

Abstract
Among primates, humans display a unique trajectory of development responsible for the many traits specific to our species. However, the inaccessibility of human and chimpanzee primary tissues has limited our ability to study human evolution. Comparative in vitro approaches using primate-derived induced pluripotent stem cells have begun to reveal species differences on the cellular and molecular levels1,2. In particular, brain organoids have emerged as a promising platform to study primate neural development in vitro3-5, although cross-species comparisons of organoids are complicated by differences in developmental timing and variability of differentiation6,7. Here, we developed a new platform to address these limitations. We first generated a panel of tetraploid hybrid stem cells by fusing human and chimpanzee induced pluripotent stem cells. We next applied this approach to study species divergence in cerebral cortical development by differentiating them into neural organoids. We found that hybrid organoids provide a controlled system for disentangling cis- and trans-acting gene expression divergence across cell types and developmental stages, revealing a signature of selection on astrocyte-related genes. In addition, we identified an up-regulation of human somatostatin receptor 2 (SSTR2), which regulates neuronal calcium signaling and is associated with neuropsychiatric disorders8,9. We discovered a human-specific response to modulation of SSTR2 function in cortical neurons, underscoring the potential of this unique platform to reveal the molecular basis of human evolution.
Comparison of human and chimpanzee induced pluripotent stem (iPS) cell-derived tissues is a valuable paradigm for identification of species-specific divergence in gene expression profiles and cellular phenotypes1,2,10. However, species differences measured in this context could be driven by non-genetic technical artifacts as well as environmental or batch effects. In addition, current stem cell and postmortem studies lack the ability to disentangle cis and trans contributions to gene expression divergence. Cis-regulatory elements, which include promoters and enhancers, only affect genes residing on the same DNA molecule and have been hypothesized to underlie the majority of morphological adaptation11; in contrast, trans-acting factors include proteins or noncoding RNAs that can regulate genes anywhere in the genome, and tend to be more constrained in evolution due to their large number of target genes.
To deconvolve these effects while simultaneously controlling for non-genetic factors, we introduce here a novel experimental approach that utilizes interspecific hybrids to uncover gene expression divergence. We first employed a cell fusion approach to generate a panel of tetraploid human-chimpanzee hybrid iPS (hyiPS) cells to map species-specific cis-regulatory landscapes. Measurement of allele specific expression (ASE) - the relative abundance of each species’ allele for a given gene - allowed us to identify the gene expression differences that are specifically driven by cis-regulatory divergence between the species. We next applied this technology to explore one of the most striking aspects of human evolution: the unique developmental trajectory of the human cerebral cortex. Recently, brain organoids – which recapitulate salient features of the developing human brain, including the generation of a diversity of cell types and gene expression patterns that resemble in vivo brain development12-15 – have allowed for interrogation of evolved differences between human and chimpanzee in vitro6,7,16,17. Due to their dynamic cellular heterogeneity, comparisons of brain organoids across species are particularly challenging, further motivating the utilization of our hybrid approach in this context. We developed a protocol to differentiate hyiPS cells into region-specific brain organoids termed hybrid cortical spheroids (hyCS), which resemble the developing cerebral cortex and contain progenitor cells, cortical neurons, and astrocytes12. Using both single-cell and bulk transcriptomics in CS after up to 200 days of in vitro differentiation, we discovered thousands of genes with divergent cis-regulation and detected evidence of selection on a set of astrocyte-related genes. We finally investigated the human upregulation of a G-protein coupled receptor gene, SSTR2, and demonstrated species-specific differences in neuronal calcium signaling following pharmacological modulation.
Fusion of human and chimpanzee iPS cells
We used polyethylene glycol (PEG)-mediated cell fusion to derive tetraploid iPS cell lines from human and chimpanzee parental lines (Fig. 1a). We co-cultured fluorescently labelled hiPS and ciPS cell lines in the presence of PEG to promote cell fusion and then extracted double-positive cells by fluorescence-activated cell sorting (Fig. 1b, Extended Data Fig. 1a). Following isolation and expansion, we verified the karyotypes of these new tetraploid cell lines (Fig. 1c-d, Extended Data Fig. 1b). We generated five hyiPS cell lines from two sets of parental lines, wherein the human and chimpanzee parental samples were matched for age and sex, yielding three male hybrid lines (XY/XY, labeled Hy1-#) and two female hybrid lines (XX/XX, labeled Hy2-#) (Supplementary Table 1). Overall, we observed stable karyotypes over time (Supplementary Table 2), with the exception of a recurrent species-agnostic gain of the long arm of chromosome 20, which has been widely reported as a common occurrence in pluripotent cell lines (Extended Data Fig. 2a-b)18,19. We found no evidence for species biased X-inactivation in any hybrid line (Extended Data Fig. 2c,d), and mitochondrial mRNAs were of human origin (Extended Data Fig. 2e).
Figure 1 |. Generation of human-chimpanzee hybrid iPS cells.
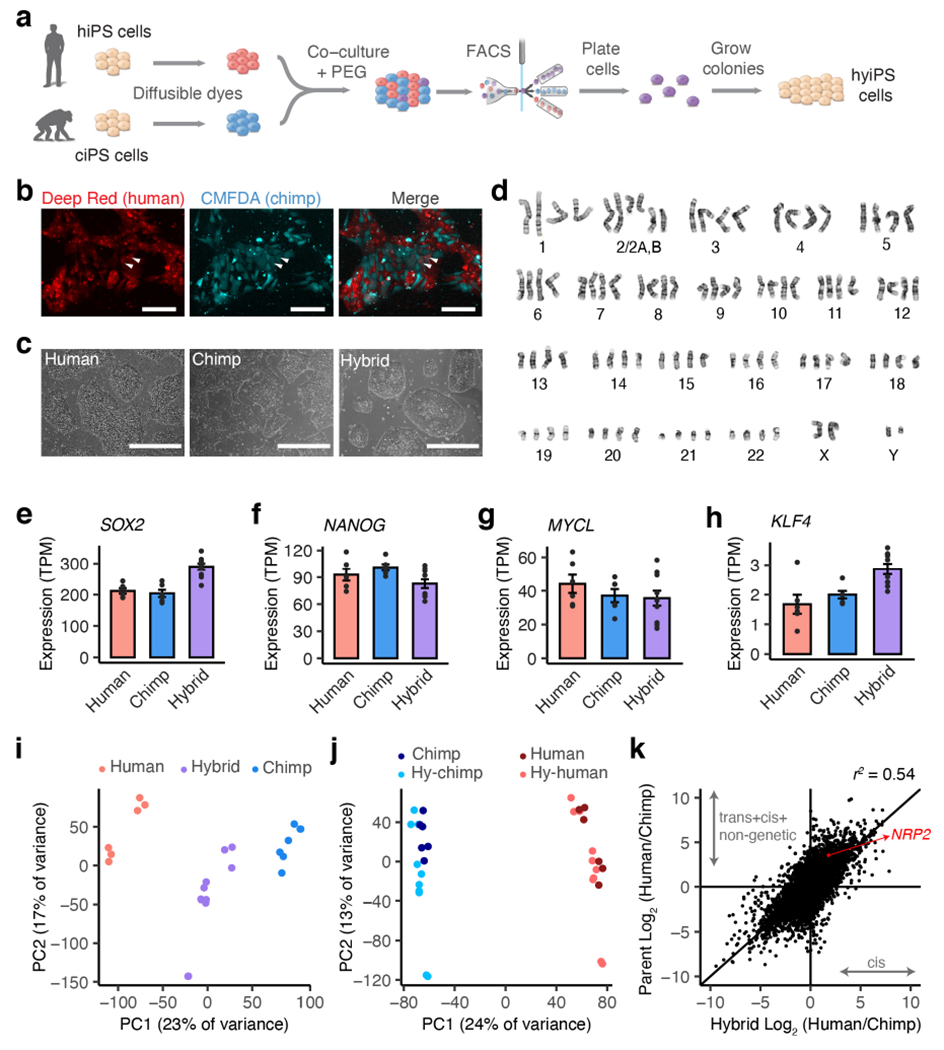
a, Generation of hybrid iPS cells. b, Fluorescent imaging of co-cultured human (H20961) and chimpanzee (C3649) iPS cells; arrows indicate putative hybrid cells; experiment was reproduced for 2 pairs of fusion cell lines to generate 5 hybrid cell lines. c, Bright field images of human (H20682), chimpanzee (C3649) and hybrid (Hy1-29) iPS cell colonies in feeder-free conditions; n= 3 human, 3 chimpanzee and 5 hybrid cell lines were cultured in this study. d, Representative karyotype for male (XY/XY) hybrid iPS cell lines. e-h, Gene expression (TPM, transcripts per million) for pluripotency markers SOX2, NANOG, MYCL, and KLF4 in human (n= 3 iPS cell lines, 2 replicates each), chimpanzee (n= 3 iPS cell lines, 2 replicates each) and hybrid (n= 5 iPS cell lines, 2 replicates each); error bars, mean ± s.e.m. i-j, Principal components plot for RNA-seq samples based on total gene expression (i) or allelic gene expression (j). k, Scatter plot showing differences in gene expression between parent cell lines (y-axis) versus between alleles in the hybrid cells (x-axis); data are from bulk RNA-seq of 3 human, 3 chimpanzee and 5 hybrid iPS cell lines, all with 2 replicates each. Scale bars, 100 μm (b), 400 μm (c).
We next performed RNA sequencing on the five hyiPS as well as three hiPS and three ciPS cell lines (including all of the parental lines; Extended Data Fig. 3a, Supplementary Table 1). The hyiPS cells showed robust expression of pluripotency markers, such as SOX2, NANOG, MYCL and KLF4 (Fig. 1e-h), and pluripotency was additionally confirmed by immunocytochemistry and PluriTest20 (Extended Data Fig. 1c-f). Principal components analysis (PCA) of all samples positioned the hyiPS cells between the hiPS and ciPS cells, indicating that gene expression in the hybrid cell lines represents an intermediate of that seen in the parental lines (Fig. 1i). We repeated this analysis after separating the hybrid sequencing reads into allelic subsets assignable to either the human or chimpanzee alleles (excluding any genes that display species-specific mapping bias, see Methods, Supplementary Table 3), and found that the hybrid allelic samples cluster closer to their species of origin than to each other, suggesting that each genome in the hyiPS cells retains a species-specific gene expression profile that is independent of ploidy (Fig. 1j, Extended Data Fig. 3b-d).
Because the human and chimpanzee alleles share the same trans-regulatory environment within every hybrid nucleus, any allele-specific expression (ASE) in the hybrid lines must be due to cis-acting changes (Supplementary Table 4). By comparing ASE with expression divergence between the parental cell lines, we determined the fraction of divergence that is explained by cis-regulatory effects on expression (Fig. 1k). For example, our hybrid data show that the ~11.6-fold higher expression of NRP2 in human cells is actually composed of a ~3.5-fold cis-regulatory divergence coupled with a ~3.3-fold difference due to trans-acting changes and non-genetic factors. Together with their parental cell lines, these hyiPS cells have the potential to disentangle the factors that drive species-specific gene expression.
Differentiation of hyiPS cells into CS
To assess divergence in gene expression during cerebral cortical development, we differentiated hyiPS cells into hyCS. We found that the species-specific gene expression profiles present in hyiPS cells are maintained throughout neural differentiation, consistent with minimal effects of the tetraploid state of these cells (Extended Data Fig. 4a,b). The hyiPS cells initially had a low success rate of differentiation; this was solved by embedding hyiPS cellular aggregates into a droplet of extracellular matrix21-23, enabling their differentiation for 200 days in vitro across three hyiPS cell lines (Fig. 2a; Extended Data Fig. 4c-e). Immunocytochemistry for progenitor cell markers SOX9, PAX6, and TBR2 identified ventricular zone-like structures which we have previously shown in human CS (hCS, Fig. 2b). We performed bulk RNA-sequencing of each cell line at four time points (Extended Data Fig. 4f,g) and further performed single cell RNA-sequencing of two hyiPS cell lines at day 150. We assessed karyotype stability over time and across individual cells, identifying three instances of chromosomal gains and no losses, suggesting that the karyotypes of hyiPS cells are largely stable during differentiation (Supplementary Table 2; Extended Data Fig. 4h-j).
Figure 2 |. Cortical differentiation of hybrid iPS cells.
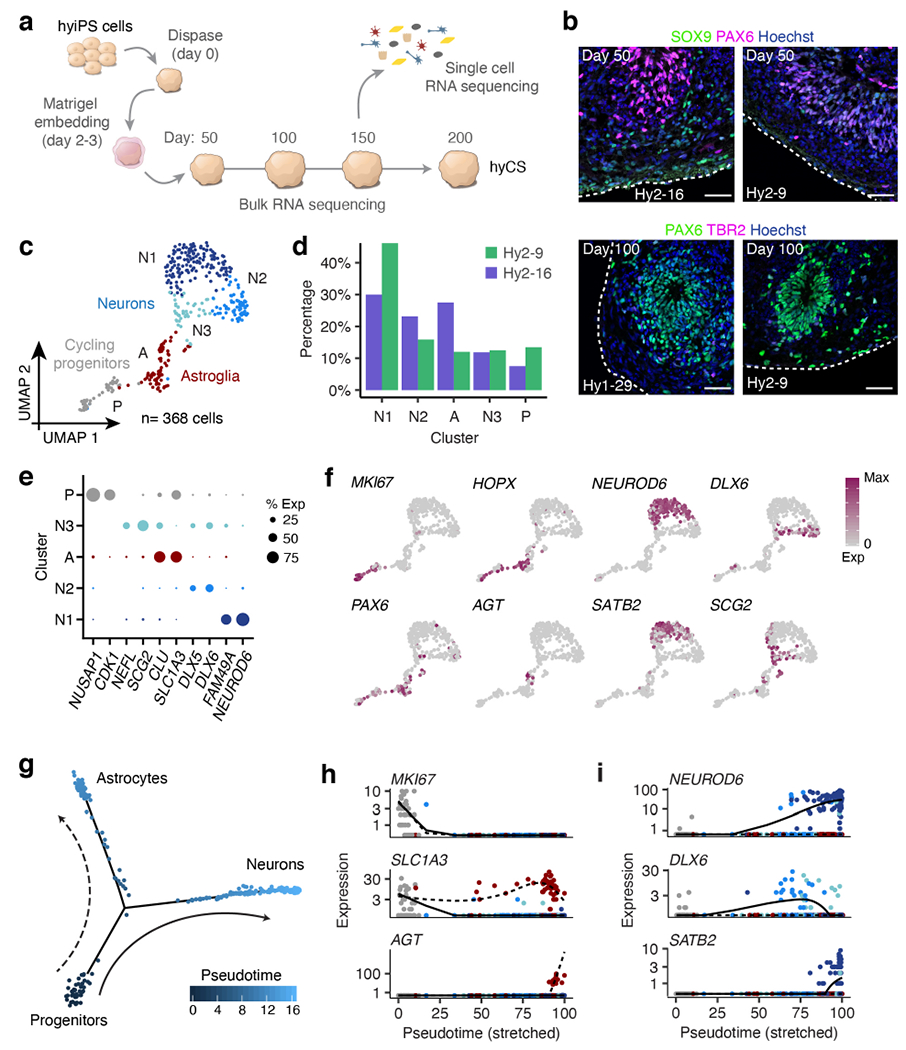
a, Generation of CS from hybrid iPS cells. b, Immunostaining of hyCS for SOX9, PAX6 and TBR2; at each time point, a maximum of 2 spheroids were fixed for immunostaining across n= 3 hybrid cell lines with n= 3 independent differentiation experiments per cell line. c, UMAP clustering of all neural cells (n= 368); clusters are identified by color and labelled by letter (A= astroglia, P= cycling progenitors, N1= glutamatergic neurons, N2= GABAergic neurons cluster 1, N3= GABAergic neurons cluster 2). d, Proportion of CS cells from each hybrid cell line in each single cell cluster (from c). e, Dotplot for expression of marker genes for each cluster in (c), size corresponds to the percent of cells in each cluster that express each gene. f, UMAP colored by expression of marker genes. g, Cell trajectory map of neural cells (n= 349) colored by pseudotime. h-i, Branch-specific expression of marker genes over pseudotime for glial cells (h) and neurons (i), colored by cell type as in (c); solid and dashed lines correspond to those in (g); lines are natural spline curves for each lineage over scaled pseudotime. Scale bars, 50 μm (b).
In our single cell data, we observed clusters of neurons, astroglia, and progenitor cells, as well as populations of mesenchymal and epithelial cells which we attribute to the extracellular matrix embedding14 (Extended Data Fig. 5a-d, Supplementary Tables 5-6). In fact, a direct comparison of gene expression between embedded and non-embedded hyCS at day 50 revealed expression of mesenchyme related genes specifically in the embedded hyCS (Extended Data Fig. 5e). Despite the introduction of these non-neural cell types, we found that gene expression profiles of the neural cells clustered well with those of cells from non-embedded CS15 (Extended Data Fig. 5f-g). We defined the neural cell clusters as: cycling progenitors, including PAX6+ radial glial cells, some of which also expressed HOPX; astroglia, including non-cycling progenitor cells and astrocytes, some of which expressed the mature astrocyte marker gene AGT; and neurons, comprised of NEUROD6+ and SLC17A7+ (VGLUT1+) glutamatergic neurons, including deep (TBR1+) and superficial (SATB2+) cortical layer markers, as well as a group of DLX5+ and DLX6+ GABA-ergic neurons (Extended Data Fig. 5h, Fig. 2c-f, Supplementary Tables 7-8). Genome wide, we did not identify any cell type-specific bias in allele specific expression (Extended Data Fig. 5i). We used trajectory mapping to linearly orient these cells according to pseudotime24 and identified two pathways of differentiation representing neurogenesis and astrogenesis, with neural progenitors residing at the root of the trajectory (Fig. 2g). While these data represent a single snapshot in development, we did observe mature astrocyte markers (AGT+) and late-born upper layer neuron markers (SATB2+) at the end of each branch, indicating a progression of differentiation consistent with in vivo cortical development25 (Fig. 2h-i).
Transcriptional changes in corticogenesis
To disentangle cis-regulatory gene expression, we differentiated three human (hiPS) and three chimpanzee (ciPS) cell lines (including all parental cell lines)1 for up to 200 days in culture and performed bulk RNA-sequencing at seven time points (Fig. 3a-b, Extended Data Fig. 6a-e; Supplementary Table 9). In cCS, we observed an enrichment for several non-neuronal cellular processes and expression of many mesenchyme-related genes, indicating a possible species difference in cell-fate specification of iPS cells (Fig. 3c, Extended Data Fig. 7a). In contrast, when we performed ASE analysis in the hybrid samples across time, we found no evidence of a bias in cell fate and further confirmed that species-specific gene expression profiles are preserved in hyCS (Fig. 3d, Extended Data Fig. 7b, Supplementary Tables 10-11).
Figure 3 |. Disentangling cis regulatory effects on gene expression in CS.
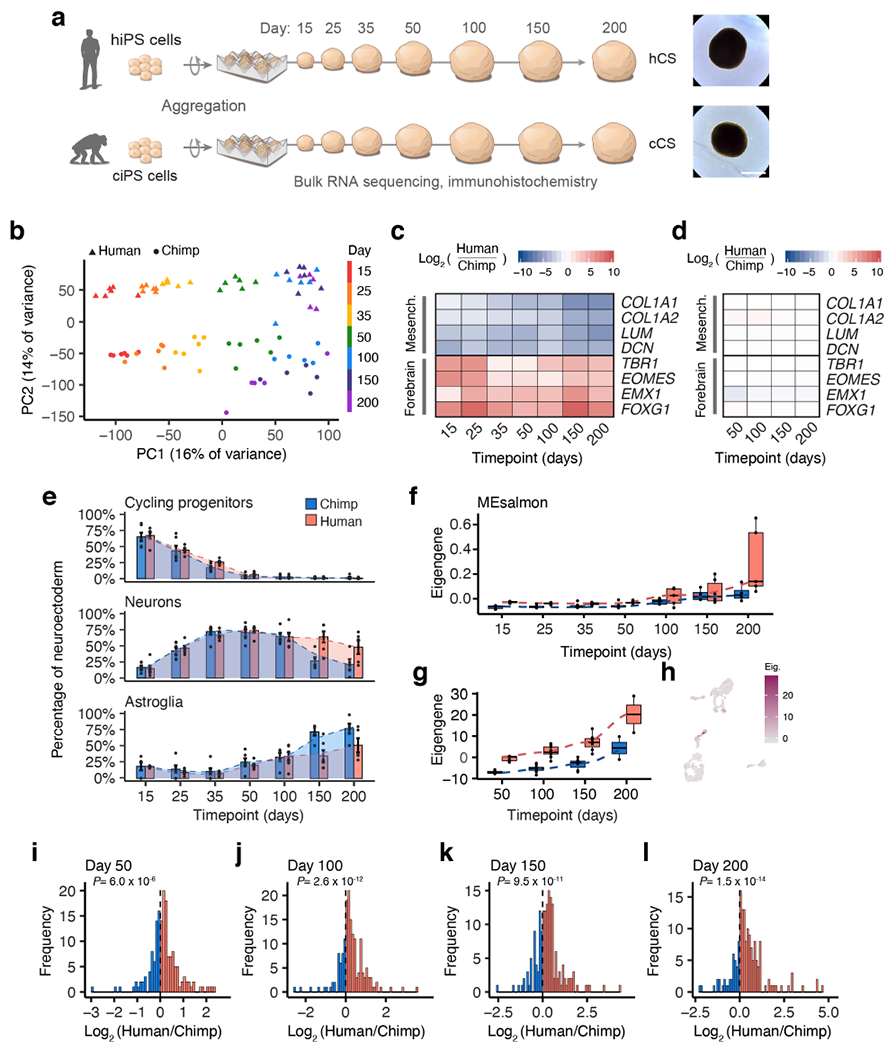
a, Generation of cortical spheroids (CS) from human and chimpanzee iPS (iPS) cells; bright field images of representative human and chimpanzee CS (lines H21792 and C3651) at day 166; images were chosen from n= 3 human and 3 chimpanzee iPS cell lines each with n= 5 spheroids imaged from a single differentiation experiment. b, Principal components plot for RNA-seq samples based on total gene expression. c, Heat map of differential expression (log2[fold-change]) between hCS and cCS for forebrain and mesenchyme-related marker genes. d, Heat map of allele specific expression (log2[fold-change]) in hybrid spheroids for forebrain and mesenchyme-related marker genes. e, Estimated cell type proportions over time in hCS and cCS, normalized as a percentage of neuroectodermal cell types; error bars, mean ± s.e.m.; curved line from LOWESS regression; in order of time points, n= 6, 6, 6, 6, 6, 6, 5 hCS and n= 6, 6, 6, 6, 5, 5, 5 cCS samples from 3 human and 3 chimpanzee iPS cell lines (1-2 replicates per cell line). f, Eigengene values for genes in the Salmon module (from weighted gene co-expression network analysis) over time in hCS and hCS; n as in (e). g, Allelic eigengene values for genes in the Salmon module over time in hyCS (see Methods); n as in (e). h, Expression of Salmon module genes (colored by eigengene rank, see Methods) in Monocle pseudotime plot; cell trajectories and pseudotime are defined in Fig. 2g. i-l, Histogram of ASE (log2[fold-change]) in all genes in the Salmon module, at each time point in bulk hyCS data; P-value from a two-sided Wilcoxon Rank Sum test, comparing module genes to all genes included in the co-expression analysis. Box plots in (f, g): center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range. Scale bars, 1 mm (a).
We next estimated the relative proportion of each cell type in our bulk RNA-sequencing samples26. This confirmed the differences in mesenchymal cell fate when deriving hCS and cCS, and additionally revealed shifts in estimated cell type proportions indicative of a slower progression from neurogenesis to astrogenesis in hCS, consistent with the neotenic delay in cortical development observed in humans27 (Fig. 3e, Extended Data Fig. 7c-h, Supplementary Table 9). To resolve these maturation trends into sets of cell type-specific genes, we used gene co-expression network analysis and identified co-expressed modules of genes28 representing proliferation, neurogenesis, and gliogenesis (Extended Data Fig. 8a-c, Supplementary Tables 12-13). We examined the allelic expression of these modules in hyCS, but found no evidence of cis-encoded differences in the expression of these genes (Extended Data Fig. 8d).
We did observe one co-expressed module of genes that showed higher expression in hCS than cCS at late time points, and which also showed strongly human-biased allelic expression in hyCS, indicative of cis-regulatory divergence (Fig. 3f,g). This module included many astrocyte-associated genes (AQP4, HEPACAM, CLU, HEPN1, S100B), and its expression was localized primarily to astrocytes in our single cell data (Fig. 3h). At each time point, we found significantly higher expression of the human alleles in this module, indicating lineage-specific selection either for up-regulation in humans or down-regulation in chimpanzees29 (Fig. 3i-l). Considering the unique features of human astrocytes30,31, these genes may have evolved to enable adaptive species-specific astrocyte functionality. Indeed, one of the most human-biased genes in this module is PMP2 (encoding Peripheral Myelin Protein 2), a gene with known human upregulation in the cerebral cortex32 that has been proposed to underlie some of the morphological differences between human and mouse cortical astrocytes33 (Extended Data Fig. 8e-g).
Functional validation of an ASE gene
We found that trends of ASE were largely shared across time points in our bulk RNA-seq data, and about 40% of effects were present only in hyCS and not in hyiPS cells (Extended Data Fig. 9a-c). We estimated that 39% of expression differences in CS can be attributed to cis-regulatory divergence (Extended Data Fig. 9d-e). Amongst genes with significant ASE, we identified 100 genes associated with neurodevelopmental disorders34 including GRIN2A (chimpanzee-biased) and SCN1A (human-biased; Extended Data Fig. 9f-i). Disease-associated ASE genes represent some of the most intriguing candidates since they suggest that some molecular changes could underlie both human-specific cortical function and dysfunction. In light of recent evidence suggesting that modulators of neuronal activity may underlie aspects of human cortical evolution35, we looked for genes that encode disease-associated regulators of neuronal development whose function could be assessed in vitro (Extended Data Fig. 9j-k, Supplementary Table 14). Somatostatin Receptor 2 (SSTR2) showed an intriguing pattern with an increasing degree of human-biased ASE as the CS mature, with expression restricted primarily to glutamatergic neurons (Extended Data Fig. 9k, Fig. 4a-b). SSTR2 encodes a G-protein coupled receptor (Gαi) for the neurotransmitter somatostatin and has an identical protein sequence between human and chimpanzee. This receptor suppresses the function of voltage-gated calcium channels36 and is thought to play a role in modulation of neural circuits37 (Fig. 4c).
Figure 4 |. Functional validation of allele specific gene expression changes.
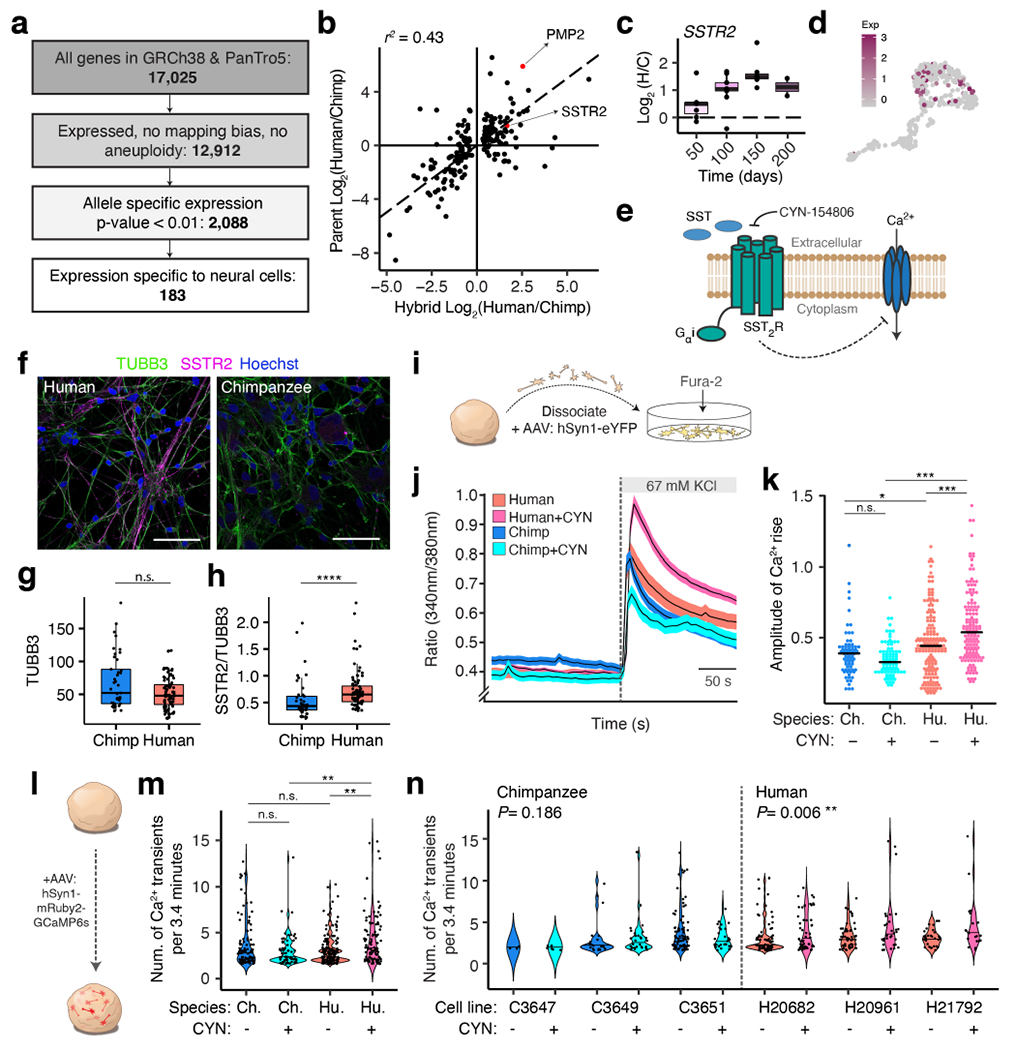
a, Hybrid ASE for SSTR2; n=7, 9, 7, 2 hyCS samples (1-2 spheroids/sample) from 3 differentiations of 3 hyiPS cell lines. b, Single-cell gene expression of SSTR2. c, SSTR2 function. d, Immunostaining for TUBB3 and SSTR2 in dissociated hCS and cCS (day 225-250); 10 images/sample. e, Fluorescence intensity (arbitrary units) of TUBB3 for (d) and Extended Data Fig. 10f; n=87 cells (hCS), 41 cells (cCS); n.s., not significant, two-sided Mann-Whitney test. f, Fluorescence intensity of SSTR2 relative to TUBB3 for (d) and Extended Data Fig. 10f; n as in (e); ****P<0.0001, two-tailed Mann-Whitney test. g, Fura-2 imaging in dissociated CS. h, Mean fluorescence ratio of Fura-2; n=164 cells from 2 lines (human), n=142 cells from 2 lines (human +CYN), n=71 cells from 1 line (chimpanzee), n=87 cells from one line (chimpanzee +CYN) dissociated between day 225-250. i, Peak amplitude (mean maximum minus baseline) for each cell in (h) upon KCl depolarization; sample sizes as in (h); *P<0.05, ***P<0.001, two-tailed Wilcoxon rank sum test; left to right P=0.087, 0.020, 4.143x10−14, 5.082x10−5. j, GCaMP6s imaging in intact CS. k, Spontaneous GCaMP6s transients; n=161 cells (3 human lines), n=110 cells (3 human lines, +CYN), n=105 cells (3 chimpanzee lines), n=87 cells (3 chimpanzee lines, +CYN) at day 130-150; **P<0.01, two-tailed Wilcoxon rank sum test; from left to right, P=0.114, 0.272, 0.00118, 0.00134. l, Data in (k) by line; left to right, n=4, 6, 21, 30, 80, 30, 78, 58, 53, 31, 30, and 21 cells/line; left to right, n=1, 1, 4, 4, 4, 4, 2, 2, 2, 2, 5, and 5 CS/line; P-values from Fisher’s method χ2(4)=6.178 (chimpanzee), χ2(6)=18.059 (human); original per-line P-values from a two-tailed Wilcoxon rank sum test. Box plots in (a, e, f): center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range. Scale bars, 10μm (d).
In hCS and cCS, SSTR2 showed an increasing degree of human-biased gene expression across time, matching the trend of allelic expression in hyCS (Extended Data Fig. 10a). SSTR2 protein showed higher abundance in hCS-derived neurons than in cCS-derived neurons (Fig. 4d-f, Extended Data Fig. 10c-g). In vivo, SSTR2 has higher expression in human than in either chimpanzee or macaque adult cortical tissue, especially in cortical layers V and VI38 (Extended Data Fig. 10b). These differences showed a striking resemblance to the pattern of SSTR2 expression in schizophrenia postmortem cortical tissue8, and expression of this gene has also been linked to Alzheimer’s dementia9.
We next tested whether the expression differences in this gene would translate into functional differences in neuronal activity between human and chimpanzee. Specifically, due to the inhibitory effect of SSTR2 on calcium signaling, we tested whether pharmacologically inhibiting this receptor would produce differential effects on intracellular calcium levels following depolarization. We used the calcium dye Fura-2 to image intracellular calcium in SYN1-labeled neurons dissociated from hCS and cCS at day 225-250 (Fig. 4g, Extended Data Fig. 10h). Following depolarization, we observed an increase in intracellular calcium in human neurons in the presence of the SSTR2 antagonist CYN-154806 (CYN; Fig. 4h,i). In contrast, application of CYN did not modulate the calcium response in chimpanzee neurons (Fig. 4h,i). In another experiment using the genetically-encoded calcium sensor GCaMP6s and stimulation by glutamate uncaging, we found that the frequency of spontaneous calcium transients in intact CS at day 130-150 increased significantly in human neurons treated with CYN, but not in chimpanzee neurons (Fig. 4j-l, Extended Data Fig. 10i). Altogether, this human-specific response was robust across multiple cell lines in distinct assays and developmental time points.
Discussion
Hybrid iPS cells provide a shared cellular context that controls for differences in gene expression resulting from both trans acting changes as well as non-genetic factors. We show that these cell lines are remarkably stable and that their tetraploidy does not interfere with species-specific transcription profiles. We used hyCS to classify differences between hCS and cCS as deriving from cis, trans, or non-genetic origins, which revealed evidence for selection on a set of astrocyte-specific genes. Our catalogue of cis-regulatory divergence during cortical development in vitro includes hundreds of genes such as SSTR2, whose ASE was strongest at timepoints representative of late gestation - a developmentally critical stage that is inaccessible in vivo. Because somatostatinergic transmission is known to modulate cortical plasticity37 and because SSTR2 expression has been associated with neuropsychiatric disease, the observed species divergence in expression may contribute to differences in both cortical function and dysfunction in the human lineage. Overall, this cellular hybrid platform represents a powerful resource for in vitro studies of human evolution, with the potential to answer questions about the origin and nature of human-specific cellular phenotypes across tissues39. Looking ahead, this cell fusion approach may provide a valuable complement to existing methods for disease modeling, wherein cell lines derived from patient and control subjects could be fused to disentangle complex polygenic disease etiologies while simultaneously controlling for experimental variability.
Methods
Culture of human and chimpanzee iPS cells
iPS cells derived from human and chimpanzee fibroblasts and reprogrammed with episomal vectors were obtained from the Gilad laboratory (see ref 1 for description and characterization of the iPS cell lines). Cultures were routinely tested for Mycoplasma and remained mycoplasma free. Three human (H20961, H21792, H20682) and three chimpanzee (C3649, C3647, C3651) iPS cell lines were thawed on Essential 8 medium (Life Technologies, A1517001) and maintained in feeder free conditions as previously described40. Briefly, cells were grown in 6-well plates coated with recombinant human vitronectin (VTN-N, Life Technologies, A14700) diluted 1:100 in Dulbecco’s phosphate-buffered saline (DPBS; Life Technologies, 14190) and passaged every four days, up to a maximum of 40 passages. Media changes were performed every day except the day after passaging or thawing. When passaging and thawing cells, media was supplemented with the ROCK inhibitor Y-27632 (10 μM, Selleckchem, S1049). Cells were cryopreserved in Essential 8 medium supplemented with ROCK inhibitor (10 μM) and 10% dimethyl sulfoxide (DMSO).
Generation of hyiPS cells
Prior to fusion, human and chimpanzee iPS cell lines were maintained in feeder free conditions as described above. Human and chimpanzee iPS cell lines were passaged four days prior to the fusion experiment. Media was changed on the following two days. On the third day, cells were labelled with diffusible dyes as follows: cells are first pre-treated with Y-27632 (5 μM, Thermo Fisher Scientific, 12-541-0) for 30 minutes. Diploid cells were dissociated with 1 mL of StemPro Accutase Cell Dissociation Reagent (Thermo Fisher Scientific, A11105-01) per well of a 6-well plate for 10 minutes at 37°C. Essential 8 medium was added to stop the dissociation and cells from each line were transferred to separate 50 mL centrifuge tubes. Cells were centrifuged at 1000 rpm for 5 minutes and the medium/Accutase was aspirated. Cells were resuspended in 10 mL of Essential 8 medium and counted using a hemocytometer, then divided into three centrifuge tubes as follows: tube 1 contained 8 million hiPS cells, tube 2 contained 8 million ciPS cells, and tube 3 contained 0.5 million hiPS cells and 0.5 million ciPS cells (for the unlabeled control condition). These tubes were centrifuged, the medium was aspirated and cells were resuspended in dye solutions (2 mL each) as follows: hiPS cells were labelled with CellTracker Deep Red (1.5 μM in DPBS, Thermo Fisher Scientific, C34565), ciPS cells were labelled with CellTracker Green CMFDA (5 μM in DPBS, Thermo Fisher Scientific, C7025) and the tube of mixed cells was resuspended in DPBS containing DMSO diluted 1:1000 only. Tubes were incubated at 37°C for 30 minutes and were resuspended every 10 minutes. Tubes were then centrifuged, dye solutions were aspirated, and cells were washed 3 times with DPBS. Cells were plated on matrigel-coated 6-well plates with 4 mL of Essential 8 medium per well with 5 μM Y-27632. Each well of the fusion plate contained 1 million hiPS cells and 1 million ciPS cells. The control plate (no fusion) contained one well of unlabeled cells (0.5 million of each species), one well of ciPS cells (1 million) only, one well of hiPS cells (1 million) only, and one co-cultured well of labelled cells (0.5 million of each species). Plates were incubated at 37°C overnight.
Fusion was performed on the following day as follows: media was aspirated from each well of the fusion plate and cells were washed twice with DPBS. Polyethylene glycol 1500 (“PEG”, Sigma-Aldrich, 10783641001) was added to each well (1 mL per well), and cells were incubated at 37°C for 2 minutes. PEG was aspirated and cells were washed 3 times with Essential 8 medium, and 4 mL of E8 medium with 5 μM Y-27632 was added. Media was changed for the cells in the control plate (E8 medium with 5 μM Y-27632).
The day after fusion, all cells were fed as normal (E8 medium with 5 μM Y-27632) and feeder cells were prepared as follows: six 10 cm dishes were coated with EmbryoMax 0.1% gelatin solution (EMD Millipore, ES-006-B) by incubating with 10 mL of this solution per plate for 30 minutes at 37°C. γ-irradiated MEF cells (ATCC, SCRC-1040) were plated at a density of 3 million cells per plate and incubated at 37°C overnight in MEF media consisting of DMEM (with high glucose/GlutaMAX supplement, Thermo Fisher Scientific, 10566-016) with 10% Fetal Bovine Serum (Thermo Fisher Scientific, 16000-044) and Penicillin-Streptomycin (1:100, Thermo Fisher Scientific, 15070-063).
The following day, the cells were first dissociated with Accutase as described above. After centrifugation and aspiration of Accutase and medium, cells were resuspended using a P1000 pipette into a solution of PBS with 0.5% Bovine Serum Albumin (Sigma-Aldrich, A9418-G), 2 mM EDTA (Thermo Fisher Scientific, 15575-020), 10 μM Y-27632, and 0.1 μg/mL DAPI (EMD Millipore, 5087410001) at a density of 0.5-1 million cells per mL in a FACS filter cap tube. Cells were then placed on ice prior to sorting. Cells positive for both Deep Red and Green CMFDA dyes and negative for DAPI (see Extended Data Fig. 2a for FACS gating strategy) were sorted into 15 mL tubes containing 2 mL of ice cold iPS medium consisting of DMEM/F12, HEPES (Thermo Fisher Scientific, 11330-032) with KnockOut Serum Replacement (KOSR; 1:5, Thermo Fisher Scientific, 10828-028), Penicillin-Streptomycin (1:100), Non-essential Amino Acids (1:100, Thermo Fisher Scientific, 11140-050), GlutaMAX Supplement (1:200, Thermo Fisher Scientific, 35050-061), 2-Mercaptoethanol (0.055 mM, Thermo Fisher Scientific, 21985-023) and FGF2 (10 ng/mL, R&D Systems, 233-FB) with 5 μM Y-27632. Cells were centrifuged at 1000 rpm for 5 minutes, medium was aspirated and cells were resuspended in iPS cell medium with 5 μM Y-27632 and plated on the prepared MEF plates at a density of 5000-10000 cells per plate.
Media was supplemented with 5 μM Y-27632 for the first 5 days after fusion, and media was changed every day until colonies were picked. When colonies became clearly visible, they were picked and transferred to the wells of a 12-well plate coated with γ-irradiated MEF cells, one colony per well. Each well was labelled with the identity of the new cell line according to the fusion pair (1 or 2) and the number of the colony picked (1 through 30), for example Hy1-30 refers to fusion 1, colony 30 (see Supplementary Table 1). Media was changed every day for the wells of the 12-well plates, and when colonies were big enough to be passaged, each line was passaged into one well of a 6-well plate. Cells were maintained on MEF cells as previously described. Karyotyping was performed by the Stanford Cytogenetics Laboratory using the GTW banding method for multiple metaphase nuclei per cell line.
Immunocytochemistry of hyiPS cells
At the time of passaging, hyiPS cells were plated onto glass coverslips placed in 24-well plates coated with recombinant human vitronectin (VTN-N, Life Technologies, A14700) diluted 1:100 in Dulbecco’s phosphate-buffered saline (DPBS; Life Technologies, 14190). When colonies had reached the desired confluency, coverslips were fixed with 4% paraformaldehyde/PBS for 10 minutes at 4°C. Coverslips were then gently washed with PBS three times to remove residual paraformaldehyde and stored at 4°C in PBS. Coverslips were transferred to a parafilm surface for staining. Blocking solution consisting of PBS with 10% normal donkey serum (NDS) and 0.3% Triton X-100 was added for 1 hour at room temperature. Primary antibodies were from the StemLite Pluripotency Antibody Kit (Cell Signaling Technology, 9656S): anti-Oct-4A (rabbit, 1:200, C30A3), anti-Sox2 (rabbit, 1:200, D6D9), anti-Nanog (rabbit, 1:200, D73G4), anti-SSEA4 (mouse, 1:200, MC813), anti-TRA-1-81 (mouse, 1:200, TRA-1-81), anti-TRA-1-60(S) (mouse, 1:200, TRA-1-60(S)). Coverslips were incubated with primary antibodies diluted in blocking solution overnight at 4°C. Coverslips were washed with PBS to remove primary antibodies, and incubated with secondary antibodies (Alexa Fluor dyes, Life Technologies) diluted 1:1000 in blocking solution for 1 hour at room temperature. Coverslips were washed with PBS and treated with Hoechst33258 (Thermo Fisher Scientific, H3569) for 3 minutes to stain nuclei. Coverslips were mounted on slides with Aqua-Poly/Mount (Polysciences, Inc., 18605-5) and imaged with a Zeiss M1 Axioscope. Images were processed with ImageJ (Fiji).
PluriTest analysis
We performed PluriTest as previously described, wherein expression data (RNA-sequencing) from bulk iPS cell samples (including all of our samples and all hiPS and ciPS cell samples from Ward et al.10 was projected onto the transcriptional profiles of several hundred validated pluripotent and non-pluripotent cell line samples. Input for this analysis was the raw fastq files from each sample with a maximum of ~15 million read pairs per sample (samples with more reads than this threshold were downsampled).
Bulk RNA sequencing
Bulk RNA samples (either a spun-down pellet of iPS cells or 2-3 CS) were snap frozen and stored at −80°C prior to RNA extraction. RNA extraction was performed using the RNeasy Mini Kit (Qiagen, 74104) with on-column DNase digestion (Qiagen, 79254). Buffer RLT was supplemented with 2-mercaptoethanol according to the manufacturer’s instructions. RNA concentrations were measured on a Nanodrop and quality was assessed using the Agilent Bioanalyzer RNA Pico assay. All samples had a RIN greater than or equal to 8.0. From each sample, 100 ng to 1 μg of total RNA was used for library preparation using the TruSeq Stranded mRNA kit (Illumina, 20020594). Libraries were prepared according to the manufacturer’s instructions. Samples were barcoded with dual-index adapters (Illumina, 20019792). Concentrations of cDNA were measured using a Qubit (HS DNA Assay, ThermoFisher Scientific, Q32851), then normalized and pooled; the quality of the pooled library was assessed with the Agilent Bioanalyzer (HS DNA assay), and in one instance a size selection gel was run to remove an adapter-dimer peak at 143bp. Libraries were then sequenced on an Illumina HiSeq instrument (either HiSeq4000 or HiSeqX) to generate 2x150bp paired-end reads.
Calling variants between GRCh38 and PanTro5
A pairwise genome alignment between GRCh38 and PanTro4 was obtained from Ensembl (ftp.ensembl.org/pub/release-84/maf/ensembl-compara/pairwise_alignments/homo_sapiens.GRCh38.vs.pan_troglodytes.CHIMP2.1.4.tar) and both single nucleotide variants and insertions/deletions (indels) were identified. A list of indel positions was formatted directly for use with Hornet/WASP (see next section). A list of variants was further filtered based on RNA-seq data (4 iPS cell samples from the Gilad lab, 2 iPS cell samples from this study, and 16 CS samples for a total of 22 bulk samples) by requiring that for a variant to be retained: (1) at least 2 reads mapped to the locus when mapping to each genome and (2) greater than 90% of the reads mapped to that locus were assigned to the correct species when mapped to each genome. This resulted in a high confidence list of phased variants (~2,800,000 loci) for each genome. Liftover (https://genome.ucsc.edu/cgi-bin/hgLiftOver) was used to convert the variant and indel coordinates from PanTro4 to those of PanTro5 when this new genome build became available.
Read mapping and transcript abundance measurement
Sequencing reads from each bulk sample were merged across sequencing lanes to produce one forward and one reverse fastq file per sample. Adapter sequences were removed with Seqprep (https://github.com/jstjohn/SeqPrep). Reads were mapped to both the human (GRCh38) and chimpanzee (PanTro5) genomes using STAR (version 2.5.1b) with the options --outSAMattributes MD NH, --outFilterMultimapNmax 1; two pass mapping was performed (with the option --sjdbFileChrStartEnd to specify the splice junctions identified in the first round of mapping) to improve alignment accuracy. Duplicate reads were identified with Samtools (version 0.1.19) and were discarded at random to avoid introducing bias. Hornet, a repackaged version of the WASP pipeline41, was used to identify and remove reads with mapping bias between the human and chimpanzee genomes (https://github.com/TheFraserLab/Hornet)42. Reads overlapping indels were also discarded. Reads were then assigned to their species of origin using the ASEr package (https://github.com/TheFraserLab/ASEr/)43, which takes mapped reads along with a file of species-specific variants and generates allele-specific read counts for each genomic feature. Unless otherwise noted, expression data presented in the figures is from reads mapped to GRCh38. Normalization of counts (TPM, FPKM, CPM) are indicated in each figure or figure legend. To account for differences in allelic read abundances across genes, normalization for sequencing depth in allelic TPM/CMP etc. calculations was performed based on the total allelic read counts for a sample, not the total of all reads per sample (and thus allelic TPM can theoretically be higher than total TPM for a given gene / allele).
Differential expression, ASE analysis, and identification of genes with mapping bias
The R package DESeq2 was used to measure differential expression between hCS and cCS samples at each time point. A log ratio test was used to compute P-values (with the options test=“LRT” and betaPrior=FALSE), with the model ~ Replicate + Species, wherein effects of different inductions are accounted for in the comparison of human and chimpanzee expression. For ASE analysis in the hyiPS cell and hyCS data, DESeq2 was used with the same parameters to measure ASE between allelic read counts instead of total read counts. For the iPS cell data, the model ~ CellLine + Species was used and for the hyCS data the model ~ Replicate +[CellLine + Species was used. For both differential expression and ASE, the default settings of DESeq2 were used to correct for multiple hypothesis testing. This was done for samples mapped on both the human (GRCh38) and chimpanzee (PanTro5) genomes. Any gene for which the log2[fold-change] values between the two mappings differed by more than 1 was removed from all subsequent analyses. This was done for each cell type separately (see Supplementary Tables S4 and S6 for genes with mapping bias in the iPS cell and CS data, respectively). Additionally, genes on chromosomes for which we identified aneuploidies, as well as the sex chromosomes, were removed from analysis, including for calculations of normalized transcript counts (chr20, X & Y for the iPS cell data, chr18, 20, X & Y for the CS data). Percentages of cis regulatory contributions mentioned in the text are calculated as abs(cis)/[abs(cis)+abs(trans/non-genetic)] where “cis” is the hybrid log2(human/chimp) and trans/non-genetic is the parental log2(human/chimp) minus hybrid log2(human/chimp). Throughout the paper, log2[fold-change] values are presented with the convention of positive values indicating higher expression in human and negative values indicating higher expression in chimpanzee, unless otherwise noted.
Statistical analysis, plotting and PCA
All statistical analyses referenced in the text and figures and all plots in the figures were created in Rstudio (v1.1.453) with R version 3.6.3 using the following packages: ggplot2, gplots, gridExtra, plyr, dplyr, reshape, scales, graphics, VennDiagram, Rtsne, Matrix, limma, and tidyverse. Additionally, statistical tests for quantification of fluorescent intensity of immunostains were computed in Prism (v8.3.1). Principal components were computed using the R command prcomp. In all cases, genes were included based on a minimal expression cutoff, which required genes to have at least 1 count per million (CPM) across all samples in the analysis. Specific statistical tests are explained in the relevant figure legend and corresponding section of the Methods.
Generation of human and chimpanzee cortical spheroids
hCS and cCS were generated as previously described40. Briefly, iPS cells were dissociated into a single cell suspension using Accutase (Innovate Cell Technologies, AT-104) and transferred to an AggreWell 800 plate (STEMCELL Technologies, 34815), with approximately 3 million cells per well. Aggrewell plates were incubated overnight (approximately 16 hours) at 37°C with 5% CO2 in Essential 8 medium supplemented with ROCK inhibitor Y-27632 (10 μM). Spheroids were resuspended by pipetting and transferred to low-attachment dishes (Corning, 3262) in Essential 6 medium (Life Technologies, A1516401) supplemented with penicillin and streptomycin (1:100, Life Technologies), Y-27632 (10 μM), dorsomorphin (2.5 μM, Sigma-Aldrich, P5499) and SB-431542 (10 μM, Tocris, 1614). Up to day 6, Essential 6 medium was changed every day and supplemented with dorsomorphin and SB-431542. After day 6, spheroids were transferred to medium consisting of Neurobasal A (Life Technologies, 10888), B-27 supplement without vitamin A (Life Technologies, 12587), penicillin and streptomycin (1:100) and GlutaMax (1:100, Life Technologies, 35050). From day 7 to 24, medium was supplemented with 20 ng/ml epidermal growth factor (R&D Systems, 236-EG) and 20 ng/ml basic fibroblast growth factor (R&D Systems, 233-FB), and from days 25 to 42 medium was supplemented with 20 ng/ml brain-derived neurotrophic factor (BDNF; Peprotech, 450-02) and 20 ng/ml NT3 (Peprotech, 450-03). Media changes were performed every day for the first 16 days, every other day until day 42, and every 4 days from day 43 onward.
Generation of hyCS
hyCS for our pilot study were generated identically to hCS and cCS described above. hyCS for the expanded data set were generated using a modified version of the protocol described in ref12. hyiPS cell lines were maintained on MEF feeder cells as previously described44. Cellular aggregates were generated by lifting intact colonies of hyiPS cells with dispase (0.35 mg/ml) and then transferred to ultra-low attachment plates (Corning, 3262) with media consisting of DMEM/F12 (1:1, Life Technologies, 11330), KnockOut Serum (20%, Life Technologies, 10828), non-essential amino acids (1 mM, Life Technologies, 11140), GlutaMax (1: 200, Life Technologies, 35050), β-mercaptoethanol (0.1 mM, Sigma-Aldrich, M3148), penicillin and streptomycin (1:100, Life Technologies, 15070), supplemented with dorsomorphin (5μM,Sigma-Aldrich), SB-431542 (10μM, Tocris), and Y-27632 (10 μM, EMD Chemicals). After 2-3 days in culture, neural aggregates were embedded in Matrigel™ matrix (Corning, 354234; Lot#6109006 was used for all experiments) as follows: Matrigel™ was first thawed at 4° C overnight and aliquoted into 1.5 mL tubes, then placed on ice. After changing media, about 50 spheroids were transferred to a new 1.5 mL tube with a P1000 pipette tip, and medium was carefully removed. Next, an ice cold P1000 pipette tip with the tip cut off was used to transfer about 0.5 mL of ice cold Matrigel™ to the tube containing the spheroids. Spheroids were immediately resuspended in Matrigel™, then dispensed onto a 100 mm cell culture plate (non-ultra-low-attachment) in small droplets (avoiding having multiple spheroids per droplet) and incubated at 37°C for 30 minutes. Then 5 mL of warmed medium was added to the plate containing Matrigel™ droplets and a cell scraper was used to dislodge the droplets, which were transferred to a low-attachment plate with a 50 mL pipette. Media supplemented with dorsomorphin and SB-431542 was changed daily as normal until day 6 of differentiation, and from there on differentiation was performed as described above for hCS and cCS. In cases where multiple spheroids were embedded in the same droplet of Matrigel™, droplets were cut apart with a #10 blade at around day 15–25 of in vitro differentiation.
Cryopreservation and sectioning of CS
CS were washed once with phosphate buffered saline (PBS), fixed in 4% paraformaldehyde/PBS overnight, then washed three times with PBS and transferred to 30% sucrose for a minimum of 72 hours. CS were then embedded in a 1:1 mix of OCT (Tissue-Tek optimum cutting temperature compound 4583, Sakura Finetek) and 30% sucrose and frozen at −80°C for at least one day. They were sliced using a cryostat (Leica, CM1860) to 15 μm sections and placed on slides. The slides were stored at −80°C until they were immunostained.
Immunocytochemistry of CS
CS sections were washed with PBS to remove residual OCT/sucrose and then blocked in a solution consisting of PBS with 10% normal donkey serum (NDS), 0.3% Triton X-100 and 1% BSA for 1 hour at room temperature. The sections were then incubated with primary antibodies diluted in a solution consisting of PBS with 2% NDS and 0.1% Triton X-100 overnight at 4°C. The following primary antibodies were used for staining: anti-PAX6 (mouse, 1:300, BioLegend, PRB-278P), anti-CTIP2 (rat, 1:300, abcam, ab18465), anti-TBR2 (mouse, 1:100, R&D Systems, MAB6166), anti-SOX9 (goat, 1:300, R&D Systems, AF3075), anti-SSTR2 (mouse, 1:100, R&D systems, MAB4224), anti-MAP2 (guinea pig, 1:200, Synaptic Systems, 188 004), and anti-TUBB3 (rabbit, 1:200, Cell Signaling TECHNOLOGY, #5568S). Sections were washed with PBS to remove primary antibodies, and incubated with secondary antibodies (Alexa Fluor dyes, Life Technologies) diluted 1:1000 in PBS with 2% NDS and 0.1% Triton X-100 for 1 hour at room temperature. Sections were washed with PBS and nuclei were stained with Hoechst 33258 (Thermo Fisher Scientific, H3569) for 5 minutes at room temperature. Sections were mounted on glass slides with Aqua-Poly/Mount (Polysciences, Inc., 18605-5) and imaged with a Leica TCS SP8 confocal microscope. Images were processed with ImageJ (Fiji).
Dissociation of hyCS for single cell RNA sequencing
hyCS were dissociated as previously described45. In brief, 2-3 CS per cell line were cut using a #10 blade and then incubated in papain enzyme solution (27.3 U/ml, Worthington), EBSS (1×, Sigma-Aldrich), 0.46% sucrose (Sigma-Aldrich), 26 mM NaHCO3 (Sigma-Aldrich), 0.5 mM EDTA (Sigma- Aldrich) at 37°C for 70 minutes in an incubator (5% CO2). The digested CS were then washed and carefully triturated in a trypsin inhibitor solution containing EBSS, 0.46% sucrose (Sigma-Aldrich), 26 mM NaHCO3 (Sigma-Aldrich), 15–30 mg trypsin inhibitor (Sigma-Aldrich). Cells were spun down and resuspended in 0.2% BSA diluted in PBS and supplemented with Y-27632 (10 μM, EMD Chemicals).
Single cells were sorted with a BD Aria Fusion instrument into 96-well plates containing 4 μl of lysis buffer consisting of 4 enzyme units (U) of RNase inhibitor (40 U/μl, Clontech, 2313B), 0.05% Triton X-100, 2.5 mM deoxynucleotide triphosphates (dNTP) and 2.5 μM Oligo-dT30VN (IDT, RNase-free HPLC purification)46. Plates were centrifuged for 2 minutes at 4°C before being stored in a −80 °C freezer until processing.
Single cell RNA sequencing via SmartSeq2
Single cell libraries were generated according to a modified version of the SmartSeq2 protocol46 as previously described15. Briefly, 96-well plates were thawed on ice and incubated at 72°C for 3 minutes to anneal the Oligo-dT30VN to polyA tails; plates were then spun down and placed on ice. Reverse transcription mixture was added at 6 μl per well so that the final solution contained 95 enzyme units SMARTScribe Reverse Transcriptase (100 U/μl, Clontech, 639538), 10 enzyme units RNase inhibitor (40 U/μl), 1 × First-Strand buffer, 5 mM dithiothreitol, (Promega, P1171), 1 M betaine (Sigma, B0300), 6 mM MgCl2 (Sigma, M1028), and 1 μM template switching oligos (TSO) (Qiagen, RNase-free HPLC purification). Reverse transcription was performed at 42°C for 90 minutes, followed by 70°C for 5 minutes.
Each well received 15 μL DNA amplification mixture consisting of 1 × KAPA HIFI Hotstart Master Mix (Kapa Biosciences, KK2602), 0.1 μM in situ polymerase chain reaction (ISPCR) oligo, and 0.56 U lambda exonuclease (5 U/μL, New England BioLabs, M0262S). cDNA was amplified using (1) 37°C 30 minutes; (2) 95°C 3 minutes; (3) 22-23 cycles of 98°C 20 seconds, 67°C 15 seconds, 72°C 4 minutes; and (4) 72°C 5 minutes. Amplified cDNA was then purified using Ampure XP beads (~0.7 volume, Beckman Coulter, A63881), and reconstituted in 20 μL elution buffer (EB). The cDNA quality and quantity were assessed using a fragment analyzer (AATI, High Sensitivity NGS Fragment Analysis Kit: 1–6,000 base pairs), and samples with concentrations below 0.03 ng/μL or abnormal peak patterns were filtered out. A total of 750 cDNA samples were pooled into two 384-well plates (0.4 μL per well) using a Mosquito X1 (TTP Labtech). Tagmentation of cDNA was performed as previously described47 (with the help of a Mosquito HTS machine for liquid transfer) using a tagmentation mixture consisting of Tn5 transposase (in-house), 5x TAPS (50 mM)-MgCl2 (25 mM) mixed 1:1 with PEG 8000 (40%) and then diluted 1:2.5 for a final volume of 1.6 μl (total 4 μl per well). Samples were incubated at 55°C for 5 minutes and the tagmentation reaction was halted with the addition of 0.4 μl of 0.1% sodium dodecylsulfate (SDS, 0.01% final) per well. PCR amplification and adapter annealing were performed by adding 2 μl of PCR mix consisting of 0.8 μl dual-index adapters (2.5 μM each), 0.08 ul KAPA HiFi DNA polymerase (non-hot-start), 0.8 μl 5x Buffer, 0.12 μl dNTPs (10 mM) and 0.2 μl water per well. Libraries were amplified using (1) 72 °C 3 minutes; (2) 95°C 30 seconds; (3) 10 cycles of 95°C 10 seconds, 55°C 30 seconds, 72°C 1 minute; and (4) 72°C 5 minutes. Libraries from each plate were pooled into one tube per plate (maximum 384 samples per pool). Amplified libraries were then purified twice using Ampure XP beads (~0.7 volume) and reconstituted in 40 μl EB. Library quality was determined with the Agilent Bioanalyzer (HS DNA assay), and libraries were sequenced on an Illumina NovaSeq instrument to generate 2x100 bp paired-end reads with an average read depth of ~2 million reads per cell.
Dimensionality reduction, clustering, and Monocle analysis
Read mapping and transcript abundance measurement for the single cell data was performed as described above for the bulk RNA-sequencing data. A matrix of total read counts per gene / per cell was assembled and used for downstream analysis with the R package Seurat48. Cells with more than 7% of reads from mitochondria or with less than 100,000 total transcripts were discarded, retaining 706 cells from the original 750. Data were log normalized with a scaling factor of 10,000 and the top 1500 most variable genes were selected based on variable stabilizing transformation and used for clustering. UMAP clustering was performed using the top 14 principle components. We defined cells as “neural” (for further analysis presented in Figure 3) if they were in the neuronal, astroglial and progenitor cell-like clusters, with the exclusion of 14 cells that displayed an expression profile more similar to mesenchymal cells, resulting in 368 neural cells. These cells were re-analyzed using the same parameters as were used for the entire data set. Similarly, co-clustering of our data (all cells) with the data from Sloan et al.15 was performed by first restricting the list of genes to those recorded in both data sets and then following the same analysis steps in Seurat (with the same filtering parameters applied to both data sets).
For construction of cell trajectories, the R package Monocle224 was used to analyze our neural cells only (n=368). Normalized read counts were converted to mRNA counts and outliers on either end of this distribution were discarded, resulting in a final data set of 348 cells. Cells were then clustered in an unsupervised manner (without user-defined marker genes), and dimensionality reduction was performed via tSNE with log normalization. Cells were then clustered via density peak clustering with a rho threshold of 6 and a delta threshold of 10. The top 1000 most variable genes across clusters were used for pseudotime ordering and dimensionality reduction was performed using the DDRTree algorithm (the default) to construct developmental trajectories.
Gene ontology enrichment tests
The gene ontology enrichment tool GOrilla49 was used for enrichment analyses. For differential expression between hCS and cCS, genes were ranked by their expression difference (log2[human/chimpanzee]) and enrichment was performed for both the top (human biased) and bottom (chimpanzee-biased) genes. For genes present in specific WGCNA modules (see below) we used an unranked list of module genes with a background set of all expressed genes that passed filtering with the WGCNA package. Reported significance values in the text are FDR q-values.
Deconvolution of bulk RNA sequencing data (CIBERSORT)
The tool CIBERSORT50 was used to estimate the proportion of five cell types (neurons, astroglia, cycling progenitor cells, mesenchymal cells and epithelial cells) in each bulk sample by defining cell type-specific gene expression profiles using our single cell data. All cells from one cell type-specific cluster (or two merged clusters in the case of neurons and mesenchymal cells) were pooled by cell line to generate two expression profiles per cell type (one from each cell line in our single cell data), then FPKM normalized. This cell profile was used as input for CIBERSORT along with the FPKM values for each bulk sample and proportions of each cell type were estimated without quantile normalization (as recommended for RNA-seq data) and 100 permutations. Calculations for percentage of neural ectoderm were computed as the percentage of a given cell type divided by the cumulative percentages of astroglia, progenitors and neurons.
Weighted gene co-expression network analysis (WGCNA)
Sets of co-expressed genes were identified in R using the WGCNA package28. We used our hCS and cCS time course data to identify gene networks, with three outlier samples excluded from the analysis (all chimpanzee, resulting in 41 human and 36 chimpanzee samples). Modules were constructed with a soft power threshold of 8, and modules were required to have 30 or more genes. Eigengenes for hCS and cCS were computed with WGCNA, and for hyCS (bulk and single cell data) we performed PCA and defined eigengenes as the value of the first PC. These eigengenes were oriented such that mean module expression was positively correlated with the eigengene value (the same method used by WGCNA).
Overlaps with SFARI
Genes from the SFARI database were downloaded directly from the Simon Foundation Autism Research Initiative website (https://www.sfari.org/resource/sfari-gene/) in December 2019.
Dissociation and monolayer culturing of hCS and cCS
hCS and cCS were dissociated and plated down for calcium imaging and immunocytochemistry experiments as previously described15. Briefly, 15–20 CS per cell line were incubated in 30 U/mL Papain (Worthington, LS 03126), 1 mM L-cystine (Sigma, C7880) and 125 U/ml DNase I (Worthington, LS002007) for 45 minutes. Following a series of Papain-inactivating washes, CS were single-cell triturated and plated down in neural media supplemented with BDNF (20 ng/mL) and NT-3 (20 ng/mL) on glass coverslips coated with poly-l-ornithine and laminin (Sigma-Aldrich) at a density of 90,000-100,000 cells per coverslip. Prior to plating, cells for these imaging experiments were first immunopanned for THY1 and HEPACAM for use in other experiments. Calcium imaging and immunocytochemistry experiments were performed within 9 and 30 days of dissociation. Coverslips were fixed for immunostaining as follows: a solution of warmed 4% PFA/4% sucrose/PBS was added to the wells with coverslips to be fixed for 20 minutes. Cells were then washed with PBS and stored in PBS at 4° C. Staining was performed as described above, and the following primary antibodies were used for staining: anti-SSTR2 (mouse, 1:100, R&D systems, MAB4224), anti-MAP2 (guinea pig, 1:200, Synaptic Systems, 188 004).
Calcium imaging on dissociated hCS and cCS
To assess the effect of SST2R modulation on calcium signaling in hCS and cCS, spheroids between days 225 and 250 of differentiation were dissociated and plated down as described above. After 2-3 days, neurons were labeled using the viral reporter AAV-DJ-hSyn1::eYFP. Calcium imaging was performed as previously described51. Briefly, cells were loaded with the ratiometric calcium indicator Fura–2 acetoxymethyl ester (1μM, Thermo Fisher Scientific, F1221) for 25 minutes at 37°C in Neurobasal-A medium containing B-27 (1:50), GlutaMAX (1:100) and Penicillin/Streptomycim (1:100). Cells were incubated for 10 min in the same medium without Fura-2 and placed in a perfusion chamber (RC-20, Warner Instruments) on the stage of an inverted fluorescence microscope (TE2000U, Nikon) equipped with an excitation filter wheel and an automated stage. Following a 4.5 minute baseline imaging in low-KCL Tyrode’s solution (5 mM KCl, 129 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 30 mM glucose, 25 mM HEPES, pH 7.4), cells were then depolarized with 67 mM KCl Tyrode’s solution (67 mM KCl, 67 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 30 mM glucose and 25 mM HEPES, pH 7.4). For testing the effect of the SST2R antagonist CYN-154806 (0.5μM, diluted in H2O), a low KCl solution containing CYN-154806 was perfused for 3.5 minutes following an initial 1 minute baseline period and depolarization was performed with 67 mM KCl Tyrode’s solution containing CYN-154806. Control conditions were treated with H2O in place of CYN. Regions of interest (ROIs) were registered based on eYFP expression and used to collect time–lapse excitation ratios (340 nm to 380 nm) using Openlab software (PerkinElmer). Peak 340 nm to 380 nm ratios following depolarization (i.e. amplitudes) were calculated and aligned using a custom-written Matlab routine. eYFP+ neurons that had an amplitude of response lower than 0.1 were excluded from the analysis. Because the amplitudes did not follow a normal distribution, two-tailed Wilcoxon Rank Sum tests were used to compare across species and conditions (Figure 5).
Calcium imaging and SST2R pharmacology in intact hCS and cCS
hCS and cCS between day 130–150 were infected with an AAV vector that expresses the red fluorophore mRuby2 and the genetically encoded calcium indicator GCaMP6s under the neuronal promoter hSyn1 (AAV1-hSyn1::mRuby2-GSG-P2A-GCaMP6s-WPRE-pA; Addgene, 50942-AAV1). Intact CS were placed in 96-well glass-bottom plates (Corning, 4580) in imaging media containing caged glutamate (MNI-caged-L-glutamate, Tocris, 1490). Experiments were performed using an environmentally controlled chamber (37°C, 5% CO2) on a Leica SP8 confocal microscope, and data was collected with LasX v3.5.5. The mRuby2 signal was used to identify virally labeled neurons. GCaMP6s and mRuby2 were imaged in 3 fields per CS at an imaging speed of 0.132 Hz. Following a baseline recording of 1.7 minutes, glutamate was uncaged using a brief laser pulse at 405 nm, followed by the administration of selective SST2R antagonist CYN-154806 (0.5μM, diluted in H2O; Tocris, 1843). After incubating for 30 minutes, and 3 fields per CS were imaged for another 1.7 minutes followed by another round of glutamate uncaging. mRuby2 expression was used for the selection of ROIs using ImageJ, from which GCaMP6 signal was extracted. Calcium spikes before and after CYN-154806 administration were detected using a custom-written Matlab routine (Matlab2019b). Briefly, mean gray values from ROI time series were transformed to relative changes in fluorescence: dF/F(t) = (F(t)-F0)/F0; where F0 represented mean gray values of the time series of each ROI. Cells that displayed slow oscillatory signals were corrected for using a high-pass filter. Calcium transients were detected as dF/F(t) crossed the threshold of 4 median absolute deviations. Only neurons that responded to glutamate uncaging (10% or more dF over baseline) with two or more calcium transients were included for the analysis. Because the frequencies did not follow a normal distribution, two-tailed Wilcoxon Rank Sum tests were used to compare across species and conditions, and the P-values for effect of the drug on each cell line were combined using Fisher’s method (Figure 5).
Extended Data
Extended Data Figure 1 |. Isolation and characterization of hyiPS cells.
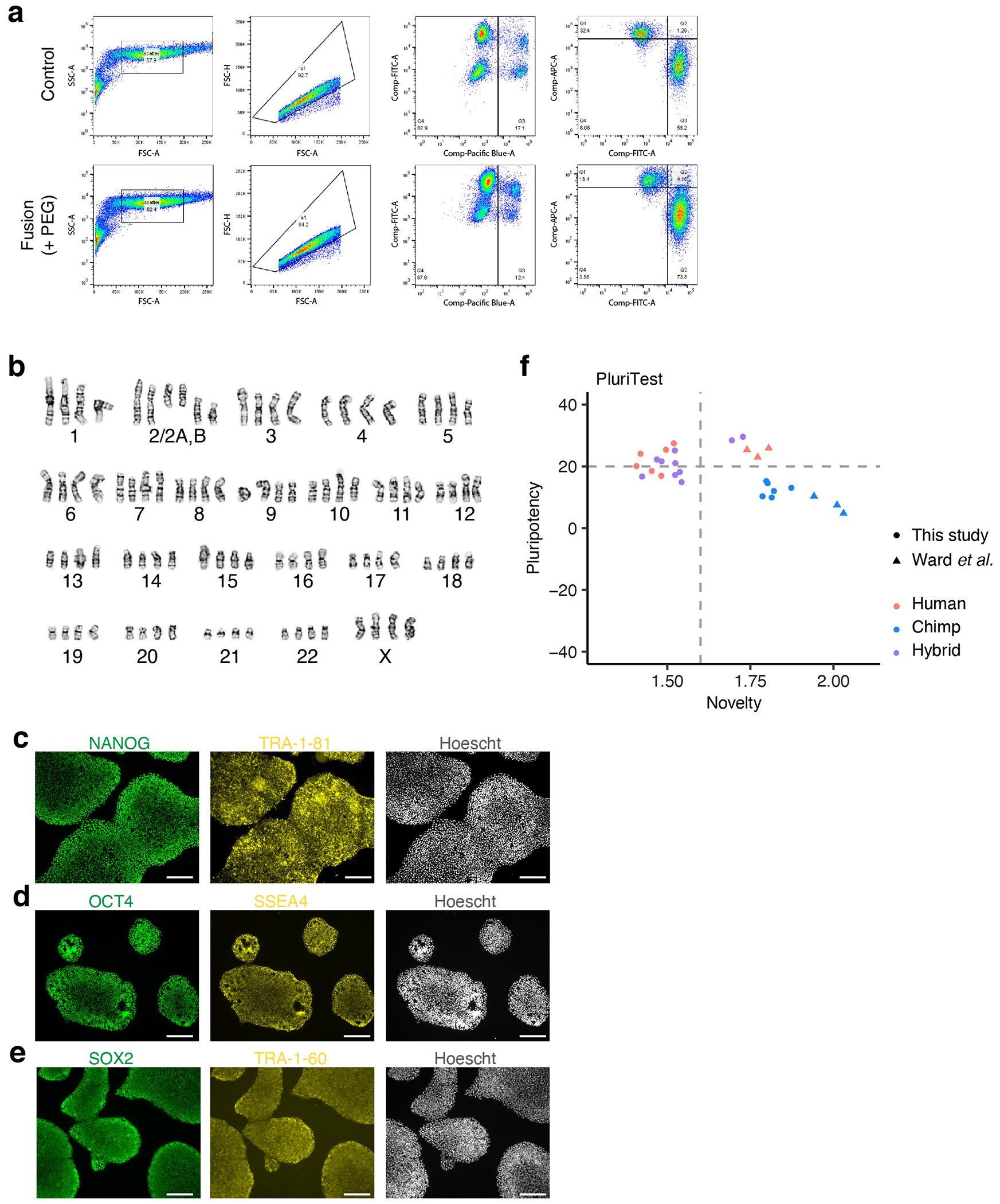
a, Fluorescence-activated cell sorting (FACS) of fused hybrid cells (representative plots for fusion of H20961 and C3649); top panel, co-cultured cells with no PEG; bottom panel, co-cultured cells with PEG; from left: initial size selection, gating out doublets, gating out dead cells, sorting for red (human) and green (chimpanzee) double positive population; FSC forward scatter, SSC side scatter, -A area, -H height; Pacific Blue measures DAPI, FITC measures Green CMFDA (chimpanzee), and APC measures Deep Red (human). b, Representative karyotype for female (XX/XX) hybrid iPS cell lines. c-e, Immunostaining for the pluripotency markers NANOG, Tra-1-81 (c), OCT4, SSEA4 (d), SOX2, Tra-1-60 (e). f, Results from PluriTest analysis of RNA-sequencing data from this study and from Ward et al.10 (see Methods); benchmarked thresholds are 20 or higher for pluripotency, 1.6 or lower for novelty (dotted lines). Scale bars, 200 μm (c-e).
Extended Data Figure 2 |. Chromosomal instability and X chromosome inactivation.
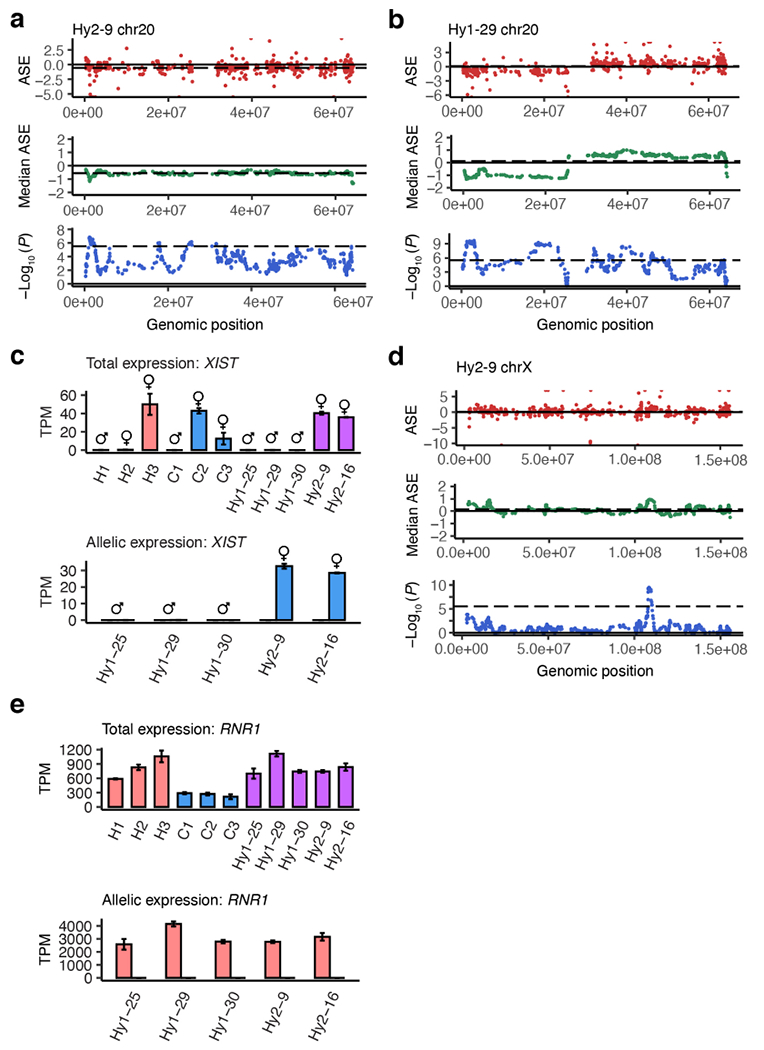
a-b, Plots showing aneuploidies on chromosome 20 indicating a gain of a chimpanzee chromosome (a) or a combined loss of the human short arm and gain of the human long arm (b); top panel, scatter plot of allele specific expression (ASE = log2[human/chimpanzee]) versus genomic location; middle panel, median ASE in a sliding window of 20 genes; bottom panel, P-values from a two-sided Wilcoxon rank sum test comparing a sliding window of 20 genes to the background of the entire genome. c, Total (top panel) and allelic (bottom panel; human allele pink, chimpanzee allele blue) expression (TPM, transcripts per million) of XIST in RNA-seq samples; symbols indicate the sex of each iPS cell line; n= 2 technical replicates per cell line. d, Plots of ASE across the X chromosome (as in a-b). e, Total and allelic expression of RNR1 (chrMT), as in (c); n= 2 technical replicates per cell line.
Extended Data Figure 3 |. RNA-sequencing of hyiPS cells.
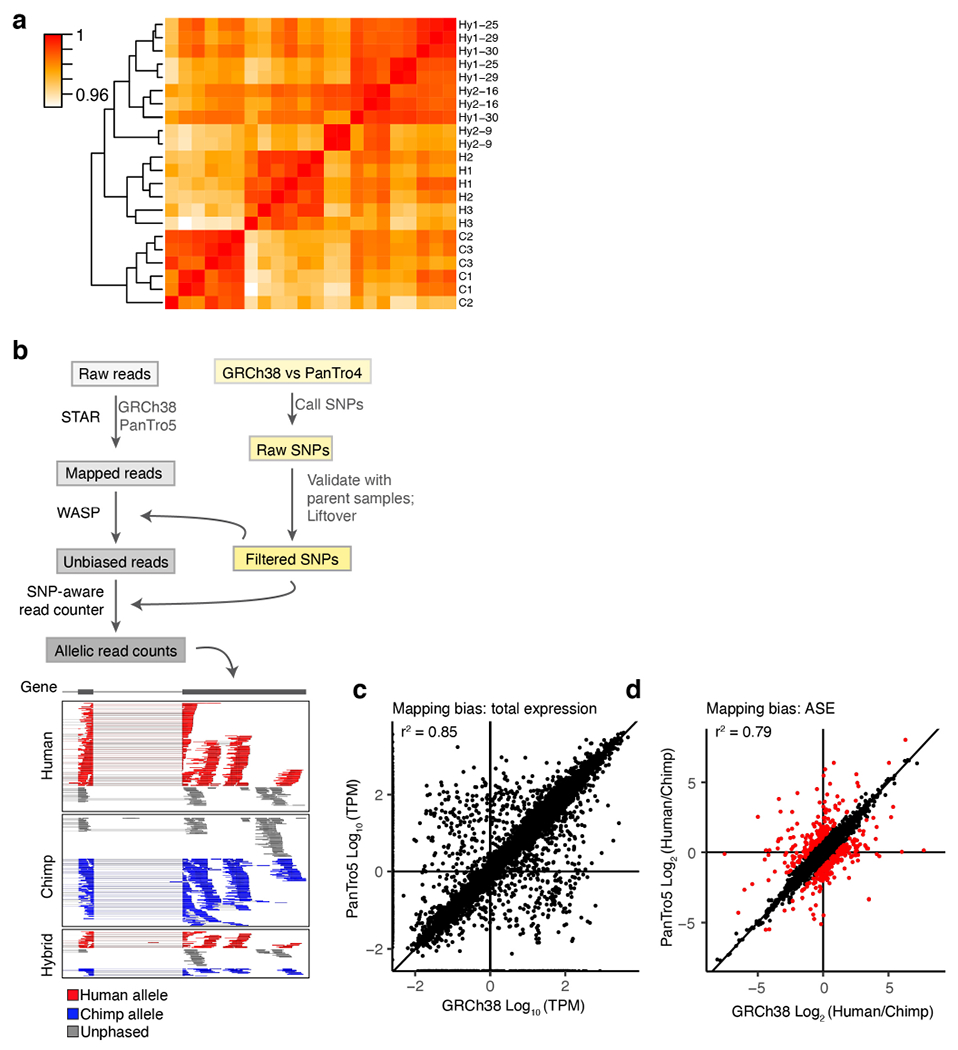
a, Heat map of correlations (Pearson’s) between RNA-seq samples from human (H1, H2, H3), chimpanzee (C1, C2, C3) and hybrid (Hy1-25, -29, -30, Hy2-9, -16) iPS cells. b, Top panel, pipeline for analysis of RNA-seq data and separation of species-specific sequencing reads; bottom panels, pileup of phased allelic reads from human, chimpanzee and hybrid RNA-seq samples for a representative gene. c, Representative scatter plot (from line Hy1-30) showing total gene expression (TPM, transcripts per million) when samples are mapped to the human genome (GRCh38, x-axis) versus the chimpanzee genome (PanTro5, y-axis); n= 1, out of 10 total hyiPS samples sequenced with similar results. d, Scatter plot of allele specific expression (ASE) in all hybrid samples when mapped to the human versus the chimpanzee genome; genes represented by the points in red are considered to have mapping bias and are eliminated from subsequent analyses; data merged from n= 10 samples from 5 hyiPS cell lines (2 replicates each).
Extended Data Figure 4 |. Generation and characterization of hyCS.
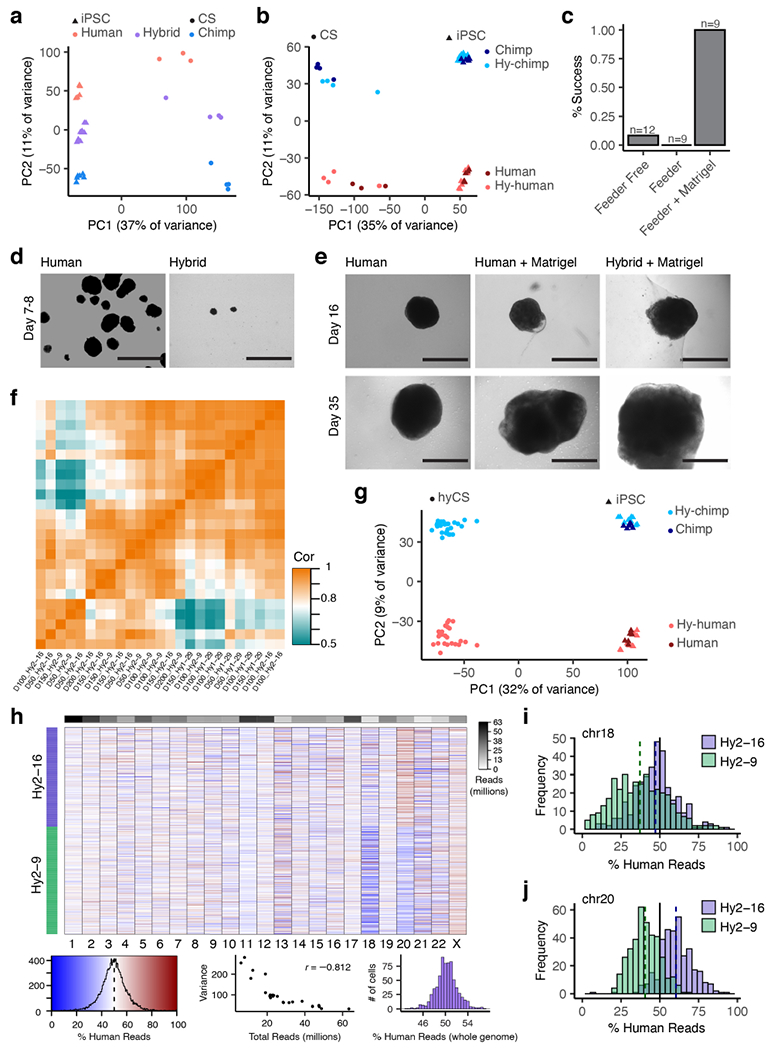
a-b, Principal components plots for iPS and CS (pilot study) RNA-seq samples based on total (a) or allelic gene expression (b). c, Rates of success of three protocols used to derive hyCS (success is defined as at least one CS from a given cell line surviving to 100 days of differentiation); n refers to the number of independent attempts to differentiate any of 3 hyiPS cell lines. d, Bright field imaging of hCS and hyCS at day 7-8 of differentiation; experiment was repeated across 3 independent differentiation experiments of 3 hyiPS and 1 hiPS cell line with similar results. e, Bright field images of Matrigel™-embedded hCS and hyCS, as well as non-embedded hCS, at days 16 and 35 of differentiation. f, Heatmap of correlations (Pearson’s) between bulk RNA-seq samples for hyCS. g, Principal components plot for iPS and hyCS (full data set) RNA-seq samples based on allelic gene expression h, Heatmap colored by the percentage of human reads in each single cell, stratified by chromosome; rows are ordered by hybrid cell line; top bar shows read depth of each chromosome across all cells; bottom left, color key and histogram for heatmap values; bottom middle, scatter plot of total read depth versus variance per chromosome, wherein fewer reads results in higher variance; bottom right, histogram showing the percent of human reads in each cell, genome-wide. i-j, Histogram of the percentage of human reads in each cell for aneuploid chromosomes 18 (i) and 20 (j), stratified by cell line. Scale bars, 1 mm (d-e).
Extended Data Figure 5 |. Single cell gene profiling of hyCS.
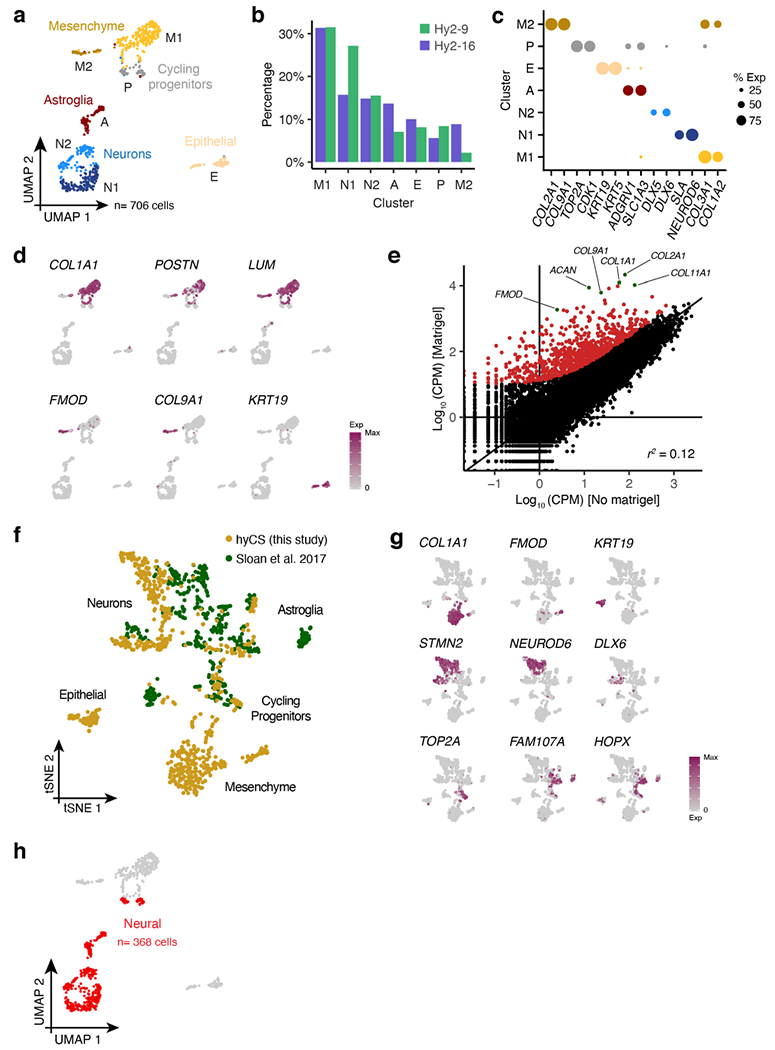
a, UMAP clustering of all cells (n= 706); clusters are identified by color and labelled by letter (A= astroglia, P= cycling progenitors, N1= glutamatergic neurons, N2= GABAergic neurons, M1= mesenchyme cluster 1, M2= mesenchyme cluster 2, E= epithelial cells). b, Proportion of cells from each hybrid cell line in each single cell cluster (from a). c, Dotplot for expression of marker genes for each cluster in (a), size corresponds to the percent of cells in each cluster that express each gene. d, UMAP colored by expression of mesenchymal and epithelial marker genes. e, Scatter plot of normalized gene expression between embedded (y-axis) and non-embedded (x-axis) hybrid (line Hy1-29) CS at day 50 of differentiation; points in red and green indicate genes whose expression is induced by the addition of Matrigel™ (see Methods). f, tSNE of all single cells from this study aggregated with cells from non-embedded spheroids in Sloan et al.15, colored by study. g, tSNE from (f) colored by expression of cell-type marker genes. h, UMAP from (a) colored according to which cells were defined as neural and used for further analysis in Fig. 3. i, Histograms of per-gene ASE, where ASE is defined as the ratio of all human reads across cells of a given cell type to all chimpanzee reads in those cells.
Extended Data Figure 6 |. Generation of hCS and cCS and RNA-sequencing.
a-b, Representative bright field images of three CS per line for three human (a) and three chimpanzee (b) cell lines at day 166. c, Immunostaining of hCS and cCS for SOX9, PAX6 and CTIP2; at each time point, a maximum of 2 spheroids were fixed for immunostaining across 3 hiPS and 3 ciPS cell lines with 4 independent differentiation experiments per cell line. d-e, Heatmap of correlations (Pearson’s) between bulk RNA-seq samples for hCS (d) and cCS (e). Scale bars, 1 mm (a-b), 50 μm (c).
Extended Data Figure 7 |. RNA-seq and cell type deconvolution in cortical spheroids.
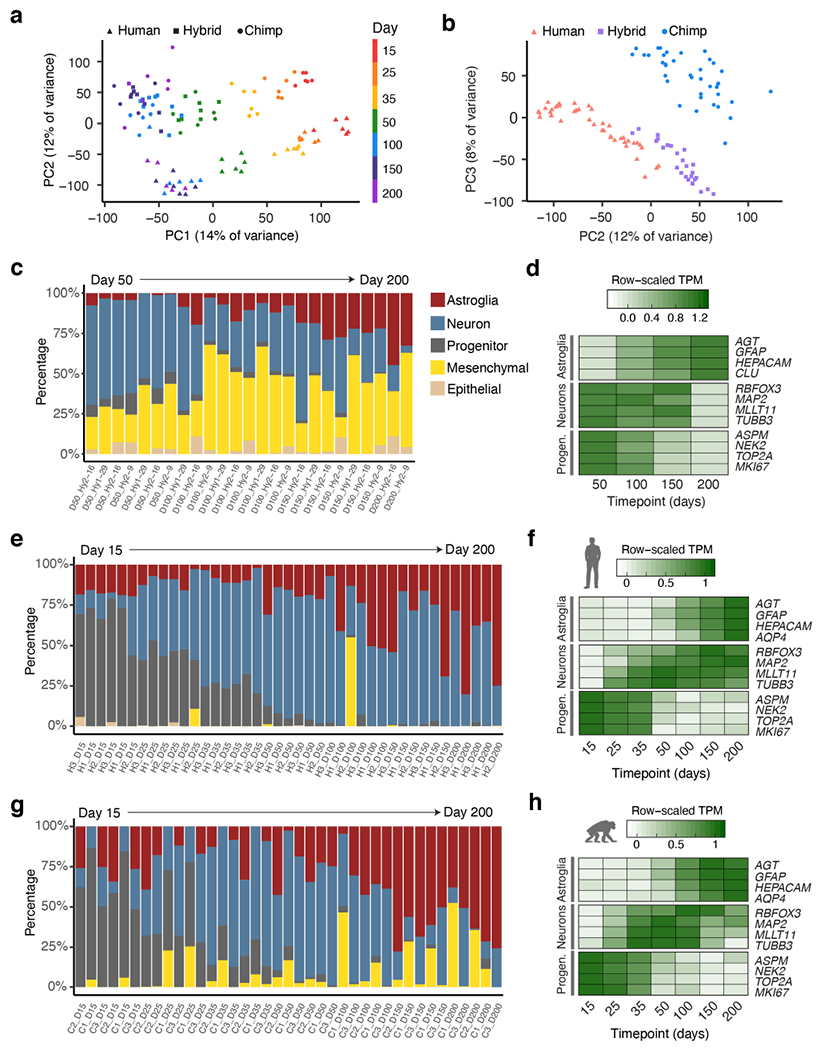
a-b, Principal components plots for RNA-seq samples based on total gene expression of parent and hybrid samples. c, e, g, Per-sample estimated cell type proportions in hyCS (c), hCS (e) and cCS (g) (see Methods). d, f, h, Normalized expression across time of cell-type specific marker genes in hyCS (d), hCS (f) and cCS (h); TPM, transcripts per million.
Extended Data Figure 8 |. Weighted gene co-expression network analysis.
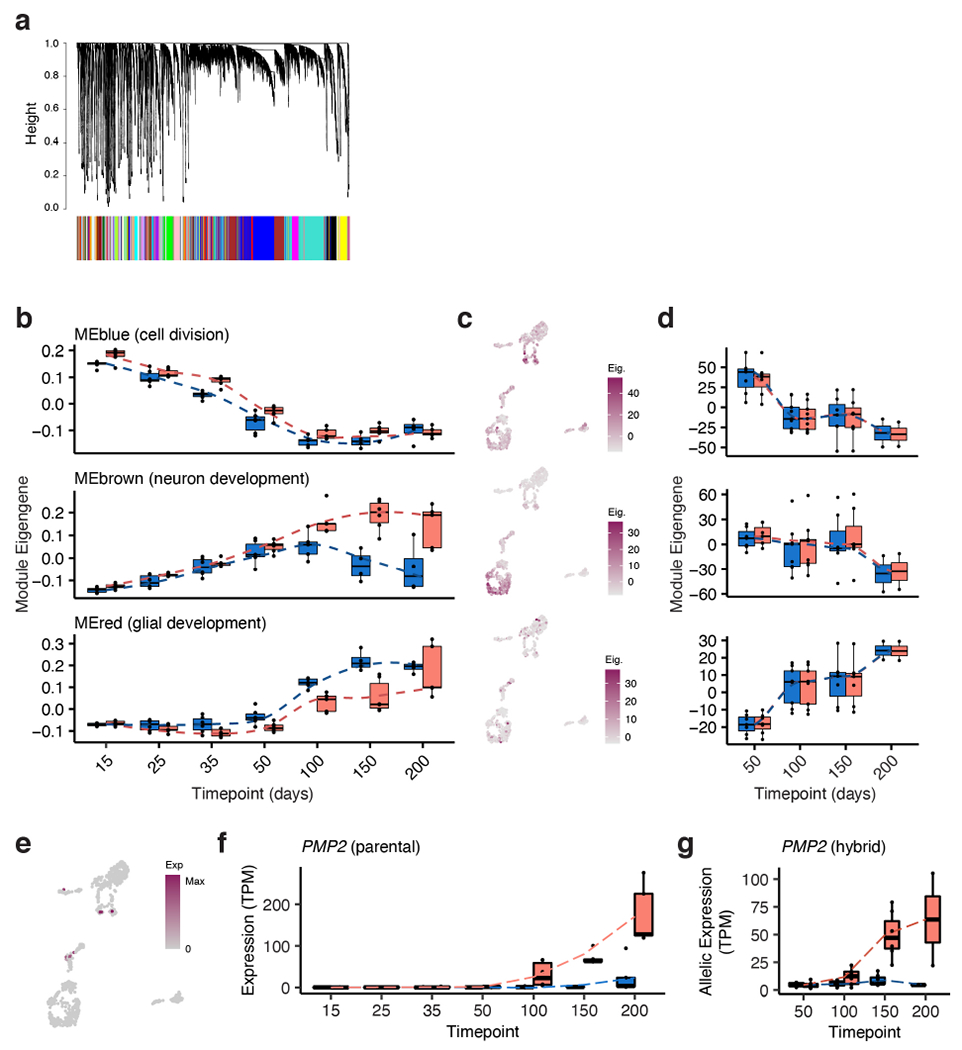
a, Dendrogram of all genes used in WGCNA; genes in the same color block belong to the same co-expressed module. b, Eigengene values for genes in the Blue, Brown and Red modules over time in hCS and cCS; chimpanzee blue, human red; in order of time points, n= 6, 6, 6, 6, 6, 6, 5 hCS and n= 6, 6, 6, 6, 5, 5, 5 cCS samples from 3 human and 3 chimpanzee iPS cell lines (1-2 replicates per cell line). c, Expression of module genes (eigengene, see Methods) in single cell data; cell clusters are defined in Extended Data Fig. 5a. d, Allelic eigengene values for genes in these modules over time in hyCS (see Methods); chimpanzee blue, human red; n as in (b). e, Single cell gene expression of PMP2. f, Expression of PMP2 in parental bulk time course; chimpanzee blue, human red; n as in (b). g, Allelic expression of PMP2 in hybrid bulk time course; chimpanzee allele blue, human allele red; n as in (b). Box plots in (b, d, f, g): center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range, dotted lines connect average values.
Extended Data Figure 9 |. Summary of ASE genes.
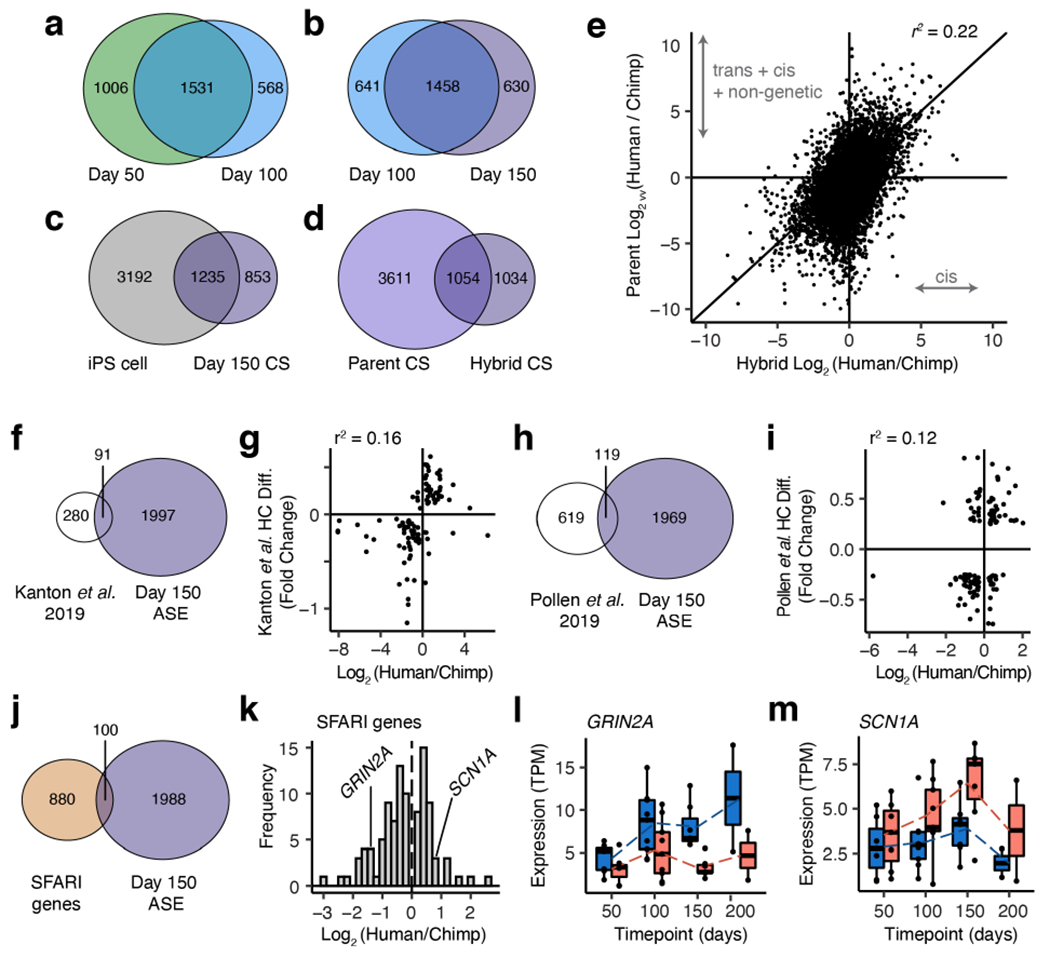
a-d, Overlap in genes with significant ASE (a,b,c; d hybrid) or differential expression (d parent) across bulk data sets; data in (d) are from day 150. e, Scatter plot showing differences in gene expression between parent lines (y-axis) versus between alleles in the hybrid (x-axis) at day 150; data are from bulk RNA-seq of 6 human, 5 chimpanzee and 7 hybrid CS samples, collected across 3 human, 3 chimpanzee and 3 hybrid iPS cell lines. f, Overlap between ASE genes and SFARI genes. g, ASE in SFARI genes from the overlapping genes in (f). h-i, Allelic expression (TPM, transcripts per million) over time in GRIN2A and SCN1A; human allele pink, chimpanzee allele blue; in order of time points n= 7, 9, 7, 2 hyCS samples (1-2 spheroids per sample) from 3 independent differentiations of 3 hyiPS cell lines. j, Filtering pipeline for prioritizing candidate genes. k, Scatter plot of hybrid ASE (x-axis) and parental differential expression (y-axis) for top candidate genes at day 150; n= 7 hyCS, 6 hCS, and 5 cCS samples (1-3 spheroids per sample) derived from 3 iPS cell lines per species and 2 independent differentiations per hiPS and ciPS cell line, n= 3 independent differentiations per hyiPS cell line. Box plots in (h, i): center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; dotted lines connect average values.
Extended Data Figure 10 |. Validation of SSTR2.
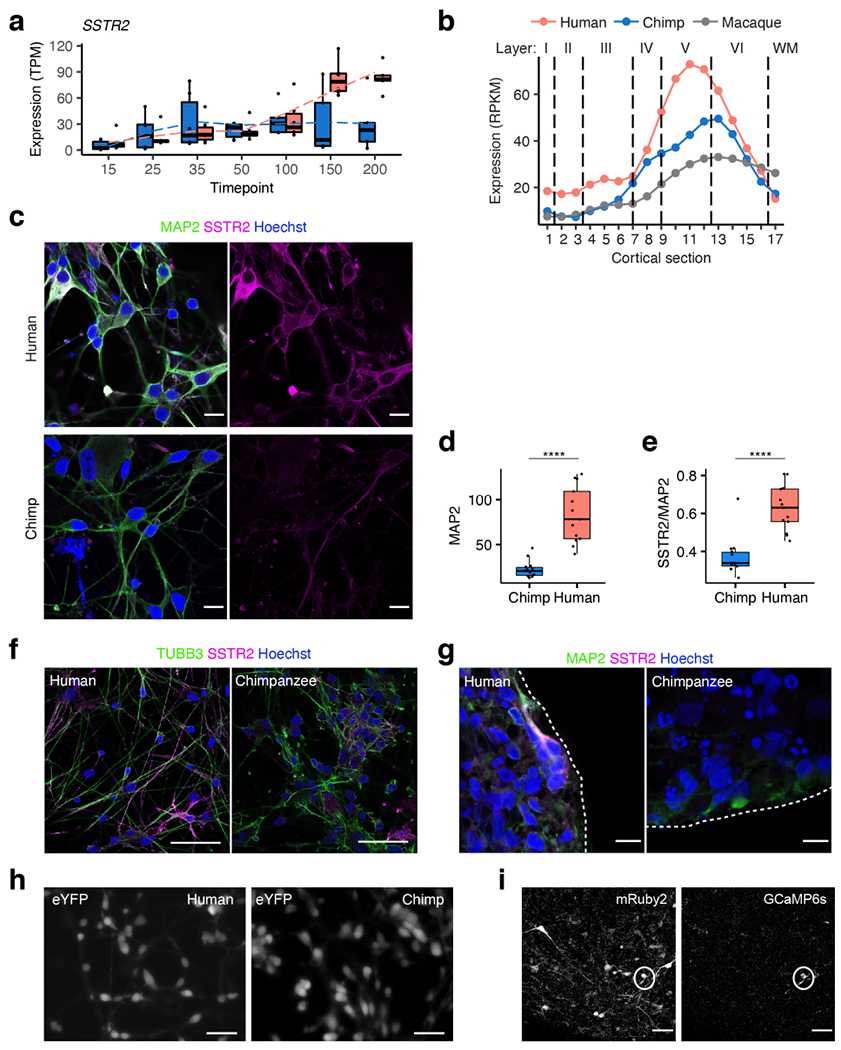
a, Expression of SSTR2 in parental bulk time course; chimpanzee blue, human red; in order of time points, n= 6, 6, 6, 6, 6, 6, 5 hCS and n= 6, 6, 6, 6, 5, 5, 5 cCS samples from 3 human and 3 chimpanzee iPS cell lines (1-2 replicates per cell line). b, Expression of SSTR2 across cortical sections in adult primate brain tissue (data from He et al.38); dotted lines indicate approximate boundaries of cortical layers; WM, white matter. c, Immunostaining for MAP2 (neuronal) and SSTR2 protein in dissociated hCS (H20682) and cCS (C3649) at day 225-250; right panels show SSTR2 only; 10 images were taken per sample and quantified. d, Quantification of fluorescence intensity (arbitrary units) of MAP2 for the images in (c); n= 13 cells for hCS, 14 cells for cCS; **** P<0.0001, two-tailed Mann-Whitney test. e, Quantification of fluorescence intensity (arbitrary units) of SSTR2 relative to MAP2 for the images in (c); n= 13 cells for hCS, 14 cells for cCS; **** P<0.0001, two-tailed Mann-Whitney test. f, Additional immunostaining for TUBB3 (neuronal) and SSTR2 in dissociated hCS (H20682) and cCS (C3649) at day 225-250; 10 images were taken per sample and quantified. g, Immunostaining for MAP2 and SSTR2 in whole hCS (H20961) and cCS (C3651) at day 160; imaging was reproduced across 3 human and two chimpanzee cell lines from one differentiation experiment with n= 3, 2, 3, 2, 3 images for lines H21792, H20682, H20961, C3649 and C3651, respectively. h, Representative still frame images of hCS (H20682) and cCS (C3649) derived neurons infected with AAV-DJ-hSyn1-eYFP; images are taken from one of the samples in Fig 4g-i; experiment was reproduced across 2 human and 1 chimpanzee cell line. i, Representative still frame images of hCS (H20682) infected with the viral vector co-encoding stable red fluorophore mRuby2 and genetically encoded calcium indicator GCaMP6s; images are taken from one of the samples in Fig. 4j-l; experiment was reproduced across 3 human and 3 chimpanzee cell lines. Box plots in (a, d, e): center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; dotted lines connect average values (a). Scale bars, 50 μm (f), 10 μm (c, g), 60 μm (h), 30 μm (i).
Supplementary Material
Supplementary Table 1 | Cell lines and assays.
Catalogue of all cell lines used in the study, details of hybrid fusion strategy, and details of how many cell lines and replicates per cell line were used in specific assays.
Supplementary Table 2 | Aneuploidies in hybrid samples.
Aneuploidies detected from RNA-sequencing data in bulk hyiPS and hyCS data.
Supplementary Table 3 | Genes with mapping bias in hyiPS cell data.
List of all genes that display mapping bias between GRCh38 and PanTro5 in data from iPS cells. LFC, log2[human/chimpanzee] from DESeq2 (see Methods).
Supplementary Table 4 | Allele specific expression in hyiPS cells (bulk RNA-seq, high-confidence genes).
Log2[human/chimpanzee] (LFC) and adjusted P-values for allele specific expression analysis in hyiPS cells. Genes have been filtered to remove those not annotated in both GRCh38 and PanTro5, genes that are too lowly expressed to estimate P-values, genes that display mapping bias, and genes on aneuploid or sex chromosomes. All values are from DESeq2 (see Methods) and data are provided for reads mapped separately to GRCh38 and PanTro5. P-values are from a likelihood ratio test and are corrected for multiple tests with the Benjamini-Hochberg method (FDR).
Supplementary Table 5 | Single cell cluster assignments (all cells).
Cluster assignments and UMAP coordinates from the R package Seurat (see Methods), as labelled in Extended Data Figure 5a, for all cells after filtering (n= 706 cells).
Supplementary Table 6 | Top genes for single cell clusters (all cells).
Top 100 cluster-specific genes per cluster for each group of cells defined in Extended Data Figure 5a. Log2[fold-change] (logFC) values and adjusted P-values are from a two-sided Wilcoxon Rank Sum test and are corrected for multiple tests within the R package Seurat (see Methods). Genes with logFC greater than 0.05 were removed.
Supplementary Table 7 | Single cell cluster assignments (neural cells).
Cluster assignments and UMAP coordinates from the R package Seurat (see Methods), as labelled in Figure 2c, for all neural cells after filtering (n= 368 cells).
Supplementary Table 8 | Top genes for single cell clusters (neural cells).
Top 100 cluster-specific genes per cluster for each group of cells defined in Figure 2c. Log2[fold-change] (logFC) values and adjusted P-values are from a two-sided Wilcoxon Rank Sum test and are corrected for multiple tests within the R package Seurat (see Methods). Genes with logFC greater than 0.05 were removed.
Supplementary Table 9 | Differential expression between hCS and cCS (bulk RNA-seq), per time point.
Log2[fold-change] (LFC) values and adjusted P-values for differential expression analysis of all genes between hCS and cCS. All values are from DESeq2 (see Methods), LFC represents log2[human/chimpanzee], and all data are from reads mapped to GRCh38. P-values are from a likelihood ratio test and are corrected for multiple tests with the Benjamini-Hochberg method (FDR).
Supplementary Table 10 | Genes with mapping bias in hyCS data (across all time points).
List of all genes that display mapping bias between GRCh38 and PanTro5 in data from hyCS. LFC, log2[human/chimpanzee] from DESeq2 (see Methods).
Supplementary Table 11 | Allele specific expression in hyCS per time point (bulk RNA-seq, high-confidence genes).
Log2[human/chimpanzee] (LFC) and adjusted P-values for allele specific expression analysis in hyCS at each time point. Genes have been filtered to remove those not annotated in both GRCh38 and PanTro5, genes that are too lowly expressed to estimate P-values, genes that display mapping bias, and genes on aneuploid or sex chromosomes. All values are from DESeq2 (see Methods) and data are provided for reads mapped separately to GRCh38 and PanTro5. P-values are from a likelihood ratio test and are corrected for multiple tests with the Benjamini-Hochberg method (FDR).
Supplementary Table 12 | WGCNA module assignments.
Gene co-expression modules (named according to color).
Supplementary Table 13 | Gene ontology enrichments for selected WGCNA modules.
List of gene ontology terms enriched in specific gene modules from WGCNA (see Methods). Top 10 significant GO terms are included for each module, except where there were less than 10 significant enrichments. P-values are computed from the hypergeometric distribution and are corrected for multiple tests (FDR).
Supplementary Table 14 | Top ASE genes at day 150.
List of top genes at day 150, shown in Extended Data Fig. 9k. LFC, log2[human/chimpanzee] from DESeq2 for reads mapped to GRCh38 (see Methods).
Acknowledgements
We thank H. Blau and G. Markov at Stanford University for advice on the hybridization experiments. We thank D. Bangs and J. Erdmann for assistance with iPS cell karyotyping. We thank R. Jones, S.D. Conley, and R. Sinha at Stanford University for assistance in constructing the single cell RNA-sequencing libraries. We thank the members of the Pasca and Fraser labs for advice and feedback on the manuscript. This work was supported by a Stanford Bio-X Interdisciplinary Initiatives Seed Grant (to S.P.P and H.B.F.), an NIH grant T32 GM007790 (supporting R.M.A), the Department of Defense National Defense Science and Engineering Graduate Fellowship (to R.M.A), the Stanford Center for Computational, Evolutionary and Human Genomics (to R.M.A.), NIH grant 2R01GM097171-05A1 (supporting H.B.F), the Stanford Medicine’s Dean’s Fellowship (to Y.M. and F.B.), the Stanford Medicine Maternal & Child Health Research Institute Postdoctoral Support Program (to Y.M. and F.B.), the American Epilepsy Society Postdoctoral Research Fellowship (to F.B.), the Stanford Wu Tsai Neurosciences Institute’s Big Idea Grants on Brain Rejuvenation and Human Brain Organogenesis (supporting S.P.P.), the Kwan Research Fund (supporting S.P.P.), the New York Stem Cell Foundation (supporting S.P.P.) and the Chan Zuckerberg Ben Barres Investigator Award (to S.P.P.). This study utilized cell lines derived from the Yerkes National Primate Research Center, which is supported by the National Institutes of Health, Office of Research Infrastructure Programs/OD [P51OD011132].
Footnotes
Competing interest declaration
Stanford University holds a patent covering the generation of region-specific brain organoids (U.S. Patent Serial No. 62/163,870;8) (S.P.P.).
Ethics statement
All experiments complied with all relevant guidelines and regulations. Approval for the derivation of human induced pluripotent stem (iPS) cell lines used in this study was granted by the University of Chicago Institutional Review Board, protocol 11-0524. Human donors in this study consented to the use of their cells (fibroblasts) to generate iPS cells for studies of evolution and cross-species comparisons, and to the generation of other cell types that would be derived from these iPS cells. Donors consented to the deposition of any resulting data from the study onto the Gene Expression Omnibus (GEO). Generation and differentiation of human-chimpanzee hybrid iPS cells was approved by the Stanford Stem Cell Research Oversight committee. The study also benefited from a consultation with the Stanford’s Benchside Ethics Consultation Service on the ethical aspects of the work. We note that these tetraploid cells are not approved for use in vivo or for attempting to generate an organism, which is unlikely biologically possible. We recommend that all future applications of these cells occur in close consultation with bioethicists.
Data and code availability statement
Raw and processed data are publicly available through the Gene Expression Omnibus under accession GSE144825. The alignment of human and chimpanzee genomes from Ensembl is available at https://ftp.ensembl.org/pub/release-84/maf/ensembl-compara/pairwise_alignments/homo_sapiens.GRCh38.vs.pan_troglodytes.CHIMP2.1.4.tar. The SFARI database is available at https://www.sfari.org/resource/sfari-gene/. All code for the described analyses of RNA-sequencing data and for making figures is publicly available at https://github.com/TheFraserLab/Agoglia_HumanChimpanzee2020. Correspondence and requests for materials should be addressed to S.P.P. (spasca@stanford.edu) and H.B.F. (hbfraser@stanford.edu).
References
- 1.Gallego Romero I et al. A panel of induced pluripotent stem cells from chimpanzees: a resource for comparative functional genomics. Elife 4, e07103, doi: 10.7554/eLife.07103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prescott SL et al. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 163, 68–83, doi: 10.1016/j.cell.2015.08.036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pasca SP The rise of three-dimensional human brain cultures. Nature 553, 437–445, doi: 10.1038/nature25032 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Muchnik SK, Lorente-Galdos B, Santpere G & Sestan N Modeling the Evolution of Human Brain Development Using Organoids. Cell 179, 1250–1253, doi: 10.1016/j.cell.2019.10.041 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qian X, Song H & Ming GL Brain organoids: advances, applications and challenges. Development 146, doi: 10.1242/dev.166074 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pollen AA et al. Establishing Cerebral Organoids as Models of Human-Specific Brain Evolution. Cell 176, 743–756.e717, doi: 10.1016/j.cell.2019.01.017 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanton S et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422, doi: 10.1038/s41586-019-1654-9 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Beneyto M, Morris HM, Rovensky KC & Lewis DA Lamina- and cell-specific alterations in cortical somatostatin receptor 2 mRNA expression in schizophrenia. Neuropharmacology 62, 1598–1605, doi: 10.1016/j.neuropharm.2010.12.029 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adori C et al. Critical role of somatostatin receptor 2 in the vulnerability of the central noradrenergic system: new aspects on Alzheimer’s disease. Acta Neuropathol 129, 541–563, doi: 10.1007/s00401-015-1394-3 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Ward MC et al. Silencing of transposable elements may not be a major driver of regulatory evolution in primate iPSCs. Elife 7, doi: 10.7554/eLife.33084 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prud’homme B, Gompel N & Carroll SB Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A 104 Suppl 1, 8605–8612, doi: 10.1073/pnas.0700488104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasca AM et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 12, 671–678, doi: 10.1038/nmeth.3415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lancaster MA et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379, doi: 10.1038/nature12517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camp JG et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A 112, 15672–15677, doi: 10.1073/pnas.1520760112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sloan SA et al. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 95, 779–790.e776, doi: 10.1016/j.neuron.2017.07.035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mora-Bermudez F et al. Differences and similarities between human and chimpanzee neural progenitors during cerebral cortex development. Elife 5, doi: 10.7554/eLife.18683 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otani T, Marchetto MC, Gage FH, Simons BD & Livesey FJ 2D and 3D Stem Cell Models of Primate Cortical Development Identify Species-Specific Differences in Progenitor Behavior Contributing to Brain Size. Cell Stem Cell 18, 467–480, doi: 10.1016/j.stem.2016.03.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amps K et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol 29, 1132–1144, doi: 10.1038/nbt.2051 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taapken SM et al. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat Biotechnol 29, 313–314, doi: 10.1038/nbt.1835 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Müller F-J et al. A bioinformatic assay for pluripotency in human cells. Nature methods 8, 315–317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin X et al. Engineering Stem Cell Organoids. Cell Stem Cell 18, 25–38, doi: 10.1016/j.stem.2015.12.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato T et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. 459, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Stingl J, Eaves CJ, Zandieh I & Emerman JT Characterization of bipotent mammary epithelial progenitor cells in normal adult human breast tissue. 67, 93–109 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Qiu X et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982, doi: 10.1038/nmeth.4402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H & Macklis JD Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci 14, 755–769, doi: 10.1038/nrn3586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen B, Khodadoust MS, Liu CL, Newman AM & Alizadeh AA Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol Biol 1711, 243–259, doi: 10.1007/978-1-4939-7493-1_12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Somel M et al. Transcriptional neoteny in the human brain. Proc Natl Acad Sci U S A 106, 5743–5748, doi: 10.1073/pnas.0900544106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. [DOI] [PMC free article] [PubMed]
- 29.Fraser HB Genome-wide approaches to the study of adaptive gene expression evolution: systematic studies of evolutionary adaptations involving gene expression will allow many fundamental questions in evolutionary biology to be addressed. Bioessays 33, 469–477, doi: 10.1002/bies.201000094 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Oberheim NA, Wang X, Goldman S & Nedergaard M Astrocytic complexity distinguishes the human brain. 29, 547–553 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Miller JA, Horvath S & Geschwind DH Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proceedings of the National Academy of Sciences 107, 12698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bozek K et al. Exceptional evolutionary divergence of human muscle and brain metabolomes parallels human cognitive and physical uniqueness. PLoS Biol 12, e1001871, doi: 10.1371/journal.pbio.1001871 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelley KW, Nakao-Inoue H, Molofsky AV & Oldham MC Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes. Nat Neurosci 21, 1171–1184, doi: 10.1038/s41593-018-0216-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basu SN, Kollu R & Banerjee-Basu S AutDB: a gene reference resource for autism research. Nucleic Acids Res 37, D832–836, doi: 10.1093/nar/gkn835 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sousa AMM, Meyer KA, Santpere G, Gulden FO & Sestan N Evolution of the Human Nervous System Function, Structure, and Development. Cell 170, 226–247, doi: 10.1016/j.cell.2017.06.036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujii Y et al. Somatostatin receptor subtype SSTR2 mediates the inhibition of high-voltage-activated calcium channels by somatostatin and its analogue SMS 201-995. FEBS Lett 355, 117–120, doi: 10.1016/0014-5793(94)01159-1 (1994). [DOI] [PubMed] [Google Scholar]
- 37.Liguz-Lecznar M, Urban-Ciecko J & Kossut M Somatostatin and Somatostatin-Containing Neurons in Shaping Neuronal Activity and Plasticity. Front Neural Circuits 10, 48, doi: 10.3389/fncir.2016.00048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He Z et al. Comprehensive transcriptome analysis of neocortical layers in humans, chimpanzees and macaques. Nat Neurosci 20, 886–895, doi: 10.1038/nn.4548 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Gokhman D et al. Human-chimpanzee fused cells reveal cis-regulation underlying skeletal evolution. Nature Genetics, accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 40.Yoon SJ et al. Reliability of human cortical organoid generation. Nat Methods 16, 75–78, doi: 10.1038/s41592-018-0255-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van de Geijn B, McVicker G, Gilad Y & Pritchard JK WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods 12, 1061–1063, doi: 10.1038/nmeth.3582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tehranchi A et al. Fine-mapping cis-regulatory variants in diverse human populations. Elife 8, doi: 10.7554/eLife.39595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Combs PA & Fraser HB Spatially varying cis-regulatory divergence in Drosophila embryos elucidates cis-regulatory logic. PLoS Genet 14, e1007631, doi: 10.1371/journal.pgen.1007631 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Birey F et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59, doi: 10.1038/nature22330 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53, doi: 10.1016/j.neuron.2015.11.013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–181, doi: 10.1038/nprot.2014.006 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Picelli S et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24, 2033–2040, doi: 10.1101/gr.177881.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502, doi: 10.1038/nbt.3192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eden E, Navon R, Steinfeld I, Lipson D & Yakhini Z GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. 10, 48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newman AM et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457, doi: 10.1038/nmeth.3337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasca SP et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 17, 1657–1662, doi: 10.1038/nm.2576 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 | Cell lines and assays.
Catalogue of all cell lines used in the study, details of hybrid fusion strategy, and details of how many cell lines and replicates per cell line were used in specific assays.
Supplementary Table 2 | Aneuploidies in hybrid samples.
Aneuploidies detected from RNA-sequencing data in bulk hyiPS and hyCS data.
Supplementary Table 3 | Genes with mapping bias in hyiPS cell data.
List of all genes that display mapping bias between GRCh38 and PanTro5 in data from iPS cells. LFC, log2[human/chimpanzee] from DESeq2 (see Methods).
Supplementary Table 4 | Allele specific expression in hyiPS cells (bulk RNA-seq, high-confidence genes).
Log2[human/chimpanzee] (LFC) and adjusted P-values for allele specific expression analysis in hyiPS cells. Genes have been filtered to remove those not annotated in both GRCh38 and PanTro5, genes that are too lowly expressed to estimate P-values, genes that display mapping bias, and genes on aneuploid or sex chromosomes. All values are from DESeq2 (see Methods) and data are provided for reads mapped separately to GRCh38 and PanTro5. P-values are from a likelihood ratio test and are corrected for multiple tests with the Benjamini-Hochberg method (FDR).
Supplementary Table 5 | Single cell cluster assignments (all cells).
Cluster assignments and UMAP coordinates from the R package Seurat (see Methods), as labelled in Extended Data Figure 5a, for all cells after filtering (n= 706 cells).
Supplementary Table 6 | Top genes for single cell clusters (all cells).
Top 100 cluster-specific genes per cluster for each group of cells defined in Extended Data Figure 5a. Log2[fold-change] (logFC) values and adjusted P-values are from a two-sided Wilcoxon Rank Sum test and are corrected for multiple tests within the R package Seurat (see Methods). Genes with logFC greater than 0.05 were removed.
Supplementary Table 7 | Single cell cluster assignments (neural cells).
Cluster assignments and UMAP coordinates from the R package Seurat (see Methods), as labelled in Figure 2c, for all neural cells after filtering (n= 368 cells).
Supplementary Table 8 | Top genes for single cell clusters (neural cells).
Top 100 cluster-specific genes per cluster for each group of cells defined in Figure 2c. Log2[fold-change] (logFC) values and adjusted P-values are from a two-sided Wilcoxon Rank Sum test and are corrected for multiple tests within the R package Seurat (see Methods). Genes with logFC greater than 0.05 were removed.
Supplementary Table 9 | Differential expression between hCS and cCS (bulk RNA-seq), per time point.
Log2[fold-change] (LFC) values and adjusted P-values for differential expression analysis of all genes between hCS and cCS. All values are from DESeq2 (see Methods), LFC represents log2[human/chimpanzee], and all data are from reads mapped to GRCh38. P-values are from a likelihood ratio test and are corrected for multiple tests with the Benjamini-Hochberg method (FDR).
Supplementary Table 10 | Genes with mapping bias in hyCS data (across all time points).
List of all genes that display mapping bias between GRCh38 and PanTro5 in data from hyCS. LFC, log2[human/chimpanzee] from DESeq2 (see Methods).
Supplementary Table 11 | Allele specific expression in hyCS per time point (bulk RNA-seq, high-confidence genes).
Log2[human/chimpanzee] (LFC) and adjusted P-values for allele specific expression analysis in hyCS at each time point. Genes have been filtered to remove those not annotated in both GRCh38 and PanTro5, genes that are too lowly expressed to estimate P-values, genes that display mapping bias, and genes on aneuploid or sex chromosomes. All values are from DESeq2 (see Methods) and data are provided for reads mapped separately to GRCh38 and PanTro5. P-values are from a likelihood ratio test and are corrected for multiple tests with the Benjamini-Hochberg method (FDR).
Supplementary Table 12 | WGCNA module assignments.
Gene co-expression modules (named according to color).
Supplementary Table 13 | Gene ontology enrichments for selected WGCNA modules.
List of gene ontology terms enriched in specific gene modules from WGCNA (see Methods). Top 10 significant GO terms are included for each module, except where there were less than 10 significant enrichments. P-values are computed from the hypergeometric distribution and are corrected for multiple tests (FDR).
Supplementary Table 14 | Top ASE genes at day 150.
List of top genes at day 150, shown in Extended Data Fig. 9k. LFC, log2[human/chimpanzee] from DESeq2 for reads mapped to GRCh38 (see Methods).
Data Availability Statement
Raw and processed data are publicly available through the Gene Expression Omnibus under accession GSE144825. The alignment of human and chimpanzee genomes from Ensembl is available at https://ftp.ensembl.org/pub/release-84/maf/ensembl-compara/pairwise_alignments/homo_sapiens.GRCh38.vs.pan_troglodytes.CHIMP2.1.4.tar. The SFARI database is available at https://www.sfari.org/resource/sfari-gene/. All code for the described analyses of RNA-sequencing data and for making figures is publicly available at https://github.com/TheFraserLab/Agoglia_HumanChimpanzee2020. Correspondence and requests for materials should be addressed to S.P.P. (spasca@stanford.edu) and H.B.F. (hbfraser@stanford.edu).