

SUMMARY
Cardiac cells generate and amplify force in the context of cardiac load, yet the membranous sheath enclosing the muscle fibers—the sarcolemma—does not experience displacement. That the sarcolemma sustains beat-to-beat pressure changes without experiencing significant distortion is a muscle-contraction paradox. Here, we report that an elastic element—the motor protein prestin (Slc26a5)—serves to amplify actin-myosin force generation in mouse and human cardiac myocytes, accounting partly for the nonlinear capacitance of cardiomyocytes. The functional significance of prestin is underpinned by significant alterations of cardiac contractility in Prestin-knockout mice. Prestin was previously considered exclusive to the inner ear’s outer hair cells; however, our results show that prestin serves a broader cellular motor function.
In brief
Zhang et al. show that the motor protein prestin (Slc26a5) is expressed in the heart, serving to amplify actin-myosin force generation in cardiomyocytes. Prestin is considered to be expressed exclusively in outer hair cells of the inner ear. However, this study demonstrates prestin’s broader cellular motor function.
Graphical Abstract
INTRODUCTION
The human heart is a durable biological motor, beating approximately 3.4 billion times in an 80-year lifespan. Actual cardiac performance originates in cardiac myocytes, which are among the few cell types that are electromotile, changing in length in response to a change in voltage. Groundbreaking work by Huxley and Hanson (1954) in the 1950s, derived from high-resolution microscopy, introduced the “sliding filament theory.” During muscle contraction, myosin, a motor protein, and the filamentous actin protein form cross-bridges, allowing myosin to slide along actin, leading to length changes and force generation (Huxley and Niedergerke, 1954).
Some muscle contraction mechanisms remain unexplained, such as how the sarcolemma, the membranous sheath enclosing the muscle fibers, accommodates ~0.2-μm-displacement length changes per sarcomere in a cardiac cycle without experiencing significant distortion—a “muscle-contraction paradox.” In muscle cells exhibiting length-tension cycles, a nonlinear amplification mechanism for force generation has been proposed (Ronceray et al., 2016). The identity of the force-element perpendicular to the z-line during sarcomere shortening, responsible for regulating cardiac power output and amplifying force, has been elusive.
Here, we invoke and test for the presence and function of a canonical non-conventional motor protein, prestin (Slc26a5), in cardiomyocytes as an amplifier of actin-myosin force generation to account for changes in nonlinear capacitance (NLC) during myocyte contraction. Prestin is a member of the mammalian solute carrier (Slc26a) gene family with distinct properties because of its molecular motor function in contrast to the predominant anion transport functions of other members (Alper and Sharma, 2013; Mount and Romero, 2004; Ohana et al., 2009). Prestin has been thought to be expressed exclusively in outer hair cells (OHCs) of the inner ear (Liberman et al., 2002; Zheng et al., 2000), where it is a direct voltage-to-force convertor and mediates electromotility of OHCs (Dallos, 2008; Dallos and Fakler, 2002; Dallos et al., 2006; He et al., 2014; Zheng et al., 2002). Prestin is part of the molecular element of cochlear sound amplification.
The motor function of prestin relies on the activities of intracellular Cl− ions, which serve as the extrinsic voltage sensor (Oliver et al., 2001, 2006) or allosteric modulators of the intrinsic voltage sensor (Rybalchenko and Santos-Sacchi, 2003; Song and Santos-Sacchi, 2010). A structural model of prestin shows a central cavity as the substrate-binding site and ion translocation pathway, suggesting the independent function of each subunit, which is also supported by available data from several reports (Bian et al., 2013; Gorbunov et al., 2014; Hallworth and Nichols, 2012; Homma et al., 2013; Wang et al., 2010). The homo-oligomeric interaction of prestin has also been reported (Bai et al., 2010; Detro-Dassen et al., 2008; Gleitsman et al., 2009; Greeson et al., 2006; Navaratnam et al., 2005).
Here, we test the hypothesis that prestin is expressed in mouse and human cardiomyocytes and serves as the cardiac mechanical amplifier. We investigated whether the genetic deletion of Slc26a5 in Prestin knockout (Slc26a5−/−) mice produces changes in in vivo and in vitro cardiac contractility. Multimodal second harmonic generation (SHG)-two-photon fluorescence (TPF) microscopy (Awasthi et al., 2016) and stimulated emission depletion (STED) microscopy analyses were further used to determine the expression and functional roles of prestin in ventricular myocytes.
RESULTS
Cloning and identification of prestin in cardiac myocytes
We successfully identified and cloned full-length prestin using reverse transcription-polymerase chain reaction (RT-PCR) from mouse atrial (ACM) and ventricular cardiomyocytes (VCM) (Figure 1A). Additionally, we identified and cloned prestin from human ventricles (hVCM, Figure 1B), the same as those found in OHCs (Liberman et al., 2002; Liu et al., 2003; Zheng et al., 2000). Primers were designed from mouse and human OHC prestin, respectively (Figures 1 and S1). A single band with the expected size of 2.2 Kb was obtained (Figure 1). Sequence analysis identified a complete open reading frame (ORF) encoding a putative protein with 744 amino acids with sequence identity between OHC-specific prestin and the cardiac clones.
Figure 1. Cloning and identification of mouse and human cardiac prestin cDNA.

(A) The amplified, full-length cardiac prestin cDNA from mouse inner ear, atrial (ACM), and ventricular cardiomyocytes (VCM) was obtained using inner ear cDNA as a positive control. The expression level of prestin is higher in mouse ventricular compared with that of atrial myocytes. The full-length ORF of the same size (~2.2 kb) was obtained from ACM and VCM.
(B) Cloning of human cardiac prestin (~2.2 kb). NTC, no template control.
(C) Expression of prestin detected by single-cell qPCR. Slc26a6, a member of the same gene family as prestin, was used as a control (*p < 0.05).
(D) Photomicrograph from smFISH using probes specific for Slc26a5. Single-ventricular myocytes were isolated from WT and compared with Slc26a5−/−.
(E) Photomicrograph from smFISH using probes specific for Slc26a6. Cells were counterstained using monoclonal anti-α-actinin2 antibodies.
To obtain additional evidence that prestin is expressed in cardiomyocytes and not in non-contractile cells in the heart, we performed single-cell qPCR (Figure 1C) and single-molecule fluorescence in situ hybridization (smFISH) (Figures 1D and 1E), using mouse isolated ventricular myocytes. Moreover, we have previously reported the expression of Slc26a6, a closely related Cl−/HCO3− transporter in the Slc26a family, in mouse and human myocytes (Kim et al., 2013; Sirish et al., 2017). Therefore, the Slc26a6 transcript was used as a positive control. Taking advantage of previously described Slc26a5−/− mice (Liberman et al., 2002) (Figure S2), we used single-cell qPCR to demonstrate the expression of Slc26a5 transcript in ventricular myocytes isolated from wild-type (WT), but not Slc26a5−/−, mice. In contrast, the expression of the Slc26a6 transcript was similar in WT and Slc26a5−/− mice (Figure 1C). The findings were further confirmed using smFISH (Figures 1D and 1E).
Ventricular myocytes isolated from Prestin-yellow fluorescence protein (YFP) knockin (KI) mice show positive YFP signals
Immunofluorescence confocal microscopic imaging shows positive staining for Slc26a5 in mouse ventricular myocytes (Figure 2A). Ventricular myocytes isolated from Slc26a5−/− mice were used as a negative control (Figure 2B). Cryosections from mouse inner ears were used as positive controls showing reactive staining in OHCs but not inner hair cells (IHCs; Figure 2C) as expected. Notwithstanding the positive and negative control experiments to ensure Slc26a5 antibody specificity, we adopted other strategies in substantiating Slc26a5 expression in cardiac myocytes. To unequivocally demonstrate that prestin is expressed in cardiac myocytes, we took advantage of the Prestin-YFP KI mouse line described previously (Yamashita et al., 2015) (Figure S3). In keeping with the antibody-specific imaging, we identified distinct YFP labeling that colocalized with α-actinin2 in Prestin-YFP KI myocytes, using anti-green fluorescence protein (GFP) antibody (Figure 2D), suggesting the localization of prestin along the transverse tubule (t-tubule) in cardiomyocytes. Ventricular myocytes isolated from WT mice were used as a negative control, showing the lack of positivity with anti-GFP antibody (Figure 2E). OHCs from the Prestin-YFP KI mice were used as positive controls (Figure 2F). We further tested the expression of prestin in skeletal and smooth muscle cells to evaluate the broader function of prestin, using tissue from Prestin-YFP KI mice (Figure S4). Indeed, we demonstrated prestin expression in skeletal muscle cells but at a relatively low level. Furthermore, prestin was not detected in smooth muscle cells.
Figure 2. Expression of prestin in cardiomyocytes.

(A) Immunofluorescence confocal microscopic imaging from mouse ventricular myocytes, demonstrating the expression of prestin (Slc26a5, middle panel, cyan). Cells were counterstained with phalloidin-fluorescein isothiocyanate (FITC; top panel, green). The merged image is shown on the bottom panel.
(B) A negative control using ventricular myocytes isolated from Slc26a5−/−. Cells were counterstained with anti-α-actinin2 antibody (green).
(C) A cryosection from the mouse organ of Corti as a positive control, demonstrating the presence of prestin (cyan) in the lateral membrane of the outer hair cells (OHCs) but not inner hair cells (IHCs) as expected. DAPI (4’,6-diamidino-2-phenylindole) was used to stain the nuclei (blue).
(D) Immunofluorescence super-resolution STED imaging from Prestin-YFP KI mouse ventricular myocytes showing the expression of prestin using anti-GFP antibody (green). Cells were counterstained using anti-α-actinin2 antibody (red).
(E) A negative control using anti-GFP antibodies in the WT control. Cells were counterstained using anti-α-actinin2 antibody (red).
(F) A cryosection from the mouse organ of Corti as a positive control from Prestin-YFP KI mice showing the presence of prestin (green) in the lateral membrane of OHCs but not IHCs. DAPI was used to stain the nuclei. All scale bars are 10 μm.
Slc26a5−/− cardiomyocytes show reduced sarcomere shortening with cardiac alternans
Prestin is distinct in the Slc26a family because of its molecular motor function, serving as a direct voltage-to-force converter (Dallos, 2008; Dallos and Fakler, 2002; Dallos et al., 2006; He et al., 2014; Zheng et al., 2002). Sarcomere-length is used as a cellular index to quantify the contraction and motor function of cardiomyocytes. We quantified sarcomere shortening in Slc26a5−/− compared with that of WT mice using IonOptix cardiac contraction measurement system. Single-myocyte measurements demonstrated a significant reduction in sarcomere shortening in Slc26a5−/− compared with that of WT myocytes (Figure 3A), with a significant increase in the incidence of cardiac alternans (Figure 3C). However, there is no significant change in Ca2+ transient (CaT) amplitude or sarcoplasmic reticulum (SR) Ca2+ load (Figures 3B, 3D, and 3E). Moreover, using SHG-TPF microscopy (Awasthi et al., 2012, 2016), we quantified single-sarcomere contraction and local Ca2+ levels in cardiomyocytes from WT and Slc26a5−/− mice (Figures 3F–3H). The technique provides multiple advantages by imaging the A-band, independent of t-tubule morphology. SHG-TPF microscopy enables microdomain contraction measurements and local Ca2+ levels simultaneously in live cardiomyocytes (Awasthi et al., 2016). Consistent with the findings using single-cell shortening, there was a significant decrease in single-sarcomere shortening, with a reduction in contraction speed (Figures 3I and 3J). In addition, there was a significant increase in the rise-time of local Ca2+, with no differences in the decay time (Figures 3H, 3K, and 3L).
Figure 3. Slc26a5−/− cardiomyocytes show reduced sarcomere shortening.

(A and B) Representative traces for sarcomere shortening and Ca2+ transients (CaT) measured using Fura-2 ratio (RFURA) from WT and Slc26a5−/− cardiomyocytes. Summary data for percentages of sarcomere shortening and peak CaT from the two groups of animals are shown in the right panels.
(C) Examples of mechanical alternans in Slc26a5−/−cardiomyocytes. Alternans were not observed in WT cardiomyocytes.
(D) There were no significant differences in the sarcoplasmic reticulum (SR) Ca2+ load between WT and Slc26a5−/− cardiomyocytes.
(E) Representative traces of SR Ca2+ load measurement of WT and Slc26a5−/− ventricular myocytes. The myocytes were first paced to steady state. The pacing was stopped for 15 s, followed by a rapid application of 20 mM of caffeine to induce SR Ca2+ release.
(F) A diagram showing quantification of sarcomere shortening during relaxed and contracted states from SHG recordings. Li is the distance between 2 adjacent A-bands within the same sarcomere, while La is the distance between the 2 A-bands across the adjacent sarcomeres. The sarcomere length is equal to the sum of Li and La and shortens during cell contraction because of the sliding of the thick filament against the thin filament.
(G) A representative example of SHG recording and the corresponding Ca2+ transients from a ventricular myocyte.
(H) Quantification of the rise time (trise) and half decay time (t1/2) from a representative Ca2+ transient.
(I–L) Summary data comparing percentages of sarcomere shortening (I), contracting speed (μm/s) (J), CaT rise time (K), and CaT half decay time (L) from WT and Slc26a5−/− cardiomyocytes. *p < 0.05. The numbers within the bar graphs represent the sample sizes.
Profound impairment of systolic and diastolic function in Slc26a5−/− mice
To test the effects of reduced sarcomere shortening in Slc26a5−/− cardiomyocytes in vivo, we performed in vivo ultrasound cardiac imaging (echocardiography) to determine cardiac function, including systolic and diastolic function, in Slc26a5−/− compared with that of WT mice. There was no evidence of cardiac hypertrophy or dilation and no significant differences in heart/body weight ratios between WT and Slc26a5−/− mice (Figure S2). Histological analyses using H&E and Picrosirius red stain of cardiac sections demonstrated no evidence of cardiac fibrosis, hypertrophy, or dilation in Slc26a5−/− mice (Figure S2). However, there was a significant reduction in the fractional shortening in Slc26a5−/− mice compared with that of WT controls (Figures 4A and 4B). A pulsed Doppler velocity profile of inflow across the mitral valve (MV) was performed to assess the diastolic function. There was a significant decrease in the ratio of the early (E) to late (A) left ventricular filling velocities (E/A ratio) in Slc26a5−/− mice compared with that of WT controls, consistent with diastolic dysfunction in the Slc26a5−/− mice (Figures 4C and 4D).
Figure 4. Echocardiographic recordings and hemodynamics monitoring demonstrate systolic and diastolic dysfunction in Slc26a5−/− mice.

(A) Representative M-mode echocardiography from WT and Slc26a5−/− mice.
(B) Summary data for fractional shortening (FS), illustrating a significant decrease in FS in knockout (KO) mice compared with that of WT.
(C) Pulsed Doppler velocity profile of inflow across the mitral valve (MV).
(D) Summary data showing a significant decrease in the E/A ratios in Slc26a5−/− compared with those of WT mice.
(E and G) Representative radial and circumferential strains in WT and Slc26a5−/− mice.
(F and H) Summary data for radial and circumferential strains showing a significant decrease in radial and circumferential strains in Slc26a5−/− mice compared with those of WT controls.
(I–P) Hemodynamic monitoring in WT and Slc26a5−/− mice showing left ventricular pressure (mmHg) (I and J), volume (μL) (K and L), and dP/dt (mmHg/s) (M and N). The pressure and volume have been calibrated. The volume calibration was performed using a cuvette (P/N 910–1049, Millar Inc.) filled with fresh heparinized 37°C mouse blood. (O) Pressure-volume relationship in WT and Slc26a5−/− mice during changes in preload. (P) Summary data for the end-systolic P-V relationship (Ees) (mmHg/μL) demonstrate reduced cardiac contractility in Slc26a5−/− mice. *p < 0.05.
A significant decrease in radial and circumferential strains in Slc26a5−/− mice
A strain refers to deformation that occurs when a force is applied and is quantified as a complex derivative of Doppler imaging. A strain is a more direct measurement of intrinsic myocardial contractility than global values of ejection fraction or fractional shortening. Positive and negative strain values indicate elongation and shortening of tissues, respectively. This quantitative analysis detects abnormalities that are not apparent visually. Indeed, Slc26a5−/− mice demonstrate a significant decrease in left ventricular radial and circumferential strain compared with that of WT animals (Figures 4E–4H).
Hemodynamic monitoring confirms a significant decrease in cardiac contractility in Slc26a5−/− mice
Because fractional shortening is load dependent, we used hemodynamic monitoring to derive the load-independent parameters of the in vivo cardiac function. We generated representative recordings of left ventricular pressure, volume, and developed pressure (dP/dt) in WT and Slc26a5−/− mice (Figures 4I, 4K, and 4M). There was a significant downward shift of the pressure-volume (P-V) loops in Slc26a5−/− mice, indicating reduced end-systolic pressure with reduced stroke volume (Figure 4O). To quantify the end-systolic P-V relationship, we obtained a series of P-V loops, by altering the preload, and derived the slope of the end-systolic P-V relationship (Ees), a load-independent measure of cardiac contractility (Figure 4P) for WT and Slc26a5−/−, respectively. Consistent with the cellular and the echocardiographic data, the end-systolic pressure (ESP; Figure 4J), maximum dP/dt (Figure 4N), and Ees (Figure 4P) in Slc26a5−/− mice were significantly reduced compared with those of WT mice. The results support a profound decrease in cardiac contractility in Slc26a5−/− mice compared with that of WT controls.
NLC properties in cardiomyocytes rendered by prestin molecules
Electromotility is the signature function of OHCs requiring prestin, which experiences membrane potential-dependent conformational changes. NLC measurement in OHCs is used to evaluate the electromotility. To establish that cardiac prestin mediates electromotility (Dallos, 2008; Dallos and Fakler, 2002; Dallos et al., 2006; He et al., 2014; Zheng et al., 2002), we recorded NLC as described previously (Huang and Santos-Sacchi, 1993; Rybalchenko and Santos-Sacchi, 2008). NLC (Cv) is determined as the total membrane capacitance (Cm) minus the linear capacitance (Cln), normalized by Cln (i.e., Cv = [Cm − Cln]/Cln) to obtain specific capacitance. NLC is plotted as a function of voltage and is fitted with the first derivative of a two-state Boltzmann function as shown in Figure 5. As positive controls, Chinese hamster ovary (CHO) cells expressing only prestin exhibit NLC (Figure 5A, left). In contrast, CHO cells expressing Slc26a6 or non-transfected cells do not demonstrate NLC (Figure 5A, middle and right panels, respectively).
Figure 5. Voltage-dependent nonlinear capacitance (NLC) in CHO cells expressing Slc26a5 and in ventricular myocytes from WT and Slc26a5−/− mice.

(A) Upper panels show representative traces of NLC recordings using voltage stair protocol ranging from −140 to +60 mV with 10-mV increments, together with a diagram of the voltage-clamp protocol. Lower panels show normalized NLC, plotted as a function of voltage, and fitted with the first derivative of a Boltzmann function describing the nonlinear charge’s movement (Santos-Sacchi, 1991) as described below. The left panel shows normalized NLC from CHO cells expressing Slc26a5. CHO cells expressing Slc26a6 and non-transfected cells were used as negative controls and are shown in the middle and right panels, respectively. As expected, CHO cells expressing Slc26a6 do not demonstrate NLC.
(B) Representative traces of capacitive currents recorded from WT and Slc26a5−/− cardiomyocytes at baseline (left panels), after a 25-μM blebbistatin (BLEB, middle panels), and after a 10-mM BDM (right panels) application.
(C) NLC obtained through correction for linear capacitance was plotted as a function of voltage and fitted with the first derivative of a Boltzmann function: Cm = Cln + Cv = Cln + (Qmaxze/kT) × exp(−ze[V − Vh]/kT)/(1 + exp[−ze{V − Vh}/kT])2, where Cm is the total capacitance of the cell, Cln is the linear capacitance, Cv is the nonlinear capacitance, V is the membrane potential, Vh is the voltage at half-maximal nonlinear charge transfer, e is the electron charge, k is Boltzmann’s constant, T is the absolute temperature, z is the valence, and Qmax is maximum nonlinear charge transfer. Salicylate (10 mM) was applied to inhibit the NLC as shown in the left panel, showing no effect on Slc26a5−/− cardiomyocytes (n = 6 for WT and n = 5 for Slc26a5−/−). Knockout of Slc26a5 results in a significant reduction and depolarization shift of NLC (Vh = −37 ± 2 mV, n = 11; and −18 ± 3 mV, n = 14 for WT and Slc26a5−/−, respectively; p < 0.05); there are no significant changes in Cln (167 ± 15 pF for WT, and 166 ± 12 pF for Slc26a5−/−, n = 14); Qmax/Cln for WT is significantly larger than that of Slc26a5−/− (10.81 ± 0.99 versus 7.74 ± 1.03 nC/μF, p < 0.05; n = 11 for WT and n = 14 for Slc26a5−/−). In the presence of 10 mM salicylate, Qmax/Cln for WT cardiomyocytes is significantly reduced (7.21 ± 1.33 nC/μF, n = 6; p < 0.05), whereas Qmax/Cln for Slc26a5−/− cardiomyocytes is not significantly altered (7.56 ± 1.41 nC/μF, n = 5). The voltage sensitivity, K (kT/ze), can be calculated based on the z values with K = 17.16 ± 1.15 mV (n = 11 for WT) and 15.20 ± 1.31 mV (n = 14 for Slc26a5−/−), p = NS. Vh for WT and Slc26a5−/− ventricular myocytes after BLEB are −37 ± 5 (n = 4) and −25 ± 3 (n = 5) mV, respectively. Vh for WT and Slc26a5−/− ventricular myocytes after BDM are −43 ± 4 (n = 3) and −32 ± 3 (n = 7) mV, respectively. NLC for WT and Slc26a5−/− at baseline are shown in dotted lines in the middle and right panels. The NLC of Slc26a5−/− myocytes at −20, −10, 0, 10, 20, 30, and 40 mV in the presence of BLEB or BDM is significantly smaller than that in the absence of the myosin inhibitors (p < 0.05).
Cardiac myocytes exhibit NLC
If cardiac myocytes express prestin, we predict prestin would contribute toward the NLC of the cells. We directly record and demonstrate NLC in ventricular myocytes (Figure 5B). In contrast to previous studies of NLC in OHCs, cardiomyocytes have large membrane capacitance. Moreover, NLC is likely to result from two processes, ion-channel gating charge movement and prestin function. Therefore, it is critical to ensure that the recorded NLC represents the voltage-dependent component of prestin. Notably, the genetic ablation of Slc26a5 results in a significant depolarization shift of NLC (Figures 5B and 5C, left panel). The calculated Vh from the two-state Boltzmann function in Slc26a5−/− was significantly different from that of the WT cardiomyocytes (Vh = −37 ± 2 [n = 11] and −18 ± 3 mV [n = 14] for WT and Slc26a5−/−, respectively; p < 0.05), suggesting a significant decrease in the sensitivity to voltages in the knockout mice. Salicylate (10 mM) was applied to inhibit the NLC in WT cardiomyocytes as shown in the left panel, showing no effect on Slc26a5−/− cardiomyocytes (Figure 5C). Prestin may amplify the actin-myosin force generation. As a feedback mechanism, the actin-myosin interaction may affect the prestin function. We, therefore, used two different excitation-contraction (EC) uncouplers, blebbistatin (BLEB) and 2,3-butanedione monoxime (BDM), to inhibit the myosin ATPase to disengage cellular excitation from contraction (Bond et al., 2013) (Figures 5B and 5C, middle and right panels). BLEB and BDM significantly restricted the dynamic range of NLC, and the amplitude was markedly reduced in Slc26a5−/− cardiomyocytes. The effects of BLEB and BDM on NLC suggest the functional coupling of myofilaments with prestin and, possibly, the regulation of ion channel gating properties by prestin in cardiomyocytes. The data further support the critical contributions of prestin to the NLC in ventricular myocytes.
DISCUSSION
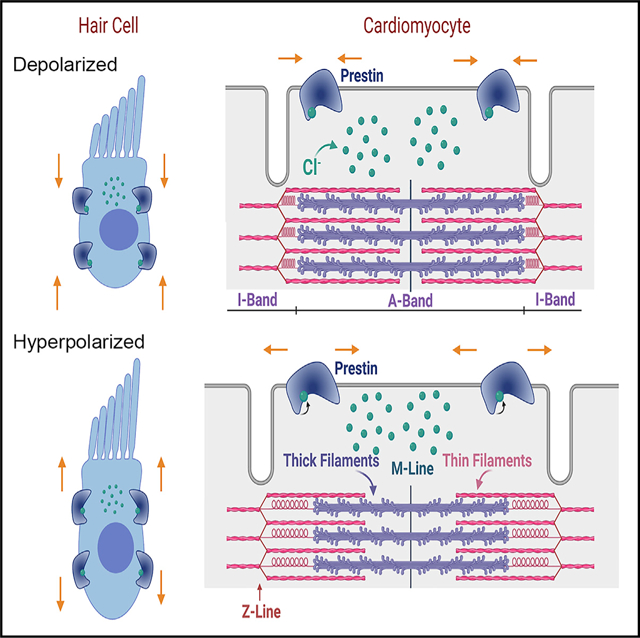
Prestin has been primarily studied as a motor protein specific to OHCs of the inner ear, consistent with the expectation that it is exclusive to the auditory system. This report alters the existing paradigm and demonstrates that prestin is also expressed prominently in cardiac myocytes. Prestin’s function in the heart addresses a previously unresolved mechanism and the paradox of muscle contraction—amplification of actin-myosin cross-bridge force generation by an elastic element, inducing a displacement that confers strain in the sarcolemma parallel to the direction of the sliding filaments, sustaining sarcolemmal distortion.
Prestin is that elastic element, expressed both in the surface sarcolemma and along the t-tubule (Figure 2). Prestin serves as a spring in parallel to the actin-myosin with a spring constant of the assembly of k = k1 (prestin) + k2 (actin-myosin) (Figures S5A and S5B). The ensemble’s force (or stress) is the sum of individual forces (i.e., F = F1 + F2) or F1/F2 = k1/k2. The strain (deformation) of the system or cell shortening (X) represents the common strain (i.e., X = X1 = X2).
We have uncovered, for the first time, two distinct components of NLC in cardiac myocytes. Apart from the expected component of NLC from voltage-dependent gating-charge movement of ion-channels in cardiomyocytes, we demonstrate an additional element of NLC resulting from the expression of prestin, representing the key signature of the protein (Ashmore, 2008; Ashmore et al., 2002; Dallos, 2008; Dallos and Fakler, 2002; Dallos et al., 2006; He et al., 2014; Liberman et al., 2002; Oliver et al., 2001; Rybalchenko and Santos-Sacchi, 2003; Song and Santos-Sacchi, 2010; Zheng et al., 2000, 2002). Indeed, we directly demonstrate that prestin contributes to the additional component of NLC in ventricular myocytes using Slc26a5−/− mice. Reports have shown that the property of NLC rendered by prestin is derived from the voltage-dependent activation of intracellular Cl− (Cli−) binding to prestin. During membrane hyperpolarization, Cli− may translocate toward the external binding sites of the protein. As a result, prestin would assume an expanded conformation. Conversely, membrane depolarization may displace bound Cl−i, inducing a contracted structure (Dallos and Fakler, 2002) (Figure S5C). Prestin may directly contribute to the cardiac motor function, working as an elastic element (Figures 3, 4, and 5). However, it is possible that prestin may regulate the function of cardiomyocytes indirectly, by modulating the gating of cardiac ion channels, affecting the excitability and Ca2+ signaling. Such modulatory effects of prestin have been reported for Kv7.4 channel (Chambard and Ashmore, 2005; Perez-Flores et al., 2020). The relatively large residual NLC observed in Slc26a5−/− myocytes reflects substantial gating charges of cardiac ion channels, including voltage-gated Na+, Ca2+, and K+ channels. Indeed, the documented depolarization shift of NLC in Slc26a5−/− compared with that of WT cardiomyocytes may reflect modulation of the cardiac ion channel function by prestin. Moreover, the rise time of calcium transient (CaT) is increased in Slc26a5−/− ventricular myocytes as quantified using SHG-TPF microscopy (Figure 3K). Therefore, additional mechanisms of prestin in cardiac motor function need to be further investigated in future studies.
Our findings support the roles of prestin in cardiac mechanical amplification serving as an elastic element to enhance the actomyosin-based sarcomere contraction system. Future studies are required to investigate the functional coupling of the two-motor systems in overall force generation in the heart.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Additional information and requests for resources and reagents should be directed to the Lead contact, Nipavan Chiamvimonvat (nchiamvimonvat@ucdavis.edu).
Materials availability
Additional information and requests for resources should be directed to and will be fulfilled by the Lead Contact. All unique reagents generated in this study are available from the Lead contact with a completed Materials Transfer Agreement.
Data and code availability
The data supporting the current study have not been deposited in a public repository because they are only used for the publication of this manuscript but are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal care and procedures were performed following the protocols approved by the Institutional Animal Care and Use Committee of the University of California, Davis, and following the National Institutes of Health guidelines. De-identified human ventricular specimens were obtained from a commercial source, per the approved UC Davis Institutional Review Board (IRB) protocol. All experiments described in the study were conducted in a blinded fashion. Specifically, the investigators who performed the in vivo analyses, physiological recordings, cell/tissue dissection, and imaging had no knowledge of the animals’ genotypes. Slc26a5−/− (KO) and Slc26a5YFP/Y.F.P. (KI) mouse models used in the study were generated previously (Liberman et al., 2002; Yamashita et al., 2015). Both male and female mice, 12–16 weeks of age were used. Slc26a5−/− mice were maintained in a mixed background of SV-129 (Taconic Biosciences) and C57BL/6J (The Jackson laboratory).
METHOD DETAILS
Molecular identification of prestin from human and mouse cardiac myocytes
Two different de-identified human ventricular specimens were obtained from a commercial source (T Cubed). Total RNA and mRNA were extracted as previously described (Kim et al., 2013). The cDNA was synthesized using the oligo-dT primer. Primers were designed based on human and mouse inner ear prestin. For amplification of full-length coding sequence of human cardiac SLC26A5, fragments 1 (1767 bp) and 2 (773 bp) were amplified using two separate sets of primers as follows (see Figures 1 and S1):
First set:
Forward: 5’-ATGGATCATGCTGAAGAAAATG-3’ (#1) Reverse: 5’-GTTGGCCATATTTGCATTTC-3’ (#2)
Second set:
Forward: 5’-CTGCTGTGATCATTGCTCTG-3’ (#3)
Reverse: 5’-CATCTATGCCTCAGGAGTGGC-3’ (#4)
Overlapping PCR was performed using fragments 1 and 2 to obtain full-length human cardiac prestin (2235 bp), then subcloned into a TA cloning vector (pCR4-TOPO) and verified using sequencing.
Slc26a5−/− and Slc26a5-YFP knockin (Slc26a5YFP/YFP) mouse models and genotyping analyses
Slc26a5−/− (KO) and Slc26a5YFP/YFP (KI) mouse models were generated previously (Liberman et al., 2002; Yamashita et al., 2015). Genotyping analyses were performed using RT-PCR of genomic DNA from wild-type (WT), Slc26a5YFP/YFP, and Slc26a5−/− mice (Figures S2 and S3). Three sets of primers were used with two forward (F) primers for WT or mutant (KO) alleles and one reverse (R) primer for both WT and mutant alleles. The primers used are as follow:
Slc26a5YFP/YFP:
Prestin-YFP-common-F: 5’-AGGAGGATATGGAGCCCAATGCCACAC-3’
Prestin-YFP-KI-R: 5’-TCGTCCTTGAAGAAGATGGTGCGCTC-3’
Prestin-YFP-WT-F: 5’-TATAAGTGCAAGAGGCCTGTTAATCTTTG-3’
WT band is 247 bp, and knockin (KI) band is 387 bp
Slc26a5−/−:
Prestin001: 5’-GCTTGATGATTGGAGGTGTG-3’
Prestin002: 5’-CTGAATGATTCCTGAAAGTAAGG-3’
Prestin003: 5’-CTGTTGTCCAAGTGCTTGCC-3’
Prestin004: 5’-GATCGCTATCAGGACATAGCG-3’
WT band is 175 bp, and KO band is 500 bp
Cardiac tissue preparation and cardiomyocyte isolation
Mice were anesthetized by intraperitoneal injection of 80 mg/kg of ketamine and 5 mg/kg of xylazine and sacrificed by exsanguination with rapid heart excision. Isolation of mouse cardiomyocytes followed the conventional enzymatic dissociation methods, as we have previously described (Kim et al., 2013). All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.), except specifically indicated.
Histological analyses
Histological analyses of cardiac tissue were performed as previously described (Sirish et al., 2016). Briefly, excised hearts were retrogradely perfused with a phosphate-buffered solution to remove blood. Fixed hearts were embedded in paraffin, and serial cardiac sections of 5 μm in thickness were taken and stained with Picrosirius Red to assess for collagen content.
Immunofluorescence confocal microscopy
Isolated cardiomyocytes were adhered to poly-L-lysine coated coverslips and fixed in 4% paraformaldehyde for 15 min. The cells were then washed 3 × 5 min with Dulbecco’s phosphate-buffered saline (DPBS) and immersed in blocking solution (10% goat serum in PBST, 0.2% Triton X-100) at room temperature for 1 hr. Primary anti-prestin antibody (H-294, Santa Cruz Biotechnology, Dallas, TX, U.S.A.) was added and incubated overnight at 4°C. Cells were then washed 3 × 15 min with PBST, 0.05% Triton X-100, and incubated with secondary antibody (Cy3 AffiniPure Goat Anti-Rabbit IgG (H+L), Jackson ImmunoResearch Laboratories, Inc. West Grove, PA, U.S.A.) for 1 hr at room temperature. The samples were further stained by phalloidin–tetramethylrhodamine B isothiocyanate (Phalloidin-TRITC) (Sigma-Aldrich). Additional primary antibodies used include monoclonal anti-α-actinin2 antibodies (A7811, Sigma-Aldrich) and rabbit anti-mouse IgG Superclonal™ Recombinant Secondary Antibody, Alexa Fluor 555 (ThermoFisher Scientific, Waltham, MA, U.S.A.). Coverslips were rewashed for 7 × 5 min with DPBS, mounted on slides using Mounting Media (Fluore-Gel with Tris Buffer, Electron Microscopy Sciences, Hatfield, U.S.A.), imaged using a Zeiss confocal LSM 700 microscope, and processed using the Zeiss Zen imaging software (Carl Zeiss, Oberkochen, Germany).
The mouse gut, limb muscle, and cochleae were harvested and fixed in 4% paraformaldehyde in PBS for 4 hours on ice for imaging of tissue samples. The tissues were rinsed three times (10 minutes per rinse) in PBS. For positive control, we used outer hair cells in the cochleae. The cochleae were immersed in 10% EDTA in PBS for 3 days. The EDTA solution was changed daily. The cochleae were rinsed three times (10 minutes per rinse) in PBS. The samples were immersed in 30% sucrose overnight for cryoprotection and embedded in OCT compound for cryo-sectioning at −20°C. Cryosections with a thickness of 10 μm were washed three times with PBS. Immunohistochemistry was performed by first blocking the samples with 10% goat serum (Jackson ImmunoResearch Laboratories Inc, West Grove, PA, USA) in PBST (PBS + 0.3% Triton X-100) for 1 hour at room temperature. For gut smooth muscle staining, the samples were incubated at 4°C overnight in PBST containing the primary antibody: rabbit anti-smooth muscle myosin heavy chain 11 (Abcam, Cambridge, MA, USA.). For cochleae staining, the samples were incubated at 4°C overnight in in PBST containing the primary anti-prestin antibody (H-294, Santa Cruz Biotechnology). The slices were washed three times with PBST and then incubated at room temperature for 2 hours with the secondary antibody: Alexa Fluor 647 Goat Anti-Rabbit IgG (H+L) for smooth muscle and Cy3 AffiniPure Goat Anti-Rabbit IgG (H+L) for cochleae (Jackson ImmunoResearch Inc.). The samples were stained with Phalloidin-TRITC and/or DAPI. Images were acquired with an Olympus FV1000 confocal microscope using 60x objective and were merged with Imaris Cell Imaging Software (Oxford Instruments, Zurich, Switzerland).
Single-molecule fluorescence in situ hybridization (smFISH)
SmFISH was performed as previously described (Lee et al., 2019; Perkins et al., 2020) in single isolated ventricular myocytes from WT and Slc26a5−/− mice using probes developed for Slc26a5 and Slc26a6. Probe hybridization closely followed the manufacturer’s instructions (Advanced Cell Diagnostics, Newark, CA, U.S.A.). Cells were treated with prechilled 4% PFA for 15 min at 4°C. The cells were dehydrated at room temperature in 50%, 70%, and 100% ethanol for 5 min each and allowed to dry for 1–2 min. Fixation and dehydration were followed by protease digestion, using Protease 4 for 30 min at room temperature. The cells were then incubated at 40°C with the following solutions: (1) target probe in hybridization buffer A for 3 h; (2) preamplifier in hybridization buffer B for 30 min; (3) amplifier in hybridization buffer B at 40°C for 15 min; and (4) label probe in hybridization buffer C for 15 min. After each hybridization step, cells were washed with wash buffer 3 times at room temperature. For fluorescent detection, the label probe was conjugated to AlexaFluor-488. Sequences of the target probes (for the specified genes), preamplifier, amplifier, and label probe are proprietary. Detailed information on the probe sequences can be obtained by signing a nondisclosure agreement provided by the manufacturer. For subsequent immunofluorescent staining, cells were counterstained using monoclonal anti-α-actinin2 antibody as described above.
Analysis of cardiac function by echocardiography
Echocardiograms using M-mode and two-dimensional (2D) measurements to assess systolic and diastolic function were performed using Vevo 2100 (FUJIFILM VisualSonics, Toronto, ON, Canada), as we have previously described (Zhang et al., 2014). The measurements represented the average of six selected cardiac cycles from at least two separate scans performed in a blinded fashion, with papillary muscles used as a point of reference for consistency in the scan level. End diastole was defined as the maximal left ventricular (LV) diastolic dimension, and end-systole was defined as the peak of posterior wall motion. Fractional shortening (FS), a surrogate of systolic function, was calculated from LV dimensions as follows: FS = ((EDD-ESD)/EDD) x100%, where EDD and ESD are LV end-diastolic and end-systolic dimension, respectively. A pulsed Doppler velocity profile of inflow across the mitral valve (MV) was performed to assess the diastolic function. The myocardial strain was quantified as a complex derivative of Doppler imaging and used as a direct measurement of intrinsic myocardial contractility.
Hemodynamic monitoring
Mice were anesthetized by intraperitoneal injection of 80 mg/kg of ketamine and 5 mg/kg of xylazine and maintained at 37°C. The arterial catheter was inserted retrogradely into the left ventricle via the carotid artery. The recording of pressure and volume was performed using Millar Pressure-Volume System MPVS-300 (Millar, Inc., Houston, TX), Power Lab, and Lab Chart 6.0 software (AD Instruments, Colorado Springs, CO, U.S.A.). The pressure and volume were calibrated before recordings. The volume calibration used fresh heparinized 37°C mouse blood and a cuvette (P/N 910–1049, Millar, Inc., Houston, TX, U.S.A.). To alter the preload, a gentle and quick abdominal compression was applied to occlude inferior vena cava.
Heterologous expression in Chinese hamster ovary (CHO) cells
Human cardiac SLC26A5 isoforms were expressed in CHO cells following the previously used protocol (Kim et al., 2013). CHO cells were cultured in F-12 media with 10% fetal bovine serum at 37°C with 5% CO2. Cardiac Slc26a5 and Slc26a6 clones were transiently transfected into cells using Lipofectamine 2000 (ThermoFisher Scientific) following the manufacturer’s instructions. The cells were plated on glass coverslips for patch-clamp recordings 2 to 3 days after transfection.
Patch-clamp recordings
Whole-cell patch-clamp recordings were performed to quantify the nonlinear capacitance (NLC) using an Axopatch 200A amplifier, Digidata 1440 digitizer, and pClamp10 software (Molecular Devices, LLC., Sunnyvale, CA, U.S.A.). All experiments were performed using 3 M KCl agar bridges. The recordings were digitally filtered at 1 kHz and digitized at 2 kHz. Borosilicate glass electrodes were pulled with a P-97 micropipette puller (Sutter Instruments, Novato, CA). The resistance of the electrodes was 1.5–3.0 MΩ when filled with the pipette solution.
For NLC recording in CHO cells, the pipette solution contained (in mM): 137.5 CsCl, 2 MgCl2, 5 D-glucose, 10 HEPES, 1 EGTA, pH 7.4 by CsOH and the bath solution contained (in mM): 20 TEA-Cl, 20 CsCl, 2 CoCl2, 96.7 NaCl, 1.47 MgCl2, 2 CaCl2, 5 D-glucose, 10 HEPES, pH 7.4 by NaOH. For NLC recordings in ventricular myocytes, pipette and bath solutions contained (in mM): 145 TEACl, 2 MgCl2, 5 D-glucose, 10 HEPES, 1 EGTA, pH 7.4 by TEA-OH. A voltage stair protocol (Figures 5A and 5B) was applied, ranging from −140 to 100 mV with 10-mV-increment with a stair duration of 20–40 ms. Data analysis was performed using Clampfit 10 software (Molecular Devices) and Origin 6.1 software (OriginLab Corp., Northampton, MA, U.S.A.). For NLC analysis, the voltage-dependent charge was fitted by a Boltzmann function, and the first derivative of the Boltzmann function was used to describe the NLC (Santos-Sacchi, 1991).
Measurement of sarcomere shortening and Ca2+ transient (CaT)
The IonOptix sarcomere detection (IonOptix LLC., Westwood, MA, U.S.A.) and fast Fourier transform (FFT) method (Jian et al., 2014) were used to measure single cardiomyocyte contraction. Instead of edge detection, the technique was preferred because the latter may lose precision if the cell’s edges move out of the focal plane during contraction. The contraction was measured using a high-speed camera (MyoCam-S, 240 to 1000 frames/s) to record the sarcomere movement. The fractional shortening was calculated as the percentage change in sarcomere length during contraction. Simultaneous Ca2+ transients (CaT) were recorded using Fura-2 dual-wavelength ratiometric method, which is more precise than using the Fluo-4 single-wavelength method. Additional analyses included assessment of sarcoplasmic reticulum (SR.) Ca2+ load, as previously described (Jian et al., 2014).
Cardiomyocytes loaded with Fura-2 were seeded in a recording chamber with pacing electrodes connected to a stimulator. Cells were paced at 0.5 Hz and continuously perfused during recording. The recording solution contained (in mM): 145 NaCl, 4 KCl, 1 CaCl2, 0.33 NaH2PO4, 1 MgCl2, 10 Glucose, 10 HEPES, and pH 7.4.
Stimulated emission depletion (STED) imaging
STED imaging was performed using a Leica TCS SP8 STED 3X microscope. Isolated mouse ventricular myocytes were fixed with 4% paraformaldehyde on No. 1.5-thickness/12-mm-round glass coverslips (Thomas Scientific, Swedesboro, NJ, USA) for 20 minutes at room temperature (RT) and washed three times with phosphate-buffered saline (PBS) containing 0.05% Triton-X. Non-specific antibody binding was blocked with 10% bovine serum albumin (BSA) in 0.1% PBS with Tween 20 PBST for 1 hour at RT. For double-labeling experiments, cells were first treated with a primary antibody (anti-GFP antibody and anti-α-actinin antibody, incubated overnight at 4°C, followed by application of secondary antibodies sequentially with Oregon Green 488 goat-anti-rabbit secondary antibody (Thermo Fisher Scientific) followed by Rhodamine goat-anti-mouse secondary antibody (Thermo Fisher Scientific) for 1-hour incubation at RT For STED imaging, ProLong Gold Antifade Mountant (Thermo Fisher Scientific) was used to minimize photobleaching. Negative controls with only antigenic peptides and secondary antibodies were performed for all experiments.
Second-harmonic generation (SHG)-two-photon fluorescence (2PF) imaging
SHG-2PF microscopy was performed as previously described (Awasthi et al., 2012, 2016). SHG is a process in which light of wavelength λ produces its second harmonic (wavelength ½λ). It passes through a noncentrosymmetric medium and occurs strongly when these macromolecules are highly ordered, as in a striated muscle. SHG was integrated with 2PF microscopy. In 2PF, laser light of wavelength λex is used to excite an electron transition at energy ½λex. After the loss of vibrational energy, a Stokes-shifted photon is emitted at λem > ½λex. The shift allows the signal from fluorophores (λem > ½λex) and harmonophores (λem = ½ λex) to be simultaneously excited, separated, and detected as previously described. In these experiments, the excitation wavelength was tuned to 940 nm. The laser power at the sample was between 10–15 mW. SHG signals were acquired by a photomultiplier tube with a 470 ± 20 nm bandpass filter in the forward direction with a 0.55 NA condenser lens. Two-photon excited fluorescence (510–590 nm) images were simultaneously collected in the epi-direction. The scanning speed was 2 μs/pixel.
We resolve sarcomere contraction from the SHG signals as previously described. Freshly isolated ventricular myocytes from WT and Slc26a5−/− were used. The bath solution contained (in mM): 145 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, 0.33 NaH2PO4, 10 D-glucose, 10 HEPES, pH 7.4 by NaOH. We denote Li as the distance between the two A-bands within the same sarcomere and La as the distance between the adjacent A-bands across the sarcomere. Li and La can be measured from a distance between the adjacent SHG-bright bands (Figure 3F). During contraction, La is shortened by the thick and thin filaments’ overlap, whereas the Li remains unchanged because the length of the thick filament within a sarcomere does not change. The sum of Li and La is equal to the sarcomere length, LSL = Li+La (e.g., the distance from Z-line to Z-line, or equivalently from M-line to M-line).
QUANTIFICATION AND STATISTICAL ANALYSIS
We estimate a sample size of 4 per experiment to detect at least 15% difference between WT and Slc26a5−/− groups with alpha = 0.05 for a two-tailed test to achieve a power of 95%, assuming the standard deviation of the differences to be 5% (SigmaStat, Systat Software Inc.). No data were excluded. Data are presented as mean ± SEM. Statistical comparisons were analyzed by t test (two-tailed). Statistical significance was considered to be achieved when p < 0.05.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-Slc26a5 | Santa Cruz Biotechnology | sc-30163 (H-294); RRID: AB_2190499 |
| Anti-pholloidin-FITC | Sigma-Aldrich | P1951; RRID:AB_2315148 |
| Anti-GFP antibody | Abcam | Ab290; RRID:AB_303395 |
| Anti-α-actinin2 antibody | Sigma-Aldrich | A7811; RRID:AB_476766 |
| Cy™3 AffiniPure Goat Anti-Rabbit IgG (H+L) secondary antibody | Jackson ImmunoResearch Inc., | AB_2338000; RRID:AB_2338000 |
| Rabbit anti-mouse IgG Superclonal™ Recombinant Secondary Antibody, Alexa Fluor 555 | ThermoFisher Scientific | A27028; RRID:AB_2536091 |
| Goat anti-mouse secondary antibody, Rhodamine | ThermoFisher Scientific | 31660; RRID:AB_228308 |
| Goat anti-rabbit IgG secondary antibody, Oregon Green 488 | ThermoFisher Scientific | O-11038; RRID:AB_2539798 |
|
| ||
| Chemicals | ||
|
| ||
| 2,3-Butanedione Monoxime (BDM.) | Sigma-Aldrich | B0753 |
| Blebbistatin | Abcam. | Ab120425 |
| Lipofectamine 2000 | Thermo Fisher Scientific | 11668019 |
|
| ||
| Software | ||
|
| ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com |
| Zeiss Zen Software | Carl Zeiss | https://www.zeiss.com/corporate/int/home.html |
| Origin 6.1 Software | OriginLab Corp. | https://www.originlab.com |
| BioRender | BioRender | https://biorender.com |
| Lab Chart 6.0 software | AD Instruments | https://www.adinstruments.com |
| pClamp10 software | Molecular Devices, LLC | https://support.moleculardevices.com/s/article/Spectranet-our-new-Customer-Care-Portal |
| Imaris Cell Imaging Software | Oxford Instruments | https://imaris.oxinst.com/ |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Slc26a5−/− knockout mouse model | Previously generated (Liberman et al., 2002) | N/A |
| Slc26a5YFP/Y.F.P. (KI) mouse model | Previously generated (Yamashita et al., 2015) | N/A |
| C57BL/6J mice | The Jackson Laboratory | https://www.jax.org/ |
| SV-129 mice | Taconic Biosciences | https://www.taconic.com/ |
Highlights.
Prestin is considered to be expressed exclusively in outer hair cells of the inner ear
The current study shows that prestin is expressed in cardiomyocytes
Prestin serves to amplify actin-myosin force generation in cardiomyocytes
Prestin serves a broader cellular motor function
ACKNOWLEDGMENTS
We thank our laboratory members for their constructive comments on the initial draft of the manuscript, Dr. Caitlin Schrein for her invaluable input, and Dr. Victor C. Lau for his assistance with data analyses. Funding: This study was supported in part by the American Heart Association beginning grant-in-aid 14BGIA18870087 and National Institutes of Health (NIH) R56 HL138392 (X.-D.Z.); NIH R01 DC016099, NIH R01 DC015252, NIH R01 DC015135, and NIH P01 AG051443 (E.N.Y.); NIH R01 HL085727, NIH R01 HL085844, NIH R01 HL137228, and NIH S10 RR033106 (N.C.); VA merit review grant I01 BX000576 and I01 CX001490 (N.C.); NIH R01 DC015010 (J.Z.); postdoctoral fellowship from NIH T32 training grant in basic & translational cardiovascular science (T32 HL086350) (C.-W.C. and P.N.T.); NIH F32 HL149288 (P.N.T.); predoctoral fellowship from NIH T32 training grant in basic & translational cardiovascular science (T32 HL086350) and NIH F31 HL136120 (H.A.L.); and American Heart Association predoctoral fellowship award 18PRE34030199 (L.R.). The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. N.C. is the Roger Tatarian Endowed Professorship holder in cardiovascular medicine and a part-time staff physician at the VA Northern California Health Care System (Mather, CA, USA).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109097.
REFERENCES
- Alper SL, and Sharma AK (2013). The SLC26 gene family of anion transporters and channels. Mol. Aspects Med. 34, 494–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore J (2008). Cochlear outer hair cell motility. Physiol. Rev. 88, 173–210. [DOI] [PubMed] [Google Scholar]
- Ashmore JF, Chambard JM, and Richmond S (2002). Cochlear transduction: from models to molecules and back again. Audiol. Neurotol. 7, 6–8. [DOI] [PubMed] [Google Scholar]
- Awasthi S, Matthews DL, Li RA, Chiamvimonvat N, Lieu DK, and Chan JW (2012). Label-free identification and characterization of human pluripotent stem cell-derived cardiomyocytes using second harmonic generation (SHG) microscopy. J. Biophotonics 5, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi S, Izu LT, Mao Z, Jian Z, Landas T, Lerner A, Shimkunas R, Woldeyesus R, Bossuyt J, Wood BM, et al. (2016). Multimodal SHG-2PF imaging of microdomain Ca2+-contraction coupling in live cardiac myocytes. Circ. Res. 118, e19–e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai JP, Surguchev A, Ogando Y, Song L, Bian S, Santos-Sacchi J, and Navaratnam D (2010). Prestin surface expression and activity are augmented by interaction with MAP1S, a microtubule-associated protein. J. Biol. Chem. 285, 20834–20843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian S, Navaratnam D, and Santos-Sacchi J (2013). Real time measures of prestin charge and fluorescence during plasma membrane trafficking reveal sub-tetrameric activity. PLoS ONE 8, e66078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond LM, Tumbarello DA, Kendrick-Jones J, and Buss F (2013). Small-molecule inhibitors of myosin proteins. Future Med. Chem. 5, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambard JM, and Ashmore JF (2005). Regulation of the voltage-gated potassium channel KCNQ4 in the auditory pathway. Pflugers Arch. 450, 34–44. [DOI] [PubMed] [Google Scholar]
- Dallos P (2008). Cochlear amplification, outer hair cells and prestin. Curr. Opin. Neurobiol. 18, 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos P, and Fakler B (2002). Prestin, a new type of motor protein. Nat. Rev. Mol. Cell Biol. 3, 104–111. [DOI] [PubMed] [Google Scholar]
- Dallos P, Zheng J, and Cheatham MA (2006). Prestin and the cochlear amplifier. J. Physiol. 576, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detro-Dassen S, Schänzler M, Lauks H, Martin I, zu Berstenhorst SM, Nothmann D, Torres-Salazar D, Hidalgo P, Schmalzing G, and Fahlke C (2008). Conserved dimeric subunit stoichiometry of SLC26 multifunctional anion exchangers. J. Biol. Chem. 283, 4177–4188. [DOI] [PubMed] [Google Scholar]
- Gleitsman KR, Tateyama M, and Kubo Y (2009). Structural rearrangements of the motor protein prestin revealed by fluorescence resonance energy transfer. Am. J. Physiol. Cell Physiol. 297, C290–C298. [DOI] [PubMed] [Google Scholar]
- Gorbunov D, Sturlese M, Nies F, Kluge M, Bellanda M, Battistutta R, and Oliver D (2014). Molecular architecture and the structural basis for anion interaction in prestin and SLC26 transporters. Nat. Commun. 5, 3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeson JN, Organ LE, Pereira FA, and Raphael RM (2006). Assessment of prestin self-association using fluorescence resonance energy transfer. Brain Res. 1091, 140–150. [DOI] [PubMed] [Google Scholar]
- Hallworth R, and Nichols MG (2012). Prestin in HEK cells is an obligate tetramer. J. Neurophysiol. 107, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He DZ, Lovas S, Ai Y, Li Y, and Beisel KW (2014). Prestin at year 14: progress and prospect. Hear. Res. 311, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma K, Duan C, Zheng J, Cheatham MA, and Dallos P (2013). The V499G/Y501H mutation impairs fast motor kinetics of prestin and has significance for defining functional independence of individual prestin subunits. J. Biol. Chem. 288, 2452–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, and Santos-Sacchi J (1993). Mapping the distribution of the outer hair cell motility voltage sensor by electrical amputation. Biophys. J. 65, 2228–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley H, and Hanson J (1954). Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature 173, 973–976. [DOI] [PubMed] [Google Scholar]
- Huxley AF, and Niedergerke R (1954). Structural changes in muscle during contraction; interference microscopy of living muscle fibres. Nature 173, 971–973. [DOI] [PubMed] [Google Scholar]
- Jian Z, Han H, Zhang T, Puglisi J, Izu LT, Shaw JA, Onofiok E, Erickson JR, Chen YJ, Horvath B, et al. (2014). Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci. Signal. 7, ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Myers R, Sihn CR, Rafizadeh S, and Zhang XD (2013). Slc26a6 functions as an electrogenic Cl−/HCO3− exchanger in cardiac myocytes. Cardiovasc. Res. 100, 383–391. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kang M, Park S, Perez-Flores MC, Zhang XD, Wang W, Gratton MA, Chiamvimonvat N, and Yamoah EN (2019). The local translation of KNa in dendritic projections of auditory neurons and the roles of KNa in the transition from hidden to overt hearing loss. Aging (Albany NY) 11, 11541–11564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC, Gao J, He DZ, Wu X, Jia S, and Zuo J (2002). Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature 419, 300–304. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Ouyang XM, Xia XJ, Zheng J, Pandya A, Li F, Du LL, Welch KO, Petit C, Smith RJ, et al. (2003). Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss. Hum. Mol. Genet. 12, 1155–1162. [DOI] [PubMed] [Google Scholar]
- Mount DB, and Romero MF (2004). The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 447, 710–721. [DOI] [PubMed] [Google Scholar]
- Navaratnam D, Bai JP, Samaranayake H, and Santos-Sacchi J (2005). N-terminal-mediated homomultimerization of prestin, the outer hair cell motor protein. Biophys. J. 89, 3345–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana E, Yang D, Shcheynikov N, and Muallem S (2009). Diverse transport modes by the solute carrier 26 family of anion transporters. J. Physiol. 587, 2179–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, He DZ, Klöcker N, Ludwig J, Schulte U, Waldegger S, Ruppersberg JP, Dallos P, and Fakler B (2001). Intracellular anions as the voltage sensor of prestin, the outer hair cell motor protein. Science 292, 2340–2343. [DOI] [PubMed] [Google Scholar]
- Oliver D, Schachinger T, and Fakler B (2006). Interaction of prestin (SLC26A5) with monovalent intracellular anions. Novartis Found. Symp. 273, 244–253; discussion 253–260, 261–244. [PubMed] [Google Scholar]
- Perez-Flores MC, Lee JH, Park S, Zhang XD, Sihn CR, Ledford HA, Wang W, Kim HJ, Timofeyev V, Yarov-Yarovoy V, et al. (2020). Cooperativity of Kv7.4 channels confers ultrafast electromechanical sensitivity and emergent properties in cochlear outer hair cells. Sci. Adv. 6, eaba1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins G, Lee JH, Park S, Kang M, Perez-Flores MC, Ju S, Phillips G, Lysakowski A, Gratton MA, and Yamoah EN (2020). Altered outer hair cell mitochondrial and subsurface cisternae connectomics are candidate mechanisms for hearing loss in mice. J. Neurosci. 40, 8556–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronceray P, Broedersz CP, and Lenz M (2016). Fiber networks amplify active stress. Proc. Natl. Acad. Sci. USA 113, 2827–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalchenko V, and Santos-Sacchi J (2003). Allosteric modulation of the outer hair cell motor protein prestin by chloride. In Biophysics of the Cochlea: from Molecules to Models, Gummer AW, ed. (World Scientific; ), pp. 116–126. [Google Scholar]
- Rybalchenko V, and Santos-Sacchi J (2008). Anion control of voltage sensing by the motor protein prestin in outer hair cells. Biophys. J. 95, 4439–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Sacchi J (1991). Reversible inhibition of voltage-dependent outer hair cell motility and capacitance. J. Neurosci. 11, 3096–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirish P, Li N, Timofeyev V, Zhang XD, Wang L, Yang J, Lee KS, Bettaieb A, Ma SM, Lee JH, et al. (2016). Molecular mechanisms and new treatment paradigm for atrial fibrillation. Circ. Arrhythm. Electrophysiol. 9, e003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirish P, Ledford HA, Timofeyev V, Thai PN, Ren L, Kim HJ, Park S, Lee JH, Dai G, Moshref M, et al. (2017). Action potential shortening and impairment of cardiac function by ablation of Slc26a6. Circ. Arrhythm. Electrophysiol. 10, e005267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, and Santos-Sacchi J (2010). Conformational state-dependent anion binding in prestin: evidence for allosteric modulation. Biophys. J. 98, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang S, Jia S, and He DZ (2010). Prestin forms oligomer with four mechanically independent subunits. Brain Res. 1333, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hakizimana P, Wu S, Hassan A, Jacob S, Temirov J, Fang J, Mellado-Lagarde M, Gursky R, Horner L, et al. (2015). Outer hair cell lateral wall structure constrains the mobility of plasma membrane proteins. PLoS Genet. 11, e1005500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Timofeyev V, Li N, Myers RE, Zhang DM, Singapuri A, Lau VC, Bond CT, Adelman J, Lieu DK, and Chiamvimonvat N (2014). Critical roles of a small conductance Ca2+-activated K+ channel (SK3) in the repolarization process of atrial myocytes. Cardiovasc. Res. 101, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shen W, He DZ, Long KB, Madison LD, and Dallos P (2000). Prestin is the motor protein of cochlear outer hair cells. Nature 405, 149–155. [DOI] [PubMed] [Google Scholar]
- Zheng J, Madison LD, Oliver D, Fakler B, and Dallos P (2002). Prestin, the motor protein of outer hair cells. Audiol. Neurotol. 7, 9–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the current study have not been deposited in a public repository because they are only used for the publication of this manuscript but are available from the corresponding author on request.