

Abstract
Background & Aims:
Clostridioides difficile toxin A (TcdA) activates the innate immune response. TcdA co-purifies with DNA. Toll like receptor 9 (TLR9) recognizes bacterial DNA to initiate inflammation. We investigated whether DNA bound to TcdA activates an inflammatory response in murine models of C difficile infection (CDI) via activation of TLR9.
Methods:
We performed studies with human colonocytes and monocytes and macrophages from wildtype and TLR9-knockout mice, incubated with TcdA or its antagonist (ODN TTAGGG), or transduced with vectors encoding TLR9 or small-interfering RNAs. Cytokine production was measured with an ELISA. We studied a transduction domain of TcdA (TcdA57–80), which was predicted by machine learning to have cell-penetrating activity and confirmed by synchrotron small-angle x-ray scattering. Intestines of CD1 mice, C57BL6J mice, and mice that express a form of TLR9 that is not activated by CpG DNA were injected with TcdA, TLR9 antagonist, or both. Enterotoxicity was estimated based on loop weight:length ratios. A TLR9 antagonist was tested in mice infected with C. difficile. We incubated human colon with an antagonist of TLR9 and measured TcdA-induced production of cytokines.
Results:
The TcdA57–80 protein transduction domain had membrane remodeling activity that allowed TcdA to enter endosomes. TcdA-bound DNA entered human colonocytes. TLR9 was required for production of cytokines by cultured cells and in human colon explants incubated with TcdA. TLR9 was required in TcdA-induced mice intestinal secretions and in the survival of mice infected by C. difficile. Even in a protease-rich environment, in which only fragments of TcdA exist, the TcdA57–80 domain organized DNA into a geometrically ordered structure that activated TLR9.
Conclusions:
Rather than inactivating DNA’s ability to binding TLR9 via binding, TcdA can in fact chaperone and organize DNA into a highly pro-inflammatory spatially periodic structure. TcdA from C difficile can bind and organize bacterial DNA to activate TLR9. TcdA and TcdA fragments remodel membranes, which allows them to access endosomes and present bacterial DNA to and activate TLR9. Rather than inactivating the ability of DNA to bind TLR9, TcdA appears to chaperone and organize DNA into an inflammatory, spatially periodic structure.
Keywords: SAXS, antibiotic-associated diarrhea and colitis, intestinal inflammation, pore formation
Introduction
Clostridioides (formerly Clostridium) difficile is an important pathogen of worldwide distribution and a frequent cause of death from severe colitis1–3. The incidence and severity of CDI have increased dramatically in Europe and North America in recent years1. The major virulence factors of C. difficile are two large protein exotoxins: toxin A (TcdA) and toxin B (TcdB), the main pathogenicity factors responsible for clinical disease. Like TcdB, TcdA glucosylates rho proteins leading to actin cytoskeletal disaggregation and cytotoxicity4. In addition to its direct cytotoxic effects, TcdA also provokes inflammatory responses including fluid secretion, immune cell influx, and tissue damage associated with clinical manifestations of CDI 5–7. TcdA contains a glucosyltransferase domain (GTD), an autocatalytic cysteine protease domain (CPD), a central transmembrane domain (TMD) and a C-terminal putative receptor binding domain (RBD) consisting of clostridial repetitive oligopeptides (CROPs)8. The GTD is known to escape from the endosome to catalyze the glycosylation of Rho-GTPases in the cytosol, leading to cytoskeletal disaggregation and cell death 4. Studies of these translocation events for both TcdA and TcdB have concentrated on the translocation domain (TD)9–15, and shown that the cholesterol content of the membrane is important16, 17. Interestingly, electrophysiology experiments have distinguished TcdA and TcdB from other pH-dependent toxins that form stable pores: both C. difficile toxins exhibit a “flickering” conductance believed to be caused by transient pore formation18. TcdA also potently activates a marked innate immune response including activation of the inflammasome, NF-κB and MAP kinases and induces a marked inflammatory response in the human colon 5, 19–21. Here we report that TcdA forms a stable complex with DNA and facilitates DNA membrane translocation. Since Toll-like receptor 9 (TLR9) recognizes intracellular CpG DNA and triggers an inflammatory response 22, we examined the role of TLR9 in CDI pathogenesis using in vitro, in vivo models and human colonic explants combined with machine learning and synchrotron X-ray diffraction structural studies.
Materials and Methods
Machine-learning based screening of TcdA for membrane-active sequences
Using a previously published Support Vector Machine (SVM) classifier from our lab23, we screened the full amino acid sequence of TcdA, from Clostridium difficile for the presence of membrane-active sequences (data not included). Candidate peptides were identified by scoring individual sequences with a variable size window. Of high scoring sequence candidates, we identified a 24 amino acid fragment in the membrane targeting N-terminal four-helix bundle, TcdA (57–80), which we refer to as TcdA57–80 here.
SAXS experiments for peptide-membrane interaction
Stock solutions of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) [sodium salt] and cholesterol (Chl) were prepared from lyophilized lipids from obtained Avanti Polar Lipids and dissolved in chloroform to a 20 mg/mL concentration. A lipid composition of DOPS/DOPE/Chl at a molar ratio of 20/70/10 was prepared from the stock solutions, and first evaporated under nitrogen before an overnight desiccation under vacuum. The remaining lipid film was resuspended to a concentration of 20 mg/mL in aqueous buffer solution at two different pH levels: 1) 140 mM NaCl + 10 mM HEPES (pH 7.4); 2) 140 mM NaCl + 10 mM sodium acetate (pH 5.0). The suspensions were incubated overnight at 37 °C. Subsequently, sonication was used to form small unilamellar vesicles (SUVs) suspensions. To select for vesicle size, the SUVs were extruded through a 0.2 μm pore Whatman nucleopore filter. The synthesized TcdA fragement, TcdA57–80, was purchased from LifeTein. The lyophilized peptide was solubilized into either the pH 7.4 or pH 5 aqueous buffer, as described above for the SUV suspensions. Solubilized peptide was mixed with the matching pH buffer SUV suspensions at specified peptide-to-lipid (P/L) charge ratio and incubated at room temperature overnight; subsequentially the samples were loaded and hermetically sealed into 1.5 mm glass capillaries (Hilgerberg GmbH, Mark-tubes). SAXS experiments were carried out at Stanford Synchrotron Radiation Lightsource (BL 4–2) with monochromatic X-rays of energy 9 keV. Samples were incubated at 37 °C and centrifuged before measurement. The scattering signature of the samples were collected using a Pilatus3 X 1M detector (pixel size 172 μm). The resulting 2D SAXS power patterns were integrated using Nika 1.5024 package for Igor Pro 6.31 and FIT2D25.
The integrated SAXS intensities I(q) vs.q were plotted using MATLAB software. The correlation peaks corresponding to each characteristic phase (lamellar, hexagonal, and cubic) were indexed according to the characteristic peak ratios permitted for each phase: Lamellar index ratios, 1:2:3; hexagonal index ratios, √1:√3:√4:√7:√9:√11:√12; and cubic (Pn3m) index ratios, √2:√3:√4. The corresponding measured Bragg peaks positions, qmeasured, were used to fit a linear regression through the scatter plot of qmeasured against their respective Miller indices. For hexagonal phases, this corresponds to fitting equation q = (4π/(a√3)) √(h2 + hk +k2) for the qmeasured vs. √(h2 + hk + k2) plot, and solving for the slope to obtain the lattice parameter a. Similarly, the cubic Pn3m phase is fitted to equation q = (2π/a) √(h2 + hk + l2) for the qmeasured vs. √(h2 + k2+ l2) plot, and solving for a. Finally, the negative Gaussian curvature present in the cubic phase was calculated using the equation <K> = 2πχ/A0a2, where χ is the Euler characteristic and A0 is the surface area per cubic unit cell for each phase. Parameter values are χ = −2 and A0 = 1.919 for Pn3m cubic phases.
Cell lines and TcdA labeling
Human intestinal cell lines HT-29 and THP-1 human monocyte cells were purchased from ATCC. Wild type and TLR9 knock-out mouse macrophage cell lines were collected as we previously described 26 and HEK293 wild type cell line were acquired from BEI Resources. TcdA was purified from C. difficile strain VPI 10463 (ATCC, Maryland, USA). Culture supernatant was fractionated by anion-exchange chromatography, and TcdA was isolated by precipitation in acetate buffer as described previously 27. TcdA was labeled using an Alexa Fluor 555 Protein Labeling kit (Molecular Probes, Inc.) according to the manufacturer’s protocol. Briefly, TcdA in TE buffer was eluted through a PD-10 column prior equilibrated with PBS to remove Tris-HCL buffer, and 0.5ml of TcdA (1.7mg) in PBS was mixed with 50μl 1M sodium bicarbonate and 1 vial of reactive dye. The mixture was stirred at room temperature for 1 hour and unlabeled free dye was removed by resin column chromatography.
DNA uptake assay
HT-29 cells were trypsinized and cell density was adjusted to 5×105 cell/ml in the complete DMEM medium, seeded on a chamber slide, and incubated for 24 hours. Bacterial source Label IT FITC Plasmid (Mirus Bio LLC, Wisconsin) was incubated with TcdA or TE buffer at room temperature for 30 minutes and diluted in DMEM to 1μg/mL plasmid with or without 100nM TcdA-Alexa 555. Cells were treated with DMEM (control), plasmid only and plasmid-TcdA mixture at 37°C for 2 hours in a 5% CO2 incubator. At the end of incubation, supernatant was aspirated and cells were washed with PBS twice, and fixed with CytoFix (BD) at 4°C for 15 minutes. After washing with PBS twice the slide was mounted with ProLong Gold antifade reagent with DAPI (Molecular Probes). The DNA uptake was observed by confocal microscopy.
Inhibition of TLR9 by its antagonist ODN TTAGGG
HT-29 cells were trypsinized, cell density was adjusted to 2×106 cell/ml in complete DMEM medium, cells seeded in a 96-well plate, and incubated for 24 hours. The medium was replaced by serum free DMEM for 12 hours after which the medium was changed again to fresh serum free medium containing TcdA, ODN TTAGGG or their mixtures for 14 hours. THP-1 cells were maintained in complete RPMI-1640 medium. Prior to experiments, cells were harvested, re-suspended in serum free RPMI-1640 medium at 7×105 cells per ml and incubated for 12 hours. Cells were then dispensed in a 96-well plate at 1×106 cells per well, and treated with TcdA, ODN TTAGGG or their mixtures diluted in serum free RPMI-1640 medium for 4 hours. At the end of treatment, the supernatants were collected for measuring IL-8 concentrations or for cell viability assay.
Transient inhibition of TLR9 with siRNA
HT-29 cells were reverse-transfected with 50nM TLR9 siRNA according to the manufacturer’s instructions. Briefly, Silencer® Select TLR9 siRNA (Sense: 5’-CUGGAAGAGCUAAACCUGATT-3’, Antisense: 5’-UCAGGUUUAGCUCUUCCAGGG-3’) or Silencer® Select Negative Control siRNA (Ambion, Inc.) were mixed with Lipofectamine RNAiMAX, and incubated for 30 minutes at room temperature. Then, in a 24-well plate, each 100μl of the complexes was mixed with 600μl HT-29 cell suspension (4×105cell/mL) to reach 50nM siRNA concentration and cells were incubated for 48 hours before treatment.
Transfection of TLR9 plasmids with an IL-8 promoter and luciferase assay
TLR9 wild type (WT), two mutants (Del_Ins2 and K51M) and pcDNA3.1 vector control plasmids are acquired from Dr. Alexander Dalpke, University of Heidelberg. To determine the activation of NF-κB/IL-8 in response to TLR9 recognition by TcdA, HEK293 wild type cells that do not express TLR9 were transfected with wild type TLR9 plasmids, each of the two TLR9 mutants, or pcDNA3.1 as a control using Lipofectamine LTX (Life Technologies, Inc.) according to the manufacturer’s instructions. Briefly, 0.75×105 cells in 0.5ml complete growth medium were seeded in a 24-well plate for 24 hours. Cells in each well were then transfected with 0.5μg TLR9 plasmid or vector, along with 0.1μg NF-κB / IL-8 for 48 hours. Then the transfected cells were serum-starved for 24 hours followed by exposure to TcdA (10nM) for 4 hours. A dual-luciferase reporter assay kit (Promega, Wisconsin) was used to measure firefly luciferase activities in cell lysate. The relative luciferase activity was then calculated by normalizing the NF-κB/IL-8 promoter-driven firefly luciferase activity of cells transfected with TLR9 plasmids against that of control cells transfected with vector.
SAXS experiments for DNA-peptide complex
A stock solution of double-stranded DNA was prepared from Escherichia coli genomic DNA (Thermo Scientific) was precipitated and resuspended in aqueous solution of 140 mM NaCL + 10 mM HEPES (pH 7.4) buffer to a concentration of 5mg/mL; the lyophilized TcdA57–80 peptide (LifeTein) was solubilized using the pH 7.4 buffer to stock concentration of 10 mg/mL. The DNA-peptide complex was formed by mixing the peptide with the DNA to the desired peptide-to-DNA (P/DNA) charge ratio. The complex was thoroughly mixed overnight and incubated at room temperature. After equilibration, the complex was transferred to a 1.5 mm quatz capillary (Hilgerberg GmbH, Mark-tubes) and hermetically sealed. SAXS measurements were perfomed at the Stanford Synchrotron Radiation Lightsource (SSRL, beamline 4–2) using monochromatic X-rays with an energy of 9 keV. The scattered X-rays were measured using a Pilatus3 X 1M detector (pixel size 172 μm). The collected 2D powder diffraction patterns were radially integrated using the Nika 1.5024 package for Igor Pro 6.31 and FIT2D25. The output curves were visualized using MATLAB software. The structure of the TcdA57–80-DNA complex was solved by calculating ratios between the measured q position, qmeasured, of the peaks; the observed peaks closely corresponded to √1:√3, which are index ratios permitted by a hexagonal columnar lattice. The lattice parameter of inter-DNA spacing, d, was calculated using a linear regression to fit equation q = (4π/(d√3)) √(h2 + hk +k2). The fit is calculated from points corresponding to qmeasured and √(h2 + hk + k2) for the Miller indices (h, k).
Experimental animals
Female CD1, C57BL/6 wild type and TLR9 mutant (C57BL/6J-Tlr9M7Btlr/Mmjax) mice were purchased from Jackson Laboratory (Bar Harbor, ME) at 8 or 12 weeks old. Mice were housed and handled according to the protocol approved by the Beth Israel Deaconess Medical Center’s institutional animal care and use committee.
Mouse ileal loop assay of TcdA enterotoxicity
Female CD1 mice of 12 weeks old were divided into four groups of 6. Each mouse was anesthetized and a closed, distal ileal loop (about 3cm in length) formed at laparotomy. The loop lumen was injected with 10μg of TcdA in 100μL with or without ODN TTAGGG (50μM) diluted in DMEM medium. ODN TTAGGG (50μM) or medium were used as controls. The closed ileal loops were harvested and their weight to length ratios determined as a measure of enterotoxicity as previously described20. The ileal loop model was also used to compare TcdA-induced enterotoxicity in C57BL/6 wild type and in TLR9 mutant female mice. Wildtype and mutant mice were cohoused for a week before ileum loop experiment. The loop lumen of the wild type and of the TLR9 mutant mice was injected with 10μg of TcdA in DMEM medium or with medium alone and loop weight-to-length ratios were determined after 4 hours (n=9 per group).
Antibiotic-associated CDI Mouse Model
The mouse antibiotic-associated CDI model was employed as previously reported 28. Briefly, 8 week old, female, C57BL6 mice (from Jackson Laboratory) were divided into two groups (n=10 per group). Mice were challenged with 1×105 cfu C. difficile (strain VPI 10463) by gavage. E6446 (60mg/kg/d) was given by gavage for five days starting on the same day of clindamycin administration. The control group received autoclaved water.
Ex vivo human colonic mucosal biopsy culture and cytokine measurement
Human colonic mucosa was obtained by forceps biopsy from the distal colon of healthy volunteers undergoing screening colonoscopy according to a protocol approved by the Committee on Clinical Investigation of Beth Israel Deaconess Medical Center. Biopsies were washed with ice-cold plain medium and immediately transferred to a 24-well plate. Each biopsy well contained 0.5ml RPMI-1640 with 100U/mL penicillin and 100μg/mL streptomycin that contained TcdA (1 or 10μg/mL) with or without ODN TTAGGG (10μM). The cultures were maintained in 95% O2 and 5% CO2 at 37°C for 24 hours. The supernatants were then harvested for cytokine measurements using Milliplex Map human cytokine/Chemokine Magnetic Bead Panel assays that measured 14 inflammatory cytokines simultaneously (i.e. IFNα2, IFNγ, IL-10, IL-1RA, IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-8, MCP-1, RANTES, TNFα and TNFβ).
Statistical analysis
Data are presented as the mean ± SEM (unless otherwise specified) of at least two individual experiments. Comparisons between two groups were analyzed using the Student’s t-test. ANOVA with post Newman-Keuls multiple comparison test was used for comparison of multiple groups. Survival results were analyzed using a Gehan-Breslow-Wilcoxon Test. A P value of less than 0.05 was considered to be statistically significant.
Results
C. difficile TcdA binds DNA and facilitates cellular entry
Upon purification of TcdA from C. difficile, we found that DNA is copurified with the toxin at a 0.06±0.02% DNA concentration (0.6μg DNA per mg toxin protein). The presence of DNA was observed in both native and recombinant toxins from multiple academic and commercial sources. DNA was extracted, followed by PCR and sequencing, and identified to be C. difficile genomic DNA and other contaminating DNAs. To directly examine the DNA binding ability of TcdA, DNA fragments with various sizes (1kb DNA ladder) were incubated with TcdA, which markedly lowered DNA marker mobility in gel electrophoresis (Figure 1A). Moreover, digestion by DNase I only leads to a 12% decrease in DNA suggesting a stable protein-DNA complex that resists DNase digestion. This indicates a capacity for stable complex formation between DNA and TcdA (or fragments thereof) that may protect DNA from enzymatic degradation.
Figure 1. C. difficile TcdA binds DNA and facilitates DNA’s cellular entry (A-E).

A) TcdA binds to DNA. Gel electrophoresis showing that the incubation of TcdA (10μg) with a 1kb DNA ladder (2μg) retarded DNA mobility. Incubation of DNA with BSA (10μg) acted as a negative control.
B) Four-helix bundle in the glucosyltransferase domain GTD. Solved 3D crystal structure of the N-terminal glucosyltransferase domain from TcdA (PDB ID: 4DMV), highlighting the position of the membrane targeting N-terminal four-helix bundle motif (yellow).
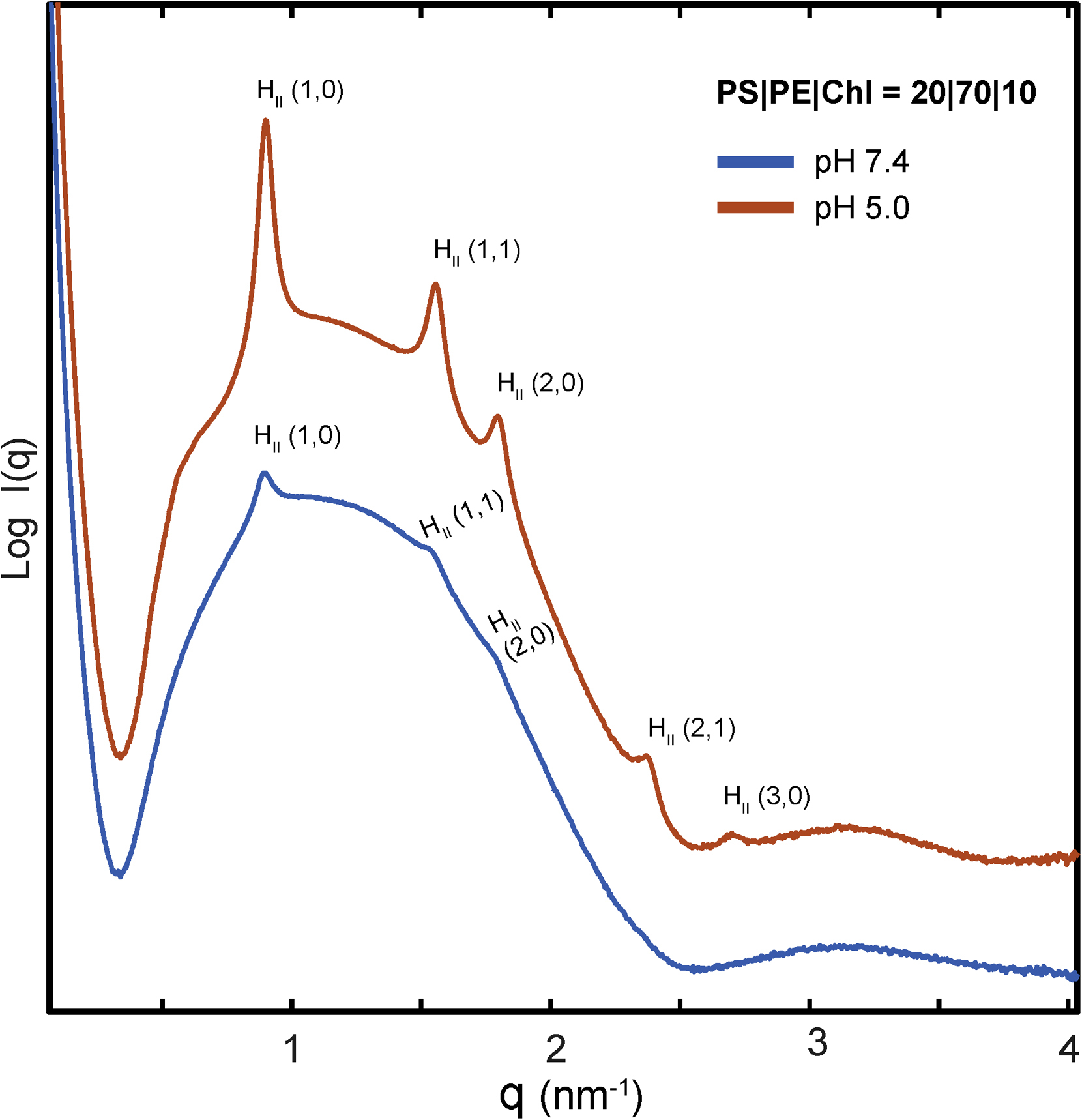
C) TcdA four-helix bundle segment, TcdA57–80, generates NGC necessary for membrane permeabilization. SAXS spectra from 20/70/10 PS/PE/Chl model membranes incubated with TcdA57–80, a 24 amino acids fragment in the TcdA N-terminal four-helix bundle, at a 1:4 peptide-to-lipid charge ratio. At both pH 7.4 and pH 5.0, TcdA57–80 induces NGC in the form of a Pn3m QII cubic phase (insert plot). The additional observed reflections for the positive curvature hexagonal (red indices) and lamellar (green indices) phases have been assigned on the curves, with periodicity of 8.0 and 5.7nm at pH 5, and 8.1 and 6.0nm at pH 7.4, respectively. No Bragg reflections were observed for the peptide solutions alone (bottom). Only hexagonal phases were for the membrane SUVs suspensions at both pHs (Supplementary Figure S1). To facilitate visualization, spectra have been manually offset vertically by a multiplicative factor.
D) Reconstruction of a Pn3m surface. 3D reconstruction depicting a Pn3m QII cubic phase, with continuous surfaces forming NGC at every point.
E) Illustration of NGC geometry. 3D rendering of the saddle-splay inherent to negative Gaussian curvature geometry (green) as superimposed on a mechanism of membrane remodeling.
F) TcdA facilitates cellular uptake of DNA. HT29 colonic epithelial cells were treated with medium (negative) or DNA-FITC (1μg/mL, green) alone or along with Alexa 555 labeled TcdA (100nM, red) for 2 hours at 37°C. Cells were fixed with Cyto-Fix at 4°C for 15 minutes and nuclei were stained with DAPI (blue). Significantly increased plasmid DNA was observed in the cytosol of TcdA-treated colonocytes (bottom right panel).
TcdA is hypothesized to undergo receptor-mediated endocytosis8, although its specific surface receptor(s) and cell entry mechanism remain heretofore elusive 29. Therefore, to systematically discover potential parallel pathways of translocation, we analyzed the entire TcdA protein sequence using a recently developed machine learning classifier23, 30, 31. Surprisingly, although recent empirical work has identified cell penetrating peptides (CPPs) like domains along the TD32, 33, the classifier identified multiple new regions of TcdA that are expected to have direct membrane remodeling activity reminiscent of CPPs, including regions far from the TD. These results suggested that TcdA is unusual in that it possesses multiple CPP domains that facilitate membrane translocation (such as that involved in cell entry and endosomal egress). We examined experimentally a 24aa peptide fragment (TcdA57–80) in the N-terminal four-helix bundle of the glycosyl-transferase domain (Figure 1B) as it has been previously described to be membrane targeting 34, 35. To investigate membrane deformations induced by TcdA57–80, we incubated the fragment with small unilamellar vesicles (SUVs) prepared from ternary phospholipid mixtures of phosphatidylserine (PS), phosphatidylethanolamine (PE), and cholesterol (Chl) at a stoichiometry of PS/PE/Chl=20/70/10, and examined the resultant peptide-membrane structure using Small Angle X-ray Scattering (SAXS) (Figure 1C). Interestingly, in addition to lamellar and inverted hexagonal phases, we observed the formation of a Pn3m bicontinuous cubic (QII) phase at both neutral pH 7.4 and at acidic pH 5, with lattice parameters of 17.87 and 21.33 nm, respectively (Figure 1C, D). This observed peptide-induced membrane remodeling is homologous to that observed for HIV TAT, the canonical CPP36: The measured Pn3m structure is rich in negative Gaussian curvature (NGC), the type of curvature that is topologically required in membrane permeation events, such as pore formation, blebbing, budding, and endocytosis (Figure 1E). This result indicates that this domain of TcdA, together with other previously reported CPPs of TcdA11 are able to mediate translocation across membranes.
To test this idea further, we examined whether TcdA facilitates cellular uptake of DNA into human colonocytes. We co-treated HT29 colonic epithelial cells with TcdA (Alexa 555-labeled) and/or bacterial plasmid DNA (FITC labeled). Confocal imaging demonstrated significantly increased DNA in the cytosol of TcdA-treated colonocytes, indicating that TcdA can facilitate DNA internalization (Figure 1F), consistent with x-ray (SAXS) and machine learning results.
TLR9 mediates TcdA-induced proinflammatory response through DNA-recognition
As it is known that TLR9 recognizes bacterial or viral DNA and leads to pro-inflammatory reactions including the production of proinflammatory cytokines 22, we examined whether cellular uptake of DNA in the presence of C. difficile TcdA results in stimulation of TLR9 pathway-associated cytokine production. We tested whether TLR9 is important in TcdA-induced cytokine production in vitro by using the TLR9 antagonist ODN TTAGGG. In THP-1 (human monocytic) cells, TcdA-induced IL-8 production was significantly suppressed by ODN TTAGGG (Figure 2A). ODN TTAGGG also inhibited TcdA-induced IL-8 production in HT29 (human colonic epithelial) cells (supplementary Figure S2). Conversely, in our control experiments, ODN TTAGGG did not inhibit IL-8 productions induced by IL-1β or TNF-α which do not act through TLR9 activation (supplementary Figure S3). We next transiently inhibited TLR9 expression in HT29 cells by transfecting the cells with an anti-TLR9 siRNA, which also significantly reduced TcdA-induced IL-8 production, compared to mock control (Figure 2B).
Figure 2. TLR9 mediates TcdA-induced proinflammatory response through DNA-recognition (A-D).

A) Antagonizing TLR9 attenuates TcdA-induced IL-8 production. THP-1 monocytes were treated with medium as a control (ctrl), 25μM ODN TTAGGG (ODN), TcdA (100nM) or TcdA together with ODN (TcdA+ODN) for 4 hours. IL-8 production was measured by ELISA. ** denotes p<0.01, Student’s t-test
B) Silencing TLR9 translation attenuates TcdA-induced IL-8 production. HT-29 cells were transfected with TLR9 siRNA (50nM) or negative control (mock) for 48 hours and then treated with TcdA for 14 hours. * denotes p<0.05, Student’s t-test
C) TLR9 knock-out mouse macrophages have less cell death in response to TcdA. Wild type (Mf-WT) or TLR9 knock-out (Mf-TLR9KO) mouse macrophages were treated with TcdA (0, 1 or 10 nM) for 24 hours. Loss of cellular viability was determined by LDH release. * denotes p<0.05, Student’s t-test
D) Wild type TLR9 overexpression results in increased NF-κB/IL-8 activation in HEK293 (TLR9 null) cells. This increase is abolished in cells transfected with TLR9 mutants that lack a functional DNA-recognition domain. HEK293 cells were transfected with vector (pcDNA3.1), TLR9 wild type (WT), or TLR9 mutants with either a deletion (Del_Ins2) or single nucleotide substitution mutation (K51M) that result in loss of DNA binding. After 48 hours cells were treated with TcdA (10nM) for 4 hours. The activation of NF-κB/IL-8 was measured by a luciferase reporter and data presented as normalized ratios. ** denotes p<0.01, Student’s t-test
To further validate the role of TLR9 in TcdA-induced cellular responses, we compared TLR9 knock-out (TLR9KO) mouse macrophages with wild type control. It has been reported that TLR9 activation may lead cells to undergo apoptosis, necrosis and pyroptosis37–39, but its role in TcdA-induced cell death is not known. We found that TLR9 knock-out macrophages were more resistant to TcdA-induced cell death as compared to wild type cells (Figure 2C).
To further examine the role of TLR9 in TcdA-induced cytokine production, we transfected HEK-293 (TLR9 null) cells with human TLR9. Overexpression of wild type TLR9 significantly increased NF-κB/IL-8 luciferase reporter signal in response to TcdA (Figure 2D). In contrast, transfection with either of two TLR9 mutants (Del_Ins2 or K51M) that cannot bind CpG DNAs 40 was associated with loss of the activation effect (Figure 2D). These findings indicate that a functioning TLR9 is essential for the signaling response induced by TcdA.
TLR9 plays a pivotal role in TcdA-induced enterotoxicity in vivo mouse models
To examine the role of TLR9 in TcdA-induced enterotoxicity in vivo, we injected the toxin into mouse ileal loops with or without the TLR9 antagonist ODN TTAGGG (Figure 3A). ODN TTAGGG significantly attenuated TcdA-induced fluid secretion (enterotoxicity), indicating chemical inhibition of TLR9 blocks in vivo response to TcdA in mouse intestine.
Figure 3. TLR9 plays a pivotal role in two in vivo CDI mouse models (A, B, C).

A) The TLR9 antagonist ODN TTAGGG inhibited C. difficile TcdA-mediated enterotoxicity in mouse ileum in vivo. CD1 mice were anesthetized and closed distal ileal loops created at laparotomy. Loops were injected with 100μL of DMEM medium alone (Ctrl) or DMEM containing ODN TTAGGG (50μM, ODN), TcdA (100μg/mL, TcdA) or TcdA (100μg/mL) plus ODN TTAGGG (50μM). Four hours after the injection, fluid secretion, an indicator of enterotoxicity, was estimated by determining loop weight-to-length ratios. Data are from a representative of three individual experiments and presented as median with 10th, 25th, 75th and 90th ranges (N=6 per group, * denotes p<0.05; NS denotes p>0.05, One-way ANOVA with multiple comparisons).
B) TLR9 mutant mice show reduced C. difficile TcdA-mediated enterotoxicity in mouse ileum in vivo. Wild type C57BL6J (WT) and mutant C57BL/6J-Tlr9M7Btlr/Mmjax (TLR9mu; expressing TLR9 that is not activated by CpG DNA) were compared using mouse ileal model of CDI. As described above in 3A, the enterotoxicity of TcdA (10μg) was assessed by loop weight-to-length ratios. Data were pooled from three independent experiments and presented as median with 10th, 25th, 75th and 90th ranges. * denotes p<0.05, ** denotes p<0.01, N=9 each group, One-way ANOVA with multiple comparisons
C) The orally active TLR9 antagonist E6446 protected mice from antibiotic-associated CDI in vivo. The murine model of antibiotic-associated CDI was employed as previously described. C57BL6 female mice received E6446 (60mg/Kg/day orally) starting one day prior to C. difficile challenge and for an additional 4 days or until death/sacrifice. Survival was recorded every 8 hours after challenge. Compared to vehicle, E6446 treatment significantly attenuated CDI in mice. * denotes p<0.05 vs. vehicle. N=10 each group. Kaplan-Meir Analysis.
In comparison to wild type mice, mice with a single nucleotide substitution that precludes TLR9 activation by CpG DNA (C57BL/6J-Tlr9M7Btlr/Mmjax, MGI Ref ID J:165701) show significantly less TcdA-induced enterotoxicity (Figure 3B), indicating that the DNA binding domain of TLR9 contributes to, at least partially, TcdA’s enterotoxicity in mouse ileum. For additional in vivo experiments we examined the role of TLR9 activation in the infectious model of antibiotic-associated CDI 28. Treatment with E6446, an orally stable TLR9 antagonist41, significantly protected mice from CDI-induced death (Kaplan-Meir Analysis: p<0.05 treated vs. control, Figure 3C), providing a 3rd line of evidence for TLR9’s importance in CDI in vivo.
TLR9 inhibition protects human colonic mucosa from TcdA-induced proinflammatory cytokine production.
Using ex vivo culture of freshly harvested human colonic mucosal biopsies, we examined the effects of the TLR9 antagonist ODN TTAGGG on TcdA-induced cytokine production. TcdA increased the concentrations of multiple pro-inflammatory cytokines in the conditioned media. The TLR9 antagonist significantly attenuated TcdA-induced proinflammatory cytokine production by the human colonic mucosal biopsies. IFNα2, a known product of TLR9 activation, was increased 2.1 fold by TcdA and this increase was prevented by antagonism of TLR9 (p<0.01). Similar inhibitory effects were also observed for other key cytokines in CDI such as TNF-α (p<0.01), TNF-β (p<0.05), MCP-1 (p<0.01) and RANTES (p<0.01) (Figure 4A). These data indicate that TLR9 activation mediates innate immune responses to C. difficile TcdA in human colon.
Figure 4. TLR9 inhibition protects human colonic mucosa from TcdA-induced proinflammatory cytokine induction while TcdA57–80 fragment organized DNA into optimal lattice spacing for TLR9 activation (A, B, C).

A) Human colonic biopsies were treated with TcdA (3nM) for 24 hours with or without ODNTTAGGG (10nM). The cytokines/chemokines released into the conditioned media were measured by immunoassay. Data were represented as mean ± SEM * denotes p<0.05; ** denotes p<0.01, NS: Not Significant;Student’s t-test.
B) TcdA57–80 fragment organizes DNA into a nanocrystalline hexagonal columnar lattice. SAXS curve of TcdA57–80 complexed with DNA (red); the formed structure generated Bragg peaks corresponding to a hexagonal lattice. These reflections are not observed from the TcdA57–80peptide alone (green), or DNA in solution by itself (violet). All samples were tested at pH 7.4. To facilitate visualization, spectra have been manually offset vertically by a multiplicative factor.
C) Diagram of a DNA hexagonal lattice presented to an array of TLR9 receptors. Illustration of the proposed DNA 3D hexagonal columnar structure organized by TcdA57–80 for TLR9 presentation.
Immune activation in the extreme case of protease-rich environments that degrade TcdA into fragments
An interesting question to consider for gut inflammation is what happens in protease-rich environments such as the colonic lumen where TcdA is partially digested into peptide fragments. We showed above that TcdA has its own protein transduction domain in addition to other CPP domains. Consistent with CPP behavior, isolated CPPs or protein transduction domains can facilitate membrane remodeling and endosomal access. Recent work has shown that it is possible for cationic amphiphilic peptides to co-assemble with dsDNA into ordered nanocrystalline complexes that drastically amplify TLR9 activation42–44. Examples include membrane-active innate immune peptides that organize into four-helix bundles44, bacterial amyloids43, and chemokines44, which all can organize and present dsDNA ligands at geometric spacings that drive TLR9 receptor clustering and multivalent binding. To mimic the protease-rich environment of the gut, we do not assume the long-term persistence of any tertiary or higher-level structure (such as the N-terminal four-helix bundle). Hence, we examined the TcdA protein transduction domain fragment described above that has been shown to have the capacity for endosomal access according to our experiments (see Figure 1 and Results above). We incubated genomic E. coli double-stranded DNA with TcdA57–80 and observed the formation of an ordered TcdA57–80-DNA complex. SAXS measurement of the complex produced Bragg reflections at q100 = 1.765 and q110 = 3.068 nm−1, indicating the formation of a columnar hexagonal DNA lattice with an inter-DNA spacing of 4.1nm (Figure 4B,C), which is in the range of optimal values that has been shown to amplify TLR9 activation in a deterministic manner: In other words, the TcdA57–80-DNA complex presents parallel DNA at an inter-DNA spacing that promotes binding of multiple TLR9s. Therefore, we find that even fragments of TcdA have the ability to enter endosomal compartments and optimally arrange DNA for TLR9 presentation and trigger a proinflammatory response.
Discussion
In this report, we demonstrate that C. difficile TcdA binds to DNA, facilitates DNA entry into human colonocytes, and organizes DNA for amplified TLR9-mediated immune activation. We find multiple cell penetrating peptide motifs in TcdA that can enhance endosomal access for the toxin. TLR9 signaling was required in TcdA-induced inflammatory responses in vitro and in vivo, and in human colonic muscosa. Finally, in the extreme case of TcdA digestion into peptide fragments in the protease-rich colonic luminal environment, we find that the TcdA57–80 protein transduction fragment capable of CPP-like membrane translocation activity can also organize bacterial DNA into a nanocrystalline complex that presents DNA ligands in a multivalent manner and thereby amplifies TLR9 activation.
We have identified putative DNA binding motifs dispersed across TcdA. Moreover, the binding to DNA appears to be electrostatic in origin and therefore quite general, since C. difficile genomic DNA, bacterial plasmid DNA and bacteriophage DNA (DNA ladders) can all bind to cationic motifs in TcdA, which is already known to bind negatively charged polymers 45. Toxins and extracellular DNA along with polysaccharides are observed as major components in the biofilm matrix of C. difficile46.
The two TcdA activities that we highlight here, the ability to remodel membranes for permeation and the ability to chaperone and organize DNA for amplified TLR9 activation, are both activities identified for antimicrobial peptides (AMP)42, 47–49. These observations suggest that TcdA is capable of mimicking components of the innate immune system to amplify inflammation.
It is indeed interesting to explore why C. difficile induces inflammation in the gut via the mechanisms described here. Although the answers for C. difficile are not known, recent studies suggest ways in which pathogens may capitalize on gut inflammation. In the case of S. Typhimurium, innate immune responses lead to production of reactive oxygen and nitrogen species. These in turn convert endogenous sulfur compounds to new respiratory electron acceptors that give S. Typhimurium a growth advantage over fermenting microbial species50. C. difficile is anaerobic, and uses different mechanisms. Recent work indicates that antibiotic-mediated disruption of native microbiota allows C. difficile to take advantage of microbiota-liberated mucosal carbohydrates51. Cognate effects from inflammation may be possible. Reduction in microbial diversity will likely also impact C. difficile’s fitness in the local ecosystem. In a larger compass, there are other cognate examples where DNA forms a cell penetrating complex that mediates pro-inflammatory outcomes. Hemozoin, an inorganic crystal produced by the protozoan parasite Plasmodium, is coated with malaria DNA that it presents to activate TLR9-mediated immune responses26. Another example is granulin, a mitogen and neurotrophic mammalian protein, which has been reported to deliver CpG-DNA to the endosome and potentiate TLR9 responses52. C. difficile TcdA, to our knowledge, is the first known major bacterial toxin that leads to DNA delivery into host cells to stimulate immune responses through TLR9. Given its widespread and promiscuous DNA binding motifs, it is conceivable that TcdA can also bind to colonic luminal DNA from the intestinal microbiota. Once the colonic epithelial barrier is disrupted by toxin, microbiota-derived DNA can be exposed to subepithelial cells (e.g. monocytes and macrophages) resulting in a robust inflammatory reaction driven by TLR9 signaling.
The observation that even fragments of TcdA (TcdA57–80) can function independently showing synergistic CPP and pro-inflammatory activities is interesting and highly suggestive of TcdA retaining these effects despite digestion by colonic luminal bacterial peptidases. Clearly, TcdA–DNA binding may not interfere with TLR9 activation; it can even amplify as shown by recent examples42, 44. Results here suggest that in addition to TcdA-DNA induced TLR9 activation, even TcdA fragments in the colonic lumen can bind and organize bacterial DNA into ordered complexes to activate DNA-dependent, TLR9-dependant, innate inflammatory responses. This TLR9-dependant pathway is apparently robust in the protease-rich environment of the gut, and exists in addition to the toxin’s known rho glucosylation and auto-protease enzymatic activities and inflammasome activation 19, 21, 53. Indeed, it is tempting to hypothesize that there are likely other TcdA fragments with similar composite activity; by comparison, there are many AMPs with cognate ability to organize DNA and activate TLR944. In recent studies, the regulation of proteases (from both the host and microflora) has been linked to pathogenesis of gastrointestinal autoimmune and inflammatory diseases54, 55. Moreover, due to the rich diversity of proteases in the gut, we hypothesize that there exists a range of pro-inflammatory mechanisms cognate to the one that we have identified, which involve entry of some toxin derivative or fragment with DNA.
The findings here introduce TcdA-DNA binding and TLR9 activation as novel pathogenic events and potential treatment targets, in Clostridioides difficile infection. Our data unambiguously demonstrate the impact of TLR9 signaling on TcdA-mediated damage and inflammation in vitro, in the mouse gut in vivo and in human colon biopsies. It is therefore interesting to consider the source of DNA used for pathological TLR9 activation. Given the abundance of bacterial DNAs in gut lumen and the promiscuous nature of electrostatic binding, it is likely that the gut commensal bacterial DNA, facilitated by CPP activity of toxin fragments, may enter gut epithelial cells, macrophages and monocytes, activate TLR9 signaling and therefore exacerbate further the inflammatory responses initiated by toxin’s enzymatically mediated, intrinsic, enterotoxicity and cytotoxicity leading to damage to the gut barrier. Recent work, however, indicates that even mammalian DNA organized in nanocrystalline complexes similar to those observed here can also activate TLR9 and this phenomenon has been implicated in autoimmune diseases42–44. This suggests that endogenous DNA from damaged epithelial cells can also contribute to TLR9 activation.
Supplementary Material
{kind=link}
{kind=link}
What you need to know:
Background and Context:
Clostridioides difficile toxin A (TcdA) activates the innate immune response. TcdA co-purifies with DNA. DNA binding to toll like receptor 9 (TLR9) activates the inflammatory response. This study investigated this pathway during C difficile infection (CDI) of mice.
New Findings:
TcdA and TcdA fragments remodel membranes, which allows them to access endosomes. TcdA fragments bound to and organized DNA for amplified activation of TLR9.
Limitations:
This study was performed in cell lines, human tissues, and mice; further studies are needed in humans.
Impact:
Rather than preventing DNA from binding TLR9, TcdA chaperones and organizes DNA into an inflammatory, spatially periodic structure.
Lay Summary:
This study identified a mechanism by which a bacterial toxin activates an inflammatory response in the intestine.
Acknowledgments
XC, XY, JdA. GCLW and CPK are involved study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; CPK, GCLW and XC obtained funding and provided study supervision; KSS, JH, IJP, HX, EUY, JH, HX provided technical support. MAG, MD, DL and JdA were involved in acquisition of data. DTG provided material support and study design.
We want to thank Dr. Alexander Dalpke from University of Heidelberg for his generous gifts of TLR9 mutant constructs. This work was supported by Irving W. and Charlotte F. Rabb Award (to XC), Crohn’s & Colitis Foundation of America (to XC), Young Investigator Award for Probiotic Research (to XC), NIH NIAID RO1 AI095256, R01 AI116596 (to CPK), NIH RO1 AI143730, NIH RO1 AI052453, NSF DMR1808459 (to GCLW), NSF GRFP DGE-1650604 (to JdA). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Footnotes
No conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kelly CP, LaMont JT. Clostridium difficile--more difficult than ever. N Engl J Med 2008;359:1932–40. [DOI] [PubMed] [Google Scholar]
- 2.Chen X, Lamont JT. Overview of Clostridium difficile infection: implications for China. Gastroenterol Rep (Oxf) 2013;1:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burke KE, Lamont JT. Clostridium difficile infection: a worldwide disease. Gut Liver 2014;8:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Just I, Selzer J, Wilm M, et al. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995;375:500–3. [DOI] [PubMed] [Google Scholar]
- 5.Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol 2011;60:1070–9. [DOI] [PubMed] [Google Scholar]
- 6.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 2005;18:247–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vedantam G, Clark A, Chu M, et al. Clostridium difficile infection: toxins and non-toxin virulence factors, and their contributions to disease establishment and host response. Gut Microbes 2012;3:121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jank T, Aktories K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 2008;16:222–9. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Wang H, Gu H, et al. Identification of an Essential Region for Translocation of Clostridium difficile Toxin B. Toxins (Basel) 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Park M, Tam J, et al. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc Natl Acad Sci U S A 2014;111:3721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larabee JL, Hauck GD, Ballard JD. Cell-penetrating peptides derived from Clostridium difficile TcdB2 and a related large clostridial toxin. The Journal of biological chemistry 2018;293:1810–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larabee JL, Bland SJ, Hunt JJ, et al. Intrinsic Toxin-Derived Peptides Destabilize and Inactivate Clostridium difficile TcdB. mBio 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamza T, Zhang Z, Melnyk RA, et al. Defective mutations within the translocation domain of Clostridium difficile toxin B impair disease pathogenesis. Pathog Dis 2016;74:ftv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orrell KE, Mansfield MJ, Doxey AC, et al. The C. difficile toxin B membrane translocation machinery is an evolutionarily conserved protein delivery apparatus. Nature Communications 2020;11:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manse JS, Baldwin MR. Binding and entry of Clostridium difficile toxin B is mediated by multiple domains. FEBS Lett 2015;589:3945–51. [DOI] [PubMed] [Google Scholar]
- 16.Giesemann T, Jank T, Gerhard R, et al. Cholesterol-dependent pore formation of Clostridium difficile toxin A. J Biol Chem 2006;281:10808–15. [DOI] [PubMed] [Google Scholar]
- 17.Papatheodorou P, Song S, López-Ureña D, et al. Cytotoxicity of Clostridium difficile toxins A and B requires an active and functional SREBP-2 pathway. Faseb j 2019;33:4883–4892. [DOI] [PubMed] [Google Scholar]
- 18.Barth H, Pfeifer G, Hofmann F, et al. Low pH-induced formation of ion channels by clostridium difficile toxin B in target cells. J Biol Chem 2001;276:10670–6. [DOI] [PubMed] [Google Scholar]
- 19.Ng J, Hirota SA, Gross O, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 2010;139:542–52, 552 e1–3. [DOI] [PubMed] [Google Scholar]
- 20.Warny M, Keates AC, Keates S, et al. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J Clin Invest 2000;105:1147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014. [DOI] [PubMed] [Google Scholar]
- 22.Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev 2008;60:795–804. [DOI] [PubMed] [Google Scholar]
- 23.Lee EY, Fulan BM, Wong GCL, et al. Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proc. Nat. Acad. Sci. USA 2016;113:13588–13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilavsky J. Nika: software for two-dimensional data reduction. Journal of Applied Crystallography 2012;45:324–328. [Google Scholar]
- 25.Hammersley AP. FIT2D: An Introduction and Overview. Internal Report No.ESRF97HA02T European Synchrotron Radiation Facility, Grenoble, 1997. [Google Scholar]
- 26.Parroche P, Lauw FN, Goutagny N, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A 2007;104:1919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sullivan NM, Pellett S, Wilkins TD. Purification and characterization of toxins A and B of Clostridium difficile. Infect Immun 1982;35:1032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Katchar K, Goldsmith JD, et al. A mouse model of Clostridium difficile-associated disease. Gastroenterology 2008;135:1984–92.( (Author names in bold designate shared co-first authorship) [DOI] [PubMed] [Google Scholar]
- 29.Tao L, Tian S, Zhang J, et al. Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells. Nat Microbiol 2019;4:1760–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silvestre-Roig C, Braster Q, Wichapong K, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019;569:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee MW, Lee EY, Lai GH, et al. Molecular Dnm1 synergistically induces membrane curvature to facilitate mitochondrial fission. ACS Central Science 2017;3:1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larabee JL, Hauck GD, Ballard JD. Cell-penetrating peptides derived from Clostridium difficile TcdB2 and a related large clostridial toxin. J Biol Chem 2018;293:1810–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larabee JL, Bland SJ, Hunt JJ, et al. Intrinsic Toxin-Derived Peptides Destabilize and Inactivate <em>Clostridium difficile</em> TcdB. mBio 2017;8:e00503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geissler B. Bacterial Toxin Effector-Membrane Targeting: Outside in, then Back Again. Frontiers in Cellular and Infection Microbiology 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geissler B, Tungekar R, Satchell KJF. Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. Proceedings of the National Academy of Sciences 2010;107:5581–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra A, Lai GH, Schmidt NW, et al. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proceedings of the National Academy of Sciences 2011;108:16883–16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krysko DV, Kaczmarek A, Krysko O, et al. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ 2011;18:1316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaiswal MK, Agrawal V, Mallers T, et al. Regulation of apoptosis and innate immune stimuli in inflammation-induced preterm labor. J Immunol 2013;191:5702–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bortoluci KR, Medzhitov R. Control of infection by pyroptosis and autophagy: role of TLR and NLR. Cell Mol Life Sci 2010;67:1643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peter ME, Kubarenko AV, Weber AN, et al. Identification of an N-terminal recognition site in TLR9 that contributes to CpG-DNA-mediated receptor activation. J Immunol 2009;182:7690–7. [DOI] [PubMed] [Google Scholar]
- 41.Franklin BS, Ishizaka ST, Lamphier M, et al. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci U S A 2011;108:3689–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt NW, Jin F, Lande R, et al. Liquid-crystalline ordering of antimicrobial peptide–DNA complexes controls TLR9 activation. Nature Materials 2015;14:696. [DOI] [PubMed] [Google Scholar]
- 43.Tursi SA, Lee EY, Medeiros NJ, et al. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLOS Pathogens 2017;13:e1006315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lande R, Lee EY, Palazzo R, et al. CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-α production in systemic sclerosis. Nature Communications 2019;10:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Braunlin W, Xu Q, Hook P, et al. Toxin binding of tolevamer, a polyanionic drug that protects against antibiotic-associated diarrhea. Biophys J 2004;87:534–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Semenyuk EG, Laning ML, Foley J, et al. Spore Formation and Toxin Production in Clostridium difficile Biofilms. PLoS One 2014;9:e87757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt NW, Wong GCL. Antimicrobial peptides and induced membrane curvature: Geometry, coordination chemistry, and molecular engineering. Current Opinion in Solid State and Materials Science 2013;17:151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee EY, Takahashi T, Curk T, et al. Crystallinity of Double-Stranded RNA-Antimicrobial Peptide Complexes Modulates Toll-Like Receptor 3-Mediated Inflammation. ACS Nano 2017;11:12145–12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee EY, Zhang C, Di Domizio J, et al. Helical antimicrobial peptides assemble into protofibril scaffolds that present ordered dsDNA to TLR9. Nature Communications 2019;10:1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winter SE, Thiennimitr P, Winter MG, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 2010;467:426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ng KM, Ferreyra JA, Higginbottom SK, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013;502:96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park B, Buti L, Lee S, et al. Granulin is a soluble cofactor for toll-like receptor 9 signaling. Immunity 2011;34:505–13. [DOI] [PubMed] [Google Scholar]
- 53.Savidge TC, Urvil P, Oezguen N, et al. Host S-nitrosylation inhibits clostridial small molecule-activated glucosylating toxins. Nat Med 2011;17:1136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biancheri P, Di Sabatino A, Corazza GR, et al. Proteases and the gut barrier. Cell and Tissue Research 2013;351:269–280. [DOI] [PubMed] [Google Scholar]
- 55.Antalis TM, Shea-Donohue T, Vogel SN, et al. Mechanisms of Disease: protease functions in intestinal mucosal pathobiology. Nature Clinical Practice Gastroenterology & Hepatology 2007;4:393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.