These results are consistent with the FACS data and clearly indicate that non-RXL motifs can act as Cy motifs on substrate.
FIG. 6.

Analysis of different Cy mutants in substrate peptides shows their ability to target substrates to cyclin-cdk complexes. Phosphorylation of different CDC6-derived substrate peptides by either cyclin E-cdk2 (black bars) or cyclin A-cdk2 (white bars) was measured. Error bars indicate standard deviations from three separate experiments.
Gradation of Cy motif activity revealed by R19W mutant.
p21N-R19W, which did not inhibit cyclin E-cdk2 in vitro (Fig. 3A), nevertheless halted cell cycle progression (percent G1 = 84.3% ± 8.7%) (Fig. 5). A peptide substrate containing an R19W Cy motif is phosphorylated 9- and 13-fold better than a peptide containing a null Cy motif by cyclin E-cdk2 and cyclin A-cdk2, respectively (Fig. 6). The most likely explanation for these results is that an R19W Cy motif contains a weak cyclin-binding activity such that p21N-R19W is unable to significantly inhibit cyclin E-cdk2 at the highest concentration tested in our in vitro kinase assays (1 μM). The cyclin-binding activity of R19W is, however, strong enough to allow p21N-R19W to inhibit cyclin E-cdk2 and suppress cell growth in vivo and to promote the phosphorylation of a substrate by cdk2 in vitro.
Differential binding of p21N Cy mutants to cyclin E-cdk2 and cyclin A-cdk2.
In Fig. 2 and 3, we showed that the deletion of the Cy motif of p21N had remarkably little effect on the ability of p21N to inhibit cyclin A-cdk2. This was puzzling considering that the importance of the Cy motif in interactions with cyclin A-cdk2 appears well established, particularly in the crystal structure of p27N complexed with cyclin A-cdk2 (21). One possible explanation was that, despite the crystal structure, the interaction of p21N with cyclin A-cdk2 was not dependent on the presence of a Cy motif, while that with cyclin E-cdk2 was absolutely dependent on the Cy motif-cyclin interaction. To test this hypothesis GST, GST-p21N, GST-p21N-ΔCy, GST-p21N-R19V, and GST-p21N-R19W were tested for their ability to interact with cyclin E and cyclin A in either the presence or absence of cdk2 (Fig. 7).
Only GST-p21N is able to pull down cyclin E alone, suggesting that, although the variant Cy motifs R19V and R19W were functional in other assays, their interactions with cyclin E were not strong enough to survive the washing conditions of a GST pull-down experiment. Consistent with this, GST-p21N-R19V was able to pull down cyclin E-cdk2 complexes while GST-p21N-ΔCy and GST-p21N-R19W failed to do so. Therefore R19V is a functional Cy motif such that its weak interaction with the cyclin, which was not detected when it was incubated with cyclin E alone, was sufficiently strong to allow the rest of p21N to form a stable complex with the cyclin E-cdk2 heterodimer. Even though p21N-R19W was unable to bind cyclin E-cdk2 in these assays, the results of both FACS and substrate phosphorylation data indicate that R19W is capable of functioning as a Cy motif but likely interacts with too low of an affinity for cyclin E to be detected in either the in vitro binding assays or in vitro kinase assays.
We also find that cyclin A alone interacts strongly with wild-type p21N and weakly with p21N-R19V. The fact that both wild-type p21N and p21N-R19V but not p21N-ΔCy bind to cyclin A suggests that the binding of p21N to cyclin A alone requires a functional Cy motif. As discussed earlier, we believe that p21N-R19W contains a partially functional Cy motif and that its inability to interact with cyclin A in this assay is consistent with the affinity of the interaction being lower than that for either the wild-type or R19V Cy motif-cyclin A interaction. This conclusion is supported by the substrate phosphorylation data from Fig. 6, in which the R19W substrate peptide is strongly phosphorylated compared to the null Cy motif peptide but still is phosphorylated less than either the wild-type or R19V peptide.
All of the Cy mutants including p21N-ΔCy are capable of binding to the cyclin A-cdk2 complex. The ability of p21N-ΔCy to pull down cyclin A-cdk2 suggests that the Cy motif-cyclin A interaction is not required for complex formation. Instead, the cdk binding site of p21N is sufficient for its association with cyclin A-cdk2 and occurs regardless of whether or not a functional Cy motif is present. This is in contrast to the association of p21N with cyclin E-cdk2, which required a functional Cy motif in order to form a stable association. The ability of p21N to interact with cyclin A-cdk2 independent of a Cy motif explains why deletion of the Cy motif had no effect on p21N′s ability to inhibit cyclin A-cdk2 kinase activity (Fig. 2 to 4).
DISCUSSION
In this study we have used an extensive mutational analysis of cdk inhibitor p21 to define the amino acid requirements of a Cy motif (Fig. 2 to 4). We have further characterized a subset of these Cy mutants to determine their effect on the ability of p21 to inhibit mammalian cell growth (Fig. 5) and their ability to target substrates to cyclin-cdk complexes (Fig. 6). We also show that the Cy motif requirement for kinase inhibition by p21 differs between cyclin E-cdk2 and cyclin A-cdk2 (Fig. 7). Through this detailed characterization of a Cy motif, we have identified a number of interesting features regarding the molecular details of the Cy motif-cyclin interaction.
Based on the sequence alignment of a large number of Cy motifs, Cy motifs have been described as having the pattern ZRXL, where Z and X are predominantly basic residues. Our results suggest that a cluster of basic residues or an arginine and leucine separated by a single amino acid (RXL) should not be the defining characteristic of a functional Cy motif. In our mutational analysis, the conserved arginine (R19 in our study) of a Cy motif can be replaced with a small hydrophobic residue such as valine or leucine with only a small effect on the ability of the mutant protein to inhibit cyclin-cdk activity. This ability to replace a polar amino acid with a hydrophobic residue with little or no loss of activity as well as the importance of having a small hydrophobic residue at residue 21 strongly emphasizes the hydrophobic character of this binding interface. In addition to emphasizing the hydrophobic nature of this binding surface, our data indicate that the amino acid sequence RXL is neither necessary nor sufficient for the formation of a functional Cy motif. The ability of VXL, LXL, or RXV to replace the wild-type RXL with only a small diminishment in p21N′s inhibitory activity for cyclin E-cdk2 suggests that RXL is not an absolute requirement for a Cy motif. In Fig. 3, we show that the mutation of wild-type Cy motif RRLFG to either RRLAA or RALAG results in a dramatic decrease in the ability of p21N to inhibit cyclin E-cdk2 even though the RXL pattern is maintained. From this data, we conclude that the defining characteristic of a functional Cy motif is not ZRXL but instead a cluster of hydrophobic residues whose context allows them to adopt the appropriate conformation for interacting with the hydrophobic substrate recognition patch on the surface of the cyclin.
Another interesting feature of cyclin-cdk recognition observed in our study is the differential dependence on the Cy motif-cyclin interaction for p21 to interact with cyclin E-cdk2 versus cyclin A-cdk2. In Fig. 7, we show that the association of p21N with cyclin E-cdk2 requires a functional Cy motif bound to cyclin E to allow subsequent binding of p21N′s cdk-binding site to the catalytic cleft of cdk2. The use of the Cy motif as the initial anchor for complex formation, which then allows the cdk-binding site to disrupt the active site of cdk2, was predicted by the high-resolution structure of p27 complexed with cyclin A-cdk2 based on the large number of cdk2 structural rearrangements required by the p27-cdk2 interaction (21). Interestingly, this appears not to be true for the association of p21N with cyclin A-cdk2. Our data indicate that cdk binding by p21N can occur independently of a functional Cy motif. Indeed, all of the Cy motif mutants tested retained their ability to bind and inhibit cyclin A-cdk2. Evidence that this differential Cy motif requirement for cyclin E-cdk2 and cyclin A-cdk2 is also present in vivo can be found in studies looking at the ability of different anti-p21 monoclonal antibodies to immunoprecipitate endogenous p21 complexes from WI-38 fibroblasts (5, 6). In this work, immunoprecipitation of p21 by CP36, an antibody that specifically recognizes the Cy motif, is able to coimmunoprecipitate cyclin A-cdk2 but not cyclin E-cdk2. The most likely explanation for these results is that the antibody disrupts both the p21Cy-cyclin E and p21Cy-cyclin A associations but that p21 is still capable of associating with cyclin A-cdk2 via its cdk-binding site whereas it is unable to bind cyclin E-bound cdk2 using this site since that association is Cy motif dependent.
Although further structural studies will be required to explain the molecular basis for this difference between cyclin A-cdk2 and cyclin E-cdk2, one possibility is that the binding of cyclin A and cyclin E to cdk2 induces different structural rearrangements in the cdk2 molecule thereby altering the interaction of cdk2 with p21 and perhaps other cellular factors. It has already been shown that the binding of a cyclin to cdk2 activates its kinase activity by inducing a number of structural rearrangements in the cdk2 protein including the remodeling of the ATP binding pocket as well as the repositioning of the T loop (9). Although it was believed that these changes only increase the overall catalytic activity of the enzyme, it is possible that these alterations in cdk structure also affect the interaction of cdk2 with p21 and other cellular factors and that the exact natures of the cdk alterations for cyclin A and cyclin E differ.
Although it is well established that different cyclin-cdk complexes including cyclin E-cdk2 and cyclin A-cdk2 have distinct substrate preferences, the molecular basis for this specificity is still unclear. Our results suggest a potential mechanism for generating specificity by targeting proteins to specific cyclin-cdk complexes. By using mutations to modulate the affinity of the Cy motif-cyclin association, we were able to create a series of p21N mutants that are able to inhibit cyclin A-cdk2 but not cyclin E-cdk2. This raises the possibility that cells could use similar mechanisms to selectively target proteins to a specific cyclin-cdk complex. We provide some experimental proof for this in Fig. 6, where we show using initial velocity measurements that an RRLFG Cy motif is a better substrate of cyclin E-cdk2 than of cyclin A-cdk2, but the situation is reversed for VRLFG and WRLFG Cy motifs.
During the mutational analysis, we were able to identify a number of p21 mutants that selectively inhibited cyclin A-cdk2. When a subset of these proteins were expressed in cells, we were able to show that the ability to inhibit cell growth correlated with the ability to inhibit cyclin E-cdk2 but not cyclin A-cdk2. Wild-type p21N and p21N-R19V were both potent inhibitors of cyclin E-cdk2 in vitro as well as potent inhibitors of cell growth in vivo. In contrast, p21N-ΔCy lost the ability to inhibit cyclin E-cdk2 in vitro as well as the ability to arrest cell growth. Since p21N-ΔCy is still capable of inhibiting cyclin A-cdk2 kinase activity in vitro, it seems likely that p21N′s ability to inhibit cell growth depends on its ability to bind and inhibit cyclin E-cdk2. Since we have not identified any mutations that confer selective inhibition of cyclin E-cdk2 but not of cyclin A-cdk2, we are unable to determine whether the inhibition of cyclin A-cdk2 is necessary for p21N′s ability to halt cell cycle progression.
Finally, our results, summarized in Table 1, show that the Cy motif-cyclin interaction can have a wider range of affinities than can be appreciated from the conserved RXL sequences identified to date. For substrate phosphorylation, the mutant Cy motifs with the weaker affinities for cyclin E are functional (compared to null Cy motifs), while for the pull-down assay on cyclin E alone only the RRLFG Cy motif is functional. For example, the R19W Cy motif produces a Cy motif-cyclin interaction that is sufficient for phosphorylation of substrates by cyclin A-cdk2 and cyclin E-cdk2 (Fig. 6) and inhibition of cyclin E-cdk2 in vivo (Fig. 5) but is not strong enough to pull down cyclin E or cyclin E-cdk2 in an in vitro binding assay (Fig. 7) or inhibit cyclin E-cdk2 in an in vitro kinase assay at the concentrations tested (Fig. 3A). Appreciation of this dynamic range of the Cy motif-cyclin interaction could be important to those trying to identify physiological substrates for cdks in diverse organisms and those seeking pharmacological inhibitors of cdks that disrupt the Cy motif-cyclin interactions.
TABLE 1.
Summary of cyclin E-Cy motif mutant interactions
| Cy motif | p21
|
Substrate promotes phosphorylation | |||
|---|---|---|---|---|---|
| Inhibition of E/K2a in vitro | Inhibition of cell growth | Association with cyclin E in vitro | Association with E/K2 in vitro | ||
| RRLFG | ++ | ++ | ++ | ++ | +++ |
| ΔCy | − | − | − | − | − |
| VRLFG | ++ | ++ | − | ++ | ++ |
| WRLFG | − | + | − | − | + |
E/K2, cyclin E-cdk2.
ACKNOWLEDGMENTS
J.A.W. and B.T.D. contributed equally to this work.
This work was supported by funds from the United States Army Medical Research and Materiel Command (DAMD 17–97-1–7314) and NIH grant CA89406. J.A.W. was supported by a predoctoral fellowship from the United States Department of Defense (National Defense Science and Engineering Graduate Fellowship).
REFERENCES
- 1.Adams P D, Li X, Sellers W R, Baker K B, Leng X, Harper J W, Taya Y, Kaelin W G., Jr Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol Cell Biol. 1999;19:1068–1080. doi: 10.1128/mcb.19.2.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams P D, Sellers W R, Sharma S K, Wu A D, Nalin C M, Kaelin W G., Jr Identification of a cyclin-cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol Cell Biol. 1996;16:6623–6633. doi: 10.1128/mcb.16.12.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown N R, Noble M E, Endicott J A, Johnson L N. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat Cell Biol. 1999;1:438–443. doi: 10.1038/15674. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Jackson P K, Kirschner M W, Dutta A. Separate domains of p21 involved in the inhibition of Cdk kinase and PCNA. Nature. 1995;374:386–388. doi: 10.1038/374386a0. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Saha P, Kornbluth S, Dynlacht B D, Dutta A. Cyclin-binding motifs are essential for the function of p21CIP1. Mol Cell Biol. 1996;16:4673–4682. doi: 10.1128/mcb.16.9.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dynlacht B D, Ngwu C, Winston J, Swindell E C, Elledge S J, Harlow E, Harper J W. Purification and analysis of CIP/KIP proteins. Methods Enzymol. 1997;283:230–244. doi: 10.1016/s0076-6879(97)83019-4. [DOI] [PubMed] [Google Scholar]
- 7.Endicott J A, Noble M E, Tucker J A. Cyclin-dependent kinases: inhibition and substrate recognition. Curr Opin Struct Biol. 1999;9:738–744. doi: 10.1016/s0959-440x(99)00038-x. [DOI] [PubMed] [Google Scholar]
- 8.Harper J W, Elledge S J, Keyomarsi K, Dynlacht B, Tsai L H, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E, et al. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeffrey P D, Russo A A, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich N P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 10.Jiang W, Hunter T. Analysis of cell-cycle profiles in transfected cells using a membrane-targeted GFP. BioTechniques. 1998;24:349–354. [PubMed] [Google Scholar]
- 11.Jiang W, Wells N J, Hunter T. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc Natl Acad Sci USA. 1999;96:6193–6198. doi: 10.1073/pnas.96.11.6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kammerer R A, Schulthess T, Landwehr R, Lustig A, Fischer D, Engel J. Tenascin-C hexabrachion assembly is a sequential two-step process initiated by coiled-coil alpha-helices. J Biol Chem. 1998;273:10602–10608. doi: 10.1074/jbc.273.17.10602. [DOI] [PubMed] [Google Scholar]
- 13.Lin X, Nelson P, Gelman I H. SSeCKS, a major protein kinase C substrate with tumor suppressor activity, regulates G1→S progression by controlling the expression and cellular compartmentalization of cyclin D. Mol Cell Biol. 2000;20:7259–7272. doi: 10.1128/mcb.20.19.7259-7272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu F, Rothblum-Oviatt C, Ryan C E, Piwnica-Worms H. Overproduction of human Myt1 kinase induces a G2 cell cycle delay by interfering with the intracellular trafficking of Cdc2-cyclin B1 complexes. Mol Cell Biol. 1999;19:5113–5123. doi: 10.1128/mcb.19.7.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma T, Zou N, Lin B Y, Chow L T, Harper J W. Interaction between cyclin-dependent kinases and human papillomavirus replication-initiation protein E1 is required for efficient viral replication. Proc Natl Acad Sci USA. 1999;96:382–387. doi: 10.1073/pnas.96.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan D O. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 17.Morgan D O. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 18.Ohtoshi A, Maeda T, Higashi H, Ashizawa S, Yamada M, Hatakeyama M. Beta3-endonexin as a novel inhibitor of cyclin A-associated kinase. Biochem Biophys Res Commun. 2000;267:947–952. doi: 10.1006/bbrc.1999.2007. [DOI] [PubMed] [Google Scholar]
- 19.Petersen B O, Lukas J, Sorensen C S, Bartek J, Helin K. Phosphorylation of mammalian CDC6 by cyclin A-cdk2 regulates its subcellular localization. EMBO J. 1999;18:396–410. doi: 10.1093/emboj/18.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts J M. Evolving ideas about cyclins. Cell. 1999;98:129–132. doi: 10.1016/s0092-8674(00)81007-7. [DOI] [PubMed] [Google Scholar]
- 21.Russo A A, Jeffrey P D, Patten A K, Massague J, Pavletich N P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 22.Saha P, Chen J, Thome K C, Lawlis S J, Hou Z H, Hendricks M, Parvin J D, Dutta A. Human CDC6/Cdc18 associates with Orc1 and cyclin-cdk and is selectively eliminated from the nucleus at the onset of S phase. Mol Cell Biol. 1998;18:2758–2767. doi: 10.1128/mcb.18.5.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saha P, Eichbaum Q, Silberman E D, Mayer B J, Dutta A. p21CIP1 and Cdc25A: competition between an inhibitor and an activator of cyclin-dependent kinases. Mol Cell Biol. 1997;17:4338–4345. doi: 10.1128/mcb.17.8.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulman B A, Lindstrom D L, Harlow E. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc Natl Acad Sci USA. 1998;95:10453–10458. doi: 10.1073/pnas.95.18.10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda D Y, Wohlschlegel J A, Dutta A. A bipartite substrate recognition motif for cyclin-dependent kinases. J Biol Chem. 2001;276:1993–1997. doi: 10.1074/jbc.M005719200. [DOI] [PubMed] [Google Scholar]
- 26.Zhu L, Harlow E, Dynlacht B D. p107 uses a p21CIP1-related domain to bind cyclin-cdk2 and regulate interactions with E2F. Genes Dev. 1995;9:1740–1752. doi: 10.1101/gad.9.14.1740. [DOI] [PubMed] [Google Scholar]