

Abstract
Natural killer (NK) cells are innate cytotoxic lymphocytes that play a key role in cancer immunosurveillance thanks to their ability to recognize and kill cancer cells. NKG2D is an activating receptor that binds to MIC and ULBP molecules typically induced on damaged, transformed or infected cells. The release of NKG2D ligands (NKG2DLs) in the extracellular milieu through protease‐mediated cleavage or by extracellular vesicle (EV) secretion allows cancer cells to evade NKG2D‐mediated immunosurveillance. In this work, we investigated the immunomodulatory properties of the NKG2D ligand MICA*008 associated to distinct populations of EVs (i.e., small extracellular vesicles [sEVs] and medium size extracellular vesicles [mEVs]). By using as model a human MICA*008‐transfected multiple myeloma (MM) cell line, we found that this ligand is present on both vesicle populations. Interestingly, our findings reveal that NKG2D is specifically involved in the uptake of vesicles expressing its cognate ligand. We provide evidence that MICA*008‐expressing sEVs and mEVs are able on one hand to activate NK cells but, following prolonged stimulation induce a sustained NKG2D downmodulation leading to impaired NKG2D‐mediated functions. Moreover, our findings show that MICA*008 can be transferred by vesicles to NK cells causing fratricide. Focusing on MM as a clinically and biologically relevant model of tumour‐NK cell interactions, we found enrichment of EVs expressing MICA in the bone marrow of a cohort of patients. All together our results suggest that the accumulation of NKG2D ligands associated to vesicles in the tumour microenvironment could favour the suppression of NK cell activity either by NKG2D down‐modulation or by fratricide of NK cell dressed with EV‐derived NKG2D ligands.
Keywords: cancer, extracellular vesicles, immune evasion, MICA, Natural Killer cells, NKG2D
1. INTRODUCTION
In the past years extracellular vesicles (EVs), including both exosomes and microvesicles, have been recognized as potent vehicles of intercellular communication due to their capacity to exchange components (i.e., proteins, lipids and nucleic acids) between cells, thereby affecting cellular behaviour (van Niel et al., 2018). Exosomes are a class of nanovesicles formed and released through the late endosomal compartment, whereas microvesicles are released from the plasma membrane with many efforts aimed at studying the biological activity of the various classes of vesicles (Mathieu et al., 2019). Mounting evidence has shown that cancer derived‐EVs have the ability to modulate both innate and adaptive immune responses and thus contribute to influence tumour progression (Kalluri, 2016).
Natural killer (NK) cells are innate cytotoxic lymphoid cells that play a key role in cancer immunosurveillance, thanks to their ability to recognize and kill cancer cells and to produce a large number of cytokines and chemokines (Chiossone et al., 2018; Morvan & Lanier, 2016). NK cell activation relies on a tight balance between activating and inhibitory signals generated by the engagement of distinct receptors. NKG2D is a C‐type lectin‐like activating receptor expressed on NK cells, γδ T cells, CD8+ T cells and some autoreactive or immunosuppressive CD4+ T cells and represents a major recognition receptor for the detection and elimination of virus‐infected, and cancer cells (Lanier, 2015). In humans, NKG2D binds to MHC class I–related chain (MIC)A, MICB, and UL16‐binding proteins (ULBP1‐6), whose expression is either null or low on normal tissues; however, they are promptly induced by different types of stress, including tumour transformation, viral and bacterial infections and autoimmune diseases (Raulet et al., 2013; Zingoni, Molfetta et al., 2018). Thus, expression of NKG2D ligands (NKG2DLs) is generally regarded as a ‘danger signal’ marking cells for immune cell attack. However, the release of NKG2DLs in the extracellular milieu through protease‐mediated cleavage or by EV secretion is a mode to control their cell surface expression and a mechanism used by cancer cells to evade NKG2D‐mediated immunosurveillance (Chitadze et al., 2013). Indeed, soluble MICA/B levels often correlate with a systemic downregulation of NKG2D surface expression, thereby impairing NKG2D‐dependent lysis of tumour cells (Salih et al., 2003; Wu et al., 2004). Concomitantly, the reduction in NKG2DL density on the tumour cell surface likely triggers less efficient killing (Ferrari de Andrade et al., 2018; Zingoni et al., 2015).
MICA is the most polymorphic NKG2DL and some polymorphisms have raised great interest since they affect MICA biology. To this regard the truncated allele MICA*008, that is highly frequent in the Caucasian population, is attached to the plasma membrane via a GPI anchor and is preferentially released associated with exosomes or secreted through exocytosis (Ashiru et al., 2010, 2013). Moreover, the presence of other NKG2DLs on EVs has been documented. As such, a number of studies have shown that NKG2DLs of both MIC and ULBP families are expressed by exosome‐like vesicles released from hematopoietic (Hedlund et al., 2011) prostate and (Lundholm et al., 2014), ovarian cancer cells (Labani‐Motlagh et al., 2015) and melanoma (López‐Cobo et al., 2018).
NK cells have long been considered essential in the immune surveillance of multiple myeloma (MM), a clonal B cell malignancy characterized by expansion of plasma cells (PCs) in the bone marrow (BM). Notably, a number of studies have shown that NK cells from patients with advanced MM often display an impairment of NK cell functions as well as a reduced expression of multiple activating receptors (Fauriat et al., 2006; Pazina et al., 2021; Vulpis et al., 2018). The activating receptor NKG2D was reported to play an important role in the NK cell–mediated recognition and killing of MM cells (Carbone et al., 2005; El‐Sherbiny et al., 2007; Soriani et al., 2009) and high levels of soluble MICA in the serum of MM patients correlate with the disease stage (Jinushi et al., 2007; Rebmann et al., 2007; Zingoni, Vulpis et al., 2018). Moreover, the NKG2D‐NKG2DL pathway has been the subject of intense research in MM targeted therapy (Abruzzese et al., 2016; Chan et al., 2018; Fionda et al., 2015; Soriani et al., 2009; Wang et al., 2020; Zingoni et al., 2016).
In this work we investigated the immunomodulatory properties of the NKG2DL MICA*008 associated with distinct populations of EVs (i.e., small extracellular vesicles [sEVs] and medium size extracellular vesicles [mEVs]) (Witwer & Théry, 2019). Interestingly, we provide novel evidence that NKG2D is specifically involved in the uptake of vesicles expressing this ligand. Our data also show that both sEVs and mEVs expressing MICA*008 are able on one hand to activate NK cells, but they also induce a sustained NKG2D downmodulation leading to an impairment of NKG2D‐mediated functions. Noteworthy our findings reveal that MICA*008 can be transferred by vesicles to NK cells causing fratricide. By using MM as model of tumour–NK cell interactions, EVs expressing MICA were detected in the BM of a cohort of MM patients thus suggesting that these vesicles play a pivotal role in the tumour microenvironment by affecting NK cell‐mediated functions at different levels.
2. RESULTS
2.1. Both small and medium EVs express MICA*008 and induce NKG2D down‐modulation
A number of studies have shown the ability of cell surface NKG2DLs to downmodulate NKG2D on NK cells (Molfetta et al., 2014; Ogasawara et al., 2004) however the capability of soluble forms of NKG2DLs (i.e., shedded vs. vesicle‐associated) to regulate NKG2D surface expression levels is still a matter of debate. To better investigate this aspect, conditioned culture media derived from ARK MM cell line transfected with MICA*008 allele or with an empty vector (Zingoni et al., 2015) were collected, depleted or not from EVs and incubated with NKL cell line. As shown in Figure 1a, NKG2D downmodulation was observed only in NK cells treated with conditioned medium comprising EVs whereas EV‐depleted medium, although it contains soluble MICA, (Figure S1) has no effect. In the same experiment, the expression of DNAM‐1 was not affected, confirming the specificity of ligand‐induced NKG2D downmodulation (Figure 1a). Both sEVs and mEVs were purified from the conditioned medium of MICA*008‐transfected ARK cell line by using an established isolation protocol that exploits their differential sedimentation properties. In particular, sEVs were recovered after high‐speed ultracentrifugation whereas intermediate‐size microvesicles were collected after a lower‐speed centrifugation. Dynamic light scattering (DLS) methodology was used to examine EV size distribution. Our results show that sEV preparations contain vesicles whose average diameter corresponded to 80 ± 12 nm, whereas that of mEVs corresponded to 120 ± 19 nm (Figure 1b). These data were confirmed by ultrastructural analysis (Figure 1c). Our findings show that the amount of total proteins recovered from EV samples was significantly higher for mEVs respect to sEVs (Figure 1d), whereas the evaluation of vesicle number by DLS demonstrated that about 107 EVs were present for each microgram of protein with no significant differences between sEVs and mEVs (Figure 1e). Canonical vesicle markers were detected by Western blot analysis showing a different content between sEVs and mEVs, as tetraspanins CD63 and CD81 were preferentially enriched in small EV preparations while calreticulin was more abundant in medium‐size EVs (Figure 1f).
FIGURE 1.
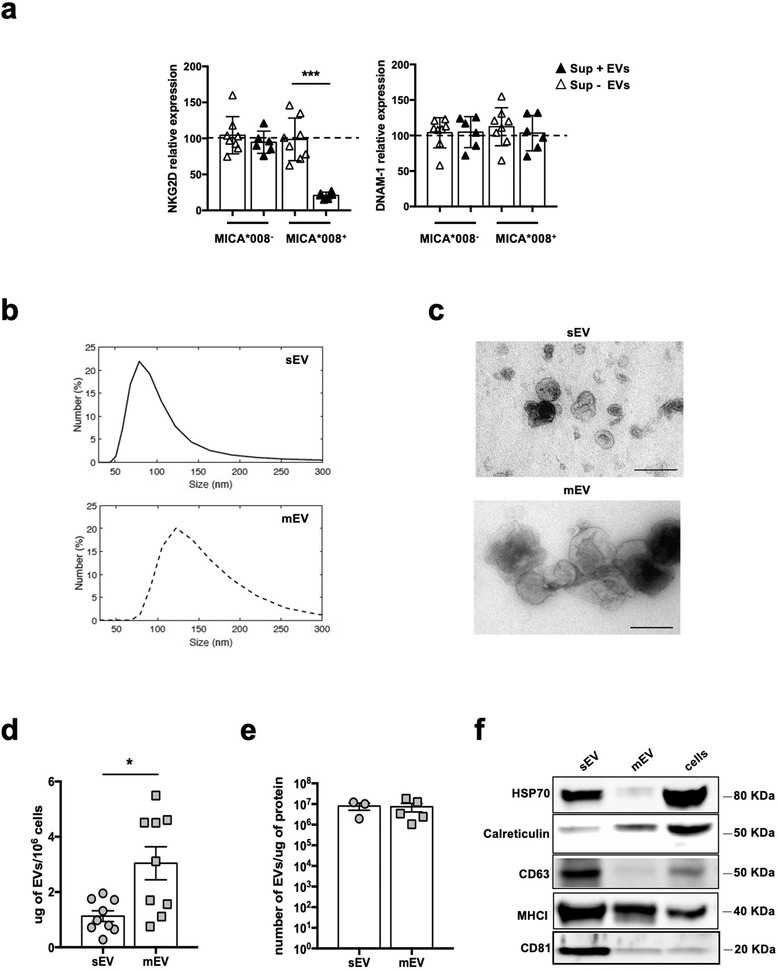
EV‐associated MICA*008 and not its soluble form induces NKG2D downmodulation. (a) Conditioned supernatants derived from ARK MICA*008 transfectants or ARK transfected with an empty vector were depleted of EVs and used to culture the NKL cell line for 24 h. NKG2D and DNAM‐1 expression was evaluated by immunofluorescence and FACS analysis. NKG2D relative expression was calculated respect to the untreated group (= 100%) represented by dashed line. Values reported represent the mean of independent experiments. (b‐f) ARK cell transfectants were cultured for 48 h in EV‐free medium and then EVs were isolated from the conditioned media as described in materials and methods. (b) Size distribution of sEVs and mEVs was analysed through dynamic light scattering (DLS). A representative experiment is shown. (c) Transmission electron microscopy (TEM) of sEVs and mEVs preparations. Bar corresponds to 200 nm. (d) The amount of total proteins recovered from EV preparations derived from 106 cells was calculated by BPA and corresponded to 1.127 μg ± 0.58 for sEVs and 3.04 μg ± 1.79 for mEVs. (e) The number of EVs/μg of protein was calculated through DLS. (f) Western blot analysis was performed on lysates (40 μg) derived from sEVs and mEVs fractions or from cell pellet, using anti‐Hsp70, anti‐calreticulin, anti‐CD63, anti‐MHC I and anti‐CD81 antibodies
The presence of MICA*008 on EVs was assessed by Western blot analysis (Figure 2a) and a specific ELISA was used to quantify the amount of MICA*008 associated to EVs; we found no significant differences between MICA*008 associated to sEVs and mEVs (Figure 2b). Finally, MICA*008 expression on the surface of EVs was also evaluated by immunofluorescence and flow cytometry (FACS) analysis, indicating that MICA was expressed on the surface of both sEVs and mEVs released by MICA*008+‐transfected ARK MM cells (Figure 2c and 2d). As shown in Figure 2e and f, both types of vesicles retain the capability to induce NKG2D downmodulation.
FIGURE 2.
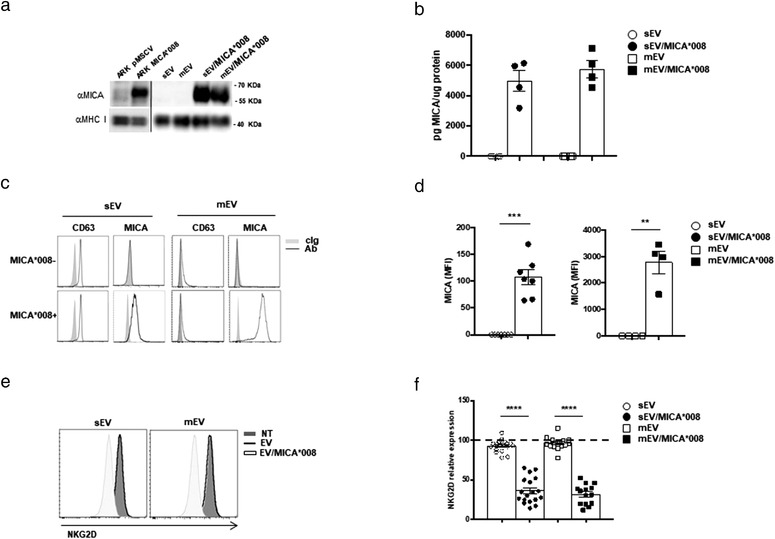
Characterization and functional activity of sEVs and mEVs expressing MICA*008. (a) Western blot analysis was performed on lysates derived from sEVs and mEVs fractions or from cell pellet of ARK transfectants, using anti‐MICA or anti‐MHC I antibodies. (b) Lysates derived fromsEVs and mEVs were assessed for the presence of MICA using a specific ELISA. Values are represented as the amount of MICA/μg of proteins and represent the mean of four independent experiments. (c) Immunofluorescence and FACS analysis of mEVs or CD63+ beads coated with sEVs and labelled with control isotypic Ig (cIg), anti‐CD63 or anti‐MICA Abs, as indicated in the figure. A representative experiment is shown. (d) Graph is referred to the mean of different experiments and values represent the mean fluorescence intensity (MFI) of MICA subtracted from the MFI value of the cIg. (e and f) The NKL cell line was incubated for 24 h with 30 μg/ml of sEVs or 20 μg/ml of mEVs; cells were collected and NKG2D expression was evaluated by immunofluorescence and FACS analysis. A representative experiment is shown. In (f) values reported represent the mean of different independent experiments. Relative expression of NKG2D was calculated considering the untreated group as 100% (dashed line)
The interaction of NKG2D with MICA on the surface of target cells promoted its internalization and degradation in the lysosomal compartment (Molfetta et al., 2014), thus we investigated the mechanism of EV‐mediated NKG2D downmodulation. At first, we asked whether the reduced surface NKG2D expression was accompanied by NKG2D internalization. As shown in Figure S2a, confocal microscopy analysis revealed NKG2D internalization upon treatment with both type of MICA*008+EVs. In addition, we found a significant reduction of NKG2D protein levels suggesting that internalized receptor complexes could be subjected to degradation (Figure S2b and c). On the other hand, the treatment of NK cells with EVs carrying MICA*008 did not affect NKG2D mRNA levels (Figure S2d).
Collectively, these results indicate that MICA*008 associated with both sEVs and mEVs and not its soluble form induce NKG2D downmodulation accompanied to receptor internalization and degradation.
2.2. NKG2D promotes the uptake of both MICA*008+ sEVs and mEVs
To evaluate if both type of vesicles were taken up by NK cells and if MICA*008 was involved, EVs were labelled with PKH26 and incubated with NK cells. Interestingly MICA*008+ vesicle uptake was significantly higher compared to the uptake of vesicles not expressing this NKG2DL (Figure 3a and 3b and Figure S3). These findings were confirmed also by confocal microscopy indicating that both type of vesicles are internalized by NK cells (Figure S4a and b). Based on our previous findings showing that NK cells take up exosomes mainly through energy‐dependent endocytosis mechanisms (Vulpis et al., 2017), we explored whether MICA*008 expressing EVs used similar ways. As shown in Figure 3c, the uptake of both sEVs and mEVs was strongly impaired when NK cells were assayed in presence of nystatin and dynasore, which implies its dependence on endocytic route(s) requiring caveolae/raft integrity. On the other hand, inhibition of micropinocytosis by EIPA, a highly conserved actin‐dependent endocytic process, and of Ca2+‐dependent mechanisms appear to be involved only in the sEV uptake. Interestingly, the presence of the specific NKG2DL MICA*008 on the surface of sEVs results in a minor sensitivity to the inhibitors and, remarkable, nystatin lacked the ability to block MICA*008+ uptake of both sEVs and mEVs. Collectively, these results indicate that MICA*008 expression correlated with an increased uptake by NK cells and also affects the mechanism of EV uptake.
FIGURE 3.
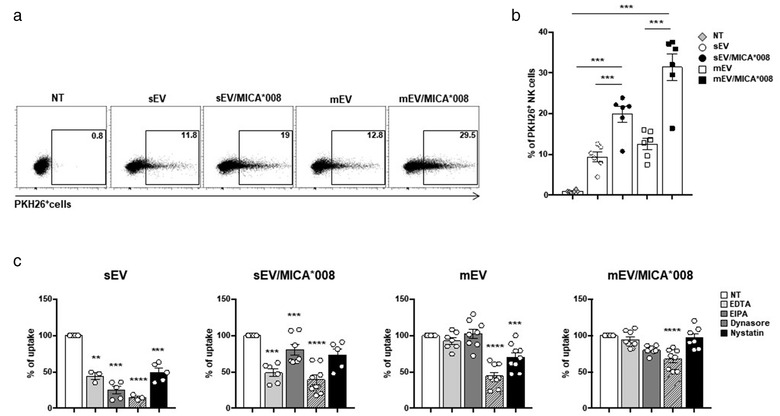
MICA*008 EVs are taken up more efficiently by NK cells. The NKL cell line was incubated for 3 h with 30 μg/ml of PKH26‐labelled sEVs or 20 μg/ml of PKH26‐labelled mEVs derived from ARK transfectants. The fluorescence of internalized EVs was evaluated by FACS analysis and measured as the percentage of PKH26+ cells. (a) One representative experiment is shown. (b) The mean of six independent experiments is shown. (c) NKL cells were pre‐treated for 1 h with EDTA (10 mM), EIPA (50 μM), dynasore (50 μM), nystatin (40 μg/ml) and then incubated for 3 h with PKH26‐labelled EVs. The fluorescence of internalized EVs was evaluated by FACS and measured as the percentage of PKH26 positive cells referred to untreated cells which were considered as 100%. Values reported represent the mean of at least three independent experiments
Next, we asked whether NKG2D could be involved in the uptake of MICA*008+ sEVs and mEVs. NK cells were thus pre‐treated with anti‐NKG2D or anti‐DNAM‐1 neutralizing antibodies before the incubation with PKH26‐labelled EVs. Interestingly, a significant reduction of MICA*008+ EV uptake was observed only in cells pre‐treated with anti‐NKG2D mAb, if compared to those incubated with anti‐DNAM‐1 mAb, strongly suggesting that NKG2D was specifically involved in the uptake of sEVs and mEVs expressing its ligand (Figure 4a). To further support this result, we examined whether NKG2D downmodulation induced by MICA*008+EVs could desensitize NK cells in the ability to capture ligand positive vesicles; NK cells were treated with MICA*008+EVs to obtain NK cells expressing low levels of NKG2D (NKG2Dlow cells) (Figure 4b and 4c). Then NKG2Dlow and NKG2Dhigh NK cell populations were incubated with PKH26‐labelled EVs. As shown in Figure 4d, a prominent reduction of MICA*008+EV uptake was observed in NKG2Dlow cells that had previously been exposed to ligand expressing vesicles. We then investigated whether downmodulation of NKG2D expression levels induced by shRNA could affect the capability of NK cells to take up NKG2DL+ EVs. To this aim, NKG2D expression was knocked down in NKL cells using lentiviral vectors containing NKG2D shRNA. As shown in Figure 4e and 4f about 70% reduction was observed at mRNA and protein level. Interestingly, NKG2D knockdown led to a mild but significant decrease of MICA*008+EV uptake (Figure 4g) demonstrating that the levels of NKG2D expression on the NK cell surface affected specifically the uptake process of ligand positive EVs.
FIGURE 4.
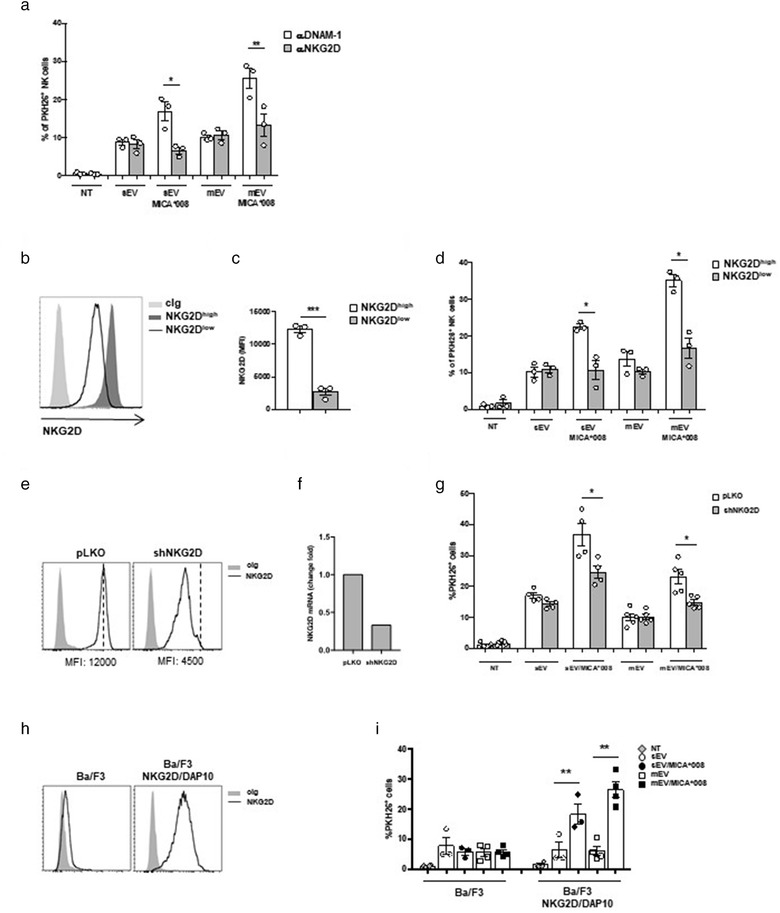
NKG2D mediates the uptake of MICA*008+ EVs. (a) NKL cells were pre‐treated with αNKG2D and αDNAM‐1 blocking mAbs and then incubated for 3 h with PKH26‐labelled sEVs (30 μg/ml) or mEVs (20 μg/ml). Data reported represent the mean of three independent experiments. Values represent the percentage of PKH26+ cells. (b and c) NKL cells were treated overnight with MICA*008+ EVs to induce NKG2D downmodulation (NKG2Dlow) or left untreated (NKG2Dhigh). (b) A representative experiment showing the different levels of NKG2D expression is shown. (c) The mean of three independent experiments is shown. (d) NKG2Dlow and NKG2Dhigh NKL cells were incubated with PKH26‐labelled EVs for 3 h as described in panel (a). Values represent the percentage of PKH26+ cells. The mean of three independent experiments is shown. (e–g) NKL cells were infected with the pLKO lentiviral vector containing a scrambled sequence or shRNA for silencing NKG2D. NKG2D expression was evaluated by (e) FACS analysis and (f) real‐time PCR. (g) NKL/pLKO and NKL/shNKG2D were incubated with PKH26‐labelled EVs for 3 h as described in panel (a). Values represent the percentage of PKH26+ cells. The mean of at least four independent experiments is shown. (h) Ba/F3 or Ba/F3 stably expressing the NKG2D/DAP10 complex were stained with cIg or anti‐NKG2D mAb and analyzed by Immunofluorescence and FACS analysis. (i) Ba/F3 or Ba/F3‐NKG2D/DAP10 were plated at 5 × 105 cells/ml and incubated for 1 h with PKH26‐labelled mEVs as described in panel (a). Values represent the percentage of PKH26+ cells. The mean of three (for sEVs) or four (for mEVs) independent experiments is shown
FIGURE 5.
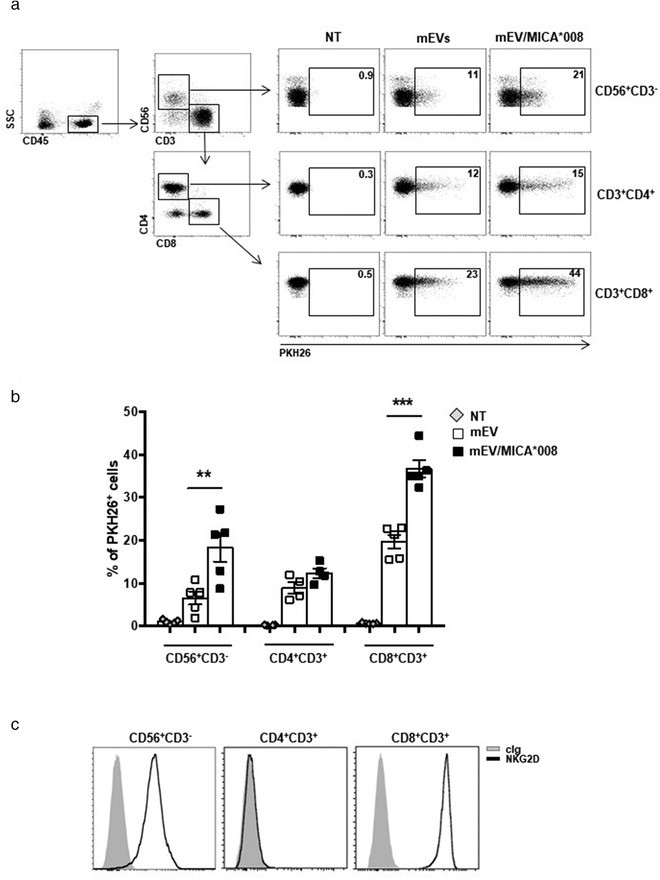
MICA*008+ EVs are preferentially uptaken by leukocyte subpopulations expressing NKG2D. Human primary peripheral blood mononuclear cells (PBMC) were incubated for 3 h with PKH26‐labelled mEVs expressing or not MICA*008. (a) Representative dot plot of different PBMC populations, by gating on CD45+CD56+CD3– for NK cells, on CD4+ and CD8+ after gating CD45+CD56–CD3+ for T lymphocytes. Representative uptake of these populations is shown. Values reported in each plot represent the percentage of PKH26+ cells. (b) The mean of at least four independent experiments is shown. (c) FACS analysis of NKG2D expression on different leukocyte populations. A representative plot is shown
These results prompted us to examine whether the ectopic expression of NKG2D could be sufficient in promoting the uptake of ligand expressing vesicles. To this purpose, the human NKG2D/DAP10 complex was stably transfected in the mouse pre‐B cell line Ba/F3 that does not express this receptor (Figure 4h). Notably, Ba/F3‐NKG2D/DAP10 represents a well‐established cellular model that was used to study the interaction of NKG2D with its cell‐surface ligands (Ogasawara et al., 2003). Interestingly our results show that the uptake of MICA*008+EVs was significantly higher only in Ba/F3 cells expressing NKG2D (Figure 4i) demonstrating that ectopic expression of NKG2D is sufficient to increase MICA*008+EV uptake.
Moreover, to mimic in vivo conditions, we determined whether NKG2DL+ mEVs could preferentially target leukocyte subpopulations expressing NKG2D. To this aim, PKH26‐labelled mEVs, expressing or not MICA*008, were incubated with PBMCs and the percentage of EV uptake was evaluated by gating on different populations including NK cells, CD4+ and CD8+ T lymphocytes as shown in Figure 5a. Interestingly, a significant increase of NKG2DL+ EV uptake was observed in NK cells and CD8+ T lymphocytes that express the cognate receptor (Figure 5a–c), whereas CD4+ T lymphocytes presented similar levels of uptake between NKG2DL+ and NKG2DL– EVs.
All together these results shed light on the importance of NKG2D/EV‐associated MICA*008 interaction in the enhancement of vesicles uptake through a specific receptor‐mediated mechanism.
2.3. MICA*008+ EVs trigger pERK and IFN‐γ production
Differently from the cleaved soluble counterparts, NKG2DL on the surface of EVs should preserve their whole‐molecule, three‐dimensional protein structure and biologic activity; thus, we assessed whether NKG2D‐mediated recognition of MICA*008 expressing EVs could trigger NK cell activation. Activation of NKG2D signalling pathway was evaluated by analysing ERK phosphorylation (Segovis et al., 2009; Sutherland et al., 2002) after 1 h of EVs incubation with NKL cells, a time point characterized by only a mild downmodulation of the receptor (Figure 6a). As shown in Figure 6b and 6c, a marked increase of ERK phosphorylation was observed in NK cells treated with MICA008* positive EVs. Importantly, to exclude that the increased ERK phosphorylation was due to pERK transfer from EVs, we evaluated pERK and total ERK levels in EV‐derived lysates. As shown in Figure S5, undetectable levels of pERK and total ERK were found confirming that the MICA*008+EV‐induced ERK phosphorylation is dependent on NKG2D signalling. Furthermore, we investigated the effect of NKG2D engagement by analysing IFN‐γ production. Interestingly, treatment of NK cells with MICA*008 positive EVs led to enhanced IFN‐γ production (Figure 6d). All together these data show that MICA*008 on the surface of EVs is able to efficiently engage NKG2D and triggers NK cell activation.
FIGURE 6.
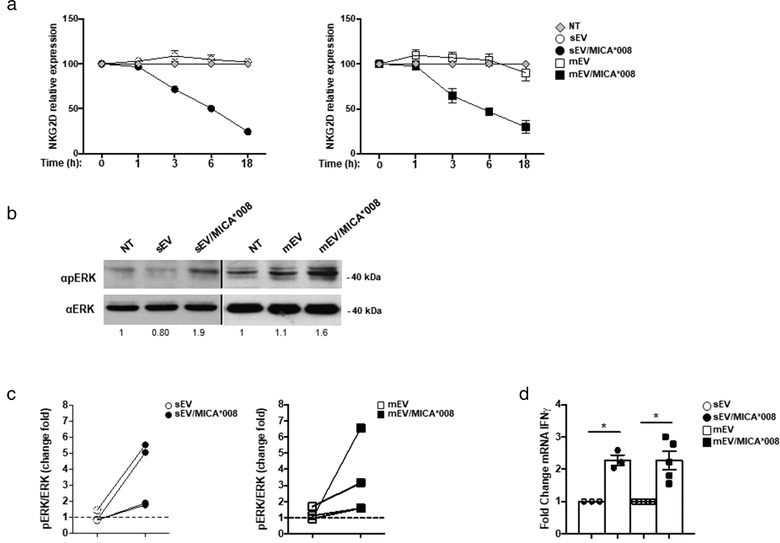
MICA*008 positive EVs engage NKG2D and trigger pERK and IFN‐γ production. (a) NKL cells were treated for different times with MICA*008+ or MICA*008– sEVs (at 30 μg/ml) or mEVs (at 20 μg/ml). Cells were harvested and NKG2D expression was evaluated by immunofluorescence and FACS analysis. Relative expression of NKG2D was calculated considering the untreated group as 100%. (b) NKL cells were treated for 1 h with MICA*008+ or MICA*008– EVs. Western blot analysis was performed on total cell lysates using ERK and phospho‐ERK (pERK) Abs. Numbers beneath each lane represent quantification of pERK by densitometry analysis normalized with ERK relative to the untreated group. (c) Quantification of immunoblots relative to different independent experiments is shown. Dashed line represents the untreated group. (d) Highly purified human primary NK cells were incubated with 30 μg/ml of sEVs or 20 μg/ml of mEVs expressing or not MICA*008 for 24 h. Real‐time PCR analysis of IFN‐γ mRNA is shown. Data, expressed as fold change units, were normalized with β‐actin and referred to MICA*008– EVs treated cells considered as calibrator. Values reported represent the mean of at least three independent experiments
2.4. Prolonged stimulation of NK cells with MICA*008 positive EVs reduces NKG2D‐mediated functions
Since NKG2D downmodulation is often associated to an impairment of NKG2D‐mediated cell functions, we asked whether MICA*008+ EVs could impair NK cells to subsequent NKG2D‐mediated engagement and triggering. To simulate a chronic and sustained NKG2DL‐mediated stimulation, NK cells were treated for 24 h with EVs expressing or not MICA*008 and then used to assess NKG2D responsiveness by measuring ERK phosphorylation upon NKG2D engagement with a specific antibody. An impairment of NKG2D‐mediated ERK phosphorylation was observed in MICA*008+EV‐pretreated NK cells (Figure 7a and 7b). Next, we investigated whether NKG2D‐mediated cytotoxicity was also affectedby performing a killing assay against P815 FcR+ target cells, in a redirected modality. Data presented in Figure 7c show that NKG2D engagement efficiently up‐regulates NK cell‐mediated lysis of P815 target cells that was suppressed using MICA*008+EV‐pretreated NK cells as effectors. Moreover, a significant reduction of cytotoxicity was also observed upon coengagement of 2B4 and NKG2D whereas the 2B4‐mediated killing was not affected. In addition, MICA*008+ EV‐pretreated NK cells were also assessed in a cytotoxic assay against the K562 sensitive target cell line the lysis of which is partially dependent on NKG2D. As shown in Figure S6, a moderate but significant reduction of the NK cell capability to kill target cells was observed. However, the impaired killing was not due to a reduction in the expression of lytic mediators as granzyme B, A and K and perforin were expressed at similar levels (Figure 7d).
FIGURE 7.
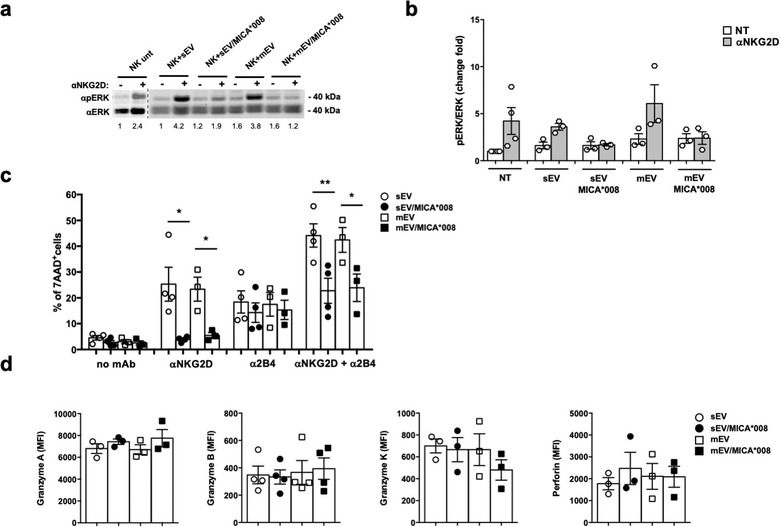
Prolonged exposure to MICA*008+ EVs impairs both NKG2D signalling pathway and NKG2D‐mediated killing. NKL cells were treated for 24 h with 30 μg/ml of sEVs or 20 μg/ml of mEVs expressing or not MICA*008. (a) Untreated or EV‐treated NKL cells were stimulated with anti‐NKG2D mAb plus goat anti‐mouse IgG for 15 min at 37°C before the lysis. Western blot analysis was performed on total cell lysates using ERK and phospho‐ERK (pERK) Abs. Numbers beneath each lane represent quantification of pERK by densitometry analysis normalized with ERK relative to the untreated group. (b) Quantification of immunoblots relative to independent experiments is shown. (c) The cytotoxic activity of EV‐treated NKL cells was evaluated by performing the 7‐AAD assay in a redirected modality against P815 FcR+ target cells loaded with different Ab as indicated in figure, at an E:T ratio of 25:1. Data reported represent the mean of at least three independent experiments. d) The NKL intracellular amount of perforin, granzyme A, B and K was evaluated through immunofluorescence and FACS analysis. The mean of at least three independent experiments is shown. Values represent the MFI of granzyme A, B, K and perforin subtracted from the MFI value of the cIg
All together these data show that MICA*008 positive EVs reduce NK cell responsiveness to subsequent NKG2D‐mediated engagement and triggering but do not influence the activity of other NK cell activating receptors (i.e., 2B4) and do not alter the levels of cytotoxic mediators.
To investigate whether NKG2D downmodulation induced by MICA*008+ EVs could occur in vivo, we relied on the fact that human MICA can bind murine NKG2D (Wiemann et al., 2005). At first we assessed whether MICA*008+EVs were effective in inducing NKG2D downmodulation ‘in vitro’ in mouse NK cells. As shown in Figure 8a, treatment of purified mouse NK cells with mEVs expressing MICA*008 determined a considerable downmodulation of the receptor. Next, mEVs expressing or not MICA were intraperitoneally (i.p.) injected into mice and NKG2D expression was evaluated after 3 and 18 h in peripheral blood (PB) and in peritoneal cavity (PC) NK cells. Our findings show that a significant reduction of NKG2D expression levels occurs after 3 h of treatment with MICA*008+mEVs in PB and PC NK cells, but downmodulation persists up to 18 h only in PC NK cells, suggesting that mEVs are rapidly cleared from blood circulation (Figure 8b). To further explore whether ‘in vivo’ induced NKG2D downmodulation could be related to a reduced capability to kill sensitive targets, we set up an ‘in vivo’ killing assay in the PC of mice injected with mEVs expressing or not MICA*008. The NK‐sensitive mouse MM cell line 5TGM1, was injected together with the NK cell resistant cell line RMA. The ratio of target (5TGM1) to control (RMA) cells was estimated 4 h after their injection. Our findings show that compared to control mEVs, injection of MICA*008+mEVs resulted in about 50% higher surviving 5TGM1 cell frequency in PC (Figure 8c). Of note, control experiments using mice depleted of NK cells by anti‐NK1.1 injection verified that 5TGM1 killing was indeed NK cell dependent and overall allow us to conclude that MICA*008+mEVs exert a marked protective effect from NK cell‐mediated killing of MM cells in vivo (Figure 8d).
FIGURE 8.
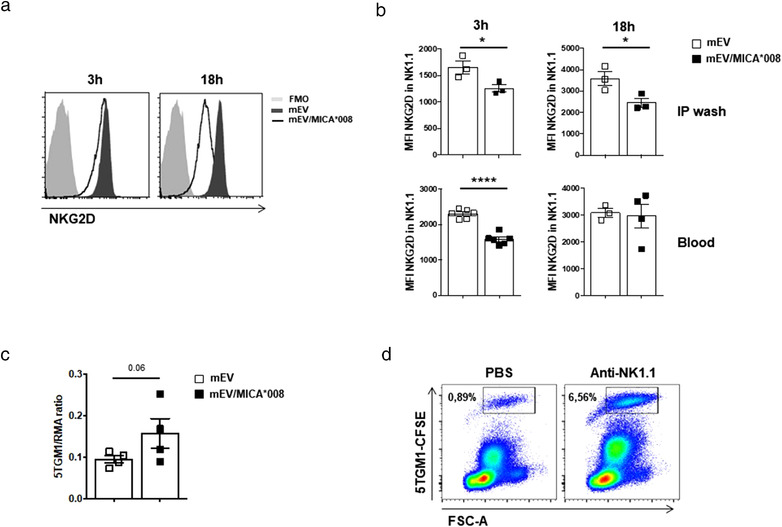
MICA*008+ mEVs induce NKG2D downmodulation ‘in vivo’. (a) Purified spleen‐derived mouse NK cells were plated at 2 × 106 cells/ml and treated with mEVs (20 μg/ml) for 3 and 18 h in the presence of IL‐15 (10 ng/ml). NKG2D expression levels were evaluated by immunofluorescence and FACS analysis. A representative experiment out of two is shown. (b) NKG2D downmodulation in vivo was assessed after i.p. injection of mEVs (50 μg) into mice. After 3 and 18 h, PB and peritoneal cells were stained with anti‐CD45.2, anti‐NK1.1, anti‐CD3 and anti‐NKG2D mAbs. NKG2D expression was measured by gating on NK.1.1+CD3–CD45+ cells. Results represent average of two independent experiments +/‐ SEM performed. (c) Host mice were i.p. treated with mEVs expressing or not MICA*008 (50 μg) 18 h before and at the time of target cell injection. Mice were i.p. injected with equal numbers (106) of target (5TGM1) and control (RMA) cells labelled with CFSE and PKH26, respectively. Cells were harvested from the PC 4 h later, stained with annexin V and the ratio of surviving target to control cells was calculated as number of annexin V– cells. Values represent average obtained from two independent experiments +/‐ SEM performed. d) In order to deplete NK1.1+ cells, anti‐NK1.1 mAb (Pk136, 100 μg/mouse) was administered to mice by i.p. injection the day before the assay. A total of 1 × 106 CFSE‐labelled live 5TGM1 cells were injected i.p. into C57BL/6 NK cell competent or NK cell depleted mice. Four hours later, mice were sacrificed and a peritoneal lavage performed. The percentage (number in gate) of CFSE‐labelled cells within the peritoneal lavage was determined by flow cytometry. One representative analysis is shown
2.5. MICA*008 can be transferred through EVs on NK cells
We observed that NKG2D downmodulation induced by both MICA*008+ sEVs and mEVs is persistent up to 72 h after treatment (Figure S7a) even after removing EVs from the culture (Figure S7b). Thus, we investigated whether beyond the direct interaction with MICA*008+EVs, other mechanisms concurred to the sustained NKG2D downmodulation. A number of studies reported that NK cells acquire cell surface proteins, including NKG2DLs, from target cells in a cell–cell contact‐dependent manner (McCann et al., 2007; Nakamura et al., 2013; Roda‐Navarro & Reyburn, 2007) but it is still unknown whether these ligands could be transferred through EVs as well. Thus, we investigated the capability of EVs carrying MICA*008 to transfer the ligand directly to NK cells. For this purpose, NK cells were exposed to MICA*008+ EVs for different times and then the presence of MICA on the surface of NK cells was assessed by immunofluorescence and FACS analysis. Interestingly, already upon 1 h incubation with MICA*008+EVs, NK cells turned to be MICA+, with no further increase after 6 h, indicating that MICA could be transferred from EVs to the NK cell surface (Figure 9a and 9b). Our results also show that MICA expression on NK cells was not related to an increase of its mRNA levels (Figure 9c). Acquisition of MICA was partially dependent on NKG2D, since a neutralizing anti‐NKG2D mAb significantly blocked the transfer (Figure 9d) suggesting that MICA could be transferred on NK cells either through NKG2D or with a mechanism independent of this receptor. As next step we examined whether NK cells that acquired MICA could induce NKG2D downmodulation. To this aim, MICA+NK cells were incubated with other CFSE‐labelled NK cells that had never seen NKG2DLs. Expression of NKG2D on CFSE+NK cells was evaluated after 24 h. As shown in Figur 9e–g, NKG2D was downmodulated, thus showing that MICA transferred to NK cells is functional and contributed to the sustained NKG2D downmodulation.
FIGURE 9.
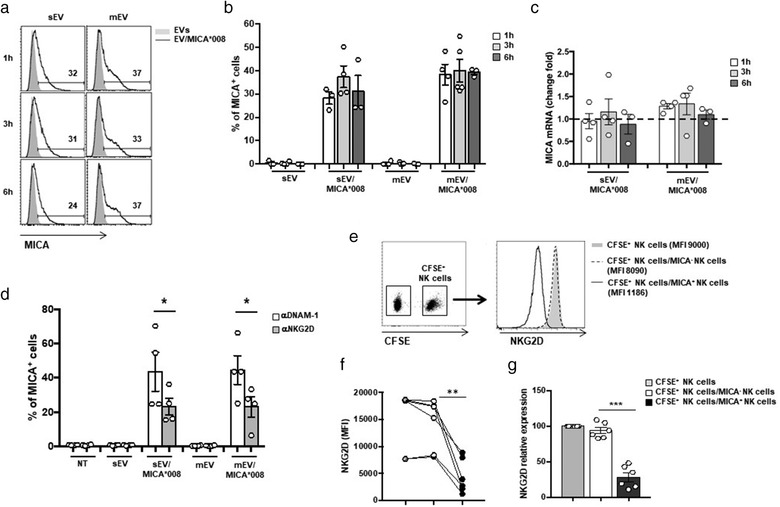
MICA*008 is transferred by EVs on NK cells. (a and b) NKL cells were incubated for different times with 30 μg/ml of sEVs and 20 μg/ml of mEVs expressing or not MICA*008 and after that MICA expression was evaluated by immunofluorescence and FACS analysis. In (a) data relative to a representative experiment are shown. Numbers above histograms represent the percentage of MICA+ cells. In (b) the mean of different independent experiments is shown. (c) NKL cells were treated as described in panel (a) and (b). Cells were harvested at different times as indicated, and real‐time PCR of MICA mRNA was performed. Data, expressed as fold change, were normalized with β‐actin and referred to MICA*008– EVs treated cells considered as calibrator (dashed line). Values reported represent the mean of at least three independent experiments. (d) NKL cells were pre‐treated with anti‐NKG2D and anti‐DNAM‐1 blocking Abs and then incubated for 3 h with 30 μg/ml of sEVs and 20 μg/ml of mEVs expressing or not MICA*008. MICA expression on NKL cells was evaluated by immunofluorescence and FACS analysis. Data reported represent the mean of four independent experiments. (e–g) NKL cells were labelled with CFSE and then cocultured with NKL cells dressed with MICA for 18 h and NKG2D expression was evaluated on CFSE+ NKL cells. A representative experiment is shown in panel (e). In (f) NKG2D MFI values relative to six independent experiments are shown. In (g) data are expressed as relative expression of NKG2D (MFI) respect to the untreated group (CFSE+ NKL cells corresponded to 100%)
2.6. Dressing of NK cells by EV‐derived MICA*008 induces fratricide
The capability of NK cells dressed with MICA‐derived from EVs to become susceptible to killing by other NK cells was assessed by using polyclonal primary NK cell cultures. Similarly to the NKL cell line, human polyclonal NK cells acquire MICA*008 by EVs (Figure 10a). Thus, autologous CFSE‐labelled NK cells were co‐cultured with target MICA*008+‐dressed NK cells and both degranulation and cytotoxic activity were evaluated. Interestingly a significant increase of CD107a degranulation marker on CFSE‐labelled NK cells was observed (Figure 10b and 10c). Moreover, cytotoxicity evaluated by 7AAD assay showed the occurrence of NK cell death (Figure 10d). These data demonstrate that acquisition of MICA*008 by EVs induces fratricide of NK cells.
FIGURE 10.
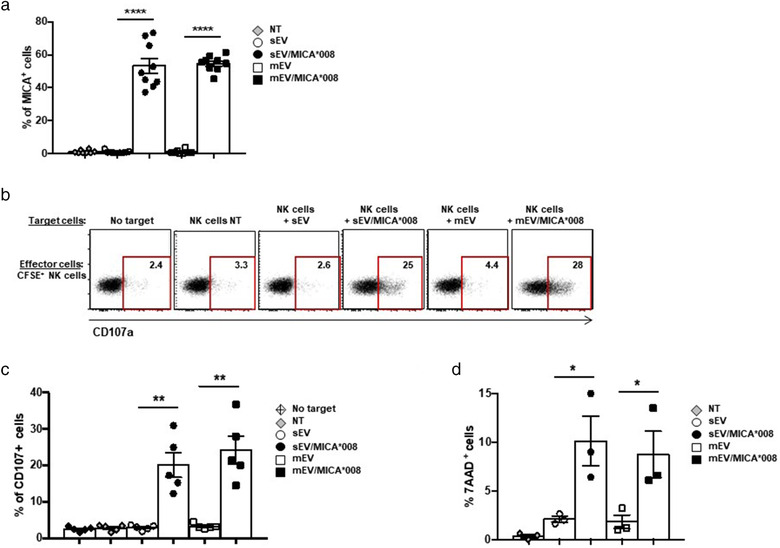
Dressing of NK cells with MICA*008 by EVs induces fratricide. (a) Human polyclonal NK cells were treated for 18 h with 30 μg/ml of sEVs and 20 μg/ml of mEVs expressing or not MICA*008. MICA expression on NK cells was evaluated by immunofluorescence and FACS analysis. Values reported represent the percentage of MICA+ cells and represent the mean of different independent experiments. (b and c) Polyclonal NK cells were labelled with CFSE and used as effectors in a CD107 degranulation assay. As target cells autologous CFSE– polyclonal NK cells treated or not with EVs were used (E:T ratio: 2.5:1). A representative experiment is shown. Values in each plot indicate the percentage of CD107+ CFSE+ NK cells. In (c) data relative to five independent experiments are shown. (d) Polyclonal NK cells were labelled with CFSE and used as effectors in a 7AAD cytotoxicity assay. As target cells autologous CFSE– polyclonal NK cells treated or not with EVs were used (E:T ratio: 12.5:1). The percentage of CFSE–7AAD+ NK cells was evaluated by immununofluorescence and FACS analysis. The mean of three independent experiments is shown
The presence of soluble MICA in cancer patient's plasma/sera has been associated to tumour progression, but it is unknown whether part of soluble MICA could be carried by vesicles. We thus investigated MICA expression on mEVs purified from the plasma derived by PB or BM of a cohort of MM cancer patients (Table 1). As shown in Figure 11a mEVs preparations were checked by electron microscopy and then analysed by immunofluorescence and FACS analysis by gating the phalloidin negative vesicle population (Figure S8). Interestingly, although the levels of MHC class I were comparable in the two pools of vesicle preparations, MICA as well as CD138 that is expressed on malignant plasma cells, were mainly found on BM‐derived EVs (Figure 11b) indicating that BM tumour microenvironment is enriched of EVs expressing MICA.
TABLE 1.
Patients characteristics
| Patient no. | Gender | Age | Stage | Monoclonal Ig | % of PC in the BM |
|---|---|---|---|---|---|
| 1 | M | 55 | Relapse | IgG‐k | 10 |
| 2 | F | 70 | Relapse | IgG‐k | 16 |
| 3 | M | 62 | Relapse | IgG‐k | 38 |
| 4 | F | 84 | Relapse | IgG‐k | 38 |
| 5 | F | 83 | Relapse | IgG‐k | 23 |
| 6 | F | 82 | Relapse | IgA‐L | 6 |
| 7 | M | 73 | Relapse | IgA‐k | 50 |
| 8 | F | 71 | Onset | Micro‐k | 38 |
| 9 | F | 80 | MGUS | IgG‐k | NV |
| 10 | F | 73 | Relapse | IgG‐L | 79 |
| 11 | M | 73 | Onset | IgG‐k | 17 |
| 12 | M | 72 | Onset | IgG‐k | 5 |
| 13 | F | 63 | Onset | IgG‐k | 20 |
| 14 | F | 83 | Onset | IgA‐l | 19 |
| 15 | M | 65 | Smoldering | IgA‐l | 6 |
| 16 | M | 72 | Relapse | IgA‐k | 13 |
| 17 | M | 62 | Smoldering | IgG‐k | 29 |
| 18 | F | 77 | Relapse | IgG‐k | 59 |
| 19 | F | 66 | Onset | IgG‐k | 14 |
Abbreviation: PC, plasma cell.
FIGURE 11.
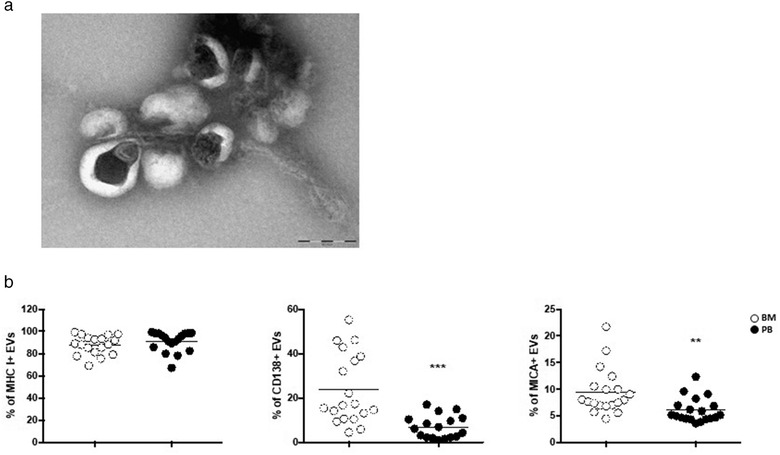
MICA is associated to BM‐derived EVs of MM patients. mEVs were isolated from peripheral blood (PB) or bone marrow (BM) aspirates of a cohort of MM patients. (a) Ultrastructural analysis of a representative sample. Bar corresponds to 200 nm. (b) Immunofluorescence and FACS analysis of mEVs using antibodies against MHC I, CD138 and MICA the expression of which is reported by percentage
3. DISCUSSION
EVs play a relevant role in intercellular communication, thanks to their ability to convey active biological molecules onto target cells (Colombo et al., 2014). Modulation of NK cell activity by EVs appears to be multi‐faceted and dependent on the identity of both the EV‐associated cargo and EV‐releasing cells (Soriani et al., 2020); as such, a number of studies have described either inhibition or activation of NK cell‐mediated functions (Borrelli et al., 2018; Clayton et al., 2008; Daßler‐Plenker et al., 2016).
Herein, we investigated how the stress‐induced NKG2D ligand MICA*008 associated to EVs could affect NK cell‐mediated functions and report that: (i) NKG2D is specifically involved in the uptake of EVs expressing its ligand; (ii) NKG2DL+ EVs induce NK cell activation after a short stimulation, but a persistent stimulation leads to NKG2D downmodulation associated to impaired NKG2D‐mediated function; (iii) NKG2DL can be transferred by vesicles to NK cells causing fratricide.
There is increasing evidence that demonstrates the existence of various types of EVs often having shared biophysical characteristics (Mathieu et al., 2019). Our study is focused on two main vesicle populations, including sEVs (<150 nm) recovered after high‐speed ultracentrifugation (100,000 × g) and intermediate‐size EVs of 150–300 nm derived after a lower speed centrifugation (13,000 × g). Our findings show that both type of vesicle populations express MICA*008 on the surface and thus are able to interact with NKG2D and induce its downmodulation. Various molecules contribute to the interaction between EVs and target cells, including integrins, immunoglobulins, proteoglycans, lectins, and the T cell immunoglobulin and mucin domain‐containing protein 4 (Tim4). These interactions appear to facilitate the endocytosis process and are important to define the selectivity of target cells (Mathieu et al., 2019). Our study reveals NKG2D as an important uptake receptor specifically involved in the capture of both sEVs and mEVs expressing its ligand and provide evidence that these vesicles specifically target leucocyte populations expressing NKG2D including CD8+ T lymphocytes and NK cells. Interestingly, specific receptor‐targeting of EV surface represents a promising strategy to be used in cancer nanomedicine (Zocchi et al., 2020). In this scenario, our results could offer an important tool to synthesize NKG2DL+ nanoparticles or alternatively generate NKG2DL+ EVs to deliver cytokines or artificially loaded short interfering RNA on populations of NKG2D expressing cells in order to enhance their anti‐tumour activity.
The presence of soluble NKG2D ligands in the sera of cancer patients has been extensively reported (Jinushi et al., 2007; Rebmann et al., 2007) and often related to a reduction of NKG2D expression levels on both NK and CD8+ T lymphocytes (Lundholm et al., 2014). However, it is still unclear whether soluble NKG2D ligands could directly induce NKG2D downregulation and/or if additional soluble factors in the sera of cancer patients like TGF‐β contribute to this effect. In addition, the method of detection of soluble NKG2D ligands does not distinguish between protease‐cleaved and vesicle‐associated NKG2D ligands.
We report herein that only MICA*008 associated to vesicles and not its soluble form retains the capability to downmodulate NKG2D. Indeed our findings show that vesicle‐deprived supernatants produced by MICA*008 transfectants are no longer able to induce NKG2D downmodulation suggesting that vesicle‐associated ligand maintains a conformation capable of engaging its cognate receptor. In line with our findings, Fernandez‐Messina and colleagues have previously shown that exosomal ULBP3 protein is much more potent in the induction of NKG2D down‐modulation than soluble ULBP2 protein (Fernández‐Messina et al., 2010). Moreover, a number of studies have shown that ‘in vitro’ treatment of NK cells with exosomal NKG2DLs derived from cancer cell lines (Hedlund et al., 2011; Labani‐Motlagh et al., 2015; Lundholm et al., 2014) or from placenta (Hedlund et al., 2009) leads to significant reduction of NKG2D expression levels; these experiments were performed using EVs expressing one or more ligands, thus it is plausible that the final effect on NKG2D downmodulation could be attributed by the cooperation between more than one ligand or alternatively one ligand could predominate over another. For instance, we found that although the long MICA allele, MICA*019, could be associated with EVs, MICA*008 is more efficient to induce NKG2D downmodulation (Figure S9).
Our findings provide novel evidence that MICA*008+EVs have the capability of inducing activation of extracellular signal‐regulated kinase (ERK) and IFN‐γ production showing that these vesicles might promote NK cell activation. To this regard, it is important to consider that NKG2D endocytosis not only leads to the reduction of receptor expression levels but also controls NKG2D mediated signal propagation (Molfetta et al., 2016; Quatrini et al., 2015). Interestingly, a previous study has shown that exosomes derived from DCs express both IL‐15Rα and the NKG2D ligand ULBP1 and induce NK cell activation (Viaud et al., 2009). Thus, we envisage that conditions aimed at inducing an upregulation of NKG2DLs on cancer cells and a transient augmentation of EV secretion could contribute to boost NK cell activity. Hedlund and colleagues have shown that oxidative stress enhances the release of NKG2D ligand‐bearing exosomes from cancer cell lines (Hedlund et al., 2011). A number of studies have shown that chemotherapeutic treatments promote exosome release in the extracellular milieu (Bandari et al., 2018; Gobbo et al., 2016; Vulpis et al., 2017, 2019; Yang et al., 2015).
On the other hand, chronic and persistent stimulation mediated by vesicles expressing MICA*008 drives sustained NKG2D downmodulation and reduction of NK cell responsiveness in vitro and in vivo subsequent to NKG2D‐mediated engagement and triggering. This hyporesponsiveness is specific so that neither the cytotoxic activity mediated by other NK cell activating receptors (i.e., 2B4) or the expression levels of cytotoxic mediators were affected. Altogether these observations suggest that increased release of vesicles in the tumour microenvironment over time determines their accumulation thus decreasing the ability of NKG2D to promote the activation of NK cells. A number of studies have shown that cancer patients affected by solid tumours (Caradec et al., 2014; Melo et al., 2015) or haematological malignancies including MM (Caivano et al., 2015; di Noto et al., 2016) possess high levels of circulating EVs. Notably, the presence of MICA+ microvesicles in the BM of MM patients allows us to suggest that these vesicles play a pivotal role in the tumour microenvironment by affecting NKG2D‐mediated functions.
Trogocytosis represents a process by which plasma membrane fragments are transferred from one cell to another during cell–cell contact. Previous evidence has shown that upon Natural Killer – Immune Synapse formation, human NK cells acquire MICA and MICB from tumour cells (McCann et al., 2007). Our findings reveal that MICA*008 can be transferred by both sEVs and mEVs to NK cells. Notably, EV‐mediated transfer of membrane proteins has also been reported in different systems and interestingly these molecules preserved their biological activity. The chemokine receptor CCR5 can be released through microvesicles and transferred to other cells thus contributing to HIV spreading (Mack et al., 2000). In addition, intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles determined the propagation of a transforming phenotype among subsets of cancer cells (Al‐Nedawi et al., 2008). Importantly, our results demonstrate that MICA transferred to NK cells by EVs is functional since it is able to engage and downmodulate its cognate receptor. Moreover, NK cells dressed with vesicle‐associated MICA*008 become susceptible to autologous NK cell lysis. Similarly, the trogocytosis‐mediated transfer of NKG2DLs causes NK cell fratricide through the NKG2D‐induced perforin pathway in vitro and in vivo (Nakamura et al., 2013). It is interesting to note that NK cell fratricide mediated by NKG2D/NKG2D ligand axis was reported in pathological ‘in vivo’ models. For example, during viral infection Ifnar−/− NK cells express increased levels of NKG2DLs and become susceptible to NK cell–mediated fratricide in a perforin‐ and NKG2D‐dependent manner (Madera et al., 2016).
In sum our findings add new insight on the dual role of NKG2DL associated to EVs in the tumour microenvironment; the NKG2DL+ EV immunostimulatory action could be proposed as therapeutic strategy aimed to boost NK cell activity in the course of chemotherapy by promoting the transient and controlled release of vesicles. However, the spreading of NKG2D ligands associated to vesicles and their chronic accumulation could restrain NK cell functions not only through NKG2D downmodulation but also by promoting NK cell fratricide revealing a new mechanism of NK suppression in cancer that may facilitate immune escape and progression.
4. MATERIALS AND METHODS
4.1. Cell lines, and human NK cell preparations
The human NK cell line NKL was cultured in RPMI 1640 medium (EuroClone, Milan, Italy) supplemented with 15% foetal bovine serum (FBS) (GIBCO, Life Technologies, Gaithersburg, MD) and recombinant human IL‐2 (200 U/ml; PeproTech, London, UK). The cDNA encoding MICA*008, was cloned in the pMSCV retroviral vector and used to transduce the human MM cell line ARK, as previously reported (Zingoni et al., 2015). The ARK transfectants, the human chronic myeloid leukaemia cell line K562, the murine BA/F3 pro‐B cell line, the murine FcR+P815 mastocytoma cell line and the RPMI 8866 cell line were maintained in RPMI 1640 medium supplemented with 15% FBS. The NK‐sensitive mouse MM cell line 5TGM1, previously shown to activate NK cells through NKG2D (Antonangeli et al., 2016), and the NK cell resistant mouse lymphoma cell line RMA were maintained in RPMI 1640 medium supplemented with 10% FBS, 2 mmol/L glutamine, 55 μmol/L of Beta‐Mercaptoethanol. All cell lines were mycoplasma free (EZ‐PCR Mycoplasma test kit; Biological Industries, Cromwell, CT). Human peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors. For some experiments, highly purified human primary NK cells were obtained from PBMCs by negative selection using magnetic beads (Miltenyi Biotec). NK cell purity was more than 95% CD56+CD3–. Polyclonal NK cell cultures were obtained by co‐culturing PBMC (1 × 105 cells/ml) with irradiated (3000 rad) RPMI 8866 cells (1 × 105 cells/ml) for 10 days at 37°C in a humified 5% CO2 atmosphere as previously described (Zingoni et al., 2000) and were routinely 90% CD16+, CD56+, CD3–, as assessed by immunofluorescence and cytofluorimetric analysis.
4.2. EV purification and recovery
EV‐free serum was obtained after centrifugation of FBS at 100,000 × g for 2–3 h in a Beckman ultracentrifuge (Beckman Coulter, Brea, CA) to remove both sEVs and mEVsfrom serum. One hundred fifty of ARK cells transduced with pMSCV or MICA*008, were cultured at high density (7 × 106 cells/ml) in 25 ml of RPMI 1640 supplemented with 15% of EV‐free FBS and antibiotics in a bioreactor (SARSTEDT, Numbrecht, Germany) for 48–72 h. sEVs purification was performed as previously reported with some modifications (Vulpis et al., 2017). Briefly, cells were harvested by centrifugation at 300 × g for 10 min. Cell‐free supernatants were then centrifuged at 2000 × g for 20 min followed by centrifugation at 12,000 × g for 40 min to remove cells, debris and EVs bigger in size than sEVs. Supernatants were filtered using a 0.22 μm filter and centrifuged at 100,000 x g for 70 min at 4°C in a Beckman ultracentrifuge to pellet exosomes. The resulting pellet was washed in a large volume of cold PBS and centrifuged again at 100,000 × g for 70 min at 4°C. Finally, sEVs were resuspended in PBS for further analyses and functional studies. mEVs were recovered during sEVs‐purification protocol through collection of the pellet obtained after centrifugation at 12,000 × g for 40 min at 4°C. This pellet was washed in a large amount of cold PBS and centrifuged again at 12,000 × g for 40 min at 4°C. The mEVs were resuspended in PBS for further analyses and functional studies. EV total protein concentration was measured by Bio‐Rad Protein Assay (BPA) using a NanoDrop spectrophotometer (Thermo Fisher Scientific). The extent of sEVs recovered by 106 cells corresponded to 1.127 μg ± 0.58, whereas the amount of mEVs corresponded to 3.04 μg ± 1.79. Thus, about 30 μg of sEVs could be obtained from about 30 × 106 cells, whereas 20 μg of mEVs could be obtained from about 10 × 106 cells.
4.3. EV purification from patients’ plasma
Plasma samples derived from PB or BM aspirates were collected through centrifugation at 1400 rpm for 10 min. Plasma samples were further diluted (1:2) in PBS and centrifuged at 2000 × g for 20 min at 4°C. Medium‐size EVs were then recovered through collection of the pellet obtained after centrifugation at 12,000 × g for 40 min at 4°C. The pellet was washed in cold PBS and centrifuged twice at 12,000 × g for 40 min at 4°C. The mEVs were resuspended in PBS for further analyses.
4.4. Ultrastructural analysis
Transmission electron microscopy (TEM) of EVs was performed as follows: briefly, EVs were fixed in 2% PFA and adsorbed on formvar‐carbon‐coated copper grids. The grids were then incubated in 1% glutaraldehyde for 5 min, washed with deionized water eight times, and then negatively stained with 2% uranyl oxalate (pH 7) for 5 min and methyl cellulose/uranyl for 10 min at 4°C. Excess methyl cellulose/uranyl was blotted off, and the grids were air‐dried and observed with a TEM (Philips Morgagni268D) at an accelerating voltage of 80 kV. Digital images were taken with Mega View imaging software.
4.5. Size experiments and evaluation of the EV number through DLS
DLS experiments were performed to measure EV size and their concentration. All the measurements were made at 25°C on a Zetasizer Nano ZS90 spectrometer (Malvern, UK) equipped with a 5 mW HeNe laser (wavelength λ D 632.8 nm) and a non‐invasive back‐scattering optical setup (NIBS). For each sample, the detected intensity was processed by a digital logarithmic correlator, which computes a normalized intensity autocorrelation function. Then, the distribution of the diffusion coefficient D was obtained by using the CONTIN method.63 D was converted into an effective hydrodynamic diameter DH through the Stokes–Einstein equation: DH D KBT/(3phD), where KBT is the system's thermal energy and h represents the solvent viscosity. Solvent‐resistant micro cuvettes (ZEN0040, Malvern, Herrenberg, Germany) have been used for experiments with a sample volume of 40 μl. EV size distribution and concentration was calculated by a recently proposed DLS‐based non‐invasive tool as described previously (Vulpis et al., 2017).
4.6. Plasmids
The pMX retroviral vectors containing cDNA encoding the human NKG2D and DAP10/FLAG sequences were kindly provided by Prof. Lewis L. Lanier (University of California, San Francisco). For knocking down NKG2D, we cloned the following sh‐NKG2D‐sequences in the pLKO lentiviral vector: NKG2D#1, 5′‐CCCAACCTACTAACAATAATT‐3′, NKG2D#2, 5′‐GCTGTATTCCTAAACTCATTA‐3′.
4.7. Virus production and in vitro transduction
For retrovirus production, the Phoenix retrovirus packaging cell line HEK293 was transfected with viral DNA (pMXpie/hDAP10/FLAG and pMXpuro/hNKG2D) and the packaging vectors (pMCMVgag‐pol and pMD2.G) using Lipofectamine 2000 (Invitrogen, San Diego, CA). With regard to lentivirus production, Phoenix cells were transfected with pLKO‐sh‐NKG2D#1 together with pLKO‐sh‐NKG2D#2, or the control vector pLKO plus psPAX2 and pMD2.G packaging plasmids. After 48 h virus containing supernatants were harvested, filtered and used for infection as follows: 1 ml viral supernatant containing Polybrene (8 mg/ml) was used to infect 3 × 105 NKL cells or BAF/3 cells for 3 h. Two infection cycles were performed. After 72 h, infected cells were grown in selection media containing puromycin at 1 μg/ml for NKL or puromycin (1 μg/ml) plus G418 (400 μg/ml) for BAF/3. To obtain a cell population expressing high levels of the NKG2D/DAP10 complex, BAF/3‐NKG2D/DAP10/FLAG transfectants were stained with anti‐FLAG mAb (clone M5, Sigma) and further sorted based on the high expression levels of FLAG/DAP10 using a BDFACSAria III (BD Bioscience) equipped with FACSDiva software (BD Biosciences version 6.1,3).
4.8. Mice
Female C57BL/6j mice (Charles River, Calco, Italy), were housed in the animal house of the Histology unit in Sapienza University under specific pathogen‐free conditions. All animal studies were designed according Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines and to national (D.lgs. 26/2014) and international law and policies (EEC Council Directive 2010/63/EU) and were approved by the Italian Ministry of Health (Health Ministry authorization 769/2015 PR and 727/2019 PR).
4.9. In vitro and in vivo effects of mEVs on mouse NK cells
Spleens C57BL/6j mice were dissociated into single cell suspensions by mechanical disruption on a 70‐μm cell strainer (Flacon, Becton Dickinson) with a rubber syringe plunger. NK cells were then enriched (80% purity) using NK Cell Isolation Kit (Miltenyi Biotech, Bergisch Gladbach, Germany). After 1 h recover in 37 °C 5%CO2 humidified incubator, purified NK cells were plated at 2 × 106 cells/ml in complete Iscove's Modified Eagle's Medium (IMEM) supplemented with 10 ng/ml of IL‐15 and treated with mEVs (20 μg/ml) for 3 and 18 h. NKG2D downmodulation in vivo was assessed after i.p. injection of mEVs (50 μg) into mice. PB was collected from the caudal vein, while peritoneal cells were collected by peritoneal lavage after mice sacrifice. Peritoneal cells and PBL were stained with fluorochrome conjugated anti‐mouse CD45.2 (104), NK1.1 (PK136), CD3 (145‐2C11), CD8 (53‐6.7) and NKG2D (CX5) mAbs (clone in brackets; Biolegend, Sand Diego, CA; Termo Fisher Scientific, Waltham, MA, USA). RBC in PBL and peritoneal cell suspensions were lysed by red blood cell lysis buffer treatment. To examine the effect of mEVs‐induced NKG2D downmodulation on the cytotoxic capacity of NK cells, in vivo cytotoxic assay based on the use of the NK cell resistant RMA cells and of the NK cell sensitive 5TGM1 cells was performed as previously described (Saudemont et al., 2010). Briefly, host mice were i.p. treated with mEVs expressing or not MICA*008 (50 μg) or with PBS 18 h before and at the time of target cell injection.
Mice were i.p. injected with equal numbers (106) of target (5TGM1) and control (RMA) cells labelled with CFSE and PKH26, respectively. Target and control cells were harvested from the PC 4 h later, stained with annexin V and the ratio of surviving target to control cells was calculated as number of annexin V– cells.
4.10. NK cell stimulation
For the functional studies NKL cells were starved for 16–18 h in 3% FBS medium without IL‐2 and then seeded at 3 × 106 cell/ml in complete medium and stimulated for different times with 30 μg/ml of sEVs or 20 μg/ml of mEVs before the assay. For stimulation with anti‐NKG2D antibody, NKL cells were incubated with saturating amount of anti‐NKG2D mAb (clone 149810, R&D Systems) for 30 min on ice. Goat anti‐mouse antibody (Jackson ImmunoResearch) was then added and incubated at 37°C for the different times as previously described (Quatrini et al., 2015). In some experiments PBMCs or polyclonal NK cells were plated at 3 × 106 cell/ml in complete medium and treated for 24–48 h with 30 μg/ml of sEVs or 20 μg/ml of mEVs.
4.11. Flow cytometry of cells and EVs
NKL cells or polyclonal NK cells were labelled with anti‐MICA/PE or anti‐MICA/APC (clone 159277, R&D Systems, Minneapolis, Minnesota), anti‐NKG2D/APC (clone 1D11), anti‐DNAM/FITC (clone DX11) or with the respective isotype control Ig (all from BD Biosciences, San Jose, CA) for 25 min at 4°C. Immunofluorescence and FACS analysis of CD63+ sEVs was performed by positive magnetic selection as previously described (Vulpis et al., 2017). Briefly, about 5–10 μg of sEVs were incubated with 20 μl of CD63+ dynabeads (Invitrogen Corporation, Carlsbad, CA) for 18–22 h at 4°C with gentle tilting. The bead‐bound sEVs were labelled with cIg/PE, cIg/APC anti‐CD63/PE (clone H5C6, BioLegend, San Diego, CA), anti‐MICA/APC (clone 159277, R&D Systems, Minneapolis, MN) for 60 min at 4°C. For the FACS analysis of mEVs, about 5–10 μg of mEVs were labelled with specific antibodies (as indicated for sEVs) in combination with Phalloidin/FITC (Invitrogen Corporation, Carlsbad, CA) for 60 min at room temperature. In regard to mEVs isolated from plasma derived from PB or BM of MM patients, 5 μg of vesicle preparations were labelled with Phalloidin/FITC in combination with anti‐MICA (clone 159277, R&D Systems), anti‐CD138 (clone MI15, BD Bioscience) and anti‐HLA‐ABC (clone W6/32, BD Bioscience). The size of mEVs was estimated by comparing the forward scatter signals with those of reference microspheres obtained from flow cytometry sub‐micron particle size reference kit (Thermo Fisher Scientific).
4.12. EVs uptake
About 100 μg of EVs diluted in PBS were incubated with the red fluorescent dye PKH26 (Sigma‐Aldrich, St Louis, MO) ). sEVs were washed twice with PBS by ultracentrifugation at 100,000 × g for 70 min, whereas mEVs were washed twice with PBS by centrifugation at 12,000 × g for 40 min. PKH26‐labelled EVs were diluted with PBS and used for uptake experiments. PKH26‐labelled EVs (30 μg/ml of sEVs or 20 μg/ml of mEVs) were incubated with NKL cells or PBMCs for 3 h. Cells were collected, washed with PBS and PBMCs were labelled with the following mAbs: anti‐CD45/PE‐Cy7 (HI30), anti‐CD56/BV421 (NCAM16.2), anti‐CD3/PerCP (SK7), anti‐CD4/FITC (SK3), anti‐NKG2D/APC (1D11) all from BD Biosciences, San Jose, CA and anti‐CD8/APC‐Vio770 (REA734, Miltenyi Biotec) for 25 min at 4°C. After PBS washing, samples were collected for immunofluorescence and FACS analysis. In a set of experiments, NKL cells were pre‐treated for 1 h with dynasore, nystatin, 5‐(N‐ethyl‐N‐isopropyl) amiloride (EIPA) and ethylenediaminetetraacetic acid disodium salt solution (EDTA) from Sigma‐Aldrich (St Louis, MO) and then incubated for 3 h with PKH26‐labelled EVs. In some experiments, before incubation with PKH26‐labelled EVs, NKL were pre‐treated with saturating amounts of anti‐NKG2D mAb ( clone 149810, R&D Systems) or anti‐DNAM1 (clone DX11, Serotec) neutralizing antibodies.
To perform confocal microscopy experiments, after 3 h incubation with PKH26‐labelled EVs, NKL cells were washed, plated on poly‐L‐lysine‐coated multichamber glass plates, let to adhere for 30 min and fixed with 4% PFA. Cells were counterstained with DAPI and coverslips were mounted using SlowFade Gold reagent (Life Technologies). Images were acquired with IX83 FV1200 MPE laser‐scanning confocal microscope equipped with FluoView 4.2 software using a 60 × /1.35 NA UPlanSAPO oil immersion objective (all from Olympus) as previously described (Zitti et al., 2017). Uptake was quantified with Fiji/ImageJ software.
4.13. NK cell degranulation and cytotoxicity assays
NKL cells were incubated with CFSE (Sigma‐Aldrich, St Louis, MO)‐labelled K562 at different Effector:Target (E:T) ratio for 4 h at 37°C. After washing with PBS/1% BSA, cells were resuspended in PBS/1% BSA and the intercalating DNA dye 7‐Aminoactinomycin D (7‐AAD) (Sigma‐Aldrich, St Louis, MO) was added for 20 min at 4°C at the concentration of 5 μg/ml. Cells were then fixed with 1% PFA (Sigma‐Aldrich, St Louis, MO) for 20 min at 4°C and at least 20,000 events in the CFSE+ gate were collected using a FACSCanto. In some experiments the 7‐AAD assay was used to measure the cytotoxic activity against P815 target cells, in the presence of saturating amounts of different mAbs (anti‐NKG2D mAb clone 149810, R&D Systems; anti‐2B4 mAb clone C1.7, Biolegend). Degranulation assay was performed as described previously (Zingoni et al., 2015). To assess intracellular amount of perforin and granzymes, NKL cells were treated with EVs for 24 h at 37°C. Cells were subsequently fixed, permeabilized, stained with the following mAbs from BD Bioscience: anti‐perforin/BV410 (δG9), anti‐granzyme A/FITC (CB9), anti‐granzyme B/FITC (GB11) and anti‐granzyme K/APC (G3H69). Autologous NK cell degranulation and cytotoxicity assays were performed using IL‐2‐treated polyclonal NK cells.
4.14. Western blot analysis and ELISA
For Western blot analysis NKL cells were lysed in 1× BASERGA lysis buffer (1%Triton X‐100, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 50 mM HEPES) plus complete protease inhibitor mixture and phosphatase inhibitors (Sigma‐Aldrich, St Louis, MO). EVs preparations were lysed in 1× RIPA lysis buffer (1% NP‐40, 0.1% SDS 50 mM Tris‐HCl pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 1 mM EDTA) plus complete protease inhibitor mixture (Sigma‐Aldrich, St Louis, MO) and phosphatase inhibitors. Protein concentration was determined with the Bio‐Rad Protein Assay (BPA). 40 to 80 μg of cell or EV extract was run on 8%–12% denaturing sodium dodecyl sulfate (SDS)‐polyacrylamide gels. Proteins were then electro‐blotted onto nitrocellulose membranes (GE Healthcare Amersham, Little Chalfont, UK) and blocked in 5% milk in TBS‐T buffer (20 mM tris‐HCl (pH 7.8), 150 mM NaCl, 0.05% Tween 20) for 1 h. Filters were then incubated with specific primary and secondary antibodies diluted in TBS‐T 1% BSA. Immunoreactive bands were visualized on the nitrocellulose membranes, using Horseradish Peroxidase (HRP)‐coupled goat anti‐rabbit Ig and the enhanced chemiluminescence kit (ECL) detection system (GE Healthcare Amersham), following the manufacturer's instructions. The following antibodies were used for Western blotting: anti‐ERK1/2 and anti pERK1/2 (T202/Y204), from Cell Signaling Technology (Danvers, MA), anti‐NKG2D (3.1.1.1) from Millipore, anti‐β‐Actin (4C2) from Sigma‐Aldrich, anti‐MICA (AMO1) from BAMOMAB (Germany), anti‐CD63 (H‐193), anti‐Hsp70/Hsc70 (W27), anti‐CD81 (H‐121), were purchased from Santa Cruz Biotechnology (CA), anti‐calreticulin from Thermo Fisher Scientific (Rockford, USA) and anti‐MHC I (clone HC10) was kindly provided from Dr P. Giacomini, Regina Elena Cancer Institute, Rome, Italy. Densitometric analysis was performed with ImageJ software. ELISA kit for the detection of MICA was from R&D System (Minneapolis, Minnesota).
4.15. RNA isolation, RT‐PCR and real‐time PCR
Total RNA from human primary purified NK cells or NKL cells was extracted using Total RNA Mini Kit (Geneaid, New Taipei City, Taiwan). Total RNA (100 ng–1 μg) was used for cDNA first‐strand synthesis using oligo‐dT (Promega, Madison, WI) in a 25 μl reaction volume. Real‐time PCR was performed using the ABI Prism 7900 Sequence Detection system (Applied Biosystems, Foster City, CA). cDNAs were amplified in triplicate using the following specific TaqMan Gene Expression Assay all conjugated with fluorochrome FAM (Applied Biosystems): IFNγ (Hs00989291_m1), MICA (Hs00792195_m1) and human β‐actin (Hs99999903_m1). NKG2D and GAPDH mRNA expression were analysed using the Power‐SYBR green mix with ROX (Applied Biosystems). Primer sequences were as follows: human NKG2D forward: 5′‐TTCAACACGATGGCAAAAGC‐3′; NKG2D reverse: 5′‐CTACAGCGATGAAGCAGCAGA‐3′; human GAPDH forward: 5′‐TCGACAGTCAGCCGCATCT‐3′; GAPDH reverse: 5′‐CCGTTGACTCCGACCTTCA‐3′. The cycling conditions were: 50°C for 10 min, followed by 40 cycles of 95°C for 30 s, and 60°C for 2 min. Data were analysed using the Sequence Detector v1.7 analysis software (Applied Biosystems). Relative expression of each gene versus the housekeeping gene was calculated according to the 2–ΔΔCt method.
4.16. Flow cytometry
Samples were acquired using a FACSCanto (BD Biosciences, San Jose, CA). Data analysis was performed using the FlowJo program.
4.17. Statistics and graphs
Error bars represent SD or where indicated SEM. Statistical analysis was performed with the Student test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All the results are presented by dot plots showing position of the individual biological replicates (Weissgerber et al., 2015).
AUTHOR CONTRIBUTION
E.V. and L.L. designed and performed experiments, analysed and interpreted the data, R.M. and R.P. achieved confocal microscope analysis, G.C. performed DLS experiments, L.M. and L.S. performed and analysed TEM images., G. Peruzzi made FACS sorting, M.T.P. and F.F. provided clinical samples and made patients' diagnosis, S.P. and A.P. processed patients' samples., L.T. and G.B. performed mouse experiments, A. Soriani and C.F. contributed with analytic tools and analyzed results; C.C., M.C. and G. Palmieri critically reviewed the manuscript; A. Santoni contributed to design research and edit the manuscript; A.Z. developed the hypothesis, designed the experimental approach and wrote the manuscript.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This work was supported by grants of Italian Association for Cancer Research (AIRC 5 × 1000 grant n.21147) and grant by Sapienza University (RM1181642771A58E and RG11916B7F06EC21). We thank the Center for Life Nano Science (CLNS) imaging facility of the Istituto Italiano di Tecnologia (IIT, Rome, Italy), where the fluorescent images were collected and FACS sorting was performed.
Vulpis, E. , Loconte, L. , Peri, A. , Molfetta, R. , Caracciolo, G. , Masuelli, L. , Tomaipitinca, L. , Peruzzi, G. , Petillo, S. , Petrucci, M.T. , Fazio, F. , Simonelli, L. , Fionda, C. , Soriani, A. , Cerboni, C. , Cippitelli, M. , Paolini, R. , Bernardini, G. , Palmieri, G. , … Zingoni, A. (2021). Impact on NK cell functions of acute versus chronic exposure to extracellular vesicle‐associated MICA: Dual role in cancer immunosurveillance. Journal of Extracellular Vesicles, 11, e12176. 10.1002/jev2.12176
Elisabetta Vulpis and Luisa Loconte equally contributed to the work.
REFERENCES
- Abruzzese, M. P. , Bilotta, M. T. , Fionda, C. , Zingoni, A. , Soriani, A. , Vulpis, E. , Borrelli, C. , Zitti, B. , Petrucci, M. T. , Ricciardi, M. R. , Molfetta, R. , Paolini, R. , Santoni, A. , & Cippitelli, M. (2016). Inhibition of bromodomain and extra‐terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell‐mediated cytotoxicity in multiple myeloma cells: Role of cMYC‐IRF4‐miR‐125b interplay. Journal of Hematology & Oncology 9, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Nedawi, K. , Meehan, B. , Micallef, J. , Lhotak, V. , May, L. , Guha, A. , & Rak, J. (2008). Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nature Cell Biology 10, 619–624. [DOI] [PubMed] [Google Scholar]
- Antonangeli, F. , Soriani, A. , Ricci, B. , Ponzetta, A. , Benigni, G. , Morrone, S. , Bernardini, G. , & Santoni, A. (2016). Natural killer cell recognition of in vivo drug‐induced senescent multiple myeloma cells. OncoImmunology 5, 10e1218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashiru, O. , Boutet, P. , Fernández‐Messina, L. , Agüera‐González, S. , Skepper, J. N. , Valés‐Gómez, M. , & Reyburn, H. T. (2010). Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Research 70, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashiru, O. , López‐Cobo, S. , Fernández‐Messina, L. , Pontes‐Quero, S. , Pandolfi, R. , Reyburn, H. T. , & Valés‐Gómez, M. (2013). A GPI anchor explains the unique biological features of the common NKG2D‐ligand allele MICA*008. Biochemical Journal 454, 295–302. [DOI] [PubMed] [Google Scholar]
- Bandari, S. K. , Purushothaman, A. , Ramani, V. C. , Brinkley, G. J. , Chandrashekar, D. S. , Varambally, S. , Mobley, J. A. , Zhang, Y. , Brown, E. E. , Vlodavsky, I. , & Sanderson, R. D. (2018). Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biology 65, 104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli, C. , Ricci, B. , Vulpis, E. , Fionda, C. , Ricciardi, M. R. , Petrucci, M. T. , Masuelli, L. , Peri, A. , Cippitelli, M. , Zingoni, A. , Santoni, A. , & Soriani, A. (2018). Drug‐induced senescent multiple myeloma cells elicit NK cell proliferation by direct or exosome‐mediated IL15 trans ‐presentation. Cancer Immunology Research 6, 860–869. [DOI] [PubMed] [Google Scholar]
- Caivano, A. , Laurenzana, I. , de Luca, L. , la Rocca, F. , Simeon, V. , Trino, S. , D'Auria, F. , Traficante, A. , Maietti, M. , Izzo, T. , D'Arena, G. , Mansueto, G. , Pietrantuono, G. , Laurenti, L. , Musto, P. , & Del Vecchio, L. (2015). High serum levels of extracellular vesicles expressing malignancy‐related markers are released in patients with various types of hematological neoplastic disorders. Tumor Biology 36, 9739–9752. [DOI] [PubMed] [Google Scholar]
- Caradec, J. , Kharmate, G. , Hosseini‐Beheshti, E. , Adomat, H. , Gleave, M. , & Guns, E. (2014). Reproducibility and efficiency of serum‐derived exosome extraction methods. Clinical Biochemistry 47, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Carbone, E. , Neri, P. , Mesuraca, M. , Fulciniti, M. T. , Otsuki, T. , Pende, D. , Groh, V. , Spies, T. , Pollio, G. , Cosman, D. , Catalano, L. , Tassone, P. , Rotoli, B. , & Venuta, S. (2005). HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 105, 251–258. [DOI] [PubMed] [Google Scholar]
- Chan, W. K. , Kang, S. , Youssef, Y. , Glankler, E. N. , Barrett, E. R. , Carter, A. M. , Ahmed, E. H. , Prasad, A. , Chen, L. , Zhang, J. , Benson, D. M. Jr , Caligiuri, M. A. , & Yu, J. (2018). A CS1‐NKG2D bispecific antibody collectively activates cytolytic immune cells against multiple myeloma. Cancer Immunology Research 6, 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiossone, L. , Dumas, P.‐Y. , Vienne, M. , & Vivier, E. (2018). Natural killer cells and other innate lymphoid cells in cancer. Nature Reviews Immunology 18, 671–688. [DOI] [PubMed] [Google Scholar]
- Chitadze, G. , Bhat, J. , Lettau, M. , Janssen, O. , & Kabelitz, D. (2013). Generation of soluble NKG2D ligands: Proteolytic cleavage, exosome secretion and functional implications. Scandinavian Journal of Immunology 78, 120–129. [DOI] [PubMed] [Google Scholar]
- Clayton, A. , Mitchell, J. P. , Court, J. , Linnane, S. , Mason, M. D. , & Tabi, Z. (2008). Human tumor‐derived exosomes down‐modulate NKG2D expression. The Journal of Immunology 180, 7249–7258. [DOI] [PubMed] [Google Scholar]
- Colombo, M. , Raposo, G. , & Théry, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual Review of Cell and Developmental Biology 30, 255–289. [DOI] [PubMed] [Google Scholar]
- Daßler‐Plenker, J. , Reiners, K. S. , van den Boorn, J. G. , Hansen, H. P. , Putschli, B. , Barnert, S. , Schuberth‐Wagner, C. , Schubert, R. , Tüting, T. , Hallek, M. , Schlee, M. , Hartmann, G. , Pogge von Strandmann, E. , & Coch, C. (2016). RIG‐I activation induces the release of extracellular vesicles with antitumor activity. OncoImmunology 5, e1219827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Sherbiny, Y. M. , Meade, J. L. , Holmes, T. D. , McGonagle, D. , Mackie, S. L. , Morgan, A. W. , Cook, G. , Feyler, S. , Richards, S. J. , Davies, F. E. , Morgan, G. J. , & Cook, G. P. (2007). The requirement for DNAM‐1, NKG2D, and NKp46 in the natural killer cell‐mediated killing of myeloma cells. Cancer Research 67, 8444–8449. [DOI] [PubMed] [Google Scholar]
- Fauriat, C. , Mallet, F. , Olive, D. , & Costello, R. T. (2006). Impaired activating receptor expression pattern in natural killer cells from patients with multiple myeloma. Leukemia 20, 732–733. [DOI] [PubMed] [Google Scholar]
- Fernández‐Messina, L. , Ashiru, O. , Boutet, P. , Agüera‐González, S. , Skepper, J. N. , Reyburn, H. T. , & Valés‐Gómez, M. (2010). Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)‐anchored NKG2D ligands. Journal of Biological Chemistry 285, 8543–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari de Andrade, L. , Tay, R. E. , Pan, D. , Luoma, A. M. , Ito, Y. , Badrinath, S. , Tsoucas, D. , Franz, B. , May, K. F. , Harvey, C. J. , Kobold, S. , Pyrdol, J. W. , Yoon, C. , Yuan, G. C. , Hodi, F. S. , Dranoff, G. , & Wucherpfennig, K. W. (2018). Antibody‐mediated inhibition of MICA and MICB shedding promotes NK cell–driven tumor immunity. Science 359, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fionda, C. , Abruzzese, M. P. , Zingoni, A. , Cecere, F. , Vulpis, E. , Peruzzi, G. , Soriani, A. , Molfetta, R. , Paolini, R. , Ricciardi, M. R. , Petrucci, M. T. , Santoni, A. , & Cippitelli, M. (2015). The IMiDs targets IKZF‐1/3 and IRF4 as novel negative regulators of NK cell‐activating ligands expression in multiple myeloma. Oncotarget 6, 23609–23630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbo, J. , Marcion, G. , Cordonnier, M. , Dias, A. M. M. , Pernet, N. , Hammann, A. , Richaud, S. , Mjahed, H. , Isambert, N. , Clausse, V. , Rébé, C. , Bertaut, A. , Goussot, V. , Lirussi, F. , Ghiringhelli, F. , de Thonel, A. , Fumoleau, P. , Seigneuric, R. , & Garrido, C. (2016). Restoring anticancer immune response by targeting tumor‐derived exosomes with a HSP70 peptide aptamer. Journal of the National Cancer Institute 108, 1–11. [DOI] [PubMed] [Google Scholar]
- Hedlund, M. , Stenqvist, A.‐C. , Nagaeva, O. , Kjellberg, L. , Wulff, M. , Baranov, V. , & Mincheva‐Nilsson, L. (2009). Human placenta expresses and secretes NKG2D ligands via exosomes that down‐modulate the cognate receptor expression: Evidence for immunosuppressive function. The Journal of Immunology 183, 340–351. [DOI] [PubMed] [Google Scholar]
- Hedlund, M. , Nagaeva, O. , Kargl, D. , Baranov, V. , & Mincheva‐Nilsson, L. (2011). Thermal‐ and oxidative stress causes enhanced release of NKG2D ligand‐bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE 6, e16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinushi, M. , Vanneman, M. , Munshi, N. C. , Tai, Y.‐T. , Prabhala, R. H. , Ritz, J. , Neuberg, D. , Anderson, K. C. , Carrasco, D. R. & (2007). MHC class I chain‐related protein A antibodies and shedding are associated with the progression of multiple myeloma 105, 1285–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. (2016). The biology and function of exosomes in cancer. Journal of Clinical Investigation 126, 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labani‐Motlagh, A. , Israelsson, P. , Ottander, U. , Lundin, E. , Nagaev, I. , Nagaeva, O. , Dehilin, E. , Baranov, V. , & Mincheva‐Nilsson, L. (2015). Differential expression of ligands for NKG2D and DNAM‐1 receptors by epithelial ovarian cancer‐derived exosomes and its influence on NK cell cytotoxicity. Tumor Biology 37, 5455–5466. [DOI] [PubMed] [Google Scholar]
- Lanier, L. L. (2015). NKG2D receptor and its ligands in host defense. Cancer Immunology Research 3, 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Cobo, S. , Campos‐Silva, C. , Moyano, A. , Oliveira‐Rodríguez, M. , Paschen, A. , Yáñez‐Mó, M. , Blanco‐López, M. C. , & Valés‐Gómez, M. (2018). Immunoassays for scarce tumour‐antigens in exosomes: Detection of the human NKG2D‐Ligand, MICA, in tetraspanin‐containing nanovesicles from melanoma. Journal of Nanobiotechnology 16, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundholm, M. , Schröder, M. , Nagaeva, O. , Baranov, V. , Widmark, A. , Mincheva‐Nilsson, L. , & Wikström, P. (2014). Prostate tumor‐derived exosomes down‐regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 9, e108925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, M. , Kleinschmidt, A. , Brühl, H. , Klier, C. , Nelson, P. J. , Cihak, J. , Plachý, J. , Stangassinger, M. , Erfle, V. , & Schlöndorff, D. (2000). Transfer of the chemokine receptor CCR5 between cells by membrane‐derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nature Medicine 6, 769–775. [DOI] [PubMed] [Google Scholar]
- Madera, S. , Rapp, M. , Firth, M. A. , Beilke, J. N. , Lanier, L. L. , & Sun, J. C. (2016). Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. Journal of Experimental Medicine 213, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu, M. , Martin‐Jaular, L. , Lavieu, G. , & Théry, C. (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nature Cell Biology 21, 9–17. [DOI] [PubMed] [Google Scholar]
- McCann, F. E. , Eissmann, P. , Önfelt, B. , Leung, R. , & Davis, D. M. (2007). The activating NKG2D ligand MHC Class I‐related chain A transfers from target cells to NK cells in a manner that allows functional consequences. The Journal of Immunology 178, 3418–3426. [DOI] [PubMed] [Google Scholar]
- Melo, S. A. , Luecke, L. B. , Kahlert, C. , Fernandez, A. F. , Gammon, S. T. , Kaye, J. , LeBleu, V. S. , Mittendorf, E. A. , Weitz, J. , Rahbari, N. , Reissfelder, C. , Pilarsky, C. , Fraga, M. F. , Piwnica‐Worms, D. , & Kalluri, R. (2015). Glypican‐1 identifies cancer exosomes and detects early pancreatic cancer. Nature 523, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molfetta, R. , Quatrini, L. , Capuano, C. , Gasparrini, F. , Zitti, B. , Zingoni, A. , Galandrini, R. , Santoni, A. , & Paolini, R. (2014). c‐Cbl regulates MICA‐but not ULBP2‐induced NKG2D down‐modulation in human NK cells. European Journal of Immunology 44, 2761–2770. [DOI] [PubMed] [Google Scholar]
- Molfetta, R. , Quatrini, L. , Zitti, B. , Capuano, C. , Galandrini, R. , Santoni, A. , & Paolini, R. (2016). Regulation of NKG2D expression and signaling by endocytosis. Trends in Immunology 37, 790–802. [DOI] [PubMed] [Google Scholar]
- Morvan, M. G. , & Lanier, L. L. (2016). NK cells and cancer: You can teach innate cells new tricks. Nature Reviews Cancer 16, 7–19. [DOI] [PubMed] [Google Scholar]
- Nakamura, K. , Nakayama, M. , Kawano, M. , Amagai, R. , Ishii, T. , Harigae, H. , & Ogasawara, K. (2013). Fratricide of natural killer cells dressed with tumor‐derived NKG2D ligand. Proceedings of the National Academy of Sciences of the United States of America 110, 9421–9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel, G. , D'Angelo, G. , & Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nature Reviews Molecular Cell Biology 19, 213–228. [DOI] [PubMed] [Google Scholar]
- di Noto, G. , Bugatti, A. , Zendrini, A. , Mazzoldi, E. L. , Montanelli, A. , Caimi, L. , Rusnati, M. , Ricotta, D. , & Bergese, P. (2016). Merging colloidal nanoplasmonics and surface plasmon resonance spectroscopy for enhanced profiling of multiple myeloma‐derived exosomes. Biosensors and Bioelectronics 77, 518–524. [DOI] [PubMed] [Google Scholar]
- Ogasawara, K. , Hamerman, J. A. , Hsin, H. , Chikuma, S. , Bour‐Jordan, H. , Chen, T. , Pertel, T. , Carnaud, C. , Bluestone, J. A. , & Lanier, L. L. (2003). Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity 18, 41–51. [DOI] [PubMed] [Google Scholar]
- Ogasawara, K. , Hamerman, J. A. , Ehrlich, L. R. , Bour‐Jordan, H. , Santamaria, P. , Bluestone, J. A. , & Lanier, L. L. (2004). NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity 20, 757–767. [DOI] [PubMed] [Google Scholar]
- Pazina, T. , Macfarlane, A. W. , Bernabei, L. , Dulaimi, E. , Kotcher, R. , Yam, C. , Bezman, N. A. , Robbins, M. D. , Ross, E. A. , Campbell, K. S. , & Cohen, A. D. (2021). Alterations of NK cell phenotype in the disease course of multiple myeloma. Cancers 13(2), 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quatrini, L. , Molfetta, R. , Zitti, B. , Peruzzi, G. , Fionda, C. , Capuano, C. , Galandrini, R. , Cippitelli, M. , Santoni, A. , & Paolini, R. (2015). Ubiquitin‐dependent endocytosis of NKG2D‐DAP10 receptor complexes activates signaling and functions in human NK cells. Science Signaling 8, ra108. [DOI] [PubMed] [Google Scholar]
- Raulet, D. H. , Gasser, S. , Gowen, B. G. , Deng, W. , & Jung, H. (2013). Regulation of ligands for the NKG2D activating receptor. Annual Review of Immunology 31, 413–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebmann, V. , Schütt, P. , Brandhorst, D. , Opalka, B. , Moritz, T. , Reza Nowrousian, M. , & Grosse‐Wilde, H. (2007). Soluble MICA as an independent prognostic factor for the overall survival and progression‐free survival of multiple myeloma patients. Clinical Immunology 123, 114–120. [DOI] [PubMed] [Google Scholar]
- Roda‐Navarro, P. , & Reyburn, H. T. (2007). Intercellular protein transfer at the NK cell immune synapse: Mechanisms and physiological significance. The FASEB Journal 21, 1636–1646. [DOI] [PubMed] [Google Scholar]
- Salih, H. R. , Antropius, H. , Gieseke, F. , Lutz, S. Z. , Kanz, L. , Rammensee, H.‐G. , & Steinle, A. (2003). Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 102, 1389–1396. [DOI] [PubMed] [Google Scholar]
- Saudemont, A. , Burke, S. , & Colucci, F. (2010). A simple method to measure NK cell cytotoxicity in vivo. Methods in Molecular Biology 612, 325–334. [DOI] [PubMed] [Google Scholar]
- Segovis, C. M. , Schoon, R. A. , Dick, C. J. , Nacusi, L. P. , Leibson, P. J. , & Billadeau, D. D. (2009). PI3K links NKG2D signaling to a CrkL pathway involved in natural killer cell adhesion, polarity, and granule secretion. The Journal of Immunology 182, 6933–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriani, A. , Zingoni, A. , Cerboni, C. , Iannitto, M. L. , Ricciardi, M. R. , di Gialleonardo, V. , Cippitelli, M. , Fionda, C. , Petrucci, M. T. , Guarini, A. , Foà, R. , & Santoni, A. (2009). ATM‐ATR‐dependent up‐regulation of DNAM‐1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK‐cell susceptibility and is associated with a senescent phenotype. Blood 113, 3503–3511. [DOI] [PubMed] [Google Scholar]
- Soriani, A. , Vulpis, E. , Cuollo, L. , Santoni, A. , & Zingoni, A. (2020). Cancer extracellular vesicles as novel regulators of NK cell response. Cytokine and Growth Factor Reviews 51, 19–26. [DOI] [PubMed] [Google Scholar]
- Sutherland, C. L. , Chalupny, N. J. , Schooley, K. , VandenBos, T. , Kubin, M. , & Cosman, D. (2002). UL16‐Binding Proteins, novel MHC Class I‐related proteins, bind to NKG2D and activate multiple signaling pathways in primary NK cells. The Journal of Immunology 168, 671–679. [DOI] [PubMed] [Google Scholar]
- Viaud, S. , Terme, M. , Flament, C. , Taieb, J. , André, F. , Novault, S. , Escudier, B. , Robert, C. , Caillat‐Zucman, S. , Tursz, T. , Zitvogel, L. , & Chaput, N. (2009). Dendritic cell‐derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL‐15Rα. PLoS ONE 4, e4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulpis, E. , Cecere, F. , Molfetta, R. , Soriani, A. , Fionda, C. , Peruzzi, G. , Caracciolo, G. , Palchetti, S. , Masuelli, L. , Simonelli, L. , D'Oro, U. , Abruzzese, M. P. , Petrucci, M. T. , Ricciardi, M. R. , Paolini, R. , Cippitelli, M. , Santoni, A. , & Zingoni, A. (2017). Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: Role of HSP70/TLR2/NF‐kB axis. OncoImmunology 6, e1279372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulpis, E. , Stabile, H. , Soriani, A. , Fionda, C. , Petrucci, M. T. , Mariggio’, E. , Ricciardi, M. R. , Cippitelli, M. , Gismondi, A. , Santoni, A. , & Zingoni, A. (2018). Key role of the CD56lowCD16low natural killer cell subset in the recognition and killing of multiple myeloma cells. Cancers 10(12), 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulpis, E. , Soriani, A. , Cerboni, C. , Santoni, A. , & Zingoni, A. (2019). Cancer exosomes as conveyors of stress‐induced molecules: new players in the modulation of NK cell response. International Journal of Molecular Sciences 20(3), 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Li, H. , Xu, W. , Pan, M. , Qiao, C. , Cai, J. , Xu, J. , Wang, M. , & Zhang, J. (2020). BCMA‐targeting bispecific antibody that simultaneously stimulates NKG2D‐enhanced efficacy against multiple myeloma. Journal of Immunotherapy 43, 175–188. [DOI] [PubMed] [Google Scholar]
- Weissgerber, T. L. , Milic, N. M. , Winham, S. J. , & Garovic, V. D. (2015). Beyond bar and line graphs: Time for a new data presentation paradigm. PLOS Biology 13,e1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann, K. , Mittrücker, H.‐W. , Feger, U. , Welte, S. A. , Yokoyama, W. M. , Spies, T. , Rammensee, H.‐G. , & Steinle, A. (2005). Systemic NKG2D down‐regulation impairs NK and CD8 T cell responses in vivo. The Journal of Immunology 175, 720–729. [DOI] [PubMed] [Google Scholar]
- Witwer, K. W. , & Théry, C. (2019). Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. Journal of Extracellular Vesicles 8, 1648167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. D. , Higgins, L. M. , Steinle, A. , Cosman, D. , Haugk, K. , & Plymate, S. R. (2004). Prevalent expression of the immunostimulatory MHC class I chain–related molecule is counteracted by shedding in prostate cancer. Journal of Clinical Investigation 114, 560–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Chen, Y. , Zhang, F. , Zhao, Q. , & Zhong, H. (2015). Increased anti‐tumour activity by exosomes derived from doxorubicin‐treated tumour cells via heat stress. International Journal of Hyperthermia 31, 498–506. [DOI] [PubMed] [Google Scholar]
- Zingoni, A. , Palmieri, G. , Morrone, S. , Carretero, M. , Lopez‐Botel, M. , Piccoli, M. , Frati, L. , & Santoni, A. (2000). CD69‐triggered ERK activation and functions are negatively regulated by CD94 /NKG2‐A inhibitory receptor. European Journal of Immunology 30, 644–651. [DOI] [PubMed] [Google Scholar]
- Zingoni, A. , Cecere, F. , Vulpis, E. , Fionda, C. , Molfetta, R. , Soriani, A. , Petrucci, M. T. , Ricciardi, M. R. , Fuerst, D. , Amendola, M. G. , Mytilineos, J. , Cerboni, C. , Paolini, R. , Cippitelli, M. , & Santoni, A. (2015). Genotoxic stress induces senescence‐associated ADAM10‐dependent release of NKG2D MIC ligands in multiple myeloma cells. The Journal of Immunology 195, 736–748. [DOI] [PubMed] [Google Scholar]
- Zingoni, A. , Vulpis, E. , Nardone, I. , Soriani, A. , Fionda, C. , Cippitelli, M. , & Santoni, A. (2016). Targeting NKG2D and NKp30 ligands shedding to improve NK cell‐based immunotherapy. Critical Reviews in Immunology 36, 445–460. [DOI] [PubMed] [Google Scholar]
- Zingoni, A. , Molfetta, R. , Fionda, C. , Soriani, A. , Paolini, R. , Cippitelli, M. , Cerboni, C. , & Santoni, A. (2018). NKG2D and its ligands: “One for all, all for one.” Frontiers in Immunology 9, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingoni, A. , Vulpis, E. , Cecere, F. , Amendola, M. G. , Fuerst, D. , Saribekyan, T. , Achour, A. , Sandalova, T. , Nardone, I. , Peri, A. , Soriani, A. , Fionda, C. , Mariggiò, E. , Petrucci, M. T. , Ricciardi, M. R. , Mytilineos, J. , Cippitelli, M. , Cerboni, C. , & Santoni, A. (2018).MICA‐129 dimorphism and soluble MICA are associated with the progression of multiple myeloma. Frontiers in Immunology 9, 926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitti, B. , Molfetta, R. , Fionda, C. , Quatrini, L. , Stabile, H. , Lecce, M. , de Turris, V. , Ricciardi, M. R. , Petrucci, M. T. , Cippitelli, M. , Gismondi, A. , Santoni, A. , & Paolini, R. (2017). Innate immune activating ligand SUMOylation affects tumor cell recognition by NK cells. Scientific Reports 7(1), 10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zocchi, M. R. , Tosetti, F. , Benelli, R. , & Poggi, A. (2020). Cancer nanomedicine special issue review anticancer drug delivery with nanoparticles: Extracellular vesicles or synthetic nanobeads as therapeutic tools for conventional treatment or immunotherapy. Cancers 12(7), 1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.