

Abstract
We take a snapshot of the recent understanding of bacterial metabolism and the bacterial‐host metabolic interplay during infection, and highlight key outcomes and challenges for the practical implementation of bacterial metabolic modelling computational tools in the pathogenesis field.
Once relegated to the supply of energy and biosynthetic precursors, it is now indubitable that metabolism mediates most of physiological processes. In the context of bacterial–host interactions where virulence is the outcome (commonly termed bacterial pathogenesis) metabolism expands far beyond its canonical role in bacterial proliferation. In addition to all sorts of recognized molecular determinants or virulence factors (toxins, flagella, translocated effectors, adhesins, invasins, etc.), bacterial pathogens are equipped with specific metabolic traits to circumvent immune defenses and antimicrobial killing, thus facilitating colonization and proliferation within their hosts. As the implementation of high‐throughput technologies elevates the pathogenesis field to the era of big data, it concurrently creates considerable challenges for our ability to interpret large data sets and identify factors that impact infectious processes. Metabolic modelling is emerging as a powerful tool allowing the integration and coherent organization of large data sets into the context of biological networks providing non‐intuitive insights on biological systems that experimental analysis alone cannot provide. Here, we take a snapshot of the recent understanding of bacterial metabolism and the bacterial–host metabolic interplay during infection, and highlight key outcomes and challenges for the practical implementation of bacterial metabolic modelling computational tools in the pathogenesis field (summarized in Fig. 1).
Fig. 1.
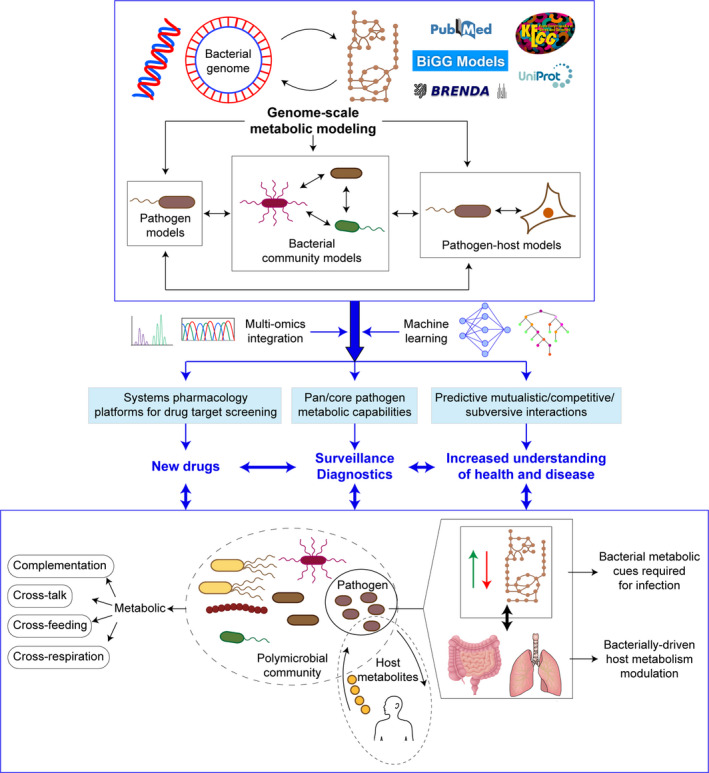
Genome‐scale metabolic network reconstructions for bacterial pathogenesis: it is time to leave a mark. Fast evolving advances in the genomics and metabolomics fields facilitate metabolic modelling of priority pathogens, of polymicrobial communities where key pathogens may have a starring role, and of host–pathogen systems. Metabolic reconstructions can yield significant benefits when combined with various layers of multi‐omics information as part of integration strategies, further enriched by the predictive potential of machine learning computational tools. Such integrative view will guide our experimental work to understand key metabolic traits in bacteria–bacteria or bacteria–host interactions where virulence is the outcome. More importantly, we foresee that such integrative view will contribute to pave the way for developing new diagnostic, treatment and surveillance procedures, seeking for their ultimate positive impact in the clinical management of bacterial infectious diseases.
Core involvement of bacterial metabolic requirements, rewiring and interactions during infection
Although the intimate association between bacterial metabolism and successful survival within the host still remains an underestimated aspect of bacterial virulence, it is clear that the understanding of specific metabolic adaptations during infection offers a multitude of new opportunities for, among others, target identification for drug development.
Bacterial metabolic requirements during infection
The increasing use of (multi)‐omic strategies on adequate in vivo models of infection greatly contributes to our understanding of pathogen metabolic requirements during infection. This is the case of in vivo transposon insertion mutagenesis coupled to next‐generation sequencing (Tn‐seq) analyses. The application of this technology during mouse intestine colonization and on piglet infection models of Campylobacter jejuni identified crucial roles for genes involved in central metabolism, nutrient acquisition or substrate utilization (De Vries et al., 2017; Gao et al., 2017). Tn‐seq screen of Streptococcus pyogenes identified genes important for bacterial fitness in human saliva involved in carbohydrate, amino acid and inorganic ion transport/metabolism (Zhu et al., 2017). Similarly, Tn‐seq analysis highlighted the importance of glucose utilization for Serratia marcescens on a murine model of bacteraemia (Anderson et al., 2017), and unravelled similar metabolic responses by enterohemorrhagic Escherichia coli (EHEC) and Vibrio cholerae during colonization of the infant rabbit colonic niche (Fu et al., 2013; Warr et al., 2019). Emphasizing this idea, another recent example using a murine model of acute pneumonia by Staphylococcus aureus identified a high proportion of genes involved in metabolic processes (tricarboxylic acid cycle (TCA), ATP production, purine and pyrimidine metabolism) as important for bacterial survival during infection (Kim et al., 2021).
Host metabolism undergoes rewiring during bacterial infection
The use of host nutrients and metabolic pathways by cytosolic and vacuolar intracellular pathogens, and the links between intracellular bacterial metabolism and the expression of virulence genes required for such intracellular replication, also keep being revealed (Eisenreich et al., 2015; Conover et al., 2016; Sprenger et al., 2018). Sophisticated modulation of the host metabolism has been reported for Mycobacterium tuberculosis which, during active disease, induces the expression of indoleamine 2,3‐dioxygenase (IDO), an enzyme involved in tryptophan catabolism (Gautam et al., 2018). In turn, S. aureus small colony variants (SCVs) were found to impair the host immunity by activating host cell glycolysis by the means of overexpressing fumC, encoding an enzyme that catalyses the degradation of the glycolysis inhibitor fumarate (Wong Fok Lung et al., 2020). Likewise, Citrobacter rodentium manipulates the host metabolism by rewiring cellular bioenergetics and cholesterol metabolism to evade innate immunity and establish a favourable gut ecosystem. Indeed, binding of C. rodentium to the gut epithelium leads, among others, to upregulation of sugar transport, aerobic glycolysis, production of phosphocreatine, activation of the cholesterol biosynthetic pathway and upregulation of cholesterol efflux proteins, resulting in higher levels of faecal cholesterol and a bloom of Proteobacteria, thus, altering the composition of the gut microbiota (Berger et al., 2017).
Bacterial metabolic rewiring upon host metabolite sensing contributes to patho‐adaptation
Pathogens adapt many methods of sensing the environment for numerous metabolites that they may encounter within the host and use them for metabolic rewiring or for translation to relevant transcriptional networks, to ultimately facilitate colonization. Notably, molecular determinants of bacterial metabolic adaptation and rewiring during infection and chronicity are major outcomes of recent elegant studies. This is the case for Salmonella typhimurium, which harvests energy by anaerobic respiration using microbiota‐derived hydrogen (H2) as an electron donor and fumarate as an electron acceptor during gut colonization by the means of a cyclic strategy: as fumarate is scarce in the gut, S. Typhimurium obtains fumarate by the C4‐dicarboxylate antiporter DcuABC‐mediated import and conversion of L‐malate and L‐aspartate. Fumarate reduction yields succinate, which is in turn exported by DcuABC in exchange for L‐aspartate and L‐malate (Nguyen et al., 2020). Likewise, as the host metabolite D‐serine influences the outcome of infection by repressing the type III secretion system of EHEC, a conserved D‐serine uptake system helps E. coli sensing levels of the metabolite by regulating its uptake from the environment in turn influencing global gene expression (Connolly et al., 2016). Even more, 1,2‐propanediol whose production by the gut microbiota is upregulated in response to murine colonization, enhances C. rodentium in vivo fitness through a fine‐tuned coordination of type III secretion system and effector expression, thus, contributing to niche‐specification within the gut (Connolly et al., 2018). Another aspect worth mentioning is the connection between the bacterial acetylome and its metabolic rewiring during infection. As shown for M. tuberculosis, a lysine acetyltransferase promotes survival by altering the flux of carbon from oxidative to reductive TCA reactions, which requires malate dehydrogenase and maintains the redox state of the NAD+/NADH pool (Rittershaus et al., 2018). For Klebsiella pneumoniae, lysine acetylation of multiple proteins involved in central metabolism, specifically the glucose 6‐phosphate dehydrogenase Zwf, drives a metabolic boost leading to greater consumption of glucose in the host airway and increased bacterial burden (Ahn et al., 2021).
Great examples of pathogen adaptation to host metabolites have also been reported, as those for the host‐derived immunometabolite itaconate, abundant in the infected lung. Itaconate induces Pseudomonas aeruginosa membrane stress resulting in downregulation of lipopolysaccharide (LPS) and upregulation of extracellular polysaccharide (EPS). In turn, itaconate‐adapted P. aeruginosa accumulate lptD mutations favouring itaconate assimilation and biofilm formation, and accumulated EPS induces itaconate production by host cells, thus, skewing the host immune response to one permissive of chronic infection (Riquelme et al., 2020). Moreover, itaconate inhibits S. aureus glycolysis and selects for strains that redirect carbon flux to fuel EPS synthesis and biofilm formation (Tomlinson et al., 2021). Therefore, both P. aeruginosa and S. aureus, by different mechanisms, adapt to the itaconate‐dominated immunometabolic response by producing biofilms associated with chronic infection of the human airway. P. aeruginosa was one of the first and still is a key subject of bacterial patho‐adaptive studies. Those include tackling its metabolic rewiring during lung colonization to show that reduction of growth rate and metabolic specialization are signatures of adaptive evolution occurring through distinct evolutionary trajectories, and highlight the importance of metabolism and its regulatory mechanisms for this bacterial ecological flexibility and chronicity within the human airways (Turner et al., 2014; La Rosa et al., 2018, 2019; Rossi et al., 2018; Perinbam et al., 2020).
Metabolic interactions in microbial communities: broadening the picture is the way to go
These outstanding single‐bacterial species studies jump into the next level of complexity when microbial communities are considered. While the explosion of microbiome analyses helps to identify individual microorganisms and microbial communities driving human health and disease, the incredibly complex metabolic interactions among microbial species in those communities pose challenging questions. Such metabolic interactions have all sorts of different outcomes resulting, for example, in the complementary utilization of central metabolism by having divergent requirements, as reported for E. coli and Proteus mirabilis during urinary tract infection (Alteri et al., 2015). Such interactions may also result in metabolic cross‐talk regulating virulence, as reported for Porphyromonas gingivalis and Streptococcus gordonii where streptococcal 4‐aminobenzoate/para‐amino benzoic acid (pABA) is required for maximal accumulation of P. gingivalis during oral polymicrobial infection (Kuboniwa et al., 2017), or in metabolite cross‐feeding and cross‐respiration also regulating virulence. This is the case of Aggregatibacter actinomycetemcomitans and S. gordonni, where the later promotes virulence of the former one by producing its preferred carbon source L‐lactate, and by enhancing the bioavailability of oxygen during infection, in turn allowing A. actinomycetemcomitans to shift from a primarily fermentative to a respiratory metabolism that enhances its fitness (Ramsey et al., 2011; Stacy et al., 2014, 2016).
Bacterial metabolic modelling claims a place in the pathogenesis field
Here, without paying closer attention to the otherwise extensive core theme of metabolic cross‐talk between host and pathogens (Olive and Sassetti, 2016; Traven and Naderer, 2019), we intend to hint at the need for complementary, synergistic and integrative approaches to contextualize the myriad of data available and probe key metabolic features governing the interactions among bacteria and/or the host during infection. We reinforce the notion that metabolic network reconstructions are crucial tools for mapping these complex interactions (Dunphy and Papin, 2018). Such reconstructions are detailed representations of cellular metabolism, which when derived from genomic annotations extend metabolism at genome‐scale. Thus, a genome‐scale metabolic model (GEM) is constructed through the systematic integration of genome annotation, ‐omics data sets and legacy knowledge such as reaction stoichiometry, and contains all metabolic reactions that can occur in a specific organism with their associated metabolites, proteins and genes. When used in combination with algorithms such as Flux Balance Analysis (FBA) (Orth et al., 2010), GEMs can predict phenotype from genotype (Gu et al., 2019; Fang et al., 2020). Since the reconstruction of the first GEM of Haemophilus influenzae (Edwards and Palsson, 1999), hundreds of novel bacterial GEMs are being generated through automatic, semi‐automatic or manual means as new genomes are sequenced. This high interest in GEMs has led in the last decade an increasing number of applications (Gu et al., 2019; Fang et al., 2020), and large community‐driven efforts towards the standardization of modelling formalisms and GEM reproducibility and reuse (Lieven et al., 2020). Today, the field is mature and ready for addressing new challenges.
Regarding bacterial pathogenesis, GEMs are promising tools to elucidate microbial metabolism in response to environmental perturbations, precise interactions among bacteria and between bacteria and their hosts. Therefore, it is not surprising that they are seen by many as critical tools to address the system level understanding of pathogens metabolism and the host–pathogen metabolic interplay (Jansma and El Aidy, 2021; Jean‐Pierre et al., 2021). In addition, GEMs can be used to test the effectiveness of treatment before clinical trials. In this regard, GEMs can be used as systems pharmacology platforms for drug target screening‐prioritization, as targeting pathways implicated in the metabolism of essential nutrients utilized during infection can interfere with microbial growth in the context of disease (Chavali et al., 2012; Mienda et al., 2018; Richelle et al., 2020). Metabolic reconstructions are, therefore, in the front of multidimensional data integration strategies by combining various layers of information into multi‐omics approaches with genomic, transcriptomic, metabolic and protein structural data sources. It is not surprising that GEMs‐based analyses have been already applied to the priority pathogens Acinetobacter baumannii, K. pneumoniae and P. aeruginosa to delineate candidates with features relevant for target selection. Even more, such computational methodologies allow the accurate prediction of the flux through each reaction in the network in response to disturbed gene expression profiles. In the context of antibiotic resistance, the integration of gene expression data as additional constraints within GEMs thus makes possible the system level understanding of the metabolic reprogramming occurring during antibiotic exposure (Bartell et al., 2017; Presta et al., 2017; Ramos et al., 2018; Zhu et al., 2018; Cesur et al., 2020).
Another key aspect is the applicability of genome‐scale metabolic network models from an epidemiological perspective, for systematic evaluation of clinical strains with hundreds of polymorphisms and strain specific metabolic configurations. As shown for E. coli, S. aureus and Salmonella, GEMs enable a systems approach to characterize the pan and core metabolic capabilities of key pathogens, to define the metabolic essence of a bacterial species, and delineate growth differences that may shed light on the adaptation process to a particular niche or colonization site during infection (Monk et al., 2013; Bosi et al., 2016; Seif et al., 2018). This approach could allow the use of personalized treatment targeted on the strain‐specific metabolic traits unravelled by modelling, paving the way for developing treatments more specific than the current ones, thus, avoiding unspecific strategies and reducing the emergence of drug resistance.
Bacterial community metabolic modelling: great tools for polymicrobial infection studies
The power of computational modelling when tackling complex microbial communities is also emerging (García‐Jiménez et al., 2021). The combined use of 16S rRNA sequencing data collected from clinical samples and in silico methods for community metabolic modelling and analysis provides a large array of novel biological insights, which have remained elusive so far. For instance, model‐based analyses were applied to predict mutualistic interactions (pathogen–pathogen and pathogen–commensal) driving community composition in polymicrobial chronic wound infections by the means of cross‐feeding of organic acids, alcohols and amino acids (Phalak and Henson, 2019). Similarly, GEMs analysis provided new insights into the metabolic determinants of pathogen dominance in the cystic fibrosis lung (Henson et al., 2019). GEMs are also great tools acting as computational scaffolds for the integration of growth and transcriptomics data from infections caused by multiple species. This notion, when applied to interrogate the growth capabilities of V. cholerae in single infections and coinfections with enterotoxigenic E. coli (ETEC), which co‐occur in a large fraction of diarrhoeagenic patients, showed that V. cholerae growth capabilities are enhanced in the presence of ETEC through cross‐fed metabolites made available to V. cholerae by ETEC (Abdel‐Haleem et al., 2020).
Integrated host–pathogen genome‐scale reconstructions
Finally, building integrated host–pathogen genome‐scale reconstructions is an ultimate goal to optimize the extraordinary capabilities of metabolic modelling in combining ‐omic as well as physiology data for mechanistic analyses of metabolism. These ideas have been developed to get further insight into M. tuberculosis or S. typhimurium host interactions (Raghunathan et al., 2009; Bordbar et al., 2010; Raghunathan and Jamshidi, 2018). Such methods keep evolving towards not only elucidating the role of the host environment on pathogen metabolism during the course of an infection, but also the impact of pathogen infection on the host within a single computational model, and improve as human metabolic network reconstructions continue to be curated.
Challenges in the integration of metabolic modelling in bacterial pathogenesis research
Although we are moving towards a widespread adoption of metabolic modelling tools in the bacterial pathogenesis field and, therefore, in the antimicrobial industry, we identify key aspects restraining their routine implementation. Our limited knowledge of the environment conditions within the host niche and its changes upon disease limits our ability to perform accurate bacterial growth simulations within different host‐mimicking media. Parallel advances in the fields or tissue metabolomics and lipidomics will surely contribute to overcome such limitations. Main limitations of GEMs include the steady state assumption, which implies that the study of temporal and evolutionary scenarios, often of critical importance in bacterial infections, is largely hampered. However, recent advances in modelling formalisms including hybrid approaches such as dynamic flux balance analysis (dFBA) and their multiple extensions are largely contributing to extend the applicability of GEMs to transient behaviours (Scott et al., 2018). In addition, the continue development of large‐scale kinetic models derived from GEMs driven by methods for the full‐scale kinetic parameter estimation such as the ORACLE framework (Miskovic and Hatzimanikatis, 2010), will provide in the short time a new generation of metabolic models suitable for the analysis of dynamic transitions and evolutionary trajectories within bacterial infections.
So far, the analysis of high‐dimensional biological data is largely limited by the lack of proper tools. However, during the last decade, developments within the field of machine learning in data visualization, deep neural networks, data fusion, model interpretation and more have resulted in new tools that hold great promise for dealing with disparate ‐omics data sets. Host–pathogen metabolism mathematical models will also be greatly benefited from machine learning, as it is becoming an unprecedented tool for the correct interpretation and exploitation of ‐omic data (Reel et al., 2021). In fact, these two computational frameworks are in the process of being combined based on their complementary characteristics and common mathematical bases (Zampieri et al., 2019). These ideas are under development in the systems metabolic engineering field, aiming to engineer production host’s biological networks for overproducing valuable materials in a sustainable manner. Recent studies are also developing new approaches that integrate the power of deep learning with metabolic modelling to allow for a mechanistic interpretation of the genetic associations discovered by machine learning. This is the case of a computational pipeline that combines machine learning with genome‐scale metabolic models to understand the systemic relationships between genetic determinants of antibiotic resistance and metabolic adaptation and rewiring mechanisms beyond annotated drug resistance genes for the opportunistic pathogen E. coli (Pearcy et al., 2021).
We foresee that metabolic modelling and deep learning will be progressively incorporated in the microbial pathogenesis research field where, complemented with comparative genomics, genetic interaction or protein structural analyses, will greatly contribute to predict the biological meaning of the variability in genetic content and metabolic capabilities across clinical isolates, infer specific selection pressures driving the outcome of the infection process, guide experimental work and ultimately guide procedures to improve the clinical management of infectious diseases.
Funding information
This work has been funded by grants from MINECO RTI2018‐096369‐B‐I00, 875/2019 from SEPAR, PC150‐151‐152 from Gobierno de Navarra to J.G and from MICIU through RobExplode PID2019‐108458RB‐I00 to J.N. CIBER is an initiative from Instituto de Salud Carlos III (ISCIII), Madrid, Spain.
Conflict of interest
None declared.
Acknowledgements
We are grateful to Nahikari López‐López for critical reading of the manuscript and help with figure making. This work has been funded by grants from MINECO RTI2018‐096369‐B‐I00, 875/2019 from SEPAR, PC150‐151‐152 from Gobierno de Navarra to J.G and from MICIU through RobExplode PID2019‐108458RB‐I00 to J.N. CIBER is an initiative from Instituto de Salud Carlos III (ISCIII), Madrid, Spain.
Microbial Biotechnology (2021) 15(1), 95–102
†These authors contributed equally to this work.
Contributor Information
Juan Nogales, Email: j.nogales@csic.es.
Junkal Garmendia, Email: juncal.garmendia@csic.es.
References
- Abdel‐Haleem, A.M. , Ravikumar, V. , Ji, B. , Mineta, K. , Gao, X. , Nielsen, J. , et al. (2020) Integrated metabolic modeling, culturing, and transcriptomics explain enhanced virulence of Vibrio cholerae during coinfection with enterotoxigenic Escherichia coli . mSystems 5: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, D. , Bhushan, G. , McConville, T.H. , Annavajhala, M.K. , Soni, R.K. , Wong Fok Lung, T. , et al. (2021) An acquired acyltransferase promotes Klebsiella pneumoniae ST258 respiratory infection. Cell Rep 35: 109196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alteri, C.J. , Himpsl, S.D. , and Mobley, H.L.T. (2015) Preferential use of central metabolism in vivo reveals a nutritional basis for polymicrobial infection. PLoS Pathog 11: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, M.T. , Mitchell, L.A. , Zhao, L. , and Mobley, L.T. (2017) Capsule production and glucose metabolism dictate fitness during Serratia marcescens bacteremia. mBio 8: e00740–e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartell, J.A. , Blazier, A.S. , Yen, P. , Thøgersen, J.C. , Jelsbak, L. , Goldberg, J.B. , and Papin, J.A. (2017) Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat Commun 8: 14631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, C.N. , Crepin, V.F. , Roumeliotis, T.I. , Wright, J.C. , Carson, D. , Pevsner‐Fischer, M. , et al. (2017) Citrobacter rodentium subverts ATP flux and cholesterol homeostasis in intestinal epithelial cells in vivo . Cell Metab 26: 738–752.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordbar, A. , Lewis, N.E. , Schellenberger, J. , Palsson, B. , and Jamshidi, N. (2010) Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol 6: 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosi, E. , Monk, J.M. , Aziz, R.K. , Fondi, M. , Nizet, V. , and Palsson, B. (2016) Comparative genome‐scale modelling of Staphylococcus aureus strains identifies strain‐specific metabolic capabilities linked to pathogenicity. Proc Natl Acad Sci USA 113: E3801–E3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesur, M.F. , Siraj, B. , Uddin, R. , Durmuş, S. , and Çakır, T. (2020) Network‐based metabolism‐centered screening of potential drug targets in Klebsiella pneumoniae at genome scale. Front Cell Infect Microbiol 9: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavali, A.K. , D’Auria, K.M. , Hewlett, E.L. , Pearson, R.D. , and Papin, J.A. (2012) A metabolic network approach for the identification and prioritization of antimicrobial drug targets. Trends Microbiol 20: 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, J.P.R. , Gabrielsen, M. , Goldstone, R.J. , Grinter, R. , Wang, D. , Cogdell, R.J. , et al. (2016) A highly conserved bacterial D‐serine uptake system links host metabolism and virulence. PLoS Pathog 12: e1005359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, J.P.R. , Slater, S.L. , O’Boyle, N. , Goldstone, R.J. , Crepin, V.F. , Ruano‐Gallego, D. , et al. (2018) Host‐associated niche metabolism controls enteric infection through fine‐tuning the regulation of type 3 secretion. Nat Commun 9: 4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover, M.S. , Hadjifrangiskou, M. , Palermo, J.J. , Hibbing, M.E. , Dodson, K.W. , and Hultgren, S.J. (2016) Metabolic requirements of Escherichia coli in intracellular bacterial communities during urinary tract infection pathogenesis. Mbio 7: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries, S.P.W. , Linn, A. , Macleod, K. , MacCallum, A. , Hardy, S.P. , Douce, G. , et al. (2017) Analysis of Campylobacter jejuni infection in the gnotobiotic piglet and genome‐wide identification of bacterial factors required for infection. Sci Rep 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy, L.J. , and Papin, J.A. (2018) Biomedical applications of genome‐scale metabolic network reconstructions of human pathogens. Curr Opin Biotechnol 51: 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, J.S. , and Palsson, B.O. (1999) Systems properties of the Haemophilus influenzae Rd metabolic genotype. J Biol Chem 274: 17410–17416. [DOI] [PubMed] [Google Scholar]
- Eisenreich, W. , Heesemann, J. , Rudel, T. , and Goebel, W. (2015) Metabolic adaptations of intracellullar bacterial pathogens and their mammalian host cells during infection (“Pathometabolism”). Metab Bact Pathog 27–58. [DOI] [PubMed] [Google Scholar]
- Fang, X. , Lloyd, C.J. , and Palsson, B.O. (2020) Reconstructing organisms in silico: genome‐scale models and their emerging applications. Nat Rev Microbiol 18: 731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. , Waldor, M.K. , and Mekalanos, J.J. (2013) Tn‐seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS‐mediated antibacterial activity in the host. Cell Host Microbe 14: 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, B. , Vorwerk, H. , Huber, C. , Lara‐Tejero, M. , Mohr, J. , Goodman, A.L. , et al. (2017) Metabolic and fitness determinants for in vitro growth and intestinal colonization of the bacterial pathogen Campylobacter jejuni . PLoS Biol 15: e2001390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Jiménez, B. , Torres‐Bacete, J. , and Nogales, J. (2021) Metabolic modelling approaches for describing and engineering microbial communities. Comput Struct Biotechnol J 19: 226–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam, U.S. , Foreman, T.W. , Bucsan, A.N. , Veatch, A.V. , Alvarez, X. , Adekambi, T. , et al. (2018) In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune‐mediated control of Mycobacterium tuberculosis . Proc Natl Acad Sci USA 115: E62–E71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, C. , Kim, G.B. , Kim, W.J. , Kim, H.U. , and Lee, S.Y. (2019) Current status and applications of genome‐scale metabolic models. Genome Biol 20: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson, M.A. , Orazi, G. , Phalak, P. , and O’Toole, G.A. (2019) Metabolic modeling of cystic fibrosis airway communities predicts mechanisms of pathogen dominance. mSystems 4: e00026–e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansma, J. , and El Aidy, S. (2021) Understanding the host‐microbe interactions using metabolic modeling. Microbiome 9: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean‐Pierre, F. , Henson, M.A. , and O’Toole, G.A. (2021) Metabolic modeling to interrogate microbial disease: a tale for experimentalists. Front Mol Biosci 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, G.‐L. , Hooven, T.A. , Norambuena, J. , Li, B. , Boyd, J.M. , Yang, J.H. , and Parker, D. (2021) Growth and stress tolerance comprise independent metabolic strategies critical for Staphylococcus aureus Infection. Mbio 12: e00814–e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboniwa, M. , Houser, J.R. , Hendrickson, E.L. , Wang, Q. , Alghamdi, S.A. , Sakanaka, A. , et al. (2017) Metabolic crosstalk regulates Porphyromonas gingivalis colonization and virulence during oral polymicrobial infection. Nat Microbiol 2: 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieven, C. , Beber, M.E. , Olivier, B.G. , Bergmann, F.T. , Ataman, M. , Babaei, P. , et al. (2020) MEMOTE for standardized genome‐scale metabolic model testing. Nat Biotechnol 38: 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mienda, B.S. , Salihu, R. , Adamu, A. , and Idris, S. (2018) Genome‐scale metabolic models as platforms for identification of novel genes as antimicrobial drug targets. Future Microbiol 13: 455–467. [DOI] [PubMed] [Google Scholar]
- Miskovic, L. , and Hatzimanikatis, V. (2010) Production of biofuels and biochemicals: in need of an ORACLE. Trends Biotechnol 28: 391–397. [DOI] [PubMed] [Google Scholar]
- Monk, J.M. , Charusanti, P. , Aziz, R.K. , Lerman, J.A. , Premyodhin, N. , Orth, J.D. , et al. (2013) Genome‐scale metabolic reconstructions of multiple Escherichia coli strains highlight strain‐specific adaptations to nutritional environments. Proc Natl Acad Sci USA 110: 20338–20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, B.D. , Cuenca V., M. , Hartl, J. , Gül, E. , Bauer, R. , Meile, S. , et al. (2020) Import of aspartate and malate by DcuABC drives H2/fumarate respiration to promote initial Salmonella gut‐lumen colonization in mice. Cell Host Microbe 27: 922–936.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive, A.J. , and Sassetti, C.M. (2016) Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol 14: 221–234. [DOI] [PubMed] [Google Scholar]
- Orth, J.D. , Thiele, I. , and Palsson, B.Ø. (2010) What is flux balance analysis? Nat Biotechnol 28: 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearcy, N. , Hu, Y. , Baker, M. , Maciel‐Guerra, A. , Xue, N. , Wang, W. , et al. (2021) Genome‐scale metabolic models and machine learning reveal genetic determinants of antibiotic resistance in Escherichia coli and unravel the underlying metabolic adaptation mechanisms. mSystems 6: e00913–e920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perinbam, K. , Chacko, J.V. , Kannan, A. , Digman, M.A. , and Siryaporn, A. (2020) A shift in central metabolism accompanies virulence activation in Pseudomonas aeruginosa . mBio 11: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phalak, P. , and Henson, M.A. (2019) Metabolic modelling of chronic wound microbiota predicts mutualistic interactions that drive community composition. J Appl Microbiol 127: 1576–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta, L. , Bosi, E. , Mansouri, L. , Dijkshoorn, L. , Fani, R. , and Fondi, M. (2017) Constraint‐based modeling identifies new putative targets to fight colistin‐resistant A. baumannii infections. Sci Rep 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan, A. , and Jamshidi, N. (2018) Integrated host‐pathogen metabolic reconstructions. Methods Mol Biol 1716: 197–217. [DOI] [PubMed] [Google Scholar]
- Raghunathan, A. , Reed, J. , Shin, S. , Palsson, B. , and Daefler, S. (2009) Constraint‐based analysis of metabolic capacity of Salmonella typhimurium during host‐pathogen interaction. BMC Syst Biol 3: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, P.I.P. , Fernández Do Porto, D. , Lanzarotti, E. , Sosa, E.J. , Burguener, G. , Pardo, A.M. , et al. (2018) An integrative, multi‐omics approach towards the prioritization of Klebsiella pneumoniae drug targets. Sci Rep 8: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey, M.M. , Rumbaugh, K.P. , and Whiteley, M. (2011) Metabolite cross‐feeding enhances virulence in a model polymicrobial infection. PLoS Pathog 7: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reel, P.S. , Reel, S. , Pearson, E. , Trucco, E. , and Jefferson, E. (2021) Using machine learning approaches for multi‐omics data analysis: a review. Biotechnol Adv 49: 107739. [DOI] [PubMed] [Google Scholar]
- Richelle, A. , David, B. , Demaegd, D. , Dewerchin, M. , Kinet, R. , Morreale, A. , et al. (2020) Towards a widespread adoption of metabolic modeling tools in biopharmaceutical industry: a process systems biology engineering perspective. npj Syst Biol Appl 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme, S.A. , Liimatta, K. , Wong Fok Lung, T. , Fields, B. , Ahn, D. , Chen, D. , et al. (2020) Pseudomonas aeruginosa utilizes host‐derived itaconate to redirect its metabolism to promote biofilm formation. Cell Metab 31: 1091–1106.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittershaus, E.S.C. , Baek, S.‐H. , Krieger, I.V. , Nelson, S.J. , Cheng, Y.‐S. , Nambi, S. , et al. (2018) A lysine acetyltransferase contributes to the metabolic adaptation to hypoxia in Mycobacterium tuberculosis . Cell Chem Biol 25: 1495–1505.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa, R. , Johansen, H.K. , and Molin, S. (2018) Convergent metabolic specialization through distinct evolutionary paths in Pseudomonas aeruginosa . Mbio 9: e00269–e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa, R. , Johansen, H.K. , and Molin, S. (2019) Adapting to the airways: metabolic requirements of Pseudomonas aeruginosa during the infection of cystic fibrosis patients. Metabolites 9: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, E. , Falcone, M. , Molin, S. , and Johansen, H.K. (2018) High‐resolution in situ transcriptomics of Pseudomonas aeruginosa unveils genotype independent patho‐phenotypes in cystic fibrosis lungs. Nat Commun 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, F. , Wilson, P. , Conejeros, R. , and Vassiliadis, V.S. (2018) Simulation and optimization of dynamic flux balance analysis models using an interior point method reformulation. Comput Chem Eng 119: 152–170. [Google Scholar]
- Seif, Y. , Kavvas, E. , Lachance, J.C. , Yurkovich, J.T. , Nuccio, S.P. , Fang, X. , et al. (2018) Genome‐scale metabolic reconstructions of multiple Salmonella strains reveal serovar‐specific metabolic traits. Nat Commun 9: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger, M. , Kasper, L. , Hensel, M. , and Hube, B. (2018) Metabolic adaptation of intracellular bacteria and fungi to macrophages. Int J Med Microbiol 308: 215–227. [DOI] [PubMed] [Google Scholar]
- Stacy, A. , Everett, J. , Jorth, P. , Trivedi, U. , Rumbaugh, K.P. , and Whiteley, M. (2014) Bacterial fight‐and‐flight responses enhance virulence in a polymicrobial infection. Proc Natl Acad Sci USA 111: 7819–7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy, A. , Fleming, D. , Lamont, R.J. , Rumbaugh, K.P. , and Whiteley, M. (2016) A commensal bacterium promotes virulence of an opportunistic pathogen via cross‐respiration. Mbio 7: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson, K.L. , Lung, T.W.F. , Dach, F. , Annavajhala, M.K. , Gabryszewski, S.J. , Groves, R.A. , et al. (2021) Staphylococcus aureus induces an itaconate‐dominated immunometabolic response that drives biofilm formation. Nat Commun 12: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traven, A. , and Naderer, T. (2019) Central metabolic interactions of immune cells and microbes: prospects for defeating infections. EMBO Rep 20: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, K.H. , Everett, J. , Trivedi, U. , Rumbaugh, K.P. , and Whiteley, M. (2014) Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet 10: e1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr, A.R. , Hubbard, T.P. , Munera, D. , Blondel, C.J. , Abel zur Wiesch, P. , Abel, S. , et al. (2019) Transposon‐insertion sequencing screens unveil requirements for EHEC growth and intestinal colonization. PLoS Pathogens 15: e1007652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Fok Lung, T. , Monk, I.R. , Acker, K.P. , Mu, A. , Wang, N. , Riquelme, S.A. , et al. (2020) Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat Microbiol 5: 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampieri, G. , Vijayakumar, S. , Yaneske, E. , and Angione, C. (2019) Machine and deep learning meet genome‐scale metabolic modeling. PLoS Comput Biol 15: e1007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L. , Charbonneau, A.R.L. , Waller, A.S. , Olsen, R.J. , Beres, S.B. , and Musser, J.M. (2017) Novel genes required for the fitness of Streptococcus pyogenes in human saliva. mSphere 2: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y. , Czauderna, T. , Zhao, J. , Klapperstueck, M. , Maifiah, M.H.M. , Han, M.L. , et al. (2018) Genome‐scale metabolic modeling of responses to polymyxins in Pseudomonas aeruginosa . Gigascience 7: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]