

Abstract
All antibiotics have to engage bacterial amphiphilic barriers such as the lipopolysaccharide-rich outer membrane or the phospholipid-based inner membrane in some manner, either by disrupting them outright and/or permeating them and thereby allow the antibiotic to get into bacteria. There is a growing class of cyclic antibiotics, many of which are of bacterial origin, that exhibit activity against Gram-negative bacteria, which constitute an urgent problem in human health. We examine a diverse collection of these cyclic antibiotics, both natural and synthetic, which include bactenecin, polymyxin B, octapeptin, capreomycin, and Kirshenbaum peptoids, in order to identify what they have in common when they interact with bacterial lipid membranes. We find that they virtually all have the ability to induce negative Gaussian curvature (NGC) in bacterial membranes, the type of curvature geometrically required for permeation mechanisms such as pore formation, blebbing, and budding. This is interesting since permeation of membranes is a function usually ascribed to antimicrobial peptides (AMPs) from innate immunity. As prototypical test cases of cyclic antibiotics, we analyzed amino acid sequences of bactenecin, polymyxin B, and capreomycin using our recently developed machine-learning classifier trained on α-helical AMP sequences. Although the original classifier was not trained on cyclic antibiotics, a modified classifier approach correctly predicted that bactenecin and polymyxin B have the ability to induce NGC in membranes, while capreomycin does not. Moreover, the classifier was able to recapitulate empirical structure–activity relationships from alanine scans in polymyxin B surprisingly well. These results suggest that there exists some common ground in the sequence design of hybrid cyclic antibiotics and linear AMPs.
Keywords: Antimicrobial peptides, Cyclic antibiotics, Bactenecin, Polymyxin, Structure-activity relationship (SAR), Machine learning
1. Introduction
It is difficult to overestimate the problem posed by antibiotic-resistant bacteria to global human health. Approximately 70% of hospital acquired infections in the United States are resistant to at least one antibiotic. Currently, over 2.8 million people acquire antibiotic-resistant infections in the U.S. each year, of which >35,000 die as a result [1]. Unfortunately, progress on antibiotic development has been slow. In the last 20 years, only two new classes of antibiotics, oxazolidinones and lipopeptides, have been approved for use, and there is already resistance to both [2–4]. Indeed, the economic dimension of the problem cannot be ignored. The costs associated with these infections in terms of annual health care costs and productivity losses are estimated to be upwards of $20 billion and $35 billion, respectively [5]. Moreover, it takes over $500 million to develop an antibiotic but typically only a few years elapse before the emergence of resistant strains, so the present version of discovery-based approach to antibiotic development is not necessarily sustainable. The growing gap between the decline of antibiotic development and the escalating emergence of bacterial drug resistance has become one of the most salient challenges for human health.
We aim to discover design principles implicit in cyclic antibiotics, a growing class of promising antibiotics with activity against recalcitrant Gram-negative bacteria. All antibiotics have to engage bacterial amphiphilic barriers by disrupting them outright and/or permeating them and thereby allowing the antibiotic to get into bacteria: these barriers include the lipopolysaccharide-rich outer membrane and the phospholipid-based inner membrane. We study a diverse collection of these cyclic antibiotics, some of bacterial origin and some synthetic, in order to identify what they have in common when they interact with bacterial membranes. These cyclic antibiotics include bactenecin, polymyxin B, octapeptin, capreomycin, and Kirshenbaum peptoids, with some of these being hybrid molecules that include peptide-based and non-peptide-based components. We find that these cyclic antibiotics virtually all have the ability to induce negative Gaussian curvature (NGC) in bacterial membranes, the type of curvature geometrically required for permeation mechanisms such as pore formation, blebbing, and budding. Interestingly, this ability to permeate bacterial membranes is a common feature of antimicrobial peptides (AMPs), which have retained antimicrobial activity despite prolonged co-evolution with bacteria and comprise a key component of the eukaryotic innate immune system [6]. We adapted our method for implementing a recently developed machine-learning classifier trained on α-helical AMP sequences [7] so that it can approximately account for non-proteinogenic amino acids and non-amino acid-based hydrophobic structural elements. Using this modified classifier, we examine three prototypical cyclic antibiotics: bactenecin, polymyxin B, and capreomycin. The first two exhibit NGC-inducing ability experimentally, while the third does not. The first and third are cyclic peptides, with the former using proteinogenic amino acids and the latter using both proteinogenic and non-proteinogenic amino acids. The second of the three is a hybrid, a lipopeptide that contains a fatty acid chain and a cyclic peptide composed of both proteinogenic and non-proteinogenic amino acids. Although the classifier was not trained specifically on cyclic antibiotics, it predicted that bactenecin and polymyxin B have the ability to induce NGC in membranes, whereas capreomycin does not, in agreement with experiments. Furthermore, the classifier was able to recapitulate published empirical structure–activity relationships from alanine scans in polymyxin B surprisingly well. Taken together, these results suggest that there exists some common ground in the sequence design of hybrid cyclic antibiotics and linear AMPs.
Before we describe our results, we provide a summary description of the various cyclic antibiotics used in this study:
Bactenecin is a 12-residue cathelicidin AMP that is found in the granules of bovine neutrophils and contains a disulfide bond, forming a cyclic structure. It is the smallest known cationic AMP and exhibits broad antimicrobial activity. Bactenecin has been shown to bind to LPS and permeabilize both outer and inner bacterial membranes [8]. Although bactenecin displays activity against both Gram-positive and Gram-negative bacteria, it has greater potency against the latter [9].
A member of the polymyxins, polymyxin B is a natural cationic lipopeptide antibiotic produced by Bacillus polymyxa and has been clinically used as a last line of defense against multidrug-resistant Gram-negative bacterial infections. Its structure consists of a peptide ring that has a peptide side chain with a fatty acid tail. Like bactenecin, polymyxin B has been shown to kill Gram-negative bacteria by binding to the LPS of their outer membrane and permeabilizing both the outer and inner membranes [10,11].
Octapeptins are a family of natural cyclic lipopeptide antibiotics isolated from Bacillus circulans that have broad antimicrobial activity against both Gram-positive and Gram-negative bacteria [12]. Despite their discovery >40 years ago, octapeptins remain largely un-characterized. The general octapeptin scaffold consists of a cyclic peptide ring linked to a D-amino acid and a fatty acid tail.
The structures of both polymyxins and octapeptins feature a cyclic peptide core that is linked to an N-terminal fatty acid chain. Both are also characterized by a hydrophobic motif in their peptide ring. As octapeptins generally possess fewer charges than polymyxins, they feature increased hydrophobicity relative to the polymyxins.
A vast number of synthetic peptidomimetic compounds have been developed to capture the conformations and functions of natural AMPs. Among these are a class of sequence-specific oligomers of N-substituted glycine called “peptoids” that feature cationic and hydrophobic side chains similar to natural peptides [13]. Kirshenbaum peptoids C3 and C124 are two potent antimicrobial cyclic peptoids that cause cell-surface damage and form pores in bacterial membranes [14,15]. However, additional antimicrobial mechanisms may also be involved.
Capreomycin is a natural polypeptide antibiotic from Streptomyces capreolus that has been used in the treatment of tuberculosis. It is believed to exert its antimicrobial effects by inhibiting ribosomal protein synthesis. For instance, studies suggest that capreomycin binds to and inhibits the 16S rRNA of the 30S ribosomal subunit in Mycobacterium tuberculosis [16,17]. Due to having similar intracellular activity and nephrotoxic side effects, capreomycin is often compared and grouped with animoglycosides. Accordingly, as aminoglycosides are unable to freely cross cell membranes and thus must enter cells via endocytosis [18–20], it is proposed that capreomycin similarly translocates through a cellular uptake pathway [16]. Indeed, recent studies have found that disruption of uptake regulators in Mycobacterium smegmatis result in greater membrane permeability and susceptibility to aminoglycosides and capreomycin [21].
2. Materials and methods
2.1. Preparation of lipid vesicles
Lyophilized phospholipids 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt) (DOPG), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (sodium salt) (DOPS), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids and dissolved in chloroform at 20 mg/mL to produce individual lipid stock solutions. Lipid compositions were prepared from the lipid stock solutions as mixtures at specified molar ratios, evaporated under nitrogen, and desiccated under vacuum overnight to form a dry lipid film. Lipid films were resuspended in aqueous 140 mM NaCl, 10 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) (pH 7.4) to a concentration of 20 mg/mL. Lipid suspensions were incubated overnight at 37 °C, sonicated until clear, and extruded through a 0.2 μm pore size Anopore membrane filter (Whatman) to form unilamellar vesicles (ULVs).
2.2. SAXS experiments
Solubilized antibiotic compounds were mixed with ULVs at specified antibiotic-to-lipid charge ratios (Ac/Lc) and hermetically sealed into quartz capillaries (Hilgenberg GmbH, Mark-tubes) for high-resolution small-angle X-ray scattering (SAXS) measurements taken at the Stanford Synchrotron Radiation Lightsource (SSRL, beamline 4–2) using monochromatic X-rays with an energy of 9 keV. A DECTRIS PILATUS3 X 1M detector (172 μm pixel size) was used to collect the scattered radiation and the resulting 2D SAXS power patterns were integrated using the Nika 1.50 [22] package for Igor Pro 6.31 and FIT2D [23].
The integrated scattering intensity I(q) was plotted against q. The phases present in each sample were determined by comparing the ratios of measured peak positions, qmeasured, with those of the permitted reflections for different crystal phases. The linear regression through points corresponding to the peaks was then used to calculate the lattice parameter, a, of each identified phase. For powder-averaged cubic phases, each peak was represented by a point having coordinates of the assigned reflection, in terms of Miller indices (h, k, l), and qmeasured. For a cubic phase, . The slope of the regression of qmeasured vs is 2π/a, which can then be used to calculate a.
2.3. Membrane activity predictions by an SVM classifier
Sequences listed in Tables 2 and 3 were evaluated for membrane disruption activity using a previously validated support vector machine (SVM) classifier [7]. The classifier outputs a probability value, P(+1), for the peptide sequence to be membrane active. (Note that the original classifier was trained with amino acid sequences only containing the 20 standard proteinogenic amino acids.) We approximated each cyclic antibiotic using a linear peptide sequence based on the standard proteinogenic amino acids. For antibiotic compounds that contain non-proteinogenic residues, each non-proteinogenic residue is approximated with a proteinogenic amino acid that best represents the original side chain (e.g., α,γ-diaminobutyric acid (Dab) was approximated with lysine). The results from the SVM classifier are presented in Fig. 3A, Tables 2 and 3.
Table 2. Peptide analogs for cyclic antibiotics and their P(+1) outputs from the SVM classifier.
Cyclic antibiotics were modeled using linear peptide sequences composed of standard proteinogenic amino acids and evaluated by the SVM classifier. As bactenecin is a peptide naturally composed of only proteinogenic amino acids, its native sequence was used.
| Antibiotic modeled | Peptide analog variant name | Peptide sequence | P(+ 1) |
|---|---|---|---|
| Bactenecin | N/A | RLCRIVVIRVCR (native) | 0.981884801 |
| Polymyxin B (without fatty acid chain) | Polymyxin B-v0 | KTKAKFLKKT | 0.008487151 |
| Polymyxin B | Polymyxin B-v1 | IKTKAKFLKKT | 0.601030492 |
| Polymyxin B | Polymyxin B-v2 | IIKTKAKFLKKT | 0.829476546 |
| Capreomycin | Capreomycin-v1 | AKASKRKA | 0.000295565 |
| Capreomycin | Capreomycin-v2 | AKASKRNA | 0.000272803 |
Table 3. SVM predictions compared with empirical observations for alanine scans of polymxin B.
Alanine scan sequences for peptide analog polymyxin B–v2 with their corresponding analogous substitutions in polymyxin B [70] (if available) and corresponding P(+1) outputs from the SVM classifier. Each alanine substitution is indicated in bold. The SVM classifier predictions agree well with the experimental antimicrobial activities of the alanine substitutions [70]. Alanine substitutions at positions 2, 5, and 9 of polymyxin B, which led to the most notable reductions in observed antimicrobial activity, correspond to peptide sequences (#2, #5, and #9, respectively) with decreased P(+1) values. Furthermore, alanine substitutions at positions 3, 7, and 10 resulted in little to no change in antimicrobial activity and correspond to peptide sequences (#3, #7, and #10, respectively) that maintain high P(+1) values.
| Peptide sequence index | Peptide sequence | Analogous substitution in polymyxin B [70] | P(+ 1) | Changes in empirically observed antimicrobial activity [70] |
|---|---|---|---|---|
| 1 | IIATKAKFLKKT | Dab1A | 0.592073833 | |
| 2 | IIKAKAKFLKKT | T2A | 0.755870297 | Reduction |
| 3 | IIKTAAKFLKKT | Dab3A | 0.869940844 | Minimal to no change |
| 4 (polymyxin B-v2) | IIKTKAKFLKKT | 0.829476546 | ||
| 5 | IIKTKAAFLKKT | Dab5A | 0.597315289 | Large reduction |
| 6 | IIKTKAKALKKT | (d-F)6(d-A) | 0.784859991 | |
| 7 | IIKTKAKFAKKT | L7A | 0.814938477 | Minimal to no change |
| 8 | IIKTKAKFLAKT | 0.645279651 | ||
| 9 | IIKTKAKFLKAT | Dab9A | 0.463844949 | Reduction |
| 10 | IIKTKAKFLKKA | T10A | 0.906324927 | Minimal to no change |
Fig. 3. Machine-learning predictions align with empirical membrane activity.
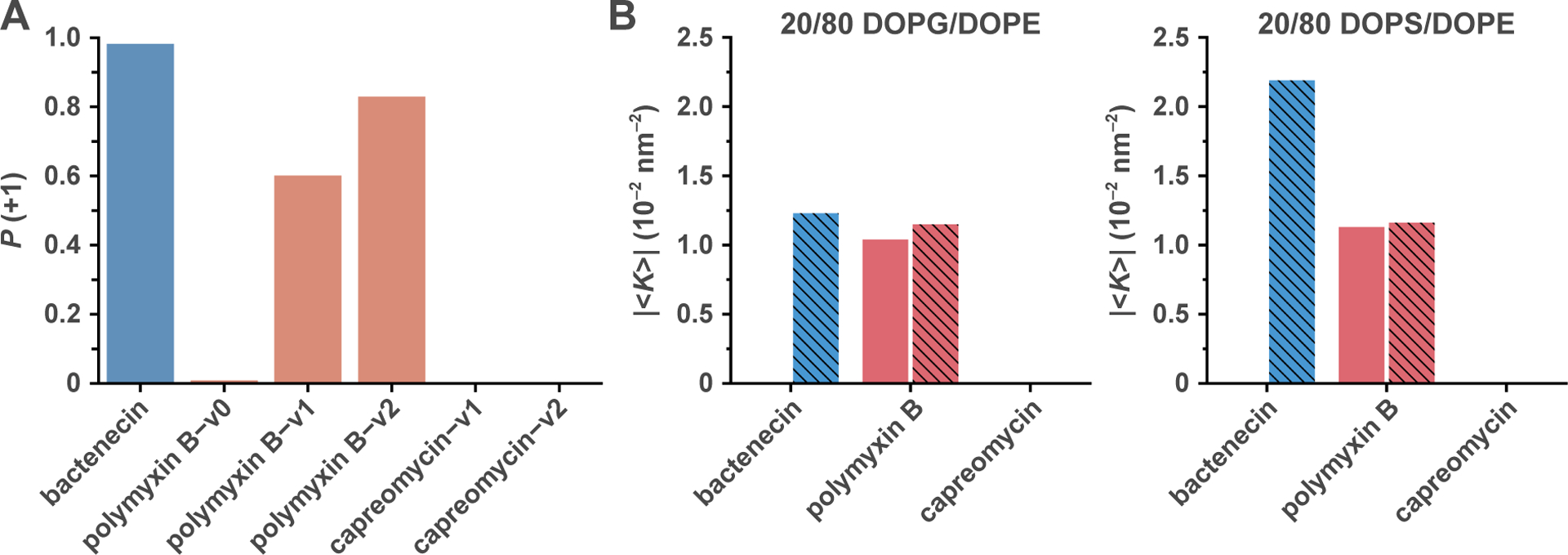
(A) The SVM classifier output P(+1) values for the peptide analogs of bactenecin, polymyxin B, and capreomycin. Polymyxin B–v0, a peptide analog representing the peptide component of polymyxin B, was not predicted to have membrane activity. However, peptide analogs polymyxin B–v1 and polymyxin B–v2, which represent both the peptide and fatty acid chain components of polymyxin B, resulted in high P (+1) values and are predicted to induce NGC. From their P(+1) values, bactenecin and polymyxin B, but not capreomycin, were predicted to induce NGC in membranes. (B) The SVM predictions are in good agreement with the observed NGC-generating ability of the compounds and suggest that the hydrophobic fatty acid chain in polymyxin B increases its membrane activity. Indeed, we observed a broader stoichiometric range (Ac/Lc = 1/2 and 1/1, A/L = 1/50 and 1/25) over which polymyxin B generates NGC, in comparison with that of bactenecin (Ac/Lc = 1/1, A/L = 1/20 only). NGC values for Ac/Lc = 1/2 and 1/1 are depicted as solid and striped bars, respectively.
3. Results and discussion
In this work, we studied experimentally seven cyclic antibiotics with potent activity against Gram-negative bacteria: bactenecin, capreomycin, polymyxin B, two octapeptin isomers, and two Kirshenbaum peptoids (Fig. 1). Specifically, we investigated whether there are any unifying features in how these cyclic antibiotics interact with membranes. Using high-resolution synchrotron SAXS, we quantitatively characterized the deformations induced by the antibiotics in model bacterial membranes. ULVs were prepared from phospholipid mixtures of DOPG, DOPS, and DOPE at distinct molar ratios to mimic the different lipid compositions of bacterial plasma membranes. Each antibiotic was incubated with ULVs at specified Ac/Lc charge ratios relative to the isoelectric point, at which the charge of the antibiotic neutralizes that of the membrane, and the resulting membrane structures were characterized using SAXS. For each antibiotic, an Ac/Lc charge ratio can be converted to a corresponding stoichiometric antibiotic-to-lipid molar ratio (A/L). At a given Ac/Lc, the corresponding A/L value depends on the net charge of the antibiotic. For the antibiotics in this study, Ac/Lc of 1/2 and 1/1 (electroneutral) correspond to A/L ranges of 1/25 to 1/15 and 1/50 to 1/30, respectively.
Fig. 1.
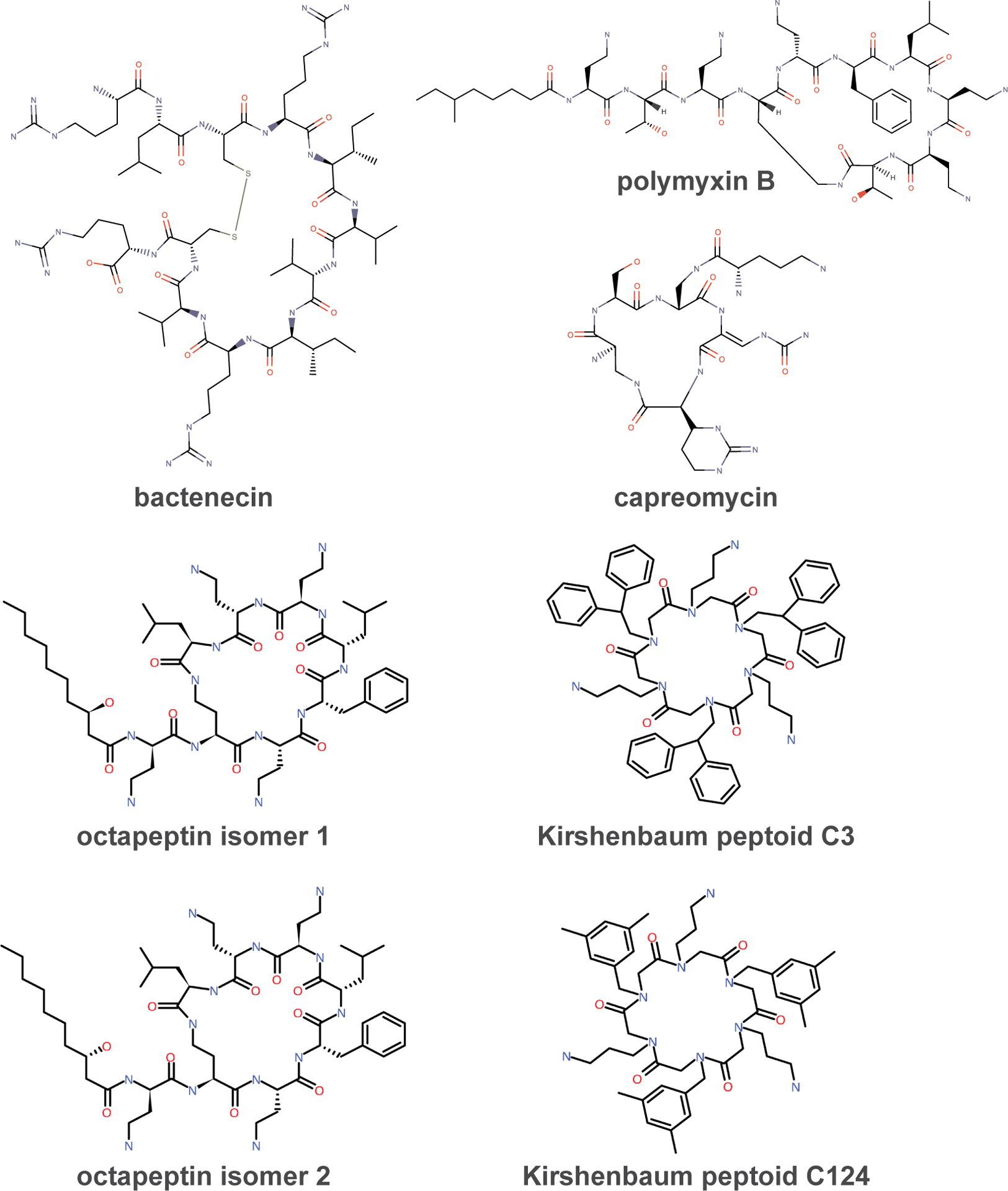
Chemical structures of the cyclic antibiotics in this study.
3.1. Cyclic antibiotics generate negative Gaussian curvature in bacterial membranes
We found that at physiologically relevant conditions, all tested antibiotics, except for capreomycin, restructured the lipid vesicles into bicontinuous cubic phases rich in NGC (Fig. 2A,B). In contrast, the control samples of ULVs only showed a broad characteristic feature consistent with the form factor of ULVs (Fig. S1A). A large number of AMPs have been recognized to exert their bactericidal effects by disrupting bacterial membranes to form pores [24–28] and induce blebbing [29,30], which can lead to membrane depolarization, leakage, and cell lysis. A topological requirement shared by such membrane-destabilizing processes is the generation of NGC in membranes. At a given point on a surface, the maximum and minimum curvatures, which are in orthogonal directions, are called the two principal curvatures, c1 and c2· Curvature is defined as c = 1/r, with r being the radius of a circle that best approximates the curve at that point. (By convention, a membrane monolayer that bends to form a convex hydrophilic surface is described as having positive curvature, whereas, a monolayer that bends to form a concave hydrophilic surface is described as having negative curvature.) Gaussian curvature, K, is the product of the principal curvatures (K = c1c2) and becomes negative when one principal curvature is positive and the other is negative. Thus, NGC characterizes a surface that bends upward along one direction and bends downward along the orthogonal direction, forming a saddle shape (Fig. 2C). NGC is the specific type of curvature geometrically required in transmembrane pores (Fig. 2D), fusion pores, and along the necks of budding vesicles. AMPs have been observed to generate NGC in a large number of studies [31–36]. NGC has been experimentally observed in other contexts where peptide–membrane or protein–membrane interactions result in membrane topological changes, such as cell-penetrating peptides (CPPs), viral fusion peptides, viral budding proteins, metaphilic peptides, and mitochondrial remodeling proteins [37–41].
Fig. 2. Cyclic antibiotics generate NGC in model bacterial membranes.
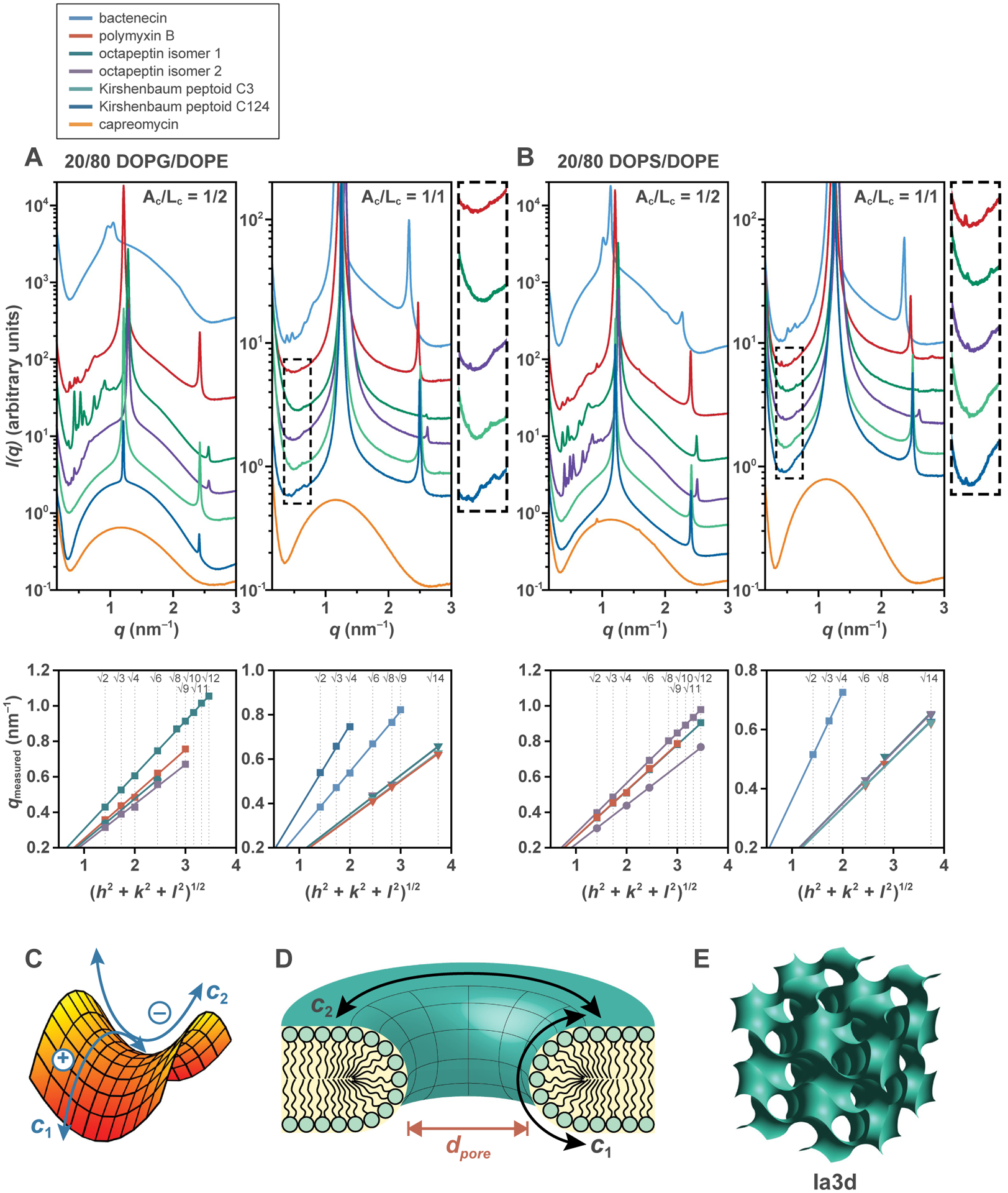
SAXS spectra from membranes incubated with each cyclic antibiotic: (A) 20/80 DOPG/DOPE at Ac/Lc = 1/2 (A/L = 1/50–1/30) (left) and Ac/Lc = 1/1 (A/L = 1/25–1/15) (right), (B) 20/80 DOPS/DOPE at Ac/Lc = 1/2 (A/L = 1/50–1/30) (left) and A c/Lc = 1/1 (A/L = 1/25–1/15) (right). Correlation peaks found in the low q region indexed to cubic phases. For clarity, expanded views of low intensity cubic reflections (dashed box regions) for Ac/Lc = 1/1 are provided alongside the spectra plots for 20/80 DOPG/DOPE and 20/80 DOPS/DOPE. Indexing of the identified cubic phases are shown below each spectra plot. Each plotted point corresponds to an indexed cubic peak, with its associated cubic lattice denoted by the marker symbol (square = Pn3m, circle = Im3m, and triangle = Ia3d). (C) NGC characterizes a saddle-shaped surface, which bends upward along one direction and bends downward along the orthogonal direction, and is defined by principal curvatures c1 and c2 that are opposite in sign. (D) NGC is found along the inner curved surface of a transmembrane pore. The directions of the principal curvatures for the curved membrane surface are indicated by black arrows. Different pore diameters (dpore) are characterized by different magnitudes of NGC. (E) Schematic of the Ia3d bicontinuous cubic phase.
Inverse bicontinuous cubic phases (QII) are among the range of lyotropic liquid-crystalline phases that can be formed by lipid systems. Of these, the Pn3m, Im3m, and Ia3d symmetries are three cubic phases most commonly observed in biological membranes [42] (Fig. 2E). Each of these cubic phases consists of two interpenetrating, but non-intersecting, aqueous volumes that are separated by a single continuous lipid bilayer. For the Pn3m, Im3m, and Ia3d, the bilayer mid-plane traces the D, P, and G minimal surfaces, respectively, and has NGC at all points on its surface [43]. For a cubic phase, the average Gaussian curvature, <K>, can be calculated from the equation <K> = (2πχ)/(A0a2), where the Euler characteristic, χ, and the dimensionless surface area per unit cell, A0, are geometric characteristics unique to each cubic phase, and a is the lattice parameter of the unit cell. Specifically, for the Pn3m, Im3m, and Ia3d cubic phases, χ = −2, −4, −8 and A0 = 1.919, 2.345, 3.091, respectively.
Membranes of many Gram-negative bacteria, such as Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa often contain high concentrations of phosphatidylethanolamine (PE) lipids [44–50]. ULVs with a representative lipid composition of 20/80 DOPG/DOPE to model Gram-negative bacterial membranes were exposed to each antibiotic compound (Ac/Lc = 1/1, A/L = 1/25–1/15) and measured using SAXS (Fig. 2A, Table 1). For bactenecin, the SAXS spectra exhibited correlation peaks with q-ratios of √2:√3:√4:√6:√8:√9, which index to a Pn3m cubic phase with a lattice parameter of 23.11 nm and average Gaussian curvature of −1.23 × 10−2 nm−2. For polymyxin B, we observed peaks with q-ratios of √6:√8:√14, corresponding to an Ia3d cubic phase (a = 37.67 nm, <K> = −1.15 × 10−2 nm−2). Octapeptin isomers 1 and 2 both generated Ia3d cubic phases with respective lattice parameters of 35.76 nm and 35.85 nm, and average Gaussian curvatures of −1.27 × 10−2 nm−2 and −1.27 × 10−2 nm−2. Kirshenbaum peptoids C3 and C124 induced Ia3d (a = 37.32 nm, <K> = −1.17 × 10−2 nm−2) and Pn3m (a = 16.67 nm, <K> = −2.36 × 10−2 nm−2) cubic phases, respectively. These six antibiotics that generated cubic phases also each induced a lamellar phase with a low repeat distance (4.80–5.40 nm), indicative of a condensed lamellar aggregate. Interestingly, the formation of such aggregates has been suggested to facilitate the development of cubic phases [51].
Table 1. Cubic phases induced by cyclic antibiotics.
The symmetries and lattice parameters of the cubic phases generated by the cyclic antibiotics in 20/80 DOPG/DOPE and 20/80 DOPS/DOPE membranes, at both Ac/Lc = 1/2 (A/L = 1/50–1/30) and 1/1 (A/L = 1/25–1/15). Empty cells indicate the absence of cubic phases.
| Antibiotic | 20/80 DOPG/DOPE | 20/80 DOPS/DOPE | ||
|---|---|---|---|---|
| Ac/Lc = 1/2 (A/L = 1/50–1/30) |
Ac/Lc = 1/1 (A/L = 1/25–1/15) |
Ac/Lc = 1/2 (A/L = 1/50–1/30) |
Ac/Lc = 1/1 (A/L = 1/25–1/15) |
|
| Bactenecin | Pn3m: 23.11 nm | Pn3m: 17.30 nm | ||
| Polymyxin B | Pn3m: 25.06 nm | Ia3d: 37.67 nm | Pn3m: 24.03 nm | Ia3d: 37.47 nm |
| Octapeptin isomer 1 | Pn3m: 20.61 nm Im3m: 26.38 nm |
Ia3d: 35.76 nm | Pn3m: 24.15 nm | Ia3d: 35.76 nm |
| Octapeptin isomer 2 | Pn3m: 28.18 nm | Ia3d: 35.85 nm | Pn3m: 22.26 nm Im3m: 28.49 nm |
Ia3d: 35.96 nm |
| Kirshenbaum peptoid C3 | Ia3d: 37.32 nm | Ia3d: 37.51 nm | ||
| Kirshenbaum peptoid C124 Capreomycin | Pn3m: 16.67 nm | Ia3d: 37.36 nm | ||
Additional PE-rich lipid compositions also produced similar results when incubated with the antibiotics (Ac/Lc = 1/1, A/L = 1/25–1/15). For 20/80 DOPS/DOPE ULVs (Fig. 2B, Table 1), the scattering spectrum for bactenecin contained peaks with q-ratios of √2:√3:√4, indicating the presence of a Pn3m cubic phase (a = 17.30 nm, <K> = −2.19 × 10−2 nm−2). Polymyxin B yielded peaks with q-ratios of √6:√8:√14, which indexed to an Ia3d cubic phase (a = 37.47 nm, <K> = −1.16 × 10−2 nm−2). Octapeptin isomers 1 and 2 both generated Ia3d cubic phases (a = 35.76 nm, <K> = −1.27 × 10−2 nm−2 and a = 35.96 nm, <K> = −1.26 × 10−2 nm−2, respectively). Similarly, the SAXS spectra of Kirshenbaum peptoids C3 and C124 exhibited peaks that indexed to Ia3d cubic phases with lattice parameters of 37.51 nm and 37.36 nm, and average Gaussian curvatures of −1.16 × 10−2 nm−2 and −1.17 × 10−2 nm−2, respectively.
Remarkably, the calculated amounts of average Gaussian curvature, <K>, on the surfaces of the cubic phases induced by these six antibiotic compounds were comparable to those induced by many AMPs [35,36]. Moreover, the capacity of these compounds to generate NGC is consistent with their ability to kill bacteria via membrane disruption, a trend that was previously identified among canonical AMPs and their analogs [35,40,52–54]. This new finding supports the generality of NGC-mediated bactericidal activity in structurally diverse natural and synthetic antimicrobial molecules. Furthermore, all seven tested antibiotic compounds did not generate NGC in low-PE membranes (data not shown), which more closely mimic those of mammalian cells [55,56]. Together, these results suggest that their preferential activity against bacterial membranes is at least in part due to membrane composition differences among cell types.
Most strikingly, we found that of the tested antibiotics, capreomycin did not generate NGC in the range of membrane compositions at physiologically realistic conditions. While this result may at first seem at odds with the behavior typical of AMPs and the other six tested compounds, capreomycin’s low membrane activity is in fact consistent with its proposed mechanism of action. Similar to the aminoglycoside class of antibiotics, capreomycin exerts its antimicrobial effects intracellularly by inhibiting protein synthesis [16,17] and is believed to gain entry into cells through a cellular uptake pathway [16,21]. Because neither of these processes would require antibiotic-mediated membrane restructuring, it is not surprising that capreomycin was not found to induce NGC and is therefore the “exception that proves the rule.” However, the precise mechanism of action for capreomycin is not fully understood, as the antibiotic is able to remain active against non-replicating dormant bacteria with significantly reduced cellular uptake [57].
3.2. Comparison of machine-learning predictions and empirical membrane activity
We previously developed a machine-learning SVM classifier trained to identify α-helical peptide sequences that have the capacity to remodel membranes via NGC generation [7]. Moreover, the SVM has been shown to be able to detect the existence of potential membrane-active regions within larger protein sequences [41,58]. The classifier exemplifies how computational methods can be used to predict the function of a peptide or protein based on its sequence-derived physicochemical properties, independent of sequence or structural homology. This type of approach can therefore promote efficient, high-throughput identification and design of new membrane-active peptide sequences with effective antimicrobial activity. To explain briefly, from a given amino acid sequence, based on 12 optimized physicochemical descriptors, the SVM outputs a score, σ, that specifies the distance of the sequence from an 11-dimensional hyperplane trained to separate known α-helical NGC-generating sequences from decoy sequences. A sequence with a high positive σ score indicates a high probability that it has the ability to induce NGC in membranes, while a large negative σ score indicates a high probability that it lacks this membrane-restructuring ability. The σ score can then be converted to a probability 0 < P(+1) < 1 that the sequence is able to induce NGC. A strong correlation was found to exist between P(+1) and the magnitude of NGC generated [7].
At present, there is no known machine-learning classifier for evaluating cyclic peptide antibiotics that are composed of both proteinogenic and non-proteinogenic amino acids. Moreover, there is not enough available data to train such a classifier. Here, we explored the possibility of adapting the SVM classifier we previously trained using natural AMP sequences to estimate the membrane activity of cyclic peptide-based antibiotic compounds via a few approximations. In alignment with their in vitro bactericidal membrane-disruptive effects, we have shown that the six cyclic antibiotics, bactenecin, polymyxin B, octapeptin isomers 1 and 2, and Kirshenbaum peptoids C3 and C124, are indeed capable of inducing NGC in model bacterial membranes. While bactenecin is a short eukaryotic peptide with a disulfide bond, polymyxin B, capreomycin, and octapeptin isomers 1 and 2 are nonribosomal polypeptide antibiotics that are composed of both standard proteinogenic amino acids and non-proteinogenic amino acids, some of which are further modified with additional functional groups. For instance, polymyxin B and octapeptins make extensive use of non-amino acid hydrocarbon-based hydrophobic tails [11,12]. Kirshenbaum peptoids C3 and C124 are synthetic peptidomimetics characterized by having different structures based on poly-N-substituted glycines, and are quite different from regular polypeptides. Therefore, to see how well the SVM trained on AMPs can capture empirical trends of cyclic antibiotics, we focus on three: bactenecin, polymyxin B, and capreomycin. The first two exhibit NGC inducing ability experimentally, while the third does not. We aimed to evaluate the antibiotic compounds with the SVM classifier by approximating each cyclic molecule using a linear peptide sequence with a composition limited to the 20 standard proteinogenic amino acids (e.g., Dab, a non-proteinogenic lysine analog, is substituted with lysine [59]), and incorporating hydrophobic residues to account for additional hydrophobic functional groups (e.g., progressive incorporation of isoleucine to approximate alkyl chains) (Table 2). For example, polymyxin B is a mixture of polymyxins, with the main components being polymyxins B1 and B2 [60], which are characterized by terminal fatty acids with hydrocarbon chains containing 8 and 7 carbons, respectively. In the peptide analog of polymyxin B, we represented the fatty acid chain by using two consecutive isoleucine residues, each of which has a 4-carbon hydrocarbon side chain. Using the SVM classifier on the peptide analogs for bactenecin, polymyxin B, and capreomycin, we found that both bactenecin and polymyxin B resulted in relatively high values of P(+1), indicating a high likelihood of inducing NGC in membranes (Fig. 3A, Table 2). On the contrary, the peptide analogs for capreomycin were not predicted to be able to generate NGC, as reflected by their low P(+1) values. Although the classifier is not trained on cyclic molecules, the SVM predictions for the peptide analogs are in surprising agreement with the observed membrane-restructuring and NGC-generating ability of the compounds (Fig. 3B).
3.3. The role of hydrophobic tails on the membrane activity of lipopeptide antibiotics: polymyxin B as a case study
For the peptide-based cyclic antibiotics, utilizing simple linear peptide analogs as inputs for the SVM yielded reasonable predictions of their membrane activity. This finding thus introduces potential new applications for the SVM as an informative tool for the analysis and understanding of existing molecules and the rational design of new ones. For instance, we can specifically examine polymyxin B, which features a cyclic peptide core that is characterized by a high proportion of cationic Dab residues and is linked to an N-terminal fatty acyl tail (Fig. 1). To probe why polymyxin B contains a hydrophobic alkyl chain in addition to a main peptide core, we modeled its peptide core using a linear peptide analog and screened it for membrane activity with the SVM classifier. The 10-amino acid long linear sequence (polymyxin B–v0) representing the peptide component of polymyxin B was not predicted to have a very high P(+1) score, in contrast with the substantially higher P(+1) values for the peptide analogs that contain additional hydrophobic residues to account for the hydrophobicity contributions from the alkyl chain (polymyxin B–v1, polymyxin B–v2) (Fig. 3A, Table 2). This suggests that the peptide core of polymyxin B alone is not capable of inducing the membrane disruption necessary for bactericidal activity, but incorporation of the hydrophobic alkyl chain will substantially raise the P(+1) values to the range in which that the molecule is likely to generate NGC and can facilitate a broader stoichiometric range over which it exhibits membrane activity (Fig. 3B). In fact, this finding is consistent with previous work that has shown that the removal of the fatty acid component of polymyxin B results in significant loss of antimicrobial activity [61–64]. Accordingly, the increase in antimicrobial activity as a result of greater hydrophobicity is a well-documented general effect in both natural AMPs and their analogs [25,65–69].
3.4. Machine-learning predictions of structure–activity relationships in polymyxin B mutants and comparison with empirical alanine scans
The contribution of each amino acid to a given peptide’s function is often inferred by performing an alanine scan on the sequence. In this manner, the peptide length is maintained while the predominant physicochemical contributions of each native amino acid are sequentially removed. Given that the SVM classifier evaluates a peptide using key learned physicochemical properties for membrane activity, it is natural to ask whether such classifications are sensitive to in silico alanine scan substitutions. As a case in point, we performed an alanine scan on the polymyxin B peptide analog, polymyxin B–v2, sequence (IIKTKAKFLKKT) starting at the first lysine. The P(+1) outputs from the classifier reflect the importance of cationic charges for membrane activity, as indicated by the decreased scores when positively charged lysines are substituted with alanines (Table 3). Interestingly, the scores agree with the previously reported in vitro results of single alanine substitutions in polymyxin B and their effects on antimicrobial activity. Specifically, alanine substitutions of cationic Dab residues at positions 5 or 9 were found to greatly reduce bactericidal activity of polymyxin B against E. coli, as demonstrated by the increased minimum inhibitory concentrations (MICs) of 16 nmol/mL and 4 nmol/mL, respectively, as compared to native polymyxin B, which has an MIC of 1 nmol/mL [70]. Our SVM classifier also predicted similar changes to antimicrobial activity, with corresponding sequences #5 and #9 having lower P(+1) values of ~0.60 and ~0.46, respectively, in comparison with ~0.83 for sequence #4 (Table 3). In addition, alanine substitution of Dab at position 3 of polymyxin B was observed to have less impact on antimicrobial activity, as evidenced by its MIC of 1 nmol/mL [70]. Our SVM prediction for sequence #3 also shows this effect, with a P(+1) value of ~0.87. Furthermore, SVM classifier predictions for alanine substitutions of other polymyxin B residues also tracked well with their experimental antimicrobial activities. Alanine substitution of threonine at position 2 led to a higher MIC [70], which is consistent with the lower P(+1) value of peptide sequence #2. Alanine substitutions of leucine and threonine at positions 7 and 10, respectively, resulted in little to no change in MIC [70] and correspond to respective peptide sequences #7 and #10, which maintain high P(+1) values. Collectively, these results indicate that our improvised SVM classifier is able to recapitulate empirical structure–activity relationships from alanine scans in polymyxin B surprisingly well.
3.5. Comparative analysis of cyclic antibiotics with prototypical linear membrane-active peptides
CPPs are peptides that are capable of efficient direct translocation across cell membranes without causing lytic membrane damage. Like AMPs, most CPPs are short, positively charged peptides that interact with negatively charged membranes [71–76]. Both AMPs and CPPs feature a high proportion of cationic residues and exert their membrane-permeating effects via mechanisms that engage NGC generation [35,37,77]. Due to their similarities, many have questioned whether AMPs and CPPs actually should be categorized as two unrelated classes [78,79]. In fact, increasing evidence indicates that some AMPs, including LL-37 and magainin 2, possess the ability to cross cell membranes [79–81], and certain CPPs, such as penetratin, display significant antimicrobial activity [82,83]. This cross-functionality suggests that AMPs and CPPs may not belong to two distinct peptide classes, but rather comprise a larger family of membrane-active peptides that exhibit a diverse range of antimicrobial activity and translocation ability.
If we consider AMPs and CPPs collectively, we can identify key behavioral trends relating to their physicochemical properties and induced membrane curvature. Studies have observed that on average, AMPs are more hydrophobic than CPPs [37,84] (Table 4). Interestingly, we found that the average magnitude of NGC, |<K>|, generated by CPPs tends to be greater than that induced by AMPs. (For uniformity, these comparisons are made typically with model membranes at lipid compositions with 20% anionic lipids and 80% PE lipids with negative intrinsic curvature.) Taken together, these trends point to an inverse relationship between hydrophobicity and |<K>| among membrane-active peptides. More specifically, we noted that AMPs, which are characterized by greater hydrophobicity, most often generate modest magnitudes of NGC, whereas CPPs typically have lower hydrophobicity and higher magnitudes of NGC. Then by comparing the hydrophobicities and NGCs of the peptide-based cyclic antibiotics (bactenecin and polymyxin B) with those of known AMPs and CPPs, we find that the antibiotics indeed are more AMP-like than CPP-like. But can these trends tell us more about the differences in behavior between AMPs and CPPs? It has been suggested that for a membrane-active peptide, increased hydrophobicity is associated with increased stability of the membrane pore, presumably as a result of having longer membrane residence times [37,85,86]. Therefore, it would make sense that AMPs, being more hydrophobic and thereby forming stable pores, are generally found to be more lytic, in contrast to CPPs, which tend to create transient pores and do not cause significant membrane damage [65,87,88]. As we have found that the level of hydrophobicity can offer temporal information about the produced membrane pore, we hypothesize that the magnitude of NGC can provide clues about the spatial character of the pore, given that additional hydrophobic insertion can have steric consequences for the curvature of the resultant pore. Previous groups have studied the effects of inserting a peptide or protein into a membrane and have found that, in response, a membrane will typically alter its thickness to match that of the inclusion in order to prevent energetically unfavorable exposure of its hydrophobic regions to a hydrophilic environment and to relieve intramembrane stresses and strains. This effect will often lead to changes in the membrane thickness and curvature by stretching, compressing, or tilting the constituent lipids [89–93]. However, we hypothesize there to be less freedom for dimensional changes in the direction normal to the membrane surface than laterally along the membrane surface due to the inherent limitations of membrane thickness changes. Thus, for membrane pores induced by AMPs or CPPs, we anticipate different magnitudes of NGC to be predominantly reflected in differences in pore size (Fig. 2D). For example, we speculate that between two given peptides, the one that induces a lower magnitude of NGC will exhibit a structural tendency to produce a larger pore. Hence, in general, we would expect larger pores for AMPs and smaller pores for CPPs. Remarkably, we discover that this is in fact consistent with empirical measurements of pore sizes for AMPs and CPPs. AMPs have been observed to create a wide range of pore sizes, with diameters of 1.3–3 nm for melittin [94,95], 2–5 nm for magainin [96,97], 2.3–3.3 nm for LL-37 [98], 2.5 nm for defensin human neutrophil peptide-2 [99], 4 nm for cecropin [100], 4.6 nm for lacticin Q [101], and 9 nm for protegrin-1 [102]. In contrast, CPPs exhibit seemingly smaller pores, with HIV-TAT producing 1.3–2 nm pores [103], 0.66 nm for polyarginine-9 [104], and no detectable pores for pep-1 [105].
Table 4. Antibiotic membrane activity relative to that of known AMPs and CPPs.
In general, AMPs are more hydrophobic than CPPs. However, we observe that the average magnitudes of NGC, |<K>|, generated by CPPs tend to be greater than that of AMPs. Together, this suggests an inverse relationship between hydrophobicity and NGC. Comparing the |<K>| induced by the cyclic antibiotics (in bold) and the average hydrophobicities of their respective peptide analogs, we see that their membrane activities are more AMP-like than CPP-like.
| Molecule | |<K>| (10−2 nm−2) 20/80 DOPG/DOPE Ac/Lc = 1/1 |
|<K>| (10−2 nm−2) 20/80 DOPS/DOPE Ac/Lc = 1/1 |
Peptide Analog Variant (for antibiotics) see Table 2 for sequences | Peptide <Hydrophobicity> (Eisenberg Consensus Scale) |
|---|---|---|---|---|
| Melittin (AMP) | 1.85 | −0.09 | ||
| Polymyxin B | 1.15 | 1.16 | Polymyxin B-v1 | −0.34 |
| Polymyxin B-v2 | −0.25 | |||
| Bactenecin | 1.23 | 2.19 | −0.29 | |
| RTD-1 (cyclic AMP) | 2.56 | −0.35 | ||
| BTD-1 (cyclic AMP) | 2.56 | −0.35 | ||
| Crp-4 (AMP) | 5.31 | −0.36 | ||
| HBD-3 (AMP) | 5.02 | −0.38 | ||
| PG-1 (AMP) | 4.62 | −0.44 | ||
| Penetratin (CPP) | 2.59 | −0.55 | ||
| HIV TAT (CPP) | 5.72 | −1.23 |
4. Conclusions
We examined a diverse collection of cyclic antibiotics, consisting of bactenecin, polymyxin B, octapeptin, capreomycin, and Kirshenbaum peptoids, in order to identify what they have in common when they interact with bacterial lipid membranes, using synchrotron X-ray diffraction and machine-learning methods. We found that all except capreomycin have the ability to induce NGC in bacterial membranes, the type of curvature geometrically required for permeation mechanisms such as pore formation, blebbing, and budding. To examine their usage of amino acids compared to that in AMPs, we analyzed amino acid sequences of bactenecin, polymyxin B, and capreomycin using a machine-learning classifier trained on α-helical AMP sequences. Although the classifier was not trained on cyclic antibiotics or peptides containing non-proteinogenic amino acids, a modified implementation correctly predicted that bactenecin and polymyxin B have the ability to induce NGC in membranes, while capreomycin does not. Moreover, a modified approach to implementing the classifier was able to recapitulate empirical structure–activity relationships from alanine scans in polymyxin B surprisingly well. Taken together, these results suggest that the sequence design of hybrid cyclic antibiotics and linear AMPs are cognate.
Supplementary Material
Acknowledgments
M.W.L. and G.C.L.W. are supported by F. Hoffmann-La Roche Ltd. G.C.L.W. acknowledges additional support from NSF DMR1808459 and NIH R01AI143730. J.d.A. is supported by the NSF GRFP DGE-1650604. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Abbreviations:
- NGC
negative Gaussian curvature
- AMP
antimicrobial peptide
- DOPG
1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- DOPS
1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- ULV
unilamellar vesicle
- Ac/Lc
antibiotic-to-lipid charge ratio
- SAXS
small-angle X-ray scattering
- SVM
support vector machine
- Dab
α,γ-diaminobutyric acid
- A/L
antibiotic-to-lipid molar ratio
- CPP
cell-penetrating peptide
- QII
inverse bicontinuous cubic phase
- PE
phosphatidylethanolamine
- MIC
minimum inhibitory concentration
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbamem.2020.183302.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].CDC, Antibiotic Resistance Threats in the United States, 2019 U.S. Department of Health and Human Services, CDC, Atlanta, GA, 2019. [Google Scholar]
- [2].Brown ED, Wright GD, Antibacterial drug discovery in the resistance era, Nature 529 (2016) 336–343. [DOI] [PubMed] [Google Scholar]
- [3].Davies J, Davies D, Origins and evolution of antibiotic resistance, Microbiol. Mol. Biol. Rev 74 (2010) 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, Ouellette M, Outterson K, Patel J, Cavaleri M, Cox EM, Houchens CR, Grayson ML, Hansen P, Singh N, Theuretzbacher U, Magrini N, Aboderin AO, Al-Abri SS, Awang Jalil N, Benzonana N, Bhattacharya S, Brink AJ, Burkert FR, Cars O, Cornaglia G, Dyar OJ, Friedrich AW, Gales AC, Gandra S, Giske CG, Goff DA, Goossens H, Gottlieb T, Guzman Blanco M, Hryniewicz W, Kattula D, Jinks T, Kanj SS, Kerr L, Kieny M-P, Kim YS, Kozlov RS, Labarca J, Laxminarayan R, Leder K, Leibovici L, Levy-Hara G, Littman J, Malhotra-Kumar S, Manchanda V, Moja L, Ndoye B, Pan A, Paterson DL, Paul M, Qiu H, Ramon-Pardo P, Rodríguez-Baño J, Sanguinetti M, Sengupta S, Sharland M, Si-Mehand M, Silver LL, Song W, Steinbakk M, Thomsen J, Thwaites GE, van der Meer JWM, Van Kinh N, Vega S, Villegas MV, Wechsler-Fördös A, Wertheim HFL, Wesangula E, Woodford N, Yilmaz FO, Zorzet A, Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis, Lancet Infect. Dis 18 (2018) 318–327. [DOI] [PubMed] [Google Scholar]
- [5].CDC, Antibiotic Resistance Threats in the United States, U.S. Department of Health and Human Services, CDC, Atlanta, GA, 2013, p. 2013. [Google Scholar]
- [6].Hancock REW, Sahl H-G, Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies, Nat. Biotechnol 24 (2006) 1551–1557. [DOI] [PubMed] [Google Scholar]
- [7].Lee EY, Fulan BM, Wong GCL, Ferguson AL, Mapping membrane activity in undiscovered peptide sequence space using machine learning, Proc. Natl. Acad. Sci 113 (2016) 13588–13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Madhongsa K, Pasan S, Phophetleb O, Nasompag S, Thammasirirak S, Daduang S, Taweechaisupapong S, Lomize AL, Patramanon R, Antimicrobial action of the cyclic peptide bactenecin on Burkholderia pseudomallei correlates with efficient membrane permeabilization, PLoS Negl. Trop. Dis 7 (2013) e2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu M, Hancock REW, Interaction of the cyclic antimicrobial cationic peptide bactenecin with the outer and cytoplasmic membrane, J. Biol. Chem 274 (1999) 29–35. [DOI] [PubMed] [Google Scholar]
- [10].Zavascki AP, Goldani LZ, Li J, Nation RL, Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review, J. Antimicrob. Chemother 60 (2007) 1206–1215. [DOI] [PubMed] [Google Scholar]
- [11].Velkov T, Thompson PE, Nation RL, Li J, Structure–activity relationships of polymyxin antibiotics, J. Med. Chem 53 (2010) 1898–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Velkov T, Roberts KD, Li J, Rediscovering the octapeptins, Nat. Prod. Rep 34 (2017) 295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang ML, Shin SBY, Benson MA, Torres VJ, Kirshenbaum K, A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents, ChemMedChem 7 (2012) 114–122. [DOI] [PubMed] [Google Scholar]
- [14].Huang ML, Benson MA, Shin SBY, Torres VJ, Kirshenbaum K, Amphiphilic cyclic peptoids that exhibit antimicrobial activity by disrupting Staphylococcus aureus membranes, Eur. J. Org. Chem 2013 (2013) 3560–3566. [Google Scholar]
- [15].Smith PT, Huang ML, Kirshenbaum K, Osmoprotective polymer additives attenuate the membrane pore-forming activity of antimicrobial peptoids, Biopolymers 103 (2015) 227–236. [DOI] [PubMed] [Google Scholar]
- [16].Reisfeld B, Metzler CP, Lyons MA, Mayeno AN, Brooks EJ, DeGroote MA, A physiologically based pharmacokinetic model for capreomycin, Antimicrob. Agents Chemother 56 (2012) 926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fu LM, Fu-Liu CS, Is Mycobacterium tuberculosis a closer relative to gram-positive or gram–negative bacterial pathogens? Tuberculosis 82 (2002) 85–90. [DOI] [PubMed] [Google Scholar]
- [18].Taber HW, Mueller JP, Miller PF, Arrow AS, Bacterial uptake of aminoglycoside antibiotics, Microbiol. Rev 51 (1987) 439–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huth ME, Ricci AJ, Cheng AG, Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection, International Journal of Otolaryngology 2011 (2011) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Poulikakos P, Falagas ME, Aminoglycoside therapy in infectious diseases, Expert. Opin. Pharmacother 14 (2013) 1585–1597. [DOI] [PubMed] [Google Scholar]
- [21].Castañeda-García A, Do TT, Blázquez J, The K+ uptake regulator TrkA controls membrane potential, pH homeostasis and multidrug susceptibility in Mycobacterium smegmatis, J. Antimicrob. Chemother 66 (2011) 1489–1498. [DOI] [PubMed] [Google Scholar]
- [22].Ilavsky J, Nika: software for two-dimensional data reduction, J. Appl. Crystallogr 45 (2012) 324–328. [Google Scholar]
- [23].Hammersley AP, FIT2D: an introduction and overview, European Synchrotron Radiation Facility Internal Report, ESRF97HA02T (1997). [Google Scholar]
- [24].Zasloff M, Antimicrobial peptides of multicellular organisms, Nature 415 (2002) 389–395. [DOI] [PubMed] [Google Scholar]
- [25].Brogden KA, Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Micro 3 (2005) 238–250. [DOI] [PubMed] [Google Scholar]
- [26].Shai Y, Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides, Biochim. Biophys. Acta Biomembr 1462 (1999) 55–70. [DOI] [PubMed] [Google Scholar]
- [27].Matsuzaki K, Sugishita K.-i., Ishibe N, Ueha M, Nakata S, Miyajima K, Epand RM, Relationship of membrane curvature to the formation of pores by magainin 2, Biochemistry 37 (1998) 11856–11863. [DOI] [PubMed] [Google Scholar]
- [28].Matsuzaki K, Why and how are peptide–lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes, Biochim. Biophys. Acta Biomembr 1462 (1999) 1–10. [DOI] [PubMed] [Google Scholar]
- [29].Kalfa VC, Jia HP, Kunkle RA, McCray PB, Tack BF, Brogden KA, Congeners of SMAP29 kill ovine pathogens and induce Ultrastructural damage in bacterial cells, Antimicrob. Agents Chemother 45 (2001) 3256–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Saiman L, Tabibi S, Starner TD, San Gabriel P, Winokur PL, Jia HP, McCray PB, Tack BF, Cathelicidin peptides inhibit multiply antibiotic-resistant pathogens from patients with cystic fibrosis, Antimicrob. Agents Chemother 45 (2001) 2838–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Staudegger E, Prenner EJ, Kriechbaum M, Degovics G, Lewis RNAH, McElhaney RN, Lohner K, X-ray studies on the interaction of the antimicrobial peptide gramicidin S with microbial lipid extracts: evidence for cubic phase formation, Biochim. Biophys. Acta Biomembr 1468 (2000) 213–230. [DOI] [PubMed] [Google Scholar]
- [32].Hickel A, Danner-Pongratz S, Amenitsch H, Degovics G, Rappolt M, Lohner K, Pabst G, Influence of antimicrobial peptides on the formation of nonlamellar lipid mesophases, Biochim. Biophys. Acta Biomembr 1778 (2008) 2325–2333. [DOI] [PubMed] [Google Scholar]
- [33].Prenner EJ, Lewis RNAH, Neuman KC, Gruner SM, Kondejewski LH, Hodges RS, McElhaney RN, Nonlamellar phases induced by the interaction of gramicidin S with lipid bilayers. A possible relationship to membrane-disrupting activity, Biochemistry 36 (1997) 7906–7916. [DOI] [PubMed] [Google Scholar]
- [34].Keller SL, Gruner SM, Gawrisch K, Small concentrations of alamethicin induce a cubic phase in bulk phosphatidylethanolamine mixtures, Biochim. Biophys. Acta Biomembr 1278 (1996) 241–246. [DOI] [PubMed] [Google Scholar]
- [35].Schmidt NW, Mishra A, Lai GH, Davis M, Sanders LK, Tran D, Garcia A, Tai KP, McCray PB, Ouellette AJ, Selsted ME, Wong GCL, Criterion for amino acid composition of Defensins and antimicrobial peptides based on Geometry of membrane destabilization, J. Am. Chem. Soc 133 (2011) 6720–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schmidt NW, Tai KP, Kamdar K, Mishra A, Lai GH, Zhao K, Ouellette AJ, Wong GCL, Arginine in α-Defensins: differential effects on bactericidal activity correspond to geometry of membrane curvature generation and peptide-lipid phase behavior, J. Biol. Chem 287 (2012) 21866–21872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mishra A, Lai GH, Schmidt NW, Sun VZ, Rodriguez AR, Tong R, Tang L, Cheng J, Deming TJ, Kamei DT, Wong GCL, Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions, Proc. Natl. Acad. Sci 108 (2011) 16883–16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yao H, Lee MW, Waring AJ, Wong GCL, Hong M, Viral fusion protein transmembrane domain adopts β-strand structure to facilitate membrane topological changes for virus–cell fusion, Proc. Natl. Acad. Sci 112 (2015) 10926–10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schmidt NW, Mishra A, Wang J, DeGrado WF, Wong GCL, Influenza virus a M2 protein generates negative Gaussian membrane curvature necessary for budding and scission, J. Am. Chem. Soc 135 (2013) 13710–13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee MW, Han M, Bossa GV, Snell C, Song Z, Tang H, Yin L, Cheng J, May S, Luijten E, Wong GCL, Interactions between membranes and “Metaphilic” polypeptide architectures with diverse side-chain populations, ACS Nano 11 (2017) 2858–2871. [DOI] [PubMed] [Google Scholar]
- [41].Lee MW, Lee EY, Lai GH, Kennedy NW, Posey AE, Xian W, Ferguson AL, Hill RB, Wong GCL, Molecular motor Dnm1 synergistically induces membrane curvature to facilitate mitochondrial fission, ACS Central Science 3 (2017) 1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Seddon JM, Templer RH, Chapter 3 - polymorphism of lipid-water systems, in: Lipowsky R, Sackmann E(Eds.), Handbook of Biological Physics, Volume 1 1995, pp. 97–160 North-Holland. [Google Scholar]
- [43].Shearman GC, Ces O, Templer RH, Seddon JM, Inverse lyotropic phases of lipids and membrane curvature, J. Phys. Condens. Matter 18 (2006) S1105. [DOI] [PubMed] [Google Scholar]
- [44].Epand RM, Epand RF, Lipid domains in bacterial membranes and the action of antimicrobial agents, Biochim. Biophys. Acta Biomembr 1788 (2009) 289–294. [DOI] [PubMed] [Google Scholar]
- [45].Epand RF, Savage PB, Epand RM, Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins), Biochim. Biophys. Acta Biomembr 1768 (2007) 2500–2509. [DOI] [PubMed] [Google Scholar]
- [46].Cronan JE, Bacterial membrane lipids: where Do we stand? Annu. Rev. Microbiol 57 (2003) 203–224. [DOI] [PubMed] [Google Scholar]
- [47].Morein S, Andersson A-S, Rilfors L, Lindblom G, Wild-type Escherichia coli cells regulate the membrane lipid composition in a window between gel and non-lamellar structures, J. Biol. Chem 271 (1996) 6801–6809. [DOI] [PubMed] [Google Scholar]
- [48].Lugtenberg EJJ, Peters R, Distribution of lipids in cytoplasmic and outer membranes of Escherichia coli K12, Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism 441 (1976) 38–47. [DOI] [PubMed] [Google Scholar]
- [49].Teixeira V, Feio MJ, Bastos M, Role of lipids in the interaction of antimicrobial peptides with membranes, Prog. Lipid Res 51 (2012) 149–177. [DOI] [PubMed] [Google Scholar]
- [50].Kondakova T, D’Heygère F, Feuilloley MJ, Orange N, Heipieper HJ, Duclairoir Poc C, Glycerophospholipid synthesis and functions in Pseudomonas, Chem. Phys. Lipids 190 (2015) 27–42. [DOI] [PubMed] [Google Scholar]
- [51].Kabelka I, Pachler M, Prévost S, Letofsky-Papst I, Lohner K, Pabst G, Vácha R, Magainin 2 and PGLa in bacterial membrane mimics II: membrane fusion and sponge phase formation, Biophys. J 118 (2020) 612–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hu K, Schmidt NW, Zhu R, Jiang Y, Lai GH, Wei G, Palermo EF, Kuroda K, Wong GCL, Yang L, A critical evaluation of random copolymer mimesis of homogeneous antimicrobial peptides, Macromolecules 46 (2013) 1908–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee MW, Chakraborty S, Schmidt NW, Murgai R, Gellman SH, Wong GCL, Two interdependent mechanisms of antimicrobial activity allow for efficient killing in nylon-3-based polymeric mimics of innate immunity peptides, Biochim. Biophys. Acta Biomembr 1838 (2014) 2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xiong M, Lee MW, Mansbach RA, Song Z, Bao Y, Peek RM, Yao C, Chen L-F, Ferguson AL, Wong GCL, Cheng J, Helical antimicrobial polypeptides with radial amphiphilicity, Proc. Natl. Acad. Sci 112 (2015) 13155–13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dawaliby R, Trubbia C, Delporte C, Noyon C, Ruysschaert J-M, Van Antwerpen P, Govaerts C, Phosphatidylethanolamine is a key regulator of membrane fluidity in eukaryotic cells, J. Biol. Chem 291 (2015) 3658–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].van Meer G, Voelker DR, Feigenson GW, Membrane lipids: where they are and how they behave, Nat Rev Mol Cell Biol 9 (2008) 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Heifets L, Simon J, Pham V, Capreomycin is active against non-replicating M. tuberculosis, Ann. Clin. Microbiol. Antimicrob 4 (2005) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yount NY, Weaver DC, Lee EY, Lee MW, Wang H, Chan LC, Wong GCL, Yeaman MR, Unifying structural signature of eukaryotic α-helical host defense peptides, Proc. Natl. Acad. Sci 116 (2019) 6944–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].De Zoysa GH, Cameron AJ, Hegde VV, Raghothama S, Sarojini V, Antimicrobial peptides with potential for biofilm eradication: synthesis and structure activity relationship studies of Battacin peptides, J. Med. Chem 58 (2015) 625–639. [DOI] [PubMed] [Google Scholar]
- [60].Orwa JA, Govaerts C, Busson R, Roets E, Van Schepdael A, Hoogmartens J, Isolation and structural characterization of polymyxin B components, J. Chromatogr. A 912 (2001) 369–373. [DOI] [PubMed] [Google Scholar]
- [61].Vaara M, Vaara T, Polycations as outer membrane-disorganizing agents, Antimicrob. Agents Chemother 24 (1983) 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Vaara M, Vaara T, Polycations sensitize enteric bacteria to antibiotics, Antimicrob. Agents Chemother 24 (1983) 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Okimura K, Ohki K, Sato Y, Ohnishi K, Uchida Y, Sakura N, Chemical conversion of natural Polymyxin B and Colistin to their N-terminal derivatives, Bull. Chem. Soc. Jpn 80 (2007) 543–552. [Google Scholar]
- [64].Sakura N, Itoh T, Uchida Y, Ohki K, Okimura K, Chiba K, Sato Y, Sawanishi H, The contribution of the N-terminal structure of Polymyxin B peptides to antimicrobial and lipopolysaccharide binding activity, Bull. Chem. Soc. Jpn 77 (2004) 1915–1924. [Google Scholar]
- [65].Wieprecht T, Dathe M, Epand RM, Beyermann M, Krause E, Maloy WL, MacDonald DL, Bienert M, Influence of the angle subtended by the positively charged Helix face on the membrane activity of amphipathic, antibacterial peptides, Biochemistry 36 (1997) 12869–12880. [DOI] [PubMed] [Google Scholar]
- [66].Dathe M, Schümann M, Wieprecht T, Winkler A, Beyermann M, Krause E, Matsuzaki K, Murase O, Bienert M, Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes, Biochemistry 35 (1996) 12612–12622. [DOI] [PubMed] [Google Scholar]
- [67].Chen Y, Mant CT, Farmer SW, Hancock REW, Vasil ML, Hodges RS, Rational design of α-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index, J. Biol. Chem 280 (2005) 12316–12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chen Y, Guarnieri MT, Vasil AI, Vasil ML, Mant CT, Hodges RS, Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides, Antimicrob. Agents Chemother 51 (2007) 1398–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yin LM, Edwards MA, Li J, Yip CM, Deber CM, Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions, J. Biol. Chem 287 (2012) 7738–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kanazawa K, Sato Y, Ohki K, Okimura K, Uchida Y, Shindo M, Sakura N, Contribution of each amino acid residue in Polymyxin B3 to antimicrobial and lipopolysaccharide binding activity, Chem. Pharm. Bull 57 (2009) 240–244. [DOI] [PubMed] [Google Scholar]
- [71].Bechara C, Sagan S, Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 587 (2013) 1693–1702. [DOI] [PubMed] [Google Scholar]
- [72].Heitz F, Morris MC, Divita G, Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics, Br. J. Pharmacol 157 (2009) 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Copolovici DM, Langel K, Eriste E, Langel Ü, Cell-penetrating peptides: design, synthesis, and applications, ACS Nano 8 (2014) 1972–1994. [DOI] [PubMed] [Google Scholar]
- [74].Koren E, Torchilin VP, Cell-penetrating peptides: breaking through to the other side, Trends Mol. Med 18 (2012) 385–393. [DOI] [PubMed] [Google Scholar]
- [75].Milletti F, Cell-penetrating peptides: classes, origin, and current landscape, Drug Discov. Today 17 (2012) 850–860. [DOI] [PubMed] [Google Scholar]
- [76].Zaro JL, Shen W-C, Cationic and amphipathic cell-penetrating peptides (CPPs): their structures and in vivo studies in drug delivery, Front. Chem. Sci. Eng 9 (2015) 407–427. [Google Scholar]
- [77].Schmidt NW, Lis M, Zhao K, Lai GH, Alexandrova AN, Tew GN, Wong GCL, Molecular basis for Nanoscopic membrane curvature generation from quantum mechanical models and synthetic transporter sequences, J. Am. Chem. Soc 134 (2012) 19207–19216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Henriques Sónia T., Melo Manuel N., Castanho Miguel A.R.B., Cell-penetrating peptides and antimicrobial peptides: how different are they? Biochem. J 399 (2006) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Splith K, Neundorf I, Antimicrobial peptides with cell-penetrating peptide properties and vice versa, Eur. Biophys. J 40 (2011) 387–397. [DOI] [PubMed] [Google Scholar]
- [80].Matsuzaki K, Murase O, Fujii N, Miyajima K, Translocation of a channel-forming antimicrobial peptide, Magainin 2, across lipid bilayers by forming a pore, Biochemistry 34 (1995) 6521–6526. [DOI] [PubMed] [Google Scholar]
- [81].Zhang X, Oglęcka K, Sandgren S, Belting M, Esbjörner EK, Nordén B, Gräslund A, Dual functions of the human antimicrobial peptide LL-37—target membrane perturbation and host cell cargo delivery, Biochim. Biophys. Acta Biomembr 1798 (2010) 2201–2208. [DOI] [PubMed] [Google Scholar]
- [82].Zhu WL, Shin SY, Antimicrobial and Cytolytic activities and plausible mode of bactericidal action of the cell penetrating peptide Penetratin and its Lys-linked two-stranded peptide, Chem. Biol. Drug Des 73 (2009) 209–215. [DOI] [PubMed] [Google Scholar]
- [83].Nekhotiaeva N, Elmquist A, Rajarao GK, Hällbrink M, Langel Ü, Good L, Cell entry and antimicrobial properties of eukaryotic cell-penetrating peptides, FASEB J. 18 (2004) 394–396. [DOI] [PubMed] [Google Scholar]
- [84].Kauffman WB, Fuselier T, He J, Wimley WC, Mechanism matters: a taxonomy of cell penetrating peptides, Trends Biochem. Sci 40 (2015) 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Matsuzaki K, Murase O, Miyajima K, Kinetics of pore formation by an antimicrobial peptide, Magainin 2, in phospholipid bilayers, Biochemistry 34 (1995) 12553–12559. [DOI] [PubMed] [Google Scholar]
- [86].Herce HD, Garcia AE, Cell penetrating peptides: how Do they Do it? J. Biol. Phys 33 (2008) 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Cornut I, Büttner K, Dasseux J-L, Dufourcq J, The amphipathic α-helix concept: application to the de novo design of ideally amphipathic Leu, Lys peptides with hemolytic activity higher than that of melittin, FEBS Lett. 349 (1994) 29–33. [DOI] [PubMed] [Google Scholar]
- [88].Dathe M, Wieprecht T, Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells, Biochim. Biophys. Acta Biomembr 1462 (1999) 71–87. [DOI] [PubMed] [Google Scholar]
- [89].Killian JA, Hydrophobic mismatch between proteins and lipids in membranes, Biochim. Biophys. Acta Rev. Biomembr 1376 (1998) 401–416. [DOI] [PubMed] [Google Scholar]
- [90].Campelo F, McMahon HT, Kozlov MM, The hydrophobic insertion mechanism of membrane curvature generation by proteins, Biophys. J 95 (2008) 2325–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ben-Shaul A, Ben-Tal N, Honig B, Statistical thermodynamic analysis of peptide and protein insertion into lipid membranes, Biophys. J 71 (1996) 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Marsh D, Lateral pressure profile, spontaneous curvature frustration, and the incorporation and conformation of proteins in membranes, Biophys. J 93 (2007) 3884–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lundbæk JA, Maer AM, Andersen OS, Lipid bilayer electrostatic energy, curvature stress, and assembly of gramicidin channels, Biochemistry 36 (1997) 5695–5701. [DOI] [PubMed] [Google Scholar]
- [94].Matsuzaki K, Yoneyama S, Miyajima K, Pore formation and translocation of melittin, Biophys. J 73 (1997) 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ladokhin AS, Selsted ME, White SH, Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: pore formation by melittin, Biophys. J 72 (1997) 1762–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Takeshima K, Chikushi A, Lee K-K, Yonehara S, Matsuzaki K, Translocation of analogues of the antimicrobial peptides Magainin and Buforin across human cell membranes, J. Biol. Chem 278 (2003) 1310–1315. [DOI] [PubMed] [Google Scholar]
- [97].Yang L, Harroun TA, Weiss TM, Ding L, Huang HW, Barrel-stave model or Toroidal model? A case study on Melittin pores, Biophys. J 81 (2001) 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lee C-C, Sun Y, Qian S, Huey W Huang, Transmembrane pores formed by human antimicrobial peptide LL-37, Biophys. J 100 (2011) 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wimley WC, Selsted ME, White SH, Interactions between human defensins and lipid bilayers: evidence for formation of multimeric pores, Protein Sci. 3 (1994) 1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Christensen B, Fink J, Merrifield RB, Mauzerall D, Channel-forming properties of cecropins and related model compounds incorporated into planar lipid membranes, Proc. Natl. Acad. Sci 85 (1988) 5072–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Yoneyama F, Imura Y, Ohno K, Zendo T, Nakayama J, Matsuzaki K, Sonomoto K, Peptide-lipid huge Toroidal pore, a new antimicrobial mechanism mediated by a Lactococcal Bacteriocin, Lacticin Q, Antimicrob. Agents Chemother 53 (2009) 3211–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Lam KLH, Wang H, Siaw TA, Chapman MR, Waring AJ, Kindt JT, Lee KYC, Mechanism of structural transformations induced by antimicrobial peptides in lipid membranes, Biochim. Biophys. Acta Biomembr 1818 (2012) 194–204. [DOI] [PubMed] [Google Scholar]
- [103].Ciobanasu C, Siebrasse JP, Kubitscheck U, Cell-penetrating HIV1 TAT peptides can generate pores in model membranes, Biophys. J 99 (2010) 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Herce HD, Garcia AE, Litt J, Kane RS, Martin P, Enrique N, Rebolledo A, Milesi V, Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides, Biophys. J 97 (2009) 1917–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Henriques ST, Quintas A, Bagatolli LA, Homblé F, Castanho MARB, Energy-independent translocation of cell-penetrating peptides occurs without formation of pores. A biophysical study with pep-1, Mol. Membr. Biol 24 (2007) 282–293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.