

Summary
Argyrins represent a family of cyclic octapeptides exhibiting promising immunomodulatory activity via inhibiting mitochondrial protein synthesis, which leads to reduced IL‐17 production by the T‐helper 17 cells. Argyrins are formed by a non‐ribosomal peptide synthetase (NRPS), originating from the myxobacterial producer strains Archangium gephyra Ar8082 and Cystobacter sp. SBCb004. In this work, a previously established heterologous production platform was employed to provide evidence of direct D‐configured amino acid incorporation by the argyrin assembly line. An adenylation domain of the argyrin NRPS was characterized and shown to have a high preference for D‐configured amino acids. Eight novel argyrin derivatives were generated via biosynthetic engineering of the heterologous production system. The system was also optimized to enable formation of methylated argyrin C and D derivatives with improved immunosuppressive activity compared with their unmethylated counterparts. Furthermore, the optimization of cultivation conditions allowed exclusive production of one major derivative at a time, drastically improving the purification process. Importantly, engineering of transcription and translation initiation resulted in a substantially improved production titre reaching 350–400 mg l−1. The optimized system presented herein thus provides a versatile platform for production of this promising class of immunosuppressants at a scale that should provide sufficient supply for upcoming pre‐clinical development.
In this study an adenylation domain of the argyrin NRPS was characterized and shown to have a high preference for D‐configured amino acids. Eight novel argyrin derivatives were generated via biosynthetic engineering of the heterologous production system. The system was also optimized to enable formation of methylated argyrin C and D derivatives with improved immunosuppressive activity compared to their unmethylated counterparts.
Introduction
Natural product‐based immunosuppressive agents have been used for decades in human medical applications, e.g., to prevent organ rejections after transplantation. Among various compound classes with immunomodulatory activity, the best known examples are cyclosporine (Canafax and Ascher, 1983), FK506 (Wallemacq and Reding, 1993) and rapamycin (Dumont and Su, 1995), the latter approved for application in humans by the FDA in 1999 (Mann, 2001). Recent results have highlighted the importance of argyrins, identifying them as non‐competitive inhibitors of the human immunoproteasome (Allardyce and Bell, 2019). They were also shown to exhibit strong immunosuppressive activity by targeting the mitochondrial elongation factor (EF‐G1) in yeast and mammalian cells, thereby reducing the IL‐17 production by the human T‐helper 17 cells (Almeida et al., 2021). In addition to the immunosuppressive activity, argyrin A induces apoptosis and blocks angiogenesis by preventing the destruction of the cyclin kinase inhibitor p27kip1 through proteasome inhibition (Nickeleit et al., 2008; Stauch et al., 2010). Those properties highlight the potential of argyrins as promising immunomodulatory drug candidates.
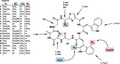
Argyrins are a family of cyclic octapeptides produced by the myxobacterial strains, Archangium gephyra Ar8082 and Cystobacter sp. SBCb004. Biosynthetically they are generated from amino acids by a non‐ribosomal peptide synthetase (NRPS) (Pogorevc et al., 2019b). NRPSs represent large multifunctional megasynthetases involved in the biosynthesis of numerous structurally diverse compounds, among them several marketed drugs, e.g., penicillin, vancomycin and cyclosporine (Finking and Marahiel, 2004; Süssmuth and Mainz, 2017). The corresponding genes, encoding the production machinery for the biosynthesis of a particular compound family, are usually clustered within the chromosome of the microbial producer strain and are referred to as the corresponding biosynthetic gene cluster (BGC). The Argyrin BGC from Cystobacter sp. SBCb004 consists of one operon including the five genes arg12345 (Pogorevc et al., 2019b). The genes arg2 and arg3 encode the two large NRPS subunits responsible for the biosynthesis of the core structure. arg1 encodes a radical S‐adenosyl‐L‐methionine (SAM)‐dependent methyltransferase catalysing the C‐methylation of the second L‐tryptophan and arg5 and arg4 encode a tryptophan‐2,3‐dioxygenase and an O‐methyltransferase responsible for oxygenation and subsequent O‐methylation of the same L‐tryptophan respectively (Fig. 1).
Fig. 1.
The argyrin core structure with highlighted amino acid substrates as they are incorporated by the corresponding NRPS modules. The structural diversity is introduced by the promiscuous NRPS modules (R1 and R2) and the highlighted tailoring enzymes Arg1, Arg4 and Arg5 (R3 and R4).
Although total synthesis approaches have already been developed for the generation of argyrins and novel derivatives (Ley and Priour, 2002; Bülow et al., 2010; Chen et al., 2014), the establishment of more efficient and cost‐effective supply routes is highly demanded to further investigate and develop this promising compound class. The BGC from Cystobacter sp. SBCb004 was recently used as a template for the development of a synthetically engineered heterologous production platform based on Myxococcus xanthus DK1622ΔmchA‐tet as host organism, allowing further exploitation of argyrins and novel derivatives thereof. This led to the identification of novel argyrin derivatives and enabled combined production titres of argyrins A and B up to 160 mg l−1 (Pogorevc et al., 2019b).
In this work, we optimized the previously reported heterologous production system by genetic engineering approaches to manipulate the transcription and translation initiation which led to an improved total argyrin production titre of 350–400 mg l−1. Importantly, we were able to direct the biosynthesis towards the production of single analogues with improved pharmaceutical properties. Coexpression of arg1 or arg451 led to heterologous production of methylated argyrins C/D, which were recently shown to exert improved immunosuppressive properties, compared with argyrins A/B (Almeida et al., 2021). Furthermore, engineering of the argyrin production profile enabled exclusive production of specific argyrin derivatives and thus drastically simplified the purification process.
Results and discussion
Argyrin production profile engineering
Production of argyrins C and D by coexpression of the radical SAM methyltransferase Arg1
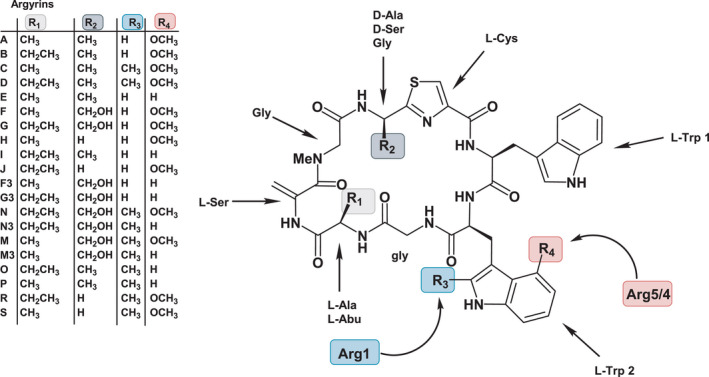
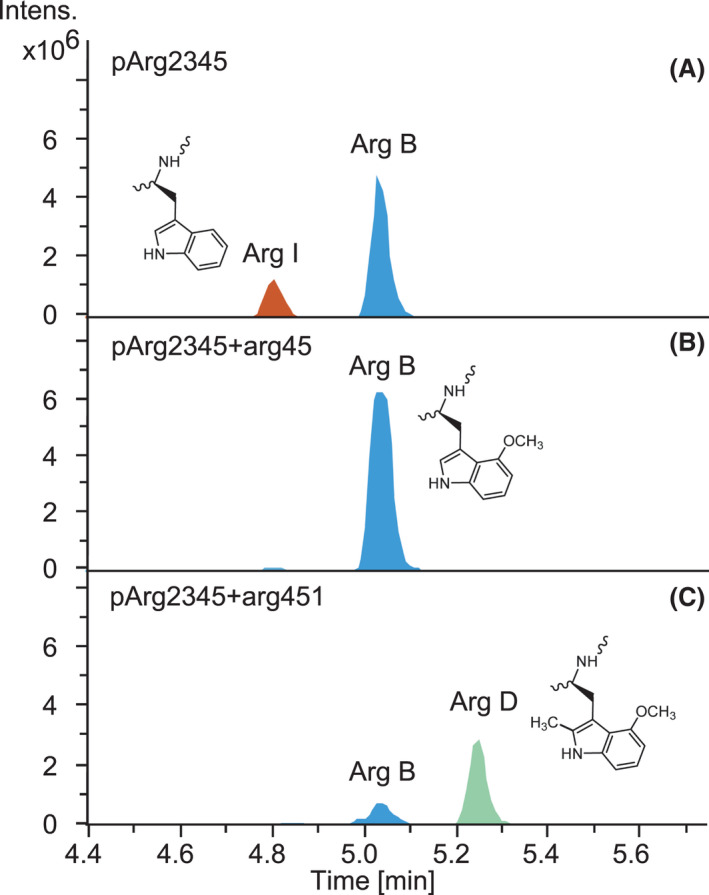
The arg1 gene was excluded from the argyrin BGC in our previous study (Pogorevc et al., 2019b) to simplify the production profile and avoid inefficient methylation by Arg1. In light of the novel immunomodulatory activities described for methylated argyrins (Almeida et al., 2021), we aimed to further improve the production of the methylated argyrin variants, such as argyrins C and D. Therefore, an expression vector for coexpression of the gene encoding the radical‐SAM methyltransferase Arg1 in the heterologous argyrin producer Mx. DK1622::pArg2345 (see Table S4 for full strain names) was designed. A previous gene miss‐annotation came to our attention which led to an extensive in silico analysis of the native producer’s (Cystobacter sp. SBCb004) genome sequence using FramePlot (Ishikawa and Hotta, 1999) and Basic Local Alignment Search Tool (BLAST) (Altschul et al., 1990) to correctly re‐annotate the arg1 gene sequence (see Experimental procedures). The identified missing piece of the arg1 gene was PCR amplified and cloned into the pGH‐arg1‐V1 plasmid obtained from a gene synthesis company. After subcloning of the reconstituted arg1 into the expression vector, the construct was integrated into the genome of Mx. DK1622::pArg2345 via homologous recombination and the production profile of Mx. DK1622::pArg2345‐arg1 was verified. HPLC‐MS analysis revealed similar production rates of argyrins A and B based on the MS peak area in the control strain and the mutant harbouring the arg1 gene copy (Fig. 2). Notably, the coexpression of arg1 led to the production of argyrins C and D as minor derivatives (Fig. 2). To investigate if the methylation efficiency can be improved to increase the conversion rate of argyrins A and B into their methylated counterparts (argyrins C and D), the production medium was supplemented with vitamin B12, an essential co‐factor for radical‐SAM enzymes (Pierre et al., 2012; Zhang and van der Donk, 2012; Kim et al., 2013). The HPLC‐MS analysis indeed revealed a full conversion of argyrins A and B into argyrins C and D respectively (Fig. 2).
Fig. 2.
Conversion of argyrins A and B into C and D after coexpression of arg1 in Mx. DK1622::pArg2345 and further improvement of argyrins C and D production by addition of vitamin B12. HPLC‐MS analysis of the fermentation broth extracts showing extracted ion chromatograms (EICs) [M + H]+ = 825.313 (Arg A), [M + H]+ = 839.329 (Arg B), [M + H]+ = 839.329 (Arg C) and [M + H]+ = 853.344 (Arg D). The full mutant names were abbreviated (see Table S4).
Argyrin production profile engineering by cultivation condition optimization
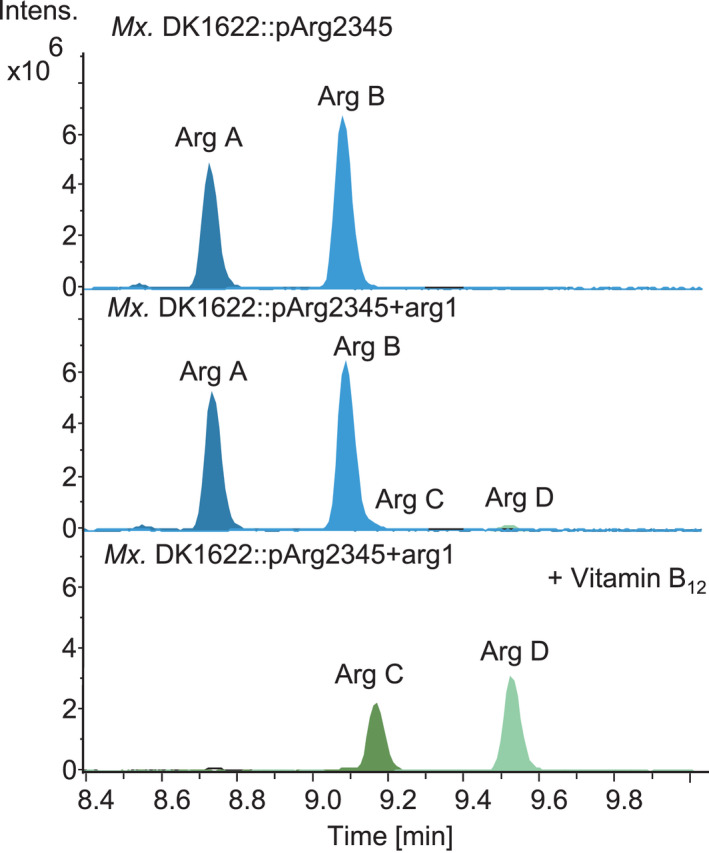
The high structural diversity of argyrins is caused by the tailoring reactions catalysed by Arg1, Arg4 and Arg5, combined with the relaxed substrate tolerance of the first and fourth modules of the NRPS. A complex mixture of numerous derivatives, produced by the native or the heterologous producers, poses a downstream processing challenge when trying to produce a specific derivative with high purity. We applied extensive medium optimization to decrease the number of produced derivatives and thus simplify the future purification process. The heterologous argyrin producer yields about the same amount of argyrins A and B when cultivated in CTT medium. In contrast, it has previously been noticed that the argyrin production profile shifts substantially in favour of argyrin A when the Mx. DK1622::pArg2345 is cultivated in M7/s4 medium, probably due to the low external supply of L‐α‐aminobutyric acid (L‐Abu) (Fig. 3A) (Pogorevc et al., 2019b). We were able to shift the production rate between argyrins A and B by cultivating the heterologous argyrin producer Mx. DK1622::pArg2345 in different media and/or by supplementing with 10mM L‐Abu. This led to a shift in the production profile towards a near exclusive production of argyrin B (Fig. 3A), supporting the hypothesis of a low external L‐Abu supply. This also indicates that the assembly line prefers incorporation of L‐Abu over L‐alanine, to yield argyrin B rather than argyrin A. To show that the shift from argyrin C to argyrin D can also be achieved by the supplementation of L‐Abu, the same cultivation experiments were performed while coexpressing the arg1 leading to a near exclusive production of argyrin D (Fig. 3B).
Fig. 3.
Argyrin production profile engineering through medium/cultivation optimization and arg1 coexpression. Variation in production of argyrins A and B in different media (A). Variation in production of argyrins C and D in different media (B). HPLC‐MS analysis of supernatant showing EICs [M + H]+ = 825.313 (Arg A), [M + H]+ = 839.329 (Arg B), [M + H]+ = 839.329 (Arg C) and [M + H]+ = 853.344 (Arg D). The full mutant names were abbreviated (see Table S4).
Evaluation of different promoter‐5’‐UTR systems and their impact on the argyrin production level
Promoters are the key regulators of gene expression that have the potential to increase or decrease secondary metabolite production levels (Ongley et al., 2013). There are only a handful of promoter systems which were successfully used for gene expression in myxobacteria, including PnptII , Ptet , Ppm and Pvan (Table S2). Most of those systems were initially developed and evaluated in different non‐myxobacterial host organisms and were later established in myxobacteria due to a lack of highly efficient myxobacterial promoters. PnptII and Pvan are commonly used for the activation of silent BGCs in terms of genome mining and also for heterologous BGC expression (Table 1). Besides the PIPTG , the Pvan is the only viable option for inducible gene expression (Iniesta et al., 2012). Notably, no other reliable inducible promoters were described for myxobacteria so far (Perlova et al., 2006). Since there was no available data for direct comparison of the gene expression or secondary metabolite production by BGCs driven by the aforementioned promoters in myxobacteria, we decided to test all commonly used promoter systems and evaluate their direct effect on the production level of argyrins with a high production titre being the main priority.
Table 1.
Heterologous production of PKS/NRPS‐derived natural products in M. xanthus.
| Compound | Native producer | Pathway type [size] | Promoter | Yield a | Ref |
|---|---|---|---|---|---|
| Alkypyrones | MCy9487 | PKS [6 kb] | Pvan | n.d. | Hug et al. (2019) |
| Argyrins A, B, I | Cystobacter sp. SBCb004 | NRPS [33 kb] | PnptII | 230 mg l−1 | Pogorevc et al. (2019b) |
| Argyrins A, B, I | Cystobacter sp. SBCb004 | NRPS [33 kb] | PnptII | 350–400 mg l−1 | This study |
| Argyrins A, B, I | Cystobacter sp. SBCb004 | NRPS [33 kb] | Pvan | 300 mg l−1 | This study |
| Argyrins A, B, I | Cystobacter sp. SBCb004 | NRPS [33 kb] | Pvan∆R | 250–300 mg l−1 | This study |
| Argyrins A, B, I | Cystobacter sp. SBCb004 | NRPS [33 kb] | Ptet | 50–100 mg l−1 | This study |
| Argyrins A, B, I | Cystobacter sp. SBCb004 | NRPS [33 kb] | Ptet∆R | 200–250 mg l−1 | This study |
| Argyrins A, B, I | Cystobacter sp. SBCb004 | NRPS [33 kb] | Ppm | 200 mg l−1 | This study |
| Bengamide | Myxococcus virescens | PKS/NRPS [25 kb] | PnptII | > 10 mg l−1 | Wenzel et al. (2015) |
| Corallopyronin A | Corallococcus coralloides | PKS/NRPS [65 kb] | PnptII | 37 mg l−1 | Sucipto et al. (2017) |
| Corallopyronin A | Corallococcus coralloides | PKS/NRPS [65 kb] | Pvan | 100 mg l−1 | Pogorevc et al. (2019a) |
| Cystobactamide Cys919‐1 | Cystobacter velatus Cbv34 | NRPS [52 kb] | Pvan | 8.1 mg l−1 | Groß et al. (2021) |
| Dawenol | Stigmatella aurantiaca | PKS [21 kb] | Native | n.d. | Oßwald et al. (2014) |
| Disorazol A2 | Sorangium cellulosum So ce12 | PKS/NRPS [58kb] | Ptet /Pcp25 | ~ 1 mg l−1 | Tu et al. (2016) |
| Epothilone | Sorangium cellulosum | PKS/NRPS [56 kb] | Native | 0.1–0.4 mg l−1 | Julien and Shah (2002); Lau et al. (2002); Oßwald et al. (2014) |
| Haliangicin | Haliangium ochraceum SMP‐2 | PKS [47.8 kb] | Native | 11 mg l−1 | Sun et al. (2016) |
| Myxochromide A | Myxococcus xanthus | PKS/NRPS [29 kb] | PnptII | ~ 500 mg l−1 | Yan et al. (2018b) |
| Myxochromide S | Stigmatella aurantiaca | PKS/NRPS [29 kb] | PnptII | > 500 mg l−1 | Fu et al. (2008) |
| Myxopyronin A | Myxococcus fulvus | PKS/NRPS [53 kb] | PnptII | 156 mg l−1 | Sucipto et al. (2017) |
| Myxothiazol | Stigmatella aurantiaca | PKS/NRPS [57 kb] | Ppm | 20 mg l−1 | Perlova et al. (2006) |
| Oxytetracycline | Streptomyces rimosus | PKS [32 kb] | Native | 10 mg l−1 | Stevens et al. (2010) |
| Pretubulysin | Cystobacter sp. | PKS/NRPS [40 kb] | Ptet | 0.2 mg l−1 | Chai et al. (2012) |
| PUFAs b | Aetherobacter fasciculatus | PKS/FAS [18 kb] | Ptet | ~ 1 mg CDW−1 | Gemperlein et al. (2014) |
| Pyxidicyclines | Pyxidicoccus fallax And48 | PKS [33 kb] | Pvan /PnptII | n.d. | Panter et al. (2018) |
| Vioprolide B, D | Cystobacter violaceus | NRPS [56 kb] | Ptet | 80 mg l−1 | Yan et al. (2018a) |
n.d., not determined.
Based on shake flask experiments.
Polyunsaturated fatty acids.
The promoter harbouring constructs were designed based on the four different promoter systems PnptII , Pvan , Ppm and Ptet . For Pvan and Ptet , we designed versions with and without repressor gene, resulting in a total of six new synthetic promoters. To minimize the translational influence of the native 5’‐untranslated regions (5’‐UTRs) downstream of each promoter, a synthetically engineered 5’‐UTR optimized for high expression in M. xanthus DK1622 was designed in silico. This synthetic 5’‐UTR was used instead of the native 5’‐UTR region in all six designed promoter harbouring constructs which were generated by DNA synthesis. A detailed overview of the promoter fragments is illustrated in Fig. S3 and described in Experimental procedures.
The original PnptII from the pArg2345‐V1‐BsaI construct was exchanged via restriction hydrolysis and DNA ligation with the six synthesized promoter sequences. The generated expression constructs with the different promoter‐5’‐UTRs (see Experimental procedures) were transferred into M. xanthus DK1622ΔmchA‐tet and integrated into the genome via homologous recombination. The cultivation of the generated strains was performed in M7/s4 medium supplemented with respective argyrin precursor amino acids to reduce the negative impact on the production caused by the lack of nutrients. It was previously shown that the argyrin production titre in amino acid supplemented M7/s4 medium increases substantially compared with the CTT or M7/s4 medium without amino acid supplementation (Pogorevc et al., 2019b). HPLC‐MS analysis of the fermentation extracts revealed that argyrins were indeed produced by all mutants, however, with substantial differences in the production levels depending on the promoter system. In this cultivation experiment, the supplementation with the amino acid mixture also increased the production of the previously described argyrin I, a derivative lacking the methoxy group introduced by the Arg5 and Arg4 enzymes (Fig. 1) (Pogorevc et al., 2019b).
Quantification of argyrins A, B and I revealed that the highest total argyrin production and an increase by about 40%, compared with the control, was achieved with the use of PnptII after exchanging the native 5’‐UTR with the synthetic one. This indicates that an efficient translation process was probably the bottleneck in the control system (Fig. 4). The expression of the argyrin BGC regulated by PtetΔR and PvanΔR resulted in higher argyrin production levels compared with the control system with PnptII and the native 5’‐UTR sequence. The lowest production was achieved by the Ppm , which, despite the optimized 5’‐UTR region, resulted in a lower argyrin production than observed in the control (Fig. 4).
Fig. 4.
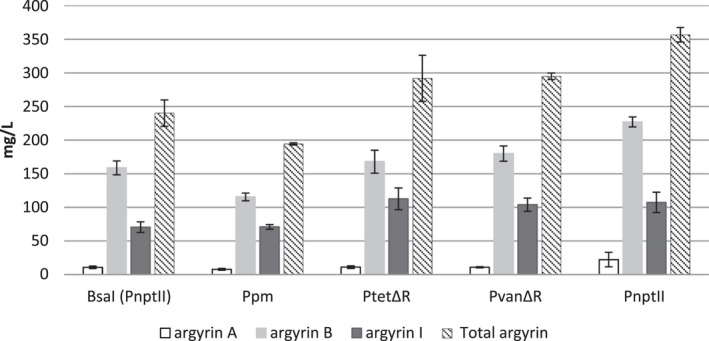
Production levels of argyrins A, B and I evaluated in heterologous argyrin producers with different promoter systems in comparison with control (PnptII‐native 5’UTR). Samples were analysed by HPLC‐MS, and the production titres of argyrins A, B and I, as well as the total argyrin amount, were quantified as described in Experimental procedures.
Second, we evaluated the production by the mutants harbouring the argyrin BGC controlled by the inducible promoter systems (Ptet and Pvan with the respective repressor genes), under induced and uninduced conditions. The versions of PtetΔR and PvanΔR without repressor were also evaluated in parallel, without addition of inducers, to allow direct comparison.
The induction of the Ptet with tetracycline did not result in increased production levels; however, the deletion of the repressor gene in PtetΔR led to a significant increase in argyrin production titre (Fig. 5). This hints that the induction of the Ptet system in M. xanthus DK1622 is not working efficiently. As expected, in case of the Pvan , the induction with 1 mM vanillate led to a near sixfold increase in production of argyrin compared with the uninduced control, reaching a similar production titre as achieved by the PvanΔR (Fig. 5). It appears that deletion of repressors in Ptet ‐ and Pvan ‐inducible promoter systems has no negative effect on the argyrin production titre. Promoters without repressors can thus be used to simplify argyrin production and eliminate the need for supplementation with inducers. Notably, argyrins do not appear to be auto‐toxic to the producer strain at the observed production titres. The attempt to evaluate the MIC at 650 mg l−1 of argyrin B resulted in argyrin precipitation and growth of M. xanthus cells on the argyrin precipitate. This correlates with our in silico analysis which showed that the exchange of S459 in the argyrin susceptible EF‐G of Pseudomonas aeruginosa with an R459 (found at this position in the EF‐G of M. xanthus) could lead to a significant clash and thereby prevent binding of argyrin B to the EF‐G of M. xanthus (Figs S5 and S6). However, the presence of repressors should be re‐evaluated for use in different, especially uncharacterized, secondary metabolite BGCs as the produced compounds could exert auto‐toxicity towards the producer strain. In such cases, inducible promoters can prove advantageous since they at least enable the detection of the auto‐toxic factor after the induction of the BGC of interest and in some cases allow production of the toxic metabolite in the presence of an adsorbent resin (Panter et al., 2018).
Fig. 5.
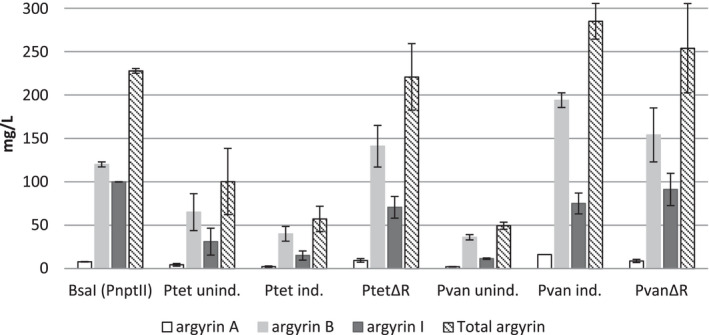
Production levels of argyrins A, B and I evaluated in heterologous argyrin producers with different promoter systems. The yields detected in mutants harbouring the argyrin BGC under the control of Ptet or Pvan were evaluated under induced and uninduced conditions. Repressor deletion mutants and control (PnptII‐native 5’UTR) were evaluated under uninduced conditions. Samples were analysed by HPLC‐MS and production titres of argyrins A, B and I were quantified as described in Experimental procedures.
From the results we conclude that the highest argyrin production titre was achieved upon expression of arg2345 under the control of the synthetic 5’‐UTR and the PnptII , with the PtetΔR and PvanΔR providing slightly lower argyrin production titres. A similar production titre was achieved with the induced Pvan in the presence of vanR. These results might serve as an orientation for future experiments to increase the production titre of other myxobacterial secondary metabolites. However, in addition to the promoter efficiency, the 5’‐UTR region can have a significant effect on protein expression (Dvir et al., 2013) influencing metabolite titres, as it was shown in this study. The improved production system generated in this study reaches a combined total production of argyrins at 350–400 mg l−1 and ranks with the most successful examples in heterologous expression studies on myxobacterial megasynthetases in M. xanthus (Table 1). We also attempted to further improve the argyrin production titre by expressing two copies of the same argyrin BGCs in one heterologous producer strain. However, the growth of the resulting mutant strains was inconsistent in these experiments and the results showed no obvious advantage of expressing two BGCs over a single BGC copy where stability of the genotype might be a complicating factor (data not shown).
Resolving the biosynthetic tailoring bottleneck
As mentioned above, supplementation of M7/s4 medium with amino acids led to a significant increase in total argyrin production including the titre of argyrin I, the non‐tailored precursor of argyrin B. This indicates that the catalytic capability of Arg5 (tryptophan‐2,3‐dioxygenase) and Arg4 (hydroxyindole O‐MT) reaches a limit where no more argyrin can be modified. To facilitate conversion of argyrin I to its methoxylated counterparts (argyrin B or D) we generated and overexpressed two separate expression constructs harbouring additional copies of the tailoring genes (arg45 or arg451 operons) regulated by P nptII in Mx. DK1622::pArg2345. The expression constructs were integrated into the host genome via the mx8 phage integrase. The HPLC‐MS analysis of the Mx. DK1622::arg2345‐arg45 production profile revealed almost complete conversion of argyrin I to argyrin B (Fig. 6). Coexpression of the arg451, on the other hand, revealed a complete conversion of argyrin I to argyrin B; however, not all argyrin B could further be methylated to argyrin D by a single copy of arg1 (Fig. 6). Additionally, after a visual inspection of the respective crude extracts we have observed a high degree of precipitation in the samples produced by the strains harbouring the arg1 gene copy. We thus assumed that argyrin D is poorly soluble in methanol. Consequently, the methanol‐dissolved crude extracts were dried and re‐dissolved in the same volume of DMSO, which resulted in no observable precipitate and indeed in a subsequent apparent increase in argyrin D was detected in the MS (Fig. S4).
Fig. 6.
Production profiles of the producer strains. Mx. DK1622::pArg2345 (A), after coexpression of arg45 (B) and after coexpression of arg451 (C). HPLC‐MS analysis of the crude extracts showing EICs [M + H]+ = 839.329 (Arg B), [M + H]+ = 809.318 (Arg I) and [M + H]+ = 853.344 (Arg D). The full names of the mutant strains were abbreviated (see Table S4).
Explaining the origin of D‐configured amino acids incorporated by the fourth module
As previously reported, the alanine (resulting in R2 = CH3 as shown in Fig. 1) and serine (R2 = CH2OH) units incorporated by the fourth module of the argyrin NRPS exhibit D‐configuration in mature argyrin derivatives. According to the currently known argyrins, alanine seems to be the preferred substrate, whereas serine and glycine (R2 = H) were only observed in minor derivatives. In most D‐amino acid‐containing non‐ribosomal peptides, L‐configured amino acids are initially activated by the A domain and subsequently epimerized by a downstream E domain or a dual condensation–epimerization domain (Marahiel and Stachelhaus, 1997; Rausch et al., 2007). This seems to also be the case in the first module of the argyrin NRPS, which activates an L‐amino acid substrate, epimerizes it by the action of the epimerization (E) domain and subsequently processes it by the downstream D‐specific C domain [DCL type according to Rausch et al. (Rausch et al., 2007)]. D‐amino acid–activating A domains are thus uncommon or poorly characterized as only a few cases have been reported thus far (Wu et al., 1987; Shi et al., 2016; Kaniusaite et al., 2021). In a recent study, five A domains located upstream of an E domain in the assembly line were shown to exert the ability to activate D‐configured amino acids in vitro (Kano et al., 2019). In contrast to the first module of the argyrin NRPS, the fourth module does not harbour an E domain for the generation of a D‐configured substrate from an A domain‐activated L‐amino acid. Thus, the origin of D‐configured amino acids at the fourth position in the argyrin structure remained unresolved and it was previously hypothesized that the downstream C domain could be responsible for their epimerization. However, this assumption could not be supported by in silico analysis according to Rausch et al. (Rausch et al., 2007) as the characteristic C domain motifs present in the C domains required for generation and/or processing of D‐configured amino acids could not be identified (Pogorevc et al., 2019b). Due to the superior solubility of D‐serine‐containing argyrin derivatives and the resulting advantageous pharmaceutical properties, the elucidation of this biosynthetic step remained of high interest.
We aimed to test if D‐configured amino acids are directly accepted by the A domain of the fourth module in the argyrin NRPS, rather than L‐configured amino acids being incorporated and subsequently epimerized. Since D‐serine‐containing argyrins are produced as minor derivatives, we assumed that supplementation of the medium with D‐serine might boost their production in case D‐serine is directly incorporated. To this end, the strains Mx. DK1622::pArg2345 and Mx. DK1622::pArg2345‐arg1 were cultivated in production media (M7/s4 supplemented with argyrin precursor amino acids) supplemented with either L‐serine or D/L‐serine. HPLC‐MS and MS/MS analyses of the culture extracts showed a significant increase in production of D‐Abu (R1 = CH2CH3) and D‐serine (R2 = CH2OH) containing argyrin derivatives G and G3, for strains cultivated in the D/L‐serine supplemented medium compared with the strains cultivated in L‐serine supplemented medium (Fig. 7 and Table S5). It is therefore likely that the D‐configured amino acids are directly incorporated by the fourth module of the argyrin NRPS, which would explain the absence of the epimerization domain in this module (Pogorevc et al., 2019b). However, the required isomerase in the natural producer remains unknown (see below). Cultivation of Mx. DK1622::pArg2345‐arg1 led to the detection of the novel argyrins N and N3 (Fig. 7), which carry an additional methyl group introduced by Arg1, compared with argyrins G and G3, as confirmed by the MS2 fragmentation (Table S5). Even though the argyrins N and N3 were not detected before, we also searched for them in the extract of the Mx. DK1622::pArg2345‐arg1 cultivated in the L‐serine supplemented medium. We indeed found very low amounts based on the MS peak intensity (Fig. 7). Cultivation in M7/s4 medium without addition of L‐Abu, but with supplementation of D‐serine, resulted in the detection of the L‐alanine (R1 = CH3) and D‐serine (R2 = CH2OH) containing argyrins M and M3 after HPLC‐MS analysis of the respective crude extracts (Table S5). Furthermore, the coexpression of arg1, medium variation and high production titres in this experiment led to increased production and detection of additional novel methylated argyrins O, P, R and S as confirmed by the MS2 fragmentation (Table S5).
Fig. 7.
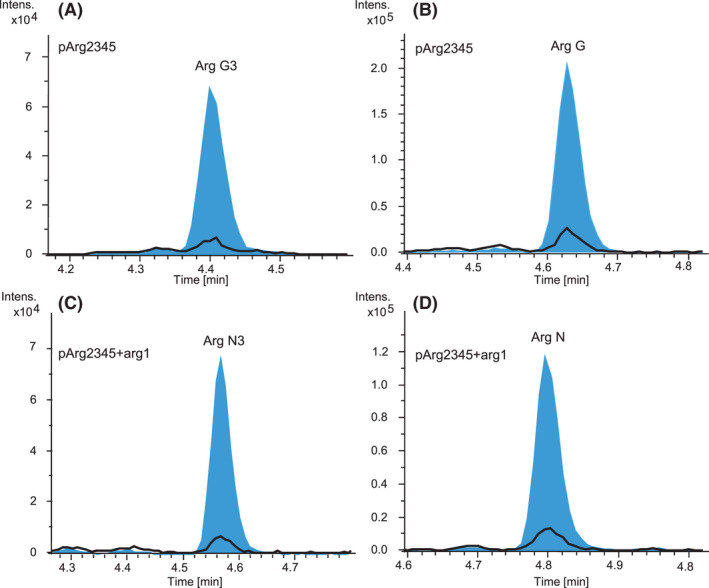
EICs of argyrin G3 [M + H]+ = 825.313 (A), argyrin G [M + H]+ = 855.324 (B), argyrin N3 [M + H]+ = 839.329 (C) and argyrin N [M + H]+ = 869.339 (D) produced by cultivation in the medium supplemented with L‐serine (black) and with D/L‐serine (blue).
To gain more insight into the specificity of the A domain responsible for the activation of the fourth module intermediates, we expressed the corresponding A‐PCP part of the Arg3 protein in E. coli. We then performed an in vitro characterization of the A domain using the malachite green phosphate assay according to the manufacturers protocol and as described by McQuade et al. (2009) as well as in vitro loading of the PCP domain and analysis by HPLC‐MS. The analysis revealed that the A domain is highly specific for D‐alanine and slightly less for D‐serine, whereas L‐configured amino acids were activated very poorly and only after an extended period of time (Fig. S8). Next, we performed in vitro loading assays by incubation of the A‐PCP bidomain with L‐alanine, L‐serine, D‐alanine, D‐serine or glycine and analysed the results by protein MS (Fig. 8). When performing the reaction with 100 µM final amino acid concentration, we observed loading of D‐alanine and D‐serine (Fig. 8C,E), whereas L‐alanine, L‐serine or glycine loading could not be observed. Interestingly, we also detected double loading of the D‐alanine (Fig. 8C), which was very surprising. This phenomenon of dual activity of an A domain, performing amino acid activation and amide bond formation, has previously been observed in coumermycin (Schmutz et al., 2003), novobiocic acid (Steffensky and Li, 2000), pacidamycin (Zhang et al., 2011) and streptothricin (Maruyama et al., 2012). In the latter case, the reaction is iterative and multiple incorporated building blocks can also be found in the final compound. This is not the case in argyrin and we can only assume that the C domain in the fourth module of the NRPS is selective for single PCP‐bound amino acids or that the kinetics of the A domain is different in vivo. When the assay was performed with higher amino acid concentration (1mM), we have also observed triple loading of D‐alanine, double loading of D‐serine and glycine as well as single loading of L‐alanine. The loading of L‐serine could also not be observed in this case (Fig. S7). The generated data are consistent with results observed in the malachite green assay, confirming that the A domain is highly D‐alanine and D‐serine selective.
Fig. 8.
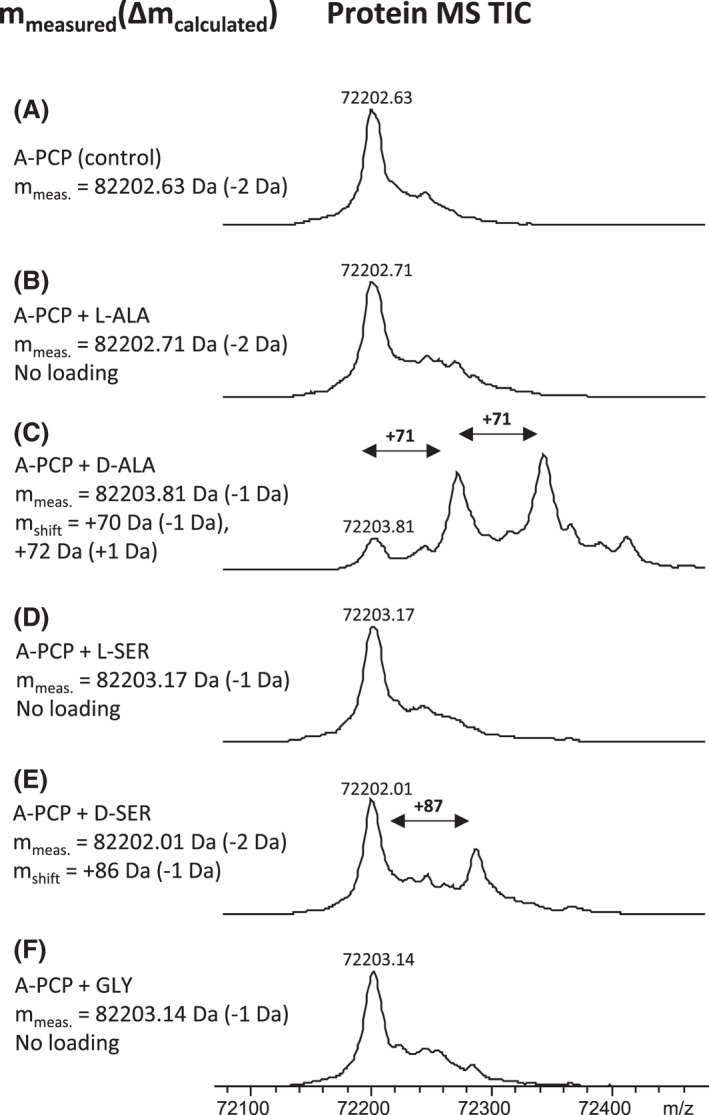
Substrate specificity of the A4 domain of the argyrin NRPS. Observed mass shifts in deconvoluted protein MS TICs reveal loading of D‐alanine (C) and D‐serine (E), onto the PCP. L‐alanine (B), L‐serine (D) and glycine (F) were not accepted by A‐PCP (control: A).
The results presented in this section indicate that the fourth module of the argyrin NRPS and the remaining downstream NRPS machinery favour incorporation and processing of D‐serine over L‐serine as long as the supply of D‐serine is sufficient. A very limited production of D‐serine‐containing argyrin derivatives (R2 = CH2OH) in the absence of the external D‐serine supply probably indicates a lack of a dedicated serine racemase in the genome of M. xanthus. This hypothesis was supported by a BLAST search in M. xanthus, using a serine racemase from Roseobacter litoralis Och 149 (Kubota et al., 2016) as the template, which found no close serine racemase homologues. The question that arises here is where the small amount of D‐serine originates from in case it is not externally added to the medium. We assume that after the depletion of D‐serine only D‐alanine is incorporated by the argyrin NRPS instead. Indeed, the supply of D‐alanine could be ensured by an alanine racemase encoded in the genome of M. xanthus DK1622, which could be confirmed by a BLAST search (GenBank accession: SDW08429.1). Alanine racemases catalyse racemization between L‐ and D‐alanine and are commonly found in bacteria as D‐alanine is an essential component of the bacterial cell wall (Strych et al., 2000; Liu et al., 2018). However, various alanine or broad‐spectrum racemases have also previously been shown to act promiscuously and catalyse L‐ to D‐serine conversion (Esaki and Walsh, 1986; Asano and Endo, 1988). The trace amounts of D‐serine‐containing argyrin derivatives that can be detected even if the medium is not supplemented by D‐serine could thus potentially originate from the medium components or from the activity of potential racemases with broad substrate spectrum present in the genome.
Conclusion
In this study we achieved conversion of argyrins A and B to their methylated counterparts, argyrins C and D, by reconstitution of the radical SAM‐dependent methyltransferase Arg1. This is especially important as argyrins C and D recently showed improved immunomodulatory activity compared with the unmethylated argyrins A and B (Almeida et al., 2021). Additionally, the cultivation conditions were adapted to allow exclusive production of specific argyrin derivatives by exploitation of the gatekeeper domain promiscuity in modules 1 and 4 of the argyrin NRPS. This broad substrate promiscuity of certain A and C domains should be studied further and could be employed in the future to, e.g. enable precursor‐directed biosynthesis for production of more and novel argyrins. By supplementing the production medium with D‐serine, we were able to shed light on the substrate preference of the fourth module of the argyrin NRPS. This experiment simultaneously resulted in an improved production of D‐serine‐containing derivatives and also led to the identification of eight novel argyrin derivatives. Furthermore, we were able to show that the A domain from the fourth module of the argyrin NRPS is indeed highly selective for D‐configured alanine and serine residues, by protein expression and in vitro characterization. In addition to increased production titres of argyrins A and B, improved production conditions in this work led to a substantial increase in the precursor product argyrin I, which could be converted into argyrin B or D by coexpression of additional arg45 or arg451 copies respectively. Notably, an expression of a single arg1 gene copy in case of the arg451 coexpression did not suffice to fully convert argyrin B to argyrin D. Therefore, an additional copy of arg1 might be added in the future to resolve this bottleneck. Furthermore, the total production of argyrin was significantly improved by implementation and optimization of different promoter systems. The library of evaluated promoters provides a valuable source of information which will be useful in future studies with the aim to optimize the heterologous production of other secondary metabolites in M. xanthus DK1622. Overall, the herein presented improved argyrin production platform enables the production of single derivatives at nearly 400 mg l−1. Since argyrins recently showed very promising potential as immunomodulators, this platform will certainly serve as a highly versatile tool to meet the supply–demand required for further preclinical testing and for production of novel argyrin derivatives in the future.
Experimental procedures
Construction and engineering of plasmids
Routine handling of nucleic acids, such as isolation of plasmid DNA, restriction endonuclease hydrolysis, DNA ligation and other DNA manipulations, was performed according to standard protocols (Sambrook and Russell, 2001). E. coli HS996 (Invitrogen, Waltham, MA, USA) was used as host for standard cloning experiments. E. coli strains were cultured in LB medium [1% tryptone, 0.5% yeast extract and 0.5% NaCl (1.5% agar)] at 30–37°C (and 200 rpm) overnight. Antibiotics were used as selection markers at the following final concentrations: 100 μg ml−1 ampicillin, 50 μg ml−1 kanamycin, 12 μg ml−1 tetracycline and 50 μg ml−1 zeocin. Transformation of E. coli strains was achieved via electroporation in 0.1‐cm‐wide cuvettes at 1250 V, a resistance of 200 Ω and a capacitance of 25 μF. Plasmid DNA was either purified by standard alkaline lysis (Sambrook and Russell, 2001), by using the GeneJet Plasmid Miniprep Kit (Thermo Fisher Scientific, Waltham, MA, USA), or the NucleoBond PC100 kit (Macherey Nagel, Düren, Germany). Restriction endonucleases, alkaline phosphatase (FastAP) and T4 DNA ligase were obtained from Thermo Fisher Scientific. Oligonucleotides used for PCR and Sanger sequencing were obtained from Sigma‐Aldrich and are listed in Table S1. PCR reactions were carried out in a Mastercycler® pro (Eppendorf SE, Hamburg, Germany) using Phusion™ High‐Fidelity, Taq DNA polymerase (Thermo Fisher Scientific) or Dream Taq DNA polymerase (Thermo Fisher Scientific) according to the manufacturer's protocol. For Taq and Dream Taq polymerase‐based PCR experiments, the initial denaturation was 5 min at 95°C, followed by 30 cycles of denaturation (30 s, 95°C), annealing (30 s, 53–64°C), elongation (1 kb min−1, 72°C) and final extension (10 min, 72°C). For Phusion™ polymerase‐based PCR experiments, the initial denaturation was 2 min at 98°C, followed by 30 cycles of denaturation (20 s, 98°C), annealing (25 s, 53–64°C) and elongation (0.5 kb min−1, 72°C); and final extension (10 min, 72°C). PCR products or DNA fragments from restriction hydrolysis were purified from agarose gel after agarose gel electrophoresis and isolated using the NucleoSpin® Gel and PCR Clean‐up (Macherey‐Nagel) or peqGold Gel Extraction (Peqlab). Red/ET recombineering experiments for plasmid modifications (Zhang et al., 1998) were performed according to the manufacturers’ protocol (Gene Bridges GmbH, Heidelberg, Germany) using the strain E. coli GB05‐red. After selection with suitable antibiotics, clones harbouring correct recombination products were identified by plasmid isolation followed by an independent hydrolysis with different endonucleases (leading to characteristic DNA band pattern in agarose‐GE). Synthetic DNA fragments were obtained from ATG:biosynthetics GmbH and delivered in pGH‐based standard cloning vectors (Table S2). Details on the construction of plasmids generated in this study are given in Table S3.
Construction of pUC18‐zeo‐Hom‐arg1B expression construct
The primers dpo‐pUC18‐F and dpo‐pUC18‐R (Table S1) were used to amplify a 731 bp DNA fragment from pUC18, containing the origin of replication (ori), via PCR using Phusion polymerase. The respective PCR fragment was ligated with the chemically synthesized zeo‐Hom‐MCS fragment after restriction hydrolysis with PmeI and SpeI restriction endonucleases, generating the pUC18‐zeo‐Hom‐MCS expression vector. The primers dpo‐arg1F and dpo‐arg1R (Table S1) were used to amplify a 238 bp arg1 extension fragment from Cystobacter sp. SBCb004 genomic DNA via PCR using Phusion polymerase. Since no obvious ribosomal binding site (RBS) sequence could be identified upstream of the arg1, an RBS from mchA [AAGGAGG (Fu et al., 2008)] was introduced via sequence extension of the forward primer (Table S1). The resulting PCR fragment was ligated into the pGH‐arg1‐V1 plasmid after restriction hydrolysis with NdeI and EcoRI, generating pGH‐arg1B, harbouring the reconstituted arg1 sequence. The pGH‐arg1‐V1 plasmid was obtained by gene synthesis and harbours a truncated version of the arg1 gene without the 5’ terminus. The 2047 bp arg1 gene fragment was released from pGH‐arg1B plasmid and inserted into previously constructed pUC18‐zeo‐Hom‐MCS expression vector by ligation after restriction hydrolysis using NdeI and BglII, yielding the pUC18‐zeo‐Hom‐arg1B expression construct (Table S3).
Construction of pUC18‐Zeo‐mx8‐PnptII‐arg45 and pUC18‐Zeo‐mx8‐PnptII‐arg451 expression constructs
The DNA fragments arg1 (2054 bp) and arg45 (2272 bp) were amplified by PCR using Phusion polymerase, pUC18‐Zeo‐Hom‐arg1 and pArg2345‐V1‐BsaI plasmids as templates and dpo‐arg1‐2F/dpo‐arg1‐2R and dpo‐arg4‐5‐F/dpo‐arg4‐5‐R as primer pairs (Table S1) respectively. The arg45 fragment was cloned into the pUC18‐Zeo‐Hom‐arg1 plasmid via restriction hydrolysis with NdeI and NsiI and DNA ligation. The generated plasmid pUC18‐Zeo‐mx8‐nptII‐arg45 and the arg1 PCR fragment were then hydrolysed with NsiI and BglII and ligated to yield the pUC18‐Zeo‐mx8‐nptII‐arg451 expression construct.
Design and construction of new synthetic promoter systems to replace the P ntpII promoter in the argyrin expression construct
To evaluate the influence of other promoter systems on the production titre of argyrin, six new expression constructs with different promoter‐5’‐untranslated region (5’‐UTR) combinations were designed. To minimize the transcriptional/translational influence of the native 5’‐UTRs downstream of each promoter, a synthetically engineered 31‐bp‐long 5’‐UTR (TACGAGAGCAAAAACGAGGAGAGGAGAAGAT) optimized for expression in M. xanthus DK1622 was designed using the RBS calculator tool (Salis and Mirsky, 2009; Espah Borujeni and Channarasappa, 2014), chemically synthesized and used to replace the native 5’‐UTR region. Each synthetic fragment consisted of specific promoter followed by the 5’‐UTR region and the first 61 bp of the downstream arg2 gene and a unique StuI restriction endonuclease recognition sequence/site (R‐site) at the 3’ end. For cloning purposes, all constructs were also flanked by a SwaI R‐site at the 5’ end, allowing integration into the pArg2345‐V1‐BsaI expression construct as outlined in the Fig. S3. To design the nptII‐synRBS‐arg2 synthetic fragment, based on the PnptII of the neomycin phosphotransferase II resistance gene from Tn5 (Beck et al., 1982), we used the same promoter sequence as originally used in the pArg2345‐V1‐BsaI expression construct. The only modification was the replacement of the 5'‐UTR region with the new, synthetic one and the introduction of the flanking restriction sites for the cloning purposes. The sequence of Pvan (Iniesta et al., 2012) was used to design the Pvan‐synRBS‐arg2 fragment and only the first 100 bp of the original Pvan promoter sequence was implemented to design the Pvan‐wo_vanR‐synRBS‐arg2 fragment with deleted repressor sequence in an attempt to make it function as a constitutive promoter system. The same approach was used in case of Ptet system (Chai et al., 2012), to design Ptet‐synRBS‐arg2 and Ptet‐wo_tetR‐synRBS‐arg2 constructs. The first 63 bp of the tet promoter sequence was used for the promoter version lacking the associated repressor gene. In case of the Ppm , originating from Pseudomonas putida, the improved version of the promoter system, generated by random mutagenesis approach (Bakke et al., 2009), was used to design the Ppm‐synRBS‐arg2 sequence. For assembly purposes, the StuI R‐site present in the promoter sequence had to be removed by exchange of a single nucleotide. The Ppm normally acts as an inducible promoter in pseudomonads; however, in M. xanthus, it was shown to work constitutively (Perlova et al., 2006). The promoter fragments with the lengths of 233 bp (Ppm ), 803 bp (Ptet ), 169 bp (PtetΔR ), 245 bp (PnptII ), 1145 bp (Pvan) and 200 bp (PvanΔR ) were released from the respective gene synthesis vectors and ligated into the pArg2345‐V1‐BsaI expression vector after restriction hydrolysis with SwaI and StuI.
Transformation and chromosomal integration of expression constructs into M. xanthus DK1622
According to a previously established electroporation procedure for M. xanthus DK1622 (Kashefi and Hartzell, 1995), the host strain Mx. DK1622::pArg2345 (Pogorevc et al., 2019b) was transformed with the generated expression construct pUC18‐zeo‐Hom‐arg1B (Table S3). M. xanthus DK1622 mutants were cultivated at 30°C in CTT medium [1% casitone, 10 mM Tris buffer pH 7.6, 1 mM KH2PO4 pH 7.6 and 8 mM MgSO4 (1.5% agar) with final pH 7.6]. For liquid cultures, the strains were grown in Erlenmeyer flasks on an orbital shaker at 180 rpm for 2–3 days. For selection of M. xanthus mutants, 50 μg ml−1 kanamycin and 50 μg ml−1 zeocin were used. After electroporation, the transformed cells were re‐suspended in 1 ml CTT medium and incubated for 6 h under constant shaking. Afterward, the transformants were mixed with 3 ml of CTT soft agar (CTT with only 7.5 g l−1 agar) and poured onto a CTT agar plate. After hardening of the soft agar layer with embedded transformant cells, the plates were incubated at 30°C for 5 days. Embedding in the soft agar prevented swarming of the transformant colonies over the agar surface, allowing the isolation of single clones and transfer onto a fresh CTT agar plate. Correct chromosomal integration of the arg1 expression construct into the downstream locus of the previously integrated argyrin BGC (Pogorevc et al., 2019b) or the argyrin BGC expression constructs into the tetR locus of the M. xanthus DK1622ΔmchA‐tet mutant via homologous recombination was verified by PCR (Fig. S1). Transformant cells (1 cm2) were scratched from the plate, re‐suspended in 50 µl of water and subsequently lysed by incubating at 95°C for 30 min prior of being added to the PCR reaction (‘colony PCR’). For each expression construct, correct chromosomal integration was confirmed using two primer combinations, revealing PCR products of the expected sizes: P5/P6 (1261 bp) and P7/P8 (1401 bp) for the arg1 expression construct and P1/P2 (1458 bp) and P3/P4 (1461 bp) for argyrin BGC expression constructs. Genomic DNA of Mx. DK1622::pArg2345 or M. xanthus DK1622ΔmchA‐tet isolated using Qiagen Puregene Core Kit A was used as negative control. The complementary experiment using primers P5/P8 or P1/P4 revealed a 1112 bp PCR products for Mx. DK1622::pArg2345 or a 1461 bp PCR product for M. xanthus DK1622ΔmchA‐tet respectively. The same primer pairs yielded no PCR product in the expression strains.
Correct chromosomal integration of plasmids integrated by the mx8 integrase was performed using two primer combinations, revealing PCR products of the expected sizes: P9/P10 (403 bp) and P11/P12 (427 bp). Genomic DNA of Mx. DK1622::pArg2345 was used as negative control. The complementary experiment using primers P9/P12 revealed a 449 bp PCR product for Mx. DK1622::pArg2345, but not for the expression strains (Fig. S2). PCR reactions were performed according to the above described conditions, using Taq polymerase; for primer sequences, see Table S1.
Cultivation conditions of argyrin producer strains
To vary the production between different argyrin derivatives (e.g. argyrins A, B, C and D), strains Mx. DK1622::pArg2345 and Mx. DK1622::pArg2345‐arg1 were cultivated in CTT and M7/s4 medium (0.5% soy flour, 0.5% corn starch, 0.2% glucose, 0.1% yeast extract, 0.1% MgSO4 x 7 H2O, 0.1% CaCl2 x 2 H2O, 1% HEPES, with final pH 7.4 and supplemented with 0.1 mg l−1 of vitamin B12 and 5 mg l−1 of FeCl3 after autoclaving) with and without supplementation of 10 mM L‐Abu. To evaluate the effect of different expression constructs on the argyrin production profile, the heterologous producers with replaced promoter were used for parallel cultivation in the M7/s4 production medium supplemented with amino acids, calculated to the final concentration of 5 mM L‐serine, 5 mM L‐cysteine, 5 mM L‐alanine, 10 mM L‐tryptophan, 10 mM glycine and 10 mM L‐Abu, before autoclaving. In case of inducible promoters (Ptet or Pvan ), the respective cultures were supplemented with 0.7 µg ml−1 tetracycline or 1 mM vanillate respectively. To verify if D‐alanine is directly activated by the argyrin NRPS, argyrin producer strains Mx. DK1622::pArg2345, Mx. DK1622::pArg2345‐arg45, Mx. DK1622::pArg2345‐arg1 and Mx. DK1622::pArg2345‐arg451 were cultivated in the M7/s4 medium supplemented with 5 mM L‐serine or D/L‐serine in addition to other argyrin substrate amino acids mentioned above. Furthermore, Mx. DK1622::pArg2345‐arg1 was also cultivated in M7/s4 medium supplemented with argyrin substrate amino acids (as described for the promoter screening experiment) and D/L‐serine without the addition of L‐Abu. Strains were inoculated from cryo stocks and grown on CTT agar plates supplemented with kanamycin 50 µg ml−1 (Mx. DK1622::pArg2345) or with kanamycin 50 µg ml−1 and zeocin 50 µg ml−1 (Mx. DK1622::pArg2345‐arg1) for several days until the plates were mostly overgrown with swarming cells. All of the cells were scraped from the plates to inoculate M7/s4 preculture medium (50 ml medium in 300 ml Erlenmeyer flask) and the seed cultures were cultivated at 30°C, 180 rpm on an orbital shaker (Infors HT) for 48 h. Five millilitre of the preculture was used to inoculate 50 ml of the M7/s4 production medium in which the strain was grown under the same conditions for 6 days. All cultures were supplemented with suitable antibiotics as mentioned above and production cultures were additionally supplemented with 4% Amberlite XAD‐16 resin. All strains were initially cultivated in biological triplicates and technical triplicates of one clone were cultivated in the follow‐up experiments. The cultivations took 6–8 days before harvesting by centrifugation at 8000 rpm for 15 min, and after the supernatant was removed the pelleted cells with Amberlite XAD‐16 resin were extracted with 50 ml of ethyl acetate twice. The extracts were filtered and dried in round bottom flasks on a rotary evaporator. The crude extracts were dissolved in 1 ml of methanol and subjected to HPLC‐MS analysis.
Analysis of the argyrin production by HPLC‐MS
Argyrin samples were routinely analysed on a Dionex Ultimate 3000 RSLC system using a Waters BEH C18 column (50 × 2.1 mm, 1.7 µm) equipped with a Waters VanGuard BEH C18 1.7 µm guard column. Separation of 1 µl sample (crude extract 1/5 dilutions in methanol) was achieved by a linear gradient from (A) H2O + 0.1% FA to (B) ACN + 0.1% FA at a flow rate of 600 µl min−1 and a column temperature of 45°C. Gradient conditions were as follows: 0–0.5 min, 5% B; 0.5–18.5 min, 5–95% B; 18.5–20.5 min, 95% B; 20.5–21 min, 95–5% B; and 21–22.5 min, 5% B. UV spectra were recorded by a DAD in the range from 200 to 600 nm. The LC flow was split to 75 µl min−1 before entering the Bruker Daltonics maXis 4G hr‐qToF mass spectrometer using the Apollo II ESI source. Mass spectra were acquired in centroid mode ranging from 150 to 2500 m/z at a 2 Hz full scan rate. Mass spectrometry source parameters were set to 500 V as end plate offset; 4000 V as capillary voltage; nebulizer gas pressure 1 bar; dry gas flow of 5 l min−1 and a dry temperature of 200°C. Ion transfer and quadrupole settings were set to Funnel RF 350 Vpp; Multipole RF 400 Vpp as transfer settings and ion energy of 5 eV as well as a low mass cut of 300 m/z as Quadrupole settings. Collision cell was set to 5.0 eV and pre pulse storage time was set to 5 µs. Spectra acquisition rate was set to 2 Hz. Calibration of the maXis4G qTOF spectrometer was achieved with sodium formate clusters before every injection to avoid mass drifts. All MS analyses were acquired in the presence of the lock masses C12H19F12N3O6P3, C18H19O6N3P3F2 and C24H19F36N3O6P3 which generate the [M + H]+ ions of 622.028960, 922.009798 and 1221.990638. The corresponding MS2 method operating in automatic precursor selection mode picks up the two most intense precursors per cycle, applies smart exclusion after five spectra and performs CID and MS/MS spectra acquisition time ramping. CID energy was ramped from 35 eV for 500 m/z to 45 eV for 1000 m/z and 60 eV for 2000 m/z. MS full scan acquisition rate was set to 2 Hz, and MS/MS spectra acquisition rates were ramped from 1 to 4 Hz for precursor Ion intensities of 10 kcts to 1000 kcts.
To quantify argyrin production levels, the following HPLC‐MS method was applied. Separation was performed on a Dionex UltiMate 3000 RSLC system equipped with a Waters reversed phase UPLC column (Acquity UPLC BEH C18 1.7 μm; 2.1 × 100 mm) using a linear gradient with solvent A (water + 0.1% formic acid) and B (acetonitrile + 0.1% formic acid) at a flow rate of 600 μl min−1 and 45°C. The gradient was initiated by a 0.5 min isocratic step with 5% B followed by an increase to 15% B within 1 min, 50% B within 11.5 min and 95% B within 1 min, which was kept for 1 min, before decreasing back to initial conditions of 5% B within 0.3 min and was kept for 1.7 min. HPLC was coupled to a Bruker Daltonics ion trap mass spec ‘Amazon speed’ system. Mass spectra were acquired in positive ionization mode with a range 200–2500 m/z at a resolution of R = 30000. Identities of the argyrins were confirmed by comparing both retention time and MS2 fragmentation pattern with authentic standards.
Quantification was performed using Bruker Daltonics Quant analysis software version 4.4. The following method was used to quantify the corresponding derivatives; argyrin I (retention time 9.83 min, MS2 fragments: 707.2; 724.2), argyrin A (retention time 10.05 min, MS2 fragments: 737.3; 754.3), argyrin B (retention time 10.60, MS2 fragments: 737.3; 754.3), argyrin C (retention time 10.71 min, MS2 fragments: 751.3; 768.3) and argyrin D (retention time 11.28 min, MS2 fragments: 751.3; 768.3).
Expression and purification of A‐PCP bidomain
To express the A‐PCP bidomain from the fourth module of the argyrin NRPS (arg3 nucleotide 1306‐3262), the respective 2014‐bp‐long DNA fragment was amplified by PCR from the pArg2345‐V1‐BsaI expression plasmid using the primer pair dpo‐A‐PCP‐TEV‐BamHI‐F2/dpo‐A‐PCP‐HindIII‐R (Table S1). The product was hydrolysed using BamHI/HindIII and ligated into similarly digested pETDuet™‐1_MXAN_3118 expression vector (unpublished data), which already contained a gene‐encoding MbtH‐like protein homologue from M. xanthus (NCBI locus tag: MXAN_RS15115), to be coexpressed [without the histidine (his) tag] together with the A‐PCP protein. The resulting pETDuet™‐1_MXAN_3118‐argM4‐A‐PCP plasmid was transformed into an E. coli BL21 (DE3) for protein expression and a single colony was picked to inoculate 100 ml LB broth containing 100 mg ml−1 ampicillin, which was incubated at 37°C overnight with shaking at 200 rpm. The overnight culture was used to inoculate 6 litres of fresh LB medium containing 100 mg ml−1 ampicillin. After growing at 37°C until OD600 of 0.5–0.6, the expression of the A‐PCP was induced by addition of 0.4 mM IPTG. The culture was further incubated at 16°C for 24 h. The next day, the cells were pelleted by centrifugation at 8000 rpm. Cell pellets were re‐suspended in 80 ml of the lysis buffer (20 mM TrisHCl pH 8, 500 mM NaCl, 0.1% triton X‐100 (V/V), 20 mM imidazole, 1 mM DTT) and lysed using a cell disruptor (Constant systems) at 30.000 psi. The cell lysate was centrifuged at 20 000 rpm for 20 min at 4°C. The supernatant was filtered using the MF‐Millipore 0.45 µm filter and afterwards loaded onto a pre‐equilibrated (lysis buffer) 5 ml Histrap HP column (GE healthcare, Chicago, IL, USA) and was washed with 20 column volumes of lysis buffer. The bound his‐tagged A‐PCP protein was eluted with 5 column volumes of buffer containing 250 mM imidazole. The eluent was injected onto a desalting column (Desalt 16/10, GE healthcare) and the chromatography was conducted using the buffer without imidazole or detergent [20 mM TrisHCl pH 8, 200 mM NaCl and 1 mM tris(2‐carboxyethyl)phosphine (TCEP)] at a flow rate of 10 ml min−1. The protein in the fractions was monitored by UV absorbance at 280 nm and the protein containing fractions were pooled. The protein containing eluate was digested with a tobacco etch virus (TEV) protease in a mass‐to‐mass ratio of 1:10 overnight at 4°C to cleave the his tag. Digested protein was then passed over a second Histrap HP column, and the flow through was collected and concentrated to around 7 ml using an Amicon Ultracel‐50 centrifugal filter unit with a molecular weight cut‐off of 50 kDa. The concentrated protein was loaded onto a gel filtration column (HiLoad 16/600 Superdex 200 pg, GE healthcare) pre‐equilibrated with the gel filtration buffer (20 mM TrisHCl pH 8, 200 mM NaCl and 1 mM TCEP). The fractions of the highest purity were pooled together and concentrated to 5 mg ml−1. The identity of the protein was confirmed using protein MS.
Malachite green assay
Characterization of the adenylation domain specificity was performed by malachite green colorimetric assay as previously described (McQuade et al., 2009). The reaction mixture contained 1.6 µl of 1 M Tris‐HCl buffer (pH 8); 3.2 µl of 5 M NaCl, 8 µl of 100 mM MgCl2; 0.5 µl of pyrophosphatase; 3.2 µl of 10 µM expressed protein (A‐PCP); 0.2 µl of 100 mM ATP and 60.9 µl of H2O. The reaction was finally started by addition of 2.4 µl of 25 mM amino acid solution. We have tested the reaction with L‐ and D‐alanine, L‐ and D‐serine and glycine. All reactions were prepared in triplicates in a 96 well plate and incubated either 1 h at 30°C or overnight at room temperature with shaking at 300 rpm. After the incubation, 20 µl of malachite green phosphate assay kit reagent a and b mixture (100 : 1) from Sigma‐Aldrich was added and the absorbance was measured at 620 nm with a TECAN Infinite® M Nano plate reader directly and again after 30 min.
Protein loading assay
The protein loading assay was performed by preparing a reaction mixture containing 10 µM expressed protein (A‐PCP), 10 mM MgCl2, 2 mM ATP, 20 mM Tris‐HCl (pH 8), 200 mM NaCl and 1 mM, 100 µM, 10 µM or 1 µM amino acid solution (L‐ or D‐alanine, L‐ or D‐serine or glycine). The mixture was incubated overnight at room temperature with shaking at 300 rpm before the samples were subjected to HPLC‐MS analysis.
Analysis of the in vitro protein reactions by HPLC‐MS
All ESI‐MS measurements were performed on a Dionex Ultimate 3000 RSLC system using an Aeris Widepore XB‐C8, 150 x 2.1 mm, 3.6 µm dp column (Phenomenex, Torrance, CA, USA). Separation of 2 µl sample was achieved by a linear gradient from (A) H2O + 0.1% FA to (B) ACN + 0.1% FA at a flow rate of 300 µl min−1 and 45°C. The gradient was initiated by a 1.0 min isocratic step at 2% B, followed by an increase to 75% B in 10 min to end up with a 3 min step at 75% B before re‐equilibration with initial conditions. UV spectra were recorded by a DAD in the range from 200 to 600 nm. The LC flow was split to 75 µl min−1 before entering the maXis 4G hr‐ToF mass spectrometer (Bruker Daltonics, Bremen, Germany) using the standard Bruker ESI source. In the source region, the temperature was set to 200°C, the capillary voltage was 4000 V, the dry‐gas flow was 5.0 l min−1 and the nebulizer was set to 1.0 bar. Mass spectra were acquired in positive ionization mode ranging from 150 to 2500 m/z at 2 Hz scan rate. Protein masses were deconvoluted by using the Maximum Entropy algorithm (Copyright 1991‐2004 Spectrum Square Associates).
Conflict of interest
None declared.
Supporting information
Fig. S1. Genotypic verification of integration of all argyrin cluster versions into the heterologous host M. xanthus DK1622 ΔmchA‐tet. Primer sets used for genotypic verification, their binding sites and amplicon sizes are illustrated. Nucleotide sequences of the primers used are listed in the Table S1. Mch stands for myxochromide.
Fig. S2. Genotypic verification of insert integration into the mx8 attb1 site of the Mx. DK1622::pArg2345. Primer sets used for genotypic verification, their binding sites and amplicon sizes are illustrated. Nucleotide sequences of the primers used are listed in the Table S1.
Fig. S3. Assembly strategy for the exchange of the promoter systems upstream of the arg2345 operon. The same strategy was applied in case of all promoter fragments. The figure shows P tet sequence as an example.
Fig. S4. Base peak chromatograms (BPCs) of the Mx. DK1622::pArg2345‐arg451 MeoH (blue) and DMSO (black) crude extracts.
Fig. S5. Clustal Omega alignment of Pseusomonas aeruginosa PAO1 and M. xanthus DK1622 FusA1 sequences. The difference S459R (numbering according to the sequence of P. aeruginosa FusA1) is highlighted with an arrow.
Fig. S6. Visualization of argyrin B and the residue S459 of the P. aeruginosa FusA1 protein binding pocket (left). If the same residue is exchanged with R459 found in the M. xanthus DK1622 FusA1 sequence, this introduces a significant clash (right). The picture is based on the co‐crystal structure (PDB Code 4FN5).
Fig. S7. Substrate specificity of the A4 domain of the argyrin NRPS. Observed mass shifts in deconvoluted protein MS TICs reveal loading of D‐alanine (C) and D‐serine (E) onto the PCP. L‐alanine (B), L‐serine (D) and glycine (F) were not accepted by the A‐PCP (control: A).
Fig. S8. Increase in absorbance relative to the control (protein without amino acid) after the malachite green assay reactions incubated at 30°C for 1 h (left) and overnight at room temperature (right).
Table S1. List of primers used in this study.
Table S2. Gene synthesis constructs obtained from ATG:biosynthetics GmbH.
Table S3. Constructs generated in this study.
Table S4. Abbreviated and full names of the M. xanthus mutants generated in this study.
Table S5. HPLC‐MS data of argyrin derivatives detected in the heterologous expression strains. Above the bold line are previously described derivatives and below the line are their methylated (R3=CH3) counterparts newly detected in this study.
Acknowledgements
This work was supported by DZIF. We would like to thank Daniel Sauer, Sophia Panter and Matej Rebek for excellent technical assistance. We would also like to thank Dr. Asfandyar Sikandar and Jan Dastbaz for excellent assistance and advice regarding protein expression and in vitro assays as well as Jan Dastbaz for providing the pETDuet™‐1_MXAN_3118 plasmid. Finally, we would like to thank Dr. Sebastian Groß for proofreading this manuscript.
Microbial Biotechnology (2021) 15(1), 353–369
Funding Information
This work was supported by DZIF.
Data Availability Statement
The sequences of the expression constructs have been deposited in the GenBank database under the following accession numbers: MZ634479‐MZ634488.
References
- Allardyce, D.J. , Bell, C.M. , and Loizidou, E.Z. (2019) Argyrin B, a non‐competitive inhibitor of the human immunoproteasome exhibiting preference for β1i. Chem Biol Drug Des 94: 1556–1567. [DOI] [PubMed] [Google Scholar]
- Almeida, L. , Dhillon‐LaBrooy, A. , Castro, C.N. , Adossa, N. , Carriche, G.M. , Guderian, M. , et al. (2021) Ribosome‐targeting antibiotics impair T cell effector function and ameliorate autoimmunity by blocking mitochondrial protein synthesis. Immunity 54: 68–83.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. , and Lipman, D.J. (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Asano, Y. , and Endo, K. (1988) Amino acid racemase with broad substrate specificity, its properties and use in phenylalanine racemization. Appl Microbiol Biotechnol 29: 523–527. [Google Scholar]
- Bakke, I. , Berg, L. , Aune, T.E. , Brautaset, T. , Sletta, H. , Tondervik, A. , and Valla, S. (2009) Random mutagenesis of the Pm promoter as a powerful strategy for improvement of recombinant gene expression. Appl Environ Microbiol 75: 2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, E. , Ludwig, G. , Auerswald, E.A. , Reiss, B. , and Schaller, H. (1982) Nucleotide sequence and exact localization of the neomycin phosphotransferase gene from transposon Tn5. Gene 19: 327–336. [DOI] [PubMed] [Google Scholar]
- Bülow, L. , Nickeleit, I. , Girbig, A.‐K. , Brodmann, T. , Rentsch, A. , Eggert, U. , et al. (2010) Synthesis and biological characterization of argyrin F. ChemMedChem 5: 832–836. [DOI] [PubMed] [Google Scholar]
- Canafax, D.M. , and Ascher, N.L. (1983) Cyclosporine immunosuppression. Clin Pharm 2: 515–524. [PubMed] [Google Scholar]
- Chai, Y. , Shan, S. , Weissman, K.J. , Hu, S. , Zhang, Y. , and Müller, R. (2012) Heterologous expression and genetic engineering of the tubulysin biosynthetic gene cluster using Red/ET recombineering and inactivation mutagenesis. Chem Biol 10: 361–371. [DOI] [PubMed] [Google Scholar]
- Chen, C.H. , Genapathy, S. , Fischer, P.M. , and Chan, W.C. (2014) A facile approach to tryptophan derivatives for the total synthesis of argyrin analogues. Org Biomol Chem 12: 9764–9768. [DOI] [PubMed] [Google Scholar]
- Dumont, F.J. , and Su, Q. (1995) Mechanism of action of the immunosuppressant rapamycin. Life Sci 58: 373–395. [DOI] [PubMed] [Google Scholar]
- Dvir, S. , Velten, L. , Sharon, E. , Zeevi, D. , Carey, L.B. , Weinberger, A. , and Segal, E. (2013) Deciphering the rules by which 5'‐UTR sequences affect protein expression in yeast. Proc Natl Acad Sci USA 110: E2792–E2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esaki, N. , and Walsh, C.T. (1986) Biosynthetic alanine racemase of Salmonella typhimurium: purification and characterization of the enzyme encoded by the alr gene. Biochemistry 25: 3261–3267. [DOI] [PubMed] [Google Scholar]
- Espah Borujeni, A. , Channarasappa, A.S. , and Salis, H.M. (2014) Translation rate is controlled by coupled trade‐offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res 42: 2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finking, R. , and Marahiel, M.A. (2004) Biosynthesis of nonribosomal peptides. Annu Rev Microbiol 58: 453–488. [DOI] [PubMed] [Google Scholar]
- Fu, J. , Wenzel, S.C. , Perlova, O. , Wang, J. , Gross, F. , Tang, Z. , et al. (2008) Efficient transfer of two large secondary metabolite pathway gene clusters into heterologous hosts by transposition. Nucleic Acids Res 36: e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemperlein, K. , Rachid, S. , Garcia, R.O. , Wenzel, S.C. , and Müller, R. (2014) Polyunsaturated fatty acid biosynthesis in myxobacteria. Different PUFA synthases and their product diversity. Chem Sci 5: 1733–1741. [Google Scholar]
- Groß, S. , Schnell, B. , Haack, P.A. , Auerbach, D. , and Müller, R. (2021) In vivo and in vitro reconstitution of unique key steps in cystobactamid antibiotic biosynthesis. Nat Commun 12: 1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug, J.J. , Panter, F. , Krug, D. , and Müller, R. (2019) Genome mining reveals uncommon alkylpyrones as type III PKS products from myxobacteria. J Ind Microbiol Biotechnol 46: 319–334. [DOI] [PubMed] [Google Scholar]
- Iniesta, A.A. , García‐Heras, F. , Abellón‐Ruiz, J. , Gallego‐García, A. , and Elías‐Arnanz, M. (2012) Two systems for conditional gene expression in Myxococcus xanthus inducible by isopropyl‐ß‐D‐thiogalactopyranoside or vanillate. J Bacteriol 194: 5875–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa, J. , and Hotta, K. (1999) FramePlot: a new implementation of the frame analysis for predicting protein‐coding regions in bacterial DNA with a high G + C content. FEMS Microbiol Lett 174: 251–253. [DOI] [PubMed] [Google Scholar]
- Julien, B. , and Shah, S. (2002) Heterologous expression of epothilone biosynthetic genes in Myxococcus xanthus . Antimicrob Agents Chemother 46: 2772–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniusaite, M. , Tailhades, J. , Kittilä, T. , Fage, C.D. , Goode, R.J.A. , Schittenhelm, R.B. , and Cryle, M.J. (2021) Understanding the early stages of peptide formation during the biosynthesis of teicoplanin and related glycopeptide antibiotics. FEBS J 288(2): 507–529. [DOI] [PubMed] [Google Scholar]
- Kano, S. , Suzuki, S. , Hara, R. , and Kino, K. (2019) Synthesis of d‐amino acid‐containing dipeptides using the adenylation domains of nonribosomal peptide synthetase. Appl Environ Microbiol 85: e00120‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashefi, K. , and Hartzell, P.L. (1995) Genetic suppression and phenotypic masking of a Myxococcus xanthus frzF‐ defect. Mol Microbiol 15: 483–494. [DOI] [PubMed] [Google Scholar]
- Kim, H.J. , McCarty, R.M. , Ogasawara, Y. , Liu, Y.‐N. , Mansoorabadi, S.O. , LeVieux, J. , and Liu, H. (2013) GenK‐catalyzed C‐6' methylation in the biosynthesis of gentamicin: Isolation and characterization of a cobalamin‐dependent radical SAM enzyme. J Am Chem Soc 135: 8093–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota, T. , Shimamura, S. , Kobayashi, T. , Nunoura, T. , and Deguchi, S. (2016) Distribution of eukaryotic serine racemases in the bacterial domain and characterization of a representative protein in Roseobacter litoralis Och 149. Microbiology (Reading Engl) 162: 53–61. [DOI] [PubMed] [Google Scholar]
- Lau, J. , Frykman, S. , Regentin, R. , Ou, S. , Tsuruta, H. , and Licari, P. (2002) Optimizing the heterologous production of epothilone D in Myxococcus xanthus . Bioeng 78: 280–288. [DOI] [PubMed] [Google Scholar]
- Ley, S.V. , Priour, A. , and Heusser, C. (2002) Total synthesis of the cyclic heptapeptide argyrin B: a new potent inhibitor of T‐cell independent antibody formation. Org Lett 4: 711–714. [DOI] [PubMed] [Google Scholar]
- Liu, S. , Wei, Y. , Zhou, X. , Zhang, K. , Peng, X. , Ren, B. , et al. (2018) Function of alanine racemase in the physiological activity and cariogenicity of Streptococcus mutans . Sci Rep 8: 5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, J. (2001) Natural products as immunosuppressive agents. Nat Prod Rep 18: 417–430. [DOI] [PubMed] [Google Scholar]
- Marahiel, M.A. , Stachelhaus, T. , and Mootz, H.D. (1997) Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem Rev 97: 2651–2674. [DOI] [PubMed] [Google Scholar]
- Maruyama, C. , Toyoda, J. , Kato, Y. , Izumikawa, M. , Takagi, M. , Shin‐ya, K. , et al. (2012) A stand‐alone adenylation domain forms amide bonds in streptothricin biosynthesis. Nat Chem Biol 8: 791–797. [DOI] [PubMed] [Google Scholar]
- McQuade, T.J. , Shallop, A.D. , Sheoran, A. , Delproposto, J.E. , Tsodikov, O.V. , and Garneau‐Tsodikova, S. (2009) A nonradioactive high‐throughput assay for screening and characterization of adenylation domains for nonribosomal peptide combinatorial biosynthesis. Anal Biochem 386: 244–250. [DOI] [PubMed] [Google Scholar]
- Nickeleit, I. , Zender, S. , Sasse, F. , Geffers, R. , Brandes, G. , Sörensen, I. , et al. (2008) Argyrin A reveals a critical role for the tumor suppressor protein p27kip1 in mediating antitumor activities in response to proteasome inhibition. Cancer Res 14: 23–35. [DOI] [PubMed] [Google Scholar]
- Oßwald, C. , Zipf, G. , Schmidt, G. , Maier, J. , Bernauer, H. S. , Müller, R. , and Wenzel, S. (2014) Modular construction of a functional artificial epothilone polyketide pathway. ACS Synth Biol 3: 759–772. [DOI] [PubMed] [Google Scholar]
- Ongley, S. , Bian, X. , Neilan, B.A. , and Müller, R. (2013) Recent advances in the heterologous expression of microbial natural product biosynthetic pathways. Nat Prod Rep 30: 1121–1138. [DOI] [PubMed] [Google Scholar]
- Panter, F. , Krug, D. , Baumann, S. , and Müller, R. (2018) Self‐resistance guided genome mining uncovers new topoisomerase inhibitors from myxobacteria. Chem Sci 9: 4898–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlova, O. , Fu, J. , Kuhlmann, S. , Krug, D. , Stewart, F. , Zhang, Y. , and Müller, R. (2006) Reconstitution of myxothiazol biosynthetic gene cluster by Red/ET recombination and heterologous expression in Myxococcus xanthus . Appl Environ Microbiol 72: 7485–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre, S. , Guillot, A. , Benjdia, A. , Sandstrom, C. , Langella, P. , and Berteau, O. (2012) Thiostrepton tryptophan methyltransferase expands the chemistry of radical SAM enzymes. Nat Chem Biol 8: 957–959. [DOI] [PubMed] [Google Scholar]
- Pogorevc, D. , Panter, F. , Schillinger, C. , Jansen, R. , Wenzel, S.C. , and Müller, R. (2019a) Production optimization and biosynthesis revision of corallopyronin A, a potent anti‐filarial antibiotic. Metab Eng 55: 201–211. [DOI] [PubMed] [Google Scholar]
- Pogorevc, D. , Tang, Y. , Hoffmann, M. , Zipf, G. , Bernauer, H.S. , Popoff, A. , et al. (2019b) Biosynthesis and Heterologous Production of Argyrins. ACS Synth. Biol 8: 1121–1133. [DOI] [PubMed] [Google Scholar]
- Rausch, C. , Hoof, I. , Weber, T. , Wohlleben, W. , and Huson, D.H. (2007) Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol Biol 7: 78–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis, H.M. , Mirsky, E.A. , and Voigt, C.A. (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 27: 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. , and Russell, D.W. (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Schmutz, E. , Steffensky, M. , Schmidt, J. , Porzel, A. , Li, S.M. , and Heide, L. (2003) An unusual amide synthetase (CouL) from the coumermycin A1 biosynthetic gene cluster from Streptomyces rishiriensis DSM 40489. Eur J Biochem 270: 4413–4419. [DOI] [PubMed] [Google Scholar]
- Shi, G. , Shi, N. , Li, Y. , Chen, W. , Deng, J. , Liu, C. , et al. (2016) d‐alanylation in the assembly of ansatrienin side chain is catalyzed by a m odular NRPS. ACS Chem Biol 11: 876–881. [DOI] [PubMed] [Google Scholar]
- Stauch, B. , Simon, B. , Basile, T. , Schneider, G. , Malek, N.P. , Kalesse, M. , and Carlomagno, T. (2010) Elucidation of the structure and intermolecular interactions of a reversible cyclic‐peptide inhibitor of the proteasome by NMR spectroscopy and molecular modeling. Angew Chem Int Ed 49: 3934–3938. [DOI] [PubMed] [Google Scholar]
- Steffensky, M. , Li, S.M. , and Heide, L. (2000) Cloning, overexpression, and purification of novobiocic acid synthetase from Streptomyces spheroides NCIMB 11891. J Biol Chem 275: 21754–21760. [DOI] [PubMed] [Google Scholar]
- Stevens, D.C. , Henry, M.R. , Murphy, K.A. , and Boddy, C. N. (2010) Heterologous expression of the oxytetracycline biosynthetic pathway in Myxococcus xanthus . Appl Environ Microbiol 76: 2861–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strych, U. , Huang, H.C. , Krause, K.L. , and Benedik, M.J. (2000) Characterization of the alanine racemases from Pseudomonas aeruginosa PAO1. Curr Microbiol 41: 290–294. [DOI] [PubMed] [Google Scholar]
- Sucipto, H. , Pogorevc, D. , Luxenburger, E. , Wenzel, S. C. , and Müller, R. (2017) Heterologous production of myxobacterial α‐pyrone antibiotics in Myxococcus xanthus . Metab Eng 44: 160–170. [DOI] [PubMed] [Google Scholar]
- Sun, Y. , Feng, Z. , Tomura, T. , Suzuki, A. , Miyano, S. , Tsuge, T. , et al. (2016) Heterologous production of the marine myxobacterial antibiotic haliangicin and its unnatural analogues generated by engineering of the biochemical pathway. Sci Rep 6: 22091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Süssmuth, R.D. , and Mainz, A. (2017) Nonribosomal peptide synthesis ‐ Principles and prospects. Angew Chem Int Ed 56: 3770–3821. [DOI] [PubMed] [Google Scholar]
- Tu, Q. , Herrmann, J. , Hu, S. , Raju, R. , Bian, X. , Zhang, Y. , and Müller, R. (2016) Genetic engineering and heterologous expression of the disorazol biosynthetic gene cluster via Red/ET recombineering. Sci Rep 6: 21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallemacq, P.E. , and Reding, R. (1993) FK506 (tacrolimus), a novel immunosuppressant in organ transplantation: clinical, biomedical, and analytical aspects. Clin Chem 39: 2219–2228. [PMC free article] [PubMed] [Google Scholar]
- Wenzel, S.C. , Hoffmann, H. , Zhang, J. , Debussche, L. , Haag‐Richter, S. , Kurz, M. , et al. (2015) Production of the bengamide class of marine natural products in myxobacteria: biosynthesis and structure‐activity relationships. Angew Chem Int Ed Engl 54: 15560–15564. [DOI] [PubMed] [Google Scholar]
- Wu, T.S. , Duncan, J. , Tsao, S.W. , Chang, C.J. , Keller, P.J. , and Floss, H.G. (1987) Biosynthesis of the ansamycin antibiotic ansatrienin (mycotrienin) by Streptomyces collinus . J Nat Prod 50: 108–118. [DOI] [PubMed] [Google Scholar]
- Yan, F. , Auerbach, D. , Chai, Y. , Keller, L. , Tu, Q. , and Hüttel, S. (2018a) Biosynthesis and heterologous production of vioprolides: Rational biosynthetic engineering and unprecedented 4‐methylazetidinecarboxylic acid formation. Angew Chem Int Ed Engl 57: 8754–8759. [DOI] [PubMed] [Google Scholar]
- Yan, F. , Burgard, C. , Popoff, A. , Zaburannyi, N. , Zipf, G. , Maier, J. , et al. (2018b) Synthetic biology approaches and combinatorial biosynthesis towards heterologous lipopeptide production. Chem Sci 9: 7510–7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Buchholz, F. , Muyrers, J.P.P. , and StewartA, A.F. (1998) A new logic for DNA engineering using recombination in E. coli . Nat Genet 20: 123–128. [DOI] [PubMed] [Google Scholar]
- Zhang, Q. , van der Donk, W.A. , and Liu, W. (2012) Radical‐mediated enzymatic methylation: a tale of two SAMS. Acc Chem Res 45: 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Ntai, I. , Bolla, M.L. , Malcolmson, S.J. , Kahne, D. , Kelleher, N.L. , and Walsh, C.T. (2011) Nine enzymes are required for assembly of the pacidamycin group of peptidyl nucleoside antibiotics. Adv Ceram Mater 133: 5240–5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Genotypic verification of integration of all argyrin cluster versions into the heterologous host M. xanthus DK1622 ΔmchA‐tet. Primer sets used for genotypic verification, their binding sites and amplicon sizes are illustrated. Nucleotide sequences of the primers used are listed in the Table S1. Mch stands for myxochromide.
Fig. S2. Genotypic verification of insert integration into the mx8 attb1 site of the Mx. DK1622::pArg2345. Primer sets used for genotypic verification, their binding sites and amplicon sizes are illustrated. Nucleotide sequences of the primers used are listed in the Table S1.
Fig. S3. Assembly strategy for the exchange of the promoter systems upstream of the arg2345 operon. The same strategy was applied in case of all promoter fragments. The figure shows P tet sequence as an example.
Fig. S4. Base peak chromatograms (BPCs) of the Mx. DK1622::pArg2345‐arg451 MeoH (blue) and DMSO (black) crude extracts.
Fig. S5. Clustal Omega alignment of Pseusomonas aeruginosa PAO1 and M. xanthus DK1622 FusA1 sequences. The difference S459R (numbering according to the sequence of P. aeruginosa FusA1) is highlighted with an arrow.
Fig. S6. Visualization of argyrin B and the residue S459 of the P. aeruginosa FusA1 protein binding pocket (left). If the same residue is exchanged with R459 found in the M. xanthus DK1622 FusA1 sequence, this introduces a significant clash (right). The picture is based on the co‐crystal structure (PDB Code 4FN5).
Fig. S7. Substrate specificity of the A4 domain of the argyrin NRPS. Observed mass shifts in deconvoluted protein MS TICs reveal loading of D‐alanine (C) and D‐serine (E) onto the PCP. L‐alanine (B), L‐serine (D) and glycine (F) were not accepted by the A‐PCP (control: A).
Fig. S8. Increase in absorbance relative to the control (protein without amino acid) after the malachite green assay reactions incubated at 30°C for 1 h (left) and overnight at room temperature (right).
Table S1. List of primers used in this study.
Table S2. Gene synthesis constructs obtained from ATG:biosynthetics GmbH.
Table S3. Constructs generated in this study.
Table S4. Abbreviated and full names of the M. xanthus mutants generated in this study.
Table S5. HPLC‐MS data of argyrin derivatives detected in the heterologous expression strains. Above the bold line are previously described derivatives and below the line are their methylated (R3=CH3) counterparts newly detected in this study.
Data Availability Statement
The sequences of the expression constructs have been deposited in the GenBank database under the following accession numbers: MZ634479‐MZ634488.