

Abstract
Transient increases in intracellular Ca2+ activate endothelium-dependent vasodilatory pathways. This process is impaired in cerebral amyloid angiopathy, where amyloid-β(1-40) accumulates around blood vessels. In neurons, amyloid-β impairs the Ca2+-permeable N-methyl-D-aspartate receptor (NMDAR), a mediator of endothelium-dependent dilation in arteries. We hypothesized that amyloid-β(1-40) reduces NMDAR-elicited Ca2+ signals in mouse cerebral artery endothelial cells, blunting dilation. Cerebral arteries isolated from 4-5 months-old, male and female cdh5:Gcamp8 mice were used for imaging of unitary Ca2+ influx through NMDAR (NMDAR sparklets) and intracellular Ca2+ transients. The NMDAR agonist NMDA (10 µmol/L) increased frequency of NMDAR sparklets and intracellular Ca2+ transients in endothelial cells; these effects were prevented by NMDAR antagonists D-AP5 and MK-801. Next, we tested if amyloid-β(1-40) impairs NMDAR-elicited Ca2+ transients. Cerebral arteries incubated with amyloid-β(1-40) (5 µmol/L) exhibited reduced NMDAR sparklets and intracellular Ca2+ transients. Lastly, we observed that NMDA-induced dilation of pial arteries is reduced by acute intraluminal amyloid-β(1-40), as well as in a mouse model of Alzheimer’s disease, the 5x-FAD, linked to downregulation of Grin1 mRNA compared to wild-type littermates. These data suggest that endothelial NMDAR mediate dilation via Ca2+-dependent pathways, a process disrupted by amyloid-β(1-40) and impaired in 5x-FAD mice.
Keywords: N-methyl-D-aspartate receptor, cerebral amyloid angiopathy, endothelium-dependent dilation, amyloid-β, endothelium Ca2+ signaling
Introduction
Proper regulation of cerebral blood flow is essential to supply oxygen and nutrients to regions of increased neuronal activity in the brain. This process, known as neurovascular coupling, is mediated by mechanisms initiated in different cell types in the brain, culminating in dilation of arterioles within the intraparenchymal microcirculation. 1 Several mechanisms ultimately lead to arteriolar dilation, however, the most robust dilatory pathways are those initiated by endothelial cells, known as endothelium-dependent dilation. In this vasodilatory paradigm, endothelial cells receive signals from the environment (partial pressure of oxygen, chemical mediators, shear stress, etc.) and generate a response that relaxes the smooth muscle layer. As a result, arterial diameter increases, leading to an increase in blood flow to regions of increased metabolic demand. Virtually all endothelium-dependent dilatory pathways (generation of nitric oxide, metabolites of arachidonic acid and endothelium-dependent hyperpolarization) rely on transient increases in intracellular Ca2+ within endothelial cells. 2
Transient increases in intracellular Ca2+ occur via Ca2+ influx through opening of Ca2+-permeable channels in the plasmalemma, and/or via release from intracellular stores. 2 Endothelial cells possess a plethora of Ca2+-permeable ion channels in the plasma membrane, including the N-methyl-D-aspartate receptor (NMDAR). 3 NMDAR are ionotropic glutamate receptors that conduct a nonselective cationic current of approximately 40 pS (depending on subunit composition), 4 and have a fractional Ca2+ permeability ∼3.6-fold higher than for Na+. 5 Molecularly, NMDA receptors often display a heterotetrameric conformation consisting of 2 GluN1 subunits and 2 GluN2 subunits (can vary between 2 A, 2B, 2 C or 2 D), although GluN3 subunits may also occur. 6 The GluN1 subunit is the ‘obligatory’ subunit of the NMDAR, as it contains the binding site for co-agonists D-serine or glycine. The GluN2 subunit contains the binding site for glutamate, and the identity of the GluN2 subunit influences a variety of biophysical properties of the NMDAR, including ion conductance, agonist affinity, resistance to Mg2+ blockade of the pore, and potential for allosteric modification.7–9 In the cerebral circulation, NMDAR activation induces dilation of middle cerebral arteries 10 and parenchymal arterioles.3,11 Further, genetic knockdown of NMDAR specifically in endothelial cells blunts neurovascular coupling in vivo. 12 Impaired neurovascular coupling responses are observed in progressive diseases linked to development of cognitive decline, such as Alzheimer’s disease and cerebral amyloid angiopathy. 13
Cerebral amyloid angiopathy is characterized by accumulation of the peptide amyloid-β(1-40) in the walls of cerebral blood vessels,14,15 and it is present in approximately 80% of Alzheimer’s disease cases. 16 Perivascular amyloid-β impairs vascular function,17,18 possibly leading to neurovascular impairments known to precede the onset of dementia in Alzheimer’s disease patients.19,20 In addition, amyloid-β reduces membrane expression and activity of NMDAR in hippocampal neurons in vitro, 21 raising the possibility that amyloid-β may exert a similar effect on NMDAR in endothelial cells. Therefore, we hypothesized that amyloid-β(1-40) inhibits NMDAR function in endothelial cells, observed as suppression of NMDAR-elicited Ca2+ signals and cerebral artery dilation.
Materials and methods
Animals
All animal procedures in this study were approved by the Institutional Animal Care and Use Committee of the University of Arizona College of Medicine (IACUC protocol 18-473), and are in accordance with the National Institutes of Health’s ‘Guide for the Care and Use of Laboratory Animals', 8th edition. All animal experiments are reported in compliance with the ARRIVE guidelines. 22 Adult (4 to 5 months of age) male and female mice were used in equal numbers. Mice were initially segregated by sex to assess possible differences. No sex differences were observed in frequency of Ca2+ events or magnitude of vasodilation to NMDA; therefore, we combined males and females for all analyses (Supplemental Figure 6).
cdh5:Gcamp8 transgenic mice were developed by the CHROMus™ initiative at Cornell University and were kindly donated by Dr. Michael Kotlikoff. These mice possess the sequence to produce the high dynamic range and fast kinetics genetically-encoded Ca2+ indicator GCaMP8 under control of the endothelial cell specific promoter cdh5. 23 cdh5:Gcamp8 transgenic mice were used for all Ca2+ imaging experiments in this study.
5x-FAD transgenic mice were originally purchased from Jackson Laboratories and are currently maintained in our breeding colony at the University of Arizona. They are a transgenic model of early-onset Alzheimer’s disease. These mice possess a genetic insertion containing 5 different mutations associated with Familial Alzheimer’s disease. Three of the mutations are associated with mutant human amyloid precursor protein: Swedish (K670N, M671L), Florida (I716V), and London (V717I), coupled with two mutations of human presenilin, M146L and L286V. Expression of transgenes is controlled by the neuronal-specific Thy1 promoter. 24
All mice are bred on a C57bl6/J background and non-transgenic wild-type littermates were used as controls.
En face arterial preparation
Transgenic cdh5:Gcamp8 mice were deeply anesthetized using isoflurane and decapitated. The brain was removed from the skull and placed into ice-cold tissue collection physiological saline solution (PSS) consisting of (in mmol/L): 140 NaCl, 5 KCl, 2 MgCl2, 10 Dextrose, 10 HEPES, pH 7.4. Brains were then transferred into a water-jacketed dissection dish connected to a circulating cold-water bath (4 °C) filled with tissue collection PSS supplemented with 0.5% bovine serum albumin (BSA, Fisher Scientific, Waltham, MA). Brains were pinned down to the dissection dish using insect pins (Fine Science Tools, Foster City, CA) with the ventral surface facing up. The basilar, middle cerebral, posterior cerebral and superior cerebellar arteries were dissected and cut longitudinally with fine spring scissors and mounted onto a Sylgard block with the endothelium-side facing up. Vessels were stretched to approximately 1.5x their diameters to introduce physiological levels of transmural stress, and the edges were pinned down using insect pins.25,26
Calcium imaging
En face arterial preparations were used for time-lapse imaging. During experiments, the arterial preparation was continuously bathed with warm (37 °C) imaging PSS consisting of (in mmol/L): 2 Ascorbic acid, 2.5 CaCl2, 119 NaCl, 4.7 KCl, 0.5 MgSO4, 1.18 KH2PO4, 10 Dextrose, 21 NaHCO3, 10 HEPES, pH 7.4, using a perfusion pump. Imaging dishes were placed on a Leica DM6 microscope with a 63x water immersion objective (numerical aperture 0.9). Time-lapse images were acquired using a high sensitivity Evolve Delta EMCDD camera (Teledyne Photometrics, Tucson, AZ) controlled by µManager software 27 in 512 × 512 pixels/fields of view at a pixel resolution of 0.290 nm. Number of cells per field of view was not significantly different between treatment groups, except in the experiments corresponding to Figure 1, where statistical significance was found between vehicle and NMDA + DAPV groups (Supplemental Figure 7).
Figure 1.
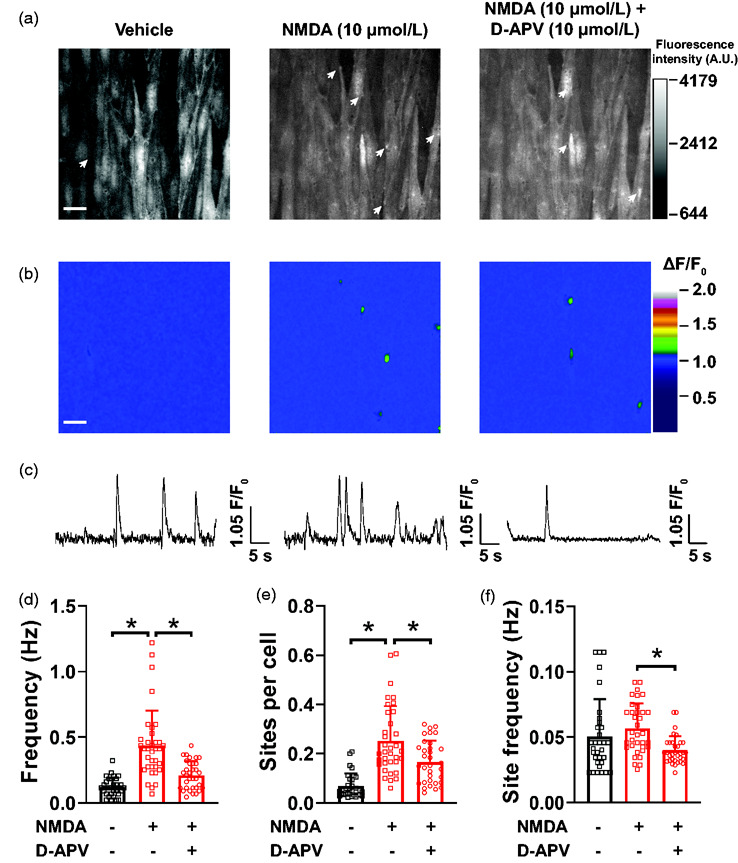
Unitary Ca2+ entry through endothelial NMDAR in pial arteries. (a) Representative maximum intensity projection of greyscale time-lapses from a field of view of endothelial cells in an en face pial artery isolated from cdh5:Gcamp8 mouse and exposed to vehicle (imaging PSS, left), NMDA (10 µmol/L, middle) and NMDA + D-APV (10 µmol/L, right). The arrows indicate sites of NMDAR sparklets. Bar = 20 µm. (b) Pseudocolored ΔF/F0 rendering of the images in (A) showing the sites of NMDAR sparklets (green) in the different recordings. Bar = 20 µm. (c) Representative line traces of fluorescence intensity (ΔF/F0) over time in individual NMDAR sites at the different treatments showing multiple openings with vehicle (left) and NMDA (middle) and the reduced NMDAR sparklets site frequency after treatment with D-APV + NMDA (right). (d-f) Summary bar graphs of whole field of view NMDAR sparklets frequency (d), sites per cell (e) and site frequency (f). All preparations in this panel were pre-treated with EGTA-AM (10 µmol/L) and CPA (30 µmol/L) to isolate Ca2+ entry events. Data are means ± SD, n = 31-35-32 fields of view from 4 different preparations isolated from 4 different mice, 2 male and 2 female. *p < 0.05, Brown-Forsythe test with a Dunnett’s T3 correction for multiple comparisons.
NMDA (10 µmol/L), dissolved in imaging PSS (vehicle), was added to the superfusate and circulated. Imaging began as soon as the NMDA solution reached the imaging dish. Preparations were then washed with imaging PSS and incubated with the NMDAR inhibitors D-APV (10 µmol/L, Tocris Biosciences, Minneapolis, MN) or MK-801 (10 µmol/L, Tocris) for 10 minutes prior to new exposure to NMDA. For experiments using amyloid-β(1-40), preparations were incubated for 30 minutes at room temperature with amyloid-β(1-40) (5 µmol/L, 28 Abcam, catalog# ab120957) in imaging PSS, then washed with fresh imaging solution at 37 °C for ∼10 minutes before recording.
For experiments recording Ca2+ influx events through NMDAR, or NMDAR sparklets, preparations were first pre-incubated for 15 minutes with EGTA-AM (10 µmol/L, Thermo Fisher Scientific, catalog# E1219) in tissue collection PSS, then treated with cyclopiazonic acid (CPA, 30 µmol/L, Tocris Bioscience, Minneapolis, MN). These treatments prevent Ca2+ release from intracellular stores, thus facilitating visualization of lower amplitude Ca2+ entry events.25,26
Time-lapse images (1000 images) from each field of view were imaged for a total duration of ∼46 seconds to visualize changes in intracellular Ca2+ concentrations in endothelial cells. Frame rate was consistent at 22–24 frames per second. Arteries were imaged no more than 2–3 times in each field of view to minimize photobleaching/light toxicity.
Analysis of Ca2+ signals
Time-lapse Ca2+ imaging files were analyzed using a custom-made software kindly provided by Dr. Mark Nelson and Dr. Adrian Bonev from the University of Vermont (SparkAn version 5.5.6.0). 29 SparkAn averages the first 10 images to define background fluorescence (F0). F0 is pseudocolored as blue, while localized increases in fluorescence (ΔF) appear on a range of warmer colors, from green to red. Individual NMDA sparklets and intracellular Ca2+ transients were analyzed to quantify event frequency (events per second, Hz), spatial spread (µm2), and peak amplitude (ΔF/F0). Attack time (1/2 time from F0 to peak amplitude), decay time (1/2 time from peak amplitude back to F0) and duration (2 × (attack time + decay time)) were assessed manually by replaying the pseudocolored images frame-by-frame and annotating start, peak, and end of individual events. Fields of view containing no events were excluded from statistical analysis because viability of cells could not be verified.
Pressure myography
5x-FAD and wild-type littermates were used for pressure myography experiments. Posterior communicating arteries (PComA) or parenchymal arterioles were carefully isolated in tissue collection PSS. Isolation of parenchymal arterioles followed a previously published protocol.30,31 After isolation, PComA or parenchymal arterioles were transferred to a custom-made pressure myograph chamber 32 filled with tissue collection PSS. One end of the vessel was cannulated onto a glass cannula (∼30–50 µm diameter at the tip); the other end was tied to the cannula in a blind-sac. The cannula was filled with vessel PSS consisting of (in mmol/L): 124 NaCl, 3 KCl, 2 MgCl2, 1.085 NaH2PO4 2H20, 26 NaHCO3, 1.8 CaCl2, 4 Dextrose. PSS was constantly oxygenated with 21% O2/5% CO2/74% N2 to maintain pH 7.4. For endothelium-denudation experiments, one end of the artery was cannulated and an air bubble was slowly passed through the intraluminal side to damage the endothelial cell layer,25,33 then the cannulas and vessels were filled with PSS and the other end was tied. The pressure myography chamber was transferred to a microscope and connected to a pressure-servo pump to control intraluminal pressure (Living Systems Instrumentation, Burlington, VT). The preparation was superfused with warm (37 °C), oxygenated vessel PSS exchanged at a rate of 3–5 mL per minute for 15 minutes at a pressure of 15 mmHg to equilibrate. Intraluminal pressure was then increased to 50 mmHg (PComA) or 40 mmHg (parenchymal arterioles) until generation of spontaneous myogenic tone. Outer and lumen diameters were recorded in real-time at 15 Hz using the IonWizard v7.3 software (IonOptix, Westwood, MA). Spontaneous myogenic tone was calculated as: Myogenic tone (%) = [1−LDT/LDP)]*100, 34 where LDT is the lumen diameter at spontaneous myogenic tone and LDP is the passive lumen diameter.
Following generation of spontaneous myogenic tone, preparations were exposed to NMDA (10 µmol/L) in the superfusing bath, which was then washed out. Preparations were then incubated with D-APV (10 µmol/L, Tocris Bioscience) for 10 minutes, followed by addition of NMDA. To elucidate the intracellular pathways involved in NMDAR-induced dilation, a subset of PComA were exposed to NMDA (10 µmol/L), which was then washed, and incubated with either Nω-Nitro-L-arginine (L-NNA, 100 µmol/L) + indomethacin (30 µmol/L, Tocris) to inhibit nitric oxide synthase and cyclooxygenases, or apamin (300 nM, Tocris) + TRAM-34 (1 µmol/L, Tocris,) to inhibit small- and intermediate-conductance Ca2+-activated K+ channels (KCa2.3 and KCa3.1, respectively), followed by another exposure to NMDA. Lastly, another subset of PComA was incubated with intraluminal amyloid-β(1-40) (5 µmol/L) for 30 minutes, then exposed to abluminal NMDA (10 µmol/L) for 10 minutes, followed by PSS wash and incubation with the KCa2.3/KCa3.1 activator NS-309 (10 µmol/L). After experimental treatment, preparations were bathed in calcium-free PSS to acquire passive diameter. Dilation data are shown as Vasodilation (%), calculated: ((LDD – LDB)/LDP) × 100, where LDD is the lumen diameter at peak dilation; LDB is lumen diameter at baseline; LDP is the passive lumen diameter.
Immunohistochemistry
5x-FAD and wild-type mice were euthanized using pentobarbital (80 mg/kg, intraperitoneal, Vortech Pharmaceuticals, Dearborn, MI). Mice were transcardially perfused with a solution of cis-diltiazem (10 µmol/L) and 1 unit/mL heparin diluted in PBS, followed by 4% paraformaldehyde. Brains were removed and post-fixed in 4% paraformaldehyde overnight at room temperature, then washed with PBS. Brains were sectioned using a Leica VT1000P vibratome (Leica Biosystems Nussloch GmbH, Nußloch, Germany) to obtain 50 µm thick coronal slices. Brain slices were permeabilized in 0.1% Triton X-100 in PBS for 2 hours, then treated with 15% formic acid in PBS for 15 minutes to enhance amyloid-β staining and subsequently washed with PBS. Slices were treated with 0.5% sodium borohydride for 15 minutes to quench autofluorescence and washed with PBS. Following washes, slices were incubated in 5% horse serum, 0.1% Triton X-100 in PBS for 1 hour, then incubated overnight with a goat polyclonal antibody against smooth muscle actin (1:500, Thermo Fisher Scientific, Waltham, MA, catalog# PA5-18292) and a rabbit monoclonal antibody against amyloid-β (1:2000, Abcam, Cambridge, MA, catalog# ab201060) diluted in 5% horse serum, 0.1% Triton X-100 in PBS. Sections were then washed with PBS prior to incubation with secondary antibody for 2 hours: donkey anti-goat AlexaFluor 594 (1:500, Jackson ImmunoResearch, West Grove, PA) and donkey anti-rabbit AlexaFluor 488 (1:500, Jackson ImmunoResearch) diluted in 2% horse serum, 0.1% Triton X-100 in PBS. Brain sections were washed with PBS and mounted on glass slides using Fluoroshield mounting media. Resulting immunolabeled slices were imaged using a Zeiss LSM880 laser scanning confocal microscope (Carl Zeiss Jena GmBH, Jena, Germany), using a 40x oil immersion objective (numerical aperture 1.3) to acquire Z-stacks of pial vessels positively labeled for smooth muscle actin. Images were processed using ImageJ to generate maximum intensity projections. No post-acquisition processing was performed in the images, and settings for laser power, PMT gain and offset were adjusted in one slice from a 5x-FAD mouse and kept constant to image the remaining 5x-FAD, wild-type and no-primary controls.
Real-time quantitative polymerase chain reaction (qPCR)
Pial arteries (basilar, cerebellar, posterior, middle and anterior cerebral arteries) mRNA was collected from wild-type and 5x-FAD mice using RNeasy Plus Micro Kit (QIAGEN, catalog# 74034) after being stored in RNA Later (QIAGEN, Redwood City, CA, catalog# 76104) following manufacturer’s instructions. mRNA was converted to cDNA using iScript™ Select cDNA Synthesis Kit (Bio-Rad, Hercules, CA, catalog# 170-8897), and 20 ng of cDNA were loaded into separate wells for qPCR amplification using Advanced Universal SYBR Green Supermix (Bio-Rad, catalog# 1725271) in an Applied Biosystems 7300 Real Time PCR System (Applied Biosystems, Foster City, CA). Samples were run in quintuplets, and glyceraldehyde 3-phosphate dehydrogenase (Gapdh) was used as endogenous control. Negative controls (no cDNA sample) were included in all experiments. Primer sequences were: Grin1 forward 5′-CCA GAT GTC CAC CAG ACT AAA G-3′, Grin1 reverse 5′-CCA TTG ACT GTG AAC TCC TCT T-3′; Gapdh forward 5′-GGA AGC CCA TCA CCA TCT T-3′, Gapdh reverse 5′-ACC AGT AGA CTC CAC GAC ATA-3′. Cycling conditions consisted of an initial step of 95 °C for 5 minutes followed by 45 cycles of 95 °C for 15 seconds, 55 °C for 30 seconds and 72 °C for 15 seconds. Amplification products were analyzed by gel electrophoresis using a 2% agarose gel stained with ethidium bromide to ensure only 1 amplicon band was present in each sample. Specificity of Grin1 primers was validated by Sanger sequencing of amplified products.
Cycle thresholds (Ct) values obtained from quintuples were averaged. Relative gene expression levels was determined by 2^ΔΔCt calculations using the difference of the averaged Ct values of the target gene and housekeeping gene (ΔCt = Ct(Gapdh) – Ct(Grin1)).
Chemicals
Unless otherwise stated all chemicals were purchased from Sigma-Aldrich, St. Louis, MO.
Data and statistical analyses
Data are expressed as means ± SD. All data analyses were performed using GraphPad Prism 9. Difference between means of 2 experimental groups were analyzed using two-tailed Student’s t-tests or Mann-Whitney test if the data did not follow a normal distribution. When 3 groups were compared, data were analyzed using one-way ANOVA or Brown-Forsythe test as a non-parametric alternative followed by Bonferroni or Dunnett’s T3 multiple comparisons tests. Amplitude (ΔF/F0) and spatial spread (µm2) of Ca2+ transients elicited by NMDAR were analyzed by constructing frequency distribution plots to identify the mode and Gaussian distribution of the events. Sample sizes were estimated based on power analyses for a sigma value of 0.05 and a desired power of 0.80.
Results
NMDAR sparklets in cerebral artery endothelial cells of cdh5:Gcamp8 mice
Unitary Ca2+ entry through single or clusters of NMDAR in endothelial cells, named NMDAR sparklets, was assessed in en face preparations of cerebral arteries incubated with EGTA-AM (10 µmol/L) and CPA (30 µmol/L) to prevent Ca2+ release from intracellular stores. Under baseline conditions (vehicle), unitary Ca2+ events were relatively rare (Figure 1, Supplemental Figure 1 and Supplemental Movie 1), as reported previously for other cationic channels.25,26 Application of the NMDAR agonist NMDA (10 µmol/L) to preparations induced an approximately 4-fold increase in unitary Ca2+ events (Figure 1, Supplemental Figure 1 and Supplemental Movie 2). The elicited events resulted from NMDAR activation, as pre-incubation with the NMDAR antagonist to the glutamate binding site, D-APV (10 µmol/L), significantly prevented induction of NMDAR sparklets (Figure 1(b) to (f), Supplemental Movie 3). Similarly, NMDAR sparklets were inhibited by the pore-blocking antagonist MK-801 (10 µmol/L, Supplemental Figure 1). The increase in frequency of NMDAR sparklets was the result of recruitment of previously silent NMDAR sparklets sites (Figure 1(a), (b) and (e)). Exposure of preparations to the NMDAR antagonists D-APV (Figure 1(d) to (f)) or MK-801 (Supplemental Figure 1) caused a reduction in both the number of active sites and site frequency. Lastly, we confirmed that the events elicited by NMDA were indeed a consequence of Ca2+ entry into the cell, as removal of extracellular Ca2+ nearly abolished NMDAR sparklets (Supplemental Figure 2).
In order to describe the biophysical properties of optically recorded NMDAR sparklets we performed frequency distribution analyses of the individual values for amplitude, attack time, decay time, total duration and spatial spread of NMDA-elicited events. A time-lapse of a typical NMDAR sparklet is shown in Supplemental Figure 3(A). Amplitudes showed a single Gaussian distribution with a peak at 1.08 ΔF/F0 (Supplemental Figure 3(B)). Attack and decay times both showed a mode of 75 ms (Supplemental Table 1), whereas total duration showed a bi-modal distribution: a mode at 300 ms and another at 500 ms (Supplemental Table 1). NMDAR sparklets were spatially restricted events, with a mode spatial spread of 8 µm2 (Supplemental Figure 3(C), Supplemental Table 1). The mode and mean values for amplitude, attack time, decay time, total duration and spatial spread are summarized on Supplemental Table 1.
NMDAR activation increases frequency of intracellular Ca2+ transients in endothelial cells.
We performed Ca2+ imaging of en face cerebral arterial preparations to test the hypothesis that NMDAR activation induces an increase in intracellular Ca2+ transients in endothelial cells of cdh5:Gcamp8 mice. Intracellular Ca2+ transients were defined as any increase in intracellular [Ca2+] without EGTA-AM or CPA to maintain intracellular Ca2+ stores. Under these conditions, intracellular Ca2+ transients consisted of: 1) low amplitude, spatially-restricted (<20 µm2) events, consistent with Ca2+ influx events; 2) subcellular higher amplitude signals with a larger spatial spread (between 20 and 100 µm2); 3) propagating Ca2+ wavelets (spreading to approximately 50% of the cytoplasm) and whole cell waves, at times spreading to neighboring cells (Supplemental Figure 4(A) and (B) and Supplemental Movie 4).
Spontaneous intracellular Ca2+ transients in endothelial cells were observed at baseline (vehicle) conditions, albeit at a low frequency (Figure 2(a), (b) and (d), Supplemental Movie 4). Incubation of preparations with NMDA (10 µmol/L) caused a significant increase in the frequency of intracellular Ca2+ transients compared to baseline (Figure 2(a), (b) and (d), Supplemental Movie 5). This increase in frequency of intracellular Ca2+ transients was a consequence of an increase in the number of active sites (Figure 2(a), (b) and (e)), as well as frequency of each individual site (Figure 2(c) and (f)). Incubation of preparations with D-APV (10 µmol/L, Figure 2) or MK-801 (10 µmol/L, Supplemental Figure 5) prevented NMDA-induced increases in intracellular Ca2+ transients frequency and recruitment of active sites. Site frequency, however, was reduced by D-APV (Figure 2 and Supplemental Movie 6) but not MK801 (Supplemental Figure 5).
Figure 2.
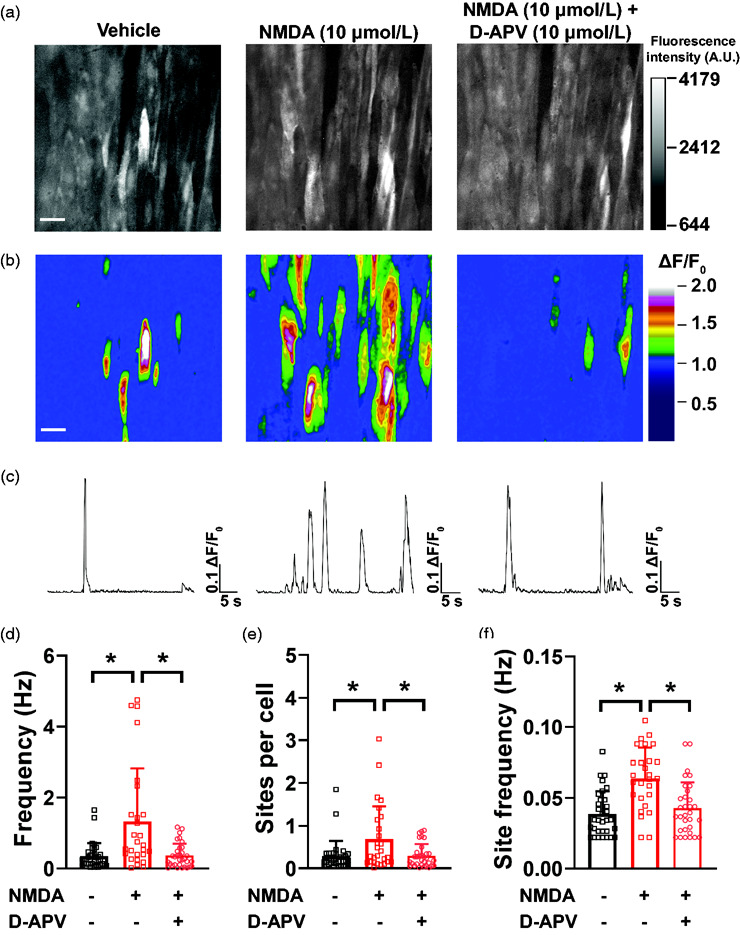
NMDA increases frequency of intracellular Ca2+ transients in cerebral artery endothelial cells. (a) Representative maximum intensity projection of greyscale time-lapses from a field of view of endothelial cells in an en face pial artery isolated from cdh5:Gcamp8 mouse and exposed to vehicle (imaging PSS, left), NMDA (10 µmol/L, middle) and NMDA + D-APV (10 µmol/L, right). Bar = 20 µm. (b) Pseudocolored ΔF/F0 rendering of the images in (A) showing the sites of intracellular Ca2+ transients (warmer colors) in the different recordings. Bar = 20 µm. (c) Representative line traces of fluorescence intensity (ΔF/F0) over time in individual sites at the different treatments showing multiple openings with NMDA (middle) and lower site frequency with vehicle (left) or after treatment with D-APV + NMDA (right). (d–f) Summary bar graphs of whole field of view frequency of intracellular Ca2+ transients (d), sites per cell (e) and site frequency (f). Data are means ± SD, n = 32-27-32 fields of view from 6 different preparations isolated from 5 mice (3 male and 2 female). *p < 0.05, Brown-Forsythe test with a Dunnett’s T3 correction for multiple comparisons.
NMDA induced PComA and parenchymal arteriole dilation via NMDAR
We then assessed whether the increase in endothelial Ca2+ signaling induced by NMDA is linked to vasodilation. We observed that exposing PComA isolated from C57bl6/J mice to NMDA (10 µmol/L) induced a transient dilation that was significantly blunted by D-APV (10 µmol/L), as well as by removal of the endothelium (Figure 3(a) and (b)). These data suggest that NMDA induces PComA dilation via activation of NMDAR in the endothelial cells. NMDAR-dependent dilation was also observed in parenchymal arterioles isolated from C57bl6/J mice (Figure 3(e)). No significant changes to myogenic tone occurred between treatment groups (Supplemental Figure 8(A) and (D)).
Figure 3.
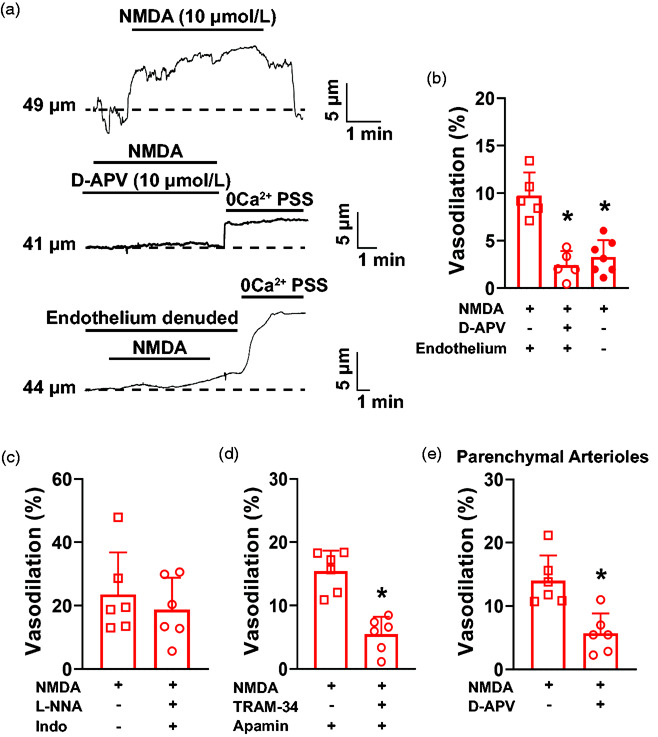
NMDA induces dilation of pial arteries from C57bl6/J mice via endothelium-dependent hyperpolarization. (a) Representative traces of the lumen diameter of an ex vivo pressurized PComA exposed to NMDA (10 µmol/L) in the absence (top) and presence (middle) of the NMDAR inhibitor D-APV (10 µmol/L). The lower trace shows absence of NMDA-induced dilation after endothelium damage caused by an air bolus. (b) Summary bar graphs showing that NMDA dilation was blunted by D-APV and by removal of the endothelium. *p < 0.05, One-way ANOVA. N = 11 arteries from 5 males and 2 females. (c) Inhibition of nitric oxide production with L-NG-nitroarginine (L-NNA, 100 µmol/L) and cyclooxygenases with indomethacin (30 µmol/L) did not affect NMDA-induced dilation of mouse cerebral PComA. N = 6 arteries from 3 mice, 2 males and 1 female. (D) KCa2.3 and KCa3.1 inhibition with apamin (300 nmol/L) and TRAM-34 (1 µmol/L), respectively, significantly blunted PComA dilation after exposure to NMDA. *p < 0.05, two-tailed Student’s t-test, N = 6 arteries from 3 mice, 1 male and 2 females. (e) Dilation of parenchymal arterioles to NMDA was significantly reduced by D-APV. *p < 0.05, two-tailed Student’s t-test, N = 6 arteries from 3 mice, 2 males and 1 female.
Next, we investigated the Ca2+-dependent intracellular pathways involved in endothelial cell NMDAR-induced dilation in PComA. We first inhibited nitric oxide synthase and cyclooxygenases with L-NNA (100 µmol/L) and indomethacin (30 µmol/L), respectively, and observed that dilation to NMDA was maintained (Figure 3(c)). We then inhibited KCa2.3/KCa3.1 with apamin (300 nM) and TRAM-34 (1 µmol/L), respectively, and observed that NMDA-induced dilation was significantly reduced (Figure 3(d)). No significant changes to myogenic tone occurred between treatment groups (Supplemental Figure 8(B) and (C)). Together, these data suggest that, in mouse PComA, NMDAR-induced dilation requires endothelial cell hyperpolarization downstream from KCa2.3/KCa3.1 activation (Supplemental Figure 9(A)).
Acute exposure to amyloid-β(1-40) decreases frequency of NMDA sparklets and NMDA-elicited intracellular Ca2+ transients
We next tested the hypothesis that amyloid-β(1-40) inhibits NMDAR activity. Cerebral arteries isolated from cdh5:Gcamp8 mice were prepared en face and incubated with a pathological concentration of amyloid-β(1-40) peptide (5 µmol/L)28,35 for 30 minutes prior to exposure to NMDA. Amyloid-β(1-40) significantly inhibited NMDAR sparklets in endothelial cells, observed as a reduction in frequency (Figure 4(b), Supplemental Movies 7 and 8), as well as the number of active sites per cell (Figure 4(a) and (c)). Site frequency at individual NMDAR sparklets sites was unchanged by amyloid-β(1-40) (Figure 4(d)).
Figure 4.
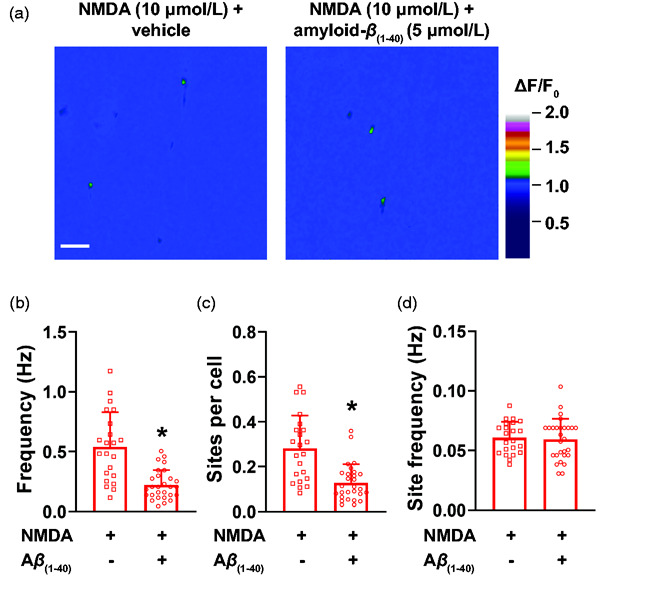
Amyloid-β(1-40) inhibits NMDAR sparklets in cerebral artery endothelial cells. (a) Representative pseudocolored images of fields of view showing sites of NMDAR sparklets (green) before (left) and after (right) incubation with amyloid-β(1-40) (Aβ(1-40), 5 µmol/L). Bar = 20 µM. (b-d) Summary bar graphs showing that amyloid-β(1-40) significantly reduces the whole field of view NMDAR sparklets frequency (b) and number of sites per cell (c), albeit it does not affect frequency of NMDAR sparklets at individual sites (d). Data are means ± SD. *p < 0.05, Mann-Whitney test, n = 21–27 fields of view from 3 different preparations isolated from 3 mice (2 male and 1 female).
Similarly, NMDA-induced intracellular Ca2+ transients were significantly inhibited by amyloid-β(1-40). We observed that incubation of en face cerebral arteries with amyloid-β(1-40) prevented NMDA from increasing frequency of intracellular Ca2+ transients (Figure 5(b) and Supplemental Movies 9 and 10), as well as the number of active sites (Figure 5(a) and (c)), without affecting site frequency (Figure 5(d)).
Figure 5.
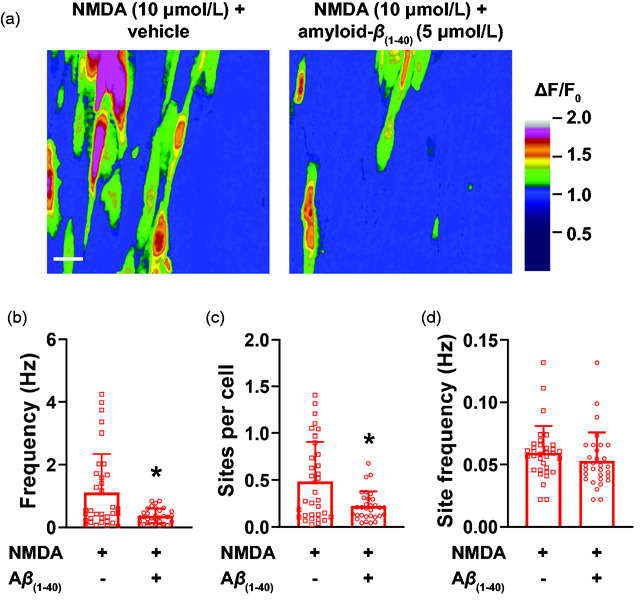
NMDAR-induced intracellular Ca2+ transients are inhibited by amyloid-β(1-40). (a) Representative ΔF/F0 pseudocolored images of fields of view showing sites of intracellular Ca2+ transients induced by NMDA before (left) and after (right) incubation with amyloid-β(1-40) (Aβ(1-40), 5 µmol/L). Bar = 20 µM. (b-d) Summary bar graphs showing that amyloid-β(1-40) significantly reduces the whole field of view frequency of intracellular Ca2+ transients induced by NMDA (B), as well as number of sites per cell (c). No differences were observed in site frequency (d). Data are means ± SD. *p < 0.05, Mann-Whitney test (n = 34-31 fields of view from 4 different preparations isolated from 4 mice (2 male and 2 female).
Acute intraluminal incubation with amyloid-β(1-40) impairs NMDAR-dependent dilation
To evaluate the acute effects of amyloid-β on NMDA-induced dilation of pial arteries, we isolated PComA from wild-type C57bl6 mice and incubated with intraluminal amyloid-β(1-40) (5 µmol/L) for 30 minutes. We observed that dilation to NMDA was blunted in PComA incubated with amyloid-β(1-40) (Figure 6), whereas dilation to the KCa2.3/KCa3.1 activator NS-309 (10 µmol/L, Tocris), the downstream mediators of NMDAR-induced dilation in our studies, was maintained (Figure 6). No significant changes to myogenic tone occurred between treatment groups (Supplemental Figure 8(A)). Together, these data reinforce our findings that acute exposure to amyloid-β(1-40) impairs endothelial cell NMDAR, thus blunting dilation to agonists of this cationic channel.
Figure 6.
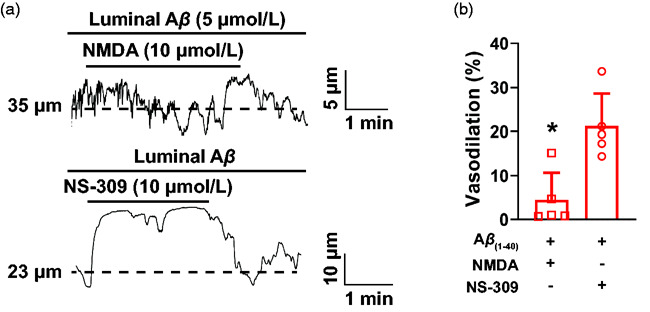
Acute exposure to amyloid-β(1-40) blunts NMDAR-induced PComA dilation. (a) Representative traces of the lumen diameter of an ex vivo pressurized PComA incubated intraluminally with amyloid-β(1-40) (5 µmol/L) and exposed to NMDA (10 µmol/L, upper trace) followed by NS-309 (10 µmol/L, lower trace). (b) Summary bar graph showing that NMDA dilation was blunted by intraluminal incubation with amyloid-β(1-40). Dilation resulting from pharmacological activation of KCa2.3/KCa3.1 with NS-309 was unaffected by amyloid-β(1-40). *p < 0.05, two-tailed Student’s t-test, N = 5 arteries from 3 mice, 2 males and 1 female.
Reduced NMDAR expression in pial arteries of 5x-FAD mice
Next, we tested the hypothesis that vascular NMDAR function is impaired in a murine model of Alzheimer’s disease, the 5x-FAD. We confirmed the presence of amyloid-β plaques around pial arteries/arterioles by immunofluorescence (Figure 7(a)), and observed that ∼45% of pial arteries from 5x-FAD showed perivascular amyloid-β accumulation (Figure 7(a) and (b)). As expected, no amyloid-β accumulated in the perivascular region of pial arteries in wild-type littermates (Figure 7(a) and (b)).
Figure 7.
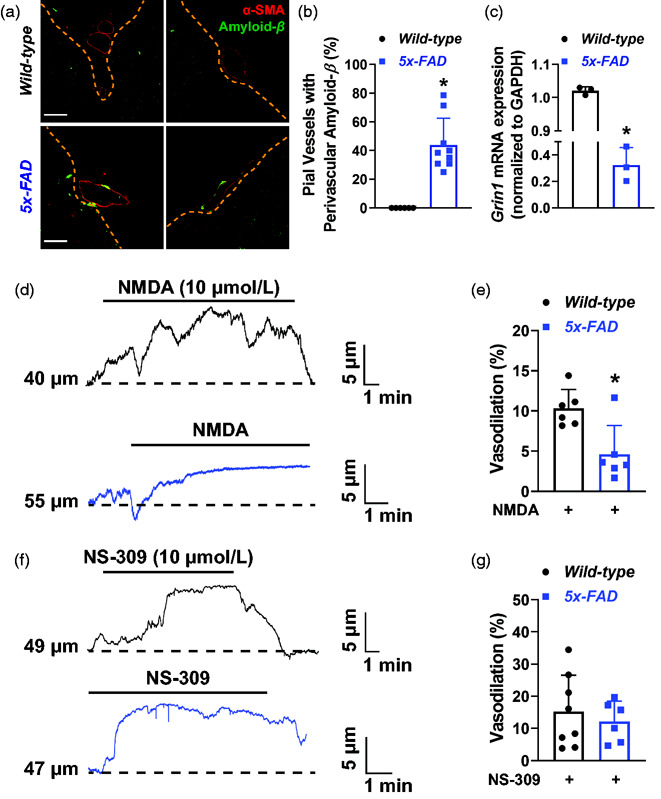
Impaired NMDA-induced dilation of pial arteries from 5x-FAD mice. (a) Representative immunofluorescence labeling of brain slices from wild-type (top) and 5x-FAD mice (bottom) showing accumulation of amyloid-β plaques (green) around pial blood vessels, identified by an α-smooth muscle actin antibody (α-SMA, red). Note the absence of amyloid-β immunoreactivity in vessels from wild-type mice. Bar = 40 µm. (b) Summary graph showing the quantification of pial blood vessels double-positive for α-SMA and amyloid-β. *p < 0.05, two-tailed Student’s t-test. N = 6 slices from 6 wild-type (3 males and 3 females) and 9 (5 males and 4 females) 5x-FAD mice. (c) qPCR data showing that Grin1 mRNA expression is significantly reduced in pial arteries of 5x-FAD mice. *p < 0.05, two-tailed Student’s t-test. A total of 15 wild-type (7 males and 8 females) and 15 5x-FAD (8 males and 7 females) were pooled into 3 distinct qPCR experiments. (d) Representative traces of the lumen diameter of pressurized PComA isolated from 5x-FAD (blue, lower) or wild-type littermates (black, upper) exposed to NMDA (10 µmol/L). (e) Summary bar graphs showing that NMDA dilation was significantly reduced in 5x-FAD. *p < 0.05, two-tailed Student’s t-test. N = 6 arteries from 6 wild-type (4 males and 2 females) and 6 arteries from 5 5x-FAD mice (3 males and 2 females). (f) Representative traces of the lumen diameter of PComA isolated from wild-type (upper trace, black) and 5x-FAD mice (lower trace, blue) showing dilation to the KCa2.3/KCa3.1 activator NS-309 (10 µmol/L). (g) Summary data showing that dilation to NS-309 was not different between 5x-FAD and wild-type littermates. N = 8 preparations from wild-type (4 males and 3 females) and 6 preparations from 5x-FAD (3 males and 2 females).
We also performed quantitative real-time PCR analysis of Grin1 mRNA, which codes for the NMDAR obligatory subunit GluN1. We observed an approximately 70% reduction in Grin1 mRNA levels in pial arteries of 5x-FAD compared to wild-type littermates (Figure 7(c)).
Impaired vasodilation to NMDA in pial arteries from 5x-FAD mice
We then expanded our experiments to study NMDA-dependent dilation in PComA isolated from 5x-FAD and wild-type littermates. Exposure of wild-type PComA to NMDA (10 µmol/L) induced dilation, as observed in our previous experiments in Figure 3. However, PComA from 5x-FAD mice showed a blunted vasodilatory response upon exposure to NMDA (Figure 7(d) and (e)). No significant changes to myogenic tone occurred between treatment groups (Supplemental Figure 8(E)). To ensure that the reduced dilation to NMDA observed in 5x-FAD was not due to impairment in downstream effectors of endothelial cell hyperpolarization, we exposed PComA from 5x-FAD mice to NS-309, and observed a similar vasodilatory response as PComA isolated from wild-type littermates (Figure 7(f) and (g)). Together, our data suggests that amyloid-β impairs endothelial cell NMDAR function, leading to improper Ca2+ handling in cerebral artery endothelial cells, consequently impairing vasodilation.
Discussion
The present study shows that activation of cerebral artery endothelial NMDAR induces transient increases in intracellular [Ca2+], observed as NMDAR sparklets and intracellular Ca2+ signals of different modalities. These NMDAR-dependent Ca2+ signals in endothelial cells are impaired by amyloid-β(1-40), the main form of amyloid-β found in perivascular plaques and cerebral amyloid angiopathy. Consequently, we report that amyloid-β(1-40) acutely disrupts NMDA-induced dilation in cerebral PComA (Supplemental Figure 9(B)). Lastly, we observed that NMDA-induced dilation of PComA is blunted in 5x-FAD mice, a transgenic mouse model of early-onset familial Alzheimer’s disease with perivascular amyloid-β accumulation. Impaired dilation to NMDA was also associated with downregulation of the mRNA coding for the obligatory GluN1 NMDAR subunit. Together, these data suggest that acute and chronic impairments in endothelial cell NMDAR function may underlie, at least in part, the cerebral microvascular dysfunction observed in cerebral amyloid angiopathy and Alzheimer’s disease. 36
Spatial and temporal matching of cerebral perfusion as a function of neuronal metabolism is necessary to maintain optimal nutrient delivery and to prevent cognitive decline. Cerebrovascular dilation involves the coordination of signals originating from different cells within the neurovascular unit, 1 with endothelial cells playing a central role. 37 Endothelial cells generate distinct molecular signals to induce vasodilation, including nitric oxide, 38 prostacyclins, 39 epoxyeicosatrienoic acids 40 and electrotonic hyperpolarization. 41 Nearly all endothelium-dependent vasodilatory pathways occur downstream from increases in intracellular [Ca2+]. 42 As such, Ca2+-permeable ion channels in the plasma membrane play a prominent role in mediating endothelium-dependent dilation, either by providing the signal necessary to activate nearby proteins and enzymes located within signaling microdomains,26,43 or by providing the initial signal to induce a more robust Ca2+ release from intracellular stores.25,44 It is in this paradigm that endothelial cell NMDAR may play an important role, as they allow Ca2+ influx upon opening and induce cerebral artery dilation.
The presence of NMDAR in the cerebral circulation remains a topic of debate. Early studies using ovine brain microcirculatory cells, most likely from capillaries, did not detect the presence of NMDAR after exposing tissue to radioactive D-APV. 45 In addition, subsequent studies suggested that NMDA, when applied through a cranial window in rabbits, does not alter the diameter of pial arterioles or isolated middle cerebral arteries. 46 Other studies showed that glutamate alone failed to dilate parenchymal arterioles from rats, 47 although it is unlikely that NMDAR would have been activated in the absence of a co-activator. Further, studies also suggest that NMDA-induced dilation requires neuronal activation in rabbits 46 and rats. 48 However, recent studies show that NMDAR is present in cerebral endothelial cells from rats and mice. In particular, Lu et al. showed that NMDAR is present in the membrane of mouse endothelial cells using transmission electron microscopy after immunogold labeling. 3 Other studies show that NMDAR activation with endogenous agonists causes dilation of isolated rat pial arteries 10 and mouse parenchymal arterioles in brain slices. 11 Endothelial cell-specific ablation of NMDAR impairs neurovascular coupling, 12 thus providing in vivo functional evidence for the role of NMDAR in dynamic blood flow regulation in the brain. Our experiments provide further confirmation of NMDAR activity in cerebral artery endothelial cells, as we show a significant increase in unitary Ca2+ influx events after exposure to NMDA, as well as dilation of PComA and parenchymal arterioles. Further, we show that these effects are receptor mediated, as blockade of NMDAR with both D-APV and MK-801 significantly inhibited NMDAR sparklets and cerebral vascular dilation.
The intracellular mechanisms underlying NMDAR-induced dilation likely involve increases in cytoplasmic [Ca2+] that activate Ca2+-dependent vasodilatory pathways. Here we show that activation of NMDAR in endothelial cells leads to an increase in optically recorded Ca2+ influx events, hereon called NMDAR sparklets. We observed that these events have low amplitude (1.08 ΔF/F0) and restricted spatial spread (8 µm2), typical of “Ca2+ sparklets”. NMDAR sparklets have a lower amplitude than influx events recorded for transient receptor potential vanilloid 3 (TRPV3 sparklets, 1.20 ΔF/F0) 49 and TRP ankyrin 1 (TRPA1 sparklets, 1.10–1.13 ΔF/F0). 25 , 43 This is consistent with the lower single-channel conductance of NMDAR at physiological conditions (∼40 pS), 4 compared to TRPV3 (∼150–200 pS) 50 and TRPA1 (∼100 pS), 51 which results in lower fractional Ca2+ entry. The events recorded in this study are indeed a result of Ca2+ influx through NMDAR, as they were virtually absent after removal of extracellular Ca2+ and were significantly reduced by D-APV and MK-801.
Due to their lower amplitude and restricted spatial spread, it is unlikely that NMDAR sparklets alone activate intracellular pathways linked to vasodilation, thus an amplification of the signal is warranted. Such is achieved by Ca2+-induced Ca2+-release events from the endoplasmic reticulum, which are higher in amplitude and have larger spatial spread. They are a consequence of opening of inositol 1,4,5-triphosphate (IP3) receptors, 52 which depends on Ca2+ and is modulated by IP3. 53 We observed that NMDA significantly increased Ca2+-induced Ca2+-release events in cerebral artery preparations, suggesting that such pathway exists in endothelial cells. Similar findings were previously reported for TRPV4 in astrocytes 54 and TRPA1 in endothelial cells.25,44 These larger intracellular Ca2+ transients are linked to vasodilation by inducing endothelial nitric oxide production and opening of KCa2.3 and KCa3.1 channels. Although previous studies highlight a role for nitric oxide in dilation induced by NMDAR,10,55 we observed that in mouse PComA, inhibition of nitric oxide and prostacyclin production did not significantly affect NMDA-induced dilation. However, we observed that inhibition of KCa2.3 and KCa3.1 channels significantly blunted NMDAR-dependent dilation. The reason underlying these differences remain unclear, although it is possible that nitric oxide generation is not as prominent for PComA dilation as endothelium-dependent hyperpolarization pathways, which are normally more prominent in the cerebral microcirculation.56,57
We also observed that pre-incubation of en face cerebral arteries with amyloid-β(1-40) reduces NMDAR sparklets, NMDA-elicited Ca2+ transients, and vasodilation. Amyloid-β(1-40) causes impairment in endothelial function, although knowledge of specific mechanisms and molecular targets remains incomplete. Initial studies showed that incubation of aortic rings with amyloid-β acutely disrupts dilation to acetylcholine via a mechanism that may involve oxidative stress. 17 Another study showed that amyloid-β(1-40), but not amyloid-β(1-42), is responsible for impaired endothelial function, observed as reduced dilation to acetylcholine, 28 a pathway linked to increases in intracellular Ca2+ transients in endothelial cells. 58 Recent studies have focused on the effects of cerebral vascular amyloidosis on ion channel function. Zhang et al. showed impaired TRPV4-dependent vasodilatory responses in cerebral arteries of a mouse model of Alzheimer’s disease, 59 without changes in KCa2.3 and KCa3.1 activity. These data suggest a possible reduction in TRPV4 sparklets or TRPV4-initiated Ca2+-induced Ca2+-release, rather than a direct effect on endothelial K+ channels. This possibility is in agreement with our findings, as we show that NMDAR sparklets, NMDA-induced intracellular Ca2+ transients and PComA dilation are reduced after incubation with amyloid-β(1-40) without direct effects on KCa2.3 and KCa3.1 activity. In addition to acute effects on NMDAR function, we observed a downregulation in Grin1 mRNA in 5x-FAD pial arteries, linked to reduced vasodilation after exposure to NMDA, without affecting dilation via KCa2.3 and KCa3.1 activity. Together, these data support deleterious effects of amyloid-β on NMDAR itself, rather than impairments in downstream vasodilatory mechanisms. Acute dysfunction and long-term downregulation of NMDAR may underlie, at least in part, the neurovascular dysfunction observed in Alzheimer’s disease models.
Limitations of the study
The limitations of our current study should be mentioned. Firstly, we were not able to directly assess if NMDAR sparklets lead to KCa2.3/KCa3.1 activation, as our indirect measurements suggest. Future studies will assess if NMDAR forms signaling complexes with KCa2.3/KCa3.1, similarly to TRPA1. 60 A second limitation is that our Grin1 mRNA expression data were collected from whole arteries homogenates, and not from an enriched endothelial cells fraction, therefore other cell types may account for the reduction in Grin1 mRNA. However, our experiments with endothelium-denuded PComA suggest that endothelial cells NMDAR are the main contributors to vasodilatory signals, thus we are confident in our interpretation. In addition, we did not elucidate the biochemical and/or molecular mechanisms of how amyloid-β(1-40) acutely impairs NMDAR in cerebral artery endothelial cells. However, a previous study showed that amyloid-β induces excessive internalization of NMDAR in cultured hippocampal neurons; 21 therefore, a similar effect could occur in endothelial cells. In addition, amyloid-β is known to induce oxidative stress in endothelial cells, 61 and oxidation of cysteine residues within the NMDAR inhibits its opening. 62 It is also possible that the reduction in intracellular Ca2+ transients is caused by alterations in intracellular Ca2+ homeostasis. IP3 receptors and the sarcoplasmic/endoplasmic reticulum ATPase, a transporter involved in maintenance of intracellular Ca2+ stores, are known to be impaired by oxidative stress.63,64 Our data suggests that amyloid-β(1-40) does impair the NMDAR, although if this loss of function is a consequence of internalization or post-translational modifications induced by oxidation remains unknown.
A final limitation is that the isolated vascular preparations used in our study may not fully recapitulate the in vivo condition. In the brain, cerebral arterioles and endothelial cells are in the center of a multicellular environment that includes astrocytes, microglia, and pericytes (the neurovascular unit), which also forms the blood-brain-barrier. Blood-brain-barrier dysfunction is a well-known feature of neurovascular disorders and seems to occur early in Alzheimer’s disease. This contributes to neurodegeneration (for reviews, see65,66) via a mechanism possibly dependent on amyloid-β-induced loss of astrocytes 67 or pericytes68,69 and microglia-dependent neuroinflammation. 70 Perivascular amyloid-β is associated with a reduction in GFAP-positive astrocytic end-feet, 71 potentially reducing sites of neurovascular coupling and impairing functional hyperemia. Additionally, pericytes are highly susceptible to amyloid-β toxicity, and loss of pericyte coverage is observed in Alzheimer’s disease patients, 72 likely increasing blood-brain-barrier permeability. Lastly, microglia exposed to amyloid-β shift to a pro-inflammatory phenotype, which can accelerate neuronal damage and progression of dementia. 73 Although our study focused on the effects of amyloid-β on the function of cerebral artery endothelial NMDAR, it is likely that, in vivo, all of these additional mechanisms are occurring simultaneously.
In summary, our data provide a new link for amyloid-β(1-40)-induced endothelial dysfunction in the cerebral circulation. As NMDAR plays a role in neurovascular coupling, its impairment may contribute to the neurovascular dysfunction observed in cerebral amyloid angiopathy and Alzheimer’s disease.
Supplemental Material
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211039592 for Amyloid-β disrupts unitary calcium entry through endothelial NMDA receptors in mouse cerebral arteries by Emily C Peters, Michael T Gee, Lukas N Pawlowski, Allison M Kath, Felipe D Polk, Christopher J Vance, Juliana L Sacoman and Paulo W Pires in Journal of Cerebral Blood Flow & Metabolism
Supplemental material, sj-pdf-2-jcb-10.1177_0271678X211039592 for Amyloid-β disrupts unitary calcium entry through endothelial NMDA receptors in mouse cerebral arteries by Emily C Peters, Michael T Gee, Lukas N Pawlowski, Allison M Kath, Felipe D Polk, Christopher J Vance, Juliana L Sacoman and Paulo W Pires in Journal of Cerebral Blood Flow & Metabolism
Acknowledgements
We would like to thank Dr. Anne McLaren Dorrance for the critical review of this manuscript. We also want to thank Dr. Mark T. Nelson and Dr. Adrian Bonev for providing the SparkAn software.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Heart, Lung and Blood Institute (R00HL140106 to PWP and 5R24HL120847 to M. Kotlikoff for the generation of cdh5:GCaMP8 mice). ECP was a recipient of a fellowship from the Undergraduate Biology Research Program (UBRP) at the University of Arizona.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions: PWP and ECP conceived and designed this study; ECP, MG, LNP, FDP, CMJ, AMK, JLS and PWP performed experiments and analyzed data; ECP wrote the initial draft of the manuscript; PWP and ECP edited the draft and wrote final version of this manuscript. All authors have read the final manuscript and agree to its submission.
Data availability: All data summarized in this manuscript is maintained in the Principal Investigator’s laboratory and is available upon reasonable request.
Supplemental materials: Supplemental material for this article is available online.
ORCID iDs
Emily C Peters https://orcid.org/0000-0001-7672-3510
Felipe D Polk https://orcid.org/0000-0001-7890-4737
Paulo W Pires https://orcid.org/0000-0001-5972-4554
References
- 1.Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 2017; 96: 17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ottolini M, Hong K, Sonkusare SK. Calcium signals that determine vascular resistance. Wiley Interdiscip Rev Syst Biol Med 2019; 11: e1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu L, Hogan-Cann AD, Globa AK, et al. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J Cereb Blood Flow Metab 2019; 39: 481–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stern P, Behe P, Schoepfer R, et al. Single-channel conductances of NMDA receptors expressed from cloned cDNAs: comparison with native receptors. Proc Biol Sci 1992; 250: 271–277. [DOI] [PubMed] [Google Scholar]
- 5.Schneggenburger R. Simultaneous measurement of Ca2+ influx and reversal potentials in recombinant N-methyl-D-aspartate receptor channels. Biophys J 1996; 70: 2165–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2013; 14: 383–400. [DOI] [PubMed] [Google Scholar]
- 7.Hogan-Cann AD, Anderson CM. Physiological roles of Non-Neuronal NMDA receptors. Trends Pharmacol Sci 2016; 37: 750–767. [DOI] [PubMed] [Google Scholar]
- 8.Zhu S, Paoletti P. Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Curr Opin Pharmacol 2015; 20: 14–23. [DOI] [PubMed] [Google Scholar]
- 9.Wyllie DJ, Livesey MR, Hardingham GE. Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 2013; 74: 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeMaistre JL, Sanders SA, Stobart MJ, et al. Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J Cereb Blood Flow Metab 2012; 32: 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stobart JLL, Lu L, Anderson HDI, et al. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 2013; 110: 3149–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogan-Cann AD, Lu P, Anderson CM. Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc Natl Acad Sci U S A 2019; 116: 10229–10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci 2004; 5: 347–360. [DOI] [PubMed] [Google Scholar]
- 14.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke 1987; 18: 311–324. [DOI] [PubMed] [Google Scholar]
- 15.Herzig MC, Winkler DT, Burgermeister P, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 2004; 7: 954–960. [DOI] [PubMed] [Google Scholar]
- 16.Ellis RJ, Olichney JM, Thal LJ, Mirra SS, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, part XV. Neurology 1996; 46: 1592–1596. [DOI] [PubMed] [Google Scholar]
- 17.Thomas T, Thomas G, McLendon C, et al. beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 1996; 380: 168–171. [DOI] [PubMed] [Google Scholar]
- 18.Price JM, Chi X, Hellermann G, et al. Physiological levels of beta-amyloid induce cerebral vessel dysfunction and reduce endothelial nitric oxide production. Neurol Res 2001; 23: 506–512. [DOI] [PubMed] [Google Scholar]
- 19.Knopman DS, Roberts R. Vascular risk factors: imaging and neuropathologic correlates. J Alzheimers Dis 2010; 20: 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith CD, Andersen AH, Kryscio RJ, et al. Altered brain activation in cognitively intact individuals at high risk for Alzheimer's disease. Neurology 1999; 53: 1391–1396. [DOI] [PubMed] [Google Scholar]
- 21.Goto Y, Niidome T, Akaike A, et al. Amyloid beta-peptide preconditioning reduces glutamate-induced neurotoxicity by promoting endocytosis of NMDA receptor. Biochem Biophys Res Commun 2006; 351: 259–265. [DOI] [PubMed] [Google Scholar]
- 22.Percie Du Sert N, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. J Cereb Blood Flow Metab 2020; 40: 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YL, Baker TM, Lee F, et al. Calcium signal profiles in vascular endothelium from Cdh5-GCaMP8 and Cx40-GCaMP2 mice. J Vasc Res 2021; 58: 159–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dinkins MB, Dasgupta S, Wang G, et al. The 5XFAD mouse model of Alzheimer's disease exhibits an age-dependent increase in anti-ceramide IgG and exogenous administration of ceramide further increases anti-Ceramide titers and amyloid plaque burden. J Alzheimers Dis 2015; 46: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pires PW, Earley S. Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. Elife 2018; 7: e35316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonkusare SK, Bonev AD, Ledoux J, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012; 336: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edelstein AD, Tsuchida MA, Amodaj N, et al. Advanced methods of microscope control using muManager software. J Biol Methods 2014; 1: e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niwa K, Younkin L, Ebeling C, et al. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A 2000; 97: 9735–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson M, Ledoux J, Taylor M, et al. Spinning disk confocal microscopy of calcium signalling in blood vessel walls. Microsc Anal (Am Ed) 2010; 24: 5–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Pires PW, Dabertrand F, Earley S. Isolation and cannulation of cerebral parenchymal arterioles. J Vis Exp 2016; (111): 53835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pires PW, Jackson WF, Dorrance AM. Regulation of myogenic tone and structure of parenchymal arterioles by hypertension and the mineralocorticoid receptor. Am J Physiol Heart Circ Physiol 2015; 309: H127–H136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westcott EB, Goodwin EL, Segal SS, et al. Function and expression of ryanodine receptors and inositol 1,4,5-trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J Physiol 2012; 590: 1849–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ralevic V, Kristek F, Hudlicka O, et al. A new protocol for removal of the endothelium from the perfused rat hind-limb preparation. Circ Res 1989; 64: 1190–1196. [DOI] [PubMed] [Google Scholar]
- 34.Pires PW, Ko EA, Pritchard HAT, et al. The angiotensin II receptor type 1b is the primary sensor of intraluminal pressure in cerebral artery smooth muscle cells. J Physiol 2017; 595: 4735–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park L, Wang G, Zhou P, et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc Natl Acad Sci U S A 2011; 108: 5063–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soontornniyomkij V, Choi C, Pomakian J, et al. High-definition characterization of cerebral β-amyloid angiopathy in Alzheimer's disease. Hum Pathol 2010; 41: 1601–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation 2013; 20: 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drouin A, Thorin-Trescases N, Hamel E, et al. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res 2007; 73: 73–81. [DOI] [PubMed] [Google Scholar]
- 39.Lombard JH, Liu Y, Fredricks KT, et al. Electrical and mechanical responses of rat middle cerebral arteries to reduced PO2 and prostacyclin. Am J Physiol 1999; 276: H509–H516. [DOI] [PubMed] [Google Scholar]
- 40.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch 2010; 459: 881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longden TA, Nelson MT. Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 2015; 22: 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev 2015; 95: 645–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan MN, Gonzales AL, Pires PW, et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci Signal 2015; 8: ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian X, Francis M, Solodushko V, Earley S, et al. Recruitment of dynamic endothelial Ca2+ signals by the TRPA1 channel activator AITC in rat cerebral arteries. Microcirculation 2013; 20: 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beart PM, Sheehan KA, Manallack DT. Absence of N-methyl-D-aspartate receptors on ovine cerebral microvessels. J Cereb Blood Flow Metab 1988; 8: 879–882. [DOI] [PubMed] [Google Scholar]
- 46.Faraci FM, Breese KR. Nitric oxide mediates vasodilatation in response to activation of N-methyl-D-aspartate receptors in brain. Circ Res 1993; 72: 476–480. [DOI] [PubMed] [Google Scholar]
- 47.Takayasu M, Dacey RG., Jr. Effects of inhibitory and excitatory amino acid neurotransmitters on isolated cerebral parenchymal arterioles. Brain Res 1989; 482: 393–396. [DOI] [PubMed] [Google Scholar]
- 48.Fergus A, Lee KS. Regulation of cerebral microvessels by glutamatergic mechanisms. Brain Res 1997; 754: 35–45. [DOI] [PubMed] [Google Scholar]
- 49.Pires PW, Sullivan MN, Pritchard HA, et al. Unitary TRPV3 channel Ca2+ influx events elicit endothelium-dependent dilation of cerebral parenchymal arterioles. Am J Physiol Heart Circ Physiol 2015; 309: H2031–H2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung MK, Lee H, Mizuno A, et al. 2-aminoethoxydiphenyl borate activates and sensitizes the heat-gated ion channel TRPV3. J Neurosci 2004; 24: 5177–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagata K, Duggan A, Kumar G, et al. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci 2005; 25: 4052–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foskett JK, White C, Cheung KH, et al. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 2007; 87: 593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor CW, Tovey SC. IP(3) receptors: toward understanding their activation. Cold Spring Harb Perspect Biol 2010; 2: a004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dunn KM, Hill-Eubanks DC, Liedtke WB, et al. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A 2013; 110: 6157–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park L, Zhou J, Koizumi K, et al. tPA deficiency underlies neurovascular coupling dysfunction by amyloid-beta. J Neurosci 2020; 40: 8160–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.You J, Johnson TD, Marrelli SP, et al. Functional heterogeneity of endothelial P2 purinoceptors in the cerebrovascular tree of the rat. Am J Physiol 1999; 277: H893–H900. [DOI] [PubMed] [Google Scholar]
- 57.Hannah RM, Dunn KM, Bonev AD, et al. Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab 2011; 31: 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taylor MS, Francis M. Decoding dynamic Ca(2+) signaling in the vascular endothelium. Front Physiol 2014; 5: 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang L, Papadopoulos P, Hamel E. Endothelial TRPV4 channels mediate dilation of cerebral arteries: impairment and recovery in cerebrovascular pathologies related to Alzheimer's disease. Br J Pharmacol 2013; 170: 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Earley S, Gonzales AL, Crnich R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-activated K+ channels. Circ Res 2009; 104: 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carrano A, Hoozemans JJ, van der Vies SM, et al. Amyloid beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal 2011; 15: 1167–1178. [DOI] [PubMed] [Google Scholar]
- 62.Lipton SA, Choi YB, Takahashi H, et al. Cysteine regulation of protein function – as exemplified by NMDA-receptor modulation. Trends Neurosci 2002; 25: 474–480. [DOI] [PubMed] [Google Scholar]
- 63.Bansaghi S, Golenar T, Madesh M, et al. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem 2014; 289: 8170–8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dremina ES, Sharov VS, Davies MJ, et al. Oxidation and inactivation of SERCA by selective reaction of cysteine residues with amino acid peroxides. Chem Res Toxicol 2007; 20: 1462–1469. [DOI] [PubMed] [Google Scholar]
- 65.Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 2018; 14: 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab 2013; 33: 1500–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blochet C, Buscemi L, Clement T, et al. Involvement of caveolin-1 in neurovascular unit remodeling after stroke: effects on neovascularization and astrogliosis. J Cereb Blood Flow Metab 2020; 40: 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen J, Xu G, Zhu R, et al. PDGFR-beta restores blood-brain barrier functions in a mouse model of focal cerebral ischemia. J Cereb Blood Flow Metab 2019; 39: 1501–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng Z, Chopp M, Chen J. Multifaceted roles of pericytes in central nervous system homeostasis and disease. J Cereb Blood Flow Metab 2020; 40: 1381–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ronaldson PT, Davis TP. Regulation of blood-brain barrier integrity by microglia in health and disease: a therapeutic opportunity. J Cereb Blood Flow Metab 2020; 40: S6–S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilcock DM, Vitek MP, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer's disease. Neuroscience 2009; 159: 1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sengillo JD, Winkler EA, Walker CT, et al. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease. Brain Pathol 2013; 23: 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maezawa I, Zimin PI, Wulff H, et al. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem 2011; 286: 3693–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211039592 for Amyloid-β disrupts unitary calcium entry through endothelial NMDA receptors in mouse cerebral arteries by Emily C Peters, Michael T Gee, Lukas N Pawlowski, Allison M Kath, Felipe D Polk, Christopher J Vance, Juliana L Sacoman and Paulo W Pires in Journal of Cerebral Blood Flow & Metabolism
Supplemental material, sj-pdf-2-jcb-10.1177_0271678X211039592 for Amyloid-β disrupts unitary calcium entry through endothelial NMDA receptors in mouse cerebral arteries by Emily C Peters, Michael T Gee, Lukas N Pawlowski, Allison M Kath, Felipe D Polk, Christopher J Vance, Juliana L Sacoman and Paulo W Pires in Journal of Cerebral Blood Flow & Metabolism