

Abstract
Bacterial protein synthesis rates have evolved to maintain preferred stoichiometries at striking precision, from the components of protein complexes to constituents of entire pathways. Setting relative protein production rates to be well within a factor of two requires concerted tuning of transcription, RNA turnover, and translation, allowing many potential regulatory strategies to achieve the preferred output. The last decade has seen a greatly expanded capacity for precise interrogation of each step of the central dogma genome-wide. Here, we summarize how these technologies have shaped the current understanding of diverse bacterial regulatory architectures underpinning stoichiometric protein synthesis. We focus on the emerging expanded view of bacterial operons, which encode diverse primary and secondary mRNA structures for tuning protein stoichiometry. Emphasis is placed on how quantitative tuning is achieved. We discuss the challenges and open questions in the application of quantitative, genome-wide methodologies to the problem of precise protein production.
Keywords: expression stoichiometry, proportional synthesis, operon mRNA isoform, differential RNA stability, differential translation
1. INTRODUCTION
In 1959—two years prior to the proposal of the operon model—Bruce Ames and Barbara Garry (4) showed that the four histidine biosynthesis enzymes in Salmonella were always produced at the same ratios, regardless of induction levels. This evidence for coordinated regulation, together with the genomic linkage of the genes encoding these enzymes, was key to François Jacob and Jacques Monod’s (82) hypothesis of polycistronic operons. At that time, it was not known that these proteins are produced in differing amounts despite being regulated and transcribed together. Such constant, nonequimolar proportions appeared to be under positive selection, as the same stoichiometry of functionally related proteins, e.g., glycolytic enzymes, was observed in cells from yeasts to mammalian tissues (131). Fast-forward to the turn of the century, when genomic techniques revealed that these constant-proportion groups constitute the foundation of gene regulatory networks, which can be decomposed into modules of coexpressed proteins that allow for a small number of regulators to control a large number of genes (53a, 144a). In essence, the synthesis rate of a protein is set in two orthogonal dimensions: the overall expression of the module it belongs to and the relative stoichiometry within the module.
Although stoichiometry is often not subject to regulation, perturbations to relative expression can be detrimental. For proteins that assemble into heteromeric complexes, excess subunits that do not have binding partners may misfold or aggregate. Some coregulated proteins play antagonistic roles with each other, making balanced production critical for their functions. For many other groups of proteins, the rationale for their precise stoichiometry remains unknown. Nevertheless, a recent comparative proteomic analysis revealed that most of them maintain the same ratios of synthesis rates across large phylogenetic distances, despite dramatic divergences in the molecular mechanisms that determine their relative rates (96). These results suggest that stoichiometric protein synthesis did not happen by chance. Rather, it is intricately optimized according to the quantitative relationship between expression and cell fitness.
Mechanistically, stoichiometric protein synthesis is often implemented at the posttranscriptional level in bacteria (Figure 1). Because many functionally related genes are organized into operons, they can be coregulated by transcription. The relative rates of protein synthesis are then set by posttranscriptional processes, such as mRNA processing, differential translation, and other RNA-based regulation. Characterizing these posttranscriptional processes at a quantitative level is thus critical for understanding the control of stoichiometric protein synthesis. It must also be noted that the idealized operon model, in which neighboring genes are exclusively cotranscribed as a single unit of polycistronic mRNA, applies to only a fraction of coregulated genes in bacteria. Neighboring genes are often punctuated by internal promoters, leaky transcription terminators, and RNA-processing sites that are also important for setting the final stoichiometry of protein synthesis. Increasingly, these transcriptional and posttranscriptional events can be better mapped throughout the genome using high-throughput technologies, revealing a diverse set of control strategies.
Figure 1.
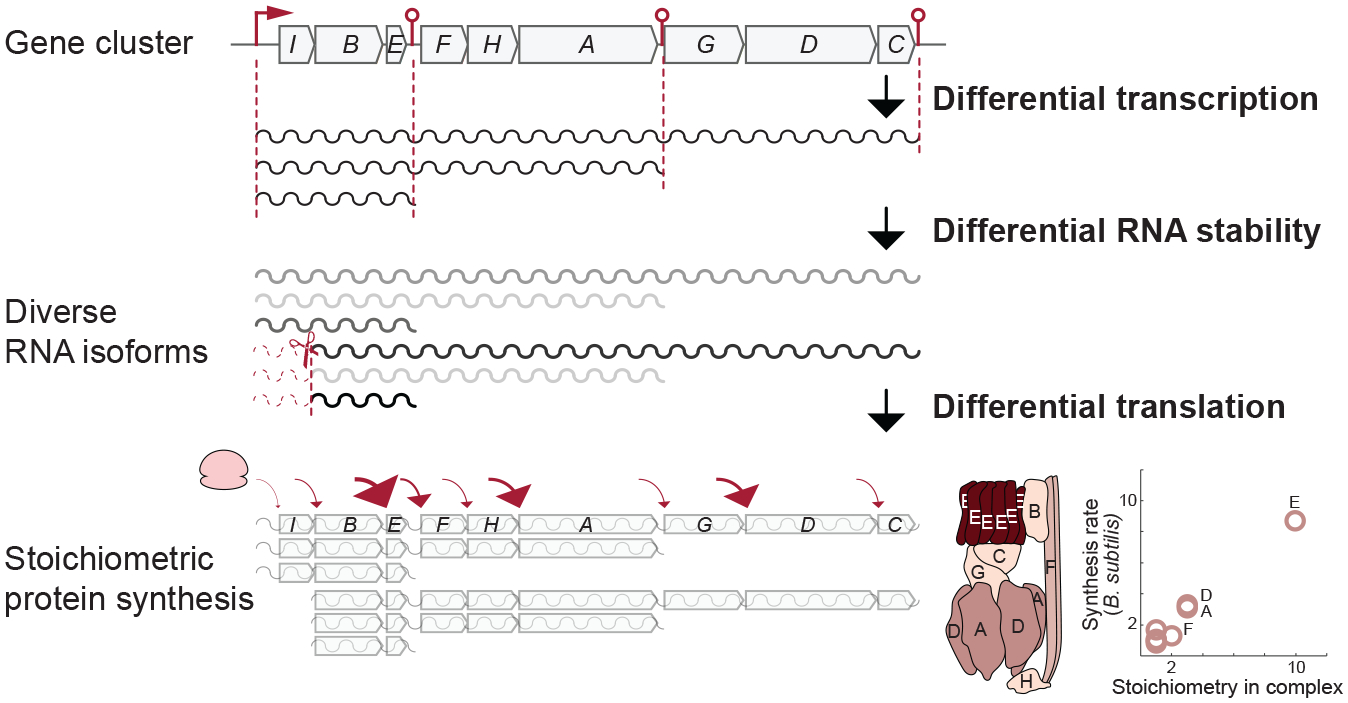
Schematic representation of the atp operon from Bacillus subtilis. Control in the expression of genes occurs at the steps of differential transcription due to leaky intrinsic terminators, followed by mRNA processing that leads to differential mRNA stability (a minor processing site in atpB is omitted for clarity; see Figure 3). The genes transcribed in the diverse polycistronic mRNA isoforms are then translated at different rates. All control steps contribute to ATP synthase subunits being produced in proportion to their stoichiometry in the complex (ribosome profiling data from Reference 96).
Here we review recent evidence for why and how bacterial cells establish quantitative control for stoichiometric protein synthesis. We start with genome-wide evidence for the requirement of precise control among coregulated genes. We then revisit the definition of operons by taking into account the diverse mRNA isoforms that one can now routinely map with high resolution using variations of RNA sequencing (RNA-seq) methods. We focus our discussion on recent progress in using these methods to understand the quantitative determinants of the stoichiometry of protein synthesis rates within operons. The stoichiometry may be further regulated by environmental signals, and Adhya (2) summarized a number of classic examples in a review. After summarizing these operon-based controls, we briefly discuss the challenges that nonproximal genes must face for concerted regulation to maintain relative stoichiometry. Finally, we close by providing a perspective on the remaining challenges that must be overcome to achieve a fully quantitative understanding of protein production in bacteria.
2. PRECISE PROTEIN SYNTHESIS CONTROL: PROPORTIONAL SYNTHESIS AND IN-PATHWAY STOICHIOMETRIES
2.1. Historical Perspective
As early as the 1960s, the study of protein synthesis in phage-infected cells provided clear evidence for differential protein synthesis from polycistronic genes (125). Despite being encoded on the same viral RNA genome, different phage proteins exhibit orders of magnitude of variation in synthesis (124). This allows the phage to produce many more copies of coat proteins than enzymes for replicating the phage. Such early evidence for gene regulatory mechanisms that tune protein synthesis stoichiometries motivated work in subsequent decades to understand the molecular basis for this control, particularly the encoding of translation efficiency (144). Stark examples of tuned protein synthesis stoichiometries continued to emerge as our understanding of bacterial physiology expanded (reviewed in 2). However, it remained unknown whether there is a broad requirement of stoichiometric protein synthesis for most operons.
2.2. Genome-Wide Quantification of Protein Synthesis
It was not until recently that tools became available to quantify the rates of protein synthesis at a genomic scale. The classic approach of metabolic labeling followed by 2D electrophoresis is not suited to simultaneously detecting thousands of proteins whose abundances span orders of magnitude, and it requires a potentially perturbative metabolic labeling step (98). mRNA levels might also be used as a proxy for protein synthesis, but such an approximation misses the greatly varying translation efficiencies observed in bacterial RNAs (100). Recent advances in mass spectrometry techniques allow increasingly accurate absolute quantification of protein synthesis, though high-precision comparisons between different proteins remain difficult (reviewed in 3, 23).
The development of ribosome profiling circumvented these challenges in globally quantifying protein synthesis. By capturing ribosome-protected mRNA footprints transcriptome-wide, ribosome profiling allows direct, genome-wide characterization of translation (80). When most ribosomes have equivalent average elongation rates and produce complete proteins, ribosome footprint density can be used as a readout of protein synthesis rates from each gene (99, 100). Ribosome profiling can be flexibly applied to diverse organisms, including a number of model and nonmodel bacteria (64, 96, 148).
2.3. Evidence for Precisely Tuned Stoichiometry
The global picture of protein synthesis provided by ribosome profiling has revealed general principles shaping proteome stoichiometry. In Escherichia coli, a comprehensive survey of obligate protein complex subunits found that they are synthesized with rates proportional to their required stoichiometry in the complex (100). This proportional synthesis indicates selective pressure against wasteful production of excess subunits, which may be more prone to aggregation and drive proteotoxicity (127). A similar principle also applies to modules for which functional constraints demand hierarchical expression, such as type II toxin-antitoxin systems or sigma–anti-sigma pairs, for which protein synthesis rates have evolved to ensure adequate amounts of the antagonist protein for robust inhibition of their partner despite active protein degradation (1, 100, 177). Proportional synthesis of protein-complex subunits appears to be widespread beyond E. coli, occurring for complexes in model gram-positive bacteria and budding yeast and for large protein complexes such as the ribosome and proteasome in higher eukaryotes (96, 160, 161).
Beyond obligate protein-complex subunits, ribosome profiling has revealed surprising requirements of relative synthesis rates for many multi-enzyme pathways that defy simple rationalization. A comparative analysis of the synthesis of proteins in cellular pathways conserved between diverse bacterial species, including evolutionarily distant E. coli and Bacillus subtilis, showed quantitatively conserved stoichiometries within cellular pathways such as translation, DNA repair, and numerous metabolic processes (96). The gene regulatory strategies that underpin these synthesis rates dramatically diverge between E. coli and B. subtilis, with widespread remodeling of operon structures and compensatory changes in translation efficiency preserving in-pathway synthesis stoichiometries (96). The divergence in gene regulatory strategies helped reveal fundamental differences in gene expression machinery between distantly related bacteria, including a lack of functional transcription-translation coupling in B. subtilis that prevents ribosome-mediated cotranscriptional regulation (83).
Convergent evolution of enzyme stoichiometries suggests that strongly preferred ratios exist for constituents of many cellular pathways, despite evidence suggesting excess capacity for enzymes (73, 87, 130). The molecular mechanisms underlying this selective pressure have been characterized experimentally for factors within the translation pathway such as the aminoacyl tRNA synthetases (129) and peptide chain release factors (95a). The extent of expression conservation over large evolutionary times underscores that relative protein synthesis rates must commonly be tuned to a degree that places strict constraints on precision at every step of the central dogma. New technologies for quantitative interrogation of gene expression can now uncover the mechanistic basis of such stoichiometric synthesis among functionally related proteins.
3. THE OPERON REVISITED: ALTERNATIVE mRNA ISOFORMS IN PROKARYOTES
3.1. Genomic Organization of Functionally Related Genes
In bacteria, proteins that take part in related cellular functions are often encoded in operons, which were originally defined as genetic units of coordinated expression (82). In the canonical view, coordinated expression is achieved via integral cotranscription of a whole gene cluster into a single polycistronic mRNA, allowing individual regulatory elements to transduce modulating signals on numerous genes at once.
Several hypotheses pertaining to the adaptive value of polycistronic transcripts have been formulated, e.g., cotranscription reduces noise in stochastic gene expression (159), facilitates complex assembly (153), or supports coregulation (82, 133). For any gene of a cotranscribed cluster, production is proportional to the RNA polymerase (RNAP) initiation frequency, with additional downstream control points contributing multiplicatively to production of each member. Hence, with cotranscription, expression changes in a functional module are actuated through control of RNAP initiation, and the orthogonal dimension of relative member stoichiometry is implemented by additional intraoperonic regulatory signals.
Importantly, operons do not always warrant equal production among cotranscribed genes within a polycistronic mRNA, and some production stoichiometries for enzymatic pathways span orders of magnitude. One way to enable such a wide range of differential protein production is diversification of mRNAs arising from a gene cluster. Indeed, in contrast to the original operon hypothesis put forth by Monod & Jacob (82), subsequent studies indicated that often there is not a unique integral mRNA per operon but rather a diverse set of isoforms (e.g., 33, 37, 71, 96, 102, 178). These isoforms not only provide different amount of templates for translation but also can possess dramatically different translation efficiencies (109a). Resolving and quantitating these isoforms genome-wide remained difficult until recently.
3.2. Mapping and Quantifying the Diversity of Primary RNA Isoforms
Variants of RNA-seq methods (recently reviewed in 76) now permit characterization of bacterial transcriptomes with unprecedented precision in both the quantification of mRNA abundance and localization of 5′ and 3′ mRNA boundaries. mRNA ends can be mapped at single-nucleotide resolution transcriptome-wide. Transcription start sites and other stable 5′ ends have been identified using various methods, such as differential RNA-seq (151), high-throughput 5′ RACE (rapid amplification of 5′ complementary DNA ends) (113), parallel assessment of ends (49), modified 5′ RACE (33), 5′-end sequencing (176), and EMOTE (exact mapping of transcript ends) (88). Stable 3′ ends of mRNAs can also be mapped at high throughput (43, 118).
In conjunction with a global readout that reports mRNA abundance, end-mapping can be used to enumerate isoforms and reveal complex transcript architectures. However, unit-by-unit quantification of identified isoforms is difficult, especially for operons with nested transcripts (33, 37). In particular, systematic biases introduced by certain molecular cloning steps can confound abundance assessments. For example, generation of the first strand of complementary DNA (cDNA) from randomly primed reverse transcription leads to a spurious depletion of signal at transcript 3′ ends (40), because fewer priming events cover these regions. Such biases preclude abundance measurement of overlapping transcripts. End-enriched RNA-seq (Rend-seq) (96), an all-in-one method that simultaneously maps 5′ and 3′ ends of transcripts and accurately quantifies mRNA abundance, partly circumvents these limitations. Rend-seq relies on sparse fragmentation of transcripts (157) followed by near-quantitative cloning of short fragments to a cDNA library (80). The resulting signal is increased by a factor of >50 at ends of mRNAs over single base pairs and is largely constant across gene bodies. From the clear demarcations of mRNA boundaries and precise abundance quantitation, individual isoform abundances can be reconstructed (96) (Figure 2).
Figure 2.
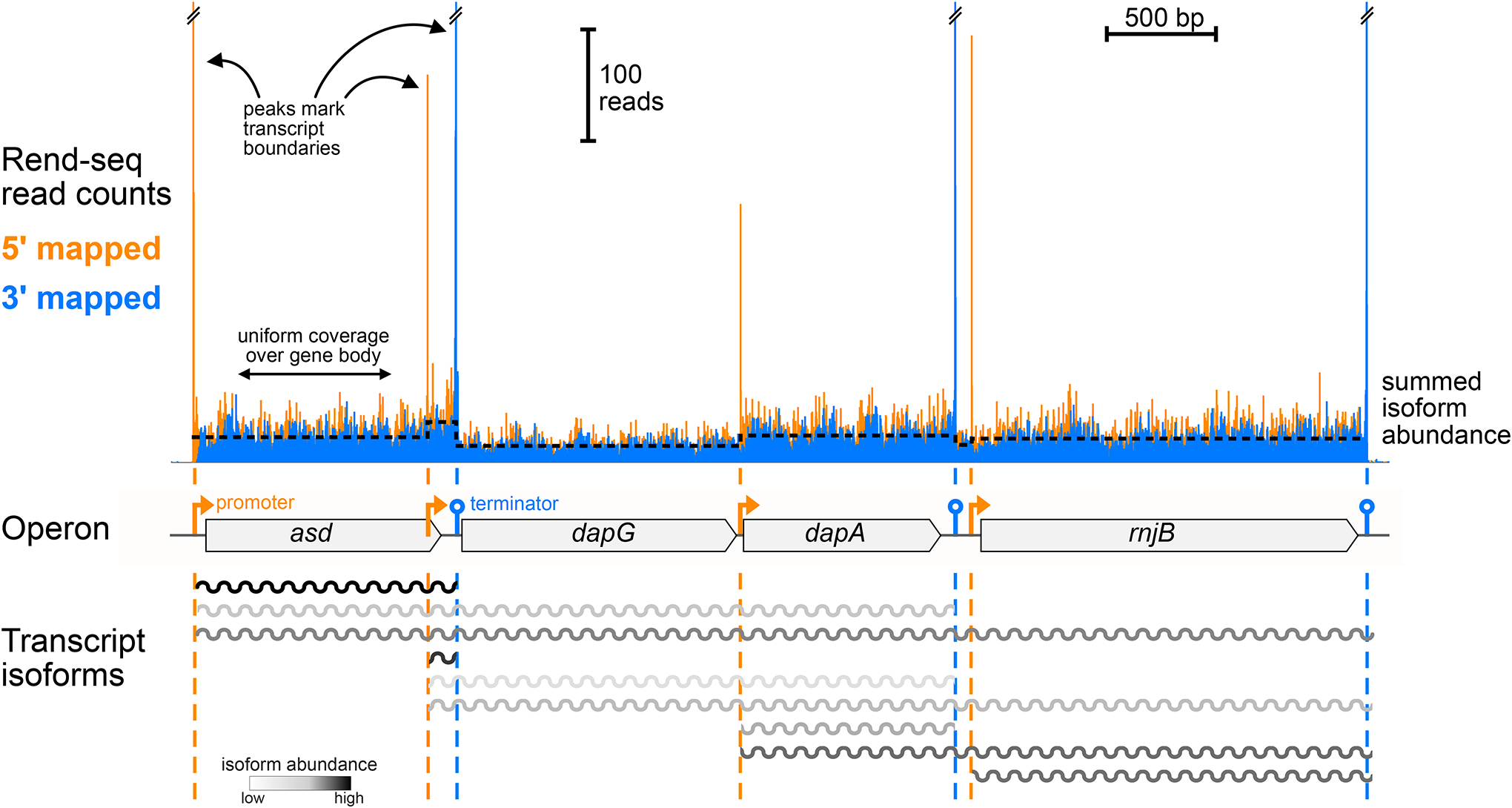
End-enriched RNA-seq (Rend-seq) data (from 96) showing coverage trace for the asd operon in Bacillus subtilis, which includes four cotranscribed genes. Peaks in 5′-mapped (orange) 3′-mapped (blue) reads mark mRNA boundaries. Four promoters can be seen, as well as three intrinsic terminators. Transcript ends were confirmed not to arise from mRNA processing by orthogonal experiments (not shown). The internal promoters and terminators lead to a complete set of nine possible mRNA isoforms, highlighting the possible complexity of transcription architecture. The read coverage between peaks can be used to infer the abundance of each isoform.
Transcription initiation and termination are major contributors to the observed mRNA isoform diversity of bacterial operons. Internal promoters are estimated to make up nearly 30% of start sites in E. coli (37). Transcription termination also plays an underappreciated role in generating diversity in operon transcripts. Programmed partial intrinsic transcription termination between genes, such as the early documented example of a leaky terminator in the σ70 operon in E. coli (25), occurs at hundreds of locations in the genomes of multiple species (37, 96). These programmed terminators act as tuning cis-elements to reduce production of genes downstream and can have activities spanning a wide dynamic range in readthrough, from a small percentage to near complete leakiness. When present in combination, internal promoters and programmed terminators can lead to a complex set of mRNAs covering a gene cluster [e.g., the asd operon in B. subtilis, with three internal promoters and two internal terminators, leading to a set of nine possible isoforms (Figure 2)].
Until recently, RNA-seq methods mostly focused on short-read approaches and did not capture long-range information in mRNAs. Consequently, possible correlations between regulatory elements were difficult to identify at a genomic scale. Techniques have now been developed to quantify full-length mRNAs on a variety of platforms (70, 84, 132, 178). Full-length approaches have already revealed intriguing isoform-specific activity of regulatory elements such as transcription terminators (178). Sequencing depth and size-dependent biases remain important bottlenecks for genome-wide characterization using these techniques.
3.3. Quantitative Determinants of Transcriptional Output
The transcription rate of an operonic gene is determined by the combined activities of upstream promoters and the intervening programmed transcription terminators. Quantitative knowledge of the determinants of these two regulatory elements is thus at the core of understanding stoichiometric expression control.
Bacterial promoters are specified by sigma factor–recognition sequences, and their activity is influenced by myriad features (reviewed in 112), including the quality of the promoter binding site; the concentration of RNAPs available to transcribe mRNAs; the number, quality, and relative positions of nearby regulatory elements [e.g., transcription factor (TF)-binding sites]; the available concentration and allosteric properties of TFs; the concentration of TF-cognate ligands; the competition between alternative sigma factors; abortive initiation; possible RNAP reinitiation; the supercoiling state of the DNA, etc.
Progress toward empirically identifying quantitative determinants of promoter activity has been driven by sequencing-based, massively parallel reporter assays (reviewed in 90) in synergy with quantitative thermodynamic models of protein-DNA interactions. This biophysical viewpoint condenses promoter complexity to a single property by positing that activity is ultimately proportional to the probability that an RNAP holoenzyme is bound to the promoter region (15). The resulting framework leads to quantitative predictions (20, 63, 171) and reduces the determination of promoter output to the identification of TF-binding sites and measurement of corresponding binding affinities. Dissection of these features is possible: Sequence-to-function relationships for RNAP and TF-binding sites have been measured at scale (8, 91, 169, 174) and binding sites precisely identified across the genome (11, 81, 168). Still, computational predictions remain imperfect (20, 63), in part due to interactions between sequence elements. For example, promoters with consensus −10 and −35 σ70 recognition sites show hard-to-predict tenfold change in activity across different surrounding sequences (169), and even binary classification of de novo DNA sequences in promoter (some activity) versus nonpromoter (no activity) categories is largely unresolved (168).
With respect to transcription termination, although much is known mechanistically about intrinsic terminators (reviewed in 135), these insights have not permitted quantitative predictions of activity. In particular, properties enabling intrinsic transcription terminators to finely punctuate operons via partial levels of RNAP readthrough are yet to be completely identified despite the simplicity of the distinctive signature of terminators: an RNA hairpin immediately followed by a stretch of multiple U nucleotides. Large numbers of intrinsic terminators’ activities have been measured both in libraries using fluorescence reporters (27, 32, 96) and sequencing (78) and in endogenous genomic contexts with RNA-seq-based approaches (84, 96, 118, 178). However, attempts to relate terminators’ biophysical properties to termination activity only capture a small fraction of the observed variance (27, 32, 96).
Quantitative prediction of terminator activity is challenging for several reasons. First, the interaction between the nascent RNAs and the RNAP is inherently kinetic (172). Second, in vivo RNA secondary structures are difficult to predict (141), especially given upstream RNA structures possibly competing with hairpin formation (27). Third, despite terminology, intrinsic terminators can be influenced by a number of proteins, such as transcription elongation factors (72, 106, 118) and sigma factors, via their possible retention through the elongation cycle (74), and by the ribosome itself, through transcription-translation coupling (101, 140). Importantly, the influence of translation on transcription termination was recently shown to be limited in B. subtilis (83), as the RNAP travels faster than ribosomes in that species. This stark difference between E. coli and B. subtilis exemplifies that regulatory rules for terminators might fundamentally differ across phylogenetic clades. Finally, RNAPs concurrently transcribing a gene could cooperate or otherwise interact (55, 89), which would modulate termination.
4. DIVERSIFYING THE TRANSCRIPTOME THROUGH RNA PROCESSING
4.1. Specifying Differential Expression Through mRNA Processing
Beyond transcriptionally generated boundaries in operonic transcripts, differential RNA stability is a common posttranscriptional mechanism to generate differential protein production from cotranscribed genes. Such events, referred to here as mRNA processing to avoid confusion with all-or-nothing, decay-initiating cleavage events, have been documented for a number of operons across many bacterial species (reviewed in 139, 165). Proper mRNA processing is required for many critical cellular functions, including cell division, for which differential stabilities following RNase E cleavage in the E. coli ftsQAZ operon set the proper ratio of cell division proteins FtsA and FtsZ required for nonfilamentous growth (25a, 162, 163).
Both exo- and endonucleases can in principle initiate processing of bacterial mRNAs. In one model, an exonuclease engages either the 5′ or 3′ end of an mRNA, processively acting until it reaches a feature that may obstruct continued degradation. In this model, the strength of the obstructing feature and differential susceptibility of transcript ends to exonucleolytic degradation control the differential abundance of coding regions. Alternatively, processing can be initiated by endonuclease cleavage within an operon, followed by differential decay of the cleavage products by exonucleases. The rate of endonuclease cleavage and exonuclease susceptibility of both ends of each cleavage product impact RNA abundances and hence protein synthesis stoichiometry in this model. The specifics of the RNA decay machinery in any particular bacterium will determine which of these routes is preferentially used and what mRNA features might specify efficient processing. In this review, we focus primarily on how mRNA processing is controlled in E. coli and B. subtilis, the two species with perhaps the best-characterized mRNA decay pathways.
Processing and decay of bacterial mRNAs are performed by a broad suite of endo- and exonucleases, and though the cohort of enzymes present in any given species can vary significantly (reviewed in 79, 117), a few common principles have emerged. At the core of the E. coli and B. subtilis mRNA decay pathways are an endonuclease (RNases E and Y, respectively), 3′–5′ decay (fulfilled by polynucleotide phosphorylase and a small number of other exonucleases that vary between species), and 5′→3′ decay activity. 5′→3′ decay is carried out by RNase J1 in B. subtilis, while in E. coli, repeated cleavage by RNase E followed by 3′–5′ exonuclease activity can fill a similar role, as RNase J1 is absent in this organism (reviewed in 36). RNases E and Y form membrane-associated oligomers and recruit a larger mRNA decay complex termed the degradosome, typically comprising exonucleases, RNA helicases, and glycolytic enzymes (reviewed in 164). Both endonucleases are thought to form these larger complexes, though RNase Y degradosomes appear to be less stable in B. subtilis than their equivalent species in E. coli, and the functional importance of such higher-order assembly in the degradosome remains unclear (164). Additional enzymes, such as RNases III and G, play a more limited role in RNA decay (34, 49, 53, 104, 156), and more comprehensive reviews of the many players in bacterial RNA decay can be found in References 10, 79, 117, and 165.
Though many examples of differential stability within operons have been characterized on a case-by-case basis, recently developed high-resolution RNA-seq techniques allow transcriptome-wide mapping of mRNA processing activity analogous to mapping of initiation and termination (43, 49, 96, 136, 151). By globally inhibiting transcription initiation with rifampicin, RNA-seq can be used to determine the individual half-lives of coding regions within each polycistronic operon (31, 44, 115). In E. coli, such an approach, combined with high-resolution mapping of 5′ and 3′ ends, revealed that dozens of operons have gene stoichiometries tuned by differential RNA stability (44). These differential decay rates appear to be shaped in part by terminal secondary structures and ribosome density along cleavage fragments. Though no global estimate of mRNA half-life based on RNA-seq is currently available in B. subtilis, Rend-seq has revealed processing events in tens of operons where endonucleolytic cleavage yields one product that is substantially more stable than the other (48). For example, multiple such cleavage events occur in the B. subtilis ATP synthase operon, shown in Figure 3. Differential stabilities are thus a common strategy to allow deviation from one-to-one stoichiometry for cotranscribed genes, despite differences in mRNA decay machinery.
Figure 3.
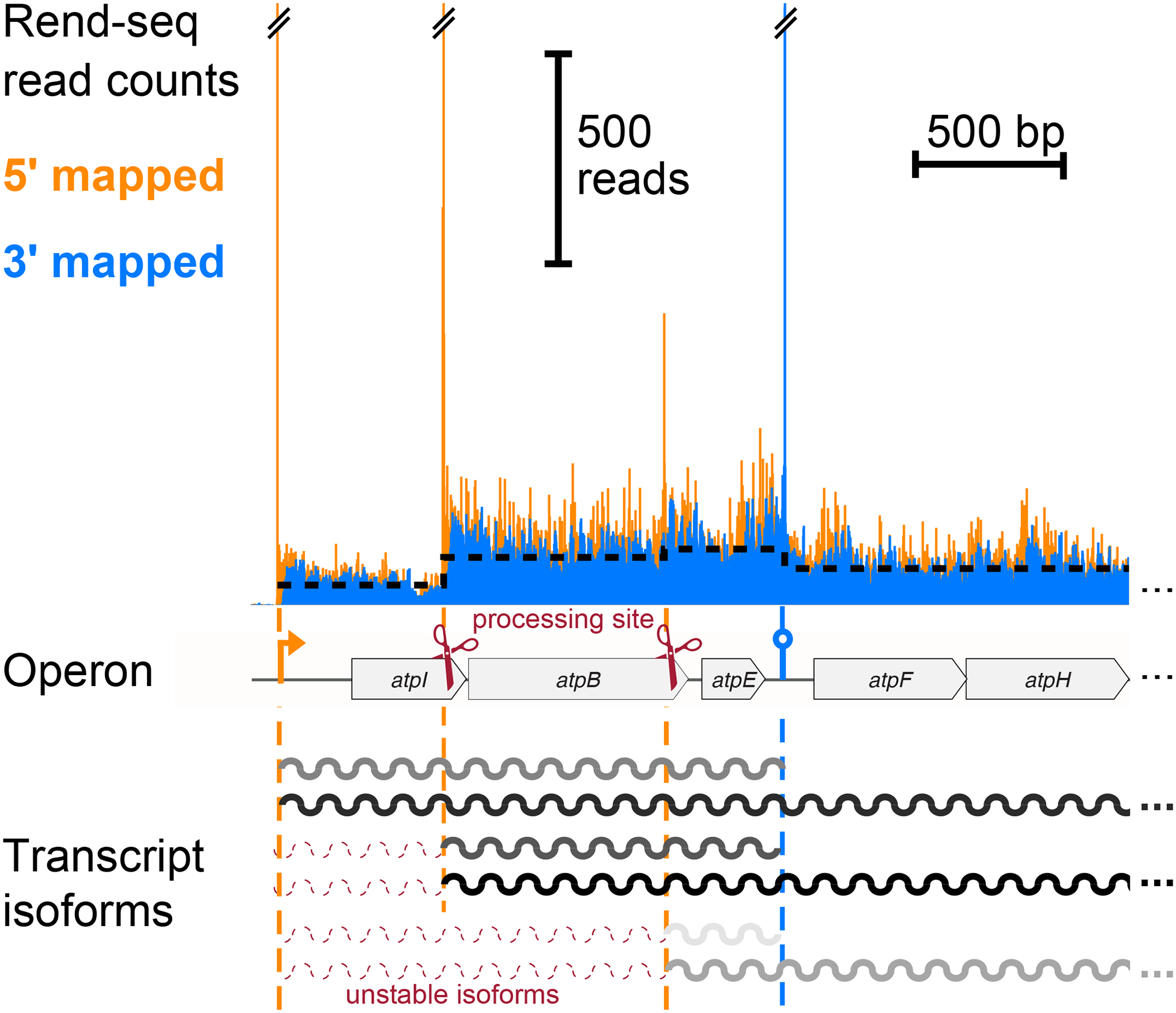
End-enriched RNA-seq (Rend-seq) data (from Reference 96) showing coverage trace for the atp operon in Bacillus subtilis, truncated to include only the first five genes. Peaks in 5′-mapped (orange) and 3′-mapped (blue) reads mark mRNA boundaries. Two RNase Y cleavage sites (scissors), one promoter, and one intrinsic terminator are shown. mRNA processing sites validated through orthogonal experiments (not shown). Darker gray indicates higher abundance of transcript isoforms (as in Figure 2), and red, dashed isoforms are rapidly degraded and therefore undetectable.
4.2. Features Encoding Differential mRNA Stabilities
To understand how differential stabilities are encoded, one must first consider what makes an mRNA an efficient substrate for processing or degradation by key players in RNA decay. Among the best-characterized decay-initiating nucleases in bacteria, RNase E is thought to degrade mRNAs through an initial cleavage (through either 5′-end-dependent or direct entry pathways) followed by sequential cleavage events in the 5′→3′ direction (allowing for 5′→3′ decay in organisms lacking a 5′→3′ exonuclease such as RNase J1) (reviewed in 10, 79, 117, 165). This activity can be inhibited by local secondary structure or other obstructions such as proteins bound to the mRNA (110, 137). Through selective capture of 5′-monophosphate-containing RNAs in a previously characterized temperature-sensitive RNase E allele, global cleavage profiles of both the E. coli and Salmonella enterica RNase E enzyme have been determined, revealing thousands of cleavage sites across the transcriptome (5, 29, 34). RNase E demonstrates a strong preference for unpaired, AU-rich substrates (110, 111), and for a U at position +2 relative to the position of cleavage, with the S. enterica RNase E recognizing a degenerate motif of the form RN↓WUU (29). RNase E can also interact with and be directed by stem-loop structures in its targets (7). Because RNase E is membrane localized, substrate proximity to the membrane is also a determinant of RNase E activity (115).
In many bacteria lacking RNase E, particularly gram-positive bacteria such as B. subtilis, RNase Y is the primary endonuclease initiating RNA processing and decay. Akin to RNase E, RNase Y is thought to be sensitive to the 5′ moiety of an mRNA (150), acting more efficiently on 5′-monophosphorylated RNAs in an end-dependent manner. However, RNase Y is also known to efficiently cleave internally within some mRNAs with a 5′ triphosphate (179). As with RNase E, RNase Y is thought to prefer unstructured, AU-rich regions, with an apparent preference for structure downstream (108, 150). In Staphylococcus aureus, transcriptome-wide mapping of RNA 5′ ends by EMOTE revealed a sequence preference for G-1 relative to the site of cleavage (88). Though RNase Y is thought to be the primary initiator of mRNA turnover in B. subtilis (53, 60, 95, 97, 165), its set of mRNA targets appears to be more limited in S. aureus and Streptococcus pyogenes (21, 88, 107), suggesting that the centrality of RNase Y in mRNA turnover may vary between organisms. The target repertoire of RNase Y in organisms more reliant on its activity, such as B. subtilis, remains to be exhaustively characterized.
Though mRNA processing and turnover are frequently initiated by endonucleases that are capable of cleaving RNA in a 5′-end-independent manner, both endonuclease and exonuclease activities can be stimulated by the presence of a 5′-monophosphate at the 5′ end of an mRNA. Such a moiety can be generated either by endonucleolytic cleavage or by the activity of RNA pyrophosphohydrolase (RppH), which is present in both E. coli and B. subtilis. RppH plays a role in decay initiation for a subset of the transcriptome, with hundreds of transcripts enriched when RppH is deleted or inactivated in both organisms (45, 65), despite evidence that B. subtilis encodes multiple enzymes that can catalyze this activity (77, 138). E. coli RppH and B. subtilis RppH have differing sequence preferences, with B. subtilis demonstrating stricter requirements, including a strong preference for a G in position +2 (62, 77). Interestingly, in E. coli, RppH is much more active against a diphosphorylated 5′ end, an apparently abundant moiety generated by an unknown phosphatase (105). In both E. coli and B. subtilis, RppH activity is strongly impeded by base-pairing at the 5′ end of an mRNA (62, 77). Downstream of either RppH or endonuclease activity, 5′ hairpins can additionally inhibit activity of RNase J1, which requires at least four unpaired nucleotides to initiate degradation (51). 3′–5′ exonucleases can be similarly impeded by 3′ hairpins (reviewed in 165). Terminal secondary structure can therefore both inhibit RNA decay initiation and stabilize decay intermediates.
The bacterial RNA decay machinery acts in consort with numerous RNA and protein factors that modulate their specificity, specifying locations of mRNA processing. Most known examples of mRNA processing in B. subtilis are dependent on a protein complex termed the Y-complex (YlbF, YmcA, and YaaT), which is known to physically interact with RNase Y (47, 48). Absence of any member of this complex ablates mRNA processing activity at tens of sites in the transcriptome (48), including the glycolytic operon (35) and the ATP synthase operon (Figure 3). In E. coli, a substantial network of small RNAs (sRNAs) contributes to mRNA processing through multiple modes of action. sRNA binding is able to direct cleavage by endonucleases, and RNase III and RNase E have both been implicated in sRNA-directed mRNA processing (9, 126). Beyond directing cleavage by endonucleases, sRNAs can inhibit exonuclease (or sequential RNase E) activity, resulting in differential stabilization of operon decay intermediates (reviewed in 9). Such a mechanism regulates glucose homeostasis in E. coli and may be a common occurrence in this organism (128).
Though we understand many of the major processes involved in differential stabilization of operonic genes, we still lack a predictive understanding of what makes an mRNA stable. Because the primary sequence determinants specifying decay initiation through endonucleases or RppH appear highly degenerate and some downstream products of RNA processing are inherently unstable, it is difficult to precisely map the determinants of differential mRNA stabilities. Secondary structure plays a role in directing RNA processing events, but precisely attributing cleavage events to secondary structural motifs is constrained by our ability to precisely predict or measure mRNA folding transcriptome-wide (141). As an additional complication, RNA decay may be concurrent with transcription and translation, the latter known to potentially stabilize mRNAs through occlusion of RNA decay machinery (165, 180). Application of massively parallel reporter assays, extensively used to study transcription and translation but not yet RNA decay, may provide the degree of control needed to dissect determinants of efficient processing at known sites. Further work coupling perturbations to the RNA decay machinery with newly developed RNA-end-mapping techniques may also continue to yield a higher-resolution map of mRNA processing across transcripts and species.
5. DIFFERENTIAL TRANSLATION EFFICIENCY TO TUNE PROTEIN PRODUCTION
For a given polycistronic mRNA isoform, each gene may be translated at a different rate. We refer to the rate of protein synthesis per copy of mRNA for a given gene as the translation efficiency (99). Different genes in the same transcript can have translation efficiencies that differ by up to 100-fold (100, 175), which is comparable to the magnitude that can be achieved by partial transcription termination. Interestingly, it is rare to find genes that have high transcription and low translation efficiency, a phenomenon that can be explained by an evolutionary trade-off between the cost and noise of expression (75). In this section, we discuss the molecular mechanisms that contribute to differential translation efficiency (99).
5.1. Autonomous Translation and Impact of Neighboring Genes
The translation efficiency of different genes in the same transcript may be dependent on or independent of each other. For many operons, every gene carries its own ribosome-binding site (RBS), which consists of a Shine-Dalgarno sequence and a start codon. Translation can thus initiate at each RBS regardless of its neighbors. There are also mechanisms that make translation of a gene dependent on translation of neighboring genes. Some of the best-characterized examples are specialized mechanisms at leader peptides whose translational stalling can alter mRNA folding and influence the accessibility of the RBS of the downstream genes (121, 122). These mechanisms have been exploited to create synthetic genes in a bicistronic context to enable controllable translation (26, 120). Because leader peptides are often not functional after their synthesis, this type of mechanism mainly serves as a way to regulate operon-wide expression and not relative stoichiometry.
Similar translational coupling between neighboring genes may be responsible for maintaining uniform translation efficiency across many ribosomal proteins encoded in the same operon. For example, ribosomal protein operons are often regulated by one of the proteins they encode, whereby an excess of unassembled subunits bind to and downregulate genes on the same mRNA (123). Coupled translation would potentially allow a single feedback regulator to control the translation of all genes stoichiometrically. Although it was originally proposed that the same ribosome would translate sequentially through such linked genes (149), more recent evidence has suggested that the dependency might be due to modulation of RBS accessibility for de novo initiation by the translation of upstream genes (30, 109, 119, 142). Still, compared to well-characterized leader peptides, the exact mechanism leading to the equimolar production rates among translationally coupled genes is less clear.
For most operons, protein production is not equimolar and requires distinct translation efficiency at each gene. As different genes are translated differentially, operon RNAs also form distinct domains of secondary structures that are separated by gene boundaries (24). RNA-folding models based on genome-wide secondary structure probing have shown that intra–open reading frame (ORF) base-pairing is generally favored over inter-ORF base-pairing, except for genes that may be translationally coupled (24, 119). Intra-ORF base-pairing is intrinsic to the evolved RNA sequence and provides a blueprint for translation efficiency, as we discuss in the following section.
5.2. Determinants of Initiation Frequency
It is generally thought that translation efficiency is determined by the initiation rate, as opposed to elongation rates, because the steady-state flux of initiating ribosomes must be equal to the flux of peptide and ribosome release (92, 99, 158). To initiate translation, the RBS must not be base-paired to other regions of the mRNA. Indeed, large-scale studies based on massively recoded reporters in E. coli have shown that translation efficiency is anticorrelated with the thermodynamic stability of RNA folding for an RBS (13, 18, 19, 26, 56, 67, 93). Ribosome profiling data for endogenous genes also showed a consistent trend (100). These results suggest that the close relationship between RNA secondary structure and translation efficiency—originally observed for the polycistronic bacteriophage RNAs (103)—is a general mechanism across the host genome.
Typically, the region of mRNA containing the RBS is less structured than other parts of the gene body, which facilitates translation initiation (12, 57, 145). Avoidance of RNA secondary structure places a constraint on codon usage, an effect that is most pronounced for the N-terminal residues for which codon substitutions strongly influence translation efficiency via RNA folding (13, 19, 56, 93). These results offer an explanation for the intriguing phenomenon that rarely used codons are enriched at the 5′ end of ORFs (167). Long-range base-pairing can also play a role in reducing the accessibility of the RBS, as synonymous codons further downstream in the gene can reduce translation efficiency if they are the reverse complement of the Shine-Dalgarno sequence (13, 85). The entirety of each coding sequence likely has evolved under this RNA folding constraint. Therefore, bacterial operons not only encode units of protein sequences but also specify the relative translation efficiency through ORF-wide secondary structures (24).
It should be noted that RNA secondary structures affect both translation efficiency and decay rates (46). Large-scale studies of the effects of gene sequence on translation have demonstrated a nearly universal correlation between protein levels and mRNA levels, making it difficult to disentangle the contribution of a synonymous variant to translation efficiency from the contribution to RNA decay (13, 18, 26, 56, 67, 93, 94). In the context of polycistronic operons, it remains to be determined how the distinct units of secondary structures and translation efficiency affect the stability of an entire mRNA.
In addition to secondary structures, the affinity of the Shine-Dalgarno sequence to the anti-Shine-Dalgarno region of the 16S rRNA is also relevant in E. coli (154). Although the correlation between the Shine-Dalgarno strength and translation efficiency is weak across endogenous E. coli genes (100), a stronger correlation can be observed once variations in secondary structure are controlled for—either using a deep mutational scanning library that only targets the Shine-Dalgarno sequence (94) or measuring the translation of orthogonal ribosomes that differ in the anti-Shine-Dalgarno region (143)). Intriguingly, orthogonal ribosomes mostly initiate at the correct start codons despite the lack of corresponding orthogonal Shine-Dalgarno sequences, again suggesting that translation initiation sites on endogenous operons are primarily hard-coded in features such as the modular secondary structures.
5.3. Roles of Elongation Kinetics
Although elongation per se does not influence the steady-state flux of protein production, it may indirectly affect translation efficiency by either triggering premature ribosome release or reducing the initiation frequency (99, 158). In the first scenario, ribosomes may be released due to stalling, collision, or programmed frameshifts (58, 59, 170). In ribosome profiling data for E. coli and B. subtilis, premature release appeared to be uncommon, except for a few specific genes that utilize this mechanism to generate truncated proteins or regulate expression (16, 39, 61, 100, 114, 166). On the other hand, a large-scale study based on randomized N-terminal coding sequence in vivo, combined with single-molecule assays in vitro, showed that ribosome stalling may occur in the first three to five codons in a context-dependent manner. Consequently, ribosomes may drop off shortly after initiation, which would be difficult to detect by ribosome profiling (170). This early ribosome drop-off may further fine-tune the initiation rates that are mainly determined by RBS accessibility.
In the second scenario, whereby elongation rates affect initiation frequency, strong ribosome pausing may lead to a pileup of trailing ribosomes that eventually occlude the RBS from further initiation. This transition occurs when the pause duration becomes longer than the time between the native initiation events (59, 99). In vivo estimates of the translation kinetics showed that the average stepping time for elongation is approximately 60 ms per codon, whereas the time between initiation events is typically a few seconds (41, 69, 86, 100). Therefore, a pause that is >20 times longer than the average elongation step time is required to create a massive ribosome pileup that can lead to RBS occlusion. Although rare codons decoded by less abundant tRNAs may have slower elongation rates, they may not cause sufficient pauses in rich media to lead to ribosome queues given that the tRNA concentration differences are typically less than 20-fold (50). Updated ribosome profiling data support this view (116). Interestingly, it has been observed that codon usage correlates with proportional synthesis, with a more biased usage for more frequently translated genes (134). This is consistent with the general trend that highly expressed genes are enriched in codons that are decoded by more abundant tRNAs. Biased codon usage is likely a consequence of evolutionary pressures to balance the supply and demand for tRNAs (54) and maximize ribosome usage (92), instead of being a driving force for translation efficiency. When the tRNA supply and demand are imbalanced because of forced expression of exogenous genes, codon choice of the overexpressed gene can become important (18). Indeed, the extent of codon influence depends on the level of overexpression (26). For native operons with cotranscription, however, ORF-specific codon usage appears to be a consequence and not a driver of translation efficiency.
6. EFFECTS OF THE BROADER CONTEXT
Although colocalization along the chromosome is common for functionally connected genes, members of pathways are sometimes not in operons. In particular, pathways involving numerous proteins, such as mRNA translation, have participating genes scattered across the chromosome. Even subunits of obligate heteromeric complexes are occasionally not contiguous (100). Placement along the chromosome can influence gene expression: A self-contained construct including a gene and its cis-regulatory elements will produce quantitatively different amounts of protein when at different genomic positions as a result of the local and long-range context. The precise requirements for stoichiometric production among proteins that span multiple distal operons suggest that the outputs of cis-regulatory elements have evolved to compensate for these position-dependent effects, through either feedback mechanisms (66) or hardwired sequence-level modulation. We briefly survey how chromosomal context affects expression.
At the simplest level, more proteins will be produced when multiple copies of the encoding gene are present in the cell. In fast-growing bacteria, single-copy genes still have different copy numbers (i.e., dosage) in the cell depending on their chromosomal position. Indeed, as one round of DNA replication initiates before completion of previous rounds, leading to multifork replication (38), genes near the origin of replication have a higher copy number compared to those close to the terminus. Quantitatively, the largest difference in gene dosage is between the origin and terminus, with a population-averaged origin-to-terminus dosage ratio higher than 3:1 in fast-growing E. coli (182). Measurements of gene expression output confirm that expression is directly proportional to chromosomal dose (17, 68, 146, 155). Importantly, the magnitude of the dosage difference can be larger than the measured stoichiometric production deviation in complexes and pathways (96, 100), so that the selective pressures sculpting cis-element activity are sensitive to these effects.
In addition to the changes in gene dosage, which gradually varies with chromosomal position, sharper local features can also influence gene expression. Concretely, gene-dosage-corrected expression typically spans a factor of 4 from position to position for insulated reporter cassettes (68, 147), or more for uninsulated ones (22). Scholz and coworkers (147) systematically investigated underlying causes of these differences, identifying AT content, binding of nucleoid-associated proteins, and proximity to high-transcription loci as features correlated with local expression propensity. Bacterial chromosomes are heavily decorated by a variety of proteins (173), and occupancy of these proteins can obstruct initiation and progression of the RNAP (reviewed in 152), leading to silencing of certain genomic regions. Furthermore, some promoters are sensitive to DNA supercoiling, which can be affected by local transcriptional activities or barriers of supercoiling spread (reviewed in 52). Additional effects involve the spatial context of a bacterial cell, with heterogeneous concentration of the central dogma machinery and specific chromosomal organization (respectively reviewed in 6 and 42). For example, the proximity of a gene locus to the inner membrane and mRNA degradation enzymes could affect its expression (115).
Overall, the nonuniform dependence of gene output on growth rate and local features raises an important question regarding how expression stoichiometry among different genes is maintained under changing environments. These results underscore that cis-regulatory elements ultimately cannot be considered in isolation and that a holistic view including local and broad genomic context effects is needed for a fully quantitative understanding.
7. CONCLUSION AND OUTLOOK
Though recent high-resolution measurements have provided insights into the regulatory landscape underpinning stoichiometric protein synthesis, our degree of predictive understanding for gene expression from these features remains relatively low. The activity of regulatory elements in bacterial operons is set by evolution to a precision at least one order of magnitude higher than what is currently achievable with the state-of-the-art synthetic biology toolbox. An aspirational challenge for the field will be to reach de novo any chosen relative expression between two exogenous genes within a factor of two or less. Currently, the best approach to achieve a prespecified stoichiometry remains to screen libraries of regulatory elements.
With limited spatial separation, the processes of bacterial gene expression are frequently highly coupled, making an understanding of transcription, translation, or RNA decay in isolation inadequate. Indeed, mounting evidence suggests that lack of modularity of regulatory elements is the norm. Careful design of experiments is critical to isolate a perturbation to only one step in gene expression and quantify any secondary alterations to transcription, translation, and mRNA stability. Further, systematic assessment of these interactions might provide a path toward integration of these concurrent processes into a coherent framework. Importantly, the gene expression machinery displays fundamental differences across bacteria, as can be seen by the limited transcription-translation coupling in Firmicutes (83). This diversity limits the species-to-species portability of regulatory elements (181), and particular care must be taken when generalizing findings across bacterial species.
Questions remain regarding how stoichiometric production is maintained in different environments. Coregulated protein production presents a challenge for systems finely tuned for proportional protein synthesis: How do the regulatory elements, shaped in sequence by evolutionary pressures toward stoichiometric synthesis, respond to fluctuating cellular demands? Regulators acting on whole operons provide one clear solution, as described for some groups of E. coli ribosomal proteins, but many pathways demonstrate more complicated operon structures or span multiple genomic loci. More comprehensive characterization of the landscape of gene regulatory networks, and in particular autoregulation, may yet reveal new buffering and feedback mechanisms robust to the fluctuating environments bacteria have evolved in.
FUTURE ISSUES.
Certain regulatory elements, such as intrinsic terminators and mRNA processing sites, are ripe for application of large-scale sequence-to-function characterization using massively parallel reporter assays.
Even for compact regulatory elements with extensive prior characterization, such as promoters, rational and model-based generation of libraries of maximally constraining elements is needed to efficiently explore the large DNA sequence space, with the particular goal of high generalizability of learned statistical or biophysical models.
The ever-increasing catalog of bacterial genomes holds potentially useful information about the regulatory code. Can evolutionary covariation analyses be used to constrain the quantitative sequence-to-function relationships of cis-elements?
Systematic quantification of regulatory elements should be undertaken in a wider diversity of bacteria to assess divergence in the regulatory code across the full bacterial phylogeny.
To what extent is expression stoichiometry conserved across steady states of growth, which are very different from rapid growth, such as in stress or nutrient-poor conditions?
What design principles allow regulatory elements for genes in the same pathway, but discontiguous on the chromosome, to be seamlessly induced or repressed across various growth conditions?
Interaction between the various processes of the central dogma, and by extension between the underlying regulatory elements controlling these processes, should be explored systematically and at high throughput using tools similar to those already available to assess sequence-to-function relationships. Examples include interaction between translation initiation (ribosome-binding site) and endonuclease cleavage (cleavage site) and between transcription initiation (promoter) and transcription termination (terminator).
The underlying causes for the precise requirement for the observed expression stoichiometry for many proteins remain obscure.
ACKNOWLEDGMENTS
We thank members of the Li lab for numerous fruitful discussions. This research was supported by NIH grant R35GM124732, the NSF CAREER Award, the Smith Odyssey Award, the Pew Biomedical Scholars Program, a Sloan Research Fellowship, the Searle Scholars Program, the Smith Family Award for Excellence in Biomedical Research, an NSF graduate research fellowship (to J.C.T.), an NIH Pre-Doctoral Training Grant (T32 GM007287, to J.C.T.), an NSERC graduate fellowship (to J.-B.L.), and an HHMI International Student Fellowship (to J.-B.L.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Ades SE, Connolly LE, Alba BM, Gross CA. 1999. The Escherichia coli σE-dependent extracytoplasmic stress response is controlled by the regulated proteolysis of an anti-σ factor. Genes Dev 13:2449–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adhya S 2003. Suboperonic regulatory signals. Sci. STKE 2003(185):e22. [DOI] [PubMed] [Google Scholar]
- 3.Aebersold R, Mann M. 2016. Mass-spectrometric exploration of proteome structure and function. Nature 537(7620):347–55 [DOI] [PubMed] [Google Scholar]
- 4.Ames BN, Garry B. 1959. Coordinate repression of the synthesis of four histidine biosynthetic enzymes by histidine. PNAS 45(10):1453–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Apirion D, Lassar AB. 1978. A conditional lethal mutant of Escherichia coli which affects the processing of ribosomal RNA. J. Biol. Chem 253(5):1738–42 [PubMed] [Google Scholar]
- 6.Bakshi S, Choi H, Weisshaar JC. 2015. The spatial biology of transcription and translation in rapidly growing Escherichia coli. Front. Microbiol 6:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandyra KJ, Wandzik JM, Luisi BF. 2018. Substrate recognition and autoinhibition in the central ribonuclease RNase E. Mol. Cell 72(2):275–85.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes SL, Belliveau NM, Ireland WT, Kinney JB, Phillips R. 2019. Mapping DNA sequence to transcription factor binding energy in vivo. PLOS Comput. Biol 15(2):e1006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barquist L, Vogel J. 2015. Accelerating discovery and functional analysis of small RNAs with new technologies. Annu. Rev. Genet 49:367–94 [DOI] [PubMed] [Google Scholar]
- 10.Bechhofer DH, Deutscher MP. 2019. Bacterial ribonucleases and their roles in RNA metabolism. Crit. Rev. Biochem. Mol. Biol 54(3):242–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belliveau NM, Barnes SL, Ireland WT, Jones DL, Sweredoski MJ, et al. 2018. Systematic approach for dissecting the molecular mechanisms of transcriptional regulation in bacteria. PNAS 115(21):E4796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bentele K, Saffert P, Rauscher R, Ignatova Z, Blüthgen N. 2013. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol 9:675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharyya S, Jacobs WM, Adkar BV, Yan J, Zhang W, Shakhnovich EI. 2018. Accessibility of the Shine-Dalgarno sequence dictates N-terminal codon bias in E. coli. Mol. Cell 70(5):894–905.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deleted in proof.
- 15.Bintu L, Buchler NE, Garcia HG, Gerland U, Hwa T, et al. 2005. Transcriptional regulation by the numbers: models. Curr. Opin. Genet. Dev 15(2):116–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blinkowa AL, Walker JR. 1990. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res 18(7):1725–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Block DHS, Hussein R, Liang LW, Lim HN. 2012. Regulatory consequences of gene translocation in bacteria. Nucleic Acids Res 40(18):8979–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boël G, Letso R, Neely H, Price WN, Wong K-H, et al. 2016. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 529(7586):358–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borujeni AE, Salis HM. 2016. Translation initiation is controlled by RNA folding kinetics via a ribosome drafting mechanism. J. Am. Chem. Soc 138(22):7016–23 [DOI] [PubMed] [Google Scholar]
- 19a.Brar GA, Weissman JS. 2015. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol 16:651–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brewster RC, Jones DL, Phillips R. 2012. Tuning promoter strength through RNA polymerase binding site design in Escherichia coli. PLOS Comput. Biol 8(12):e1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broglia L, Lécrivain A-L, Renault TT, Hahnke K, Ahmed-Begrich R, et al. 2020. An RNA-seq based comparative approach reveals the transcriptome-wide interplay between 3′-to-5′ exoRNases and RNase Y. Nat. Commun 11:1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryant JA, Sellars LE, Busby SJW, Lee DJ. 2014. Chromosome position effects on gene expression in Escherichia coli K-12. Nucleic Acids Res 42(18):11383–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buccitelli C, Selbach M. 2020. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet 21(10):630–44 [DOI] [PubMed] [Google Scholar]
- 24.Burkhardt DH, Rouskin S, Zhang Y, Li G-W, Weissman JS, Gross CA. 2017. Operon mRNAs are organized into ORF-centric structures that predict translation efficiency. eLife 6:e22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burton ZF, Gross CA, Watanabe KK, Burgess RR. 1983. The operon that encodes the sigma subunit of RNA polymerase also encodes ribosomal protein S21 and DNA primase in E. coli K12. Cell 32(2):335–49 [DOI] [PubMed] [Google Scholar]
- 25a.Cam K, Rome G, Krisch HM, Bouché JP. 1996. RNase E processing of essential cell division genes mRNA in Escherichia coli. Nucleic Acids Res 24(15):3065–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cambray G, Guimaraes JC, Arkin AP. 2018. Evaluation of 244,000 synthetic sequences reveals design principles to optimize translation in Escherichia coli. Nat. Biotechnol 36(10):1005–15 [DOI] [PubMed] [Google Scholar]
- 27.Cambray G, Guimaraes JC, Mutalik VK, Lam C, Mai Q-A, et al. 2013. Measurement and modeling of intrinsic transcription terminators. Nucleic Acids Res 41(9):5139–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deleted in proof.
- 29.Chao Y, Li L, Girodat D, Förstner KU, Said N, et al. 2017. In vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol. Cell 65(1):39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chemla Y, Peeri M, Heltberg ML, Eichler J, Jensen MH, et al. 2020. A possible universal role for mRNA secondary structure in bacterial translation revealed using a synthetic operon. Nat. Commun 11(1):4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, Shiroguchi K, Ge H, Xie XS. 2015. Genome‐wide study of mRNA degradation and transcript elongation in Escherichia coli. Mol. Syst. Biol 11:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y-J, Liu P, Nielsen AAK, Brophy JAN, Clancy K, et al. 2013. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 10(7):659–64 [DOI] [PubMed] [Google Scholar]
- 33.Cho B-K, Zengler K, Qiu Y, Park YS, Knight EM, et al. 2009. The transcription unit architecture of the Escherichia coli genome. Nat. Biotechnol 27(11):1043–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clarke JE, Kime L, Romero AD, McDowall KJ. 2014. Direct entry by RNase E is a major pathway for the degradation and processing of RNA in Escherichia coli. Nucleic Acids Res 42(18):11733–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Commichau FM, Rothe FM, Herzberg C, Wagner E, Hellwig D, et al. 2009. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol. Cell. Proteom 8(6):1350–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Condon C 2007. Maturation and degradation of RNA in bacteria. Curr. Opin. Microbiol 10:271–78 [DOI] [PubMed] [Google Scholar]
- 37.Conway T, Creecy JP, Maddox SM, Grissom JE, Conkle TL, et al. 2014. Unprecedented high-resolution view of bacterial operon architecture revealed by RNA sequencing. mBio 5(4):e01442–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper S, Helmstetter CE. 1968. Chromosome replication and the division cycle of Escherichia coli Br. J. Mol. Biol 31(3):519–40 [DOI] [PubMed] [Google Scholar]
- 39.Craigen WJ, Caskey CT. 1986. Expression of peptide chain release factor 2 requires high-efficiency frameshift. Nature 322(6076):273–75 [DOI] [PubMed] [Google Scholar]
- 40.Creecy JP, Conway T. 2015. Quantitative bacterial transcriptomics with RNA-seq. Curr. Opin. Microbiol 23:133–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai X, Zhu M, Warren M, Balakrishnan R, Patsalo V, et al. 2016. Reduction of translating ribosomes enables Escherichia coli to maintain elongation rates during slow growth. Nat. Microbiol 2:16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dame RT, Rashid F-ZM, Grainger DC. 2020. Chromosome organization in bacteria: mechanistic insights into genome structure and function. Nat. Rev. Genet 21(4):227–42 [DOI] [PubMed] [Google Scholar]
- 43.Dar D, Shamir M, Mellin JR, Koutero M, Stern-Ginossar N, et al. 2016. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science 352(6282):aad9822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dar D, Sorek R. 2018. Extensive reshaping of bacterial operons by programmed mRNA decay. PLOS Genet 14(4):e1007354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deana A, Celesnik H, Belasco JG. 2008. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451:355–58 [DOI] [PubMed] [Google Scholar]
- 46.Del Campo C, Bartholomäus A, Fedyunin I, Ignatova Z. 2015. Secondary structure across the bacterial transcriptome reveals versatile roles in mRNA regulation and function. PLOS Genet 11(10):e1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeLoughery A, Dengler V, Chai Y, Losick R. 2016. Biofilm formation by Bacillus subtilis requires an endoribonuclease-containing multisubunit complex that controls mRNA levels for the matrix gene repressor SinR. Mol. Microbiol 99(2):425–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeLoughery A, Lalanne J-B, Losick R, Li G-W. 2018. Maturation of polycistronic mRNAs by the endoribonuclease RNase Y and its associated Y-complex in Bacillus subtilis. PNAS 115:E5585–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DiChiara JM, Liu B, Figaro S, Condon C, Bechhofer DH. 2016. Mapping of internal monophosphate 5′ ends of Bacillus subtilis messenger RNAs and ribosomal RNAs in wild-type and ribonuclease-mutant strains. Nucleic Acids Res 44(7):3373–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong H, Nilsson L, Kurland CG. 1996. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol 260(5):649–63 [DOI] [PubMed] [Google Scholar]
- 51.Dorléans A, de la Sierra-Gallay IL, Piton J, Zig L, Gilet L, et al. 2011. Molecular basis for the recognition and cleavage of RNA by the bifunctional 5′−3′ exo/endoribonuclease RNase J. Structure 19:1252–61 [DOI] [PubMed] [Google Scholar]
- 52.Dorman CJ. 2006. DNA supercoiling and bacterial gene expression. Sci. Prog 89(Part 3–4):151–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Durand S, Gilet L, Bessières P, Nicolas P, Condon C. 2012. Three essential ribonucleases—RNase Y, J1, and III—control the abundance of a majority of Bacillus subtilis mRNAs. PLOS Genet 8:e1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53a.Eisen MB, Spellman PT,Brown PO,Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. PNAS 95(25):14863–68. Erratum. 1999. PNAS 96(19):10943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elf J, Nilsson D, Tenson T, Ehrenberg M. 2003. Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300(5626):1718–22 [DOI] [PubMed] [Google Scholar]
- 55.Epshtein V, Nudler E. 2003. Cooperation between RNA polymerase molecules in transcription elongation. Science 300(5620):801–5 [DOI] [PubMed] [Google Scholar]
- 56.Espah Borujeni A, Borujeni AE, Cetnar D, Farasat I, Smith A, et al. 2017. Precise quantification of translation inhibition by mRNA structures that overlap with the ribosomal footprint in N-terminal coding sequences. Nucleic Acid Res 45:5437–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eyre-Walker A, Bulmer M. 1993. Reduced synonymous substitution rate at the start of enterobacterial genes. Nucleic Acids Res 21(19):4599–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farabaugh PJ. 1996. Programmed translational frameshifting. Annu. Rev. Genet 30:507–28 [DOI] [PubMed] [Google Scholar]
- 59.Ferrin MA, Subramaniam AR. 2017. Kinetic modeling predicts a stimulatory role for ribosome collisions at elongation stall sites in bacteria. eLife 6:e23629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Figaro S, Durand S, Gilet L, Cayet N, Sachse M, Condon C. 2013. Bacillus subtilis mutants with knockouts of the genes encoding ribonucleases RNase Y and RNase J1 are viable, with major defects in cell morphology, sporulation, and competence. J. Bacteriol 195:2340–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flower AM, McHenry CS. 1990. The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. PNAS 87(10):3713–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foley PL, Hsieh P-K, Luciano DJ, Belasco JG. 2015. Specificity and evolutionary conservation of the Escherichia coli RNA pyrophosphohydrolase RppH. J. Biol. Chem 290(15):9478–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Forcier TL, Ayaz A, Gill MS, Jones D, Phillips R, Kinney JB. 2018. Measuring cis-regulatory energetics in living cells using allelic manifolds. eLife 7:e40618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fremin BJ, Sberro H, Bhatt AS. 2020. MetaRibo-Seq measures translation in microbiomes. Nat. Commun 11(1):3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frindert J, Zhang Y, Nübel G, Kahloon M, Kolmar L, et al. 2018. Identification, biosynthesis, and decapping of NAD-capped RNAs in B. subtilis. Cell Rep 24:1890–901.e8 [DOI] [PubMed] [Google Scholar]
- 66.Garmendia E, Brandis G, Hughes D. 2018. Transcriptional regulation buffers gene dosage effects on a highly expressed operon in Salmonella. mBio 9:e01446–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goodman DB, Church GM, Kosuri S. 2013. Causes and effects of N-terminal codon bias in bacterial genes. Science 342(6157):475–79 [DOI] [PubMed] [Google Scholar]
- 68.Goormans AR, Snoeck N, Decadt H, Vermeulen K, Peters G, et al. 2020. Comprehensive study on Escherichia coli genomic expression: Does position really matter? Metab. Eng 62:10–19 [DOI] [PubMed] [Google Scholar]
- 69.Gorochowski TE, Chelysheva I, Eriksen M, Nair P, Pedersen S, Ignatova Z. 2019. Absolute quantification of translational regulation and burden using combined sequencing approaches. Mol. Syst. Biol 15(5):e8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grünberger F, Knüppel R, Jüttner M, Fenk M, Borst A, et al. 2020. Exploring prokaryotic transcription, operon structures, rRNA maturation and modifications using Nanopore-based native RNA sequencing. bioRxiv 2019.12.18.880849. 10.1101/2019.12.18.880849 [DOI] [Google Scholar]
- 71.Güell M, van Noort V, Yus E, Chen W-H, Leigh-Bell J, et al. 2009. Transcriptome complexity in a genome-reduced bacterium. Science 326(5957):1268–71 [DOI] [PubMed] [Google Scholar]
- 72.Gusarov I, Nudler E. 2001. Control of intrinsic transcription termination by N and NusA: the basic mechanisms. Cell 107:437–49 [DOI] [PubMed] [Google Scholar]
- 73.Hackett SR, Zanotelli VRT, Xu W, Goya J, Park JO, et al. 2016. Systems-level analysis of mechanisms regulating yeast metabolic flux. Science 354(6311):aaf2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harden TT, Wells CD, Friedman LJ, Landick R, Hochschild A, et al. 2016. Bacterial RNA polymerase can retain σ70 throughout transcription. PNAS 113(3):602–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hausser J, Mayo A, Keren L, Alon U. 2019. Central dogma rates and the trade-off between precision and economy in gene expression. Nat. Commun 10(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hör J, Gorski SA, Vogel J. 2018. Bacterial RNA biology on a genome scale. Mol. Cell 70:785–99 [DOI] [PubMed] [Google Scholar]
- 77.Hsieh P-K, Richards J, Liu Q, Belasco JG. 2013. Specificity of RppH-dependent RNA degradation in Bacillus subtilis. PNAS 110(22):8864–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hudson AJ, Wieden H-J. 2019. Rapid generation of sequence-diverse terminator libraries and their parameterization using quantitative Term-Seq. Synth. Biol 4(1):ysz026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hui MP, Foley PL, Belasco JG. 2014. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet 48:537–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324(5924):218–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ireland WT, Beeler SM, Flores-Bautista E, McCarty NS, Röschinger T, et al. 2020. Deciphering the regulatory genome of Escherichia coli, one hundred promoters at a time. eLife 9:e55308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jacob F, Monod J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol 3:318–56 [DOI] [PubMed] [Google Scholar]
- 83.Johnson GE, Lalanne J-B, Peters ML, Li G-W. 2020. Functionally uncoupled transcription-translation in Bacillus subtilis. Nature 585:124–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ju X, Li D, Liu S. 2019. Full-length RNA profiling reveals pervasive bidirectional transcription terminators in bacteria. Nat. Microbiol 4(11):1907–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kelsic ED, Chung H, Cohen N, Park J, Wang HH, Kishony R. 2016. RNA structural determinants of optimal codons revealed by MAGE-Seq. Cell Syst 3(6):563–71.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kennell D, Riezman H. 1977. Transcription and translation initiation frequencies of the Escherichia coli lac operon. J. Mol. Biol 114(1):1–21 [DOI] [PubMed] [Google Scholar]
- 87.Keren L, Hausser J, Lotan-Pompan M, Vainberg Slutskin I, Alisar H, et al. 2016. Massively parallel interrogation of the effects of gene expression levels on fitness. Cell 166(5):1282–94.e18 [DOI] [PubMed] [Google Scholar]
- 88.Khemici V, Prados J, Linder P, Redder P. 2015. Decay-initiating endoribonucleolytic cleavage by RNase Y is kept under tight control via sequence preference and sub-cellular localisation. PLOS Genet 11:e1005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim S, Beltran B, Irnov I, Jacobs-Wagner C. 2019. Long-distance cooperative and antagonistic RNA polymerase dynamics via DNA supercoiling. Cell 179:106–19.e16 [DOI] [PubMed] [Google Scholar]
- 90.Kinney JB, McCandlish DM. 2019. Massively parallel assays and quantitative sequence-function relationships. Annu. Rev. Genomics Hum. Genet 20:99–127 [DOI] [PubMed] [Google Scholar]
- 91.Kinney JB, Murugan A, Callan CG Jr., Cox EC. 2010. Using deep sequencing to characterize the biophysical mechanism of a transcriptional regulatory sequence. PNAS 107(20):9158–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klumpp S, Dong J, Hwa T. 2012. On ribosome load, codon bias and protein abundance. PLOS ONE 7(11):e48542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324(5924):255–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuo S-T, Jahn R-L, Cheng Y-J, Chen Y-L, Lee Y-J, et al. 2020. Global fitness landscapes of the Shine-Dalgarno sequence. Genome Res 30(5):711–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Laalami S, Bessières P, Rocca A, Zig L, Nicolas P, Putzer H. 2013. Bacillus subtilis RNase Y activity in vivo analysed by tiling microarrays. PLOS ONE 8:e54062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95a.Lalanne J-B, Parker DG, Li G-W. 2021. Spurious regulatory connections dictate the expression-fitness landscape of translation factors. Mol. Syst. Biol 17:e10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lalanne J-B, Taggart JC, Guo MS, Herzel L, Schieler A, Li G-W. 2018. Evolutionary convergence of pathway-specific enzyme expression stoichiometry. Cell 173(3):749–61.e38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lehnik-Habrink M, Schaffer M, Mäder U, Diethmaier C, Herzberg C, Stülke J. 2011. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol. Microbiol 81:1459–73 [DOI] [PubMed] [Google Scholar]
- 98.Lemaux PG, Herendeen SL, Bloch PL, Neidhardt FC. 1978. Transient rates of synthesis of individual polypeptides in E. coli following temperature shifts. Cell 13:427–34 [DOI] [PubMed] [Google Scholar]
- 99.Li G-W. 2015. How do bacteria tune translation efficiency? Curr. Opin. Microbiol 24:66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li G-W, Burkhardt D, Gross C, Weissman JS. 2014. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157:624–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li R, Zhang Q, Li J, Shi H. 2016. Effects of cooperation between translating ribosome and RNA polymerase on termination efficiency of the Rho-independent terminator. Nucleic Acids Res 44(6):2554–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li S, Dong X, Su Z. 2013. Directional RNA-seq reveals highly complex condition-dependent transcriptomes in E. coli K12 through accurate full-length transcripts assembling. BMC Genomics 14:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lodish HF. 1968. Bacteriophage f2 RNA: control of translation and gene order. Nature 220(5165):345–50 [DOI] [PubMed] [Google Scholar]
- 104.Luciano DJ, Hui MP, Deana A, Foley PL, Belasco KJ, Belasco JG. 2012. Differential control of the rate of 5′-end-dependent mRNA degradation in Escherichia coli. J. Bacteriol 194(22):6233–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Luciano DJ, Vasilyev N, Richards J, Serganov A, Belasco JG. 2017. A novel RNA phosphorylation state enables 5′ end-dependent degradation in Escherichia coli. Mol. Cell 67(1):44–54.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mandell ZF, Oshiro RT, Yakhnin AV, Vishwakarma R, Kashlev M, et al. 2021. NusG is an intrinsic transcription termination factor that stimulates motility and coordinates gene expression with NusA. eLife 10:e61880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marincola G, Schäfer T, Behler J, Bernhardt J, Ohlsen K, et al. 2012. RNase Y of Staphylococcus aureus and its role in the activation of virulence genes. Mol. Microbiol 85:817–32 [DOI] [PubMed] [Google Scholar]
- 108.Marincola G, Wolz C. 2017. Downstream element determines RNase Y cleavage of the saePQRS operon in Staphylococcus aureus. Nucleic Acids Res 45:5980–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McCarthy JEG, Gualerzi C. 1990. Translational control of prokaryotic gene expression. Trends Genet 6:78–85 [DOI] [PubMed] [Google Scholar]
- 109a.McCormick DM, Lalanne J-B, Lan TCT, Rouskin S, Li G-W. 2021. Sigma factor dependent translational activation in Bacillus subtilis. RNA In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McDowall KJ, Kaberdin VR, Wu SW, Cohen SN, Lin-Chao S. 1995. Site-specific RNase E cleavage of oligonucleotides and inhibition by stem-loops. Nature 374(6519):287–90 [DOI] [PubMed] [Google Scholar]
- 111.McDowall KJ, Lin-Chao S, Cohen SN. 1994. A+U content rather than a particular nucleotide order determines the specificity of RNase E cleavage. J. Biol. Chem 269(14):10790–96 [PubMed] [Google Scholar]
- 112.Mejía-Almonte C, Busby SJW, Wade JT, van Helden J, Arkin AP, et al. 2020. Redefining fundamental concepts of transcription initiation in bacteria. Nat. Rev. Genet 21(11):699–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mendoza-Vargas A, Olvera L, Olvera M, Grande R, Vega-Alvarado L, et al. 2009. Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLOS ONE 4(10):e7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Meydan S, Klepacki D, Karthikeyan S, Margus T, Thomas P, et al. 2017. Programmed ribosomal frameshifting generates a copper transporter and a copper chaperone from the same gene. Mol. Cell 65:207–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moffitt JR, Pandey S, Boettiger AN, Wang S, Zhuang X. 2016. Spatial organization shapes the turnover of a bacterial transcriptome. eLife 5:e13065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mohammad F, Green R, Buskirk AR. 2019. A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife 8:e42591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mohanty BK, Kushner SR. 2016. Regulation of mRNA decay in bacteria. Annu. Rev. Microbiol 70:25–44 [DOI] [PubMed] [Google Scholar]
- 118.Mondal S, Yakhnin AV, Sebastian A, Albert I, Babitzke P. 2016. NusA-dependent transcription termination prevents misregulation of global gene expression. Nat. Microbiol 1:15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mustoe AM, Busan S, Rice GM, Hajdin CE, Peterson BK, et al. 2018. Pervasive regulatory functions of mRNA structure revealed by high-resolution SHAPE probing. Cell 173(1):181–95.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mutalik VK, Guimaraes JC, Cambray G, Lam C, Christoffersen MJ, et al. 2013. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods 10:354–60 [DOI] [PubMed] [Google Scholar]
- 121.Nakatogawa H, Ito K. 2001. Secretion monitor, SecM, undergoes self-translation arrest in the cytosol. Mol. Cell 7(1):185–92 [DOI] [PubMed] [Google Scholar]
- 122.Narayanan CS, Dubnau D. 1985. Evidence for the translational attenuation model: ribosome-binding studies and structural analysis with an in vitro run-off transcript of ermC. Nucleic Acids Res 13:7307–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nomura M, Gourse R, Baughman G. 1984. Regulation of the synthesis of ribosomes and ribosomal components. Annu. Rev. Biochem 53:75–117 [DOI] [PubMed] [Google Scholar]
- 124.Oeschger MP, Nathans D. 1966. Differential synthesis of bacteriophage-specific proteins in MS2-infected Escherichia coli treated with actinomycin. J. Mol. Biol 22(2):235–47 [DOI] [PubMed] [Google Scholar]
- 125.Ohtaka Y, Spiegelman S. 1963. Translational control of protein synthesis in a cell-free system directed by a polycistronic viral RNA. Science 142:493–97 [DOI] [PubMed] [Google Scholar]
- 126.Opdyke JA, Fozo EM, Hemm MR, Storz G. 2011. RNase III participates in GadY-dependent cleavage of the gadX-gadW mRNA. J. Mol. Biol 406(1):29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Oromendia AB, Dodgson SE, Amon A. 2012. Aneuploidy causes proteotoxic stress in yeast. Genes Dev 26(24):2696–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Papenfort K, Sun Y, Miyakoshi M, Vanderpool CK, Vogel J. 2013. Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 153:426–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parker DJ, Lalanne J-B, Kimura S, Johnson GE, Waldor MK, Li G-W. 2020. Growth-optimized aminoacyl-tRNA synthetase levels prevent maximal tRNA charging. Cell Syst 11(2):121–30.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, et al. 2016. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165(6):1493–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pette D, Luh W, Buecher T. 1962. A constant-proportion group in the enzyme activity pattern of the Embden-Meyerhof chain. Biochem. Biophys. Res. Commun 7:419–24 [DOI] [PubMed] [Google Scholar]
- 132.Pitt ME, Nguyen SH, Duarte TPS, Teng H, Blaskovich MAT, et al. 2020. Evaluating the genome and resistome of extensively drug-resistant Klebsiella pneumoniae using native DNA and RNA Nanopore sequencing. Gigascience 9:giaa002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Price MN. 2005. Operon formation is driven by co-regulation and not by horizontal gene transfer. Genome Res 15:809–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Quax TEF, Wolf YI, Koehorst JJ, Wurtzel O, van der Oost R, et al. 2013. Differential translation tunes uneven production of operon-encoded proteins. Cell Rep 4(5):938–44 [DOI] [PubMed] [Google Scholar]
- 134a.Rackham O, Chin JW. 2005. A network of orthogonal ribosome·mRNA pairs. Nat. Chem. Biol 1:159–66 [DOI] [PubMed] [Google Scholar]
- 135.Ray-Soni A, Bellecourt MJ, Landick R. 2016. Mechanisms of bacterial transcription termination: All good things must end. Annu. Rev. Biochem 85:319–47 [DOI] [PubMed] [Google Scholar]
- 136.Redder P 2018. Mapping 5′-ends and their phosphorylation state with EMOTE, TSS-EMOTE, and nEMOTE. Methods Enzymol 612:361–91 [DOI] [PubMed] [Google Scholar]
- 137.Richards J, Belasco JG. 2019. Obstacles to scanning by RNase E govern bacterial mRNA lifetimes by hindering access to distal cleavage sites. Mol. Cell 74(2):284–95.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Richards J, Liu Q, Pellegrini O, Celesnik H, Yao S, et al. 2011. An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol. Cell 43(6):940–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rochat T, Bouloc P, Repoila F. 2013. Gene expression control by selective RNA processing and stabilization in bacteria. FEMS Microbiol. Lett 344(2):104–13 [DOI] [PubMed] [Google Scholar]
- 140.Roland KL, Powell FE, Turnbough CL Jr. 1985. Role of translation and attenuation in the control of pyrBI operon expression in Escherichia coli K-12. J. Bacteriol 163(3):991–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. 2014. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505(7485):701–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Saito K, Green R, Buskirk AR. 2020. Ribosome recycling is not critical for translational coupling in Escherichia coli. eLife 9:e59974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Saito K, Green R, Buskirk AR. 2020. Translational initiation in E. coli occurs at the correct sites genome-wide in the absence of mRNA-rRNA base-pairing. eLife 9:e55002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sampson LL, Hendrix RW, Huang WM, Casjens SR. 1988. Translation initiation controls the relative rates of expression of the bacteriophage lambda late genes. PNAS 85(15):5439–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144a.Sastry AV, Gao Y, Szubin R, Hefner Y, Xu S, et al. 2019. The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat. Commun 10:5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Scharff LB, Childs L, Walther D, Bock R. 2011. Local absence of secondary structure permits translation of mRNAs that lack ribosome-binding sites. PLOS Genet 7(6):e1002155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schmid MB, Roth JR. 1987. Gene location affects expression level in Salmonella typhimurium. J. Bacteriol 169(6):2872–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Scholz SA, Diao R, Wolfe MB, Fivenson EM, Lin XN, Freddolino PL. 2019. High-resolution mapping of the Escherichia coli chromosome reveals positions of high and low transcription. Cell Syst 8(3):212–25.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schrader JM, Zhou B, Li G-W, Lasker K, Childers WS, et al. 2014. The coding and noncoding architecture of the Caulobacter crescentus genome. PLOS Genet 10(7):e1004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schümperli D, McKenney K, Sobieski DA, Rosenberg M. 1982. Translational coupling at an intercistronic boundary of the Escherichia coli galactose operon. Cell 30(3):865–71 [DOI] [PubMed] [Google Scholar]
- 150.Shahbabian K, Jamalli A, Zig L, Putzer H. 2009. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 28:3523–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, et al. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464(7286):250–55 [DOI] [PubMed] [Google Scholar]
- 152.Shen BA, Landick R. 2019. Transcription of bacterial chromatin. J. Mol. Biol 431:4040–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shieh Y-W, Minguez P, Bork P, Auburger JJ, Guilbride DL, et al. 2015. Operon structure and cotranslational subunit association direct protein assembly in bacteria. Science 350(6261):678–80 [DOI] [PubMed] [Google Scholar]
- 154.Shine J, Dalgarno L. 1974. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. PNAS 71(4):1342–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sousa C, de Lorenzo V, Cebolla A. 1997. Modulation of gene expression through chromosomal positioning in Escherichia coli. Microbiology 143(Part 6):2071–78 [DOI] [PubMed] [Google Scholar]
- 156.Stead MB, Marshburn S, Mohanty BK, Mitra J, Pena Castillo L, et al. 2011. Analysis of Escherichia coli RNase E and RNase III activity in vivo using tiling microarrays. Nucleic Acids Res 39(8):3188–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stern-Ginossar N, Weisburd B, Michalski A, Le VTK, Hein MY, et al. 2012. Decoding human cytomegalovirus. Science 338(6110):1088–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Subramaniam AR, Zid BM, O’Shea EK. 2014. An integrated approach reveals regulatory controls on bacterial translation elongation. Cell 159(5):1200–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Swain PS. 2004. Efficient attenuation of stochasticity in gene expression through posttranscriptional control. J. Mol. Biol 344(4):965–76 [DOI] [PubMed] [Google Scholar]
- 160.Taggart JC, Li G-W. 2018. Production of protein-complex components is stoichiometric and lacks general feedback regulation in eukaryotes. Cell Syst 7(6):580–89.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Taggart JC, Zauber H, Selbach M, Li G-W, McShane E. 2020. Keeping the proportions of protein complex components in check. Cell Syst 10(2):125–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Takada A, Nagai K, Wachi M. 2005. A decreased level of FtsZ is responsible for inviability of RNase E-deficient cells. Genes Cells 10(7):733–41 [DOI] [PubMed] [Google Scholar]
- 163.Tamura M, Lee K, Miller CA, Moore CJ, Shirako Y, et al. 2006. RNase E maintenance of proper FtsZ/FtsA ratio required for nonfilamentous growth of Escherichia coli cells but not for colony-forming ability. J. Bacteriol 188:5145–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tejada-Arranz A, de Crécy-Lagard V, de Reuse H. 2020. Bacterial RNA degradosomes: molecular machines under tight control. Trends Biochem. Sci 45(1):42–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Trinquier A, Durand S, Braun F, Condon C. 2020. Regulation of RNA processing and degradation in bacteria. Biochem. Biophys. Acta Gene Regul. Mech 1863:194505. [DOI] [PubMed] [Google Scholar]
- 166.Tsuchihashi Z, Kornberg A. 1990. Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. PNAS 87(7):2516–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, et al. 2010. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141(2):344–54 [DOI] [PubMed] [Google Scholar]
- 168.Urtecho G, Insigne KD, Tripp AD, Brinck M, Lubock NB, et al. 2020. Genome-wide functional characterization of Escherichia coli promoters and regulatory elements responsible for their function. bioRxiv 2020.01.04.894907. 10.1101/2020.01.04.894907 [DOI] [Google Scholar]
- 169.Urtecho G, Tripp AD, Insigne KD, Kim H, Kosuri S. 2019. Systematic dissection of sequence elements controlling σ70 promoters using a genomically encoded multiplexed reporter assay in Escherichia coli. Biochemistry 58(11):1539–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Verma M, Choi J, Cottrell KA, Lavagnino Z, Thomas EN, et al. 2019. A short translational ramp determines the efficiency of protein synthesis. Nat. Commun 10(1):5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vilar JMG, Leibler S. 2003. DNA looping and physical constraints on transcription regulation. J. Mol. Biol 331(5):981–89 [DOI] [PubMed] [Google Scholar]
- 172.von Hippel PH, Yager TD. 1991. Transcript elongation and termination are competitive kinetic processes. PNAS 88(6):2307–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vora T, Hottes AK, Tavazoie S. 2009. Protein occupancy landscape of a bacterial genome. Mol. Cell 35(2):247–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vvedenskaya IO, Zhang Y, Goldman SR, Valenti A, Visone V, et al. 2015. Massively systematic transcript end readout, “MASTER”: transcription start site selection, transcriptional slippage, and transcript yields. Mol. Cell 60(6):953–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wikström PM, Wikström PM, Lind LK, Berg DE, Björk GR. 1992. Importance of mRNA folding and start codon accessibility in the expression of genes in a ribosomal protein operon of Escherichia coli. J. Mol. Biol 224:949–66 [DOI] [PubMed] [Google Scholar]
- 176.Wurtzel O, Dori-Bachash M, Pietrokovski S, Jurkevitch E, Sorek R. 2010. Mutation detection with next-generation resequencing through a mediator genome. PLOS ONE 5(12):e15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yamaguchi Y, Park J-H, Inouye M. 2011. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet 45:61–79 [DOI] [PubMed] [Google Scholar]
- 178.Yan B, Boitano M, Clark TA, Ettwiller L. 2018. SMRT-Cappable-seq reveals complex operon variants in bacteria. Nat. Commun 9(1):3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yao S, Richards J, Belasco JG, Bechhofer DH. 2011. Decay of a model mRNA in Bacillus subtilis by a combination of RNase J1 5′ exonuclease and RNase Y endonuclease activities. J. Bacteriol 193:6384–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yarchuk O, Jacques N, Guillerez J, Dreyfus M. 1992. Interdependence of translation, transcription and mRNA degradation in the lacZ gene. J. Mol. Biol 226(3):581–96 [DOI] [PubMed] [Google Scholar]
- 181.Yim SS, Johns NI, Park J, Gomes AL, McBee RM, et al. 2019. Multiplex transcriptional characterizations across diverse bacterial species using cell-free systems. Mol. Syst. Biol 15(8):e8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zheng H, Bai Y, Jiang M, Tokuyasu TA, Huang X, et al. 2020. General quantitative relations linking cell growth and the cell cycle in Escherichia coli. Nat. Microbiol 5(8):995–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]