

Summary
The term multicentric Castleman disease (MCD) encompasses a spectrum of conditions that share some overlapping clinicopathological manifestations. The fundamental pathogenetic mechanism involves dysregulated cytokine activity, causing systemic inflammatory symptoms as well as lymphadenopathy. Some of the histological changes in lymph nodes resemble the histology of unicentric Castleman disease (UCD). However, based on current knowledge, the use of this shared nomenclature is unfortunate, since these disorders differ in pathogenesis and prognosis. In Kaposi sarcoma-associated herpesvirus (KSHV)-associated MCD, cytokine overactivity is caused by viral products, which can also lead to atypical lymphoproliferations and potential progression to lymphoma. In idiopathic MCD, the hypercytokinemia can result from various mechanisms, which ultimately lead to different constellations of clinical presentations and varied pathology in lymphoid tissues. The authors review the evolving concepts and definitions of the various conditions under the eponym of multicentric Castleman disease.
Key words: Castleman disease, Kaposi sarcoma-associated herpesvirus, human herpesvirus type 8, interleukin-6, TAFRO syndrome
Multicentric Castleman Disease (MCD) - Juan Rosai and the evolution of the concept
The term Castleman disease began with a single case discussed at the recurring clinicopathological conferences (CPC’s) of the Massachusetts General Hospital and published in 1954 1. It was an example of what we now consider unicentric Castleman disease (UCD). Ultimately two histological and clinical variants were identified 2. One subtype had prominent hyalinization, regressive changes in follicles, with increased vascularity (hyaline vascular type), while the second had an increase in plasma cells and was more often associated with systemic symptoms (plasma cell type). However, both forms presented with localized mass lesions.
In 1980 Glauco Frizzera, working with Juan Rosai, presented a paper at the USCAP meeting, then known as the United States and Canadian Branch of the International Academy of Pathology, describing 10 patients with a multicentric lymphoid disorder with histological features resembling UCD. They expanded on their observations in a manuscript published in 1983, describing a systemic lymphoproliferative disorder with morphological features of Castleman’s disease 3. Their pivotal description mainly pertained to lymph nodes, but splenomegaly was present in most of the 15 patients, and the authors also reported the histological findings in the spleen from 4 cases.
All patients presented with constitutional symptoms including fever, night sweats, weight loss and generalized lymphadenopathy. An important clue to the pathogenesis was that two patients had Kaposi sarcoma. Other clinical features included anemia, thrombocytopenia, and polyclonal hypergammaglobulinemia. The clinical evolution of the disease was variable, with four patients having a fatal outcome in 5-14 months, while other patients had a chronic, indolent course, with remissions and exacerbations. At the time of publication, six patients were still alive after 39-156 months. The causes of death were multifactorial, with five patients dying of sepsis, and one developing lymphoma.
Based on what we know today, it is likely that many of the reported patients had Human Herpesvirus 8 (HHV8) [also known as Kaposi sarcoma-associated herpesvirus (KSHV)]-associated MCD, while others with more protracted and chronic disease may have had idiopathic MCD (iMCD). Rosai and colleagues also noted similarities to “collagen vascular diseases” 3, and speculated on similarities to what was known at the time as angioimmunoblastic lymphadenopathy (AIL) and T-zone lymphoma. Their landmark article helped set the stage for the discovery of KSHV/HHV8 in patients with Kaposi sarcoma in 1984, and was an important step in defining the clinical and pathological features of the various conditions included in the concept of MCD 4. MCD is not a single disease entity, but the overlapping features of vascular proliferation, plasmacytosis, and immune dysfunction may be related to similarities in pathophysiology as we will discuss below.
KSHV associated MCDHHV8: an etiologic agent for multiple hematologic diseases
HHV8, also known as KSHV, was first isolated from Kaposi sarcoma in 1984 by Chang et al. 5. Subsequently, it was determined to be causally related to MCD 6. Uncontrolled HHV8 infection causes approximately 50% of MCD cases and is ubiquitous in HIV-associated Castleman disease. (Fig. 1) HHV8 is also the etiologic agent of primary effusion lymphoma (PEL) 7. Latent HHV8 infection is overwhelmingly common. HHV8 seroprevalence ranges from 6% to 50%, depending on the geographic regions and subpopulations 8. However, HHV8 remains in a dormant state in most cases. Only a minority of infected individuals develop HHV8-associated MCD and other diseases years or even decades later, emphasizing the role of cofactors in determining the development of such diseases. In this respect, immunodeficiency is the primary risk factor for developing HHV8-associated diseases, with HIV infection being the most common underlying immunocompromised state.
Figure 1.
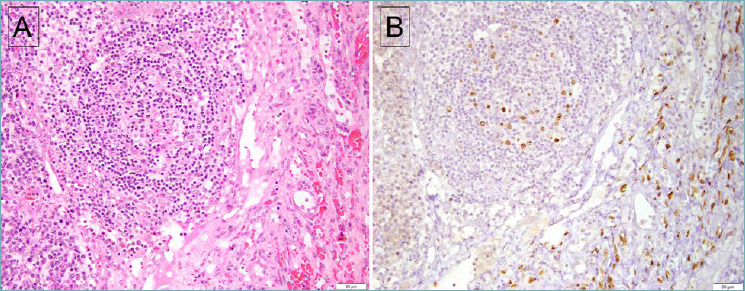
Lymph node involved by both MCD and KS. (A) The follicle shows regressive changes, with an adjacent spindle cell proliferation associated with extravasated red blood cells. (B) Staining for LANA-1 is positive in both lymphoid cells within the follicle as well as the spindle cells of KS. However, the spindle cells of KS are negative for vIL-6 (not shown).
Like all other members of the herpesvirus family, HHV8 establishes lifelong infection in the host. Due to its large, complex genome, this virus can sustain chronic infection through skillful immune evasion and low-copy replication by frequent switching between latent and lytic replication cycles. Lymphoid and endothelial cells serve as major latent reservoirs of HHV8 9, although HHV8 can also infect and persist in other types of cells, such as monocytes, dendritic cells, and epithelial cells. During latency, a large portion of the HHV8 genome is kept silenced through multiple epigenetic modifications 10. Transcription is limited to a few latency-associated genes, including the latency-associated nuclear antigen-1 (LANA-1), the most abundantly expressed protein consistently detected in all HHV8-infected tumors. The restricted expression of viral antigens aids the virus in avoiding recognition by the host immune system while allowing for long-term viral persistence 11,12. Therefore, the human immune system can suppress the infection yet never eliminate this pathogen. Under appropriate conditions, the latently infected cells can be induced to enter the lytic cycle. Some of the well-established factors that activate lytic replication of HHV8 include immune suppression and co-pathogenic infections, as well as cellular stress, hypoxia, and inflammation. The lytic phase is characterized by the expression of a highly ordered cascade of genes that ensures efficient replication of virions 13-15.
It should be noted that the expression of HHV8 genes does not always follow the latency-versus-lytic-replication paradigm. Certain viral genes that are typically expressed during lytic cycles can be activated by host transcriptional machinery, independent of full lytic activation 16. For instance, the expression of HHV8-encoded viral IL-6 (vIL-6) was shown to be induced by the X-box binding protein-1 (XBP-1), a transcription factor highly expressed in the B-cell lineage 17. The extra layer of regulation adds to the heterogeneity of viral gene expression profiles among different HHV8-associated diseases. For example, in Kaposi sarcoma, most tumor cells have latent infection, while lytic proteins are expressed in a small percentage of cells. A greater proportion of cells in PEL express lytic proteins, while MCD demonstrates the highest frequency of lytic replication (up to 25%) 18-20. Conceivably, the heterogeneity in viral gene expression profiles contributes to the wide clinicopathological spectrum of HHV8-associated diseases, ranging from subclinical to progressive disease, from localized to systemic illness, and from reactive hyperplasia or benign scarring to overt malignancy.
Pathogenesis of HHV8-associated MCD
Speculation abounds over whether MCD is an autoimmune, infectious, reactive, or clonal disease 21. Despite some early obstacles and lingering controversies, a new conceptual framework of MCD pathogenesis has been proposed: both the nodal and systemic manifestations are reactive changes to elevated levels of IL-6 and, to a lesser extent, other circulating factors in the cytokine and chemokine storm 21. MCD can be further subcategorized based on the presence of HHV8 into HHV8-associated MCD and HHV8-negative iMCD. Although the underlying mechanisms responsible for the cytokine storm in iMCD cases remains hypothesis-generating, the prominent pathogenic role of HHV8 is undisputed in HHV8-associated MCD cases. In these patients, immunosuppression enables HHV8 to escape host immune control. The increased lytic replication triggers antiviral signaling cascades that lead to excessive production of human IL-6 (hIL-6) and other cytokines, including IL-10, tumor necrosis factor-α (TNF-α), and IL-1.
Notably, the HHV8 genome is known to encode for homologs of several human regulatory proteins involved in cell proliferation, survival, and immune response 22,23. In particular, the production of vIL-6, a “pirate” cytokine with optimized and unregulated IL-6 functions, is thought to be a critical disease driver of HHV8-associated MCD, and the circulating level of vIL-6 correlates with disease activity 24,25. Although vIL-6 shares only 25% sequence identity with and demonstrates lower signaling potency than its human counterpart, hIL-6 23,26,27, it is capable of stimulating all of the known hIL-6-induced signaling pathways in a similar manner 28,29. Furthermore, vIL-6 functions in a wider variety of cell types since it is able to signal merely by engaging the ubiquitously expressed gp130 subunit of IL-6 receptor. In contrast, signaling of hIL-6 requires the full IL-6 receptor (both gp130 and gp80) 28, 30.
Both vIL-6 and hIL-6 play a pleiotropic role in MCD: they stimulate the proliferation of B cells and plasmablasts, activate angiogenesis, and mediate systemic inflammatory symptoms 31-33. Consequently, these mechanisms lead to the characteristic pathologic changes and clinical manifestations, described in greater detail in the following section. Foremost is hIL-6, which exerts a potent chemotactic activity and attracts plasmablasts to cluster around small vessels, producing an environment that is more conducive to cross-infection and paracrine signaling among these cells. This process amplifies the plasmablasts and is followed by massive viral replication, cell lysis, and excessive cytokine production, which reciprocally augments cell-to-cell transmission and cell proliferation. The ultimate outcome of such a vicious circle is an exponential escalation of hIL-6 and vIL-6, both locally and systemically.
At the cellular level, the binding of hIL-6 and vIL-6 to IL-6 receptors results in activation of JAK/STAT and MAPK pathways 34. Both pathways serve a key role in orchestrating the expansion of the two infective compartments – plasmablasts and lymphovascular endothelial cells. The effects of cytokines on local target-cell populations are manifested by lymphoid hyperplasia and, consequently, increased immunoglobulin production. The effects are also manifested by activation of endothelial cells and vascular endothelial growth factor (VEGF) production, resulting in neo-angiogenesis with the formation of penetrating vessels. Additionally, the lytic replication of HHV8 leads to the destruction of endothelial cells, resulting in hyalinized scars and temporary lymph node swelling.
At the systemic level, both hIL-6 and vIL-6 exert a potent proinflammatory effect; their cooperative effect with other cytokines released into the circulation during disease flares results in the systemic inflammatory response syndrome 24,35,36. In addition, these cytokines can deregulate host immunity against co-infecting pathogens, such as HIV and EBV. Human herpesvirus 8 and the co-infecting microbes can build a mutually beneficial relationship to promote expansion and enhance the pathogenic activity of one another through multifaceted interactions, such as the direct interplay between microbial products and indirect modulation of the microenvironment. The undesirable synergistic interactions not only accelerate the progression of HHV8-associated MCD, but also aggravate the pathologies associated with the coinfections 37,38. Hence, the development and severity of HHV8-associated MCD are deeply influenced by host factors and complementing viral infections 39.
Advances in the understanding of the pathogenesis of HHV8-associated MCD have provided useful insights into new therapeutic strategies. Agents targeting the essential cytokine pathways, such as antibodies against IL-6/IL-6R and antagonists of the IL-1 receptor, have been proven effective in alleviating systemic manifestations 32,40. Targeting both infected cellular reservoirs (lymphoid and endothelial cells) is emerging as a novel therapeutic concept. For example, anti-CD20 and anti-CD19 strategies can comprehensively destroy the lymphoid reservoir of HHV8 and have been shown to improve patient outcomes of HHV8-associated MCD 41. While multimodal approaches targeting both lymphoid and endothelial compartments have not been translated into routine clinical practice, promising preclinical results are expected to pave the way for successful implementation of this approach in the near future 42.
Characterizing pathology and clinical presentations of HHV8-associated MCD
HHV8-positive MCD is assigned as a separate group of MCD owing to its viral etiology, variable clinical course, and high likelihood of resulting in HHV8-positive lymphomas. Patients with MCD typically present with systemic symptoms that are often severe and can be life-threatening without proper treatment 24. Clinical manifestations of HHV8-associated MCD mainly fall into three categories. First and foremost, this disease often demonstrates episodic exacerbation of systemic inflammatory response, which includes constitutional symptoms, organomegaly, cytopenia, multiple organ dysfunction, elevated levels of acute-phase proteins, hypergammaglobulinemia, and hypoalbuminemia. The hemophagocytic syndrome can occur in up to 50% of HHV8-associated MCD cases. Second, hypoalbuminemia may lead to prominent fluid overload, manifesting as edema, ascites, pleural or pericardial effusions, and seizures. Finally, patients may suffer from symptoms related to complications or comorbidities, such as HIV (in 82% of patients )43 and other infections, Kaposi sarcoma (in 48% of patients) 6, lymphoma, and paraneoplastic pemphigus. It is important to bear in mind that none of the symptoms are exclusively discriminatory for MCD; they all overlap with symptoms of other diseases, including viral infection, rheumatic or vasculitic disease, and malignancies. A lymph node biopsy is of the utmost importance in establishing the correct diagnosis, since current pathological diagnostic criteria of MCD are based on histologic findings in lymph nodes, although extranodal sites can also be involved. It is of interest that severe inflammatory symptoms with similar laboratory findings as seen in HHV8-associated MCD can be observed in a subset of patients that have no significant nodal disease (KSHV inflammatory cytokine syndrome or KICS) 44,45, as well as in patients that are starting anti-retroviral therapy as part of their immune reconstitution (KSHV-IRIS) 46.
Lymph nodes involved in HHV8-associated MCD show regressive changes in follicles and increased vascularity (Fig. 2). The nodes typically reveal relatively preserved lymph node architecture, with involuted follicles and penetrating venules. The interfollicular zone is characterized by vascular proliferation and prominent plasmacytic infiltrates. A unique and invariable feature of this form of MCD is the presence of HHV8-infected “plasmablasts”, located primarily in the mantle zones but also seen randomly in the interfollicular area. In some cases, these plasmablasts expand to form larger collections, previously described as microlymphomas 47-49, which may invade or replace the germinal centers (GC). These cells are characteristically medium to large with vesicular nuclei, one or more nucleoli, and amphophilic cytoplasm. Like normal plasma cell precursors, they are uniformly positive for MUM1; variably express CD20 and CD79a; lack PAX5, BCL6, and CD138; and are negative for EBV. The plasmablasts exclusively express IgM and lambda light chain; however, they show a polyclonal or oligoclonal pattern of immunoglobulin gene rearrangement at the DNA level 50.
Figure 2.
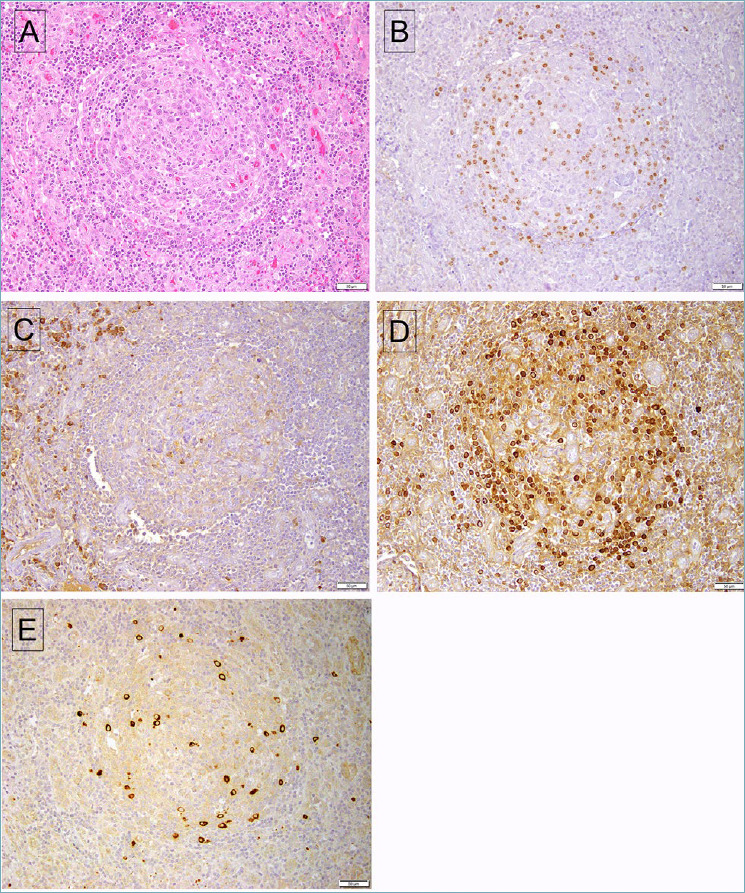
HHV8-associated MCD involving lymph node. (A) The follicle shows an attenuated mantle cuff, with increased vascularity most evident in the interfollicular area. (B) A stain for LANA-1 shows positive cells mainly at the periphery of the affected follicle. (C) A stain for kappa light chain shows positivity in interfollicular plasma cells, while the HHV8-infected plasma cells (D) stain for lambda light chain. (E) The HHV8-positive cells also express vIL-6.
Human herpesvirus 8 exemplifies a pathogen that can alter immunoglobulin diversity by reprogramming its host cells. The enigmatic restriction of HHV8 infection to IgM lambda-expressing B cells is a long-recognized feature of MCD; however, understanding of the underlying mechanisms remains incomplete. It is thought to be driven by the viral manipulation of human signaling pathways. The plasmablasts appear to originate from pre-GC naïve B cells, as they do not harbor somatic mutations in the rearranged immunoglobulin genes 49. It has been postulated that HHV8 viral latent products may be able to transform IgM-positive naïve B cells into plasmablasts in the absence of GC reactions, possibly by mediating signaling events that normally occur in GC reactions. Thus, the infected plasmablasts lack class switching recombination and remain IgM-positive. Furthermore, HHV8-encoded proteins, such as vFLIP, can act as an NF-κB activator; NF-κB signaling is essential for generating lambda light chain-expressing B cells following unsuccessful kappa gene rearrangement in normal B-cell development 51. Thus, HHV8 likely reinduces immunoglobulin rearrangement in the originally kappa-expressed cells through induction of human NF-κB transcriptional activity.
Relationship of HHV8-associated MCD with other HHV8-associated lymphoproliferative disorders
It has been well-established that HHV8-associated MCD is associated with a heightened risk for non-Hodgkin lymphoma 47,48,52. The incidence of non-Hodgkin lymphoma in patients with HIV-associated MCD is reported to be about 15-fold higher than that in the general HIV-infected population 47. The most common type of non-Hodgkin lymphomas arising in the background of MCD is designated as HHV8-positive diffuse large B-cell lymphoma (DLBCL), not otherwise specified in the WHO classification. Human herpesvirus 8-positive DLBCL is characterized by destruction of nodal architecture and sheets of plasmablastic cells, displaying an immunophenotype that is virtually identical to that of the HHV8-infected plasmablasts in MCD; in particular, these cells also strongly express IgM with lambda light-chain restriction 50. However, the development of frank lymphoma is characterized by clonal immunoglobulin gene rearrangements, in contrast to the polyclonal or oligoclonal nature of the plasmablasts in MCD. These observations have led to the conclusion that HHV8-positive DLBCL likely represents a selective clonal expansion of HHV8-infected plasmablasts following polyclonal activation. One convincing piece of evidence for this hypothesis comes from the observation that, in rare cases, there was a sequential progression of MCD with individual plasmablasts to “microlymphomas” and finally HHV8-positive DLBCL. Upon closer examination, additional genetic abnormalities were found in the “microlymphomas” 53. In this respect, it is important to note that although the so-called “microlymphomas” pose the potential to transition to overt lymphoma, only a fraction of these cases progresses to frank lymphoma. Additionally, a clear clonal relationship has not been established between the “microlymphoma” and concomitant or subsequent lymphoma 26. Therefore, such cases are probably best regarded as a variant of MCD or no more than a noncommitted precursor of lymphoma.
Synchronous or metachronous PEL represents another HHV8-associated malignancy in patients with MCD. Primary effusion lymphoma typically presents as effusions in the absence of a tumor mass. The neoplastic cells are pleomorphic with features of immunoblastic, plasmablastic, or anaplastic cells. While EBV infection is not a prerequisite for PEL development, the vast majority of PEL cases are coinfected with EBV that exhibits restricted gene expression 54; the small subset of PEL without coinfection of EBV is usually seen in immunocompetent patients 50. In comparison to HHV8-positive DLBCL, it is not as well-established whether PEL arising in the setting of MCD originates from HHV8-infected plasmablasts. Conceptually, the transformation of a node-based disease into an effusion-based lymphoma seems counterintuitive.
Indeed, MCD and PEL are different in many aspects: First, the phenotype of PEL differs from that of the plasmablasts in MCD in that PEL is often positive for activation markers and occasionally expresses T-cell markers but usually lacks surface and cytoplasmic immunoglobulin but. Second, PEL cells show evidence of rearranged immunoglobulin genes and high levels of somatic mutations 55. They also exhibit a gene expression profile between that of DLBCL and plasma cells 56,57. These features seem to indicate that PEL is derived from a transition stage between antigen-selected GC B cells and terminally differentiated plasma cells, in contrast to HHV8-associated MCD, which likely originates from naïve B cells. This model fits best with current knowledge of immunophenotypic and genetic features of PEL, but an alternative pathway is also considered. According to the second scenario, both MCD and PEL originate from the same HHV8-infected subtype of B cells, while additional pathogenic factors in PEL influence the disease genetics, phenotype, and manifestations. One important cooperative factor to consider is coinfecting EBV, which is known to imitate GC biology and could conceivably be responsible for clonal evolution toward a post-GC phenotype 58,59. Furthermore, the discovery of lambda-restricted HHV8-infected plasmablastic cells in body fluids introduces a novel concept of a “liquid form” of MCD 60 and lays a foundation for the transformation of HHV8-associated MCD into an effusion-based lymphoma.
Germinotropic lymphoproliferative disorder (GLPD)
Besides PEL, coinfection by HHV8 and EBV has been identified in germinotropic lymphoproliferative disorder (GLPD). This entity was first described in 2002 61; so far, fewer than 20 cases have been reported in the literature 62. In sharp contrast to PEL, as well as the other HHV8-associated diseases, GLPD affects mainly immunocompetent individuals, presents as localized lymphadenopathy without obvious systemic symptoms, and has a favorable clinical course. This finding leads to an intriguing question: Why do the same oncogenic factors (HHV8 and EBV) diverge into two diseases that have such vastly different outcomes (PEL and GLPD)? Speculations on possible mechanisms include host immune status and microenvironment (effusion vs. GC), which may influence key aspects of lymphomagenesis. The unanswered questions highlight gaps in knowledge that must be filled in by future research; genomic and epigenomic comparison of different HHV8-related lymphoproliferative disorders may be an attractive research direction.
Histologically, GLPD is characterized by aggregates of plasmablasts coinfected by HHV8 and EBV that preferentially colonize GCs but may also extend into the interfollicular regions (Fig. 3). The plasmablasts are positive for MUM-1 but lack expression of CD45, CD138, and B-cell markers. Like MCD, the plasmablastic cells show monotypic light chain expression but are polyclonal at the molecular level. The lymph nodes often show features resembling Castleman disease, such as atrophic and hyalinized follicles, vascular proliferation, and marked plasmacytosis 63,64.
Figure 3.
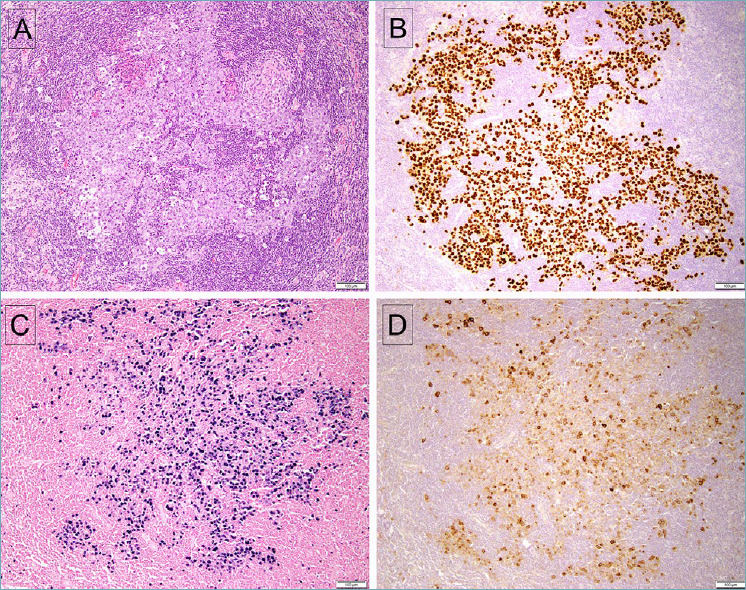
Germinotropic lymphoproliferative disorder. (A) The affected follicle shows irregular cords and sheets of atypical large lymphoid cells. (B) The lesional cells are positive for LANA-1, and also positive for EBV, with EBER in situ hybridization (C). D. The affected cells are also positive for vIL-6.
The distinction between GLPD and HHV8-MCD requires the integration of clinical and pathological data. Namely, GLPD is coinfected by HHV8 and EBV. In this regard, GLPD is different from MCD, which is almost always EBV-negative. Second, in GLPD, plasmablasts primarily infiltrate GC, whereas in MCD, they involve the mantle zones. Third, GLPD shows monotypic kappa or lambda light chains, while MCD is only positive for IgM lambda. Lastly, localized involvement in immunocompetent individuals is more compatible with a diagnosis of GLPD.
Idiopathic MCD HHV8-negative MCD and other Castleman-like conditions
Approximately half or more of the MCD cases are negative for HHV8, mostly in HIV-negative patients. The etiology of the HHV8-negative MCD is largely unknown, and these cases have been referred to as iMCD 65. Among them, a subgroup of patients presents with a characteristic constellation of symptoms/signs including thrombocytopenia, anasarca, fever, reticulin fibrosis/renal dysfunction, and organomegaly, which gives rise to the acronym TAFRO syndrome. This subset of iMCD is now considered a specific entity (TAFRO-iMCD) with clinical and histologic presentations distinct from other non-TAFRO iMCD cases, although both entities are driven by cytokine hypersecretion. Additionally, an MCD-like presentation can also be caused by paraneoplastic mechanisms, such as in POEMS (polyneuropathy, organomegaly, endocrinopathy, M-proteins and skin changes) syndrome. Several other conditions, such as lymphoid malignancies and IgG4-related disease, may mimic MCD in both the clinical presentation and histopathology, adding further complexity in differential diagnosis.
TAFRO syndrome and TAFRO-associated iMCD (TAFRO-iMCD)
TAFRO syndrome is a systemic inflammatory disorder first reported in Japan 66, and was later also described in Caucasian patients 67. The Japanese TAFRO Syndrome Research Team proposed the diagnostic criteria of TAFRO syndrome in 2015, which was further updated in 2019 68,69. The diagnostic criteria include clinical and laboratory parameters, as well as disease conditions to be excluded. The pathological findings of TAFRO syndrome often resemble those of Castleman disease. However, a lymph biopsy is not always available in patients of TAFRO syndrome, often due to anasarca, bleeding tendency, or the minor extent of lymphadenopathy. Thus, lymph node biopsy was listed only as a minor diagnostic criterion. Despite some overlapping features, many clinical manifestations are distinct from those of iMCD, and TAFRO syndrome is considered a distinct disorder by many investigators. To distinguish from non-TAFRO iMCD, another research group proposed separate diagnostic criteria for TAFRO syndrome-associated iMCD (TAFRO-iMCD) 70, in which the characteristic histopathological findings of iMCD in lymph nodes are essential for diagnosis. Applying these diagnostic criteria, iMCD is divided into two categories: TAFRO-associated iMCD (TAFRO-iMCD) and iMCD not otherwise specified (iMCD-NOS). It is noteworthy that those cases of TAFRO syndrome without proven iMCD by lymph node biopsy share similar clinical, laboratory and prognostic features as TAFRO-iMCD, while distinct from iMCD-NOS 71, suggesting that TAFRO syndrome and TAFRO-iMCD defined by different diagnostic criteria represent the same clinical entity, and both require prompt diagnosis and intensive treatment.
In addition to those disease-defining symptoms/signs such as thrombocytopenia, anasarca, fever, reticulin fibrosis/renal dysfunction, and organomegaly, TAFRO-iMCD exhibits additional clinical and laboratory features that are distinct from iMCD-NOS, such as hypogammaglobulinemia (in contrast to hypergammaglobulinemia in iMCD-NOS), higher neutrophil counts, elevated transaminases, alkaline phosphatase and γ-glutamyl transpeptidase, and higher C-reactive protein levels 65,71,72. Patients with TAFRO-iMCD usually have more aggressive clinical course, with significantly longer lengths of hospitalization. Based on the severity classification proposed by the Castleman Disease Collaborative Network (CDCN), the severe iMCD cases often present with the TAFRO-iMCD subtype and have higher mortality rate especially during the first few months 71,73-75.
The lymphadenopathy in TAFRO-iMCD is usually milder than in iMCD-NOS. Approximately 40% of patients do not develop radiographically enlarged lymph nodes 72. Histologically, the involved lymph nodes usually exhibit fewer plasma cells than seen in other forms of iMCD. There is often marked vascular proliferation in the interfollicular areas, and the germinal centers are often atrophic with increased vessels lined by plump endothelial cells with enlarged nuclei and less hyalinization (Fig. 4). Bone marrow biopsies often show megakaryocyte hyperplasia with slight atypia such as multiple and separated nuclei. Mild loose reticulin fibrosis and occasional megakaryocytic emperipolesis are also common findings, while significant plasmacytosis is not observed 70,76.
Figure 4.
Idiopathic MCD with features of TAFRO. (A) The follicle shows regressive changes with markedly increased vascularity in the paracortex. (B) Staining for Factor VIII highlights the prominent vascularity.
The distinct clinical presentation suggests that TAFRO-iMCD may have a unique pathogenesis different from iMCD-NOS. For example, IL-6, which is a hallmark cytokine that is elevated in iMCD, is often only mildly elevated in TAFRO-iMCD. The common clinical features associated with IL-6 hypersecretion, such as thrombocytosis and hypergammaglobulinemia, are not observed in TAFRO-iMCD, in contrast to the iMCD-NOS patients 70, suggesting that elevated serum IL-6 might not be the primary pathogenesis driving the inflammatory responses in TAFRO-iMCD. A recent plasma proteomic study identified a distinct proteomic profile in TAFRO-iMCD versus iMCD-NOS, further supporting the notion that these iMCD subtypes may be diverse chemokines/cytokines driving the symptomatology 77. Another genetic study by next generation sequencing using a target panel of ~500 genes further shed light in the genetic basis of TAFRO-iMCD. A somatic MAP2K2 (MEK2) mutation and a germline RUNX1 mutation were identified in two patients with TAFRO-iMCD. In both patients, ERK was significantly activated, suggesting a potential role of the MAPK signaling in the pathogenesis of TAFRO-iMCD 78.
iMCD-NOS
International evidence-based consensus criteria were proposed for iMCD 4. According to this scheme, the diagnosis of iMCD requires enlarged lymph nodes with histopathologic features of CD, plus at least two clinical and/or laboratory features as minor criteria. Five main pathologic features were highlighted, namely regressed germinal centers, follicular dendritic cell (FDC) prominence, vascularity, hyperplasic germinal centers and plasmacytosis. iMCD cases exhibit a spectrum of histopathologic features. On one end of the spectrum is the hypervascular” histopathologic subtype that shows features resembling the unicentric hyaline vascular Castleman disease, with regressed germinal centers and prominence of FDC, while the “plasmacytic” subtype lies on the other end of the spectrum, typified by marked plasmacytosis but with residual hyperplastic germinal centers (Fig. 5). Many patients actually show a “mixed” subtype. As discussed above, most TAFRO-iMCD cases demonstrate hypervascular or mixed histopathology, but there are also iMCD-NOS patients with similar histopathology that do not have the TAFRO clinical manifestations.
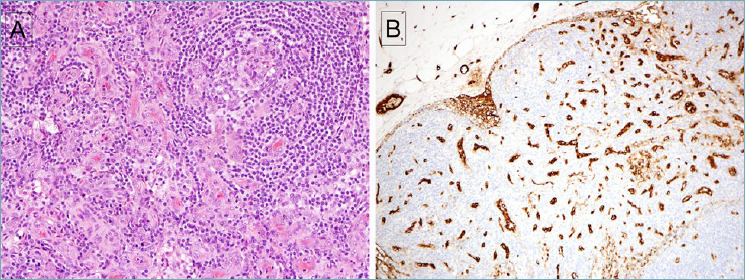
Figure 5.
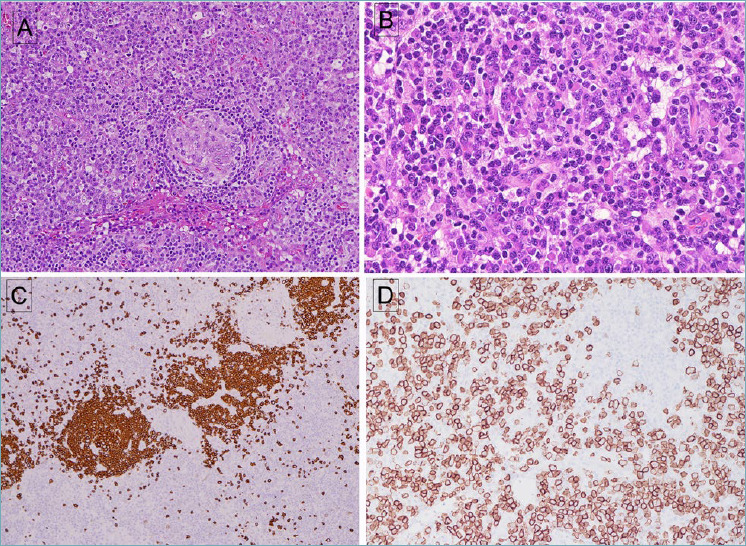
Idiopathic MCD, plasma cell type. (A) A small follicle with attenuated mantle cuff is surrounded by numerous mature plasma cells. (B) Plasma cells have mature nuclear features, with some cells containing Russell-body inclusions. (C) A stain for CD20 is positive in follicles but negative in plasma cells. Some increase in vascularity within the follicles is evident. (D) Abundant plasma cells are positive for CD138. Plasma cells were polytypic for kappa and lambda (not shown).
The histopathologic subtyping of iMCD into hypervascular, plasmacytic, and mixed subtypes provides a diagnostic scheme to encompass the heterogeneity in the histopathologic presentation of iMCD. Early data from the randomized controlled trial of siltuximab, an anti-IL-6 therapy, showed that all responders had either plasmacytic or mixed histopathologic subtypes, while none of the patients who achieved a durable response to siltuximab were classified as hypervascular subtype by central review 79. Based on the data, the National Comprehensive Cancer Network (NCCN) had issued guidance recommending first-line siltuximab therapy for iMCD, except for patients with the hypervascular histopathology 80. However, recent data from CDCN showed that the histopathologic subtypes are often inconsistently assigned among pathologists, with only 23% concordance rate in three pathologic reviews at local site, central review and a CDCN expert panel in the study 81. This inconsistency has limited the clinical utility of the histopathologic iMCD subtyping. Additionally, the real-world data showed that severe iMCD patients, including cases of TAFRO-iMCD and iMCD-NOS of hypervascular subtype, may respond to anti-IL-6 therapy. Therefore, currently there is insufficient evidence to guide treatment based solely on the iMCD histopathologic subtype 81.
As in HHV8-associated MCD, iMCD is also characterized by proinflammatory hypercytokinemia, in particular IL-6. The pathophysiologic significance of IL-6 in iMCD has been confirmed by the efficacy of anti-IL-6 therapy. Currently the anti-IL-6 monoclonal antibody (siltuximab or tocilizumab) with or without corticosteroids, is the preferred first-line therapy for iMCD, as recommended by the international consensus treatment guideline proposed by CDCN 75. While the anti-IL-6 therapy represented a significant breakthrough in the treatment of iMCD, a substantial portion of patients remain refractory. For those patients, the second-line therapy includes rituximab in combination with immunomodulatory agent and steroid. Cytotoxic chemotherapy is generally reserved for patients with severe iMCD who fail to respond to the initial therapies. The resistance to anti-IL-6 therapy suggests that additional pathways may underlie the pathogenesis of iMCD and may serve as important targets for future iMCD therapies. Studies have shown that the mTOR signaling may be one potential target. Activation of mTORC1 has been found in iMCD lymph nodes by immunohistochemical studies for pS6, p4EBP1, and p70S6K. A proteomic signature of increased mTORC1 signaling was also detected in serum from iMCD patients by gene set enrichment analysis 82. Another serum-based proteomic study also identified PI3K/Akt/mTOR pathway activation in anti-IL-6-refractory TAFRO-iMCD cases, and the administration of sirolimus, an mTOR inhibitor, was able to induce remission in these patients 83,84. These data provide the rationale for therapeutic targeting of mTOR pathway in iMCD, and clinical trials of sirolimus for anti-IL-6-resistant iMCD are currently underway 85,86.
Despite the progress in understanding cytokine and signal pathway activation in iMCD, the underlying genetic mechanisms that drive the diseases processes have not been elucidated. There have been few studies investigating the genetic abnormalities. A recent whole-exome sequencing study on 22 patients of iMCD identified somatic alterations in five genes (NCOA4, DARS2, MTCL1, RABPE1 and DNAH11), which are associated with unfavorable clinical outcomes 87. Among them, NCOA4 mutation was identified in 5 of 22 iMCD patients (23%), including 4 patients (18%) showing a same L261F mutation. Comparison of the mutation frequencies across different cancers has revealed that iMCD has the highest incidence of NCOA4 mutations, and the NCOA4 L261F mutation has not been reported in other cancers, suggesting that this genetic mutation might play an essential role in the pathogenesis of iMCD. NCOA4 encodes the nuclear receptor co-activator 4, which is a co-activator of a variety of nuclear receptors. Structural modeling predicts that the L261F mutation results in instability of the protein and may change the conformation and phenotype. However, further studies are needed to confirm the roles of NCOA4 in iMCD. Additionally, alterations in genes involved in chromatin organization, including SETD1A, ASH1L, KMT2E and DNMT3A, have also been found in a subset of iMCD patients 88. Another unanswered question is the cell types that are responsible for driving the iMCD pathogenesis and producing the cytokines. A subset of iMCD patients responds to rituximab, supporting B cells as an important contributor at least in some iMCD patients. Other cell types, including other lymphocytes, plasma cells, monocytes, endothelial cells, and follicular dendritic cells are likely also involved, as B-cell depletion is not effective in all patients 89.
POEMS syndrome
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-proteins and skin changes) is a paraneoplastic syndrome due to an underlying monoclonal plasma cell neoplasm. The clinical manifestations are thought to be caused by hypersecretion of proinflammatory and angiogenic cytokines, including vascular endothelial growth factor that is abundantly present in the plasma cells (both clonal and polyclonal) in patients of POEMS syndrome 90. Chan et al. working with Juan Rosai, described some of the distinctive vascular lesions seen in POEMS 91. Patients usually present with sclerotic bone lesions, rather than the typical osteolytic lesions seen in multiple myeloma. Most patients have a λ light chain-expressing plasma cell clone in their bone marrow, with highly restrictive usage of two λ variable (V) domains (IGLV1-40 and IGLV1-44) 92. Half of patients show lymphoid aggregates in the bone marrow biopsy, with distinctive rimming by plasma cells. Megakaryocyte hyperplasia and clustering is also a frequent finding 93.
Up to 30% of POEMS patients to have lymphadenopathy with Castleman-like histology, and the presence of Castleman disease is one of the major criteria for the diagnosis of POEMS syndrome 94,95. Thus, all patients presenting with iMCD, especially the plasmacytic histologic subtype, should be carefully surveyed to exclude the possibility of POEMS syndrome, since the treatment is entirely different from that of iMCD, and requires eradication of the culprit plasma cell clone.
Other disease conditions with histopathology mimicking iMCD
A variety of neoplastic and non-neoplastic conditions are known to exhibit a Castleman-like histomorphology, causing diagnostic challenges. Both Hodgkin and non-Hodgkin lymphomas may display Castleman-like features. Classic Hodgkin lymphoma can show hyaline sclerosis or florid plasmacytosis reminiscent of Castleman disease 96. These Castleman-like histologic features likely represent a nonspecific immune response to the immunologic stimuli in the tumor microenvironment. In some cases, the Castleman-like histology is caused by cytokine-producing lymphoma cells, as reported in cases of intravascular large B-cell lymphoma secreting IL-6 97. Angioimmunoblastic T-cell lymphoma often shows atrophic germinal centers and proliferation of high endothelial venules, which may mimic the hyaline vascular or hypervascular subtype of iMCD. A rare variant of follicular lymphoma can show Castleman-like morphology, including neoplastic follicles with onionskin-like mantle zones and penetrating hyalinized blood vessels, but features of follicular lymphoma are usually evident to make the correct diagnosis 98. Additionally, various non-neoplastic conditions, such as acquired immunodeficiency syndrome, syphilis and autoimmune disorders may also give rise to a Castleman-like morphology. Another differential diagnosis of iMCD is IgG4-related disease, which is a systemic inflammatory disorder characterized by sclerosing inflammation rich in IgG4-expressing plasma cells. iMCD patients may have an elevated serum IgG4 level, while some cases of IgG4-related disease may show Castleman-like morphology. Both conditions present with systemic lymphadenopathy with extranodal involvement, and sometimes the differentiation can be difficult 99. In general, patients of IgG4-related disease tend to be older than patients of iMCD. The affected organs overlap between two conditions, but the presence of pancreatitis or sialo-dacryoadenitis suggests IgG4-related disease. Histologically, both conditions may be rich in plasma cells, but the plasma cells are often arranged in sheets in iMCD, while more commonly admixed with lymphocytes in IgG4-related disease. Serum IgG4 levels or absolute number of IgG4-positie cells in tissue are not useful for discriminating between the two conditions; the serum IgG4/IgG ratio and the ratio of IgG4/IgG-positive cells in tissue are more reliable discriminators 100.
Concluding remarks
In this article, we reviewed the evolving concepts and definitions of the various conditions under the eponym of MCD and summarize current knowledge regarding the histopathology and pathogenesis of lesions within the MCD spectrum. The current belief is that both the nodal and systemic manifestations are reactive changes to elevated levels of IL-6 and other circulating factors in the cytokine and chemokine storm; the hypercytokinemia can result from various mechanisms, which ultimately leads to different constellations of clinical presentations and pathological features.
Despite growing knowledge about the clinicopathological features of these conditions and the underlying dysregulated cytokine activity, diagnosis and accurate classification of MCD remains challenging. The clinical presentation is highly heterogeneous, and the pathological findings are not specific. Secondly, these patients are at a high risk of developing lymphoproliferative disorders, which can greatly confound the differential diagnoses. Thirdly, a lymph biopsy is not always available, which has sparked the interest of investigators to explore alternative diagnostic approaches. Additionally, our understanding of the underlying molecular mechanisms that drive the diseases processes is still at its infancy. Genomic and epigenomic characterization of MCD and related lymphoproliferative disorders may represent an attractive future research area that potentially leads to advances in diagnosis and therapy.
Figures and tables
Acknowledgements
This work was supported by the intramural research program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
Author’s contribution
All authors contributed to drafting of the manuscript and review and editing of the final text. The figures were provided by Drs. Pittaluga and Jaffe.
Ethical consideration
All studies were in compliance with an IRB approved protocol and institutional guidelines for the study of human tissues.
References
- 1.Castleman B, Towne VW. Case records of the Massachusetts General Hospital; weekly clinicopathological exercises; founded by Richard C. Cabot. N Engl J Med 1954;251:396-400. https://doi.org/10.1056/NEJM195409022511008. 10.1056/NEJM195409022511008 [DOI] [PubMed] [Google Scholar]
- 2.Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 1972;29:670-683. doi:10.1002/1097-0142(197203)29:3<670::aid-cncr2820290321>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 3.Frizzera G, Banks PM, Massarelli G, et al. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease. Pathological findings in 15 patients. Am J Surg Pathol 1983;7:211-231. https://doi.org/10.1097/00000478-198304000-00001 10.1097/00000478-198304000-00001 [DOI] [PubMed] [Google Scholar]
- 4.Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017;129:1646-1657. Epub 2017 Jan 13. https://doi.org/10.1182/blood-2016-10-746933 10.1182/blood-2016-10-746933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994;266:1865-1869. https://doi.org/10.1126/science.7997879 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 6.Soulier J, Grollet L, Oksenhendler E, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995;86:1276-1280. [PubMed] [Google Scholar]
- 7.Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186-1191. https://doi.org/10.1056/NEJM199505043321802 10.1056/NEJM199505043321802 [DOI] [PubMed] [Google Scholar]
- 8.Rohner E, Wyss N, Trelle S, et al. HHV-8 seroprevalence: a global view. Syst Rev 2014;3:11. https://doi.org/10.1186/2046-4053-3-11. 10.1186/2046-4053-3-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brousset P, Cesarman E, Meggetto F, et al. Colocalization of the viral interleukin-6 with latent nuclear antigen-1 of human herpesvirus-8 in endothelial spindle cells of Kaposi’s sarcoma and lymphoid cells of multicentric Castleman’s disease. Hum Pathol 2001;32:95-100. https://doi.org/10.1053/hupa.2001.21131 10.1053/hupa.2001.21131 [DOI] [PubMed] [Google Scholar]
- 10.Gunther T, Grundhoff A: The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog 2010;6:e1000935. https://doi.org/10.1371/journal.ppat.1000935 10.1371/journal.ppat.1000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guito J, Lukac DM: KSHV reactivation and novel implications of protein isomerization on lytic switch control. Viruses 2015;7:72-109. https://doi.org/10.3390/v7010072 10.3390/v7010072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes DJ, Wood JJ, Jackson BR, et al. NEDDylation is essential for Kaposi’s sarcoma-associated herpesvirus latency and lytic reactivation and represents a novel anti-KSHV target. PLoS Pathog 2015;11:e1004771. https://doi.org/10.1371/journal.ppat.1004771 10.1371/journal.ppat.1004771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pantry SN, Medveczky PG: Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus replication. Semin Cancer Biol 2009;19:153-157. https://doi.org/10.1016/j.semcancer.2009.02.010. 10.1016/j.semcancer.2009.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toth Z, Maglinte DT, Lee SH, et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 2010;6:e1001013. https://doi.org/10.1371/journal.ppat.1001013 10.1371/journal.ppat.1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu X, Shahir AM, Sha J, et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi’s sarcoma-associated herpesvirus replication. J Virol 2014;88:4466-4479. Epub 2014 Feb 5. https://doi.org/10.1128/JVI.03326-13 10.1128/JVI.03326-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang H, Dittmer DP, Shin YC, et al. Role of Notch signal transduction in Kaposi’s sarcoma-associated herpesvirus gene expression. J Virol 2005;79:14371-14382. https://doi.org/10.1128/JVI.79.22.14371-14382.2005 10.1128/JVI.79.22.14371-14382.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu D, Wang V, Yang M, et al. Induction of Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Viral Interleukin-6 by X-Box Binding Protein 1. J Virol 2016;90:368-378. https://doi.org/10.1128/JVI.01192-15 10.1128/JVI.01192-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katano H, Sato Y, Kurata T, et al. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology 2000;269:335-344. https://doi.org/10.1006/viro.2000.0196 10.1006/viro.2000.0196 [DOI] [PubMed] [Google Scholar]
- 19.Parravicini C, Chandran B, Corbellino M, et al. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am J Pathol 2000;156:743-749. https://doi.org/10.1016/S0002-9440(10)64940-1 10.1016/S0002-9440(10)64940-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan X, Wang H, Nicholas J: Human herpesvirus 8 interleukin-6 (vIL-6) signals through gp130 but has structural and receptor-binding properties distinct from those of human IL-6. J Virol 1999;73:8268-8278. https://doi.org/10.1128/JVI.73.10.8268-8278.1999 10.1128/JVI.73.10.8268-8278.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fajgenbaum DC: Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood 2018;132:2323-2330. https://doi.org/10.1182/blood-2018-05-848671. 10.1182/blood-2018-05-848671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coscoy L. Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Nat Rev Immunol 2007;7:391-401. https://doi.org/10.1038/nri2076 10.1038/nri2076 [DOI] [PubMed] [Google Scholar]
- 23.Moore PS, Boshoff C, Weiss RA, et al. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996;274:1739-1744. https://doi.org/10.1126/science.274.5293.1739 10.1126/science.274.5293.1739 [DOI] [PubMed] [Google Scholar]
- 24.Polizzotto MN, Uldrick TS, Wang V, et al. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood 2013;122:4189-4198. https://doi.org/10.1182/blood-2013-08-519959 10.1182/blood-2013-08-519959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aoki Y, Tosato G, Fonville TW, et al. Serum viral interleukin-6 in AIDS-related multicentric Castleman disease. Blood 2001;97:2526-2527. https://doi.org/10.1182/blood.v97.8.2526 10.1182/blood.v97.8.2526 [DOI] [PubMed] [Google Scholar]
- 26.Neipel F, Albrecht JC, Ensser A, et al. Human herpesvirus 8 encodes a homolog of interleukin-6. J Virol 1997;71:839-842. https://doi.org/10.1128/JVI.71.1.839-842.1997 10.1128/JVI.71.1.839-842.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aoki Y, Jones KD, Tosato G. Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. J Hematother Stem Cell Res 2000;9:137-145. https://doi.org/10.1089/152581600319351 10.1089/152581600319351 [DOI] [PubMed] [Google Scholar]
- 28.Osborne J, Moore PS, Chang Y: KSHV-encoded viral IL-6 activates multiple human IL-6 signaling pathways. Hum Immunol 1999;60:921-927. https://doi.org/10.1016/s0198-8859(99)00083-x 10.1016/s0198-8859(99)00083-x [DOI] [PubMed] [Google Scholar]
- 29.Boulanger MJ, Chow DC, Brevnova E, et al. Molecular mechanisms for viral mimicry of a human cytokine: activation of gp130 by HHV-8 interleukin-6. J Mol Biol 2004;335:641-654. https://doi.org/10.1016/j.jmb.2003.10.070 10.1016/j.jmb.2003.10.070 [DOI] [PubMed] [Google Scholar]
- 30.Li H, Nicholas J. Identification of amino acid residues of gp130 signal transducer and gp80 alpha receptor subunit that are involved in ligand binding and signaling by human herpesvirus 8-encoded interleukin-6. J Virol 2002;76:5627-5636. https://doi.org/10.1128/jvi.76.11.5627-5636.2002 10.1128/jvi.76.11.5627-5636.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheller J, Chalaris A, Schmidt-Arras D, et al. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011;1813:878-888. https://doi.org/10.1016/j.bbamcr.2011.01.034 10.1016/j.bbamcr.2011.01.034 [DOI] [PubMed] [Google Scholar]
- 32.Nishimoto N, Terao K, Mima T, et al. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008;112:3959-3964. https://doi.org/10.1182/blood-2008-05-155846 10.1182/blood-2008-05-155846 [DOI] [PubMed] [Google Scholar]
- 33.Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood 1989;74:1360-1367. [PubMed] [Google Scholar]
- 34.Kagan P, Sultan M, Tachlytski I, et al. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS One 2017;12:e0176173. https://doi.org/10.1371/journal.pone.0176173 10.1371/journal.pone.0176173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oksenhendler E, Carcelain G, Aoki Y, et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected patients. Blood 2000;96:2069-2073. [PubMed] [Google Scholar]
- 36.Bower M, Veraitch O, Szydlo R, et al. Cytokine changes during rituximab therapy in HIV-associated multicentric Castleman disease. Blood 2009;113:4521-4524. https://doi.org/10.1182/blood-2008-12-197053 10.1182/blood-2008-12-197053 [DOI] [PubMed] [Google Scholar]
- 37.Suda T, Katano H, Delsol G, et al. HHV-8 infection status of AIDS-unrelated and AIDS-associated multicentric Castleman’s disease. Pathol Int 2001;51:671-679. https://doi.org/10.1046/j.1440-1827.2001.01266.x 10.1046/j.1440-1827.2001.01266.x [DOI] [PubMed] [Google Scholar]
- 38.Pinzone MR, Berretta M, Cacopardo B, et al. Epstein-barr virus- and Kaposi sarcoma-associated herpesvirus-related malignancies in the setting of human immunodeficiency virus infection. Semin Oncol 2015;42:258-271. https://doi.org/10.1053/j.seminoncol.2014.12.026 10.1053/j.seminoncol.2014.12.026 [DOI] [PubMed] [Google Scholar]
- 39.Quinlivan EB, Zhang C, Stewart PW, et al. Elevated virus loads of Kaposi’s sarcoma-associated human herpesvirus 8 predict Kaposi’s sarcoma disease progression, but elevated levels of human immunodeficiency virus type 1 do not. J Infect Dis 2002;185:1736-1744. https://doi.org/10.1086/340652 10.1086/340652 [DOI] [PubMed] [Google Scholar]
- 40.Kurzrock R, Voorhees PM, Casper C, et al. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res 2013;19:3659-3670. https://doi.org/10.1158/1078-0432.CCR-12-3349 10.1158/1078-0432.CCR-12-3349 [DOI] [PubMed] [Google Scholar]
- 41.Uldrick TS, Polizzotto MN, Aleman K, et al. Rituximab plus liposomal doxorubicin in HIV-infected patients with KSHV-associated multicentric Castleman disease. Blood 2014;124:3544-3552. https://doi.org/10.1182/blood-2014-07-586800 10.1182/blood-2014-07-586800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stary G, Kohrgruber N, Herneth AM, et al. Complete regression of HIV-associated multicentric Castleman disease treated with rituximab and thalidomide. AIDS 2008;22:1232-1234. https://doi.org/10.1097/QAD.0b013e3282fa75ce 10.1097/QAD.0b013e3282fa75ce [DOI] [PubMed] [Google Scholar]
- 43.Oksenhendler E, Boutboul D, Fajgenbaum D, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years, Br J Haematol 2018;180:206-216. https://doi.org/10.1111/bjh.15019 10.1111/bjh.15019 [DOI] [PubMed] [Google Scholar]
- 44.Polizzotto MN, Uldrick TS, Wyvill KM, et al. Clinical features and outcomes of patients with symptomatic kaposi sarcoma herpesvirus (kshv)-associated inflammation: prospective characterization of KSHV Inflammatory Cytokine Syndrome (KICS). Clin Infect Dis 2016;62:730-738. https://doi.org/10.1093/cid/civ996 10.1093/cid/civ996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uldrick TS, Wang V, O’Mahony D, et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin Infect Dis 2010;51:350-358. https://doi.org/10.1086/654798 10.1086/654798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Connick E, Kane MA, White IE, et al. Immune reconstitution inflammatory syndrome associated with Kaposi sarcoma during potent antiretroviral therapy. Clin Infect Dis 2004;39:1852-1855. https://doi.org/10.1086/426078 10.1086/426078 [DOI] [PubMed] [Google Scholar]
- 47.Oksenhendler E, Boulanger E, Galicier L, et al. High incidence of Kaposi sarcoma-associated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood 2002;99:2331-2336. https://doi.org/10.1182/blood.v99.7.2331 10.1182/blood.v99.7.2331 [DOI] [PubMed] [Google Scholar]
- 48.Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 2000;95:1406-1412. [PubMed] [Google Scholar]
- 49.Du MQ, Liu H, Diss TC, et al. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood 2001;97:2130-2136. https://doi.org/10.1182/blood.v97.7.2130 10.1182/blood.v97.7.2130 [DOI] [PubMed] [Google Scholar]
- 50.Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised Fourth Edition. Edited by Lyon, France, International Agency for Research on Cancer; 2017. [Google Scholar]
- 51.Derudder E, Cadera EJ, Vahl JC, et al. Development of immunoglobulin lambda-chain-positive B cells, but not editing of immunoglobulin kappa-chain, depends on NF-kappaB signals. Nat Immunol 2009;10:647-654. https://doi.org/10.1038/ni.1732 10.1038/ni.1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weisenburger DD, Nathwani BN, Winberg CD, et al. Multicentric angiofollicular lymph node hyperplasia: a clinicopathologic study of 16 cases. Hum Pathol 1985;16:162-172. https://doi.org/10.1016/s0046-8177(85)80065-4 10.1016/s0046-8177(85)80065-4 [DOI] [PubMed] [Google Scholar]
- 53.Chadburn A, Said J, Gratzinger D, et al. HHV8/KSHV-Positive Lymphoproliferative Disorders and the Spectrum of Plasmablastic and Plasma Cell Neoplasms: 2015 SH/EAHP Workshop Report-Part 3. Am J Clin Pathol 2017;147:171-187. https://doi.org/10.1093/ajcp/aqw218 10.1093/ajcp/aqw218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nador RG, Cesarman E, Chadburn A, et al. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996;88:645-656. [PubMed] [Google Scholar]
- 55.Fan W, Bubman D, Chadburn A, et al. Distinct subsets of primary effusion lymphoma can be identified based on their cellular gene expression profile and viral association. J Virol 2005;79:1244-1251. https://doi.org/10.1128/JVI.79.2.1244-1251.2005 10.1128/JVI.79.2.1244-1251.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klein U, Gloghini A, Gaidano G, et al. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood 2003;101:4115-4121. https://doi.org/10.1182/blood-2002-10-3090 10.1182/blood-2002-10-3090 [DOI] [PubMed] [Google Scholar]
- 57.Jenner RG, Maillard K, Cattini N, et al. Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc Natl Acad Sci U S A 2003;100:10399-10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Panagopoulos D, Victoratos P, Alexiou M, et al. Comparative analysis of signal transduction by CD40 and the Epstein-Barr virus oncoprotein LMP1 in vivo. J Virol 2004;78:13253-13261. https://doi.org/10.1128/JVI.78.23.13253-13261.2004 10.1128/JVI.78.23.13253-13261.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minamitani T, Ma Y, Zhou H, et al. Mouse model of Epstein-Barr virus LMP1- and LMP2A-driven germinal center B-cell lymphoproliferative disease. Proc Natl Acad Sci U S A 2017;114:4751-4756. Epub 2017 Mar 28. https://doi.org/10.1073/pnas.1701836114 10.1073/pnas.1701836114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ascoli V, Calabro ML, Giannakakis K, et al. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8-associated polyclonal body cavity effusions that mimic primary effusion lymphomas. Int J Cancer 2006;119:1746-1748; author reply 1749-1750. https://doi.org/10.1002/ijc.21965 10.1002/ijc.21965 [DOI] [PubMed] [Google Scholar]
- 61.Petzer AL, Gunsilius E, Hayes M, et al. Low concentrations of STI571 in the cerebrospinal fluid: a case report. Br J Haematol 2002;117:623-625. https://doi.org/10.1046/j.1365-2141.2002.03523.x 10.1046/j.1365-2141.2002.03523.x [DOI] [PubMed] [Google Scholar]
- 62.Ghoneima A, Cooke J, Shaw E, et al. Human herpes virus 8-positive germinotropic lymphoproliferative disorder: first case diagnosed in the UK, literature review and discussion of treatment options. BMJ Case Rep 2020;13: e231640. https://doi.org/10.1136/bcr-2019-231640 10.1136/bcr-2019-231640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhavsar T, Lee JC, Perner Y, et al. KSHV-associated and EBV-associated Germinotropic Lymphoproliferative Disorder: New Findings and Review of the Literature. Am J Surg Pathol 2017;41:795-800. https://doi.org/10.1097/PAS.0000000000000823 10.1097/PAS.0000000000000823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zanelli M, Fraternali Orcioni G, Zizzo M, et al. HHV-8- and EBV-positive germinotropic lymphoproliferative disorder. Ann Hematol 2019;98:2439-2441. Epub 2019 Aug 3. https://doi.org/10.1007/s00277-019-03773-0 10.1007/s00277-019-03773-0 [DOI] [PubMed] [Google Scholar]
- 65.Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol 2016;3:e163-175. Epub 2016 Mar 17. https://doi.org/10.1016/S2352-3026(16)00006-5 10.1016/S2352-3026(16)00006-5 [DOI] [PubMed] [Google Scholar]
- 66.Kawabata H, Takai K, Kojima M, et al. Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly : a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop 2013;53:57-61. https://doi.org/10.3960/jslrt.53.57 10.3960/jslrt.53.57 [DOI] [PubMed] [Google Scholar]
- 67.Louis C, Vijgen S, Samii K, et al. TAFRO Syndrome in caucasians: a case report and review of the literature. Front Med (Lausanne) 2017;4:149. https://doi.org/10.3389/fmed.2017.00149 10.3389/fmed.2017.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Epub 2016 Mar 18. Int J Hematol 2016;103:686-692. https://doi.org/10.1007/s12185-016-1979-1 10.1007/s12185-016-1979-1 [DOI] [PubMed] [Google Scholar]
- 69.Masaki Y, Kawabata H, Takai K, et al. 2019 Updated diagnostic criteria and disease severity classification for TAFRO syndrome. Int J Hematol 2020;111:155-158. Epub 2019 Nov 28. https://doi.org/10.1007/s12185-019-02780-1 10.1007/s12185-019-02780-1 [DOI] [PubMed] [Google Scholar]
- 70.Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol 2016;91:220-226. https://doi.org/10.1002/ajh.24242 10.1002/ajh.24242 [DOI] [PubMed] [Google Scholar]
- 71.Fujimoto S, Sakai T, Kawabata H, et al. Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol 2019;94:975-983. Epub 2019 Jun 21. https://doi.org/10.1002/ajh.25554 10.1002/ajh.25554 [DOI] [PubMed] [Google Scholar]
- 72.Nishimura Y, Hanayama Y, Fujii N, et al. Comparison of the clinical characteristics of TAFRO syndrome and idiopathic multicentric Castleman disease in general internal medicine: a 6-year retrospective study. Intern Med J 2020;50:184-191. https://doi.org/10.1111/imj.14404 10.1111/imj.14404 [DOI] [PubMed] [Google Scholar]
- 73.Sakashita K, Murata K, Takamori M: TAFRO syndrome: current perspectives. J Blood Med 2018;9:15-23. https://doi.org/10.2147/JBM.S127822 10.2147/JBM.S127822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV- and HHV-8-negative Castleman disease. Blood 2017;129:1658-1668. Epub 2017 Jan 18. https://doi.org/10.1182/blood-2016-11-748855 10.1182/blood-2016-11-748855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018;132:2115-2124. Epub 2018 Sep 4. https://doi.org/10.1182/blood-2018-07-862334 10.1182/blood-2018-07-862334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang HW, Pittaluga S, Jaffe ES. Multicentric Castleman disease: Where are we now? Semin Diagn Pathol 2016;33:294-306. Epub 2016 May 16. https://doi.org/10.1053/j.semdp.2016.05.006 10.1053/j.semdp.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pierson SK, Stonestrom AJ, Shilling D, et al. Plasma proteomics identifies a ‘chemokine storm’ in idiopathic multicentric Castleman disease. Am J Hematol 2018;93:902-912. Epub 2018 May 16. https://doi.org/10.1002/ajh.25123 10.1002/ajh.25123 [DOI] [PubMed] [Google Scholar]
- 78.Yoshimi A, Trippett TM, Zhang N, et al. Genetic basis for iMCD-TAFRO. Oncogene 2020;39:3218-3225. Epub 2020 Feb 12. https://doi.org/10.1038/s41388-020-1204-9 10.1038/s41388-020-1204-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 2014;15:966-974. Epub 2014 Jul 17. https://doi.org/10.1016/S1470-2045(14)70319-5 10.1016/S1470-2045(14)70319-5 [DOI] [PubMed] [Google Scholar]
- 80.Abramson JS. Diagnosis and management of Castleman disease. J Natl Compr Canc Netw 2019;17:1417-1419. https://doi.org/10.6004/jnccn.2019.5037 10.6004/jnccn.2019.5037 [DOI] [PubMed] [Google Scholar]
- 81.Fajgenbaum DC, Wu D, Goodman A, et al. Insufficient evidence exists to use histopathologic subtype to guide treatment of idiopathic multicentric Castleman disease. Am J Hematol 2020;95:1553-1561. https://doi.org/10.1002/ajh.25992 10.1002/ajh.25992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arenas DJ, Floess K, Kobrin D, et al. Increased mTOR activation in idiopathic multicentric Castleman disease. Blood 2020;135:1673-1684. https://doi.org/10.1182/blood.2019002792 10.1182/blood.2019002792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fajgenbaum DC, Langan RA, Japp AS, et al. Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6-blockade-refractory idiopathic multicentric Castleman disease. J Clin Invest 2019;129:4451-4463. https://doi.org/10.1172/JCI126091 10.1172/JCI126091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stern RM, Berliner N. Targeting the mTOR pathway in idiopathic multicentric Castleman disease. J Clin Invest 2019;129:4086-4088. https://doi.org/10.1172/JCI131332 10.1172/JCI131332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koga T, Hagimori N, Takemori S, et al. Randomized, double-blind, placebo-controlled, parallel-group trial of sirolimus for tocilizumab-resistant idiopathic multicentric Castleman disease: study protocol for clinical trial. Medicine (Baltimore) 2020;99:e20710. https://doi.org/10.1097/MD.0000000000020710 10.1097/MD.0000000000020710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vandenbroucke JP: In defense of case reports and case series. Ann Intern Med 2001;134:330-334. https://doi.org/10.7326/0003-4819-134-4-200102200-00017 10.7326/0003-4819-134-4-200102200-00017 [DOI] [PubMed] [Google Scholar]
- 87.You L, Lin Q, Zhao J, et al. Whole-exome sequencing identifies novel somatic alterations associated with outcomes in idiopathic multicentric Castleman disease. Br J Haematol 2020;188:e64-e67. Epub 2019 Dec 21. https://doi.org/10.1111/bjh.16330 10.1111/bjh.16330 [DOI] [PubMed] [Google Scholar]
- 88.Butzmann A, Kumar J, Sridhar K, et al. A Review of Genetic Abnormalities in Unicentric and Multicentric Castleman Disease. Biology (Basel) 2021;10: 251. https://doi.org/10.3390/biology10040251 10.3390/biology10040251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fajgenbaum DC, Shilling D. Castleman Disease pathogenesis. Hematol Oncol Clin North Am 2018;32:11-21. https://doi.org/10.1016/j.hoc.2017.09.002 10.1016/j.hoc.2017.09.002 [DOI] [PubMed] [Google Scholar]
- 90.Wang C, Huang XF, Cai QQ, et al. Remarkable expression of vascular endothelial growth factor in bone marrow plasma cells of patients with POEMS syndrome. Leuk Res 2016;50:78-84. Epub 2016 Sep 26. https://doi.org/10.1016/j.leukres.2016.09.017 10.1016/j.leukres.2016.09.017 [DOI] [PubMed] [Google Scholar]
- 91.Chan JK, Fletcher CD, Hicklin GA, et al. Glomeruloid hemangioma. A distinctive cutaneous lesion of multicentric Castleman’s disease associated with POEMS syndrome. Am J Surg Pathol 1990;14:1036-1046. https://doi.org/10.1097/00000478-199011000-00005 10.1097/00000478-199011000-00005 [DOI] [PubMed] [Google Scholar]
- 92.Bender S, Javaugue V, Saintamand A, et al. Immunoglobulin variable domain high-throughput sequencing reveals specific novel mutational patterns in POEMS syndrome. Blood 2020;135:1750-1758. https://doi.org/10.1182/blood.2019004197 10.1182/blood.2019004197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dao LN, Hanson CA, Dispenzieri A, et al. Bone marrow histopathology in POEMS syndrome: a distinctive combination of plasma cell, lymphoid, and myeloid findings in 87 patients. Blood 2011;117:6438-6444. Epub 2011 Mar 8. https://doi.org/10.1182/blood-2010-11-316935 10.1182/blood-2010-11-316935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dispenzieri A. POEMS syndrome: 2021 update on diagnosis, risk-stratification, and management. Am J Hematol 2021;96:872-888. Epub 2021 May 31. https://doi.org/10.1002/ajh.26240 10.1002/ajh.26240 [DOI] [PubMed] [Google Scholar]
- 95.Khouri J, Nakashima M, Wong S. Update on the diagnosis and treatment of POEMS (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Gammopathy, and Skin Changes) syndrome: a review. JAMA Oncol 2021;7:1383-1391. https://doi.org/10.1001/jamaoncol.2021.0586 10.1001/jamaoncol.2021.0586 [DOI] [PubMed] [Google Scholar]
- 96.Zarate-Osorno A, Medeiros LJ, Danon AD, et al. Hodgkin’s disease with coexistent Castleman-like histologic features. A report of three cases. Arch Pathol Lab Med 1994;118:270-274. [PubMed] [Google Scholar]
- 97.Shiroshita K, Kikuchi T, Okayama M, et al. Interleukin-6-producing Intravascular large B-cell lymphoma with lymphadenopathy mimicking the histology of multicentric Castleman disease. Intern Med 2020;59:3061-3065. Epub 2020 Aug 4. https://doi.org/10.2169/internalmedicine.5046-20 10.2169/internalmedicine.5046-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pina-Oviedo S, Miranda RN, Lin P, et al. Follicular lymphoma with hyaline-vascular Castleman-like features: analysis of 6 cases and review of the literature. Hum Pathol 2017;68:136-146. Epub 2017 Sep 2. https://doi.org/10.1016/j.humpath.2017.08.024 10.1016/j.humpath.2017.08.024 [DOI] [PubMed] [Google Scholar]
- 99.Sato Y, Kojima M, Takata K, et al. Multicentric Castleman’s disease with abundant IgG4-positive cells: a clinical and pathological analysis of six cases. J Clin Pathol 2010;63:1084-1089. Epub 2010 Oct 24. https://doi.org/10.1136/jcp.2010.082958. 10.1136/jcp.2010.082958 [DOI] [PubMed] [Google Scholar]
- 100.Otani K, Inoue D, Fujikura K, et al. Idiopathic multicentric Castleman’s disease: a clinicopathologic study in comparison with IgG4-related disease. Oncotarget 2018;9:6691-6706. https://doi.org/10.18632/oncotarget.24068 10.18632/oncotarget.24068 [DOI] [PMC free article] [PubMed] [Google Scholar]