

Abstract
Cells utilize ubiquitin-mediated proteolysis to regulate the activity of numerous proteins involved in signal transduction, cell cycle control, and transcriptional regulation. For a number of transcription factors, there appears to be a direct correlation between transcriptional activity and protein instability, suggesting that cells use targeted destruction as one method to down-regulate or attenuate gene expression. In this report we demonstrate that retinoid X receptors (RXRs) which function as versatile mediators of nuclear hormone-dependent gene expression are marked for destruction upon binding agonist ligands. Interestingly, when RXR serves as a heterodimeric partner for retinoic acid (RAR) or thyroid hormone (TR) receptors, binding of agonists by RAR or TR leads to degradation of both the transcriptionally active RAR or TR subunits as well as the transcriptionally inactive RXR subunit. Furthermore, using a series of mutants in the ligand-dependent activation domain (activation function 2), we demonstrate that agonist-stimulated degradation of RXR does not require corepressor release, coactivator binding, or transcriptional activity. Taken together, the data suggest a model for targeted destruction of transcription factors based on structural or conformational signals as opposed to functional coupling with gene transcription.
Proper regulation of gene expression is an essential feature of eukaryotic development and cellular homeostasis. To this end organisms have evolved a number of mechanisms to tightly control the activity of the trans-acting factors that regulate gene expression at the transcriptional level. Members of the nuclear hormone receptor superfamily illustrate one well-studied example of tightly regulated transcriptional control (for a review, see reference 40). Nuclear receptors are sequence-specific DNA-binding proteins that regulate target genes in response to the direct binding of small lipophilic ligands. Like many transcription factors, nuclear receptors are modular with separable DNA-binding and ligand-binding domains (LBD). Ligand binding to receptors initiates a conformational change throughout the LBD that disrupts interactions with corepressors and promotes interactions with coactivators (for a review see reference 22). A conserved helix near the carboxy terminus (helix 12) occupies unique positions when structures of unliganded, agonist-occupied, and antagonist-occupied LBDs are compared (for reviews, see references 22 and 43). Importantly, mutagenesis experiments indicate that helix 12, also referred to as the activation function 2 (AF-2) helix, is necessary for ligand-dependent transactivation by nuclear receptors (21). Recent work indicates the AF-2 helix contributes an essential surface to the formation of an agonist-dependent hydrophobic pocket that serves as a binding site for coactivators (14, 18, 24, 41, 46, 64). The alternative positions occupied by the AF-2 helix in the unliganded or antagonist-occupied conformations preclude the formation of the coactivator pocket and often favor the binding of corepressors (26, 44, 49). Thus, by modulating protein-protein interactions, a conformational change induced by hormones or other small molecule ligands is translated into a transcriptional response.
In contrast to the wealth of knowledge regarding the induction of transcription by nuclear receptors and other transcription factors, far less is known about how cells turn off transcription factors once activated. Nonetheless, inappropriate transcription or too much transcription factor activity is often detrimental, and indeed misregulation of several transcription factors has been shown to contribute to oncogenesis (for reviews, see references 27, 31, and 62). Recent work indicates that the half-lives of many transcription factors, including nuclear receptors, are controlled by ubiquitin-dependent proteolysis. Interestingly, agonist binding appears to decrease receptor half-life (2, 5, 12, 23, 32, 37, 45, 70), suggesting that one mechanism to shut off or attenuate receptor-mediated transcription is by targeting transcriptionally active receptors for destruction. A correlation between protein stability and transcriptional activity has also been made for other transcription factors (30, 42, 52). Based on these observations, several investigators have suggested that interactions between transcription factors and coactivators or other components of the transcriptional machinery may serve as the signal that targets active transcription factors for ubiquitin-mediated proteolysis.
Retinoid X receptors (RXRs) play important roles in numerous nuclear receptor-dependent signaling pathways. Not only can RXR function as a homodimer, but this receptor also serves as an obligate heterodimeric partner for many other receptors, including those for retinoic acid (RARs), thyroid hormone (TRs), vitamin D, prostanoids (peroxisome proliferator-activated receptor [PPAR]), oxysterols, bile acids, xenobiotics, and several orphan receptors (39). In this study, we used receptor-specific synthetic ligands and a series of AF-2 domain mutations to examine the stability of RXR homo- and heterodimers. Activation of one subunit of an RXR-dependent heterodimer leads to degradation of the entire dimeric complex, indicating that the complex is recognized as a single entity by the degradation machinery. Strikingly, we show that although receptors must assume an active conformation to signal destruction, transcriptional activity or interaction with cofactors is not required.
MATERIALS AND METHODS
Plasmids and inhibitors.
The receptor and reporter vectors used in this study have been previously described (19, 54–57, 63). RXR point mutants were generated by PCR using oligonucleotides containing the desired mutation or by using a QuickChange site-directed mutagenesis kit (Stratagene). FLAG-tagged RAR403 was generated by introducing two copies of the FLAG epitope (DYKDDDDK) at the amino terminus of RAR403. The proteasome inhibitors N-acetyl-Leu-Leu-Nlc-CHO (ALLN) and MG132 were purchased from BIOMOL. The kinase inhibitors PD98059 and AG-490 were purchased from Calbiochem.
Cell culture and transfection.
CV1 cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. Prior to transfection, cells were seeded in 10-cm-diameter plates (4 × 105 cells/plate) for Western blotting experiments or 48-well plates (1.5 × 104 cells/well) for luciferase assays in DMEM supplemented with 10% charcoal-resin-split fetal bovine serum. After 12 to 16 h of growth at 37°C, cells were transfected with the DOTAP transfection reagent as instructed by the manufacturer (Roche Molecular Biochemicals). For Western blots, cells were transfected with 10 μg of RXR expression plasmids. When heterodimers with RAR and TR were examined, 5 μg of each expression plasmid was transfected. To determine the effect of proteasome activity on RXR transactivation, each well was transfected with 36 ng of the UASGx4-tk-luc reporter, 36 ng of pCMX-GAL4-hRXRα LBD (amino acids 222 to 462) or pCMX-GAL4 (amino acids 1 to 147), and as an internal control 60 ng of pCMX-β-galactosidase. For functional analysis of RXR homodimers, each well was transfected with 36 ng of the CRBPII-tk-luc reporter, 36 ng of pCMX-hRXRα, or the appropriate RXR mutant and as an internal control 60 ng of pCMX-β-galactosidase. For two-hybrid assays, each well was transfected with 36 ng of UASGx4-tk-luc reporter, 36 ng of the pCMXGAL4-SRC-1 (amino acids 381 to 891), pCMXGAL4-GRIP1 (amino acids 322 to 1121), pCMXGAL4-TRAP220 (amino acids 637 to 656), or pCMXGAL4-SMRT (amino acids 2004 to 2517) fusion (9, 57, 58), 36 ng of VP16-RXR LBD (wild type or mutant; amino acids 222 to 462), and as an internal control 60 ng of pCMX-β-galactosidase. After 5 h at 37°C, the medium was removed, the cells were washed once, and 200 μl of fresh medium was added with or without the ligands described in the figure legends. Cells were harvested after an additional 36 h of growth at 37°C. Luciferase activity of each sample was normalized by the level of β-galactosidase activity. Each transfection was carried out in duplicate and repeated at least three times.
Nuclear extract preparation and Western blotting.
Nuclear extracts were prepared from transfected CV1 cells as described by Schreiber et al. (53). For Western blots, 10 μl of each sample was resolved on sodium dodecyl sulfate (SDS)–10% gels, transferred to polyvinylidene difluoride membranes, and probed with the appropriate antibodies. Anti-human RXRα (hRXRα) (sc-774; 0.1 μg/ml; Santa Cruz Biotechnology), anti-TATA-binding protein (TBP (E4151; 285 ng/ml; Promega), anti-mouse RXRβ (MA3-812); 10 μg/ml; (Affinity Bioreagents) anti-hRARα (sc-551; 0.1 μg/ml; Santa Cruz Biotechnology), anti-hTRβ (MA1-215; 2.0 μg/ml; Affinity Bioreagents), anti-FLAG M5 (F4042; Sigma), and anti-progesterone receptor (PR) (11). For all experiments, an identical gel was stained with Coomassie blue to ensure that equal amounts of total protein were loaded in each lane.
Immunoprecipitation and pulse-chase analysis.
CV1 cells were cultured in DMEM supplemented with 10% fetal bovine serum. Prior to transfection, cells were seeded in 10-cm-diameter plates (4 × 105 cells/plate) and transfected with 10 μg of pCMX-hRXRα. After 36 h, cells were labeled with [35S]methionine (100 μCi/ml) for 4 h (Fig. 3A) or 45 min (Fig. 3B). After labeling, cells were either directly lysed or chased with a 200-fold excess of unlabeled methionine in the presence or absence of LGD1268. Upon completion of the experiment, medium was removed, and cells were washed twice with 5 ml of ice-cold phosphate-buffered saline, then scraped off the plate in 1.0 ml of phosphate-buffered saline, and transferred to an Eppendorf tube. Cells were pelleted for 1.0 min at 14,000 × g at 4°C and and then lysed in 50 μl of lysis buffer (50 mM Tris [pH 8.8], 5 mM EDTA, 2.0% SDS, 10 mM dithiothreitol, 100 μM sodium vanadate, 10 mM sodium fluoride, Complete protease inhibitors, EDTA free [Roche Molecular Biochemicals]). Following lysis, the extract was diluted with 950 μl of dilution buffer (20 mM Tris [pH 8.8], 150 mM NaCl, 2 mM EDTA, 100 μM sodium vanadate, 10 mM sodium fluoride, Complete protease inhibitors, EDTA free [Roche Molecular Biochemicals]) and passed through a 25-gauge needle to shear DNA. The extract was pelleted for 10 min at 14,000 × g at 4°C, and the supernatant was transferred to a new tube. To clear the supernatant, 15 μl of protein A/G-agarose (sc-2003; Santa Cruz Biotechnology) was added, and the samples were incubated with gentle rocking for 1 h at 4°C. Following this incubation, the beads were pelleted, the cleared supernatants were transferred to new tubes, 2 μg of anti-hRXRα (sc-774; Santa Cruz Biotechnology) prebound to 10 μl of protein A/G-agarose was added, and the mixture was incubated with gentle rocking for 3 h at 4°C. The beads were then pelleted and washed three times for 10 min at room temperature with 1.0 ml of high-salt buffer (20 mM Tris [pH 8.8], 500 mM NaCl, 2 mM EDTA, 0.2 mM dithiothreitol, 100 μM sodium vanadate, 10 mM sodium fluoride, Complete protease inhibitors, EDTA free [Roche Molecular Biochemicals]), followed by a quick rinse with 1.0 ml of 10 mM Tris (pH 8.8). Bound protein was eluted with 10 μl of SDS-gel sample buffer.
FIG. 3.

Agonists decrease the half-life of RXR. (A) CV1 cells were transfected with an expression plasmid for hRXRα and incubated in the absence (lane 2) or presence (lane 3) of 1.0 μM LGD1268. After 36 h, cells were labeled with [35S]methionine for 4 h in the continued absence or presence of LGD1268, and RXR was immunoprecipitated from whole-cell extracts as described in Materials and Methods. Lane 1, 14C-molecular weight markers; lane 2, [35S]methionine-labeled in vitro-translated (IVT) RXR; lanes 3 and 4, immunoprecipitated samples. (B) CV1 cells were transfected with an expression plasmid for hRXRα and incubated in the absence of ligands for 36 h. Cells were pulsed for 45 min with [35S]methionine and chased in absence (lanes 3 and 5) or presence (lanes 4 and 6) of LGD1268 for the times noted. RXR was immunoprecipitated from whole-cell extracts as described in Materials and Methods. Lane 1, [35S]methionine-labeled in vitro-translated RXR. (C) CV1 cells were transfected with an expression plasmid for hRXRα and incubated for 36 h in the absence of ligands to allow expression of RXR. LGD1268 (1.0 μM) was then added, and nuclear extracts were prepared at the times after ligand addition indicated and examined by Western blotting using anti-RXR
Immunoprecipitation-Western blot analysis.
CV1 cells were cultured in DMEM supplemented with 10% fetal bovine serum. Prior to transfection, cells were seeded in 10-cm plates (4 × 105 cells/plate) and transfected with 10 μg of pRSV-mRXRβ (38) or pRSV as a negative control. After transfection, cells were incubated with and without LGD1268 and MG132 as described in the legend to Fig. 4C. Upon completion of the experiment, ubiquitinated proteins were immunoprecipitated as described above, using 2 μg of antiubiquitin polyclonal antibody (sc-9133; Santa Cruz Biotechnology) prebound to 10 μl of protein A/G. Bound protein was eluted with 10 μl of SDS-gel sample buffer. Precipitated mouse RXRβ was detected by Western blotting as described above.
FIG. 4.

Ligand-dependent degradation of RXR requires proteasome activity and is independent of protein synthesis. CV1 cells were transfected with an expression plasmid for hRXRα and incubated for 36 h in the absence of ligands to allow expression of RXR. (A) After 36 h, cells were pretreated for 30 min in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of cycloheximide (10 μg/ml). Following the 30-min pretreatment, LGD1268 (1.0 μM) was then added (lanes 2 and 4), and the cells were incubated for an additional 2 h in the presence of cycloheximide as indicated. RXR protein levels were examined by Western blotting with anti-RXR and anti-TBP antibodies. The numbers under the RXR blot indicate the percentage of RXR relative to the untreated control (lane 1). Quantitation was done using a Storm 840 PhosphorImager (Molecular Dynamics). (B) After 36 h, cells were pretreated for 30 min in the absence (lanes 1 and 2) or presence of 100 μM of the proteasome inhibitor ALLN (lanes 3 and 4) or MG132 (lanes 5 and 6). Following the 30-min pretreatment, LGD1268 (1.0 μM) was then added (lanes 2, 4, and 6), and the cells were incubated for an additional 6 h in the presence of proteasome inhibitors as indicated. RXR levels were analyzed as described for panel A. (C) CV1 cells were transfected with either an empty expression vector (lane 1) or an expression vector for mouse RXRβ (lanes 2 to 5), and cells were incubated for 36 h in the absence of ligands to allow expression of RXR. After 36 h, cells were pretreated for 30 min in absence (lanes 2 and 3) or presence (lanes 1, 4, and 5) of 10 μM MG132. Following the 30-min pretreatment, LGD1268 (1.0 μM) was then added (lanes 3 and 5), and the cells were incubated for an additional 6 h in the presence of MG132 as indicated. Upon completion of the incubation, cells were lysed and whole-cell extracts were immunoprecipitated (IP) with a polyclonal antibody to ubiquitin (Ub) as described in Materials and Methods. Proteins bound to the beads were eluted, and RXR protein levels were examined with a monoclonal antibody against mouse RXRβ.
RESULTS
Ubiquitin-mediated degradation of RXR.
To examine the influence of ligands on the stability of RXR, CV1 cells were transfected with an hRXRα expression plasmid and treated with the RXR-specific agonist LGD1069 (also known as bexarotene or Targretin) (3) or LGD1268 (4). After incubation with ligands, RXR protein levels were examined by Western blotting. As shown in Fig. 1A and B, treatment with either agonist results in a >90% decrease in RXR levels (lanes 1 to 3). Dose-response studies indicate that the 50% effective concentrations for receptor degradation and transactivation are similar (approximately 5 nM [Fig. 1C and D]) (4). In addition, similar results are obtained when endogenous CV1 RXR protein is examined in the absence of transfection (Fig. 2). Since the Western blots in Fig. 1 and 2 utilized high-salt extracts from isolated nuclei, we were concerned that the decrease in RXR proteins levels may represent relocalization of RXR to the cytoplasm or to a subnuclear compartment that prevents quantitative extraction. To address this issue, cells were treated with LGD1268 for 36 h as in Fig. 1 and labeled with [35S]methionine, and RXR was immunoprecipitated from whole-cell extracts made by lysis with 2% SDS. The results of the immunoprecipitation in Fig. 3A clearly show a dramatic decrease in RXR levels in whole-cell extracts from LGD1268-treated cells (compare lanes 3 and 4). Pulse-chase immunoprecipitation analysis and time course experiments using Western blotting indicate that the half-life for ligand-dependent degradation is approximately 2 h, compared to 4 h in the absence of ligand (Fig. 3B and C) (47). Consistent with effects on RXR stability, ligand-dependent destruction of RXR does not require new protein synthesis, and the effects of ligand on relative receptor stability are similar in the presence and absence of protein synthesis (Fig. 4A, compare lanes 1 and 2 with lanes 3 and 4). Furthermore, treatment of cells with the proteasome inhibitor ALLN or MG132 blocks degradation (Fig. 4B), and immunoprecipitation-Western blot experiments demonstrate a ligand-stimulated ubiquitination of RXR (Fig. 4C, compare lanes 4 and 5). Taken together, the data shown in Fig. 1 to 4 indicate that binding of agonists to RXR induces degradation of the activated receptor via the ubiquitin/proteasome pathway.
FIG. 1.

RXR agonists decrease the amount of transfected RXR in CV1 cells. (A and B) CV1 cells were transfected with an expression plasmid for hRXRα and incubated for 36 h in the absence (lane 1) or presence of 1.0 μM RXR-specific agonist LGD1069 (lane 2) or LGD1268 (lane 3). After incubation with ligands, nuclear extracts were prepared and examined by Western blotting using anti-RXR and anti-TBP antibodies. (A) Western blots. (B) Coomassie blue-stained gel demonstrating that equal amounts of total protein are present in all lanes. (C) CV1 cells were transfected with an expression plasmid for hRXRα and incubated for 36 h in the absence (lane 1) or presence (lanes 2 to 5) of the RXR-specific agonist LGD1268 at the concentrations noted. RXR and TBP levels were analyzed as described for panel A. (D) CV1 cells were transfected with hRXRα along with the CRBPII-tk-luc reporter and a β-galactosidase expression plasmid. Following transfection, cells were incubated in the presence of different concentrations of LGD1268. After 36 h, luciferase activity was determined and normalized by β-galactosidase activity. EC50, 50% effective concentration.
FIG. 2.
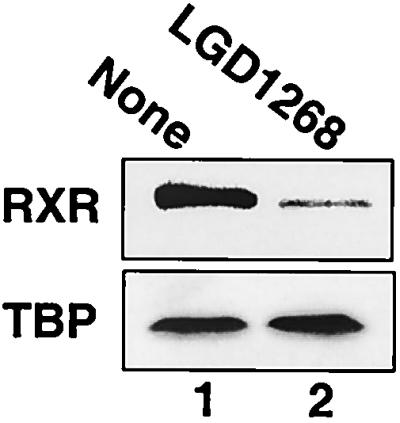
Ligand-dependent degradation of endogenous RXR. CV1 cells were cultured for 24 h in the absence (lane 1) or presence (lane 2) of 1.0 μM LGD1268. After incubation with ligands, nuclear extracts were prepared and examined by Western blotting using anti-RXR and anti-TBP antibodies.
Since treatment of cells with proteasome inhibitors stabilizes RXR in the presence of agonists, we sought to determine whether increasing RXR protein levels influences transcriptional activity. To this end, the effect of the proteasome inhibitor MG132 on the activity of a GAL4-RXR LBD fusion was examined (Fig. 5). As shown in Fig. 5A, the GAL4-RXR LBD fusion is also degraded in an agonist-dependent fashion, indicating that the LBD itself is sufficient to signal destruction. Interestingly, in the presence of MG132, the response to the RXR agonist LGD1268 is reduced by 65% (Fig. 5B), while the constitutive activity of GAL4(1–147) assayed on the same reporter is not significantly altered (Fig. 5C). Similar effects of proteasome inhibitors on the activity of the estrogen and thyroid hormone receptors have recently been observed (12, 37). Thus, proteasome activity appears to be a general requirement for maximum transactivation by nuclear receptors (see Discussion).
FIG. 5.

Proteasome activity is required for RXR transactivation. (A) CV1 cells were transfected with an expression plasmid for GAL4-hRXRα LBD (lanes 1 and 2) or full-length hRXRα (lanes 3 and 4) and incubated for 36 h in the absence (lanes 1 and 3) or presence of 1.0 μM LGD1268 (lanes 2 and 4). After incubation with ligands, RXR protein levels were examined by Western blotting with anti-RXR and anti-TBP antibodies. (B and C) CV1 cells were transfected with a reporter with four GAL4 binding sites (UASGx4-tk-luc), a β-galactosidase expression plasmid, and a construct expressing a GAL4-hRXRα LBD fusion (amino acids 222 to 462) (B) or GAL4(1-147) (C). After transfection, cells were cultured for 24 h in the absence of ligands to allow expression of the GAL4 constructs. After 24 h, cells were incubated for 16 h with vehicle (bar 1), 10 μM MG132 (bar 2), 1.0 μM LGD1268 (bar 3), or 10 μM MG132 plus 1.0 μM LGD1268 (bar 4); luciferase activity was determined and normalized by β-galactosidase activity. The activity relative to that observed with the reporter alone is expressed. The numbers listed above bars 3 and 4 are the fold inductions by LGD1268 in the absence (bar 3) or presence (bar 4) of MG132.
Ligand-dependent degradation of RXR requires agonist activity.
In contrast to agonist-dependent degradation, administration of the RXR homodimer antagonist LG100754 (8, 34), which binds RXR specifically, has little or no effect on RXR levels (Fig. 6A). LG100754, however, does competitively inhibit agonist-dependent degradation (Fig. 6B). The failure of an antagonist to induce degradation supports the hypothesis that transcriptional activity serves as a signal for ubiquitin-mediated proteolysis (12, 23, 37, 42, 52). To confirm this conclusion, the relative stability of an RXR mutant that has the essential helix 12 deleted (RXR443) was examined. Although this mutant cannot activate transcription or interact with coactivators, it binds LGD1268 with little change in affinity (54, 55). Consistent with the results observed with the antagonist LG100754, removing the AF-2 domain (helix 12) from RXR inhibits destruction (Fig. 6C). Interestingly, in the presence of LGD1268 the amount of RXR443 appears to slightly increase (Fig. 6C, compare lanes 3 and 4). This ligand-dependent stabilization most likely results from the overall compaction of the LBD that occurs upon ligand binding (6, 7, 51, 68). Similar stabilization by agonists is readily observed in vitro when partial protease protection experiments are used to probe ligand-dependent conformational changes (29, 36, 57).
FIG. 6.

Agonist activity and helix 12 are required for ligand-stimulated degradation. (A) CV1 cells were transfected with an expression plasmid for hRXRα, and cells were incubated for 36 h in the absence (lane 1) or presence of 1.0 μM LGD1268 (RXR-selective agonist; lane 2) or 100 nM LG100754 (RXR-specific antagonist; lane 3). After incubation with ligands, RXR protein levels were examined by Western blotting with anti-RXR and anti-TBP antibodies. (B) CV1 cells were transfected with an expression plasmid for hRXRα and incubated for 36 h in the absence (lane 1) or presence of 10 nM LGD1268 (RXR-selective agonist; lane 2), 1.0 μM LG100754 (RXR-specific antagonist; lane 3), 10 nM LGD1268 plus 1.0 μM LG100754 (lane 4). RXR levels were analyzed as described for panel A. (C) CV1 cells were transfected with an expression plasmid for hRXRα (lanes 1 and 2) or the helix 12 (AF-2) deletion mutant RXR443 (lanes 3 and 4). Cells were incubated for 36 h in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of 1.0 μM LGD1268. RXR levels were analyzed as described for panel A.
Inhibition of MKK destabilizes RXR.
Phosphorylation has been shown to serve as a positive signal for the degradation of several proteins, including IκB (10), cyclin D1 (16), cyclin E (67), β-catenin (48), and recently PR (35). In the case of PR, mitogen-activated protein (MAP) kinase phosphorylation of a single site in the amino-terminal domain (serine 294) is required for agonist-dependent degradation (35). Since RXR is also a substrate for MAP kinase (1), we examined the effect of kinase inhibitors on RXR stability (Fig. 7). Inhibition of MAP kinase signaling using the MAP kinase kinase (MKK) inhibitor PD98059 reduces the level of RXR, mimicking the RXR agonist LGD1268 (Fig. 7A, lanes 1 to 3). The combination of PD98059 and LGD1268 appears to act synergistically (lane 4). The decrease in RXR levels observed upon MKK inhibition contrasts the observations made for PR which is stabilized by PD98059 even in the presence of the agonist R5020 (Fig. 5B) (35). Furthermore, the effect of PD98059 is specific, as AG-490, a tyrosine kinase inhibitor, has no effect on RXR levels (Fig. 7C).
FIG. 7.

Inhibition of MKK activity decreases RXR levels. (A) CV1 cells were transfected with an expression plasmid for hRXRα and incubated for 36 h in the absence of ligands to allow expression of RXR. After 36 h, cells were pretreated for 30 min in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of 100 μM MKK inhibitor PD98059. Following the 30-min pretreatment, LGD1268 (1.0 μM) was then added (lanes 2 and 4), and the cells were incubated for an additional 6 h in the presence or absence of PD98059 as indicated. Upon completion of the experiment, RXR protein levels were examined by Western blotting. (B) Same as panel A except an expression plasmid for human PR-B and the synthetic PR agonist R5020 (20 nM) were used. (C) Same as panel A except the tyrosine kinase inhibitor AG-490 was used in place of PD98059. (D) Same as panel A except the RXR AF-2 deletion mutant RXR443 was used and treatment with LG1268 alone was omitted. RXR443 is stable in the presence of LGD1268 (Fig. 6C).
The results of Fig. 7A indicate that RXR can be destabilized by either inhibition of MKK activity or agonist binding. Interestingly, MKK inhibition also reduces the levels of the RXR AF-2 deletion mutant, RXR443 (Fig. 7D, compare lanes 1 and 2). However, in contrast to the wild-type receptor, the effect of the combination of PD98059 and LGD1268 on RXR443 is no different from the effect observed with PD98059 alone (compare lanes 2 and 3), again demonstrating that helix 12 is required for the destabilizing effect of agonist binding. Taken together, the results suggest two independent mechanisms to regulate RXR stability, one dependent on and the other independent of the integrity of helix 12.
Both subunits of RXR-dependent heterodimers are destroyed.
To examine the stability of RXR-dependent heterodimers constructs expressing RXR and hRARα were transfected into CV1 cells and treated with RAR-specific (TTNPB) (66) and RXR-specific (LGD1268) agonists (Fig. 8A). Interesting, when the RAR subunit is activated by TTNPB, both RAR and RXR levels are decreased (Fig. 8A, compare lanes 1 and 3), indicating that both subunits of the dimeric complex are degraded. The quantitatively weaker effect of the RXR agonist LGD1268 on RAR levels (lane 2, approximately 50% decrease) most likely arises from the weaker binding of RXR ligands to RXR-RAR heterodimers (19, 33, 65). To further define the mechanisms controlling heterodimer stability, dimers formed between a stabilized RXR (RXR443) and RAR were examined. Similar to the wild-type RXR-RAR heterodimer, treatment with the RAR agonist TTNPB reduces the levels of both subunits of the RXR443-RAR heterodimer (Fig. 8B, compare lanes 1 and 3), including the RXR helix 12 deletion that is stable as a RXR homodimer (Fig. 6C). In contrast, when a transcriptionally inactive dominant-negative RAR helix 12 deletion mutant (RAR403) (13) is paired with wild-type RXR, both subunits of the RXR-RAR403 heterodimer are now resistant to TTNPB treatment (Fig. 8C). A FLAG-tagged RAR403 construct was used for this experiment because the 403 deletion removes the epitope recognized by the RAR antibody. Placement of the FLAG epitope at the amino terminus of wild-type RAR has no effect on ligand-dependent degradation (I. Schulman, unpublished data) (Fig. 5). Thus, only a single transcriptionally active subunit is necessary to target the dimeric complex for degradation. When activation is blocked by a dominant-negative subunit, the complex is stabilized. To support the above conclusions, we again turned to the RXR-specific ligand LG100754. Although LG00754 antagonizes RXR homodimers (Fig. 6A) (8, 34), this ligand activates RXR-RAR heterodimers via a unique mechanism dependent on RAR's helix 12 that we have termed the phantom ligand effect (34, 56). Thus, in contrast to LG100754's lack of effect on the stability of RXR homodimers, we would predict this ligand to mark RXR-RAR heterodimers for destruction. As shown in Fig. 8 (compare lanes 1 and 4), the predicted results are observed, indicating that the relative stability of RXR in the presence of LG100754 is dependent on dimerization status.
FIG. 8.

Ligand binding destabilizes both subunits of RXR-RAR heterodimers. CV1 cells were transfected with equal amounts of the expression plasmids encoding hRARα and hRXRα and incubated for 36 h in the absence (lane 1) or presence of 1.0 μM LGD1268 (RXR specific; lane 2) 100 nM TTNPB (RAR specific, lane 3), or 100 nM LG100754 (RXR specific: lane 4). After incubation with ligands, RAR and RXR protein levels were examined by Western blotting with anti-RAR or anti-RXR antibodies. The RAR antibody does not recognize the RAR403 AF-2 deletion mutant; therefore, a construct with two copies of the FLAG epitope was used and detected with anti-FLAG antibodies. For each blot, all four samples were run on the same gel. The positions of lanes 3 and 4, however, were reversed in the figure for clarity of presentation.
To extend the observation that activation of a single subunit targets RXR-dependent heterodimers for destruction, we determined the influence of receptor-specific ligands on RXR-TR heterodimers (Fig. 9A). Western blot analysis of CV1 cells transfected with RXR and human TRβ indicate that, as observed with RXR-RAR, both the TR and RXR subunits are degraded in response to T3 (TR specific; lane 3) or LGD1268 (RXR specific; lane 2). Similar results have been observed for RXR-PPARγ heterodimers (23). To eliminate concerns that the heterodimer stability experiments utilize overexpressed receptors, we used a nontransfected cell culture system to examine RXR-TR stability in response to T3. The GH1 cell line, a pituitary-derived cell line that has been used to study induction of the growth hormone gene by RXR-TR heterodimers (15, 20, 61), was cultured in the absence or presence of T3. After 24 h, nuclear extracts were prepared and RXR-TR levels were determined by gel shift analysis (Fig. 9B). Consistent with the transfection results, treatment with T3 results in a >90% decrease in RXR-TR DNA binding activity as determined by PhosphorImager analysis (compare lanes 2 and 3).
FIG. 9.

Both subunits of RXR-TR heterodimers are destabilized by ligand binding to TR. (A) CV1 cells were transfected with equal amounts of expression plasmids for hRXRα and hTRβ and incubated for 36 h in the absence (lane 1) or presence of 1.0 μM LGD1268 (RXR specific; lane 2), 100 nM T3 (TR specific; lane 3), or 1.0 μM LGD1268 plus 100 nM T3 (lane 4). After incubation with ligands, protein levels were examined by Western blotting with anti-TR antibodies, anti-RXR antibodies, and anti-TBP antibodies. (B) GH1 cells were cultured for 24 h in the absence (lane 2) or presence (lane 3) of 10 nM T3. Nuclear extracts were prepared and equal amounts of total protein were used to examine binding to a 32P-labeled probe derived from the palindromic TR element in the growth hormone gene. Lane 1 contains baculovirus-expressed RXR and TR.
Transcriptional activity and coactivator interactions are not required for ligand-dependent degradation.
The data on the stability of RXR homo- and heterodimers described above present a paradox. On one hand, the observations are consistent with the idea that transcriptional activity and coactivator interactions are necessary for ligand-dependent degradation of RXR homo- and heterodimers. Nevertheless, at least when dimerized with a transcriptionally active partner, an RXR mutant (RXR443) incapable of directly activating transcription can be targeted for destruction. To further examine the correlation between transcriptional activity and protein stability, five point mutations in the AF-2 domain of RXR were examined. All five mutants bind ligand with wild-type affinities (54); nevertheless, their ability to activate transcription is compromised (Fig. 10A). Two of the mutants (E453K and E456K [Fig. 10B, lanes 9 to 12]) are stable in the presence of the RXR agonist LGD1268, a result consistent with the hypothesis that stability correlates with transcriptional activity. Surprisingly, however, the other three transcriptionally inactive mutants are still degraded in a ligand-dependent manner (Fig. 10B, lanes 3 to 8). We further compared wild-type RXR with the M454A/L455A mutant to determine if the kinetics of ligand-stimulated degradation were significantly altered. However, as shown in Fig. 10C, the time courses of ligand-stimulated degradation are similar for both transcriptionally active and inactive receptors.
FIG. 10.

Transcriptional activity is not required for ligand-stimulated degradation. (A) CV1 cells were transfected with equal amounts of plasmids for hRXRα or the RXR AF-2 mutants noted indicated along with the CRBPII-tk-luc reporter and a β-galactosidase expression plasmid. Following transfection, cells were incubated in the absence (open bars) or presence (black bars) of 1.0 μM LGD1268. After 36 h, luciferase activity was determined and normalized by β-galactosidase activity. Relative activity compared to the reported alone is expressed. The fold induction (+LGD1268/−LGD1268) for each receptor is reported over the black bars. (B) CV1 cells were transfected with equal amounts of plasmids for hRXRα or the RXR AF-2 mutants indicated and incubated for 36 h in the absence (lanes 1, 3, 5, 7, 9, and 11) or presence (lanes 2, 4, 6, 8, 10, and 12) of 1.0 μM LGD1268. After incubation with ligands, RXR protein levels were examined by Western blotting with anti-RXR antibodies. (C) CV1 cells were transfected with expression plasmids for wild-type hRXRα or the M454A/L455A mutant and incubated for 36 h in the absence of ligands to allow expression of RXR. LGD1268 (1.0 μM) was then added, and nuclear extracts were prepared at the times after ligand addition indicated. RXR protein levels were examined by Western blotting with anti-RXR antibodies.
To characterize the functional activity of the AF-2 mutants in greater detail, mammalian two-hybrid analysis was used to examine interactions with coactivators and corepressors. As expected, little or no interaction is observed between the AF-2 mutants and the coactivators steroid receptor coactivator 1 (Fig. 11A), glucocorticoid receptor-interacting protein 1 (Fig. 10B), TR-associated protein 220 (Fig. 10C), TBP (54), or SUG1 (Schulman, unpublished). Thus, coactivator interaction is not required for ligand-stimulated degradation. Although RXR by itself interacts poorly, if at all, with corepressors (Fig. 11D), deletion of the RXR helix 12 allows a robust receptor-corepressor interaction to be observed (55, 69). Since deletion of helix 12 also stabilizes RXR in the presence of agonists (Fig. 6C), we hypothesized that the stability of the E453K and E456K mutants could result from increased interactions with corepressors. As shown in Fig. 11D, however, this hypothesis is incorrect. While two of the AF-2 mutants (L451A and M454A/L455A) do exhibit increased interaction with the silencing mediator of retinoid and thyroid receptors (Fig. 11D), the stable E453K and E456K mutants behave like the wild-type receptor. The results of Fig. 10 and 11 clearly separate ligand-dependent degradation from ligand-dependent regulation of transcription, indicating that transcriptional activity and corepressor-coactivator interactions per se cannot be necessary to target RXR for destruction.
FIG. 11.

Coactivator and corepressor interactions are not required for ligand-stimulated degradation. In a mammalian two-hybrid assay, CV1 cells were transfected with constructs expressing a GAL4-coactivator or- corepressor fusion, VP16-RXR LBD fusions (wild type or mutant), a reporter with four GAL4 binding sites (UASGx4-tk-luc), and a β-galactosidase expression plasmid. After transfection, cells were cultured for 36 h in the absence (white bars) or presence of 100 nM LGD1268 (black bars). Luciferase activity was then determined and normalized by β-galactosidase activity. The activity relative to that observed with GAL4-fusion plus the empty VP16 vector is reported. (A) human steroid receptor coactivator 1 (SRC-1; amino acids 381 to 891); (B) mouse glucocorticoid receptor-interacting protein 1 (GRIP1; amino acids 322 to 1121); (C) human TR-associated protein 220 (TRAP220; amino acids 637 to 656). (D) human silencing mediator of retinoid and thyroid receptors (SMRT; amino acids 2004 to 2517).
DISCUSSION
In this report we demonstrate that binding of agonists to RXR not only leads to positive transactivation but also signals a rapid destruction of active receptors. By the apparent coupling of transactivation of target genes with destruction of the triggering receptor, cells have built a fail-safe mechanism to ensure that transcription will persist only in the continued presence of the signal inducer (hormone or small molecule). By serving as an obligate DNA-binding partner for numerous other receptors, RXR can influence a wide array of hormonal signaling pathways (39). Examination of both RXR-RAR and RXR-TR heterodimers indicates that in response to activation of one subunit, the entire heterodimeric complex is targeted for destruction. Kopf et al. arrived at a similar conclusion for RXR-RAR heterodimers (32). The concurrent destruction of both dimeric subunits whether transcriptionally active or not contrasts with the results observed for oligomeric complexes of stable and unstable variants of the yeast α2 repressor. In the case of α2, only the unstable subunits of the complex are degraded (25). The targeted destruction of both subunits of RXR-dependent heterodimers has implications for the global regulation of nuclear receptor signaling within cells. Reduction of RXR levels by activation of a particular heterodimeric partner may decrease the availability of RXR for other dimeric partners. Thus, the hormone-dependent destruction of a common essential subunit provides an additional mechanism for cross talk among seemingly independent hormonal signaling pathways. Additionally, the observation that MAP kinase signaling can influence RXR stability suggests the possibility that cell surface receptors can directly and differentially (compare the effects of PD98059 on PR and RXR [Fig. 7A and B]) influence nuclear receptor-dependent gene expression.
Although treatment with proteasome inhibitors increases the quantity of RXR in cells, transcriptional activity is reduced. Similar observations have been made for the estrogen receptor (37) and TR (12), suggesting that proteasome activity is generally required for nuclear receptor activity. Two nonexclusive mechanisms to explain the proteasome requirement come to mind. First, degradation of a labile repressor may be required for transactivation. Second, ubiquitination of receptors and/or cofactors may directly inhibit their transcriptional activity independent of degradation as has recently been reported for the yeast transcription factor MET4 (28). Importantly, our studies have shown only that overexpressed RXR is directly ubiquitinated. Thus, we cannot rule out the possibility that endogenous RXR is degraded via a ubiquitin-independent pathway. Identification of the sites of ubiquitination on nuclear receptors and the creation of mutants that are unable to be tagged for destruction will help to distinguish between these and other mechanisms.
A number of recent studies have demonstrated a correlation between transcriptional activity and protein stability for nuclear receptors as well as other transcription factors (5, 12, 23, 32, 37, 42, 45, 52, 70). Indeed, point mutations in the activation domains of c-Myc and VP16 that reduce transactivation increase relative protein stability (42, 52). Similarly, liganding RXR with an antagonist or blocking the agonist-mediated conformational change by removing helix 12 results in an increase in receptor stability. Nevertheless, the observation that the same stable RXR helix 12 deletion mutant is degraded when dimerized with an active partner prompted a more detailed examination of the relationship between transcriptional activity and protein stability. Interestingly, three transcriptionally inactive AF-2 mutants, two in helix 12 and one in helix 3, are still degraded in a ligand-dependent manner. Our results thus indicate that agonist binding and helix 12 are necessary and sufficient to signal receptor degradation. Transcriptional activity, corepressor interaction, or coactivator binding is not required.
One mechanism consistent with the ability to separate transcription and receptor stability would be that receptors in the holo or active conformation are recognized for ubiquitin-mediated proteolysis, independent of their transcriptional activity. Thus, we would suggest the relatively unstable transcriptionally inactive point mutants are competent to assume a conformation that resembles the active form and signals degradation. These mutants nonetheless fail to activate transcription because amino acids critical for interactions with coactivators have been altered. At first glance, this transcription-independent mechanism is at odds with the results for other constitutively active transcription factors such as c-Myc and VP16 for which transcriptional activity and stability correlate (42, 52). However, the activation domains of many constitutively active transcription factors appear to be relatively unstructured in solution, and it is only upon interaction with coactivators that an ordered structure is achieved (50, 60). In a similar fashion, helix 12 of nuclear receptors appears to be relatively flexible, capable of assuming multiple conformations in the absence of ligand. Therefore, we suggest that for nuclear receptors binding of ligand, and for constitutively active transcription factors interaction with coactivators, functionally serves to drive transcription factors into a conformation that is favorably recognized by the degradation machinery.
Interestingly, mutation of either glutamic acid 453 or 456 to lysine produces receptors that are relatively stable in the presence of agonists. An explanation for the stability of these two mutants consistent with the conformational change hypothesis described above would be to propose that these amino acid changes do not allow the proper repositioning of helix 12 upon agonist binding. A recent crystal structure of the RXR LBD bound to the agonist 9-cis retinoic acid, however, indicates that both E453 and E456 appear to be surface exposed (17). Therefore, the possibility that E453 and E456 contribute to a surface that mediates direct interaction with the degradation machinery cannot be ruled out. A similar mutation of a glutamic acid residue in helix 12 of PPARγ also results in a stable receptor (23).
These studies examining RXR mutants and RXR-dependent heterodimers raise important questions about the recognition of agonist-bound receptors by the ubiquitin-mediated proteolytic machinery. First, what factor(s) recognizes liganded receptors and targets them for destruction? Although proper positioning of helix 12 is apparently required, our data strongly suggest that recognition is not via the typical LxxLL-receptor interaction that has been defined for nuclear receptor coactivators (14, 24, 41, 46, 59). Second, it would be of interest to know how the transcriptionally silent subunits of heterodimers are targeted for degradation. The observation that even a stabilized RXR mutant is degraded in the context of a transcriptionally active RXR-RAR heterodimer clearly supports the conclusion that both subunits of the dimer are recognized as a single functional entity. In conclusion, the ability to genetically separate transcriptional activity from receptor stability suggests an additional level of cellular control beyond ligand-mediated transcriptional control and provides the basis for novel pharmacological approaches to receptor regulation.
ACKNOWLEDGMENTS
We thank M. Manchester and D. Chakravarti for comments on the manuscript and the medicinal chemistry department at Ligand Pharmaceuticals for providing LGD1069, LGD1268, and LG100754.
REFERENCES
- 1.Adam-Stitah S, Penna L, Chambon P, Rochette-Egly C. Hyperphosphorylation of the retinoid X receptor alpha by activated c-Jun NH2-terminal kinases. J Biol Chem. 1999;274:18932–18941. doi: 10.1074/jbc.274.27.18932. [DOI] [PubMed] [Google Scholar]
- 2.Alarid E T, Bakopoulos N, Solodin N. Proteasome-mediated proteolysis of estrogen receptor: a novel component in autologous down-regulation. Mol Endocrinol. 1999;13:1522–1534. doi: 10.1210/mend.13.9.0337. [DOI] [PubMed] [Google Scholar]
- 3.Boehm M F, Zhang L, Badea B A, White S A, Mais D E, Berger E, Suto C M, Goldman M E, Heyman R A. Synthesis and structure-activity relationships of novel retinoid X receptor-selective retinoids. J Med Chem. 1994;37:2930–2941. doi: 10.1021/jm00044a014. [DOI] [PubMed] [Google Scholar]
- 4.Boehm M F, Zhang L, Zhi L, McClurg M R, Berger E, Wagoner M, Mais D E, Suto C M, Davies P J A, Heyman R A, Nadzan A M. Design and synthesis of potent retinoid X receptor selective ligands that induce apoptosis in leukemia cells. J Med Chem. 1995;38:3146–3155. doi: 10.1021/jm00016a018. [DOI] [PubMed] [Google Scholar]
- 5.Boudjelal M, Wang Z, Voorhees J J, Fisher G J. Ubiquitin/proteasome pathway regulates levels of retinoic acid receptor gamma and retinoid X receptor alpha in human keratinocytes. Cancer Res. 2000;60:2247–2252. [PubMed] [Google Scholar]
- 6.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 7.Bourguet W, Vivat V, Wurtz J M, Chambon P, Gronemeyer H, Moras D. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol Cell. 2000;5:289–298. doi: 10.1016/s1097-2765(00)80424-4. [DOI] [PubMed] [Google Scholar]
- 8.Canan Koch S S, Dardashti L J, Hebert J J, Croston G E, Flatten K S, Heyman R A, Nadzan A M. Identification of the first retinoid X receptor homodimer antagonist. J Med Chem. 1996;39:3229–3234. doi: 10.1021/jm960311d. [DOI] [PubMed] [Google Scholar]
- 9.Chen J D, Evans R M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Hagler J, Palombella V J, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 11.Clemm D L, Sherman L, Boonyaratanakornkit V, Schrader W T, Weigel N L, Edwards D P. Differential hormone-dependent phosphorylation of progesterone receptor A and B forms revealed by a phosphoserine site-specific monoclonal antibody. Mol Endocrinol. 2000;14:52–65. doi: 10.1210/mend.14.1.0413. [DOI] [PubMed] [Google Scholar]
- 12.Dace A, Zhao L, Park K S, Furuno T, Takamura N, Nakanishi M, West B L, Hanover J A, Cheng S. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proc Natl Acad Sci USA. 2000;97:8985–8990. doi: 10.1073/pnas.160257997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Damm K, Heyman R A, Umesono K, Evans R A. Functional inhibition of retinoic acid response by dominant negative retinoic receptor mutants. Proc Natl Acad Sci USA. 1993;90:2989–2993. doi: 10.1073/pnas.90.7.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darimont B D, Wagner R L, Apriletti J W, Stallcup M R, Kushner P J, Baxter J D, Fletterick R J, Yamamoto K R. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis K D, Berrodin T J, Stelmach J E, Winkler J D, Lazar M A. Endogenous retinoid X receptors can function as hormone receptors in pituitary cells. Mol Cell Biol. 1994;14:7105–7110. doi: 10.1128/mcb.14.11.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diehl J A, Zindy F, Sherr C J. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 17.Egea P F, Mitschler A, Rochel N, Ruff M, Chambon P, Moras D. Crystal structure of the human RXRalpha ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 2000;19:2592–2601. doi: 10.1093/emboj/19.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng W, Ribeiro R C, Wagner R L, Nguyen H, Apriletti J W, Fletterick R J, Baxter J D, Kushner P J, West B L. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280:1747–1749. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- 19.Forman B M, Umesono K, Chen J, Evans R M. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 20.Glass C K, Franco R, Weinberger C, Albert V R, Evans R M, Rosenfeld M G. A c-erb-A binding site in rat growth hormone gene mediates trans-activation by thyroid hormone. Nature. 1987;329:738–741. doi: 10.1038/329738a0. [DOI] [PubMed] [Google Scholar]
- 21.Glass C K, Rose D W, Rosenfeld M G. Nuclear receptor coactivators. Curr Opin Cell Biol. 1997;9:222–232. doi: 10.1016/s0955-0674(97)80066-x. [DOI] [PubMed] [Google Scholar]
- 22.Glass C K, Rosenfeld M G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 23.Hauser S, Adelmant G, Sarraf P, Wright H M, Mueller E, Spiegelman B M. Degradation of the peroxisome proliferator-activated receptor gamma is linked to ligand-dependent activation. J Biol Chem. 2000;275:18527–18533. doi: 10.1074/jbc.M001297200. [DOI] [PubMed] [Google Scholar]
- 24.Heery D M, Kalkhoven E, Hoare S, Parker M G. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 25.Hochstrasser M, Varshavsky A. In vivo degradation of a transcriptional regulator: the yeast alpha 2 repressor. Cell. 1990;61:697–708. doi: 10.1016/0092-8674(90)90481-s. [DOI] [PubMed] [Google Scholar]
- 26.Hu X, Lazar M A. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- 27.Hunter T. Oncoprotein networks. Cell. 1997;88:333–346. doi: 10.1016/s0092-8674(00)81872-3. [DOI] [PubMed] [Google Scholar]
- 28.Kaiser P, Flick K, Wittenberg C, Reed S I. Regulation of transcription by ubiquitination without proteolysis: Cdc34/SCF(Met30)-mediated inactivation of the transcription factor Met4. Cell. 2000;102:303–314. doi: 10.1016/s0092-8674(00)00036-2. [DOI] [PubMed] [Google Scholar]
- 29.Keidel S, LeMotte P, Apfel C. Different agonist- and antagonist-induced conformational changes in retinoic acid receptors analyzed by protease mapping. Mol Cell Biol. 1994;14:287–298. doi: 10.1128/mcb.14.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim T K, Maniatis T. Regulation of interferon-gamma-activated STAT1 by the ubiquitin-proteasome pathway. Science. 1996;273:1717–1719. doi: 10.1126/science.273.5282.1717. [DOI] [PubMed] [Google Scholar]
- 31.Kinzler K W, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 32.Kopf E, Plassat J L, Vivat V, de The H, Chambon P, Rochette-Egly C. Dimerization with RXRs and phosphorylation modulate the retinoic acid- induced degradation of RARα and RARγ and through the ubiquitin-proteasome pathway. J. 2000. Biol. Chem. [DOI] [PubMed] [Google Scholar]
- 33.Kurokawa R, DiRenzo J, Boehm M, Sugarman J, Gloss B, Rosenfeld M G, Heyman R A, Glass C K. Regulation of retinoid signalling by receptor polarity and allosteric control of ligand binding. Nature. 1994;371:528–531. doi: 10.1038/371528a0. [DOI] [PubMed] [Google Scholar]
- 34.Lala D S, Mukherjee R, Schulman I G, Canan-Koch S S, Dardashti L J, Nadzan A M, Croston G E, Evans R M, Heyman R A. Activation of specific RXR heterodimers by an antagonist of RXR homodimers. Nature. 1996;383:450–453. doi: 10.1038/383450a0. [DOI] [PubMed] [Google Scholar]
- 35.Lange C A, Shen T, Horwitz K B. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leid M. Ligand-induced alteration of the protease sensitivity of retinoid X receptorα. J Biol Chem. 1994;269:14175–14181. [PubMed] [Google Scholar]
- 37.Lonard D M, Nawaz Z, Smith C L, O'Malley B W. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939–948. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- 38.Mangelsdorf D J, Borgmeyer U, Heyman R A, Zhou J Y, Ong E S, Oro A E, Kakizuka A, Evans R M. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992;6:329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- 39.Mangelsdorf D J, Evans R M. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 40.Mangelsdorf D J, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Kastner P, Mark M, Chambon P, Evans R M. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McInerney E M, Rose D W, Flynn S E, Westin S, Mullen T M, Krones A, Inostroza J, Torchia J, Nolte R T, Assa-Munt N, Milburn M V, Glass C K, Rosenfeld M G. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12:3357–3368. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molinari E, Gilman M, Natesan S. Proteasome-mediated degradation of transcriptional activators correlates with activation domain potency in vivo. EMBO J. 1999;18:6439–6447. doi: 10.1093/emboj/18.22.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr Opin Cell Biol. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 44.Nagy L, Kao H Y, Love J D, Li C, Banayo E, Gooch J T, Krishna V, Chatterjee K, Evans R M, Schwabe J W. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 1999;13:3209–3216. doi: 10.1101/gad.13.24.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nawaz Z, Lonard D M, Dennis A P, Smith C L, O'Malley B W. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci USA. 1999;96:1858–1862. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nolte R T, Wisely G B, Westin S, Cobb J E, Lambert M H, Kurokawa R, Rosenfeld M G, Willson T M, Glass C K, Milburn M V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 47.Nomura Y, Nagaya T, Hayashi Y, Kambe F, Seo H. 9-cis-Retinoic acid decreases the level of its cognate receptor, retinoid X receptor, through acceleration of the turnover. Biochem Biophys Res Commun. 1999;260:729–733. doi: 10.1006/bbrc.1999.0969. [DOI] [PubMed] [Google Scholar]
- 48.Orford K, Crockett C, Jensen J P, Weissman A M, Byers S W. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem. 1997;272:24735–24738. doi: 10.1074/jbc.272.40.24735. [DOI] [PubMed] [Google Scholar]
- 49.Perissi V, Staszewski L M, McInerney E M, Kurokawa R, Krones A, Rose D W, Lambert M H, Milburn M V, Glass C K, Rosenfeld M G. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999;13:3198–3208. doi: 10.1101/gad.13.24.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radhakrishnan I, Perez-Alvarado G C, Parker D, Dyson H J, Montminy M R, Wright P E. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 51.Renaud J-P, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Crystal structure of the RAR-γ ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 52.Salghetti S E, Muratani M, Wijnen H, Futcher B, Tansey W P. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc Natl Acad Sci USA. 2000;97:3118–3123. doi: 10.1073/pnas.050007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schreiber E, Matthias P, Muller M M, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schulman I G, Chakravarti D, Juguilon H, Romo A, Evans R M. Interactions between the retinoid X receptor and a conserved region of the TATA-binding protein mediate hormone-dependent transactivation. Proc Natl Acad Sci USA. 1995;92:8288–8292. doi: 10.1073/pnas.92.18.8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulman I G, Juguilon H, Evans R M. Activation and repression by nuclear hormone receptors: hormone modulates an equilibrium between active and repressive states. Mol Cell Biol. 1996;16:3807–3818. doi: 10.1128/mcb.16.7.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulman I G, Li C, Schwabe J W R, Evans R M. The phantom ligand effect: allosteric control of transcription by the retinoid X receptor. Genes Dev. 1997;11:299–308. doi: 10.1101/gad.11.3.299. [DOI] [PubMed] [Google Scholar]
- 57.Schulman I G, Shao G, Heyman R A. Transactivation by retinoid X receptor-peroxisome proliferator-activated receptor gamma (PPARγ) heterodimers: intermolecular synergy requires only the PPARγ hormone-dependent activation function. Mol Cell Biol. 1998;18:3483–3494. doi: 10.1128/mcb.18.6.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shao G, Heyman R A, Schulman I G. Three amino acids specify coactivator choice by retinoid X receptors. Mol Endocrinol. 2000;14:1198–1209. doi: 10.1210/mend.14.8.0495. [DOI] [PubMed] [Google Scholar]
- 59.Shiau A K, Barstad D, Loria P M, Cheng L, Kushner P J, Agard D A, Greene G L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 60.Uesugi M, Nyanguile O, Lu H, Levine A J, Verdine G L. Induced alpha helix in the VP16 activation domain upon binding to a human TAF. Science. 1997;277:1310–1313. doi: 10.1126/science.277.5330.1310. [DOI] [PubMed] [Google Scholar]
- 61.Umesono K, Giguere V, Glass C K, Rosenfeld M G, Evans R M. Retinoic acid and thyroid hormone induce gene expression through a common responsive element. Nature. 1988;336:262–265. doi: 10.1038/336262a0. [DOI] [PubMed] [Google Scholar]
- 62.Weinberg R A. How cancer arises. Sci Am. 1996;275:62–70. doi: 10.1038/scientificamerican0996-62. [DOI] [PubMed] [Google Scholar]
- 63.Wen D X, Xu Y F, Mais D E, Goldman M E, McDonnell D P. The A and B isoforms of the human progesterone receptor operate through distinct signaling pathways within target cells. Mol Cell Biol. 1994;14:8356–8364. doi: 10.1128/mcb.14.12.8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Westin S, Kurokawa R, Nolte R T, Wisely G B, McInerney E M, Rose D W, Milburn M V, Rosenfeld M G, Glass C K. Interactions controlling the assembly of nuclear-receptor heterodimers and co-activators. Nature. 1998;395:199–202. doi: 10.1038/26040. [DOI] [PubMed] [Google Scholar]
- 65.Wiebel F F, Gustafsson J-A. Heterodimeric interaction between retinoid X receptor α and orphan nuclear receptor OR1 reveals dimerization-induced activation as a novel mechanism of nuclear receptor activation. Mol Cell Biol. 1997;17:3977–3986. doi: 10.1128/mcb.17.7.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willhite C C, Dawson M I, Reichert U. Receptor-selective retinoid agonists and teratogenic activity. Drug Metab Rev. 1996;28:105–119. doi: 10.3109/03602539608993994. [DOI] [PubMed] [Google Scholar]
- 67.Won K A, Reed S I. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996;15:4182–4193. [PMC free article] [PubMed] [Google Scholar]
- 68.Wurtz J-M, Bourguet W, Renaud J-P, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 69.Zhang J, Hu X, Lazar M A. A novel role for helix 12 of retinoid X receptor in regulating repression. Mol Cell Biol. 1999;19:6448–6457. doi: 10.1128/mcb.19.9.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu J, Gianni M, Kopf E, Honore N, Chelbi-Alix M, Koken M, Quignon F, Rochette-Egly C, de The H. Retinoic acid induces proteasome-dependent degradation of retinoic acid receptor alpha (RARα) and oncogenic RARα fusion proteins. Proc Natl Acad Sci USA. 1999;96:14807–14812. doi: 10.1073/pnas.96.26.14807. [DOI] [PMC free article] [PubMed] [Google Scholar]