

Abstract
Background
Fabry disease (FD), an X-linked lysosomal storage disorder caused by a deficiency in alfa-galactosidase A (α-Gal A) activity due to mutations in the GLA gene, has a prevalence of 0–1.69% in patients undergoing haemodialysis; however, its prevalence in patients with chronic kidney disease (CKD) Stages 1–5 is unknown.
Methods
Serum α-Gal A activity analysis and direct sequencing of GLA were used to screen for FD in 2122 male patients with CKD, including 1703 patients with CKD Stage 5D and 419 with CKD Stages 1–5. The correlation between serum α-Gal A activity and confounding factors in patients with CKD Stages 1–5 was evaluated.
Results
FD prevalence rates in patients with CKD Stage 5D and CKD Stages 1–5 were 0.06% (1/1703) and 0.48% (2/419), respectively. A patient with CKD Stage 5D exhibited a novel GLA mutation, p.Met208Arg, whereas two patients with CKD Stages 1–5 had c.370delG and p.Met296Ile. p. Met208Arg caused moderate structural changes in the molecular surface region near the substituted amino acid residue but did not affect the catalytic residues Asp170 and Asp231 in α-Gal A. Serum α-Gal A activity in patients with CKD Stages 1–5 was inversely correlated with age (P < 0.0001) but directly correlated with estimated glomerular filtration rate (P < 0.0001).
Conclusions
FD prevalence was much higher in male patients with CKD Stages 1–5 than in those with CKD Stage 5D. FD screening in patients with CKD Stages 1–5 may improve patient survival, decreasing the number of patients with CKD Stage 5D.
Keywords: Fabry disease, screening, chronic kidney disease, haemodialysis, nephropathy
KEY LEARNING POINTS
What is already known about this subject?
Screenings for Fabry disease (FD) have shown that FD prevalence in male patients undergoing haemodialysis is 0–1.69%.
Screenings for FD in male patients with chronic kidney disease (CKD) Stages 1–5 have shown that FD prevalence is 0.59–1.80%.
Although some screenings for FD in patients with CKD Stages 1–5 have been conducted, FD prevalence in Japan remains unclear.
What this study adds?
The FD prevalence was much higher in male patients with CKD Stages 1–5 [0.48% (2/419)] than in those with CKD Stage 5D [0.06% (1/1703)]. This is the first report comparing FD prevalence between male patients with CKD Stage 5D and those with CKD Stages 1–5.
We found a novel mutation in patients with FD, p.Met208Arg, which causes moderate structural changes in the molecular surface region near the substituted amino acid residue but does not affect the catalytic residues in alfa-galactosidase A (α-Gal A).
The global FD prevalence in male patients with CKD Stage 5D could be 0.22% (63/28 931) (Table 3), while the total FD prevalence rate in patients with CKD Stages 1–5 could be 0.69% (11/1601).
What impact this may have on practice or policy?
Screening for FD is valuable and necessary to diagnose FD in patients with CKD Stages 1–5 and avoid progression of complications.
Gene mutation analysis is strongly recommended to identify the cause of pathological FD and exclude the functional variants if α-Gal A activity is low.
Although there is lack of clear evidence regarding the efficacy of enzyme replacement therapy in ameliorating kidney disease progression, FD screening of patients with CKD Stages 1–5 could lead to better patient survival and therefore decrease the number of FD patients with CKD Stage 5D.
INTRODUCTION
Fabry disease (FD) is an X-linked lysosomal storage disorder that leads to the accumulation of globotriaosylceramide (Gb3) in cells with decreased alfa-galactosidase A (α-Gal A) activity [1]. Poor clearance of Gb3 stored in the kidney and heart contributes to tissue damage, leading to conditions such as end-stage kidney disease (ESKD) and heart failure [2, 3]. Based on the presence or absence of characteristic symptoms, age of onset, sex and gene mutations, FD phenotypes are classified into three main categories: classic FD males, late-onset FD males and FD females.
Screenings for FD revealed that the FD prevalence in male patients undergoing haemodialysis (HD) is 0–1.69% [4, 5]. To date, although several studies on FD screenings have included female patients, α-Gal A activity in heterozygous FD is sometimes within the normal range. Notably, Sakuraba et al. [6] reported that the average values of serum and leukocyte α-Gal A activity in female patients with FD are approximately half of the control mean. Therefore female patients with FD cannot be clearly identified through screening based on the measurement of α-Gal A activity. Thus only male patients were included in this study of FD screening to avoid obtaining high false-negative results. Furthermore, although some screenings for FD in patients with chronic kidney disease (CKD) Stages 1–5 have been conducted [7, 8], FD prevalence in Japan is not yet known.
In this study, male patients with CKD Stage 5D and those with CKD Stages 1–5 were screened for FD by measuring serum α-Gal A activity followed by GLA gene analysis. Leukocyte α-Gal A assay and measurement of plasma globotriaosylsphingosine (lyso-Gb3) were performed to determine the diagnosis. We also compared the FD prevalence in these patients and evaluated the correlation between serum α-Gal A activity and confounding factors in patients with CKD Stages 1–5.
MATERIALS AND METHODS
Patients and data collection
The workflow of this study is summarized in Figure 1. From June 2014 to August 2018, a total of 2122 male patients with CKD Stages 1–5D were recruited, including 1703 patients undergoing HD or peritoneal dialysis in 43 dialysis hospitals and clinics and 419 patients with CKD Stages 1–5 in Kurume University Hospital (Figure 1). Patients with CKD Stage 5D were dialysed for 4–5 h with high-flux dialysers three times a week. Fasting blood was drawn from the antecubital vein of patients with CKD Stages 1–5 to determine blood urea nitrogen (BUN) and serum creatinine (SCr) levels. Spot urine test was performed to measure the level of proteinuria (g/g Cr). Additionally, estimated glomerular filtration rate (eGFR) was calculated using the following formula: eGFR = 0.741 × 175 × SCr−1.154 × age−0.203 × (0.742 if female) [9, 10]. To avoid a high rate of false-negative results, female patients were excluded from this screening. CKD was defined according to the criteria stated in the Kidney Disease: Improving Global Outcomes (2012) Clinical Practice Guidelines. Written informed consent was obtained from all patients and the study protocol was approved by the Institutional Ethics Committee of Kurume University School of Medicine, Japan (no. 13296). This work was conducted in accordance with the Helsinki Declaration of 1975, as revised in 2013, and was registered with the University Hospital Medical Information Network clinical trials database (UMIN000040203).
FIGURE 1.

Study workflow.
Measurement of α-Gal A activity in serum and leukocytes
α-Gal A activity in serum and leukocytes was measured as described previously [6, 11]. In brief, serum samples obtained at the start of dialysis were centrifuged at 3000 rpm for 10 min, then kept at 4°C for 24 h. The serum samples were stored at –30°C until α-Gal A activity measurement. α-Gal A activity was measured using a substrate solution containing 5 mmol/L 4-methylumbelliferyl α-D-galactopyranoside (Calbiochem, La Jolla, CA, USA) as a substrate and 117 mmol/L N-acetyl-d-galactosamine (Sigma-Aldrich, St. Louis, MO, USA) as a specific inhibitor of α-N-acetylgalactosaminidase in 0.1 mol/L citrate−phosphate buffer, pH 4.6 [12]. In the first screening, 20 μL of serum was mixed with 40 μL of the substrate solution in a 96-well plate. The mixture was then incubated at 37°C for 4 h, after which the reaction was stopped by adding 200 μL of 0.2 mol/L glycine buffer, pH 10.7; the released 4-methylumbelliferone was measured using a Wallac 1420 ARVO MX Multilabel Counter (PerkinElmer, Waltham, MA, USA) at excitation and emission wavelengths of 355 and 460 nm, respectively. When the serum α-Gal A activity was <1.5 nmol/h/mL, the leukocyte α-Gal A activity was measured. In the second examination, 10 μL of leukocyte homogenate (10 μg protein) was mixed with 40 μL of the substrate solution in a 1.5-mL microtube and the mixture was incubated at 37°C for 30 min. Then the reaction was stopped by adding 950 μL of 0.2 mol/L glycine buffer, pH 10.7, and the released 4-methylumbelliferone was measured using a spectrofluorometer (F2700; Hitachi, Tokyo, Japan) at excitation and emission wavelengths of 365 and 450 nm, respectively.
GLA gene analysis
GLA gene analysis was performed for subjects that exhibited serum α-Gal A activity <1.5 nmol/h/mL (mean 1.75 SD) [6]. In brief, genomic DNA was purified from leukocytes using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions and all seven exons, intron/exon boundaries and specific intronic regions containing IVS4 + 919 of the GLA gene were amplified by polymerase chain reaction (PCR) using appropriate primers [6, 13]. Then the PCR fragments were directly sequenced. The Fabry database (http://fabry-databese.org/) and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/) were assessed for variant filtering and classification.
Measurement of lyso-Gb3 concentration in plasma
Lyso-Gb3 concentration in plasma was measured using liquid chromatography–tandem mass spectrometry as described previously [14]. Briefly, an LCMS-8040 triple quadrupole mass spectrometer (Shimadzu, Kyoto, Japan) equipped with an electrospray ionization interface was used in the positive ion mode and the multiple reaction monitoring (MRM) conditions were optimized with an automatic MRM optimization function. In the MRM mode, the following transitions were monitored using a stable isotope labelled lyso-Gb3 as an internal standard: m/z 786.8→282.3 for lyso-Gb3 and m/z 790.8→286.2 for the labelled lyso-Gb3. Serum and leukocyte α-Gal A assays, plasma lyso-Gb3 measurement and GLA gene analysis were performed in the research laboratory at the Department of Clinical Genetics, Meiji Pharmaceutical University, Tokyo, Japan.
Statistical analysis
To compare the α-Gal A activity between patients with CKD Stage 5D and CKD Stages 1–5, a Wilcoxon rank test was performed, as the α-Gal A activity was not normally distributed. To determine the correlation between α-Gal A activity and age, BUN, Cr, eGFR or proteinuria, regression analysis was conducted. All statistical analyses were performed using JMP Pro version 14 software (SAS Institute, Cary, NC, USA). Data are presented as mean ± SD. P-values <0.05 were considered statistically significant.
RESULTS
Serum α-Gal A activity
Male patients with CKD Stage 5D and CKD Stages 1–5 were distributed according to their serum α-Gal A activity (Figure 2). Most patients with CKD Stage 5D and CKD Stages 1–5 had serum α-Gal A activities of 4–<4.5 (Figure 2A) and 3.5–<4.0 (Figure 2B) nmol/h/mL, respectively. However, 10 patients with CKD Stages 5D and 4 with CKD Stages 1–5 showed serum α-Gal A activity <1.5 nmol/h/mL (Figure 3).
FIGURE 2.

Distribution of patients with (A) CKD Stage 5D and (B) CKD Stages 1–5according to their serum α-Gal A activity.
FIGURE 3.
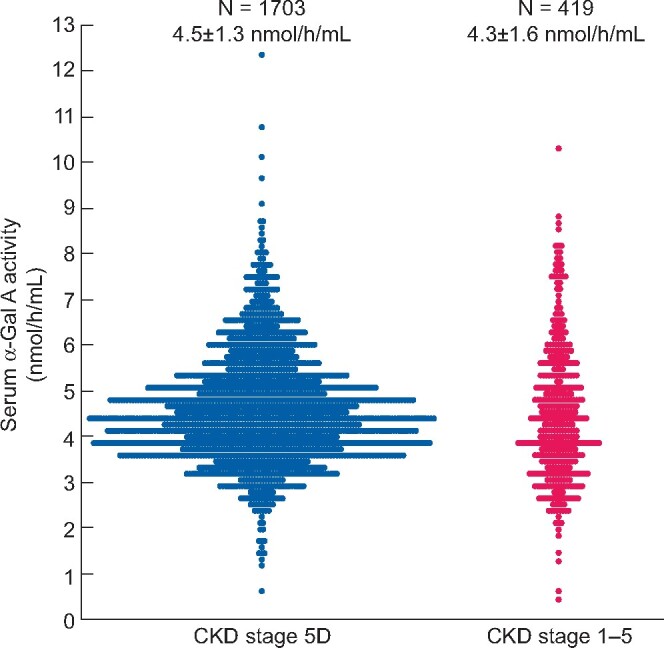
Serum α-Gal A activity in patients with CKD Stage 5D and CKD Stages 1–5.
Screening of FD in HD patients
We evaluated serum α-Gal A activity in 1703 male patients with CKD Stage 5D. The mean serum α-Gal A activity was 4.5 ± 1.3 nmol/h/mL and 10 patients undergoing dialysis showed low serum α-Gal A activity (Table 1). These patients had ESKD due to chronic glomerulonephritis (n = 3), diabetic nephropathy (n = 4) or unknown aetiologies (n = 3). Among them, two patients refused further examination and eight patients were tested for leukocyte α-Gal A activity, plasma lyso-Gb3 and gene mutations. Patient 1 showed very low serum and leukocyte α-Gal A activities (0.2 nmol/h/mL and 0.2 nmol/h/mg protein, respectively; normal range 1.6–14.6 nmol/h/mL and 20–62 nmol/h/mg protein, respectively) and elevated plasma lyso-Gb3 level (54 nmol/L; normal range 0.14–0.75 nmol/L) (Table 1).
Table 1.
Serum and leukocyte α-Gal A activity, plasma lyso-Gb3 concentration and GLA gene variants in 10 patients with CKD Stage 5D
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
|
Serum α-Gal A (nmol/h/mL) |
0.2 | 1.0 | 1.1 | 0.8 | 1.4 | 0.9 | 1.4 | 1.3 | 1.1 | 1.2 |
| Leukocyteα-Gal A (nmol/h/mg protein) | 0.2 | 25 | NA | 18 | NA | 19 | 20 | 21 | 12 | 12 |
| Plasma lyso- Gb3 (nmol/L) | 54 | 0.27 | NA | 0.13 | NA | 0.46 | 0.34 | 0.14 | 0.54 | 0.34 |
| GLA variant | p.Met208Arg | p.Glu66Gln | NA | p.Glu66Gln | NA | p.Glu66Gln | p.Glu66Gln | p.Glu66Gln | p.Glu66Gln | p.Glu66Gln |
| ACMG classification | PS3, PM2, PM5, PP2, PP4 | BS2, BS3 | – | BS2, BS3 | – | BS2, BS3 | BS2, BS3 | BS2, BS3 | BS2, BS3 | BS2, BS3 |
NA, not applicable.
Furthermore, seven patients had the p.Glu66Gln allele, which is known as a functional GLA variant [15, 16] (Table 1). The FD prevalence in patients with CKD Stage 5D was 0.06%, which corroborated the previous report [11]. In a 49-year-old patient undergoing HD (Patient 1), a novel GLA missense mutation, p. Met208Arg (ATG→AGG), was detected; when this patient was 26 years old, proteinuria was detected; at 39 years of age the kidney function of the patient decreased (BUN 55 mg/dL, SCr 6.4 mg/dL); and at 44 years of age the patient started undergoing dialysis. However, typical FD findings, such as angiokeratoma, acroparesthesia, hypohidrosis and corneal opacities, were absent in this patient. These findings were compatible with the late-onset type of FD. Furthermore, the mother of the patient had been admitted to a psychiatric hospital due to bipolar disorder and was diagnosed with FD based on genetic analysis. Enzyme replacement therapy (ERT) (agalsidase-β 1 mg/kg, 50 mg every 2 weeks) was initiated intravenously during HD sessions. Moreover, the older brother of the patient was diagnosed with proteinuria and left ventricular hypertrophy (LVH). His SCr level was within the normal range (0.97 mg/dL), proteinuria level was slightly high (1.47 g/gCr) and mulberry bodies and cells were noted. Additionally, leukocyte α-Gal A activity was <0.1 nmol/h/mg protein and plasma lyso-Gb3 was 32 nmol/L. Kidney biopsy demonstrated vacuolization and foamy changes in podocytes, while electron microscopy revealed abundant lamellar bodies in the podocyte cytoplasm.
Predicted p.Met208Arg-induced structural change in α-Gal A
To predict p.Met208Arg-induced structural changes in α-Gal A, we performed in silico structural analysis, as described previously [17]. Briefly, a structural model of the mutant α-Gal A protein was generated by homology modelling using the molecular modelling software TINKER (http://dasher.wustl.edu/tinker/). The crystal human α-Gal A structure (PDB: 1R46) [18] was used as a template and energy minimization was performed. The root mean square gradient was set at 0.05 kcal/mol·Å. To determine the effect of the mutation, the mutant α-Gal A model was superimposed on the wild-type α-Gal A structure based on the Cα atoms using the least square mean fitting algorithm. An atom was considered as affected by the mutation when the position of the atom in a mutant differed from that in the wild-type structure by >0.15 Å, based on the total root mean square distance.
Figure 4 shows the affected atoms in the 3D α-Gal A structure. The mutation is located on the surface of the (β/α)8-barrel domain in human α-Gal A. Moreover, p. Met208Arg caused moderate structural change on the molecular surface region near the substituted amino acid residue but did not affect the catalytic residues Asp170 and Asp231 in α-Gal A.
FIGURE 4.

Colouring of the atoms in the three-dimensional structure of α-Gal A induced by p.Met208Arg. The backbone of α-Gal A is represented as a ribbon. The affected atoms are indicated as small spheres and their colours show the distance between the mutant and wild-type variants as follows: blue <0.15 Å, cyan ≥0.15–<0.30 Å, green ≥0.30–<0.45 Å, yellow ≥0.45–<0.60 Å, orange ≥0.60–<0.75 Å and red ≥0.75 Å. The substituted and catalytic residues are presented as a Corey–Pauling–Koltun model and blue spheres, respectively.
Screening of FD in patients with CKD Stages 1–5
We screened for FD in 419 male patients with CKD Stages 1–5. The characteristics of the patients were as follows: mean age 60.9 ± 16.1 years, mean BUN 40.3 ± 32.9 mg/dL, SCr 3.5 ± 3.0 mg/dL and mean eGFR 35.2 ± 30.2 mL/min/1.73 m2. Additionally, the mean proteinuria level of the patients was 4.2 ± 4.3 g/g Cr and the serum α-Gal A activity was 4.3 ± 1.6 nmol/h/mL. In total, four patients showed serum α-Gal A activity ˂1.5 nmol/h/mL; gene analysis of these patients revealed that Patient 2 had a c.370delG mutation (a classic FD variant), Patient 3 had a p.Glu66Gln mutation (a functional variant) and Patient 4 had a p.Met296Ile mutation (a late-onset variant) (Table 2). Patient 1 was excluded from gene analysis because he did not present with any typical clinical manifestation of FD, including high leukocyte α-Gal A activity. Patient 2 exhibited typical symptoms of FD and was administrated agalsidase-α at a dose of 0.2 mg/kg. However, his kidney function gradually decreased. Patient 3 did not show any typical symptoms of FD. Patient 4 exhibited nephrotic syndrome, with completely diminished proteinuria due to prednisolone administration, as recently reported [19]. FD prevalence in male patients with CKD Stages 1–5 [0.48% (2/419)] was much higher than in those with CKD Stage 5D [0.06% (1/1703)].
Table 2.
Serum and leukocyte α-Gal A activity, plasma lyso-Gb3 concentration and GLA gene variants in four patients with CKD Stages 1–5
| Patient no. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Age (years) | 61 | 40 | 63 | 67 |
| Serum α-Gal A (nmol/h/mL) | 0.9 | <0.1 | 1.1 | 0.2 |
| Leukocyte α-Gal A (nmol/h/mg protein) | 54 | <0.1 | 7 | 1.0 |
| Plasma lyso-Gb3 (nmol/L) | NA | 262 | NA | 7.4 |
| GLA variant | NA | c.370delG | p.Glu66Gln | p.Met296Ile |
| ACMG classification | – | PVS1, PS3, PM2 | BS2, BS3 | PS1, PS3, PM2, PP1 |
NA, not applicable.
α-Gal A activities in patients with CKD Stages 1–5 except for FD and functional variants were slightly lower than in those with CKD Stage 5D (4.3 ± 1.5 and 4.5 ± 1.2 nmol/h/mL, respectively; P = 0.004). Furthermore, the serum α-Gal A activity in the patients with CKD Stages 1–5, except for the four aforementioned patients, was inversely correlated with age (R2 = 0.082, P < 0.0001), BUN (R2 = 0.128, P < 0.0001) and SCr (R2 = 0.129, P < 0.0001), but directly correlated with eGFR (R2 = 0.210, P < 0.0001) (Figure 5A–D). Conversely, serum α-Gal A activity and proteinuria levels were not significantly correlated (Figure 5E).
FIGURE 5.

The correlation between serum α-Gal A activity and (A) age, (B) BUN, (C) SCr, (D) eGFRand (E) proteinuriain patients with CKD Stages 1–5 except for FD.
DISCUSSION
This is the first report comparing FD prevalence between male patients with CKD Stage 5D and those with CKD Stages 1–5. FD prevalence was much higher in male patients with CKD Stages 1–5 [0.48% (2/419)] than in those with CKD Stage 5D [0.06% (1/1703)]. In addition, we identified a novel mutation of FD, p.Met208Arg. The clinical findings of the patient are consistent with those of late-onset variants.
Although several screenings for FD have been conducted among male patients undergoing HD, there is a notable difference in the FD prevalence (Table 3). The FD prevalence observed in this study was consistent with that reported by Maruyama et al. [35]. However, functional GLA variants are sometimes included in the analysis of FD prevalence rate [25, 29]. Here we detected p.Glu66Gln in seven patients with CKD Stage 5D whose α-Gal A activity was <1.5 nmol/h/mL. According to the American College of Medical Genetics (ACMG) classification, p.Met208Arg and p.Glu66Glu are considered to be pathogenic and benign variants, respectively. FD is known as a congenital metabolic disease, and the diagnosis of this disease should involve both analysis of genetic variants and detection of biochemical and symptomatic abnormalities. Since a patient with the p.Met208Arg variant showed an extreme decrease in α-Gal A activity, with massive accumulation of Gb3 and lyso-Gb3 in the kidney and plasma, this variant is pathogenic. Lee et al. [15] reported that considerable residual α-Gal A activity was shown both in the leukocytes of p. Glu66Gln patients (19.0–30.3% of normal activity) and in transiently overexpressed COS-7 cells (43.8 ± 3.03% of normal activity). Furthermore, as the allele frequency of p.Glu66Gln determined in 833 unrelated Korean individuals was remarkably high, at 1.046%, they concluded that p.Glu66Gln is a functional polymorphism rather than a pathogenic mutation. Later, Sueoka et al. [43] confirmed that the plasma lyso-Gb3 level, a biomarker of FD, in patients with p.Glu66Gln was apparently lower than in FD patients and that there was no difference between the p.Glu66Gln patients and the healthy subjects. In this study, none of the patients with the p.Glu66Gln allele showed any typical clinical manifestations of FD. Doi et al. [11] eliminated 8 of 10 patients with low α-Gal A activity and the p.Glu66Gln gene variant from their prevalence analysis. Therefore the handling of functional or partially functional alleles such as p.Glu66Gln requires attention and if the α-Gal A activity is low, gene mutation analysis is strongly recommended to identify the cause of pathological FD.
Table 3.
Summary of the recent screening for FD in male patients with CKD Stage 5D

|

|
The screening of patients with CKD Stage 5D led to the identification of a novel GLA gene missense mutation, p. Met208Arg. Structural analysis revealed that p.Met208Arg causes moderate structural changes on the molecular surface but does not affect the catalytic residues in α-Gal A. Thus a folding defect, followed by enzyme degradation, could be involved in the pathogenesis of this disease. Although it is difficult to assess whether the structural changes cause phenotypic changes of the ACMG variant, it has been postulated that the small structural changes on the molecular surface region of α-Gal A may cause a late-onset variant rather than the classic variant [44, 45]. Notably, the patient harbouring the GLA gene mutation had kidney dysfunction as well as LVH, but showed no subjective symptoms of typical FD, suggesting that it might be a late-onset variant. Interestingly, his older brother was also diagnosed with FD, although his kidney function was within the normal range despite harbouring the same mutation. Related adult males with FD can have different kidney phenotypes despite the same classic genotype [46], owing to different epigenetic and environmental factors and/or the coexistence of other kidney disease. Thus further genetic and/or epigenetic research is warranted. Moreover, two patients could not be examined for gene mutation; however, they were speculated to possess the p.Glu66Gln variant because they have not-so-low α-Gal A activity levels and low plasma lyso-Gb3 levels.
In patients with CKD Stages 1–5, the FD prevalence was 0.48% (2/419), including one classic and one late-onset phenotype, corroborating a previous report from Taiwan [0.59% (6/1012)] that included two classic and four late-onset phenotypes from the high-risk CKD population [8]. However, that study excluded patients with diabetic kidney disease (DKD) and autosomal dominant polycystic kidney disease (ADPKD). FD may coexist with other kidney diseases such as ADPKD [47], DKD [38], amyloidosis [48] and minimal change nephrotic syndrome [19], hence we included all CKD patients regardless of the kidney disease.
To date, the FD prevalence rates in patients with CKD Stage 5D and CKD Stages 1–5 have not been compared. In this study, the FD prevalence in patients with CKD Stage 5D was much lower than in patients with CKD Stages 1–5. The risk of mortality in FD patients with CKD Stage 5D may be much higher than in those with CKD Stages 1–5. Indeed, the European Renal Association–European Dialysis and Transplant Association registry demonstrated that the survival rate of FD patients undergoing dialysis was 41% at 5 years [49], whereas there was a 10% risk of mortality by 50 years of age in non-dialysis FD patients [50]. Therefore the number of FD patients with CKD Stage 5D could be reduced due to death, which is one of the explanations. Another explanation is that the progression of kidney dysfunction in FD may be much slower than in other kidney diseases. In fact, Schiffmann et al. [50] reported that the mean rate of eGFR decline in male FD patients is 2.93 mL/min/1.73 m2/year [50]; however, it is relatively faster in patients with other types of CKD, including DKD [51], leading to increased prevalence of FD in patients with CKD Stages 1–5. ERT has been shown to improve patients’ quality of life and possibly ameliorate the progression of typical Fabry cardiomyopathy, even in patients undergoing HD [52]. Therefore, although there is lack of clear evidence regarding the effectiveness of ERT in ameliorating kidney disease progression, early screening for FD is recommended in patients with CKD Stages 1–5 to improve patient survival, thereby decreasing the number of FD patients with CKD Stage 5D.
Serum or plasma α-Gal A activity in patients undergoing HD can be higher [21], unchanged [25, 30] or lower [33] than in healthy subjects. In this study, the serum α-Gal A activity in patients with CKD Stages 1–5 was slightly lower than in those with CKD Stage 5D. Moreover, serum α-Gal A activity may fluctuate because of differences in confounding factors such as age, sex and kidney function. In patients with CKD Stages 1–5, serum α-Gal A activity might be influenced by ageing and progression of kidney dysfunction. Since the measurement of leukocyte α-Gal A activity may be more accurate than that of serum α-Gal A activity, future studies should measure leukocyte α-Gal A activity to clarify the precise correlation.
To date, 25 screenings for FD in patients with CKD Stages 5D and 2 screenings for FD in patients with CKD Stages 1–5 have been reported (Tables 3 and 4). The phenotypes of the FD patients were determined according to their manifestations and family history as follows: classic, late onset, functional variants and unknown. In Japan, the prevalence of p.Glu66Gln is higher than in other countries. Here we present for the first time the FD prevalence in males, excluding possible functional polymorphisms. While the global FD prevalence in male patients with CKD Stage 5D may be 0.22% (63/28 931) (Table 3), it may be only 0.14% (18/12 641) in Japan. In Turkey, Taiwan and Japan, the total FD prevalence rate in patients with CKD Stages 1–5 could be 0.69% (11/1601) (Table 4) [7, 8]. In Japan, total FD prevalence in both types of CKD patients was lower than in other countries, which may be ascribed to the different racial and genetic backgrounds.
Table 4.
Summary of the recent screening for FD in male patients with CKD Stages 1-5

|
Nevertheless, there are several limitations to this study. First, the patient sample size was small and the target population for the CKD Stages 1–5 study was much smaller than the CKD Stage 5D population, consequently the FD prevalence may not be significant. Second, we recruited patients only from the south coast of Japan. Third, we did not examine female patients with CKD. Fourth, we could not evaluate the differences in confounding factors between male patients with CKD Stages 1–5 and CKD Stage 5D because the data could not be obtained from patients with CKD Stage 5D. Therefore a large and nationwide clinical screening for FD in both male and female CKD patients is required to confirm the accurate FD prevalence.
In conclusion, the FD prevalence in male patients with CKD Stages 1–5 was much higher than in those with CKD Stage 5D. Additionally, we identified a novel missense mutation, p. Met208Arg, in GLA that corresponds to the late-onset FD phenotype. Although symptoms and extra-kidney complications are important for early diagnosis of FD, it is sometimes misdiagnosed. Indeed, three FD patients were not diagnosed with FD before the screening in this study. Therefore early screening for FD is recommended in patients with CKD Stages 1–5 to improve patient survival, which may decrease the number of FD patients at CKD Stage 5D.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the following investigators: Yoshifumi Wada, Wada Naika Cardiovascular Clinic, Ryotaro Ando, Sugi Cardiovascular Hospital; Michiaki Usui, Usui Clinic; Michio Chiba, Chiba Naika Junkankika; Atsuo Moriyama, Moriyama Naika; Hiroshi Miyazaki, Miyazakinaika Medical Clinic; Motoko Tanaka, Akebono Clinic; Shuji Iida, Iida Clinic; Shunichi Imadachi, Imadachi Naika Clinic; Keisuke Kono, Imamura Hospital; Keiichiro Uemura, Uemura Urological Clinic; Kazuhiro Sonoda, Saiseikai Hita Hospital; Makio Nagano, Omuta City Hospital; Syumon Kasuga, Kasuga Clinic, Masako Onzuka, Kano Clinic; Hideharu Tanaka, Kurume Ekimae Clinic; Atsuko Ohara, Yame General Hospital; Yoshimi Takamiya, Yokokura Hospital; Shoji Sakai, Shimonoseki City Hospital; Tetsuya Tajiri, Jinseikai Clinic Otsu; Tetsuya Tajiri, Jinseikai Clinic Kurokami; Tetsuya Tajiri, Jinseikai Clinic Nagamine; Tetsuya Tajiri, Jinseikai Clinic Hikarinomori; Harumichi Higashi, St Mary's Hospital; Yoshiyuki Tomiyoshi, Takagi Hospital; Sakuya Ito, Tanushimaru Central Hospital; Yasuyuki Toyama, Toyama Naika; Yoshiharu Hori, Chikugo Clinic; Akiko Nagata, Nagata Hospital; Yasuhiko Nakano, Nakano Naika Junkankika; Hirosaburo Naganuma, Naganuma Clinic; Yoshifumi Nakamura, Nakamura Clinic; Toru Sanai, Fukumitsu Hospital; Kenkichi Majima, Majima Naika Junkankika; Hiroshi Matsuo, Matsuo Naika Clinic, Noboru Matsuoka, Matsuoka Naika Clinic; Yoshinobu Muto, Muto Naika Clinic; Yosuke Nakayama, Munakata Suikokai General Hospital; Akihiko Muraishi, Muraishi Junkankika Naika; Takuro Yamashita, Yamashita Urological Clinic; Nobuyuki Yoshitake, Yoshitake Urological Clinic; Atsuro Tajiri, JCHO Kurume General Hospital.
FUNDING
This research was partly supported by Japan Society for the Promotion of Science, Grants-in-Aid for Welfare and Scientific Research (C) (no. 19k08693) (K.F) and (no. 17K10058) (S.S.).
AUTHORS’ CONTRIBUTIONS
All the authors were involved in the conception of the study, the acquisition and interpretation of patient data and drafting of the article. M.N., Y.K., Y.N., R.S. and K.F. prepared the manuscript. Y.K., N.N. and A.N. diagnosed FD from the kidney biopsy specimens and corrected the samples. T.To., T.Ts. and H.S. measured α-Gal A activity and lyso-Gb3 and analysed GLA mutations. S.S. analysed the structural change of α-Gal A. All authors agreed to be accountable for all aspects of the study and to ensure that questions related to the accuracy or integrity of any part of the study are appropriately investigated and resolved.
CONFLICT OF INTEREST STATEMENT
K.F. has received personal fees from Sumitomo Dainippon Pharma and Sanofi Japan outside of the submitted work. H.S. has received personal fees and grants from Sumitomo Dainippon Pharma and Sanofi Japan outside of the submitted work. T.To. has received a grant from Sanofi Japan outside of the submitted work. The authors have no conflicts of interest directly relevant to the content of this article.
DATA AVAILABILITY STATEMENT
The data in this article are available from the corresponding author upon reasonable request.
Contributor Information
Akiko Nagata, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
Makoto Nasu, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
Yusuke Kaida, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
Yosuke Nakayama, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
Yuka Kurokawa, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
Nao Nakamura, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
Ryo Shibata, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
Takuma Hazama, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
Takahiro Tsukimura, Department of Functional Bioanalysis, Meiji Pharmaceutical University, Tokyo, Japan.
Tadayasu Togawa, Department of Functional Bioanalysis, Meiji Pharmaceutical University, Tokyo, Japan.
Seiji Saito, Department of Medical Management and Informatics, Hokkaido Information University, Hokkaido, Japan.
Hitoshi Sakuraba, Department of Clinical Genetics, Meiji Pharmaceutical University, Tokyo, Japan.
Kei Fukami, Department of Medicine, Division of Nephrology, Kurume University School of Medicine, Fukuoka, Japan.
REFERENCES
- 1. Brady RO, Gal AE, Bradley RM et al. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med 1967; 276: 1163–1167 [DOI] [PubMed] [Google Scholar]
- 2. Breunig F, Weidemann F, Beer M et al. Fabry disease: diagnosis and treatment. Kidney Int 2003; 63(Suppl 84): S181–S185 [DOI] [PubMed] [Google Scholar]
- 3. Møller AT, Jensen TS. Neurological manifestations in Fabry’s disease. Nat Clin Pract Neurol 2007; 3: 95–106 [DOI] [PubMed] [Google Scholar]
- 4. Andrade J, Waters PJ, Singh RS et al. Screening for Fabry disease in patients with chronic kidney disease: limitations of plasma α-galactosidase assay as a screening test. Clin J Am Soc Nephrol 2008; 3: 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bekri S, Enica A, Ghafari T et al. Fabry disease in patients with end-stage renal failure: the potential benefits of screening. Nephron Clin Pract 2005; 101: c33–c38 [DOI] [PubMed] [Google Scholar]
- 6. Sakuraba H, Tsukimura T, Togawa T et al. Fabry disease in a Japanese population-molecular and biochemical characteristics. Mol Genet Metab Rep 2018; 17: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turkmen K, Guclu A, Sahin G et al. The prevalence of Fabry disease in patients with chronic kidney disease in Turkey: the TURKFAB study. Kidney Blood Press Res 2016; 41: 1016–1024 [DOI] [PubMed] [Google Scholar]
- 8. Lin CJ, Chien YH, Lai TS et al. Results of Fabry disease screening in male pre-end stage renal disease patients with unknown etiology found through the platform of a chronic kidney disease education program in a northern Taiwan medical center. Kidney Blood Press Res 2018; 43: 1636–1645 [DOI] [PubMed] [Google Scholar]
- 9. Imai E, Horio M, Nitta K et al. Modification of the Modification of Diet in Renal Disease (MDRD) study equation for Japan. Am J Kidney Dis 2007; 50: 927–937 [DOI] [PubMed] [Google Scholar]
- 10. Levey AS, Coresh J, Greene T et al. Using standardized serum creatinine values in the Modification of Diet in Renal Disease study equation for estimating glomerular filtration rate. Ann Intern Med 2006; 145: 247–254 [DOI] [PubMed] [Google Scholar]
- 11. Doi K, Noiri E, Ishizu T et al. High-throughput screening identified disease-causing mutants and functional variants of α-galactosidase A gene in Japanese male hemodialysis patients. J Hum Genet 2012; 57: 575–579 [DOI] [PubMed] [Google Scholar]
- 12. Mayes JS, Scheerer JB, Sifers RN et al. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry's disease. Clin Chim Acta 1981; 112: 247–251 [DOI] [PubMed] [Google Scholar]
- 13. Takata T, Okumiya T, Hayashibe H et al. Screening and detection of gene mutations in Japanese patients with Fabry disease by non-radioactive single-stranded conformation polymorphism analysis. Brain Dev 1997; 19: 111–116 [DOI] [PubMed] [Google Scholar]
- 14. Sakuraba H, Togawa T, Tsukimura T et al. Plasma lyso-Gb3: a biomarker for monitoring fabry patients during enzyme replacement therapy. Clin Exp Nephrol 2018; 22: 843–849 [DOI] [PubMed] [Google Scholar]
- 15. Lee BH, Heo SH, Kim GH et al. Mutations of the GLA gene in Korean patients with Fabry disease and frequency of the E66Q allele as a functional variant in Korean newborns. J Hum Genet 2010; 55: 512–517 [DOI] [PubMed] [Google Scholar]
- 16. Watanabe H, Goto S, Miyashita A et al. Role of the p.E66Q variant of GLA in the progression of chronic kidney disease. Clin Exp Nephrol 2015; 19: 225–230 [DOI] [PubMed] [Google Scholar]
- 17. Saito S, Ohno K, Sakuraba H. Comparative study of structural changes caused by different substitutions at the same residue on α-galactosidase A. PLoS One 2013; 8: e84267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human α-galactosidase. J Mol Biol 2004; 337: 319–335 [DOI] [PubMed] [Google Scholar]
- 19. Fujisawa H, Nakayama Y, Nakao S et al. Effectiveness of immunosuppressive therapy for nephrotic syndrome in a patient with late-onset Fabry disease: a case report and literature review. BMC Nephrol 2019; 20: 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Utsumi K, Kase R, Takata T et al. Fabry disease in patients receiving maintenance dialysis. Clin Exp Nephrol 2000; 4: 49–51 [Google Scholar]
- 21. Nakao S, Kodama C, Takenaka T et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int 2003; 64: 801–807 [DOI] [PubMed] [Google Scholar]
- 22. Linthorst GE, Hollak CE, Korevaar JC et al. α-Galactosidase A deficiency in Dutch patients on dialysis: a critical appraisal of screening for Fabry disease. Nephrol Dial Transplant 2003; 18: 1581–1584 [DOI] [PubMed] [Google Scholar]
- 23. Kotanko P, Kramar R, Devrnja D et al. Results of a nationwide screening for Anderson-Fabry disease among dialysis patients. J Am Soc Nephrol 2004; 15: 1323–1329 [DOI] [PubMed] [Google Scholar]
- 24. Ichinose M, Nakayama M, Ohashi T et al. Significance of screening for Fabry disease among male dialysis patients. Clin Exp Nephrol 2005; 9: 228–232 [DOI] [PubMed] [Google Scholar]
- 25. Tanaka M, Ohashi T, Kobayashi M et al. Identification of Fabry’s disease by the screening of α-galactosidase A activity in male and female hemodialysis patients. Clin Nephrol 2005; 64: 281–287 [DOI] [PubMed] [Google Scholar]
- 26. Merta M, Reiterova J, Ledvinova J et al. A nationwide blood spot screening study for Fabry disease in the Czech Republic haemodialysis patient population. Nephrol Dial Transplant 2007; 22: 179–186 [DOI] [PubMed] [Google Scholar]
- 27. Porsch DB, Nunes AC, Milani V et al. Fabry disease in hemodialysis patients in southern Brazil: prevalence study and clinical report. Ren Fail 2008; 30: 825–830 [DOI] [PubMed] [Google Scholar]
- 28. Terryn W, Poppe B, Wuyts B et al. Two-tier approach for the detection of alpha-galactosidase A deficiency in a predominantly female haemodialysis population. Nephrol Dial Transplant 2008; 23: 294–300 [DOI] [PubMed] [Google Scholar]
- 29. Fujii H, Kono K, Goto S et al. Prevalence and cardiovascular features of Japanese hemodialysis patients with Fabry disease. Am J Nephrol 2009; 30: 527–535 [DOI] [PubMed] [Google Scholar]
- 30. Gaspar P, Herrera J, Rodrigues D et al. Frequency of Fabry disease in male and female haemodialysis patients in Spain. BMC Med Genet 2010; 11: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wallin EF, Clatworthy MR, Pritchard NR. Fabry disease: results of the first UK hemodialysis screening study. Clin Nephrol 2011; 75: 506–510 [DOI] [PubMed] [Google Scholar]
- 32. Nishino T, Obata Y, Furusu A et al. Identification of a novel mutation and prevalence study for Fabry disease in Japanese dialysis patients. Ren Fail 2012; 34: 566–570 [DOI] [PubMed] [Google Scholar]
- 33. Kalkan Uçar S, Sozmen E, Duman S et al. Alpha-galactosidase A activity levels in Turkish male hemodialysis patients. Ther Apher Dial 2012; 16: 560–565 [DOI] [PubMed] [Google Scholar]
- 34. Okur I, Ezgu F, Biberoglu G et al. Screening for Fabry disease in patients undergoing dialysis for chronic renal failure in Turkey: identification of new case with novel mutation. Gene 2013; 527: 42–47 [DOI] [PubMed] [Google Scholar]
- 35. Maruyama H, Takata T, Tsubata Y et al. Screening of male dialysis patients for Fabry disease by plasma globotriaosylsphingosine. Clin J Am Soc Nephrol 2013; 8: 629–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Herrera J, Miranda CS. Prevalence of Fabry’s disease within hemodialysis patients in Spain. Clin Nephrol 2014; 81: 112–120 [DOI] [PubMed] [Google Scholar]
- 37. Sayilar EI, Ayar Y, Yavuz M. Prevalence of Fabry disease among Turkish dialysis patients: data from hemodialysis centers in Bursa province. Clin Nephrol 2016; 85: 165–172 [DOI] [PubMed] [Google Scholar]
- 38. Saito O, Kusano E, Akimoto T et al. Prevalence of Fabry disease in dialysis patients: Japan Fabry disease screening study (J-FAST). Clin Exp Nephrol 2016; 20: 284–293 [DOI] [PubMed] [Google Scholar]
- 39. Silva CA, Barreto FC, Dos Reis MA et al. Targeted screening of Fabry disease in male hemodialysis patients in Brazil highlights importance of family screening. Nephron 2016; 134: 221–230 [DOI] [PubMed] [Google Scholar]
- 40. Veloso VSP, Ataides TL, Canziani MEF et al. A novel missense GLA mutation (p.G35V) detected in hemodialysis screening leads to severe systemic manifestations of Fabry disease in men and women. Nephron 2018; 138: 147–156 [DOI] [PubMed] [Google Scholar]
- 41. Moiseev S, Fomin V, Savostyanov K et al. The prevalence and clinical features of Fabry disease in hemodialysis patients: Russian nationwide Fabry dialysis screening program. Nephron 2019; 141: 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nagata A, Nasu M, Kaida Y et al. Screening of Fabry disease in patients with chronic kidney disease in Japan. Nephrol Dial Transplant 2021 [DOI] [PMC free article] [PubMed]
- 43. Sueoka H, Ichihara J, Tsukimura T et al. Nano-LC-MS/MS for quantification of lyso-Gb3 and its analogues reveals a useful biomarker for Fabry disease. PLoS One 2015; 10: e0127048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saito S, Ohno K, Sese J et al. Prediction of the clinical phenotype of Fabry disease based on protein sequential and structural information. J Hum Genet 2010; 55: 175–178 [DOI] [PubMed] [Google Scholar]
- 45. Sugawara K, Ohno K, Saito S et al. Structural characterization of mutant alpha-galactosidases causing Fabry disease. J Hum Genet 2008; 53: 812–824 [DOI] [PubMed] [Google Scholar]
- 46. Mignani R, Moschella M, Cenacchi G et al. Different renal phenotypes in related adult males with Fabry disease with the same classic genotype. Mol Genet Genomic Med 2017; 5: 438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pisani A, Riccio E, Cianciaruso B et al. Simultaneous multicystic kidney and Anderson-Fabry disease: 2 separate entities or same side of the coin. J Nephrol 2011; 24: 806–808 [DOI] [PubMed] [Google Scholar]
- 48. Taguchi K, Moriyama A, Kodama G et al. The coexistence of multiple myeloma-associated amyloid light-chain amyloidosis and Fabry disease in a hemodialysis patient. Intern Med 2017; 56: 841–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tsakiris D, Simpson HK, Jones EH et al. Report on management of renale failure in Europe, XXVI, 1995. Rare diseases in renal replacement therapy in the ERA-EDTA registry. Nephrol Dial Transplant 1996; 11(Suppl 7): 4–20 [DOI] [PubMed] [Google Scholar]
- 50. Schiffmann R, Warnock DG, Banikazemi M et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant 2009; 24: 2102–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chapter 2: definition, identification, and prediction of CKD progression. Kidney Int Suppl 2011; 3: 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pisani A, Spinelli L, Sabbatini M et al. Enzyme replacement therapy in Fabry disease patients undergoing dialysis: effects on quality of life and organ involvement. Am J Kidney Dis 2005; 46: 120–127 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data in this article are available from the corresponding author upon reasonable request.