

Abstract
One of the most oft-employed methods for C–C bond formation involving the coupling of vinyl-halides with aldehydes catalyzed by Ni and Cr (Nozaki–Hiyama–Kishi, NHK) has been rendered more practical using an electroreductive manifold. Although early studies pointed to the feasibility of such a process those precedents were never applied by others due to cumbersome setups and limited scope. Here we show that a carefully optimized electroreductive procedure can enable a more sustainable approach to NHK, even in an asymmetric fashion on highly complex medicinally relevant systems. The e-NHK can even enable non-canonical substrate classes, such as redox-active esters, to participate with low loadings of Cr when conventional chemical techniques fail. A combination of detailed kinetics, cyclic voltammetry, and in situ UV-vis spectroelectrochemistry of these processes illuminates the subtle features of this mechanistically intricate process.
GRAPHICAL ABSTRACT

INTRODUCTION
The venerable Nozaki–Hiyama–Kishi (NHK) reaction, first discovered in 19771 and formalized in 19862 is one of the most useful and reliable C–C bond-forming reactions in organic synthesis (Figure 1).3 It generally involves the cross-coupling of an alkenyl halide with an aldehyde through the use of stoichiometric Cr and catalytic Ni to afford an allylic alcohol product.4 Versions of this reaction that are catalytic in Cr have emerged using a stoichiometric metal reducing agent,5 and asymmetry can be induced using chiral sulfonamide-based ligands.6 Applications of the NHK reaction are manifold and have emerged in natural product total synthesis,7 medicinal chemistry, and even process chemistry settings. In fact, the commercial route to the FDA-approved, natural product-inspired medicine Halaven utilizes this reaction more than once.8 Given the enabling nature of this reaction and its documented utility in industry, a more sustainable variant was pursued to avoid the use of the often superstoichiometric quantities of reducing agents employed to render the reaction catalytic in Cr. In theory, the most inexpensive path to achieving this is through electroreductive means, and three reports point to this proof of concept as highlighted in Figure 1B.9–11 Grigg showed that a Pd-variant of the NHK could be achieved in a divided-cell setup,9 whereas Tanaka10 and Durandetti11 demonstrated galvanostatic means for achieving NHK reactions with Ni catalysts. The issues with these early studies were the poor substrate scope, difficult setups (divided cell or reference electrodes), and the use of expensive electrodes such as Pt. We thus undertook a clean-slate approach to evaluate if the shortcomings of the prior art could be addressed to overcome these limitations, which have so far prevented any application of these electrochemical methods. In this work an electroreductively driven approach to the NHK reaction is reported that exhibits a broad substrate scope for real-world substrates and is operationally simple and demonstrably scalable. Exemplified herein is the ability to employ not only alkenyl halides in complex settings (both racemic and with asymmetric induction) but also redox-active esters.12, 13 The latter transformation previously required four equivalents of Cr salts and could not be rendered catalytic through conventional chemical strategies.14 Finally, insights are provided into both the canonical NHK and electrochemical variant through a combination of detailed kinetics, cyclic voltammetry, and in situ UV-vis spectroelectrochemical studies.
FIGURE 1.
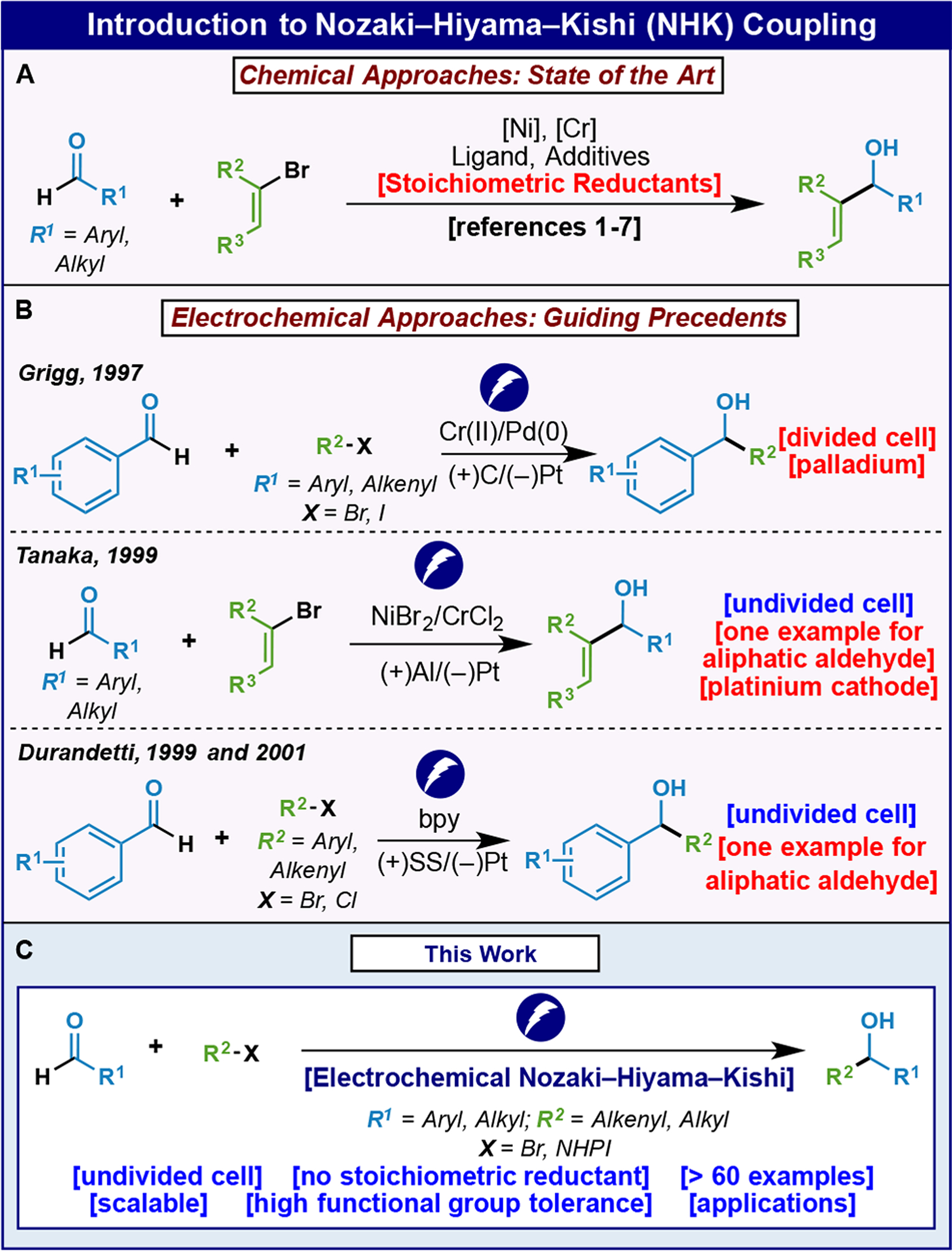
Introduction to Nozaki–Hiyama–Kishi coupling.
ELECTROCHEMICAL NOZAKI–HIYAMA–KISHI COUPLING: DEVELOPMENT AND SCOPE
Optimization studies began using alkyl aldehyde 1 and alkenyl bromide 2. In its final manifestation a 62% isolated yield of allylic alcohol 3 could be obtained using only 20 mol% Cr (Table 1, Top). Operationally, the setup uses a simple commercial potentiostat, and basic precautions to limit oxygen and water content are employed (reactions conducted with an Ar balloon). As the NHK reaction historically consists of numerous reagents and additives, several iterative rounds of optimization were conducted to arrive at the final protocol. At the outset, known literature conditions (Table 1, entries 1–4) were applied to these substrates leading to low isolated yields with no remaining starting materials. Although the conditions of Durandetti and Perichon11b delivered the highest yield (entry 3, 12% under the same conditions that led to similar yields of exact products reported in that paper), they were not a basis of our further optimization due to the experimentally onerous requirement for a pre-electrolysis of a stainless-steel anode (along with carefully weighing to determine the amount of metal removed) followed by exchange with an iron anode. Similarly, entries 1 and 2 were problematic due to the requirement of slow syringe pump addition of the aryl halide along with the need for careful control of potential (reference electrode). Although the yield was low, the Tanaka conditions10 (entry 4) benefitted from a more practical procedure with a simple undivided cell setup and were therefore chosen as a framework from which to optimize. The development of a synthetically useful e-NHK reaction required exploration of five main parameters: (1) oxophilic additives, (2) ligands for Ni, (3) Ni source, (4) Cr source, and (5) electrochemical parameters (electrode, electrolyte, current). A summation of this study (for a more extensive summary, see SI) is outlined in Table 1 (entries 5–18). The use of Cp2ZrCl2 (0.5 equiv) was found to be superior to other common oxophilic additives such as silyl chlorides (entries 5 and 6). A ligand screen demonstrated that phenanthrolines performed better than bipyridines, and that n-Bu substitution at 2 and 2ʹ (L1) further improved the reactivity (entries 7–10). The Ni and Cr sources also played a vital role in reaction efficiency with NiCl2·glyme and CrCl2 proving optimal (entries 11–13). Electrochemically, an inexpensive Al-based sacrificial anode and Ni-foam cathode provided higher yield and both the electrolyte (TBAB) and current settings (constant potential, undivided cell, no reference electrode) were optimum (entries 14–18).
TABLE 1.
Development and optimization of the e-NHK coupling.

|
0.2 mmol.
Isolated yield.
Yields determined by crude 1H NMR using 1,3,5-trimethoxybenzene as the internal standard.
Cr(II)/Ni(II) are generated from stainless steel anode (304L, Fe/Cr/Ni = 72/18/10).
With a concrete set of conditions in hand, the scope of e-NHK was evaluated as depicted in Table 2 with direct comparison in nearly all cases to past precedent (Tanaka, Table 1, entry 4). Aside from operational complexities of past e-NHK methods, the demonstrated use of alkyl aldehydes was severely limited as well as the range of usable alkenyl halides. In this work a variety of both coupling partners could be employed—encompassing a broad range of functional groups such as esters (8, 11, and 21), heterocycles (11, 16, and 17), ethers (10, 12, 20, and 23), alkynes (10), alkyl chlorides (9 and 19), sulphones (22), and imides (25). The range of suitable aldehydes was also expanded to include α,β-unsaturated (31) and aryl systems containing cyclopropanes (37), saturated heterocycles (33), and alkyl chlorides (32).
TABLE 2.
Scope of the e-NHK coupling. Yields of isolated products are indicated in each case; yields in parentheses are from reactions under Tanaka’s condition determined by crude 1H NMR using 1,3,5-trimethoxybenzene as the internal standard. TBAB = tetra-n-butylammonium bromide. Standard reaction conditions: aldehyde (1.0 equiv), alkenyl bromide (2.0 equiv), NiCl2·glyme (2 mol%), L1 (3 mol%), CrCl2 (20 mol%), Cp2ZrCl2 (0.5 equiv), TBAB (0.1 M), DMF (0.08 M), Al(+)/Ni foam(−), Ecell = 2V, 4F/mol.

|
ASYMMETRIC VARIANT
Kishi’s breakthrough discovery on the use of ligands to induce asymmetry was also translated to this electrochemical variant as depicted in Scheme 1.6 In this case, a stainless steel anode proved ideal in terms of conversion and enantiocontrol, which was modest (4.5:1) but on par with that observed in the purely chemical version (cf 44). The main difference in the experimental setup involves a pre-coordination event of the ligand to the Cr salt and proton sponge (1 h) followed by addition to a solution of the remaining components and subsequent electrolysis. In contrast to prior reports, the electrochemical variant described herein does not require 2–3 equivalents of a metal reducing agent (Mn) and a full equivalent of Zr.6d
SCHEME 1. Enantioselective version of the e-NHK coupling.
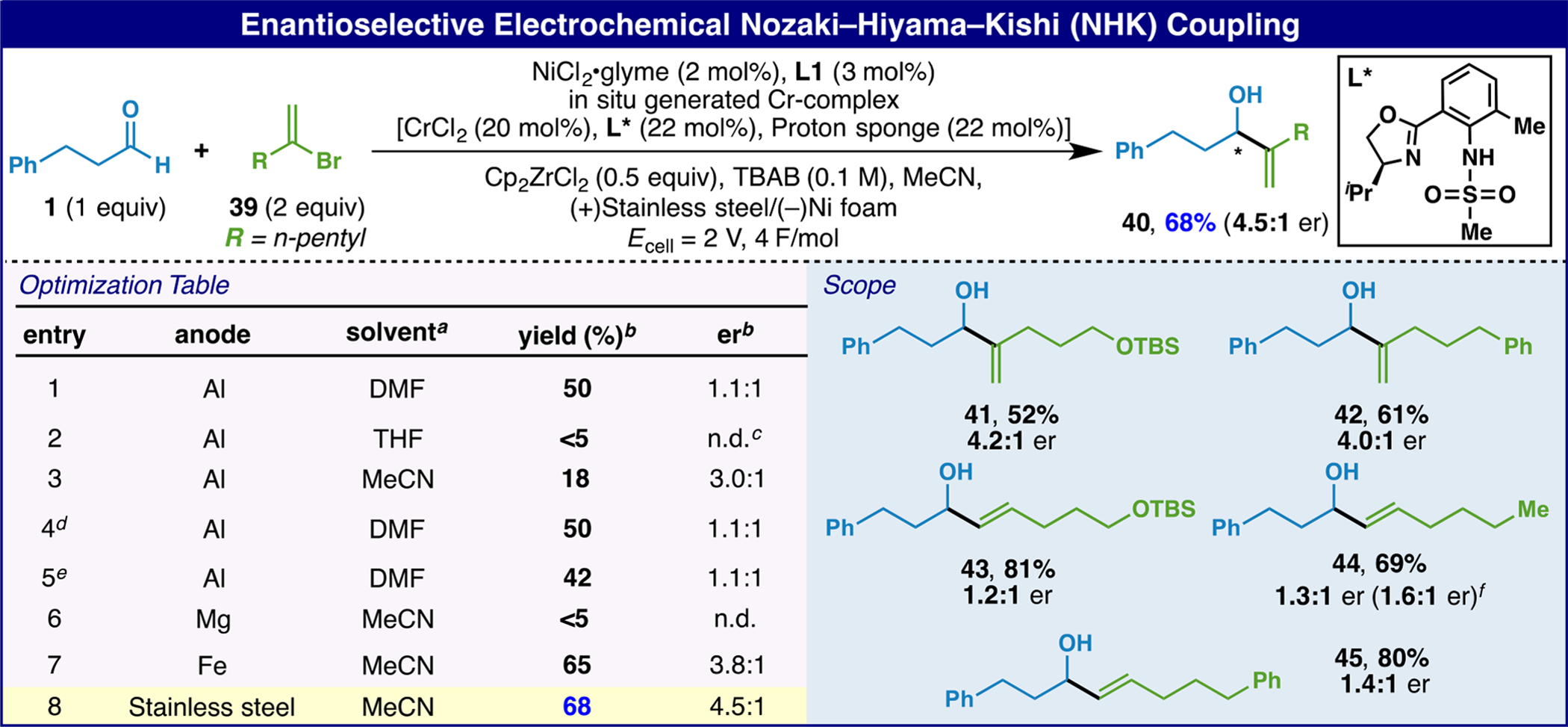
aUsed for both Cr-complex formation and electrochemical reaction. bYields determined by crude 1H NMR using CH2Br2 as the internal standard; er determined by Mosher ester analysis. cNot determined. dDIPEA was used for Cr-complex formation instead of proton sponge. eCH3CN was used for Cr-complex formation; DMF/CH3CN (4:1 v/v). fer reported by Kishi et al. using stoichiometric CrCl2–L* complex.
With viable conditions for both the classical and asymmetric NHK variants, five real-world examples of the latter manifold were pursued using internal Eisai intermediates, some of which are relevant to Halaven itself (Scheme 2).8a–c The chemoselectivity of these transformations is notable and bodes well for adoption on a larger scale. Thus, aldehyde 46 could be employed to deliver allylic alcohol 47a or 47b as the major product as controlled by the ligand choice (Scheme 2A). Similarly, aldehyde 48 could be engaged by alkenyl bromide 49 bearing homoallylic chloride to furnish either diastereomer as the major product (Scheme 2B). The complex sugar-derived aldehyde 5 could be easily alkenylated to controllably provide diastereomer 52a or 52b based on the stereochemistry of the ligand employed (Scheme 2C). The preparation of 52a was accomplished on gram-scale demonstrating the robustness and scalability of the e-NHK protocol. Even when both components were complex, oxygen-rich architectures, a serviceable yield of product was obtained with complete control of diastereoselectivity (Scheme 2D). Notably, this example demonstrates the use of enol triflates in the e-NHK reaction. Finally, a macrocyclizing e-NHK reaction was successful on aldehyde 56, delivering Halaven intermediate 57 in good yield (Scheme 2E).
SCHEME 2. Applications of the e-NHK coupling.
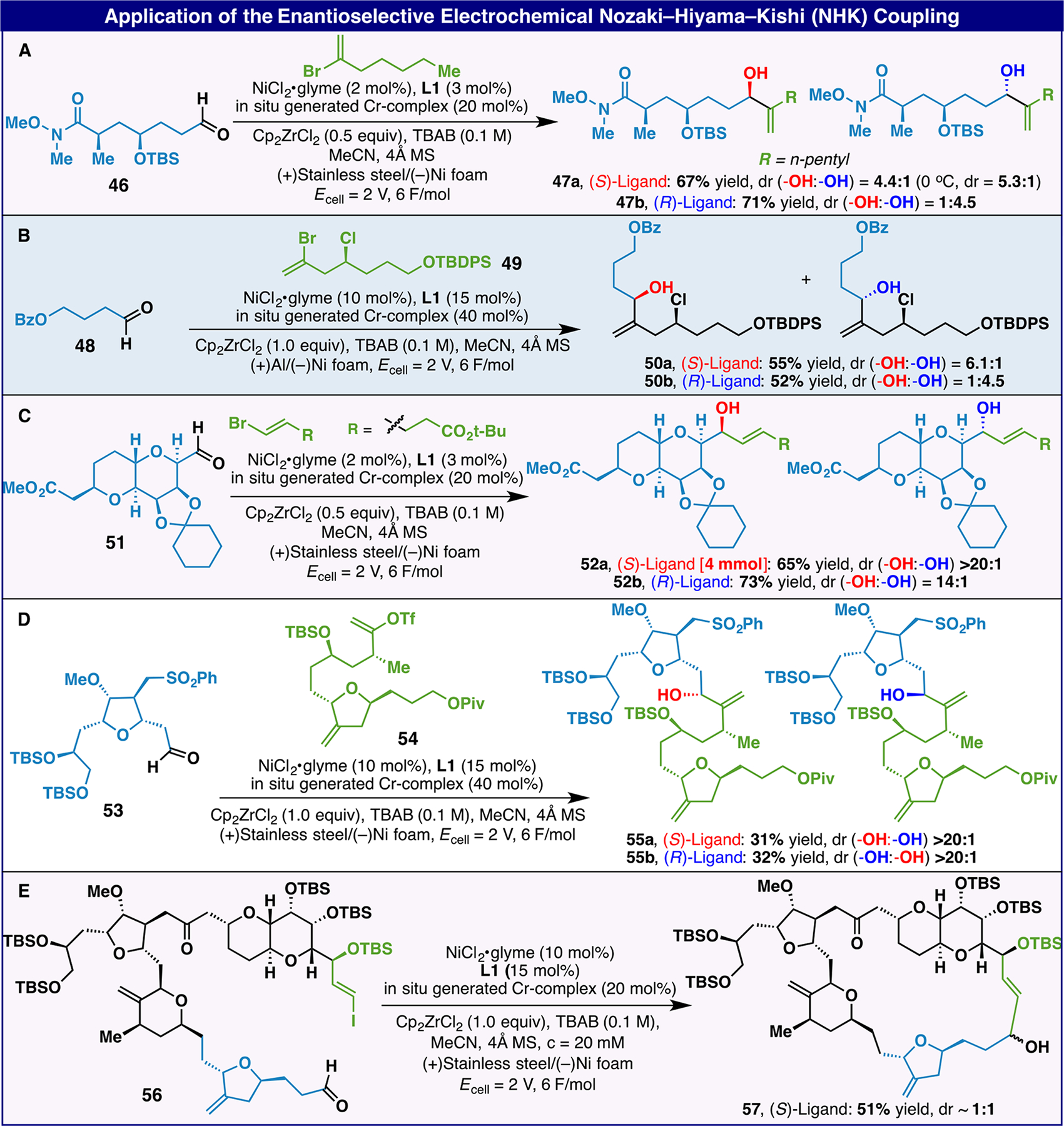
Yields of isolated products are indicated in each case. See SI for reaction conditions. (A to E) Application of the e-enantioselective NHK to five real-world examples including some that are relevant to the synthesis of Halaven.
ELECTROCHEMICAL DECARBOXYLATIVE NOZAKI–HIYAMA–KISHI COUPLING
Attention then turned to a recent disclosure using redox-active esters (RAEs) in the NHK reaction (Scheme 3). In that work,14 all four equivalents of Cr were needed to achieve good conversion (Scheme 3A, entry 1). Despite multiple attempts, the use of exogenous metal reducing agents with lower Cr levels proved unsuccessful (entry 2). In stark contrast to conventional NHK reactions (vide supra), this problem was singularly addressed using electroreductive conditions, which in optimized form delivered a good yield of adduct 85 from aldehyde 59 and RAE 58. In this transformation, a silyl chloride was found to be optimal rather than a Zr-based oxophilic additive (entries 3–5). The use of CrCl3 rather than CrCl2, a lower current (2.5 mA vs 5 mA, entry 7) and a Ni-foam cathode (entry 8) proved essential. The scope of this coupling was found to be identical to the chemical variant and in nearly all cases provided a similar conversion to that reported previously using only 20 mol% Cr vs four equivalents and only one equivalent of RAE vs two. In a few cases however (Scheme 3C), chemical conditions proved superior (81, 83, and 84).
SCHEME 3. E-decarboxylative NHK coupling.
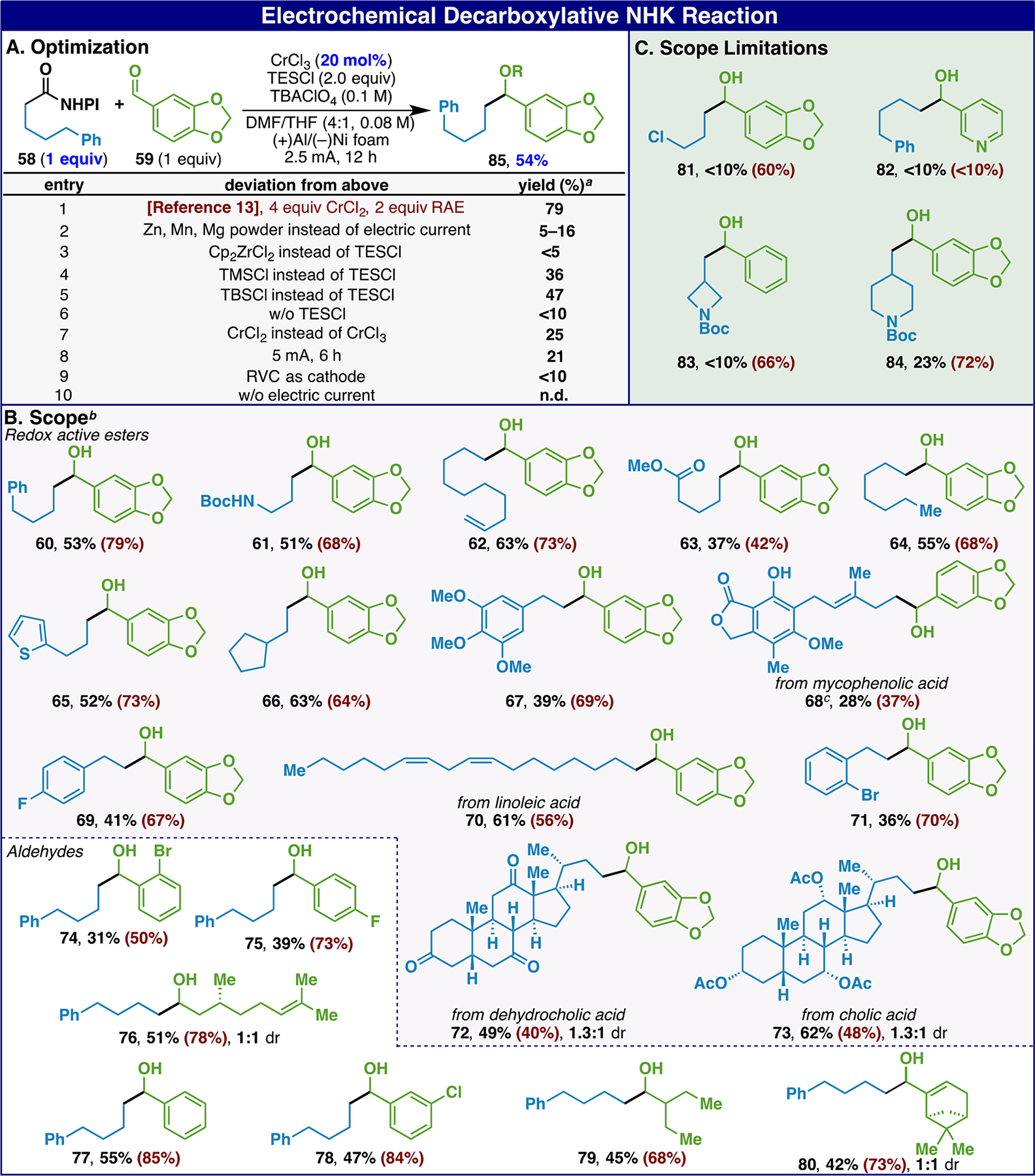
a0.2 mmol; yields determined by crude 1H NMR using 1,3,5-trimethoxybenzene as the internal standard. bIsolated after desilylation (TBAF, THF) of the crude mixture. cIsolated as corresponding triethylsilyl ether.
KINETICS INVESTIGATION OF BULK ELECTROLYSIS
From a mechanistic standpoint, the classical NHK reaction has been evaluated only sporadically and thus in-depth study was pursued in this work. Figure 2B outlines the main mechanistic observations supported by kinetics, cyclic voltammetry, and UV-vis spectroelectrochemistry. The proposed classical NHK mechanism adapted to the electrochemical protocol is shown in Figure 2A (left). The proposed mechanism involves a complex set of interconnected cycles: low-valent Ni activates the vinyl halide in one cycle, while Cr turns over the Ni catalyst and is in turn reduced by an external chemical reductant. Cr participates in a second cycle involving transmelation of the vinyl-Ni(II) species, followed by aldehyde addition and trapping of the resulting Cr alkoxide species to provide the product. These cycles may be envisioned as a set of connected cogs, meaning that the overall rate will be controlled by the slowest cog (cycle). To our knowledge no detailed kinetics studies have been performed. We undertook kinetic studies15 of both the classical NHK and the optimized e-NHK to develop further comparative insight into the mechanism. As shown in Figure 2C, both protocols show a slight induction period, but the e-NHK reaction proceeds nearly twice as rapidly as the classical reaction using a chemical reductant. In addition, the e-NHK reaction shows overlay in the “same excess” protocol of reaction progress kinetic analysis (RPKA, see SI), indicating no erosion of electrode efficiency over the course of the reaction. Further studies probed the effect of concentrations of reactants, Ni, Cr, or Zr, as well as the effect of changing current (for e-NHK) and Mn reductant (for classical NHK) (see SI). For both protocols, little change in rate was observed with changes in any of these variables, with the exception of the concentration of Cr and, in the case of e-NHK, current. These findings suggest that the Cr(III) reduction step controls the overall rate of the complex set of reactions. This reveals an advantage of the e-NHK over the classical reaction: the rate of the Cr reduction step may be increased by increasing the current, while increasing the concentration of the chemical reductant failed to substantially alter the reaction rate. As discussed above, the e-NHK protocol allows coupling of redox-active esters that fail in the classical NHK protocol. This striking difference is demonstrated by comparing the reaction progress profiles of the two protocols for the reaction of RAE 58 with aldehyde 59 as shown in Figure 2C. According to the proposed mechanism shown in Figure 2A (right), it is hypothesized that the success of the e-NHK decarboxylative variant can be attributed to this rapid and selective Cr-reduction in contrast to chemical reducing agents that lead to mostly decomposition of the RAE rather than productive coupling. As shown in Figure 2F, reactions initiated with CrCl3, which is relatively insoluble in DMF/THF, exhibit an induction period that decreases with increasing current. After the initial induction period, reaction rate becomes impervious to the magnitude of the current, and the reaction exhibits zero-order kinetics in [58] and positive order kinetics in both [Cr] and [59]. This supports the proposal that the reactions at the electrode are rapid and the rate-determining step becomes the reaction between the Cr-alkyl species and the aldehyde (See SI for details).
FIGURE 2.
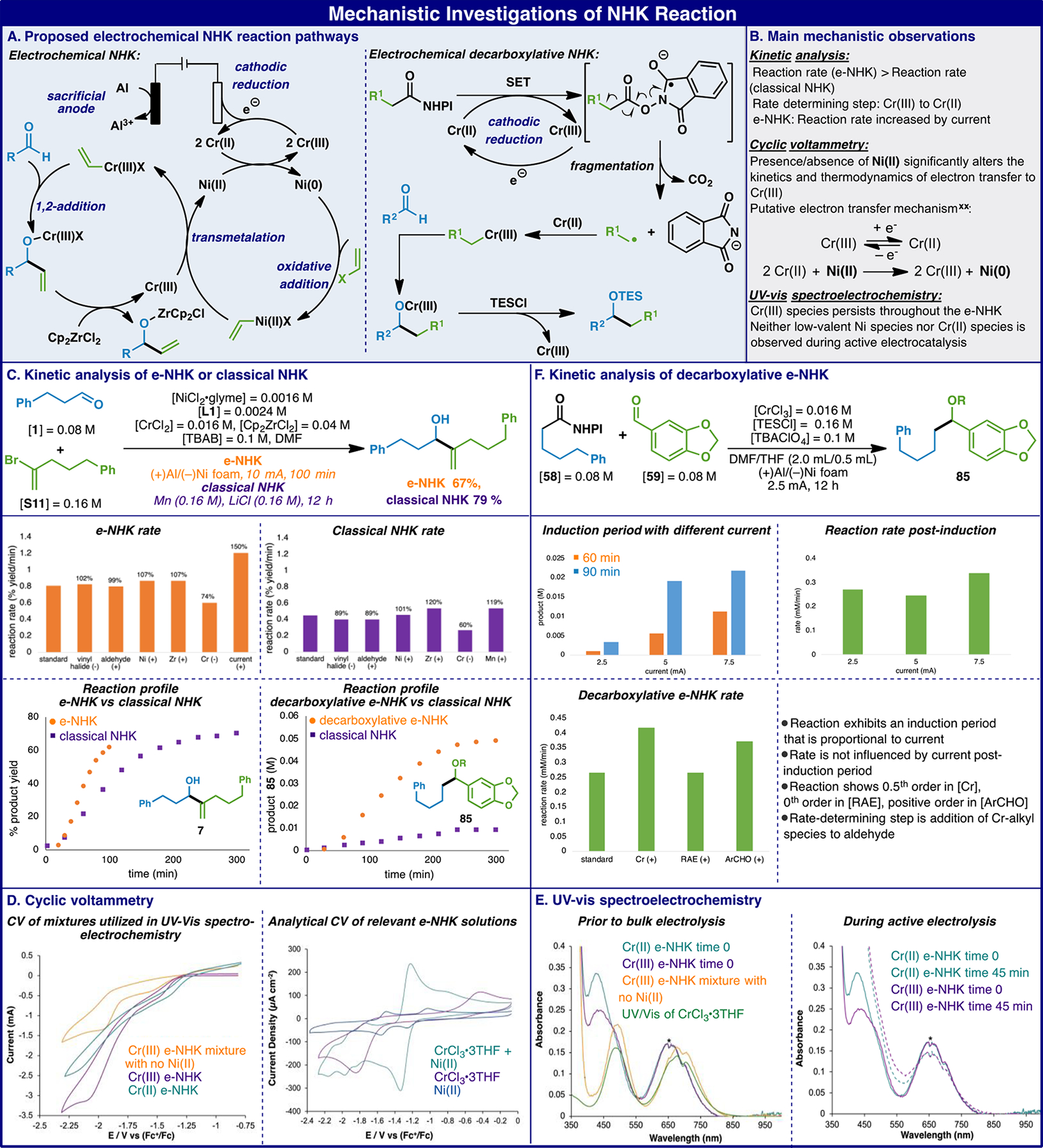
Proposed mechanism and mechanistic studies.
Figure 2C middle: Comparison of the dependence of reaction rates for classical NHK (purple bars, right) and e-NHK (orange bars, left) on concentrations of 1, S11, NiCl2·glyme, CrCl2, Cp2ZrCl2, and Mn (for classical NHK) and current (for e-NHK). The designations (+) and (−) indicate a reaction carried out with an increase or decrease, respectively, in the concentration of the noted variable. Percent deviation of rate from standard conditions is given above each bar. See Supporting Information for details and complete kinetic profiles. Fig 2C bottom left: Comparison of reaction profiles for classical NHK (purple squares) with e-NHK (orange circles) for the reaction to form product 7 (see Table 1) using 0.08 M aldehyde 1 and 0.16 M vinyl bromide S11 in DMF. [NiCl2·glyme] = 0.0016 M; [L1] = 0.0024 M; [CrCl2] = 0.016 M; [Cp2ZrCl2] = 0.04 M. For classical NHK: Mn powder = 0.16 M; [LiCl] = 0.16 M. For e-NHK: [TBAB] = 0.1 M; electrodes: (+)Al/(−)Ni foam; constant current at 10 mA. Fig 2C bottom right: Comparison of reaction profiles for classical decarboxylative NHK (purple squares) with electrochemical decarboxylative NHK (orange circles) for the reaction to form product 85 (see Scheme 3) using 0.08 M aldehyde 59 and 0.08 M redox active ester 58 in DMF/THF (see Scheme 3). [CrCl3] = 0.016 M; [TESCl] = 0.16 M. For classical NHK: Mn powder = 0.16 M. For e-NHK: [TBAClO4] = 0.1 M; electrodes: (+)Al/(−)Ni foam; constant current at 2.5 mA. Figure 2D left: Cyclic voltammetry of the e-NHK reaction under conditions for spectroelectrochemical studies, prior to bulk electrolysis. (Black): Cr(III)-based e-NHK mixture that does not contain the Ni(II). (Teal): Cr(II)-based e-NHK. (Plum): Cr(III)-based e-NHK. All experiments contain [Cr] = 0.016 M, [NiCl2·glyme] = 0.0016 M, [L4] = 0.0024 M, [Cp2ZrCl2] = 0.04 M, [1] = 0.08 M, [S11] = 0.16 M, [TBAPF6] = 0.1 M, DMF, a Ni working electrode, an Al counter electrode. CV data acquired with 0.025 V/s scan rate. Figure 2D right: CrCl3·3THF: [CrCl3·3THF] = 2 mM. CrCl3·3THF+ Ni(II) catalyst: [CrCl3·3THF] = 2 mM, [NiCl2·glyme] = 0.2 mM, [L4] = 0.3 mM. Ni(II) catalyst: [NiCl2·glyme] = 0.2 mM, [L4] = 0.3 mM. All CV experiments were run in DMF with [TBAPF6] = 0.1 M and acquired with a scan rate of 100 mV/s, GC working electrode, and Al counter electrode. All potentials referenced to Fc+/0. Figure 2E left. [S11] = 160 mM, [1] = 80 mM, [CrCl2] = 16 mM, [Cp2ZrCl2] = 40 mM, [NiCl2·glyme] = 1.6 mM, [L4] = 2.4 mM (teal), [S11] = 160 mM, [1] = 80 mM, [CrCl3·3THF] = 16 mM, [Cp2ZrCl2] = 40 mM, [NiCl2·glyme] = 1.6 mM, [L4] = 2.4 mM (plum), [S11] = 160 mM, [1] = 80 mM, [CrCl3·3THF] = 16 mM, [Cp2ZrCl2] = 40 mM, [NiCl2·glyme] = 0 mM, [L4] = 0 mM (tangerine), and [CrCl3·3THF] = 16 mM (clover). Figure 2E right: UV-Vis of: Cr(II)-based e-NHK mixture prior to bulk electrolysis (solid teal), Cr(II)-based e-NHK mixture after an applied potential of −1.5 V vs Fc+/0 for 15 minutes and then an applied potential −2.0 V vs Fc+/0 for 30 minutes (dashed teal), Cr(III)-based e-NHK mixture prior to bulk electrolysis (solid plum), Cr(III) based e-NHK mixture after an applied potential of −1.5 V vs Fc+/0 for 15 minutes and then an applied potential −2.0 V vs Fc+/0 for 30 minutes (dashed plum). Absorbance data were baseline-corrected by taking the absorbance at 900 nm to be zero. *indicates portions of the UV-vis absorbance data that have signal saturation inherent to the detector/light source used for these experiments. Figure 2F middle left: Comparison of induction period with different current. [58] = 0.08 M, [59] = 0.08 M, [CrCl3] = 0.016 M, [TESCl] = 0.16 M, [TBAClO4] = 0.1 M, with 2.5 mA, 5 mA and 7.5 mA, respectively. Figure 2F middle right: reaction rate after removing induction period. Figure 2F bottom left: Comparison of the dependence of reaction rates on concentration of 58, 59, and CrCl3.
ELECTROANALYTICAL INVESTIGATIONS
To further elucidate the e-NHK electron transfer processes and support the critical steps of the proposed mechanism, cyclic voltammetry (CV) and UV-vis spectroelectrochemistry were employed. Previous work by Berkessel and coworkers demonstrated the utility of both of these electroanalytical techniques in characterizing species relevant to NHK chemistry;16 thus, we were inspired to apply this approach to examine the e-NHK. To confirm whether or not the e-NHK had voltammetry consistent with an electrocatalytic mechanism, we performed a series of analytical CV measurements. CV of the e-NHK reaction mixture using CrCl2 pre-catalyst did not display significant catalytic current for a potential sweep from 0 V to −3.5 V vs Fc+/0 (Figure S2). We suspected that the rapid chemical reduction of Ni(II) (NiCl2·glyme and L4) by CrCl2 obfuscated the voltammetry. When CrCl3·3THF was used as the Cr source, however, a large cathodic current was observed at −2.0 V vs. Fc+/0 (Figure 2D Left, plum).
To further identify potential species associated with this catalytic wave, we acquired analytical CVs of the individual e-NHK reaction components and then sequentially combined components of the e-NHK reaction towards the complete reaction mixture. The formal potential of CrCl3·3THF in DMF was determined to be −1.06 V vs Fc+/0 with a peak-to peak separation, ΔEp, of 1.1 V at a scan rate of 100 mV/s, which suggests slow electron transfer due to a large inner-sphere reorganization energy (Figure 2D right, plum).Upon addition of Ni(II) to a solution of CrCl3·3THF, the main redox process shifts to a half-wave potential of −1.2 V vs Fc+/0 and exhibits greater reversibility (ΔEp = 0.130 V at 100 mV/s) (Figure 2D right, teal). While we cannot yet assign the nature of this redox-active species, the influence of Ni(II) on the redox behavior suggests the presence of a potential interaction between the two metal species during electrocatalysis, which is in accord with prior electrochemical studies.16 Introduction of Cp2ZrCl2 and alkyl aldehyde 1 to the mixture of Cr(III) and Ni(II) did not significantly change the CV of the Cr(III)/Ni(II) mixture (Figure S4, clover vs teal line). In contrast, addition of alkenyl bromide S11 to a solution of Cr(III), Ni(II), and Cp2ZrCl2 yielded a significant cathodic current at −2.0 V vs Fc+/0, which has an onset potential at a cathodic wave that is only present when Ni(II) is in solution (Figure S7, blue). Thus, we hypothesize that this catalytic current originates from the electrocatalytic, low-valent Ni homocoupling of the alkenyl bromide, a reaction precedented by Nédélec and coworkers employing similar electrolysis conditions.17 Next we investigated the concentration dependencies of the catalytic wave and observed a linear correlation between the peak catalytic current and both [CrCl3·3THF] and [Ni(II)] (Figures S9 and S10).20 These electroanalytical results differ from the observed kinetics of the reaction under bulk electrolysis. We note that the analytical CV studies require 8-fold more dilute concentrations, which can alter the kinetics of the e-NHK and shift the Ni catalyst resting state from a kinetically saturated species to another elementary step that has a rate dependence on [Ni]. In electrocatalysis, a linear relationship between [catalyst] and catalytic current is expected for an electrocatalytic mechanism based upon ErC’i (i.e. reversible electron transfer followed by an irreversible, catalytic chemical step).18 The concentration dependence data for Cr(III) and Ni(II) are indeed consistent with an e-NHK mechanism that proceeds first by reversible cathodic electron transfer to Cr(III) followed by irreversible electron transfer from the resulting Cr(II) to the Ni(II) catalyst, which would regenerate Cr(III) and form a proposed low-valent Ni species that oxidatively adds to 1 for cross-coupling, as suggested by the reaction pathways in Figure 2A. These results and those discussed below point to a potential Cr(III) resting redox state.
After characterizing the catalytic current for the e-NHK system, we next attempted to observe the proposed Cr(III) catalyst resting state during bulk electrolysis by utilizing a combination of ex situ UV-vis spectroscopy and in situ UV-vis spectroelectrochemistry. The ligand field transitions of pseudo-octahedral Cr(III) and high-spin, pseudo-octahedral Cr(II) in ~Oh symmetry provide excellent handles for determining active components in the e-NHK mixture. For example, the UV-vis spectrum of CrCl3·3THF dissolved in DMF exhibits two bands with λmax = 677 nm and 487 nm. These bands can be attributed to 4A2g → 4T2g, 4T1g transitions, respectively (Figure S23). Of note, a Fano antiresonance is observed on the lower-energy ligand field transition (~695 nm); this feature reflects the spin-orbit interaction between a spectroscopically spin-forbidden ligand field excited state (likely specific orbital components of either the 2Eg or 2T1g states) with a vibrationally broadened spin-allowed ligand field excited state.21,22 Antiresonances have been observed for various other six-coordinate Cr(III) complexes.19,23 For Cr(II), we observe a band at λmax = 843 nm, which can be assigned as the 5Eg → 5T2g transition (Figure S21). We initially monitored the reduction of Cr(III) to Cr(II) in the absence of other components, confirming that one-electron reduction decreased absorbance due to Cr(III) and increased absorbance due to Cr(II) (Figure S28). We then probed the complete CrCl3·3THF-based e-NHK reaction mixture during active electrocatalysis. Both ligand field transitions for Cr(III) persisted at open circuit potential (OCP) (Figure 2E Left, plum) and while holding the potential at both −1.5 V vs Fc+/0 or −2.0 V vs Fc+/0 (Figure 2E Right, plum), suggesting persistence of Cr(III) during electrolysis. Interestingly, both absorptions for Cr(III) slightly blue-shift and increase in intensity relative to an independently prepared CrCl3·3THF sample (Figure 2E Left, clover) as well as a non-electrolyzed CrCl3·3THF-based e-NHK mixture that excluded Ni(II) catalyst (Figure 2E Left, tangerine). One possibility for this shift is a change in the inner-sphere coordination environment during redox cycling of Cr(III/II) or an interaction between Ni and Cr(III). However, a definitive conclusion cannot be drawn from these experiments, and a future study will be directed at structural and spectroscopic characterization of potential metal-metal interactions.
We then performed UV-vis spectroelectrochemistry of the e-NHK mixture using CrCl2 as the Cr source, despite the absence of catalytic current in the analytical CV of this system. Interestingly, upon addition of the Ni(II) catalyst to the CrCl2 e-NHK mixture at OCP, the Cr(II)-based 5E → 5T2 transition immediately disappeared with concomitant appearance of Cr(III) ligand field transitions (Figure 2E Left, Teal). The spectra suggest that CrCl2 rapidly reduces Ni(II) to a catalytically active low-valent Ni complex. This is in agreement with the lack of any detectable buildup of Cr(II) species during active electrocatalysis for either the Cr(II)-based e-NHK or the Cr(III)-based e-NHK reaction mixture. Temporal monitoring of the bulk electrolysis for both the Cr(II)- and Cr(III)- based e-NHK mixtures also revealed decreases in absorbance for Cr(III) ligand field transitions (Figure 2E Right). The change in [Cr(III)] at the beginning of bulk electrolysis could be associated with either the induction period of the reaction or the partial positive order in [Cr] observed during the bulk electrolysis kinetic studies. Additionally, these in situ UV-vis spectra of e-NHK bulk electrolysis support minimal buildup of a putative low-valent Ni species during active electrocatalysis.
Taken together, the results from these electroanalytical studies illustrate the following: (1) the thermodynamic and kinetic redox properties of the Cr(III) are significantly different in the presence of Ni(II), (2) the e-NHK electron transfer processes likely proceed first by electrochemically reversible cathodic electron transfer to Cr(III) followed by electrochemically irreversible electron transfer from Cr(II) to the Ni(II) catalyst, (3) a Cr(III) species persists throughout the duration of the e-NHK, which could correspond to a putative resting state preceding rate-determining electron transfer observed under bulk electrolysis conditions, and (4) there does not appear to be an appreciable buildup of Cr(II) or low-valent Ni species by UV-vis spectroscopy during active electrocatalysis.
CONCLUSION
In summary, the NHK reaction, one of the most oft-employed methods for C–C bond formation, particularly in natural product total synthesis, has been rendered more sustainable24 using electroreductive conditions. Inspired by early proof-of-concept work in this area, a careful choice of ligand, Cr and Ni sources, and optimization of electrochemical parameters allow the practitioner to avoid the use of superstoichiometric metallic reducing agents and dramatically expand the scope of those original reports. Application to Kishi’s asymmetric variant as well as multiple realistic substrate classes is also demonstrated. Most importantly, the RAE-variant of the NHK reaction, which cannot be rendered catalytic in Cr using exogenous chemical reducing agents, can be uniquely enabled electrochemically. Finally, a combination of detailed kinetics, CV, and in situ UV-vis spectroelectrochemical studies teach new lessons about this useful transformation and may aid in the invention and improvement of other electroreductive processes with wide utility for synthesis.
Supplementary Material
ACKNOWLEDGMENT
Financial support for this work was provided by NIH (GM-118176), NSF (CCI Phase 1 grant 1740656 and Phase II grant 2002158), and Fujian Juhong Trade & Business Co. Ltd (Y.G.). We thank Eisai for their generous donation of compounds S24, S25, S26, S27, 53, 56, and L*. Authors are grateful to Dr. Dee-Hua Huang and Dr. Laura Pasternack (Scripps Research) for assistance with nuclear magnetic resonance (NMR) spectroscopy, to Dr. Jason Chen, Brittany Sanchez and Emily Sturgell (Scripps Automated Synthesis Facility) for assistance with HPLC, HRMS and LCMS analysis. Glovebox for anaerobic procedures provided through NIH grant 1S10OD025208. RGH acknowledges financial support from Caltech and the Dow Next Generation Educator Fund.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. The Supporting Information contains all experimental procedures, analysis, and com-pound characterization data.
REFERENCES
- (1).Okude Y; Hirano S; Hiyama T; Nozaki H Grignard-Type Carbonyl Addition of Allyl Halides by Means of Chromous Salt. A Chemospecific Synthesis of Homoallyl Alcohols. J. Am. Chem. Soc 1977, 99, 3179–3181. [Google Scholar]
- (2).(a) Jin H; Uenishi J; Christ WJ; Kishi Y Catalytic Effect of Nickel(II) Chloride and Palladium(II) Acetate on Chromium(II)-Mediated Coupling Reaction of Iodo Olefins with Aldehydes. J. Am. Chem. Soc 1986, 108, 5644–5646. [Google Scholar]; (b) Takai K; Tagashira M; Kuroda T; Oshima K; Utimoto K; Nozaki H Reactions of Alkenylchromium Reagents Prepared from Alkenyl Trifluoromethanesulfonates (Triflates) with Chromium(II) Chloride under Nickel Catalysis. J. Am. Chem. Soc 1986, 108, 6048–6050. [DOI] [PubMed] [Google Scholar]
- (3).For selected reviews, see:; (a) rstner A Carbon − Carbon Bond Formations Involving Organochromium(III) Reagents. Chem. Rev 1999, 99, 991–1045. [DOI] [PubMed] [Google Scholar]; (b) Avalos M; Babiano R; Cintas P; Jimenez JL; Palacios JC Synthetic Variations Based on Low-Valent Chromium: New Developments. Chem. Soc. Rev 1999, 28, 169–177. [Google Scholar]; (c) Wessjohann LA; Scheid G Recent Advances in Chromium(II)- and Chromium(III)-Mediated Organic Synthesis. Synthesis 1999, 1999, 1–36. [Google Scholar]; (d) Lumbroso A; Cooke ML; Breit B Catalytic Asymmetric Synthesis of Allylic Alcohols and Derivatives and Their Applications in Organic Synthesis. Angew. Chem., Int. Ed 2013, 52, 1890–1932. [DOI] [PubMed] [Google Scholar]; (e) Takai K Addition of Organochromium Reagents to Carbonyl Compounds. Org. React 2004, 253–612. [Google Scholar]; (f) Takai K Organochromium Reagents. In Comprehensive Organic Synthesis, 2nd ed.; Knochel P, Molander GA, Eds.; Elsevier Science: Oxford, U.K., 2014; Vol. 1, pp 159–203. [Google Scholar]; (g) Hargaden GC; Guiry PJ The Development of the Asymmetric Nozaki–Hiyama–Kishi Reaction. Adv. Synth. Catal 2007, 349, 2407–2424. [Google Scholar]; (h) Tian Q; Zhang G Recent Advances in the Asymmetric Nozaki–Hiyama–Kishi Reaction. Synthesis 2016, 48, 4038–4049. [Google Scholar]; (i) Gil A; Albericio F; Álvarez M Role of the Nozaki–Hiyama–Takai–Kishi Reaction in the Synthesis of Natural Products. Chem. Rev 2017, 117, 8420–8446. [DOI] [PubMed] [Google Scholar]
- (4).Kochi JK; Davis DD Reduction of Organic Halides by Chromium(II). Mechanism of the Formation of Benzylchromium Ion. J. Am. Chem. Soc 1964, 86, 5264–5271. [Google Scholar]
- (5).(a) Fürstner A; Shi N A Multicomponent Redox System Accounts for the First Nozaki–Hiyama–Kishi Reactions Catalytic in Chromium. J. Am. Chem. Soc 1996, 118, 2533–2534. [Google Scholar]; (b) Fürstner A; Shi N Nozaki–Hiyama–Kishi Reactions Catalytic in Chromium. J. Am. Chem. Soc 1996, 118, 12349–12357. [Google Scholar]; (c) Kuroboshi M; Tanaka M; Kishimoto S; Goto K; Tanaka H; Torii S Ni/Cr/Al Multi-Metal Redox-Mediated Alkenylation of Aldehydes. Tetrahedron Lett 1999, 40, 2785–2788. [Google Scholar]
- (6).(a) Wan Z-K; Choi H.-w.; Kang F-A; Nakajima K; Demeke D; Kishi Y Asymmetric Ni(II)/Cr(II)-Mediated Coupling Reaction: Stoichiometric Process. Org. Lett 2002, 4, 4431–4434. [DOI] [PubMed] [Google Scholar]; (b) Choi H.-w.; Nakajima K; Demeke D; Kang F-A; Jun H-S; Wan Z-K; Kishi Y Asymmetric Ni(II)/Cr(II)-Mediated Coupling Reaction: Catalytic Process. Org. Lett 2002, 4, 4435–4438. [DOI] [PubMed] [Google Scholar]; (c) Kurosu M; Lin M-H; Kishi Y Fe/Cr- and Co/Cr-Mediated Catalytic Asymmetric 2-Haloallylations of Aldehydes. J. Am. Chem. Soc 2004, 126, 12248–12249. [DOI] [PubMed] [Google Scholar]; (d) Namba K; Kishi Y New Catalytic Cycle for Couplings of Aldehydes with Organochromium Reagents. Org. Lett 2004, 6, 5031–5033. [DOI] [PubMed] [Google Scholar]; (e) Namba K; Cui S; Wang J; Kishi Y A New Method for Translating the Asymmetric Ni/Cr-Mediated Coupling Reactions from Stoichiometric to Catalytic. Org. Lett 2005, 7, 5417–5419. [DOI] [PubMed] [Google Scholar]; (f) Liu S; Kim JT; Dong C-G; Kishi Y Catalytic Enantioselective Cr-Mediated Propargylation: Application to Halichondrin Synthesis. Org. Lett 2009, 11, 4520–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Guo H; Dong C-G; Kim D-S; Urabe D; Wang J; Kim JT; Liu X; Sasaki T; Kishi Y Toolbox Approach to the Search for Effective Ligands for Catalytic Asymmetric Cr-Mediated Coupling Reactions. J. Am. Chem. Soc 2009, 131, 15387–15393. [DOI] [PubMed] [Google Scholar]; (h) Zhang Z; Huang J; Ma B; Kishi Y Further Improvement on Sulfonamide-Based Ligand for Catalytic Asymmetric 2-Haloallylation and Allylation. Org. Lett 2008, 10, 3073–3076. [DOI] [PubMed] [Google Scholar]; (i) Liu X; Li X; Chen Y; Hu Y; Kishi Y On Ni Catalysts for Catalytic, Asymmetric Ni/Cr-Mediated Coupling Reactions. J. Am. Chem. Soc 2012, 134, 6136–6139. [DOI] [PubMed] [Google Scholar]
- (7).For selected total syntheses, see:; (a) Armstrong RW; Beau J-M; Cheon SH; Christ WJ; Fujioka H; Ham W-H; Hawkins LD; Jin H; Kang SH; Kishi Y; Martinelli MJ; McWhorter WW Jr.; Mizuno M; Nakata M; Stutz AE; Talamas FX; Taniguchi M; Tino JA; Ueda K; Uenishi J; White JB; Yonaga M Total Synthesis of a Fully Protected Palytoxin Carboxylic Acid. J. Am. Chem. Soc 1989, 111, 7525–7530. [Google Scholar]; (b) Armstrong RW; Beau JM; Cheon SH; Christ WJ; Fujioka H; Ham W-H; Hawkins LD; Jin H; Kang SH; Kishi Y; Martinelli MJ; McWhorter WW Jr.; Mizuno M; Nakata M; Stutz AE; Talamas FX; Taniguchi M; Tino JA; Ueda K; Uenishi J; White JB; Yonaga M Total Synthesis of Palytoxin Carboxylic Acid and Palytoxin Amide. J. Am. Chem. Soc 1989, 111, 7530–7533. [Google Scholar]
- (8).(a) Chase CE; Fang FG; Lewis BM; Wilkie GD; Schnaderbeck MJ; Zhu X Process Development of Halaven®: Synthesis of the C1–C13 Fragment from D-(−)-Gulono-1,4-lactone. Synlett 2013, 24, 323–326. [Google Scholar]; (b) Austad BC; Benayoud F; Calkins TL; Campagna S; Chase CE; Choi H.-w.; Christ W; Costanzo R; Cutter J; Endo A; Fang FG; Hu Y; Lewis BM; Lewis MD; McKenna S; Noland TA; Orr JD; Pesant M; Schnaderbeck MJ; Wilkie GD; Abe T; Asai N; Asai Y; Kayano A; Kimoto Y; Komatsu Y; Kubota M; Kuroda H; Mizuno M; Nakamura T; Omae T; Ozeki N; Suzuki T; Takigawa T; Watanabe T; Yoshizawa K Process Development of Halaven®: Synthesis of the C14–C35 Fragment via Iterative Nozaki–Hiyama–Kishi Reaction–Williamson Ether Cyclization. Synlett 2013, 24, 327–332. [Google Scholar]; (c) Austad BC; Calkins TL; Chase CE; Fang FG; Horstmann TE; Hu Y; Lewis BM; Niu X; Noland TA; Orr JD; Schnaderbeck MJ; Zhang H; Asakawa N; Asai N; Chiba H; Hasebe T; Hoshino Y; Ishizuka H; Kajima T; Kayano A; Komatsu Y; Kubota M; Kuroda H; Miyazawa M; Tagami K; Watanabe T Commercial Manufacture of Halaven®: Chemoselective Transformations En Route to Structurally Complex Macrocyclic Ketones. Synlett 2013, 24, 333–337. [Google Scholar]; (d) Yu MJ; Zheng W; Seletsky BM From Micrograms to Grams: Scale-Up Synthesis of Eribulin Mesylate. Nat. Prod. Rep 2013, 30, 1158–1164. [DOI] [PubMed] [Google Scholar]; (e) Bauer A Story of Eribulin Mesylate: Development of the Longest Drug Synthesis. Top. Heterocycl. Chem 2016, 44, 209–270. [Google Scholar]
- (9).Grigg R; Putnikovic B; Urch CJ Electrochemically Driven Catalytic Pd(0)/Cr(II) Mediated Coupling of Organic Halides with Aldehydes. The Nozaki–Hiyama–Kishi Reaction. Tetrahedron Lett 1997, 38, 6307–6308. [Google Scholar]
- (10).Kuroboshi M; Tanaka M; Kishimoto S; Tanaka H; Torii S Electrochemical Regeneration of Chromium(II). Alkenylation of Carbonyl Compounds. Synlett 1999, 69–70. [Google Scholar]
- (11).(a) Durandetti M; Périchon J; Nédélec J-Y Nickel- and Chromium-Catalysed Electrochemical Coupling of Aryl Halides with Arenecarboxaldehydes. Tetrahedron Lett 1999, 40, 9009–9013. [Google Scholar]; (b) Durandetti M; Nédélec J-Y; Périchon J An Electrochemical Coupling of Organic Halide with Aldehydes, Catalytic in Chromium and Nickel Salts. The Nozaki−Hiyama−Kishi Reaction. Org. Lett 2001, 3, 2073–2076. [DOI] [PubMed] [Google Scholar]
- (12).For selected examples on the use of RAEs to form C-C bond, see:; (a) Cornella J; Edwards JT; Qin T; Kawamura S; Wang J; Pan C-M; Gianatassio R; Schmidt M; Eastgate MD; Baran PS Practical Ni-Catalyzed Aryl−Alkyl Cross-Coupling of Secondary Redox-Active Esters. J. Am. Chem. Soc 2016, 138, 2174–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A General Alkyl-Alkyl Cross-Coupling Enabled by Redox-Active Esters and Alkylzinc Reagents. Science 2016, 352, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang J; Qin T; Chen T-G; Wimmer L; Edwards JT; Cornella J; Vokits B; Shaw SA; Baran PS Nickel-Catalyzed Cross-Coupling of Redox-Active Esters with Boronic Acids. Angew. Chem., Int. Ed 2016, 55, 9676–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Toriyama F; Cornella J; Wimmer L; Chen T-G; Dixon DD; Creech G; Baran PS Redox-Active Esters in Fe-Catalyzed C−C Coupling. J. Am. Chem. Soc 2016, 138, 11132–11135. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sandfort F; O’Neill MJ; Cornella J; Wimmer L; Baran PS Alkyl−(Hetero)aryl Bond Formation via Decarboxylative Cross-Coupling: A Systematic Analysis. Angew. Chem., Int. Ed 2017, 56, 3319–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Edwards JT; Merchant RR; McClymont KS; Knouse KW; Qin T; Malins LR; Vokits B; Shaw SA; Bao D-H; Wei FL; Zhou T; Eastgate MD; Baran PS Decarboxylative Alkenylation. Nature 2017, 545, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Smith JM; Qin T; Merchant RR; Edwards JT; Malins LR; Liu Z; Che G; Shen Z; Shaw SA; Eastgate MD; Baran PS Decarboxylative Alkynylation. Angew. Chem., Int. Ed 2017, 56, 11906–11910. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wang J; Lundberg H; Asai S; Martín-Acosta P; Chen JS; Brown S; Farrell W; Dushin RG; O’Donnell CJ; Ratnayake AS; Richardson P; Liu Z; Qin T; Blackmond DG; Baran PS Kinetically Guided Radical-Based Synthesis of C(sp3)−C(sp3) Linkages on DNA. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E6404–E6410. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Chen T-G; Barton LM; Lin Y; Tsien J; Kossler D; Bastida I; Asai S; Bi C; Chen JS; Shan M; Fang H; Fang FG; Choi H.-w.; Hawkins L; Qin T; Baran PS Building C(sp3)-Rich Complexity by Combining Cycloaddition and C-C Cross-Coupling Reactions. Nature 2018, 560, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Ni S; Garrido-Castro AF; Merchant RR; deGruyter JN; Schmitt DC; Mousseau JJ; Gallego GM; Yang S; Collins MR; Qiao JX; Yeung K-S; Langley DR; Poss MA; Scola PM; Qin T; Baran PS A General Amino Acid Synthesis Enabled by Innate Radical Cross-Coupling. Angew. Chem., Int. Ed 2018, 57, 14560–14565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc 2016, 138, 5016–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Jamison CR; Overman LE Fragment Coupling with Tertiary Radicals Generated by Visible-Light Photocatalysis. Acc. Chem. Res 2016, 49, 1578–1586. [DOI] [PubMed] [Google Scholar]; (m) Liu X-G; Zhou C-J; Lin E; Han X-L; Zhang S-S; Li Q; Wang H Decarboxylative Negishi Coupling of Redox-Active Aliphatic Esters by Cobalt Catalysis. Angew. Chem., Int. Ed 2018, 57, 13096–13100. [DOI] [PubMed] [Google Scholar]; (n) Chen T-G; Zhang H; Mykhailiuk PK; Merchant RR; Smith CA; Qin T; Baran PS Quaternary Centers by Nickel-Catalyzed Cross-Coupling of Tertiary Carboxylic Acids and (Hetero)Aryl Zinc Reagents. Angew. Chem., Int. Ed 2019, 58, 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Wang J; Cary BP; Beyer PD; Gellman SH; Weix DJ Ketones from Nickel-Catalyzed Decarboxylative, Non-Symmetric Cross-Electrophile Coupling of Carboxylic Acid Esters. Angew. Chem., Int. Ed 2019, 58, 12081–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Proctor RSJ; Davis HJ; Phipps RJ Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419–422. [DOI] [PubMed] [Google Scholar]; (q) Fu M-C; Shang R; Zhao B; Wang B; Fu Y Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide. Science 2019, 363, 1429–1434. [DOI] [PubMed] [Google Scholar]
- (13).For selected electrochemical transformations of RAEs, see:; (a) Li H; Breen CP; Seo H; Jamison TF; Fang Y-Q; Bio MM Ni-Catalyzed Electrochemical Decarboxylative C−C Couplings in Batch and Continuous Flow. Org. Lett 2018, 20, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koyanagi T; Herath A; Chong A; Ratnikov M; Valiere A; Chang J; Molteni V; Loren J One-Pot Electrochemical Nickel-Catalyzed Decarboxylative Sp2 -Sp3 Cross-Coupling. Org. Lett 2019, 21, 816–820. [DOI] [PubMed] [Google Scholar]; (c) Liu Y; Xue L; Shi B; Bu F; Wang D; Lu L; Shi R; Lei A Catalyst-Free Electrochemical Decarboxylative Cross-Coupling of N-Hydroxyphthalimide Esters and N-Heteroarenes towards C(sp3)−C(sp2) Bond Formation. Chem. Commun 2019, 55, 14922–14925. [DOI] [PubMed] [Google Scholar]; (d) Niu K; Song L; Hao Y; Liu Y; Wang Q Electrochemical Decarboxylative C3 Alkylation of Quinoxalin-2(1H)-ones with N-Hydroxyphthalimide Esters. Chem. Commun 2020, 56, 11673–11676. [DOI] [PubMed] [Google Scholar]; (e) Lian F; Xu K; Meng W; Zhang H; Tan Z; Zeng C Nickel-Catalyzed Electrochemical Reductive Decarboxylative coupling of N-Hydroxyphthalimide Esters with Quinoxalinones. Chem. Commun 2019, 55, 14685–14688. [DOI] [PubMed] [Google Scholar]; (f) Chen X; Luo X; Peng X; Guo J; Zai J; Wang P Catalyst-Free Decarboxylation of Carboxylic Acids and Deoxygenation of Alcohols by Electro-Induced Radical Formation. Chem. - Eur. J 2020, 26, 3226–3230. [DOI] [PubMed] [Google Scholar]
- (14).Ni S; Padial NM; Kingston C; Vantourout JC; Schmitt DC; Edwards JT; Kruszyk MM; Merchant RR; Mykhailiuk PK; Sanchez BB; Yang S; Perry MA; Gallego GM; Mousseau JJ; Collins MR; Cherney RJ; Lebed PS; Chen JS; Qin T; Baran PS A Radical Approach to Anionic Chemistry: Synthesis of Ketones, Alcohols, and Amines. J. Am. Chem. Soc 2019, 141, 6726–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For selected reviews on reaction progress kinetic analysis, see:; (a) Blackmond DG Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem., Int. Ed 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]; (b) Mathew JS; Klussmann M; Iwamura H; Valera F; Futran A; Emanuelsson EAC; Blackmond DG Investigations of Pd-Catalyzed ArX Coupling Reactions Informed by Reaction Progress Kinetic Analysis. J. Org. Chem 2006, 71, 4711–4722. [DOI] [PubMed] [Google Scholar]; (c) Blackmond DG Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- (16).Harnying W; Kaiser A; Klein A; Berkessel A Cr/Ni-Catalyzed Vinylation of Aldehydes: A Mechanistic Study on the Catalytic Roles of Nickel and Chromium. Chem. - Eur. J 2011, 17, 4765–4773. [DOI] [PubMed] [Google Scholar]
- (17).Cannes C; Condon S; Durandetti M; Périchon J; Nédélec J-Y Nickel-Catalyzed Electrochemical Couplings of Vinyl Halides: Synthetic and Stereochemical Aspects. J. Org. Chem 2000, 65, 4575–4583. [DOI] [PubMed] [Google Scholar]
- (18).Bard AJ; Faulkner LR Electrochemical Methods Fundamentals and Applications; 2nd ed. John Wiley & Sons: Nashville, TN: 2000. [Google Scholar]
- (19).Taktak O; Souissi H; Souha K Electronic Structure and Fano Antiresonance of Chromium Cr(III) Ions in Alkali Silicate Glasses. Journal of Luminescence 2015, 161, 368–373. [Google Scholar]
- (20). We note that the [Ni(II)] has a significantly larger influence on the magnitude of the e-NHK catalytic current than the [Cr(III)] or the [alkenyl bromide]. See Supporting Information for more details.
- (21).Fano U Effects of Configuration Interaction on Intensities and Phase Shifts. Phys. Rev 1961, 124, 1866–1878. [Google Scholar]
- (22).Sturge MD; Guggenheim HJ; Pryce MHL; Antiresonance in the Optical Spectra of Transition-Metal Ions in Crystals. Phys. Rev. B 1970, 2, 2459–2471. [Google Scholar]
- (23).Lempicki A; Andrews L; Nettel SJ; McCollum BC; Solomon EI Spectroscopy of Cr3+ in Glasses: Fano Antiresonances and Vibronic “Lamb Shift”. Phys. Rev. Lett 1980, 44, 1234–1237. [Google Scholar]
- (24).Kawamata Y; Baran PS Electrosynthesis: Sustainability Is Not Enough. Joule 2020, 4, 701–704. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.