

Summary
To understand the conditions necessary to initiate and terminate seizures, optogenetically induced hippocampal seizures were investigated with LFP, fMRI, and optogenetic inhibition. During afterdischarge (AD) induction using optogenetics, simultaneous LFP recordings identified that the stimulations with earlier ictal onset times were more likely to result in ADs and were more difficult to curtail with optogenetic inhibition. These results were generalizable across two different initiation sites, the dorsal and ventral hippocampus. fMRI during optogenetic induction of AD showed that ADs initiated from the dorsal or ventral hippocampus exhibited distinct brain-wide seizure networks. Short duration seizures initiated in the dorsal and ventral hippocampus were unilateral and bilateral respectively, while longer duration seizures from either site recruited broader, bilateral networks. Interestingly, when optogenetic inhibition is ineffective at stopping seizures, the network activity spreads more extensively but largely overlaps with the network activity associated with seizures that can be stopped. These results provide novel insights on how seizures can be generated and inhibited, which has significant implications for optimizing seizure interventions.
Introduction
Targeted therapies such as deep brain stimulation (DBS) (Fisher et al., 2010) or more recently optogenetics (Wykes et al., 2016) promise to revolutionize the treatment of refractory epilepsy by interfering with specific neuronal circuits involved in the generation and propagation of seizures. However, while considered to be a goal of many experimental epilepsy therapies, it is unknown whether local inhibition at the seizure origin is sufficient to curtail seizure activity or whether rapid seizure propagation across a wider network necessitates a multi-site approach (Tung et al., 2018). Furthermore, because of our poor understanding of seizure networks in animal models, there has been limited development towards a multi-focal approach. A greater understanding of seizure propagation pathways is a necessary step towards enabling targeted therapies for treating refractory patients.
Prior to the advent of optogenetics, investigations into seizure mechanisms were primarily limited to pharmacological agents, which lack both temporal control and cell-type specificity. Optogenetics employs genetic targeting which can be used to disentangle the role of different cell types and networks in seizure maintenance (Bohannon and Hablitz, 2018; Choy et al., 2017; Ellender et al., 2014; Karnani et al., 2016; Lu et al., 2016; Sessolo et al., 2015; Shiri et al., 2016; Yekhlef et al., 2015). Most investigations into using optogenetics for seizure control have focused on spontaneous seizure models (Krook-Magnuson et al., 2015; Krook-Magnuson et al., 2013; Krook-Magnuson et al., 2014; Paz et al., 2013; Wykes et al., 2012), which on the one hand afford an opportunity to study ictogenesis and how different brain states relate to intervention efficacy (Ewell et al., 2015). But on the other hand, without temporal control of seizure onset, it is challenging to define the exact origin of the seizures or employ advanced imaging methods such as optogenetic functional MRI (ofMRI) (Desai et al., 2011; Lee et al., 2010) or calcium imaging (Khoshkhoo et al., 2017). Therefore, guided by our previous studies (Duffy et al., 2015; Weitz et al., 2015), we circumvent this problem by inducing seizure-like afterdischarges (ADs) through optogenetic stimulation of excitatory CAMKII neurons. The advantages of using optogenetics over electrical stimulation include the ability to precisely control origin of seizure in terms of cell type, location, and temporal firing pattern as well as the possibility of artifact-free recording during the stimulation period (Osawa et al., 2013).
Using a model of optogenetically induced afterdischarges, we first investigate whether we can use LFP to predict whether trials progressed to AD by taking advantage of the stimulation-artifact free nature of these recordings. Next, we ask whether it is possible to suppress ADs using spatially localized optogenetic inhibition at the seizure originating focus and whether it is possible predict the success and failure of optogenetic inhibition aimed at suppressing ADs. Finally, using simultaneous LFP-fMRI, we investigated the seizure network of ADs that could and could not be suppressed using local inhibition in both the dorsal and the ventral hippocampus.
Results
Optogenetic seizure initiation in the ventral hippocampus
To induce afterdischarges, CAMKII positive excitatory neurons in ventral hippocampus were optogenetically stimulated (Fig. S1). Initially, we calibrated the stimulation parameters required to induce an AD in each subject by gradually increasing the light power delivered to the VH until the threshold for AD induction was found (Fig. 1a,b). A mean light power of 5.5±1.1 mW was required for induction of ADs under dexmedetomidine. Optogenetics permits stimulation-artifact free LFP recordings (Osawa et al., 2013) which enabled us to estimate the seizure onset time i.e. the onset of large amplitude spiking during stimulation. We hypothesized that onset time would be predictive of the probability of generating an afterdischarge. Examples of onset time estimation are shown in Fig. S2. Briefly, a sliding window of the evoked response was subtracted from the data to reveal large amplitude spiking, which was identified using a thresholding approach.
Figure 1: More progressed ADs with earlier ictal onset time are more likely to be sustained.
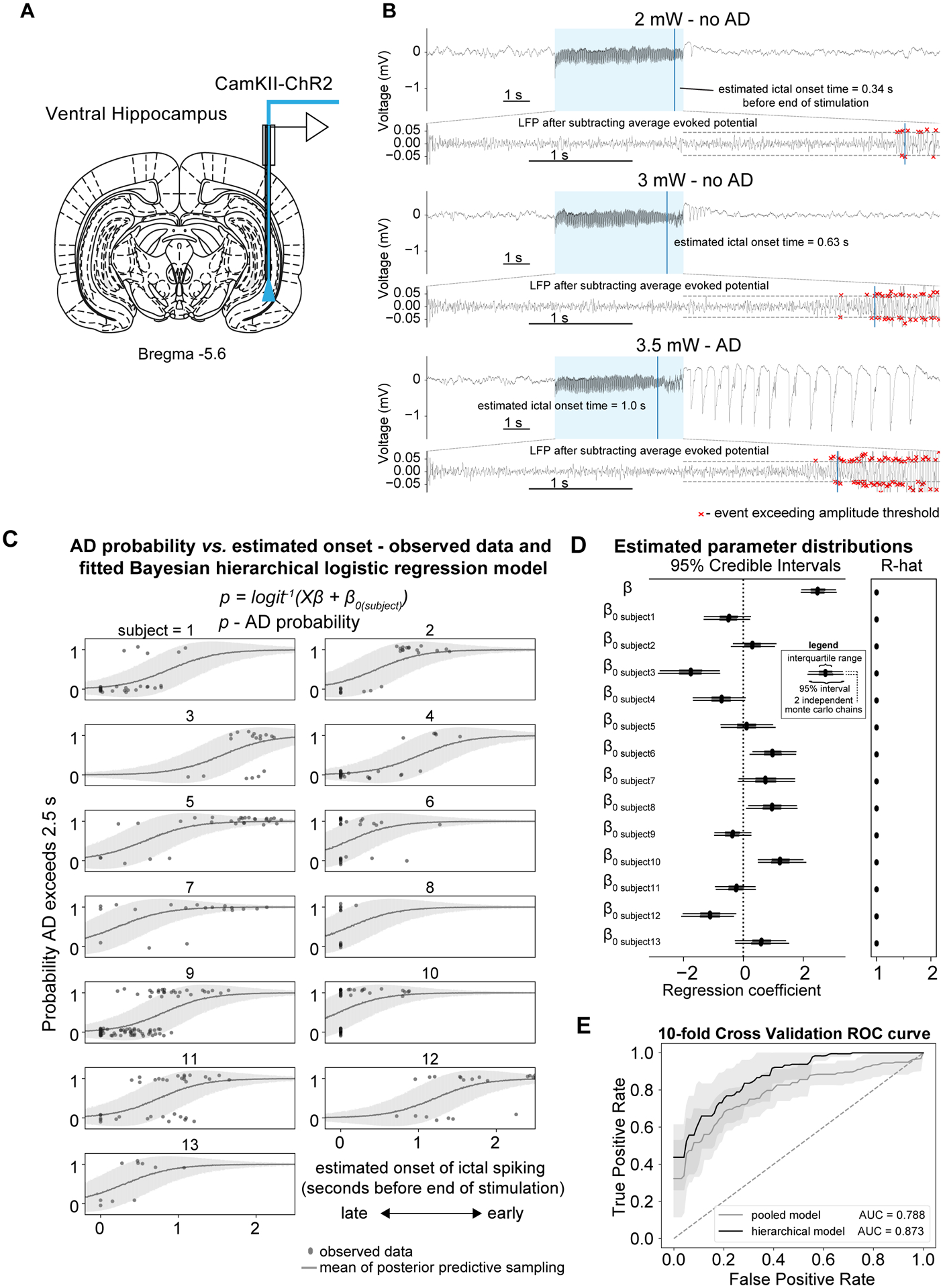
(A) Optrodes were implanted into the ventral hippocampus for electrophysiology and optogenetic excitation. (B) Example LFP traces from a single subject and session illustrating that ADs are not induced at lower light intensities e.g. 2 and 3 mW (top and middle panel respectively) but stimulating ChR2 positive neurons with more intense light e.g. 3.5 mW is able to induce ADs (lower panel). The AD that was sustained had an earlier estimated ictal onset time. The lower sub-panels illustrate how onset time is calculated by subtracting the sliding window evoked potential. (C) AD probability vs. ictal onset time as modelled using a hierarchical Bayesian logistic regression model where intercepts were allowed to vary across subjects. (excluding trials from sessions where the seizure threshold was already known i.e. fMRI sessions, n=13, AD-206 trials, noAD-192 trials). The line shows the mean±SD of the samples from the posterior predictive distribution of the binary outcome variable e.g. AD or no AD and the individual points show the observed data. (D) Left panel - Forest plot of estimated parameter distributions showing the interquartile range and 5th and 95th percentiles for 2 independently run Monte Carlo chains. Ictal onset time was determined to be a significant predictor of ADs as the 95% highest posterior density (HPD) parameter distribution did not contain zero. Right panel – Gelman-Rubin convergence statistic (r-hat) indicating convergence of Monte Carlo chains. Values here were less than 1.1 indicating good convergence. (E) 10-fold cross validation demonstrating good predictive performance of the hierarchical model compared to the pooled model.
Predicting successful generation of afterdischarges
To determine the relationship between the ictal onset time and the severity of the resulting ADs, we compared this ictal onset time to the probability of successful AD generation, which was defined here as those longer than 2.5s in duration (measured from the end of the stimulation period). Visually, trials with sustained ADs were associated with an earlier ictal onset time, compared to stimulations that did not persist for longer than the duration threshold (Fig. 1b). To test the reliability of this observation, we first compared the onset times between trials with sustained ADs to subthreshold trials shorter than the 2.5s threshold (noAD-206 trials, AD-192 trials, n=13) (Fig. 1c), and found that trials which progressed to AD had significantly earlier ictal onset times compared to those which did not. This relationship was clear in most subjects, however in some subjects e.g. 6 and 8, there was frequently no detectable spiking prior to the end of the stimulation period. As different subjects are known to have different propensities for ADs, the AD probability was modeled using a hierarchical Bayesian model with ictal onset time as the explanatory variable and the intercepts (β0) allowed to vary across subjects (Fig. 1c,d). Figure 1c shows the observed data for individual subjects alongside the mean±SD of the posterior predictive samples. The data generated from the model reproduces the pattern of the observed data well, suggesting a good model fit. Earlier onset significantly predicted the occurrence of ADs, indicated by the ictal onset regression coefficient and their 95% Highest-Posterior Density (HPD) uncertainty intervals being greater than zero. The Gelman-Rubin convergence statistic (R-hat) was less than 1.1 for all parameters indicating the sampling procedure converged well.
We assessed the sensitivity and specificity of the varying intercept model on held-out data and using a receiver operating characteristic (ROC) analysis. A ROC area under curve (AUC) of 0.87 was obtained using a 10-fold cross validation (Fig. 1e), while the pooled model yielded an AUC of 0.79, demonstrating the advantage of using a hierarchical model over a model where the data was pooled across subjects. False positive classification occurred occasionally, typically where the LFP exhibited early onset spiking but ADs did not persist for longer than the 2.5 s threshold (Fig. S2b). False negatives were typically associated with increased amplitude in time with the stimulation frequency but without detectable spiking (Fig. S2c). After having established that it was possible to reliably invoke ADs using optogenetic stimulation and having identified that ictal onset time provided a predictive marker of afterdischarge generation, we then investigated the possibility of interrupting ADs by silencing local neuronal populations at the site of AD initiation.
Expression of both ChR2 and eNpHR3.0 in overlapping cell populations
To determine the effectiveness of inhibiting local neuronal populations in interrupting ADs in the VH, we targeted the hSyn promoter to express eNpHR3.0 in all neuronal subtypes. Co-expression of both inhibitory and excitatory opsins was confirmed from histological sections taken from rats co-injected with both AAV5-ChR2 under the CAMKII promoter and AAV5-eNpHR3.0 under the hSyn promoter. Although we expected that the efficiency of transduction could suffer upon simultaneous administration of the 2 different viruses, confocal data suggested that it was possible to express both opsins in overlapping cell populations, with ChR2 predominantly being expressed in axons and dendrites, while eNpHR3.0 was more strongly expressed within the soma (Fig. S1a,b). As expected, rats injected with the hSyn-eNpHR3.0 virus expressed the inhibitory opsin in both CAMK expressing excitatory neurons and GAD67 positive inhibitory neurons (Fig. S1c).
Optimization of the light intensity needed for optogenetic inhibition
To determine the approximate light intensity needed for inhibition of local ventral hippocampal neurons using eNpHR3.0, we assessed spiking at the local site with multi-unit recordings (MUA). A combined optrode for light delivery and MUA recordings was constructed such that the tip of the tungsten electrode lay approximately 0.65 mm below the optical fiber tip (Fig. S3a,b). As expected, delivering continuous 589 nm orange light to activate eNpHR3.0 was effective at inhibiting spontaneous neuronal firing in the ventral hippocampus in rats expressing hSyn-eNpHR3.0 (Fig. S3c,d).
We then tested the capacity of eNpHR3.0 inhibition to suppress optogenetic stimulation by activating eNpHR3.0 and ChR2 simultaneously. We found that at lower light intensities e.g. <10 mW (1100 mW/mm2) eNpHR3.0 inhibition did not prevent ADs despite inhibition (Fig. S3e). Where higher light intensities were used e.g. above 18 mW (2079 mW/mm2), this was effective at preventing the occurrence of afterdischarges in rats expressing eNpHR3.0 under the hSyn promoter (9 trials in 3 animals) and therefore a slightly higher intensity - 21 mW (2425 mW/mm2) - was used to maximize the probability of seizure suppression across the rest of the experiments. In addition to testing the effectiveness of inhibition using hSyn-eNpHR3.0, we also tested CAMKII-eNpHR3.0, but found it to be less effective at inhibiting spontaneous firing and afterdischarges (Fig. S3f–k), potentially due to the lower virus titer available or selective targeting of excitatory neurons. For this reason, we focused on inhibition of hSyn-positive neurons for seizure suppression.
Interruption of seizure-like afterdischarges in the ventral hippocampus using eNpHR3.0
First, we tested the possibility of interrupting ADs in the VH using local inhibition immediately following the seizure-generating stimulation (Fig. 2a,b). As indicated by the examples in Fig. 2c, we found that in certain cases, seizure-like activity could be interrupted within the 5 s period of optogenetic inhibition, but large amplitude spiking still occurred for a few seconds during the early period of successful inhibition. Therefore, to capture the relative success and failure of inhibition, we binarized the data with AD thresholds at 2.5s and 5s. At both thresholds, the effect of inhibition was robust and effectively decreased the AD probability (Fig. 2d). The distribution of the AD durations with and without inhibition are shown in Fig. 2e. The median AD duration without inhibition was 10.1±4.2 s and with inhibition was shortened to 0.83±8.6. The AD duration was significantly reduced in 4 of 6 animals (p<0.05, paired t-test) upon optogenetic inhibition (Fig. 2e).
Figure 2: Local optogenetic inhibition was able to curtail seizure-like afterdischarges.
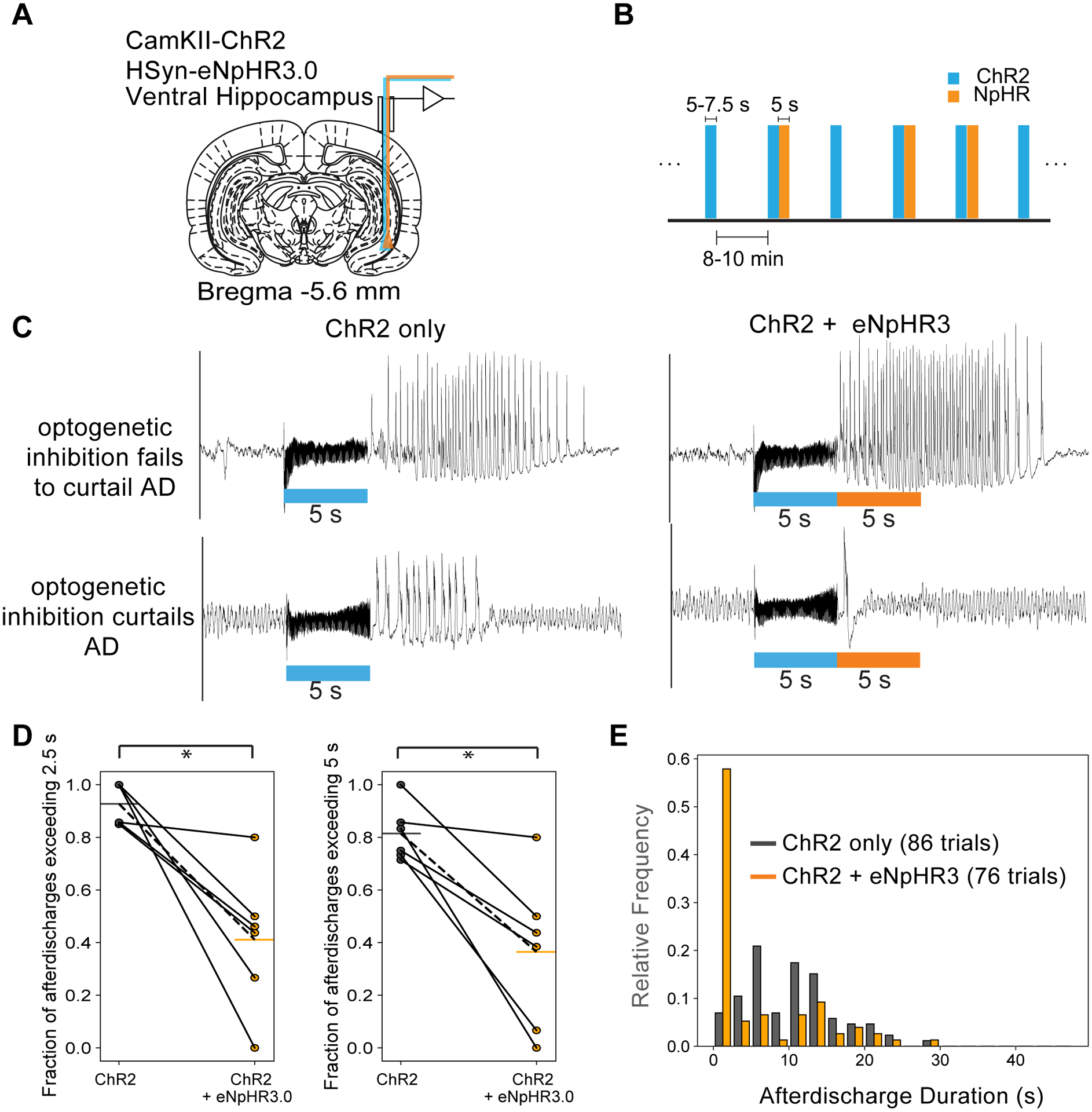
(A) Ventral hippocampus was targeted for electrophysiology and optogenetic excitation and inhibition. (B) Experimental design. ChR2 excitation only and ChR2 followed by NpHR3.0 inhibition blocks were randomized and applied with a 10 min inter-stimulus interval. Example LFP traces from rats expressing hSyn-eNpHR3.0 illustrating examples where optogenetic inhibition was considered to have failed to curtail ADs (top panels) and where optogenetic inhibition was considered to have succeeded in curtailing ADs (lower panels). The traces on the left and right panels are from the same recording sessions. The left panels are from stimulations consisting of the control condition (ChR2 only) and the right panels are from where the optogenetic inhibition was applied immediately following the ChR2 blue light stimulation. These examples illustrate that ADs from sessions where the optogenetic inhibition fails to curtail the AD appeared longer and more severe. (D) Proportion of ADs exceeding 2.5/5 s in duration for the two different conditions – the control condition (ChR2 only) vs. the optogenetic inhibition condition (ChR2 + eNpHR3). (E) Histograms of AD duration for the two different stimulation conditions. This metric was included as AD duration can be highly variable across different sessions and subjects. * p<0.05 represents based on paired t-tests for n=6 subjects.
We aimed to test whether failure of optogenetic inhibition to curtail ADs was related to onset time (Fig. 3a,b). Therefore, we used a linear hierarchical Bayesian model to investigate the relationship between the success and failure of inhibition and seizure progression (Fig. 3c), with intercepts allowed to vary across subjects to account for within subject correlation and the fact that different subjects have different propensities for ADs (Fig. 3c,d).
Figure 3: More progressed ADs cannot be interrupted using optogenetic inhibition.
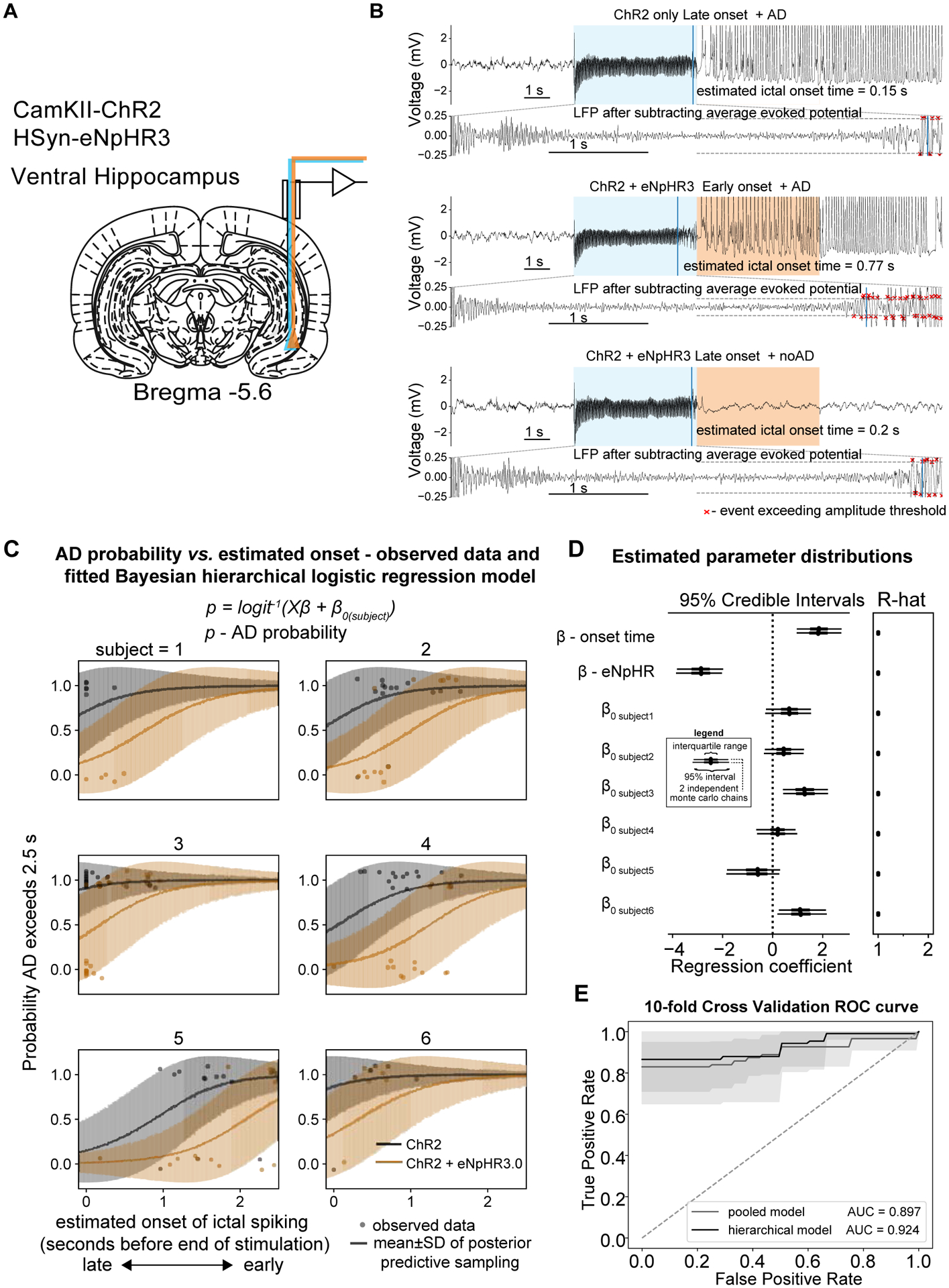
Given the relationship between ictal onset time and AD probability, the onset time was used as a covariate in a subsequent regression analysis. (A) Ventral hippocampus was targeted for electrophysiology and optogenetic excitation and inhibition. (B) Examples LFP traces indicating stimulation condition and ictal onset time from a single session. Upper panel − ChR2 only and late onset ictal activity with a sustained AD. Middle panel − ChR2 + eNpHR3.0 and early onset ictal activity with a sustained AD. Lower panel − ChR2 + eNpHR3.0 and late onset ictal activity without after discharge. (C) Modelling using a Bayesian hierarchical (random intercept) logistic regression. i.e. the log odds of the AD probability logit(p) was explained by a linear combination of the stimulation condition and ictal onset time along with a subject specific intercept term. The line shows the mean±SD of the samples from the posterior predictive distribution of the binary outcome variable e.g. AD or noAD and the individual points show the observed data. (D) Left panel - Forest plot of estimated parameter distributions showing the interquartile range and 5th and 95th percentiles for 2 independently run Monte Carlo chains. Ictal onset time and stimulation condition (ChR2 vs. ChR2 + eNpHR3.0) were both determined to be significant predictors of ADs as their 95% highest posterior density (HPD) parameter distributions did not contain zero. Right panel – Gelman-Rubin convergence statistic (r-hat) indicating convergence of Monte Carlo chains. Values here were less than 1.1 indicating good convergence. (E) 10-fold cross validation demonstrating good predictive performance of the hierarchical model compared to the pooled model. All panels include n=6 rats.
Results from the model suggested that earlier onset time again predicted ADs and that optogenetic inhibition significantly reduced AD probability (both onset time and stimulation condition were significant predictors because 95% HPDs did not contain zero). The samples drawn from the posterior distribution fit the observed data well (Fig. 3d) and demonstrate how more progressed (earlier onset) ADs were less likely to be curtailed. In addition, the hierarchical varying intercept model demonstrated excellent capacity to predict ADs on held out data and yielded an AUC of 0.92 on a 10-fold cross validation (Fig. 3e), compared to 0.9 for the pooled logistic regression model.
Interruption of seizure-like afterdischarges in the dorsal hippocampus using eNpHR3.0
To determine if ictal onset time also provided a predictive marker of AD inhibition in the dorsal hippocampus, we applied the same experimental paradigm used in the ventral hippocampus experiments to ADs initiated from the DH (Fig. S4a). Co-injection of the 2 viruses to the DH also led to robust expression of the 2 opsins (Fig. S1a). We also investigated the impact of stimulation duration as it was necessary to increase the stimulation duration to drive ADs in the presence of local inhibition in the DH. ADs elicited with shorter duration stimulation periods (5 s) were interrupted much more readily compared to those elicited with longer duration stimuli (6–7.5 s) i.e. ChR2 stimulation periods greater than 5 s in duration were not readily curtailed (Fig. S4a–e). There was a strong linear relationship (Fig. S4h, p<0.05, t-test) between stimulation duration and ictal onset time (which was measured in relation to the end of the stimulation period) and therefore, as expected, ictal onset time was significantly earlier in trials which were not curtailed using optogenetic inhibition (Fig. S4i, p<0.05, paired t-test) further highlighting its utility as a marker of AD inhibition.
Spread of activity in ADs initiated from the dorsal vs. ventral hippocampus
After having shown that it was possible to curtail ADs in either the dorsal or ventral hippocampus, we characterized to what extent the spread of seizure activity differed between these two different initiation sites. We mapped the relationship between the seizure network activity on fMRI and AD duration to explore how propagation patterns differed in ADs induced in the DH compared to the VH. A binary variable was used to describe voxels were described as either active (if they exceeded the p<0.001 statistical threshold) or not active and a logistic function was fitted across all subjects and trials as a function of the AD duration as illustrated in Fig. 4a. The inflection point of this fitted curve provides an estimation of the average activation time at each voxel during an AD and thus was used to visualize the spread of seizure activity over the course of ADs originating in different regions. Among the trials which resulted in ADs, there was no difference between the activation time maps with and without optogenetic inhibition (Fig. S5), therefore these data were combined. These maps exhibited distinct patterns in seizures initiated from the dorsal hippocampus compared to the ventral hippocampus. ADs originating from the VH displayed early activation in short duration ADs in the ipsilateral VH, septum and ipsilateral prefrontal cortex (Fig. 4b). The average activation time in the contralateral VH was 17±2 s later than the ipsilateral hippocampus, while the contralateral PFC was also 18±6 s later than the VH. However, in the dorsal hippocampus it was significantly later (27±5 s) demonstrating that the DH was only active during long duration ADs.
Figure 4: Voxel-wise activation time maps comparing the spread of seizure activity between ADs originating in the dorsal and ventral hippocampi.
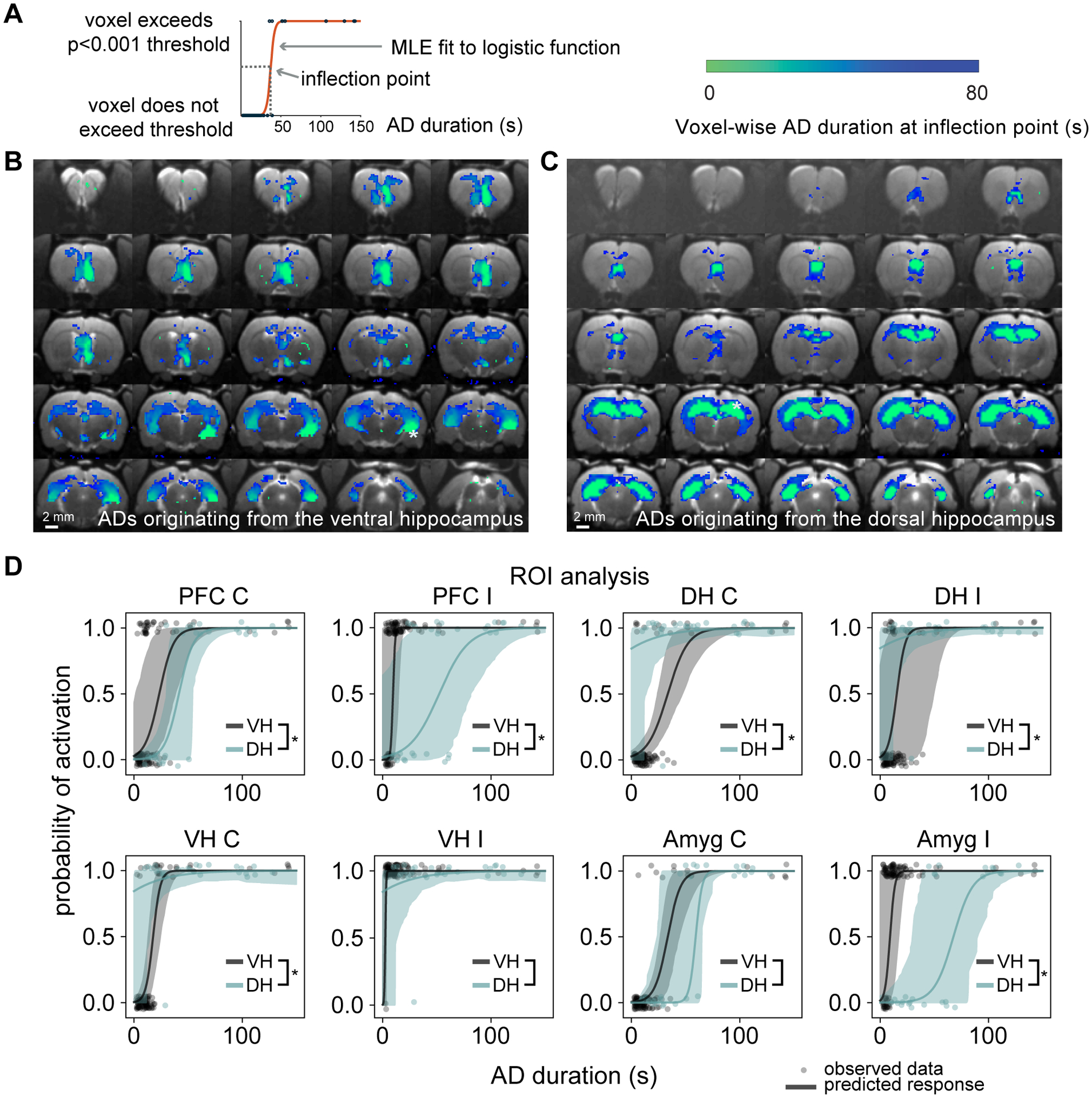
(A) A logistic function was fitted to each of the individual trials (blue dots) which progressed to AD and the inflection point was used as an estimate of the AD duration at which 50% of trials displayed activation for each voxel. (B) Voxel-wise relationship between AD duration and fMRI activation for focal VH ADs and (C) focal DH ADs. Distinct patterns were observed for seizures originating from the ventral hippocampus compared to the dorsal hippocampus. Activation is present in the ipsilateral PFC for short duration ADs, whereas the inflection point is slightly later for the contralateral VH and PFC. However, both the ipsilateral and contralateral DH are only activated in longer duration ADs. In seizures originating from the DH, both the ipsi- and contralateral DH and VH are activated in short duration ADs, whereas longer duration ADs may start to involve cortical regions. (D) ROI-wise analysis. Regions were considered to be activated if their volume of activation exceeded 10% of the maximum volume within each subject. Observed data are represented as a binary response variable with “1” representing the region as active and “0” representing not active. The response predicted by the random intercept logistic regression is displayed as the mean ± 95% bootstrapped confidence intervals. * denotes that the inflection point is significantly different at a level of p<0.05, based on hypothesis testing using parametric bootstrapping (VH: n=6 rats and DH: n=3 rats)
The activation time map generated from ADs originating from the DH (Fig. 4c) indicated that short duration ADs involve both ipsilateral and contralateral hippocampi with a significant spread of activation toward the temporal pole. Longer duration ADs spread to the PFC as well as other cortical regions. These data support the idea that contrary to ADs elicited from the VH, focal DH seizures quickly spread bilaterally to the entire hippocampus, whereas focal VH ADs did not involve the DH until much later and instead spread to the amygdala and PFC. Quantitatively, the inflection points in different ROIs were compared using a random-intercept model (Fig. 4d). Activation of the ipsilateral amygdala and both the right and left PFC were significantly earlier in ADs initiated from the VH compared to the DH (27±5 s and 29±9 s respectively), while activation of the contralateral hippocampus was 26±3 s earlier in the DH. After mapping the network activation for ADs initiated in the DH and VH, we then looked to understand the seizure network that was able to sustain activity in the presence of local inhibition, by investigating the differences between two different scenarios, where seizure activity sustained despite local inhibition and where it was curtailed. Furthermore, the analysis was restricted to ADs that were < 25 s in duration, to understand the networks capable of sustaining seizure activity during the early stages of seizure maintenance.
Simultaneous LFP and fMRI of afterdischarges in the ventral hippocampus
We next used whole brain fMRI imaging to investigate which regions were involved in ADs that could not be curtailed using local inhibition (Fig. 5a,b). A typical activation map from a trial which was curtailed by optogenetic inhibition displayed only local activation alongside activation of the septum and very limited regions within the prefrontal cortex (Fig. S6a). While a single, short duration (16 s) AD from the same session that sustained despite optogenetic inhibition involved activity across a large volume in the anterior-posterior direction of the VH as well as the lateral septum, hypothalamus, anterior thalamus and prefrontal cortex (Fig. S6b). There appeared to be involvement of the anterior amygdala although susceptibility artifacts arising from the air-tissue interface surrounding the ear canal limited a more thorough assessment of the posterior amygdala. Within the hippocampus, activity was restricted to the ventral region and did not spread to the dorsal hippocampus. Where ADs were not sustained, the LFP (Fig. 4c) displayed high amplitude spiking following the end of the stimulation period. Where ADs persisted, the fMRI response within the ipsilateral PFC, amygdala and PFC was also prolonged (Fig. 5d). The fixed-effects group level activation maps (Fig. 5e,f) were in accordance with the single trial examples, but in addition, there was activation across the anterior, medial-dorsal, ventral-medial and dorsal-lateral subregions of the thalamus. These findings were consistent across subjects as illustrated by plotting the fraction significant activation across regions (Fig. 5f,g).
Figure 5: fMRI activation maps for ADs originating from the ventral hippocampus indicates that ADs that did not progress were limited to the hypothalamus, amygdala and PFC.
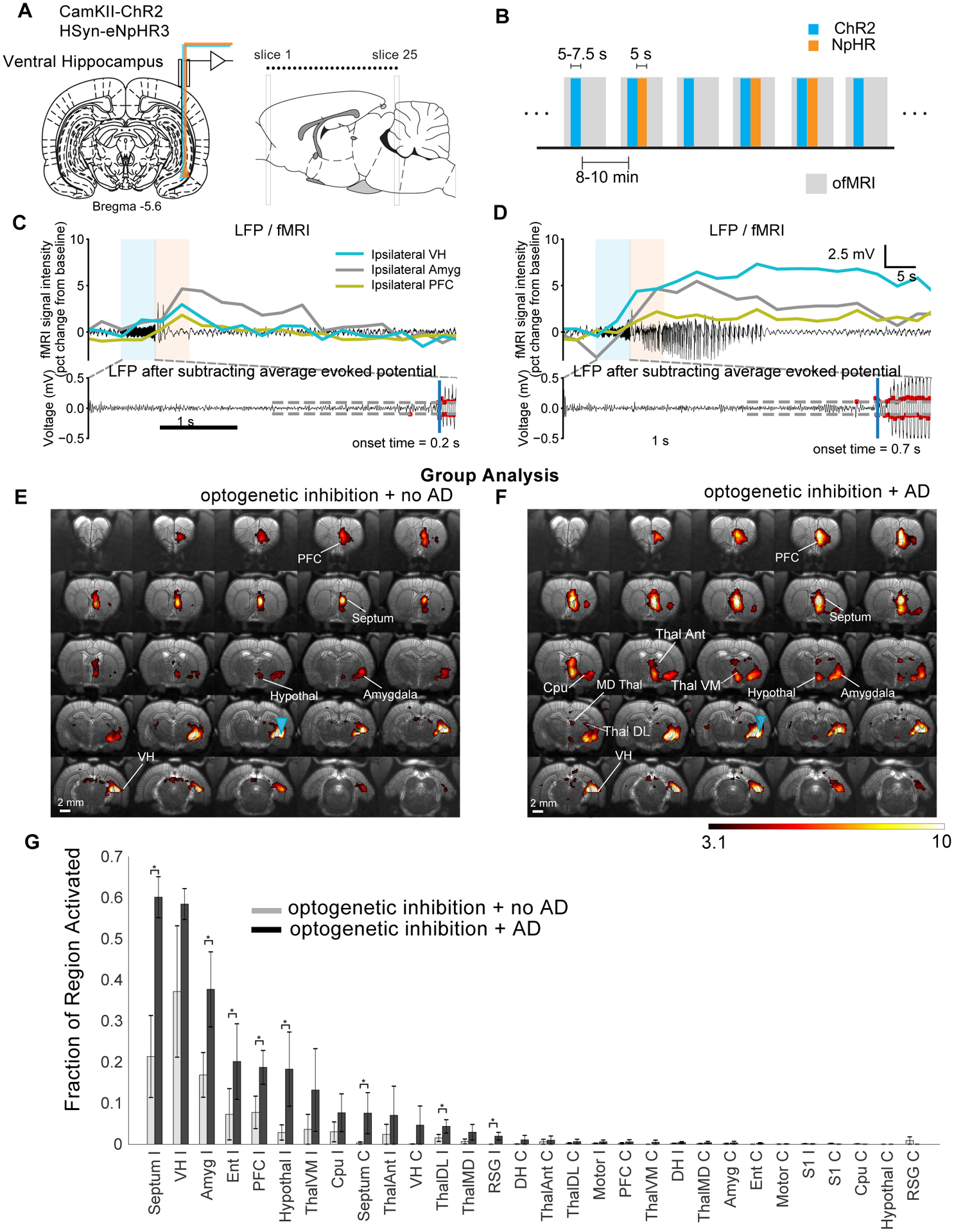
(A) Optrodes were implanted into the ventral hippocampus for electrophysiology and optogenetic excitation and inhibition. (B) Within session fMRI stimulation paradigm involved randomly alternating between trials which did and did not involve eNpHR3.0. (C and D) Examples of simultaneously acquired LFP and fMRI signal from different ROIs. Fixed-effects group-level analysis (n=3) for trials that did not (E) and did (F) progress to ADs despite optogenetic inhibition. (G) Fraction of region activated with and without successful optogenetic inhibition of AD. Single trial T-statistic maps are thresholded at p<0.001, uncorrected, whereas group-level activation maps are shown at a threshold of p<0.0001, uncorrected. Data are represented as mean ± SEM. Abbreviations are as follows: Amyg (amygdala), Cpu (caudate putamen), DH (dorsal hippocampus), Ent (entorhinal cortex), PFC (prefrontal cortex), RSG (retrosplenial granular cortex), ThalDL (dorsomedial thalamus), ThalMD, (mediodorsal thalamus), ThalVM (ventromedial thalamus). * indicates the fixed-effects difference between AD vs. no AD at a significance level of 0.05.
Simultaneous LFP and fMRI of afterdischarges in the dorsal hippocampus
Where ADs originating in the DH were curtailed using optogenetics, single-trial fMRI typically displayed activation across a large volume in the anterior posterior direction of the dorsal hippocampus (Fig. S6c). Where ADs persisted, activation spread bilaterally across the hippocampus and dorsal septum (Fig. S6d) with a greater spread of activation towards the temporal pole of the hippocampus. For trials that were curtailed, large amplitude spiking that commenced towards the end of the ChR2 stimulation did not continue for longer than the 2.5 s threshold (Fig. 6c). Where ADs did persist, there exhibited a prolonged fMRI response across the entire hippocampus, with a delayed response in the VH compared to the DH (Fig. 6d). The group-level fixed-effects analysis supported the results from the trial examples. Specifically, where ADs were not sustained (Fig. 6e), there was spread of activity towards the temporal pole of the hippocampus ipsilaterally but remained highly restricted within the DH on the contralateral side. Where ADs persisted, activation typically spread bilaterally throughout the entire hippocampus as well as the retrosplenial cortex (Fig. 6f). Quantitatively, 51±15% of the contralateral DH ROI was activated when ADs persisted compared to 10±4%, when they did not (Fig. 6g).
Figure 6: fMRI activation maps with dorsal hippocampus stimulation and inhibition shows that ADs did not progress were limited to the anterior dorsal hippocampus.
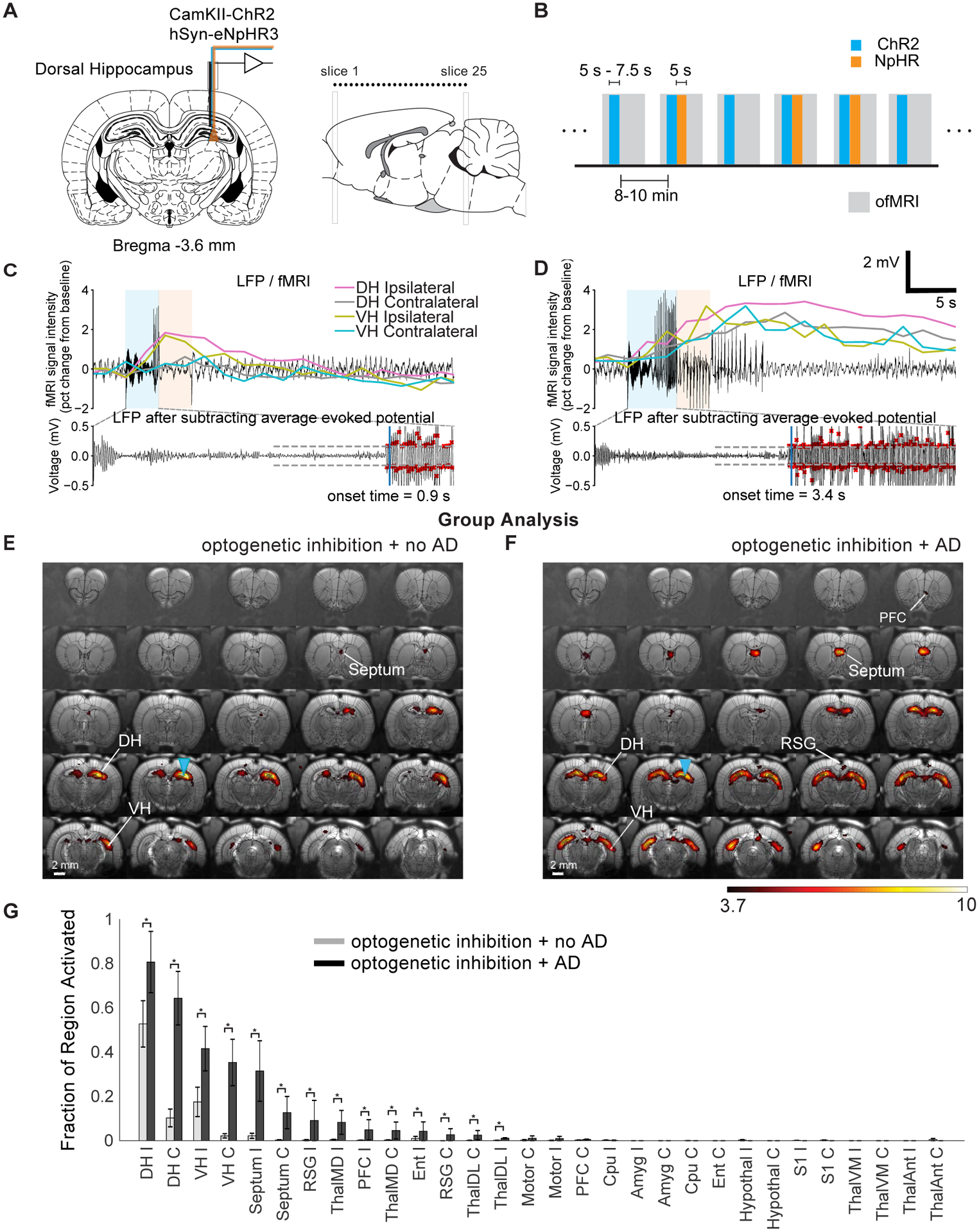
(A) Optrodes were implanted into the dorsal hippocampus for electrophysiology and optogenetic excitation and inhibition. (B) Within session fMRI stimulation paradigm involved randomly alternating between trials which did and did not involve eNpHR. (C and D) Simultaneously acquired LFP and fMRI signal from different ROIs. (E and F) Fixed-effects group-level (t-statistic) analysis (n=3) for trials that did not (E) and did (F) progress to ADs despite optogenetic inhibition. (G) Fraction of region activated with and without successful optogenetic inhibition of ADs. T-statistic maps are thresholded at p<0.0001, uncorrected. Data are represented as mean ± SEM. Abbreviations are as follows: Amyg (amygdala), Cpu (caudate putamen), DH (dorsal hippocampus), Ent (entorhinal cortex), PFC (prefrontal cortex), RSG (retrosplenial granular cortex), ThalDL (dorsomedial thalamus), ThalMD, (mediodorsal thalamus), ThalVM (ventromedial thalamus). * indicates the fixed-effects difference between AD vs. no AD at a significance level of 0.05.
Effect of optogenetic inhibition and high light intensity on the fMRI signal
To rule out any fMRI artifacts related to optogenetic inhibition and to demonstrate the effectiveness of optogenetic inhibition at suppressing local activation, we characterized the effect of the inhibition itself on the fMRI response. A block-design stimulation paradigm was used for detection efficiency (Fig. 7a,b) and the rats implanted with optrodes with the dorsal hippocampus were used because in the VH group, the stimulation site was partially obscured by susceptibility artifacts arising from the air-tissue interface of the ear canals.
Figure 7: Optogenetic inhibition applied during optogenetic stimulation dramatically attenuates the fMRI response, while applied by itself optogenetic inhibition does not alter the CBV weighted fMRI signal.
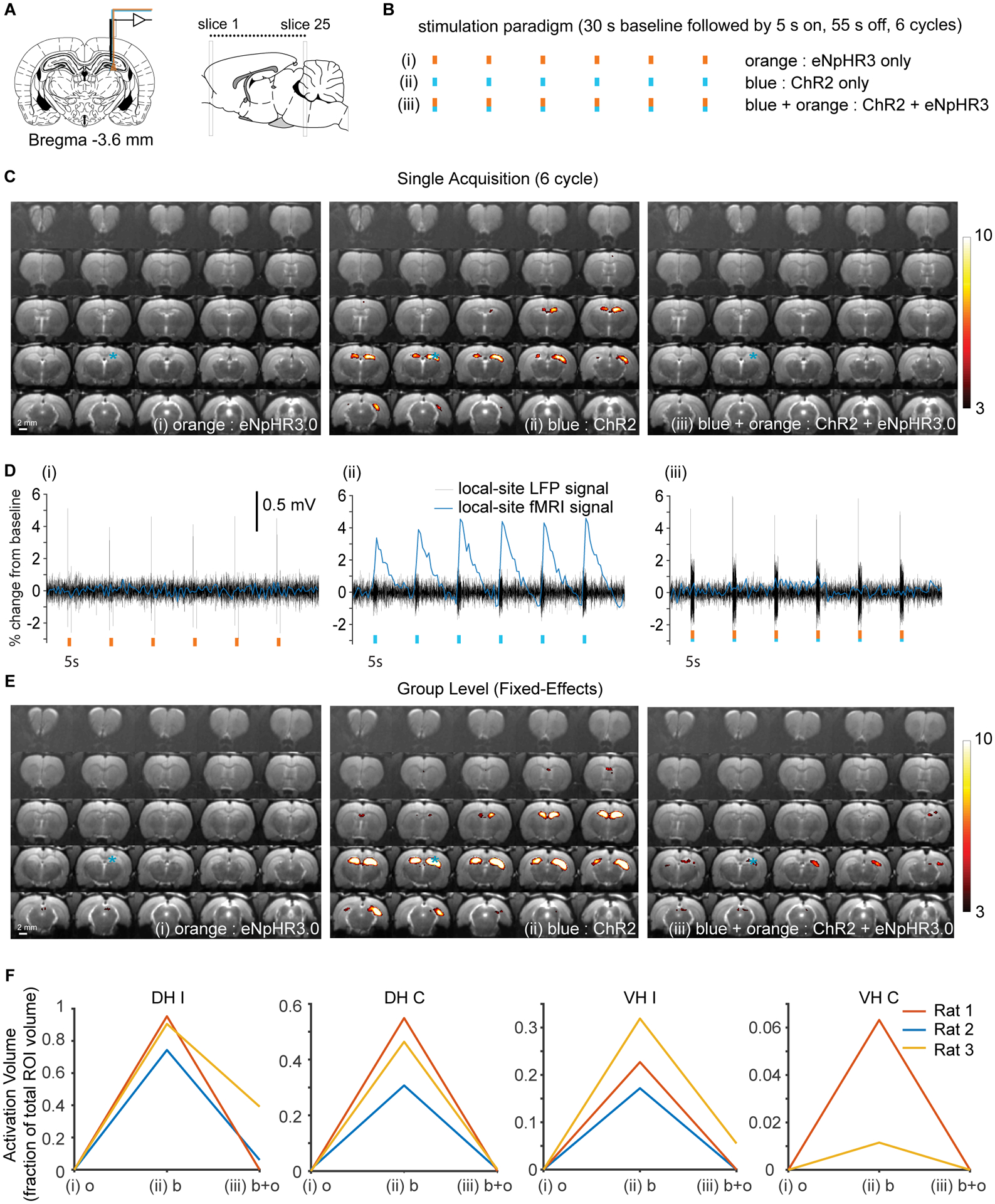
(A) Optrodes were implanted into the dorsal hippocampus for electrophysiology and optogenetic excitation and inhibition. (B) Three separate stimulation paradigms were compared in each animal using simultaneous LFP-ofMRI. (i) Optogenetic inhibition applied on its own (eNpHR3 only). (ii) Optogenetic stimulation of ChR2 using blue light only (ChR2 only). (iii) Optogenetic stimulation and inhibition applied at the same time (ChR2 + eNpHR3 together). (C) Typical activation maps from a single trial and single subject for the three different conditions listed above. (D) Time courses for fMRI and LFP for the example data shown in (C). Note that local optogenetic inhibition applied during activation of ChR2 almost completely eliminated the fMRI response, whereas the LFP response to ChR2 stimulation remained intact. (E) Fixed-effects fMRI analysis activation maps generated from across all trials and all 3 subjects. (F) Activation volumes for the three different subjects plotted for the 4 different regions that are activated. Note that only very limited activation occurred in one rat when halorhodopsin and ChR2 were activated simultaneously using blue and orange light. Abbreviations: DH I – dorsal hippocampus ipsilateral, DH C - dorsal hippocampus contralateral, VH I – ventral hippocampus ipsilateral, VH C – ventral hippocampus contralateral. All activation maps are shown at p< 0.001, uncorrected. E to change to orange, blue then blue + orange
First, in order to show that we could detect CBV-weighted fMRI signal changes, we stimulated ChR2 expressing CAMKII-positive neurons in the dorsal hippocampus with low light intensity 0.7 mW (81 mW/mm2) and observed a strong fMRI response in the ipsilateral and contralateral DH (Fig. 7c,ii). Given the light intensity that we used for the orange laser 21 mW (2420 mW/mm2) was much higher than that used for the blue laser, we expected a large volume of optogenetic inhibition to overwhelm the volume of excitation and the CBVw fMRI response to be diminished upon switching the blue and orange laser on at the same time. Our results supported this hypothesis and as in the example shown in Fig. 7c,iii, the optogenetic inhibition completely eliminated the fMRI signal. As we hypothesized, optogenetic inhibition alone did not evoke any observable change in fMRI signal (Fig. 7d,i), but did cause some deflection of the LFP at the onset and end of the stimulation pulse. Despite optogenetic inhibition, the effect of the ChR2 stimulation was evident on the LFP trace (Fig. 7d,iii), which displayed an evoked response at the stimulation frequency during stimulation periods. The dramatically reduced activation volumes upon simultaneous optogenetic inhibition were consistent in all 3 rats (Fig. 7e,f).
Discussion
Our results revealed how local inhibition of neuronal populations using optogenetics could reduce the probability of evoked ADs originating in the dorsal or ventral hippocampus. To understand the extent to which halorhodopsin was able to suppress ADs, a multivariate hierarchical Bayesian logistic regression was used to model the AD probability using ictal onset time and the stimulation condition as predictors. Results suggested that onset time predicted the success and failure of optogenetic inhibition to curtail ADs. Simultaneous LFP-fMRI was used to map the seizure networks responsible for sustaining seizure activity in the presence of local inhibition. Distinct networks were identified for ADs originating from the VH compared to those generated from the DH. Interestingly, these networks were largely similar for ADs that could be curtailed compared to those that could not while activity spread more extensively in those that could not be curtailed.
Our statistical model based on the ictal onset time suggests that the effectiveness of local optogenetic inhibition to curtail ADs is related to their progression, with earlier onset ADs being less sensitive to local inhibition. The hypothesis that earlier onset ADs were more difficult to curtail was supported both in the VH and the DH and was further supported by the observation that ADs elicited in the DH with shorter duration stimulation periods (5 s) were interrupted much more readily compared to those elicited with longer duration stimuli (6–7.5 s), suggesting that local inhibition rapidly became ineffective as seizures spread beyond their focus. LFP-fMRI was used to visualize the spread of seizure activity where ADs could not be stopped using eNpHR3.0 to those in which they were curtailed locally. Our data suggests that focal seizures elicited from the DH that were interrupted locally, reliably involved both hippocampi. Where ADs could not be curtailed, they did not necessarily involve extrahippocampal regions but rather spread further towards the temporal pole.
ADs elicited from the VH that could be optogenetically disrupted, frequently involved limited ipsilateral activation of the septum, amygdala, prefrontal cortex and hypothalamus. Short-duration ADs that were not curtailed using local optogenetic inhibition spread extensively throughout these regions as well as to the thalamus. The network activity involved in ADs that could and could not be curtailed was therefore largely similar but different in extent, while ADs initiated from the DH were dramatically different from those initiated from the VH.
By observing the relationship between fMRI activation and AD duration, we identified highly distinct patterns of activity in focal DH ADs compared to focal VH ADs. ADs elicited from the dorsal hippocampus involved the contralateral dorsal hippocampus from the start and rapidly spread towards the ventral hippocampus. Whereas focal VH ADs typically remained unilateral, but gradually spread to the contralateral ventral hippocampus and then to the septal pole. The DH and VH are considered to be functionally and genetically disparate while exhibiting distinct connectivity to other regions (Fanselow and Dong, 2010). The VH is most prominently connected to the hypothalamus, amygdala and prefrontal cortex, while the DH has dominant projections to the retrosplenial cortex (Fanselow and Dong, 2010; Jay and Witter, 1991; Meibach and Siegel, 1977; Pitkanen et al., 2000). This connectivity is largely reflected in our data where we found seizures initiated from the VH exhibited early activation of the hypothalamus, septum, prefrontal cortex and amygdala, while the retrosplenial cortex was the primary extrahippocampal region involved in focal DH afterdischarges. It is surprising that shorter duration (< 25 s) ADs from the VH were predominantly unilateral and for seizures longer than this, activation in the contralateral hemisphere was evident firstly in the ventral hippocampus and septum, suggesting that seizure activity might spread to the contralateral hemisphere via the ventral hippocampal commissure. Afterdischarges initiated from the dorsal hippocampus activated the contralateral DH from the start in all trials. These data are consistent with other fMRI studies which show that direct electrical stimulation of the DH leads to bilateral activation of the hippocampus (Moreno et al., 2015), except in the case of significant short-term plasticity which leads to extrahippocampal spread.
Our results are also consistent with our previous optogenetic fMRI study showing that stimulation of the dorsal hippocampus leads to activation patterns limited to the hippocampal formation and retrosplenial cortex whereas stimulation of the intermediate hippocampus causes much more widespread activation of the PFC (Weitz et al., 2015). In line our data, Motelow et al. found that electrically induced focal DH seizures in naïve rats leads to fMRI activation limited to both hippocampi, the septal nuclei, and the anterior hypothalamus (Motelow et al., 2015). Contrary to their work however, we did not observe the widespread negative fMRI signal throughout the thalamus and cortex. Differences could be explained by different methods of AD induction including cell type specific stimulation, different fMRI contrast mechanisms, and anesthetic regimens. The work by Toyoda et al. indicates that spontaneous seizures in the pilocarpine model, rapidly spread from the VH to the contralateral VH or to the ipsilateral DH (Toyoda et al., 2013). However, our results indicate that in the non-epileptic brain, ADs initiated from the VH remain unilateral except in the case of longer and more severe ADs which spread to and possibly are then sustained by the contralateral VH. The current study also provides evidence that in the non-epileptic brain, shorter duration afterdischarges initiated in the VH did not spread to the dorsal hippocampus. It would be interesting to compare these results by using LFP-fMRI in a model of chronic epilepsy to determine whether seizure activity spreads more rapidly across these circuits.
In our study, after the AD threshold was found we achieved around a 80% success rate for inducing ADs, which is in line with that reported by Khoshkhoo et al. (Khoshkhoo et al., 2017). One limitation of the current study is an inability to induce ADs with a 100% success rate. This is particularly relevant due to the necessity of inducing ADs at a level which was just above the threshold for seizure induction but has also been found to be a problem in other studies (Khoshkhoo et al., 2017) which needed to vary the light power and frequency in order to achieve consistent seizure induction using optogenetics. Excluding the trials that were clearly below the seizure-threshold ensured that most of the included trials would have progressed to ADs in the absence of optogenetic interference. However, a more reproducible method of inducing ADs could not be used since using a stimulus significantly above the threshold for AD induction would also result in severe ADs that cannot be curtailed using local inhibition. Another limitation relates to the difficulty in separating the fMRI response from the stimulation period to that of the post-stimulus seizure activity. Separating the two responses could enable us to more precisely localize the regions involved during the very early phases of seizure maintenance. However, the fMRI response is known to exhibit non-linearity, particularly during spiking activity (Pouliot et al., 2012), which prevented us from taking this approach.
Using CBV weighted fMRI proved to be highly advantageous compared to BOLD in that activation was readily detected in single trials without the need for averaging. Furthermore, using CBV weighted fMRI typically improves detection sensitivity and contrast to noise ratio by at least a factor of 2 over BOLD fMRI (Mandeville et al., 2004) and confers more confidence that the activation maps are representative of the true extent of ictal activity. Another advantage of our protocol was the use of short duration (5 s) optogenetic stimulation to drive relatively short duration ADs (median = 10.1 s) which were therefore typically much shorter than ADs or spontaneous seizures in SE models, which last around a minute (Toyoda et al., 2013). There were two reasons why this was advantageous. First, shorter ADs are likely to induce much less of a kindling effect. Evidence for this pertains to the fact that we did not observe any motor seizures even after a single animal had experienced multiple ADs and this made it possible to study ADs repeatedly and reproducibly in the same animal, even when ADs originated from the VH which is known to be kindled relatively rapidly. Second, using relatively mild, short-duration ADs resulted in seizure activity that was limited to just a few regions beyond the initiation site making it possible to map the regions involved early in the course of ictal activity using fMRI, which otherwise lacks the time resolution necessary for investigating the rapid spread of seizure activity.
Finally, previous studies found evidence of heating artifacts on fMRI (Christie et al., 2013; Schmid et al., 2017) or evidence of light-evoked vessel dilation (Rungta et al., 2017). Notably, despite the highly intense (2420 mW/mm2 5 s period of continuous light at 589 nm, we found no evidence of such artifacts. Unlike the aforementioned studies, our experiments were carried out several weeks after the initial surgery using chronically implanted fibers. This means that blood from the surgical procedure - which absorbs light strongly – may have had time to be cleared. Despite this, we have previously shown it is possible to drive heating-related artifacts on fMRI using chronically implanted fibers and a 20 s period of blue light at 2560 mW/mm2 (Duffy et al., 2015). However, it is worth noting that it was with a much longer stimulation duration than we used in the current study and also that blue light will have a significantly higher degree of absorption in brain tissue compared to orange light, leading to enhanced local heating (Stujenske et al., 2015).
In conclusion, we showed that it was possible to curtail optogenetically induced ADs using local optogenetic inhibition and that failure to curtail ADs was explained well by ictal onset time. ADs that could be suppressed locally exhibited similar but less wide spread seizure networks compared to those that could not be inhibited. Our results provide important insights for future investigations into targeted epilepsy therapies and furthers our understanding of the spread of seizure activity from the dorsal and ventral hippocampus.
Materials and Methods
Surgical procedure for virus injection and implantation of optrodes
Animal husbandry and experimental protocols were in strict accordance with the National Institutes of Health (NIH) and Stanford University’s Institutional Animal Care and Use Committee (IACUC) guidelines. Animals were housed under environmentally controlled conditions, a 12 hour light-dark cycle with food and water provided ad libitum.
The surgical procedure was designed to drive expression of both eNpHR3.0 and ChR2 in the ventral hippocampus in overlapping neuronal populations by administering via co-injection of 2 viruses of the same serotype (Kim et al., 2013). For AD induction, CAMKII neurons were targeted for ChR2 expression. For the inhibitory opsin, eNpHR3.0, we tested both the human synapsin (hSyn) promoter and also the CAMKII promoter for their capacities to interrupt ADs. Adult male Sprague-Dawley rats (n=18 in total) were obtained from Charles River Laboratories and initially 13 rats were used with eNpHR3.0 expression driven by the hSyn promoter in n=6 rats and the CAMKII promoter in n=5 rats. Under our experimental conditions, hSyn-eNpHR3.0 was more effective at curtailing ADs than CAMKII-eNpHR3.0, therefore only the hSyn group data was analyzed, except for in Fig. 1, where both groups were included as only ChR2 stimulations (without optogenetic inhibition) were examined. Both groups also received viruses encoding for ChR2 under the CAMKII promoter. 1.5 μl AAV-5 eYFP-CAMKII (titer = 8.5×1012 vg/ml) and 1.5 μl AAV-5-eNpHR3.0-mCherry viruses were obtained from the University of North Carolina (UNC) vector core at the highest titers that were available at the time (titer = 6.7×1012 vg/ml for hSyn targeting and 4.7×1012 vg/ml for CAMKII targeting).
Rats were anesthetized using 5% isoflurane in pure oxygen and then maintained on 2–3% throughout the duration of the surgical procedure. Viruses encoding the eNpHR3.0 and ChR2 genes were mixed in a 1:1 ratio (v/v) and injected into the right hippocampus (VH coordinates = AP:−5.6 mm, LR: 5.7 mm, DV:6 mm from dura) using a 33 gauge needle attached to a Hamilton syringe. A syringe pump (Micro 4, World Precision Instruments, FL) was used to ensure a constant rate (150 nl/min) of administration. MRI compatible carbon fiber optrodes, constructed with 0.22 numerical aperture, 105 μm diameter step-index multimodal optical fiber (ThorLabs, Newton, NJ) as described previously (Duffy et al., 2015), were inserted so that the tip of the electrode and fiber resided just above the injection site. Before implantation, the optrodes were checked to ensure that the percentage of light transmission was greater than 70% and light transmission to the brain was estimated to be 70% of the input intensity. Two brass screws were inserted above the cerebellum to anchor the dental cement and to serve as ground and reference electrodes. Finally, the electrode wires were soldered to a DF13 connector (Hirose, Japan) and all components were secured to the skull using light-curable dental cement. Additional subjects (n=2 for the hSyn-eNpHR3.0 group and n=2 for the CAMKII-eNpHR3.0 group) in were used for multi-unit recording and therefore optrodes were not implanted. Sustained release buprenorphine (1 mg/kg, s.c.) was given pre-operatively to alleviate pain and discomfort due to the procedure. Local administration of lidocaine (4%) and bupivacaine (0.25%) was also administered pre- and post-operatively. To allow time for viral-induced protein expression, experiments were performed at least 6 weeks following the surgical procedure.
Following initial experiments that indicated that eNpHR3.0 expression under the hSyn promoter was more effective at curtailing ADs compared to experiments targeting CAMKII, an additional group of (n=3) rats where the hSyn-eNpHR3.0 and hSyn-ChR2 viruses were co-injected into the dorsal hippocampus (DH coordinates = AP:−3.6 mm, LR: 3.2 mm, DV:2.2 mm from dura) were included for comparison with the VH targeting experiments.
Electrophysiology recordings
LFP recordings outside of the MRI environment were performed using a differential amplifier (DP304, Warner Instruments) and a data acquisition system (USB 6259, National Instruments). Multiunit recordings were performed using the OpenEphys system and a sharpened tungsten electrode (0.5 kOhm impedance, 120 μm diameter, AM systems) attached to a 105 μm diameter optical fiber.
Estimation of ictal onset time
Ictal onset time was estimated during the ChR2 stimulation period by subtracting the averaged evoked potential to produce an estimate of the underlying stimulation independent LFP (Fig. S2). First, each 25 ms stimulation epoch was zero centered and an average evoked potential at each epoch was generated using the sliding-window mean evoked potential averaged over 8 consecutive epochs. This average evoked potential was subtracted from the initial mean centered data and this was used to detect underlying ictal activity using a simple thresholding approach. Events were defined every time the LFP after subtracting the evoked potential exceeded an empirical threshold (4.5× the standard deviation of the baseline signal, where the baseline was defined as the period between 500–2500 ms as no obvious ictal activity occurred during this period). Finally, the ictal onset time was defined as time at which 5 events occurred within a 500 ms period.
Anesthesia regimen for induction of seizure-like afterdischarges
Rats were sedated using a bolus (0.05 mg/kg), followed by a continuous intravenous (i.v.) infusion (0.1 mg/kg/h) of dexmedetomidine via a 24-gauge catheter inserted into one of the lateral tail veins. Using this anesthetic regimen, we have previously shown that it is possible to reproducibly induce ADs (Duffy et al., 2015). At the end of each session, atipamazole (0.5 mg/kg, s.c.) was given to partially reverse the effects of dexmedetomidine.
Induction of seizure-like afterdischarges
For selectively stimulating ChR2 expressing cells to induce ADs, we used a 473 nm (blue light) diode-pumped solid-state laser (Laserglow Technologies, Toronto, Canada). A train of 200 pulses (7.5 ms in duration) with a frequency of 40 Hz was generated using a Master-9 (AMPI, Israel) pulse stimulator. The duration of the pulse train was 5 s. To find appropriate parameter thresholds for AD induction, light intensity was gradually increased by stimulating every 2 minutes with an increased intensity in steps of 0.35 mW (40 mW/mm2 per pulse at the fiber tip) starting at 0.7 mW (80 mW/mm2). If no ADs were induced at the level of 1200 mW/mm2, then during MRI imaging sessions the duration of the pulse train was increased up to 7.5 in steps of 0.5 s. After the threshold was found, an 8 to 10-minute interstimulus interval between consecutive stimulations was used to abate any post-ictal refractory effects.
For selectively hyperpolarizing eNpHR3.0 expressing cells, a 589 nm (orange light) diode-pumped solid-state laser (Laserglow Technologies, Toronto, Canada) was used. The orange light was delivered continuously for 5 s to the same location as the blue light (through the same optical fiber). The light was switched on and off using a mechanical shutter (Uniblitz, Vincent Associates, NY) triggered with the Master-9 to enable stable light intensity and precise timing. The orange light was applied immediately after the blue light was switched off. This condition was compared to a control condition in which no orange light was applied. For each individual session, a random permutation of the set containing 5 orange light stimulations and 5 control conditions was used to generate equal numbers of each condition within sessions. If no ADs were elicited during a control stimulation, then the light intensity was increased by 40 mW/mm2 until ADs could be induced.
fMRI data acquisition
All data acquisition was performed using the 7T horizontal-bore system (Bruker BioSpec 70/30) at the Stanford Center for Innovation in In-Vivo Imaging (SCi3). An 86 mm diameter 2-channel volume coil was used for RF excitation with a 20 mm single-loop surface coil as the RF receiver. Similar to the recording sessions outside of the scanner, during MRI acquisitions, rats were sedated using a bolus (0.05 mg/kg, s.c.) of dexmedetomidine followed by a continuous infusion (0.1 mg/kg, i.v.) via a cannula inserted into a lateral tail vein. A single bolus of Feraheme (15 mg/kg, i.v.) was used for cerebral blood volume (CBV) weighted imaging for the enhanced contrast to noise ratio (Mandeville et al., 1998) and microvascular sensitivity (Zhao et al., 2006) that this technique offers in comparison to BOLD fMRI. fMRI acquisition was carried out approximately 15 min post contrast agent injection using a 4-shot segmented spiral readout with the following acquisition parameters: TR=0.75 ms, TE = 9 ms, flip angle = 30, field-of-view = 32 × 32 mm, matrix = 70 × 70, slice thickness = 0.6 mm, number of repetitions = 130, number of dummy scans = 4.
Simultaneous LFP recordings and processing
LFP recordings concurrent with the fMRI data acquisition were carried out using a 16 channel BrainAmp ExG MR amplifier (Brain Products, Germany) with a low pass filter of 1000 Hz and a sampling rate of 5000 Hz. Gradient artifacts were removed using Niazy et al.’s implementation of average artifact subtraction (AAS) (Allen et al., 2000; Niazy et al., 2005) modified for segmented MRI sequences and adapted so that template estimation excluded the highly periodic stimulation periods or episodes of high-amplitude spiking. Additionally, a low pass filter cutoff of 70 Hz was employed as the final stage of the procedure to diminish the effects of any remaining artifacts.
Inclusion criteria for optogenetic inhibition experiments
For experiments investigating the effects of optogenetic inhibition, it is important to only include trials where the stimulation is sufficient for AD induction. For this reason, a strict quality control criterion was used for optogenetic inhibition experiments. Where there was no evidence of AD following blue light stimulation only trials (trials without optogenetic inhibition) i.e. where the duration was less than 1 s, these trials were excluded along with all preceding trials up until a blue light stimulation only trial that results in an AD of greater than 1 s. This criterion ensured that only trials that were above the threshold for seizure induction were included.
fMRI data analysis and statistics
fMRI data was analyzed using SPM12 (http://www.fil.ion.ucl.ac.uk/spm). The data were first smoothed using a Gaussian kernel of 0.5 mm at full-width at half-maximum. This was followed by motion correction using a 6-parameter rigid registration. The images first underwent manual brain masking and then were aligned to a common space using a 12-parameter affine registration as implemented in the NiftyReg software package (Modat et al., 2014; Ourselin et al., 2001). This common template was segmented into regions of interest by registration to an atlas (Valdés-Hernández et al., 2011) followed by manual correction. A general linear model (GLM) was used to generate activation maps. This entailed searching for optimal parameters for a custom gamma distribution function. We used a single gamma distribution, as there is no evidence of an undershoot using CBVw fMRI (Mandeville, 2012) and estimated the shape and scale parameters using Levenberg-Marquardt non-linear least squares combined with grid search to ensure the absence of local minima (Fig. S7a–f) (Liu et al., 2017). The fMRI data used for estimating gamma parameters was collected from the same animals from the block-design trials. This custom gamma distribution was convolved with the stimulation period together with the afterdischarge period to model the activation across both periods as a single rectangle function. The high-pass cut-off value was set to 400 s, while serial correlations were modeled using an autoregressive AR(1) model and estimation was performed using the restricted maximum likelihood method.
Single subject, single trial t-statistic activation maps were thresholded at a level of p<0.001, uncorrected for multiple comparisons. For the fixed-effects group level analysis, a stricter threshold of p<0.0001 (uncorrected for multiple comparisons) was applied. For the ROI-wise analysis, AD vs. no-AD were compared using a 2-sample t-test across trials. For the comparison between the dorsal and ventral hippocampus, a voxel-wise inflection point map was generated as a measure of the average onset time by using a (pooled) fixed-effects model and maximum-likelihood estimation. There was no difference between the activation time maps with and without optogenetic inhibition (Fig. S5), therefore these data were combined for the results displayed in Fig. 4. For the ROI analysis in Fig. 4, a region was considered to be active if the volume of activation was greater than 10% of the maximum activation volume in each subject. A random-intercept model with subjects as random effects was fitted using lme4 in R (Bates et al., 2014). The inflection point in the VH was compared to the DH in each region using parametric bootstrapping (Bates et al., 2014) and a significance level of 0.05. For region-wise analyses, the parcellation used for segmentation is shown in Fig. S7g. Activation times were reported as the median ± standard deviation but displayed in Fig. 4 as the median ± 5th and 95th percentiles.
Histology
Rats were perfuse-fixed using phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). Tissue was left in 4% PFA overnight before being cryoprotected in 20% sucrose solution. Sections were cut at 50 μm and mounted on glass slides for imaging under a confocal microscope (Zeiss LSM 880).
Statistical analysis of afterdischarges
Unless otherwise stated, hypothesis testing between groups was carried out using paired t-tests at a significance level of 0.05. Afterdischarges were considered to be sustained if their duration exceeded 2.5 seconds. This threshold was chosen to allow time for optogenetic inhibition to take effect and because conditioning on ADs that exceeded the 5 s period of optogenetic inhibition, the AD duration was not significantly shorter for trials that included optogenetic inhibition, suggesting that inhibition had little effect if ADs were severe or progressed enough to exceed the period of optogenetic inhibition. The median AD duration without inhibition was 11 s, more than 4× greater than the duration threshold.
Afterdischarge probability (Fig. 1 and Fig. 3) was modeled using hierarchical Bayesian logistic regression with intercepts allowed to vary across subjects. Hierarchical models are useful where the observations are grouped e.g. by subject. Instead of pooling the data and fitting a single regression model, or fitting a separate regression model to each subject, using a hierarchical model allows information to be shared across subjects i.e. estimations for each subject are partially based on data from other subjects, while subjects with a lower number of samples are more influenced by the population average. In the hierarchical model, AD probability (p) was modeled as: p = logit−1(Xβ + β0subject), where the intercept for each subject is assumed to arise from a common distribution: . In Fig. 1, which displays data from ChR2 stimulation only, the input matrix (X) included ictal onset time as the only explanatory variable. Fig. 3 also included trials with optogenetic inhibition, therefore X was expanded to include activation of eNpHR3.0 as a binary variable. Weakly informative priors were used (Gelman et al., 2008) as these are typically preferred over noninformative priors which can lead to improper posterior distributions (Gelman, 2006). Normal distributions centered on zero and with a variance of 1 were used as priors for the mean parameters and half-Cauchy distributions with a scale factor equal to 5 were used for the variance parameters. PyMC3 (Salvatier et al., 2015) and the No-U-Turn sampler were used for sampling from the posterior, as it is known to be more efficient than traditional Markov Chain Monte Carlo methods (Hoffman and Gelman, 2011). Finally, the 95% highest posterior density was used to test the significance of the corresponding β parameter value for each input variable.
Supplementary Material
Acknowledgements
The authors would like to thank all the Lee Lab members for their help with the experiments and writing of the manuscript. This work was supported by NIH/NINDS R01NS087159, NIH/NIA R01NS091461, NIH/NINDS RF1AG047666, and NIH/NIMH RF1MH114227.
Footnotes
Declarations of Interests
J.H.L. is a founder of LVIS.
References:
- Allen PJ, Josephs O, and Turner R (2000). A method for removing imaging artifact from continuous EEG recorded during functional MRI. NeuroImage 12, 230–239. [DOI] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, and Walker S (2014). Fitting linear mixed-effects models using lme4. arXiv preprint arXiv:14065823. [Google Scholar]
- Bohannon AS, and Hablitz JJ (2018). Optogenetic dissection of roles of specific cortical interneuron subtypes in GABAergic network synchronization. The Journal of physiology 596, 901–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy M, Duffy BA, and Lee JH (2017). Optogenetic study of networks in epilepsy. Journal of neuroscience research 95, 2325–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie IN, Wells JA, Southern P, Marina N, Kasparov S, Gourine AV, and Lythgoe MF (2013). fMRI response to blue light delivery in the naive brain: implications for combined optogenetic fMRI studies. Neuroimage 66, 634–641. [DOI] [PubMed] [Google Scholar]
- Desai M, Kahn I, Knoblich U, Bernstein J, Atallah H, Yang A, Kopell N, Buckner RL, Graybiel AM, Moore CI, et al. (2011). Mapping brain networks in awake mice using combined optical neural control and fMRI. Journal of neurophysiology 105, 1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy BA, Choy M, Chuapoco MR, Madsen M, and Lee JH (2015). MRI compatible optrodes for simultaneous LFP and optogenetic fMRI investigation of seizure-like afterdischarges. Neuroimage 123, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Raimondo JV, Irkle A, Lamsa KP, and Akerman CJ (2014). Excitatory Effects of Parvalbumin-Expressing Interneurons Maintain Hippocampal Epileptiform Activity via Synchronous Afterdischarges. The Journal of Neuroscience 34, 15208–15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewell LA, Liang L, Armstrong C, Soltesz I, Leutgeb S, and Leutgeb JK (2015). Brain State Is a Major Factor in Preseizure Hippocampal Network Activity and Influences Success of Seizure Intervention. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 15635–15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, and Dong HW (2010). Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65, 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R, Salanova V, Witt T, Worth R, Henry T, Gross R, Oommen K, Osorio I, Nazzaro J, Labar D, et al. (2010). Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia 51, 899–908. [DOI] [PubMed] [Google Scholar]
- Gelman A (2006). Prior distributions for variance parameters in hierarchical models (comment on article by Browne and Draper). Bayesian analysis 1, 515–534. [Google Scholar]
- Gelman A, Jakulin A, Pittau MG, and Su Y-S (2008). A weakly informative default prior distribution for logistic and other regression models. The Annals of Applied Statistics 2, 1360–1383. [Google Scholar]
- Hoffman MD, and Gelman A (2011). The No-U-Turn Sampler: Adaptively Setting Path Lengths in Hamiltonian Monte Carlo.
- Jay TM, and Witter MP (1991). Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. The Journal of comparative neurology 313, 574–586. [DOI] [PubMed] [Google Scholar]
- Karnani MM, Jackson J, Ayzenshtat I, Hamzehei Sichani A, Manoocheri K, Kim S, and Yuste R (2016). Opening Holes in the Blanket of Inhibition: Localized Lateral Disinhibition by VIP Interneurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 3471–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshkhoo S, Vogt D, and Sohal VS (2017). Dynamic, Cell-Type-Specific Roles for GABAergic Interneurons in a Mouse Model of Optogenetically Inducible Seizures. Neuron 93, 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Ash RT, Ceballos-Diaz C, Levites Y, Golde TE, Smirnakis SM, and Jankowsky JL (2013). Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualising and manipulating neuronal circuits in vivo. The European journal of neuroscience 37, 1203–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, and Soltesz I (2015). In vivo evaluation of the dentate gate theory in epilepsy. The Journal of physiology 593, 2379–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Oijala M, and Soltesz I (2013). On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun 4, 1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Szabo GG, Armstrong C, Oijala M, and Soltesz I (2014). Cerebellar Directed Optogenetic Intervention Inhibits Spontaneous Hippocampal Seizures in a Mouse Model of Temporal Lobe Epilepsy. eNeuro 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Durand R, Gradinaru V, Zhang F, Goshen I, Kim DS, Fenno LE, Ramakrishnan C, and Deisseroth K (2010). Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature 465, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Duffy BA, Bernal-Casas D, Fang Z, and Lee JH (2017). Comparison of fMRI analysis methods for heterogeneous BOLD responses in block design studies. Neuroimage 147, 390–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhong C, Wang L, Wei P, He W, Huang K, Zhang Y, Zhan Y, Feng G, and Wang L (2016). Optogenetic dissection of ictal propagation in the hippocampal-entorhinal cortex structures. Nat Commun 7, 10962–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandeville JB (2012). IRON fMRI measurements of CBV and implications for BOLD signal. Neuroimage 62, 1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandeville JB, Jenkins BG, Chen YC, Choi JK, Kim YR, Belen D, Liu C, Kosofsky BE, and Marota JJ (2004). Exogenous contrast agent improves sensitivity of gradient-echo functional magnetic resonance imaging at 9.4 T. Magnetic resonance in medicine 52, 1272–1281. [DOI] [PubMed] [Google Scholar]
- Mandeville JB, Marota JJ, Kosofsky BE, Keltner JR, Weissleder R, Rosen BR, and Weisskoff RM (1998). Dynamic functional imaging of relative cerebral blood volume during rat forepaw stimulation. Magnetic resonance in medicine 39, 615–624. [DOI] [PubMed] [Google Scholar]
- Meibach RC, and Siegel A (1977). Efferent connections of the hippocampal formation in the rat. Brain research 124, 197–224. [DOI] [PubMed] [Google Scholar]
- Modat M, Cash DM, Daga P, Winston GP, Duncan JS, and Ourselin S (2014). Global image registration using a symmetric block-matching approach. Journal of medical imaging (Bellingham, Wash) 1, 024003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno A, Morris RGM, and Canals S (2015). Frequency-Dependent Gating of Hippocampal–Neocortical Interactions. Cerebral Cortex. [DOI] [PubMed] [Google Scholar]
- Motelow JE, Li W, Zhan Q, Mishra AM, Sachdev RN, Liu G, Gummadavelli A, Zayyad Z, Lee HS, Chu V, et al. (2015). Decreased subcortical cholinergic arousal in focal seizures. Neuron 85, 561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niazy RK, Beckmann CF, Iannetti GD, Brady JM, and Smith SM (2005). Removal of FMRI environment artifacts from EEG data using optimal basis sets. NeuroImage 28, 720–737. [DOI] [PubMed] [Google Scholar]
- Osawa S, Iwasaki M, Hosaka R, Matsuzaka Y, Tomita H, Ishizuka T, Sugano E, Okumura E, Yawo H, Nakasato N, et al. (2013). Optogenetically induced seizure and the longitudinal hippocampal network dynamics. PloS one 8, e60928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ourselin S, Roche A, Subsol G, Pennec X, and Ayache N (2001). Reconstructing a 3D structure from serial histological sections. Image and Vision Computing 19, 25–31. [Google Scholar]
- Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng K, Deisseroth K, and Huguenard JR (2013). Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nature neuroscience 16, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Pikkarainen M, Nurminen N, and Ylinen A (2000). Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Annals of the New York Academy of Sciences 911, 369–391. [DOI] [PubMed] [Google Scholar]
- Pouliot P, Tremblay J, Robert M, Vannasing P, Lepore F, Lassonde M, Sawan M, Nguyen DK, and Lesage F (2012). Nonlinear hemodynamic responses in human epilepsy: a multimodal analysis with fNIRS-EEG and fMRI-EEG. Journal of neuroscience methods 204, 326–340. [DOI] [PubMed] [Google Scholar]
- Rungta RL, Osmanski B-F, Boido D, Tanter M, and Charpak S (2017). Light controls cerebral blood flow in naive animals. Nat Commun 8, 14191–14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatier J, Wiecki T, and Fonnesbeck C (2015). Probabilistic Programming in Python using PyMC.
- Schmid F, Wachsmuth L, Albers F, Schwalm M, Stroh A, and Faber C (2017). True and apparent optogenetic BOLD fMRI signals. Magnetic resonance in medicine 77, 126–136. [DOI] [PubMed] [Google Scholar]
- Sessolo M, Marcon I, Bovetti S, Losi G, Cammarota M, Ratto GM, Fellin T, and Carmignoto G (2015). Parvalbumin-Positive Inhibitory Interneurons Oppose Propagation But Favor Generation of Focal Epileptiform Activity. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 9544–9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiri Z, Manseau F, Levesque M, Williams S, and Avoli M (2016). Activation of specific neuronal networks leads to different seizure onset types. Annals of neurology 79, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stujenske JM, Spellman T, and Gordon JA (2015). Modeling the Spatiotemporal Dynamics of Light and Heat Propagation for In Vivo Optogenetics. Cell reports 12, 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda I, Bower MR, Leyva F, and Buckmaster PS (2013). Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. The Journal of Neuroscience 33, 11100–11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung JK, Shiu FH, Ding K, and Gross RE (2018). Chemically activated luminopsins allow optogenetic inhibition of distributed nodes in an epileptic network for non-invasive and multi-site suppression of seizure activity. Neurobiology of disease 109, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés-Hernández PA, Sumiyoshi A, Nonaka H, Haga R, Aubert-Vásquez E, Ogawa T, Iturria-Medina Y, Riera JJ, and Kawashima R (2011). An in vivo MRI template set for morphometry, tissue segmentation, and fMRI localization in rats. Frontiers in neuroinformatics 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz AJ, Fang Z, Lee HJ, Fisher RS, Smith WC, Choy M, Liu J, Lin P, Rosenberg M, and Lee JH (2015). Optogenetic fMRI reveals distinct, frequency-dependent networks recruited by dorsal and intermediate hippocampus stimulations. Neuroimage 107, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, Hashemi KS, Walker MC, Schorge S, and Kullmann DM (2012). Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Science translational medicine 4, 161ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykes RC, Kullmann DM, Pavlov I, and Magloire V (2016). Optogenetic approaches to treat epilepsy. Journal of neuroscience methods 260, 215–220. [DOI] [PubMed] [Google Scholar]
- Yekhlef L, Breschi GL, Lagostena L, Russo G, and Taverna S (2015). Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. Journal of neurophysiology 113, 1616–1630. [DOI] [PubMed] [Google Scholar]
- Zhao F, Wang P, Hendrich K, Ugurbil K, and Kim SG (2006). Cortical layer-dependent BOLD and CBV responses measured by spin-echo and gradient-echo fMRI: insights into hemodynamic regulation. Neuroimage 30, 1149–1160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.