

Abstract
Mitapivat (AG-348) is a novel, first-in-class oral small molecule allosteric activator of the pyruvate kinase enzyme. Mitapivat has been shown to significantly upregulate both wild-type and numerous mutant forms of erythrocyte pyruvate kinase (PKR), increasing adenosine triphosphate (ATP) production and reducing levels of 2,3-diphosphoglycerate. Given this mechanism, mitapivat has been evaluated in clinical trials in a wide range of hereditary hemolytic anemias, including pyruvate kinase deficiency (PKD), sickle cell disease, and the thalassemias. The clinical development of mitapivat in adults with PKD is nearly complete, with the completion of two successful phase III clinical trials demonstrating its safety and efficacy. Given these findings, mitapivat has the potential to be the first approved therapeutic for PKD. Mitapivat has additionally been evaluated in a phase II trial of patients with alpha- and beta-thalassemia and a phase I trial of patients with sickle cell disease, with findings suggesting safety and efficacy in these more common hereditary anemias. Following these successful early-phase trials, two phase III trials of mitapivat in thalassemia and a phase II/III trial of mitapivat in sickle cell disease are beginning worldwide. Promising preclinical studies have additionally been done evaluating mitapivat in hereditary spherocytosis, suggesting potential efficacy in erythrocyte membranopathies as well. With convenient oral dosing and a safety profile comparable with placebo in adults with PKD, mitapivat is a promising new therapeutic for several hereditary hemolytic anemias, including those without any currently US Food and Drug Administration (FDA) or European Medicines Agency (EMA)–approved drug therapies. This review discusses the preclinical studies, pharmacology, and clinical trials of mitapivat.
Keywords: hemolytic anemia, hereditary spherocytosis, mitapivat, pyruvate kinase activator, pyruvate kinase deficiency, sickle cell disease, thalassemia
Introduction
As the final enzymatic step of the Embden–Meyerhof glycolytic pathway, the pyruvate kinase enzyme catalyzes the conversion of phosphenolpyruvate to pyruvate, resulting in the generation of adenosine triphosphate (ATP). It is one of just two ATP-generating enzymes in this pathway (and the net ATP yield of glycolysis prior to pyruvate kinase is zero as two early steps require ATP). There are four pyruvate kinase isoforms in mammals (red cell, liver, muscle-1, and muscle-2) encoded by two genes (PKLR and PKM). While most human cells are capable of aerobic metabolism of glucose and therefore able to generate considerable additional ATP from the citric acid cycle and oxidative phosphorylation, erythrocytes lack the metabolic machinery required for aerobic metabolism. Therefore, erythrocytes are largely reliant on anaerobic glycolysis for ATP production. As ATP is critical for erythrocyte cellular maintenance and survival, its deficiency leads to premature and pathophysiologic red cell destruction in the form of hemolytic anemia and ineffective erythropoiesis. This is exemplified by the clinical manifestations of an entire family of glycolytic enzyme defects, which result in a wide spectrum of chronic, lifelong hemolytic anemias. The most common of these, and the most common congenital nonspherocytic hemolytic anemia worldwide, is pyruvate kinase deficiency (PKD). 1 Other erythrocyte disorders, such as sickle cell disease and the thalassemias, may result in a state of increased stress and energy utilization such that the normal but limited erythrocyte ATP production adequate in normal physiologic circumstances is no longer adequate, causing premature cell death. 2 , 3 Therefore, therapeutics capable of augmenting erythrocyte ATP production may be useful in a broad range of hemolytic anemias with diverse pathophysiologies (Figure 1).
Figure 1.

Rationale for use of mitapivat in three hereditary hemolytic anemias for which human clinical trials demonstrating efficacy and/or safety have been performed.
Mitapivat (AG-348) is a first-in-class, oral small molecule allosteric activator of the pyruvate kinase enzyme.4 Erythrocyte pyruvate kinase (PKR) is a tetramer, physiologically activated in allosteric fashion by fructose bisphosphate (FBP). Mitapivat binds to a different allosteric site from FBP on the PKR tetramer, allowing for the activation of both wild-type and mutant forms of the enzyme (in the latter case, allowing for activation even in many mutant PKR enzymes not induced by FBP). 4 Given this mechanism, it holds promise for use in both pyruvate kinase deficient states (PKD in particular) and other hemolytic anemias without defects in PK but greater erythrocyte energy demands. Mitapivat has been granted orphan drug designation by the US Food and Drug Administration (FDA) for PKD, thalassemia, and sickle cell disease and by the European Medicines Agency (EMA) for PKD. Several clinical trials evaluating the use of mitapivat to treat PKD, thalassemia, and sickle cell disease have been completed, are ongoing, and are planned. This review will briefly discuss the preclinical data and the pharmacology for mitapivat, before examining in depth the completed, ongoing, and officially announced clinical trials evaluating mitapivat for a wide range of hereditary hemolytic anemias.
Preclinical studies and pharmacology of mitapivat
Preclinical studies
Interest in pyruvate kinase activators was initially focused on potential utility for oncologic applications. 5 In a 2012 report, Kung and colleagues described experiments with an activator of PKM2 intended to manipulate metabolic vulnerabilities of cancer cells that could be exploited with certain cancer therapies. 6 Mitapivat (originally AG-348, Agios Pharmaceuticals, Cambridge, MA, USA) was subsequently recognized as a potent activator of PKR. Mitapivat is a sulfonamide drug taken orally as mitapivat sulfate. The chemical structure of mitapivat is illustrated in Figure 2. Early biochemical studies performed in recombinant wild-type PKR and a variety of mutant PKR proteins demonstrated augmentation of enzyme activity by approximately two- to sixfold. 7 In mice with wild-type PKR, administration of mitapivat resulted in increased PKR activity, increased ATP, and decreased 2,3-diphosphoglycerate (2,3-DPG). 7 In vitro studies examining blood samples from humans with PK deficiency demonstrated increased PKR activity of up to 3.4-fold and increased ATP levels of up to 2.4-fold following exposure to mitapivat. 4
Figure 2.
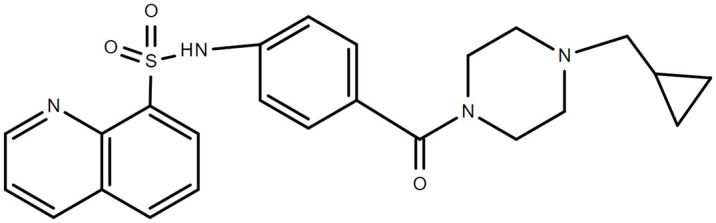
Chemical structure of mitapivat.
Pharmacokinetic studies of mitapivat performed in rats, dogs, and monkeys demonstrated rapid oral absorption, good oral bioavailability, and a high volume of distribution at steady state. 8
Preclinical studies of mitapivat in thalassemia and sickle cell disease have also been performed. In an ex vivo treatment study of erythrocytes from patients with beta-thalassemia, mitapivat was found to increase PKR activity and ATP levels. 9 In a beta-thalassemia mouse model (Hbbth3/+ mice), mitapivat ameliorated ineffective erythropoiesis, anemia, and iron overload. 2 In sickle cell disease, an ex vivo treatment study of mitapivat was performed to evaluate its effect on PKR properties, metabolism, and sickling behavior. 3 At baseline, reduced PKR activity and thermostability were observed in patients with sickle cell disease. PKR activity increased substantially (mean increase of 129%) following treatment with mitapivat. Increases of a similar magnitude were seen in mean ATP levels, and PKR thermostability also improved. 2,3-DPG levels declined 17%, p50 decreased 5%, and a significant 9% decrease in the point of sickling (the specific pO2 at which erythrocytes start to sickle) was also seen after treatment with mitapivat. 3
Mitapivat may also reduce hemolysis in patients with erythrocyte cytoskeletal defects. In a mouse model of hereditary spherocytosis, treatment with mitapivat over 6 months resulted in improvement of anemia with reduced reticulocyte count, reductions in markers of hemolysis such as bilirubin and lactate dehydrogenase, a decrease in the spleen weight to mouse weight ratio, reduced hepatic and splenic iron overload, and a reduction in the proportion of phosphatidylserine positive erythrocytes. 10 If confirmed in humans, these findings suggest a potential therapeutic potential for mitapivat in erythrocyte membranopathies in addition to what has already been demonstrated in enzymopathies, hemoglobinopathies, and thalassemias.
Pharmacokinetic and pharmacodynamic studies in humans
Two phase I randomized, placebo-controlled, double-blind studies in healthy volunteers aged 18–60 years were performed to assess the pharmacokinetics, pharmacodynamics, and safety of mitapivat. 11 In a single ascending dose study, 12 sequential cohorts of eight subjects each were randomized 2:6 to receive a single dose of either oral placebo or mitapivat (30, 120, 360, 700, 1400, or 2500 mg). In a multiple ascending dose study, six sequential cohorts of eight subjects each were randomized 2:6 to receive placebo or mitapivat administered every 12 h or every 24 h for 14 days. Mitapivat was safe in healthy volunteers, with no deaths or serious treatment-emergent adverse events (TEAEs) in either study, and only one grade 3+ TEAE (abnormal liver function tests after receiving 21 doses of 700 mg mitapivat every 12 h in one subject). TEAEs were more commonly reported in patients randomized to higher doses of mitapivat (⩾700 mg) and were most commonly low-grade headache, nausea, or vomiting. Mitapivat had good oral bioavailability and was absorbed well in the fasted and fed states. Cmax and area under the curve (AUC) increased with increasing dose, though not proportionally at higher doses. Steady state was reached after approximately 1 week in patients receiving 60 mg mitapivat every 12 h. 11
With regard to pharmacodynamics, a single dose of mitapivat resulted in minimal increases in ATP blood levels, but did decrease 2,3-DPG levels within 3 h, which took approximately 120 h to return to baseline. 11 In the multiple ascending dose study, the maximum ATP increase from baseline on day 14 was 60%, and ATP increases for doses above 60 mg every 12 h were not dose-proportional (suggesting a plateau of the stimulatory effect beyond this dose). The maximum decrease from baseline in 2,3-DPG on day 14 was 47%. 11
Based on these studies, the terminal half-life of mitapivat was estimated at 3–6 h. 11 It is primary eliminated via hepatic metabolism, metabolized by multiple cytochrome P450 (CYP) enzymes, including CYP3A4 (predominantly) as well as CYP1A2, CYP2C8, and CYP2C9. 11 Mitapivat has been shown to induce CYP3A4 and CYP2B6. Importantly, it is also a mild-to-moderate inhibitor of the aromatase enzyme, an off-target effect that has potential implications for its use in the long-term treatment of patients with hereditary hemolytic anemias; this will be discussed in greater detail in subsequent sections.
Clinical trials of mitapivat in PKD
PKD background
PKD is a rare autosomal recessive congenital anemia, with a prevalence approximated at between 1 in 20,000 and 1 in 300,000 persons (and possibly higher in malaria-endemic regions).1,12,13 It is a disease of considerable genetic diversity, as over 350 mutations resulting in PKD, primarily missense mutations, have been identified in the PKLR gene.14,15 Diagnosis is accomplished via enzymatic activity measurements and/or molecular testing.16,17 Patients with PKD have a broad spectrum and burden of disease, ranging from asymptomatic incidentally discovered mild anemia to severe anemia and lifelong transfusion-dependence from birth.18,19 In addition to the symptoms and quality of life impacts of chronic anemia, including reduced energy, limited exercise tolerance, cognitive effects, and fatigue, 20 patients also may suffer from chronic complications of lifelong hemolysis and ineffective erythropoiesis, including iron overload, extramedullary erythropoiesis, gallstones, osteopenia and osteoporosis, endocrinopathies, delayed puberty, and leg ulcers, among other complications.21,22 There are no FDA- or EMA-approved drug therapies for PKD. Splenectomy can improve the hemolytic anemia and modestly improve hemoglobin in approximately half of patients. 23 Hematopoietic stem cell transplantation is a high-risk option in patients with severe transfusion-dependent disease, functionally trading PKD and its complications for transplant-related morbidity (primarily graft-versus-host disease) and a risk of mortality. 24 Most patients are managed with supportive care alone, receiving folic acid supplementation and red cell transfusion (given primarily to improve symptoms, not based on a specific hemoglobin threshold) in addition to management of PKD complications (i.e. iron chelators, bisphosphonates, etc.). 23
Completed, ongoing, and planned clinical trials of mitapivat in PKD are summarized inTables 1 and 2, and described in detail in the following sections.
Table 1.
Completed clinical trials evaluating mitapivat for the treatment of hereditary hemolytic anemias.
| Study | Patient number (n) | Design, location | Study population | Major results |
|---|---|---|---|---|
| Yang et al. 11 (NCT04000165) | (n = 48 (SAD) (n = 48 (MAD) |
Phase I SAD and MAD, The United States | Healthy subjects | Mitapivat safe, with AEs more frequent at doses ⩾700 mg Pharmacokinetics favorable with low variability Dose-dependent changes in blood glycolytic intermediates consistent with glycolysis activation (increased ATP, reduced 2,3-DPG) |
| Grace et al. 25 (DRIVE-PK, NCT02476916) | Mitapivat (n = 52) | Phase II, North America and Europe | Adults with PKD who were not regularly transfused | Mitapivat safe and well-tolerated, with mild headache, insomnia, and nausea as most common AEs reported PK/PD parameters similar to healthy subjects 50% of patients had Hgb increase ⩾1.0 g/dl from baseline; improvement not seen in patients with two non-missense mutations or two R479H mutations Markers of hemolysis and erythropoiesis improved |
| Al-Samkari et al. 26 (ACTIVATE, NCT03548220) | Mitapivat (n = 40) Placebo (n = 40) |
Phase III randomized, Worldwide | Adults with PKD who were not regularly transfused with at least one non-R479H missense mutation | Met primary efficacy endpoint: mitapivat superior to placebo in achieving Hgb improvement ⩾1.5 g/dl (40% versus 0%) Met all secondary efficacy endpoints: improvement in average hemoglobin, lactate dehydrogenase, bilirubin, haptoglobin, reticulocyte percentage, and PKD-specific PRO measures (PKDD and PKDIA), all significantly greater in mitapivat arm than placebo arm Excellent safety profile; no patients on mitapivat discontinued treatment for any reason, including AEs; most common AEs in mitapivat arm were nausea and headache, and both were more common in placebo-treated patients PKDD and PKDIA underwent successful internal validation in this study |
| Glenthoj et al. 27 (ACTIVATE-T, NCT03559699) | Mitapivat (n = 27) | Phase III nonrandomized, Worldwide | Adults with PKD who were regularly transfused with at least one non-R479H missense mutation | Met primary efficacy endpoint: mitapivat reduced transfusion burden ⩾33% in 37% of enrolled patients Annualized number of RBC transfusions declined 39% 22% of patients rendered transfusion-free No AEs leading to treatment discontinuation |
| Kuo et al. 28 (NCT03692052) | Mitapivat (n = 20) | Phase II, The United States, Canada, and Europe | Adults with alpha- or beta-thalassemia who were not regularly transfused | Met primary efficacy endpoint: 16 patients (11/15 with beta-thalassemia and 5/5 with alpha-thalassemia) achieved Hgb increase ⩾1.0 g/dl Hemolytic and erythropoietic markers improved Responses were sustained with continued treatment Mitapivat well-tolerated with safety profile similar to prior studies |
| Xu et al. 29 (NCT04610866) | Mitapivat (n = 17) | Phase I MAD, The United States | Adults with sickle cell disease (HbSS) | Mitapivat safe and well-tolerated Mean hemoglobin change of +1.2 g/dl with mitapivat 50 mg twice daily Hemolytic markers improved Decreased mean 2,3-DPG and p50 and increased ATP in dose-dependent fashion |
AEs, adverse events; ATP, adenosine triphosphate; 2,3-DPG, 2.3-diphosphoglycerate; MAD, multiple ascending dose; PKD, pyruvate kinase deficiency; PK/PD, pharmacokinetic/pharmacodynamic; PKDD, pyruvate kinase deficiency diary; PKDIA, pyruvate kinase deficiency impact assessment; PRO, patient-reported outcome; SAD, single ascending dose.
Table 2.
Currently ongoing and planned clinical trials evaluating mitapivat for the treatment of hereditary hemolytic anemias.
| Study | Design, location | Study population |
|---|---|---|
| AG-348-011 (NCT03853798) | Phase III open-label extension for patients participating in ACTIVATE and ACTIVATE-T, Worldwide | Adults with PKD with at least one non-R479H missense mutation |
| ENERGIZE 30 (NCT04770753) | Phase III randomized, Worldwide | Adults with alpha- or beta-thalassemia who are not regularly transfused |
| ENERGIZE-T 30 (NCT04770779) | Phase III randomized, Worldwide | Adults with alpha- or beta-thalassemia who are regularly transfused |
| RISE UP (NCT05031780) | Phase II/III | Patients with sickle cell disease |
| ESTIMATE31 (EudraCT 2019-003438-18) | Phase II open-label, MAD, the Netherlands | Patients with sickle cell disease |
| ACTIVATE-KidsT (NCT05144256) | Phase III randomized, Worldwide | Children with PKD |
PKD, pyruvate kinase deficiency; MAD, multiple ascending dose.
Phase II DRIVE-PK study
Following encouraging preclinical and phase I studies, the phase II DRIVE-PK study evaluated the safety and efficacy of mitapivat in adults with PKD who were not regularly transfused, defined as having had three or fewer units of red cells transfused in the 12 months prior to initiating treatment with mitapivat (and no transfusions in the 4 months prior to treatment). 25 Fifty-two anemic (hemoglobin <12 g/dl in men or <11 g/dl in women) adults (38% female) were enrolled and randomized to receive mitapivat 50 mg twice daily or 300 mg twice daily for a 24-week core study period, with an optional long-term extension to follow. The primary study objective was assessment of safety and the side-effect profile. Patients were closely followed for potential acute and subacute toxicities for mitapivat with laboratory testing, electrocardiography, and physical examination, and had interval dual energy X-ray absorptiometry (DEXA) scanning performed to monitor for potential changes in bone density. Monitoring with DEXA was done to monitor for potential deleterious impacts of the off-target aromatase inhibition of the drug on bone mineral density, as well as potential positive on-target effects on bone mineral density from a reduction in ineffective erythropoiesis and erythron expansion. Secondary objectives included characterization of mitapivat pharmacokinetics and pharmacodynamics and clinical efficacy (measured by changes in hemoglobin and hemolysis markers).
In DRIVE-PK, mitapivat was well-tolerated, with mild headache (24 patients), insomnia (22 patients), and nausea (21 patients) being the most common adverse events reported. 25 The vast majority of these events resolved within a week of drug initiation. Serious TEAEs felt potentially related to mitapivat occurring in more than one patient included hypertriglyceridemia in four patients and rebound hemolysis in two patients. In terms of efficacy, 26 patients (50%) had a hemoglobin increase from baseline of ⩾1.0 g/dl, with a mean maximum increase of 3.4 g/dl (range = 1.1–5.8 g/dl). The median time to hemoglobin increase was just 10 days, and improvements were durable in the vast majority of patients who continued treatment. A clear relationship between underlying genotype and hemoglobin improvement was noted, such that patients with two drastic, non-missense mutations (i.e. indels, nonsense mutations) or two copies of the R479H mutation (a founder mutation prevalent in the American Amish community) did not respond, and patients with two non-R479H missense mutations were most likely to respond. In addition, a clear relationship and positive correlation was observed between the level of PKR protein in erythrocytes at baseline and hemoglobin response. Markers of hemolysis including reticulocyte count, indirect bilirubin, and haptoglobin all improved in patients exhibiting a hemoglobin response. Pharmacokinetics and pharmacodynamics in patients with PK deficiency were similar as what was observed in prior phase I studies of healthy volunteers.
Given the off-target aromatase inhibition of mitapivat and the high rate of osteopenia and osteoporosis in patients with PKD, 32 the impact of mitapivat on bone mineral density, (positive, negative, or none at all) is critical to discern given the expectation for long-term and/or indefinite treatment. Mitapivat could also have a positive impact on bone mineral density via reversal of erythron expansion through reduction of hemolysis. An analysis of long-term data from DRIVE-PK and its extension, including patients treated for up to 56 months, found that bone mineral density was largely stable over time in adults with PKD receiving mitapivat. 33 Although studies with even longer follow-up are needed to truly appreciate any potential impact, given the natural history of progressively worsening bone mineral density in these patients, stability alone is promising.
Phase III ACTIVATE study
Although the full manuscript describing the final results of the ACTIVATE study is yet to be published, the results for this study have been published in abstract form. Therefore, data from the published abstract are described in this section. 26
ACTIVATE was an international, phase III, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of mitapivat in adults with PKD who were not regularly transfused, defined as patients with four or fewer transfusion episodes (days in which a red cell transfusion was received) in the preceding 12 months. To qualify, patients required two or more documented mutant PKLR alleles, at least one of which needed to be a non-R479H missense mutation (in recognition of the nonresponding genotypes in DRIVE-PK). Patients were required to have a greater degree of anemia than in DRIVE-PK, with a baseline hemoglobin of <10.0 g/dl irrespective of sex. In addition, patients with a splenectomy in the preceding year or a history of any prior hematopoietic stem cell transplant were excluded. Eligible patients were randomized 1:1 to mitapivat or matching placebo, entering a 12-week individualized dose-escalation period (5 mg twice daily to 20 mg twice daily to 50 mg twice daily, with dose escalation generally indicated if a patient had not yet reached a normal hemoglobin for sex) followed by a 12-week fixed dose period. Patients completing the study were then eligible to enter an open-label extension study, which is currently ongoing.
The primary endpoint of ACTIVATE was a hemoglobin response, defined as a ⩾1.5 g/dl increase in hemoglobin from baseline sustained at two or more scheduled assessments during the fixed dose period (week 16, 20, or 24 of the study). Secondary endpoints included the average change from baseline in hemoglobin, reticulocytes, and markers of hemolysis (bilirubin, lactate dehydrogenase, and haptoglobin) at weeks 16, 20, and 24, as well as the change from baseline to week 24 in two PKD-specific health-related quality-of-life patient-reported outcome (PRO) measures, the pyruvate kinase deficiency diary (PKDD), and the pyruvate kinase deficiency impact assessment (PKDIA). These two PRO measures are novel instruments developed specifically to assess health-related quality of life in PKD, 34 and they underwent internal validation in the ACTIVATE trial.
A total of 80 patients were enrolled. While one patient randomized to placebo left the study prior to initiating study drug, no patients in either arm discontinued treatment after beginning study drug. The population was balanced between the mitapivat and placebo arms, with similar mean age, sex breakdown, and racial/ethnic breakdown in both groups. Although the patients in the ACTIVATE study were not transfusion-dependent, they nonetheless had a high burden of disease (as is common in non-transfusion-dependent patients with PKD), including high rates of iron overload and prior receipt of splenectomy. Approximately two-thirds of patients enrolled had two missense mutations, and one-third had one missense mutation and one non-missense mutation. Baseline rates of disease complications were similar in the two study arms.
Mitapivat met the primary endpoint in the ACTIVATE study, with 16 patients (40%) in the mitapivat arm achieving a hemoglobin response versus 0 patients (0%) in the placebo arm. In addition, the study met all of the secondary efficacy endpoints, with an average change in hemoglobin from baseline to the fixed dose period of +1.62 g/dl in the mitapivat arm versus −0.15 in the placebo arm, as well as significant improvements in LDH, bilirubin, haptoglobin, and reticulocyte percentage. Improvement in all of these markers occurred relatively rapidly with dose escalation during the dose-escalation period and was maintained over time. Significant improvement in both PRO measures, the PKDD and PKDIA, was also observed in the mitapivat arm as compared with the placebo arm.
As the first randomized controlled trial of mitapivat and only such trial to date, safety data in ACTIVATE are of particular interest. Here, mitapivat also performed very well. The most common TEAEs in the mitapivat arm were nausea and headache, both of which were actually more common in patients receiving placebo than receiving mitapivat. Importantly, no TEAEs led to treatment discontinuation.
Phase III ACTIVATE-T study
Although the full manuscript describing the final results of the ACTIVATE-T study is yet to be published, the results for this study have been published in abstract form. Therefore, data from the published abstract are described in this section. 27
ACTIVATE-T was an international, phase III, single-arm, open-label study evaluating the efficacy and safety of mitapivat in adults with PKD who were regularly transfused, defined as patients with six or more transfusion episodes in the preceding 12 months. As in ACTIVATE, patients required two or more documented mutant PKLR alleles, at least one of which being a non-R479H missense mutation, and they could not have had a splenectomy in the preceding year. Eligible patients began with a 16-week individualized mitapivat dose-escalation period (5 mg twice daily to 20 mg twice daily to 50 mg twice daily) followed by a 24-week fixed dose period. Patients completing the study were then eligible to enter an open-label extension study, which is currently ongoing. Of note, transfusions were strictly protocolized on ACTIVATE-T. Each patient had an individualized hemoglobin transfusion threshold established with a set number of red cell units to be transfused when this threshold was met, both calculated according to individual historical transfusion requirements in the year prior to enrollment. Red cell transfusions could only be administered per protocol if a patient reached their individualized hemoglobin transfusion threshold.
The primary endpoint of ACTIVATE-T was a reduction in transfusion burden, defined as a 33% reduction in transfusion requirements during the 24-week fixed dose period as compared with the subject’s historical transfusion burden standardized to 24 weeks. Secondary endpoints included the proportion of transfusion-free responders (defined as no transfusions during the fixed dose period) and annualized number of RBC units transfused.
A total of 27 patients were enrolled, of which 20 completed the study, 6 discontinued treatment, and 1 was lost to follow-up. For the purposes of statistical analysis, patients discontinuing treatment and lost to follow-up were considered nonresponders for the primary endpoint. ACTIVATE-T met its primary endpoint, with 10 patients (37%) achieving a reduction in transfusion burden of ⩾33%. In terms of secondary endpoints, the annualized number of RBC units transfused declined by 39%, and six patients (22%) were free of transfusions during the fixed dose period. Mitapivat was also well-tolerated in transfusion-dependent patients, with no TEAEs leading to discontinuation of treatment.
Following the success of the ACTIVATE and ACTIVATE-T studies evaluating mitapivat in adults, a study of mitapivat for pediatric PKD is now planned.
Clinical trials of mitapivat in thalassemia and sickle cell disease
Completed, ongoing, and planned clinical trials of mitapivat in thalassemia and sickle cell disease are summarized in Tables 1 and 2 and described in detail in the following sections.
Phase II study of mitapivat in non-transfusion-dependent alpha- or beta-thalassemia
Although the full manuscript describing the final results of the phase II study of mitapivat in non-transfusion-dependent thalassemia is yet to be published, the results for this study have been published in abstract form. Therefore, data from the published abstract are described in this section. 28
A phase II, open-label, multicenter study of mitapivat in alpha- and beta-thalassemia has been completed. This study enrolled 20 adults with non-transfusion-dependent thalassemia (beta-thalassemia, hemoglobin E/beta-thalassemia, or hemoglobin H disease) with a baseline hemoglobin of ⩽10 g/dl. Enrolled patients began with a 24-week core period, treated with mitapivat 50 mg twice daily with potential dose escalation to 100 mg twice daily after 6 weeks, and could enter an open-label extension after the 24-week core period. The primary endpoint of the study was a hemoglobin response, defined as an increase in hemoglobin from baseline of ⩾1.0 g/dl at any time between weeks 4 and 12 of the study. A total of 15 patients with beta-thalassemia (2 with HbE/beta-thalassemia) and 5 patients with alpha-thalassemia were enrolled. All patients were dose-escalated to mitapivat 100 mg twice daily at week 6. The study met its primary endpoint, with 16 patients (80%) achieving a hemoglobin response, including 11 of the patients with beta-thalassemia and all 5 of the patients with alpha-thalassemia. This response was sustained in eight of the beta-thalassemia patients and all five alpha-thalassemia patients with ongoing treatment. Improvements in hemoglobin were seen irrespective of the severity of baseline anemia, and improvements in markers of erythropoiesis and hemolysis were also observed. Mitapivat was well-tolerated in this study, with a safety profile similar to prior mitapivat studies. One patient developed grade 3 renal impairment leading to treatment discontinuation, although this was ultimately adjudicated as unrelated to mitapivat.
On the strength of these results, two international, phase III, randomized, placebo-controlled studies of mitapivat in thalassemia are planned: the ENERGIZE study, evaluating mitapivat in non-transfusion-dependent patients with thalassemia, and the ENERGIZE-T study, evaluating mitapivat in transfusion-dependent patients with thalassemia. 30
Phase I and II studies of mitapivat in sickle cell disease
Although the full manuscript describing the final results of the phase I study of mitapivat in sickle cell disease is yet to be published, the results for this study have been published in abstract form. Therefore, data from the published abstract are described in this section. 29
This phase I multiple ascending dose study of mitapivat in sickle cell disease, which completed in August 2021, enrolled a total of 17 patients, of which 16 were evaluable for response. Adults with sickle cell disease (HbSS) and a baseline hemoglobin ⩾7.0 g/dl without transfusions or erythropoietin therapy in the preceding 3 months were eligible. Stable doses of hydroxyurea and/or l-glutamine were permitted. Enrolled patients received either three or four ascending doses of mitapivat (5, 20, 50, and 100 mg twice daily) for 2 weeks each. The primary endpoint was safety and tolerability, and secondary endpoints included changes in hemoglobin, hemolytic markers, 2,3-DPG and ATP levels, and markers of Hb S polymerization (i.e. p50). In this study mitapivat was safe and well-tolerated, with just one serious TEAE possibly attributable to study drug (a vaso-occlusive crisis while the drug was being tapered). The mean change in hemoglobin at the 50 mg twice daily dose was +1.2 g/dl (range = −0.3 to +2.9 g/dl), which returned to baseline after the drug was tapered. Nine of 16 patients achieved a hemoglobin response (improvement by ⩾1.0 g/dl relative to baseline at any dose level) Hemolytic markers including lactate dehydrogenase, total bilirubin, reticulocytes, and aspartate aminotransferase similarly improved with mitapivat and normalized after its discontinuation. Mean 2,3-DPG levels decreased and ATP levels increased in a dose-dependent fashion, and decreases in p50 were also observed.
Preliminary results of the ongoing phase II ESTIMATE study have also been published in abstract form. 34 This open-label study is enrolling patients with sickle cell disease aged 16 years or older. Data on six enrolled subjects have been published, demonstrating no serious adverse events and overall comparable results thus far to the aforementioned phase I study.
Given the promising findings of both studies, the RISE UP study, a phase II/III trial of mitapivat in patients with sickle cell disease, is planned.
Conclusion
Mitapivat is a promising, first-in-class allosteric activator of pyruvate kinase with documented safety and efficacy across a wide spectrum of hereditary hemolytic anemias, including PKD, alpha- and beta-thalassemia, and sickle cell disease. Preclinical work suggests potential efficacy for erythrocyte membranopathies as well. Its mechanism of action allows it the potential of broad efficacy across a number of hemolytic states and conditions of ineffective erythropoiesis. It has been safe and well-tolerated in all completed human studies thus far, most notably in a phase III randomized trial in PKD. While improvements in hemoglobin, transfusion requirements, and markers of hemolysis and hematopoiesis are now well-documented with mitapivat treatment, time will tell if it is effective to halt or even reverse many of the morbid complications of chronic hemolysis, such as osteopenia and osteoporosis, iron overload, and extramedullary hematopoiesis. In addition, there are other important questions yet to be answered, including the efficacy and safety of mitapivat in the pediatric population and the potential for possible TEAEs related to long-term use of mitapivat over many years or decades as is required to maintain the drug effect. In particular, the off-target aromatase inhibition that thus far has appeared clinically insignificant in adults may be more relevant in developing children. Furthermore, mitapivat has yet to be examined in randomized trials in patients with thalassemia and sickle cell disease. To address these questions and others, additional trials in thalassemia, sickle cell disease, and pediatric PKD are now ongoing or planned, and long-term extension studies are ongoing in adults with PKD and thalassemia.
Footnotes
Authors’ Note: Hanny Al-Samkari is the recipient of the Harvard KL2/Catalyst Medical Research Investigator Training Award and the American Society of Hematology Scholar Award. Artwork in Figure 1 was reproduced and modified from Servier Medical Art (https://smart.servier.com/) in accordance with the Creative Commons license CC BY 3.0 (permission given for use and adaptation for any purpose, medium, or format).
Author contributions: Hanny Al-Samkari wrote the first draft of the manuscript and contributed to concept and design, data collection, data analysis, creation of tables and figures, critical revision of the manuscript, and final approval. Eduard J. van Beers contributed to concept and design, critical revision of the manuscript, and final approval.
Conflict of interest statement: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Hanny Al-Samkari: Consultancy (Agios, Dova/Sobi, Argenx, Rigel, Novartis, Moderna, Forma), Research funding (Agios, Dova, Amgen). Eduard J. van Beers: Consultancy and Research Funding (Agios).
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics approval statement: Ethics approval was not required for this review.
Informed consent statement: Informed consent was not required for this review.
ORCID iD: Hanny Al-Samkari  https://orcid.org/0000-0001-6175-1383
https://orcid.org/0000-0001-6175-1383
Contributor Information
Hanny Al-Samkari, Division of Hematology, Massachusetts General Hospital, Harvard Medical School, Zero Emerson Place, Suite 118, Office 112, Boston, MA 02114, USA.
Eduard J. van Beers, Universitair Medisch Centrum Utrecht, Utrecht, The Netherlands
References
- 1. Al-Samkari H, Van Beers EJ, Kuo KHM, et al. The variable manifestations of disease in pyruvate kinase deficiency and their management. Haematologica 2020; 105: 2229–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matte A, Federti E, Kung C, et al. The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a beta-thalassemia mouse model. J Clin Invest 2021; 131: e144206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rab MAE, Bos J, van Oirschot BA, et al. Decreased activity and stability of pyruvate kinase in sickle cell disease: a novel target for mitapivat therapy. Blood 2021; 137: 2997–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rab MAE, Van Oirschot BA, Kosinski PA, et al. AG-348 (mitapivat), an allosteric activator of red blood cell pyruvate kinase, increases enzymatic activity, protein stability, and ATP levels over a broad range of PKLR genotypes. Haematologica 2021; 106: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang J, Walsh MJ, Brimacombe KR, et al. ML265: a potent PKM2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft model. Probe Reports from the NIH Molecular Libraries Program, Bethesda, MD, 2010. [PubMed] [Google Scholar]
- 6. Kung C, Hixon J, Choe S, et al. Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem Biol 2012; 19: 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kung C, Hixon J, Kosinski PA, et al. AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency. Blood 2017; 130: 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y, Kosinski P, Histen G, et al. Preclinical pharmacokinetic/pharmacodynamic relationships for AG-348, an investigational small-molecule activator of pyruvate kinase. Haematologica 2015; 100: 298–299. [Google Scholar]
- 9. Rab MAE, van Oirschot BA, van Straaten S, et al. Decreased activity and stability of pyruvate kinase in hereditary hemolytic anemia: a potential target for therapy by AG-348 (Mitapivat), an allosteric activator of red blood cell pyruvate kinase. Blood 2019; 134: 3506. [Google Scholar]
- 10. Matte A, Anand W, Federti E, et al. The pyruvate kinase activator mitapivat ameliorates anemia and prevents iron overload in a mouse model of hereditary spherocytosis (abstract). Blood 2020; 136: 29. [Google Scholar]
- 11. Yang H, Merica E, Chen Y, et al. Phase 1 single- and multiple-ascending-dose randomized studies of the safety, pharmacokinetics, and pharmacodynamics of AG-348, a first-in-class allosteric activator of pyruvate kinase R, in healthy volunteers. Clin Pharmacol Drug Dev 2019; 8(2): 246–259. [DOI] [PubMed] [Google Scholar]
- 12. Beutler E, Gelbart T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood 2000; 95: 3585–3588. [PubMed] [Google Scholar]
- 13. Secrest MH, Storm M, Carrington C, et al. Prevalence of pyruvate kinase deficiency: a systematic literature review. Eur J Haematol 2020; 105(2): 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bianchi P, Fermo E, Lezon-Geyda K, et al. Genotype-phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am J Hematol 2020; 95: 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bianchi P, Fermo E. Molecular heterogeneity of pyruvate kinase deficiency. Haematologica 2020; 105: 2218–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Al-Samkari H, Addonizio K, Glader B, et al. The pyruvate kinase (PK) to hexokinase enzyme activity ratio and erythrocyte PK protein level in the diagnosis and phenotype of PK deficiency. Br J Haematol 2021; 192(6): 1092–1096. [DOI] [PubMed] [Google Scholar]
- 17. Bianchi P, Fermo E, Glader B, et al. Addressing the diagnostic gaps in pyruvate kinase deficiency: consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am J Hematol 2019; 94(1): 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Al-Samkari H, van Beers EJ, Morton DH, et al. Characterization of the severe phenotype of pyruvate kinase deficiency. Am J Hematol 2020; 95: E281–E285. [DOI] [PubMed] [Google Scholar]
- 19. Boscoe AN, Yan Y, Hedgeman E, et al. Comorbidities and complications in adults with pyruvate kinase deficiency. Eur J Haematol 2021; 106(4): 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Al-Samkari H, van Beers EJ, Morton DH, et al. Health-related quality of life and fatigue in children and adults with pyruvate kinase deficiency. Blood Adv 2021. Epub ahead of print 1 September 2021. DOI: 10.1182/bloodadvances.2021004675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grace RF, Bianchi P, van Beers EJ, et al. Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood 2018; 131: 2183–2192. [DOI] [PubMed] [Google Scholar]
- 22. van Beers EJ, van Straaten S, Morton DH, et al. Prevalence and management of iron overload in pyruvate kinase deficiency: report from the Pyruvate Kinase Deficiency Natural History Study. Haematologica 2019; 104(2): e51–e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grace RF, Mark Layton D, Barcellini W. How we manage patients with pyruvate kinase deficiency. Br J Haematol 2019; 184(5): 721–734. [DOI] [PubMed] [Google Scholar]
- 24. van Straaten S, Bierings M, Bianchi P, et al. Worldwide study of hematopoietic allogeneic stem cell transplantation in pyruvate kinase deficiency. Haematologica 2018; 103(2): e82–e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grace RF, Rose C, Layton DM, et al. Safety and efficacy of mitapivat in pyruvate kinase deficiency. N Engl J Med 2019; 381: 933–944. [DOI] [PubMed] [Google Scholar]
- 26. Al-Samkari H, Galacteros F, Glenthoj A, et al. ACTIVATE: a phase 3, randomized, multicenter, double-blind, placebo-controlled study of mitapivat in adults with pyruvate kinase deficiency who are not regularly transfused (abstract). HemaSphere 2021; 5: S270. [Google Scholar]
- 27. Glenthoj A, van Beers E, Al-Samkari H, et al. ACTIVATE-T: a phase 3, open-label, multicenter study of mitapivat in adults with pyruvate kinase deficiency who are regularly transfused (abstract). HemaSphere 2021; 5: S271. [Google Scholar]
- 28. Kuo KH, Layton DM, Lal A, et al. Results from a phase 2, open-label, multicenter study of the oral pyruvate kinase inhibitor mitapivat in adults with non-transfusion-dependent alpha- or beta-thalassemia (abstract). HemaSphere 2021; 5: S267. [Google Scholar]
- 29. Xu JZ, Conrey A, Frey I, et al. Mitapivat (AG-348) demonstrates safety, tolerability, and improvements in anemia, hemolysis, oxygen affinity, and hemoglobin S polymerization kinetics in adults with sickle cell disease: A phase 1 dose escalation study (abstract). Blood 2021; 138: 10. [Google Scholar]
- 30. Kuo KH, Layton DM, Al-Samkari H, et al. ENERGIZE and ENERGIZE-T: two phase 3, randomized, double-blind, placebo-controlled studies of mitapivat in adults with non-transfusion-dependent or transfusion-dependent alpha- or beta-thalassemia. HemaSphere 2021; 5: PB1805. [Google Scholar]
- 31. van Dijk M, Rab MAE, Rijneveld AW, et al. Safety and efficacy of mitapivat (AG-348), an oral activator of pyruvate kinase R, in subjects with sickle cell disease: A phase 2, open-label study (ESTIMATE) (abstract). Blood 2021; 138: 2047. [Google Scholar]
- 32. Al-Samkari H, Grace RF, Glenthoj A, et al. Early-onset osteopenia and osteoporosis in patients with pyruvate kinase deficiency (abstract). HemaSphere 2021; 5: EP692. [Google Scholar]
- 33. Al-Samkari H, van Beers E, Barcellini W, et al. Bone mineral density is stable in adults with pyruvate kinase deficiency receiving long-term treatment with mitapivat (abstract). HemaSphere 2021; 5: EP696. [Google Scholar]
- 34. Salek S, Boscoe AN, Piantedosi S, et al. Development of the pyruvate kinase deficiency diary and pyruvate kinase deficiency impact assessment: disease-specific assessments. Eur J Haematol 2020; 104(5): 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]