

ABSTRACT
Background
Rapid advances in neuroimaging technologies in the exploration of the living human brain also apply to movement disorders. However, the accurate diagnosis of Parkinson's disease (PD) and atypical parkinsonian disorders (APDs) still remains a challenge in daily practice.
Methods
We review the literature and our own experience as the Movement Disorder Society–Neuroimaging Study Group in Movement Disorders with the aim of providing a practical approach to the use of imaging technologies in the clinical setting.
Results
The enormous amount of articles published so far and our increasing recognition of imaging technologies contrast with a lack of imaging protocols and updated algorithms for differential diagnosis. The distinctive pathological involvement in different brain structures and the correlation with imaging findings obtained with magnetic resonance, positron emission tomography, or single‐photon emission computed tomography illustrate what qualitative and quantitative measures may be useful in the clinical setting.
Conclusion
We delineate a pragmatic approach to discuss imaging technologies, updated imaging algorithms, and their implications for differential diagnoses in PD and APDs.
Keywords: Parkinson's disease, multiple system atrophy, progressive supranuclear palsy, atypical parkinsonisms, imaging, MRI, PET
Innovations in neuroimaging have enabled an accelerated understanding of movement disorders. They have been instrumental in reflecting the pathological changes that occur in Parkinson's disease (PD) and atypical parkinsonian disorders (APDs). 1
These advancements have generated great expectations; however, a consensus approach on how to use them in everyday practice is lacking. The increasing recognition of imaging biomarkers contrast with PD diagnostic accuracy, which remains suboptimal, particularly among nonexperts compared with diagnosis performed by movement disorders experts (73.8% vs. 83.9%) 2 and in early disease (26% during the first year of symptoms onset). 3
The clinical application of advanced imaging technologies is still limited, and their standardization will take time given the multidimensionality of these diseases. However, newer imaging technologies are emerging, and with them emerges the need to determine which method, singly or in combination, can be used in daily clinical practice.
Needless to say, this translational value is urgently needed because at the moment the only approved approach is the use of a marker of the dopamine transporter with 123I‐fluoropropyl 2‐carbomethoxy‐3‐(4‐iodophenyl) tropane (CIT) with single‐photon emission computed tomography (DAT‐SPECT) in the differential diagnosis of degenerative from nondegenerative parkinsonism, although other markers for presynaptical dopaminergic function are used in clinical practice.
Here we intend to provide a clinical and pragmatic approach to the use of imaging techniques to improve the diagnosis of PD and APDs.
Imaging Techniques Available in Clinical Practice in Parkinsonisms
Parkinson's Disease
One of the key pathological structures affected by the neurodegenerative process in PD is the substantia nigra (SN). However, routine magnetic resonance imaging (MRI) shows a relative lack of structural pathology in T1 and T2 1.5 T MRI at the brainstem compared with other neurodegenerative diseases such as Alzheimer's disease (AD), where atrophy can be clearly evident at certain stages of disease. 4
Currently, PD diagnosis is made clinically, and the new Movement Disorder Society (MDS) criteria, which maintain the centrality of the motor syndrome, are designed to be applicable without the need for ancillary diagnostic testing. 5 However, in the diagnostic work‐up of PD, routine 1.5 T MRI is indicated with the main purpose of ruling out an underlying secondary cause for the disorder rather than making a specific diagnosis (Fig. 1). 6
FIG 1.
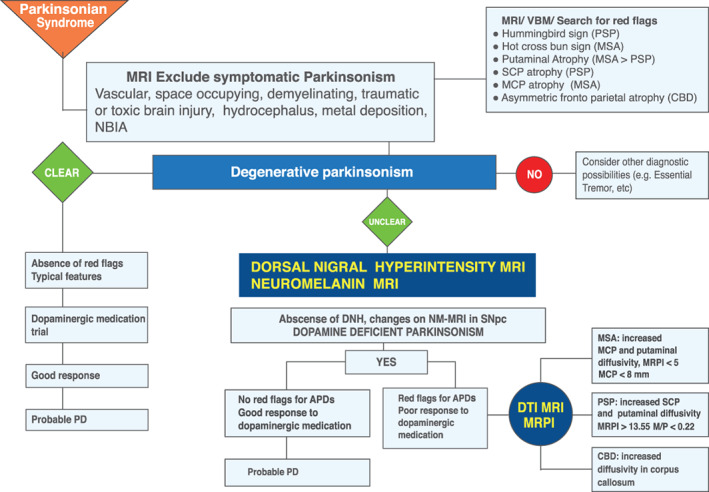
MR imaging diagnosis algorithm. APDs, atypical parkinsonian disorders; CBD, corticobasal degeneration; DNH, dorsal nigral hyperintensity; MCP, middle cerebellar peduncle; MR, magnetic resonance; MRI, magnetic resonance imaging; MRPI, Magnetic Resonance Parkinsonism Index; MSA, multiple system atrophy; NM, neuromelanin; PD, Parkinson's disease; PSP, progressive supranuclear palsy; SCP, superior cerebellar peduncle; NBIA, Neurodegeneration with Brain Iron Accumulation.
If there are no contraindications for undergoing a brain MRI, it is recommended to perform 1 at least in the course of the disease, and the protocol should include T1‐weighted, T2 flair, diffusion‐weighted imaging (DWI) and susceptibility weighted imaging (SWI), both in the sagittal and transversal planes. 7
Although MRI is not approved as a biomarker for the differential diagnosis of parkinsonian syndromes, 6 in clinical practice and in certain contexts such as unclear cases or cases without good responses to dopaminergic treatment, MRI can be considered for this purpose combined with clinical diagnostic criteria.
If distinctive signs are present, 1.5 T MRI can provide clues to the differential diagnosis of PD and APDs, showing atrophy and signal changes of the putamen and T2‐hyperintensity of the pons (the “hot cross bun” sign) and middle cerebellar peduncles (MCP) in multiple system atrophy (MSA), midbrain atrophy relative to the pons (the “hummingbird” sign) in progressive supranuclear palsy (PSP), or asymmetric dorsal frontal or parietal atrophy in corticobasal degeneration (CBD). The limitation here is sensitivity because approximately half of the patients with APDs will not present these signs, especially early in the disease. 8
Brain morphometry does not have a practical impact in the daily diagnostic work‐up of PD and is probably used more in research or in clinical trials. Morphometric studies allow the visualization of brain volume, shape, and surface as well as the segmentation of the basal ganglia (BG). In PD, cortical thinning was observed in the orbitofrontal, ventrolateral prefrontal, and occipitoparietal regions as well as a small decrease in striatal volume in some studies. 9
As knowledge advances, more sophisticated imaging will become available, as is the case in high‐field and ultra–high field MRI, and in the future, methods will become available that integrate a single hybrid scanner MRI and positron emission tomography (PET) or automated imaging analysis. 1
High‐Field MRI
Dorsal Nigral Hyperintensity
Currently, the advent of higher field MRI reflects SN neurodegenerative changes. The SN pars compacta (SNpc) is located dorsally and contains dopaminergic neurons distributed in calbindin‐rich zones or a matrix and also in 5 calbindin‐poor zones, known as nigrosomes. 10 Using high‐resolution SWI, nigrosome‐1 can be seen as a hyperintense ovoid area, called the dorsal nigral hyperintensity (DNH) sign, which has the appearance of a “swallow tail” (Fig. 2). 11
FIG 2.

MR imaging in PD and PSP. DNH, dorsal nigral hyperintensity; HC, healthy control; MCP, middle cerebellar peduncle; MR, magnetic resonance; MRI, magnetic resonance imaging; MRPI, Magnetic Resonance Parkinsonism Index; NM, neuromelanin; PD, Parkinson's disease; PSP, progressive supranuclear palsy; SN, substantia nigra; SNc, substantia nigra pars compacta; SCP, superior cerebellar peduncle; SWI, susceptibility weighted imaging; AC‐PC, anterior commissure ‐ posterior commissure; MSA‐P, Multiple system atrophy‐ parkinsonian type; M/P, midbrain‐to‐pons ratio.
Several studies using 3 T MRI with an optimized sequence have shown the absence of the DNH signal in most patients with PD but not in healthy controls (HCs), revealing 100% sensitivity and 95% specificity in distinguishing PD from HCs, 12 although it is unable to differentiate PD from APDs because the DNH signal loss is also seen in patients with APDs. A recent meta‐analysis suggested that the visual assessment of DNH could provide excellent differential diagnostic accuracy for PD compared with HCs. 13
Although there is no definitive consensus as to the use of DNH imaging in clinical practice, and it is not approved for the diagnosis or differential diagnosis of PD, the wide availability of 3 T scanners and the use of 3 T MRI in the clinic has led to an increased use of this technique. In this regard, DNH imaging may be considered in the differential diagnosis of degenerative from nondegenerative parkinsonisms or from essential tremor (ET), and in this context its use may be comparable to DAT‐SPECT (Figs. 2 and 5).
FIG 5.
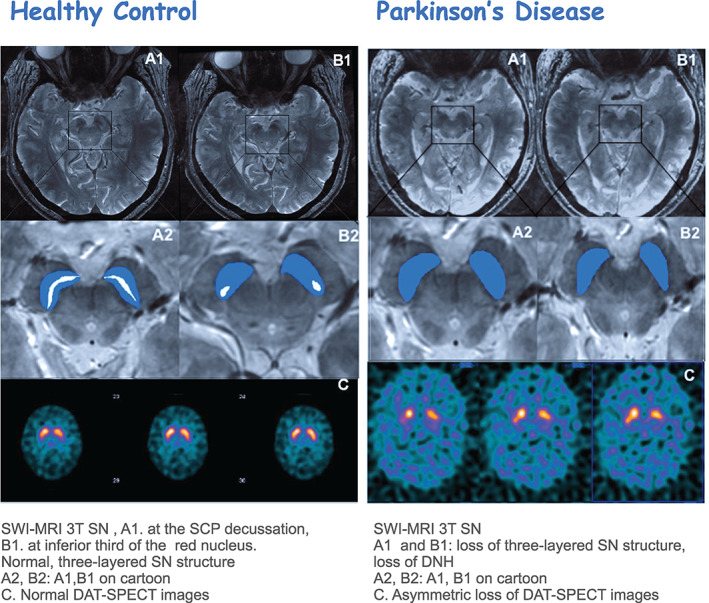
Multimodal imaging SWI MRI 3T‐ DAT SPECT. DAT‐SPECT, dopamine transporter single‐photon emission computed tomography; DNH, dorsal nigral hyperintensity; PET, positron emission tomography; SCP, superior cerebellar peduncle; SN, substantia nigra; SWI, susceptibility weighted imaging.
However, more studies are required to demonstrate if this technique can be equivalent to DAT‐SPECT. 14 The lack of standardization of imaging protocols including spatial resolution and imaging planes may limit its use 15 as well as the relative low sensitivity of MRI compared with nuclear imaging techniques such as PET or SPECT, although 3 T MRI avoids the risks associated with ionizing radiation. 16
The correlation between DNH abnormality and nigrostriatal dopaminergic dysfunction was analyzed in a few studies, as in a study of PD carriers of the G2019S, Leucine‐rich repeat kinase 2 (LRRK2) mutation, in whom the loss of DNH predicted the ipsilateral striatal DAT signal abnormality with a sensitivity of 87.5% and a specificity of 83.6%. 17
The ultra–high field MRI (7 T and higher) in brain imaging provides improvement in contrast and spatial resolution, and it will probably find its way to be integrated into clinical practice. However, at this moment, it still remains a research tool because of the technical and safety aspects that need further development. 16
Iron Imaging
The iron content in the SN increases in PD by about 30% in histological studies compared with HCs. 18 Although patients with APDs have also increased iron content in the SN, they present more prominent subcortical iron deposition in the striatum than in PD or HCs. 19
Iron‐sensitive MRI has been applied to identify iron‐related signal changes for the diagnosis and differential diagnosis of PD in research studies.
In clinical practice, the use of iron imaging is limited to the use of SWI, which is purely a qualitative method, for visual inspection and detection of paramagnetic iron as exemplified by a decreased signal in the putamen in patients with APDs (Fig. 3 )). 12
FIG 3.
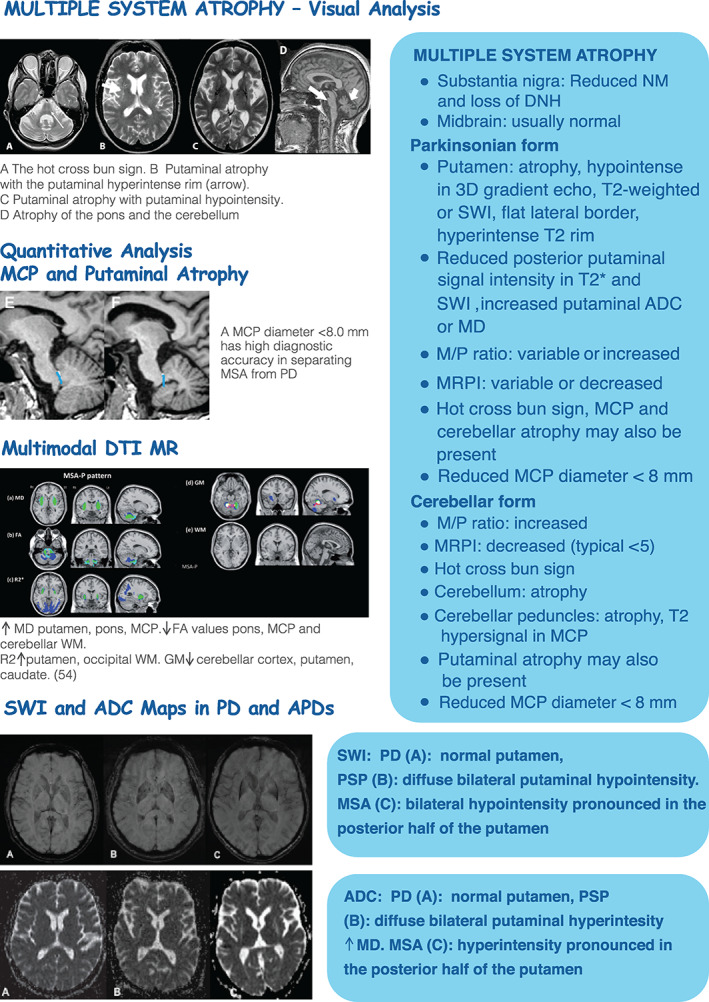
MR imaging in MSA and APDs. ADC, apparent diffusion coefficient; APDs, atypical parkinsonian disorders; DNH, dorsal nigral hyperintensity; FA, fractional anisotropy; HC, healthy control; MCP, middle cerebellar peduncle; MD, mean diffusivity; MR, magnetic resonance; MRI, magnetic resonance imaging; MRPI, Magnetic Resonance Parkinsonism Index; MSA, multiple system atrophy; NM, neuromelanin; PD, Parkinson's disease; PSP, progressive supranuclear palsy; SWI, susceptibility weighted imaging; VM, white matter; GM, gray matter; M/P, midbrain‐to‐pons ratio.
Results on subcortical topographical iron deposition between patients with MSA and PSP vary among studies; however, they are more prevalent and severe in PSP compared with MSA‐P, except in the putamen that contains more iron in MSA‐P, presenting as hypointense signal changes with atrophy and flattening of lateral border of the posterior putamen, which together are quite specific for Multiple system atrophy‐ parkinsonian type (MSA‐P). 20 In PD, the increased iron content in nigrosome‐1 produces the loss of the DNH signal that can be observed in 3 T MRI.
Atypical Parkinsonisms
Progressive Supranuclear Palsy
Midbrain and Superior Cerebellar Peduncle Atrophy
PSP is an APDs challenging to distinguish from PD. 21 Traditionally the clinical diagnostic criteria of PSP rely on the demonstration of a vertical supranuclear gaze palsy plus postural instability and falls within the first year of symptom onset. However, considering the phenotypic heterogeneity of PSP, the new MDS diagnostic criteria identify 4 functional domains (ocular motor dysfunction, postural instability, akinesia, and cognitive dysfunction) as clinical predictors of PSP. Within each of them, 3 clinical features are proposed defining the diagnostic criteria, stratified by 3 degrees of diagnostic certainty. 22
Brain imaging is relevant to demonstrate predominant midbrain atrophy and to rule out alternative diagnosis. In patients with PSP with vertical supranuclear gaze palsy, midbrain atrophy is observed as the “hummingbird” sign and the “morning glory” sign (Fig. 2). Using voxel‐based morphometry, patients with PSP also show atrophy in the thalamus (TH) and insula. 8
Recently, 2 sets of measures have been found to be useful to differentiate PSP from PD and MSA: the midbrain‐to‐pons ratio with a cut‐off value <0.21 and the MR Parkinsonism Index (MRPI) calculated by pons area/midbrain area × MCP diameter/superior cerebellar peduncle (SCP) diameter, with a cut‐off value ≥12.9 versus MSA‐P, and 13.6 versus PD and HCs, which is significantly higher in PSP compared with HC.
A recent version, the MRPI 2.0 includes the measurement of the third ventricle width and has a cut‐off value ≥2.91. The MRPI differentiates Progressive Supranuclear Palsy—Parkinsonism Predominant (PSP‐P) from PD with a sensitivity and a specificity of 73.5% and 98.1%, respectively, whereas the MRPI 2.0 shows a higher sensitivity (100%) and similar specificity (94.3%) in differentiating these 2 groups. 23
This index may well become extremely useful in clinical practice, as a recent report from the MDS‐PSP Study Group indicated that the MRPI and the midbrain‐to‐pons ratio were the most reliable biomarkers for the diagnosis of PSP‐RS, supporting a clinical diagnosis at the individual level in both the early and late stages of the disease. 22
In clinical practice, this pattern of brain atrophy in PSP is also supported by a recently published meta‐analysis, which found that the presence of reduced midbrain and SCP diameters were useful to separate PSP from PD. 24
Multiple System Atrophy
Putaminal and MCP Atrophy
MSA is an adult‐onset, sporadic, progressive neurodegenerative disease characterized by variable severity of parkinsonian features, cerebellar ataxia, autonomic failure, urogenital dysfunction, and corticospinal signs. 25
The underlying neuropathological changes that occur in MSA can be visualized using regional volume changes that reflect significant brain atrophy in the putamen, MCP, cerebellum, pons, and medulla oblongata. 24 , 26 In daily practice and integrated with clinical criteria, structural imaging can assist in the diagnosis and differential diagnosis of MSA, as exemplified by the demonstration of putaminal, pontine, and MCP atrophy in MRI in both Multiple system atrophy ‐ cerebellar subtype (MSA‐C) and MSA‐P. The signal increases on T2‐weighted sequences, which include the “hot cross bun” sign, the “MCP” sign (hyperintensity in the MCP), and the “putaminal rim” sign (hyperintense signal in the dorsolateral margin of the putamen), have high positive predictive values for the diagnosis of MSA. However, it has to be taken into account that the signal abnormalities are more intense at 3 T than at 1.5 T, which could increase false positives for MSA, and the “hot cross bun sign” can occasionally be found in nondegenerative parkinsonism and in spinocerebellar ataxias (Fig 3). 27
Conventional MRI demonstrated high specificity for distinguishing MSA from PD and HCs; however, its sensitivity can be suboptimal in the early stages of the disease, where patients with MSA‐P may not present putaminal or infratentorial changes at 1.5 T, 8 although in up to 30% of patients with MSA, putaminal MRI abnormalities may precede clinical diagnostic criteria for about 1 year and more. 28
Other quantitative measures available in clinical practice include brainstem area and cerebellar peduncle widths that can be manually obtained. An MCP diameter <8.0 mm has good diagnostic accuracy in separating MSA from PD 29 but seems to also discriminate MSA from PSP with fairly acceptable diagnostic accuracy (Figs.3) . 24
Diffusion Magnetic Resonance Imaging (dMRI)
Although DWI estimates water diffusion through the application of magnetic field gradient pulses, diffusion tensor imaging (DTI) requires the application of strong diffusion gradients to quantify the degree of fractional anisotropy (FA) and the global movement of the water molecules (apparent diffusion coefficient [ADC] and mean diffusivity [MD]), which are useful for assessing white matter integrity and tractography. 30
In clinical practice, DWI can be used to increase the sensitivity of MRI as a diagnostic tool to detect changes in brain tissue integrity, which produce a decrease in the number of barriers that restrict the movement of water, causing the ADC to increase in brain areas where neurodegeneration occurs. 31 Nonetheless, it has some limitations because it is a quantitative method that requires experience and a normative database for its interpretation.
In parkinsonisms, DWI is useful for the discrimination of MSA from PD and HCs even in the early stages because the increase in putaminal diffusivity is directly visible in the ADC or MD maps, 32 although PD with longer disease duration and concomitant white matter changes, as well as PSP, may also present increased putaminal diffusivity (Fig. 3), hence additional measures such as volume changes or increased diffusivity within the posterior fossa need to be introduced to reliably separate MSA from PSP. 33 In addition, abnormal diffusion metrics in the MCP can be found in MSA and in the SCP in PSP. 34
Molecular Imaging Studies—PET and SPECT Studies
Presynaptic Dopaminergic Imaging
Molecular imaging gives us the opportunity to assess neurotransmitter function or metabolic activity in vivo, or neuropathology as in the case of abnormal protein deposition. Presynaptic dopaminergic imaging allows a better understanding of disease pathophysiology and can sometimes aid in the process of differential diagnosis.
From a clinical perspective, presynaptic dopaminergic PET and SPECT tracers are mainly used to differentiate degenerative parkinsonism from nondegenerative conditions at the diagnostic phase. For the differentiation of PD from other forms of parkinsonism, the combination of a presynaptic dopamine, dopaminergic (DA) tracer with Fluorodeoxyglucose (FDG)‐PET (studying glucose metabolism) can shed light into differential diagnoses (Figs 4, 5 and 6) 35 , 36
FIG 4.
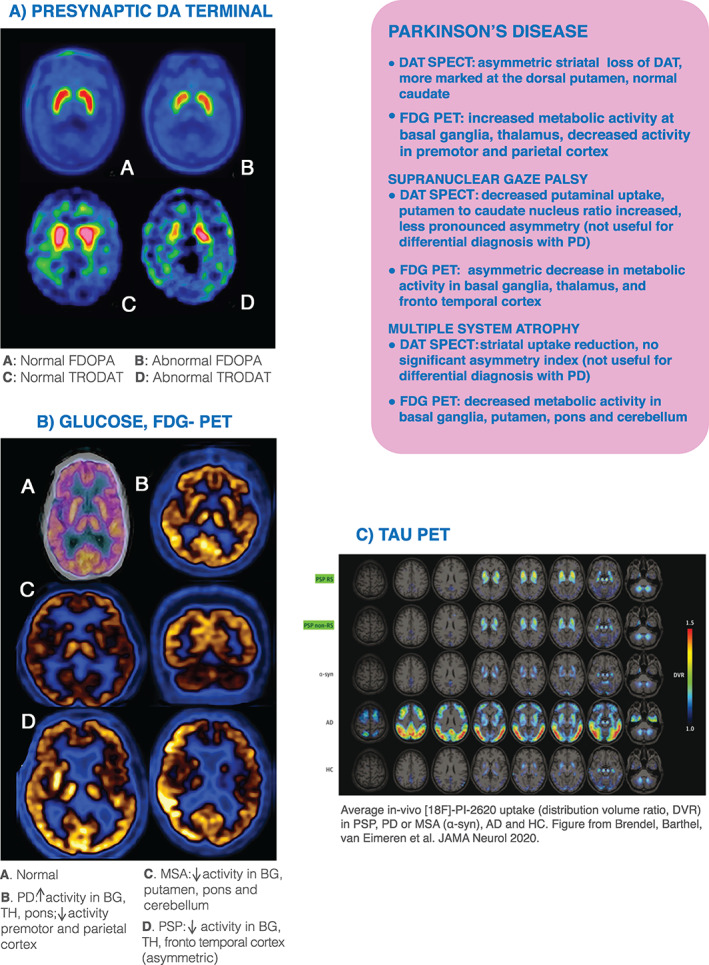
PET imaging of DA, Glucose and Tau protein aggregation. AD, Alzheimer's disease; APDs, atypical parkinsonian disorders; BG, basal ganglia; DAT‐SPECT, dopamine transporter single‐photon emission computed tomography; FDOPA, [18F]fluoro‐L‐dopa uptake; HC, healthy control; MR, magnetic resonance; MSA, multiple system atrophy; PD, Parkinson's disease; PET, positron emission tomography; PSP, progressive supranuclear palsy; TH, thalamus; FDG:18 Fluorodeoxyglucose; DA, dopaminergic; RS, Richardson´s Syndrome; TRODAT, technetium‐labeled dopamine transporter ligand. Fig 4C is reprinted from Brendel et al. 71
FIG 6.
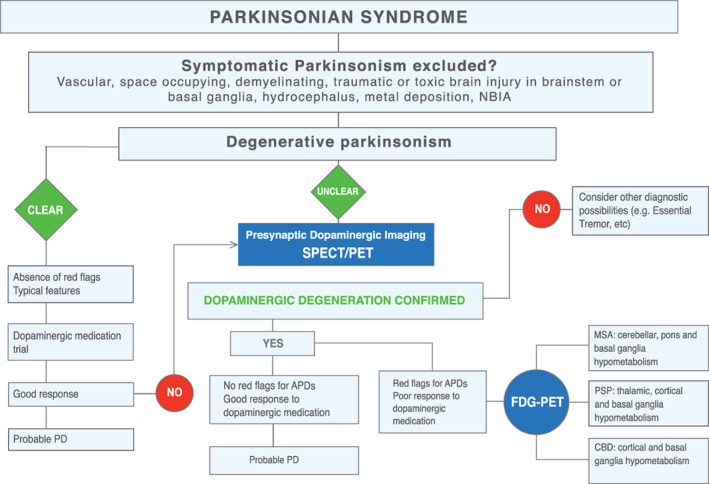
PET imaging diagnosis algorithm. APDs, atypical parkinsonian disorders; CBD, corticobasal degeneration; MSA, multiple system atrophy; PD, Parkinson's disease; PET, positron emission tomography; PSP, progressive supranuclear palsy; SPECT, single‐photon emission computed tomography; NBIA: Neurodegeneration with Brain Iron Accumulation.
Use of PET and SPECT Tracers
Using PET, DA synthesis can be measured through [18F]fluoro‐L‐dopa uptake and decarboxylation into fluorodopamine by L‐aromatic amino acid decarboxylase. 37 DA storage into synaptic vesicles can be assessed with [11C] or [18F]dihydrotetrabenazine, having the advantage of being less prone to compensatory changes. 38 , 39
Several tracers have been used to study DAT availability with PET, such as [11C]CFT: 2‐beta‐carbomethoxy‐3‐beta‐(4‐fluorophenyl)tropane [11C]CFT, [11C]RTI‐32: 11C carboxymethoxy‐3B‐aryltropan [11C]RTI‐32, [11C]methylphenidate, or [18F]FP‐CIT.
With SPECT (Fig. 4 and5), presynaptic DAT function has been mainly investigated with the 123I‐linked tropane derivatives FP‐CIT (ioflupane) and beta‐CIT or Technetium‐99m ‐labeled dopamine transporter ligand ([99mTc]TRODAT‐1).
The mechanism of the decreased striatal DAT binding remains unclear as the binding deficit does not seem to associate well with axonal or somal loss of nigrostriatal neurons in PD. 40 , 41
DAT‐SPECT imaging has been approved by both the US Food and Drug Administration (FDA) and the European Medicines Agency for the differentiation of parkinsonism from ET, and the normal functional neuroimaging of the presynaptic dopaminergic imaging has been included as an absolute exclusion criterion in the last version of the MDS Clinical Diagnostic Criteria for PD. 5
Glucose Metabolism
The cerebral glucose metabolism is associated with the integrity and synaptic activity of neurons (Fig. 4). This condition makes [18F]FDG‐PET studies especially relevant in neurodegenerative diseases because they can identify “patterns of brain hypometabolism.” 42 They offer the clinicians the advantage of being at hand in the majority of clinical centers and a strong “imaging–clinical” correlation where the reciprocity with the clinical symptoms is more significant than with the underlying pathology, as demonstrated by Granert et al, who built a topological map to localize a single subject's disease status according to “PD‐typical” or “AD‐typical,” observing a strong correlation between the cognitive and motor patterns. 43
According to studies with group comparisons, PD is associated with a pattern of increased activity in the BG, TH, and pons (and often cerebellum) combined with reduced activity in the premotor and parietal cortex. However, on an individual basis and at first glance/visual analysis, [18F]FDG‐PET may also not show major abnormalities in PD. 44 , 45 On the other hand, APDs present a more consistent pattern, even on an individual basis. MSA is associated with impaired glucose metabolism in the BG, putamen, pons, and cerebellum and PSP in the BG and thalamic and cortical areas (Figs. 4 ,and 6), 46 therefore the clinical utility of [18F]FDG‐PET as a means of discriminating PD from APDs has been supported by the latest criteria for PSP and MSA. 22 , 25
As opposed to the advantage of [18F]FDG PET studies that reliably differentiate PD from APDs, presynaptic dopaminergic terminal imaging is not sufficient alone for differential diagnosis because the pattern of reduced tracer binding is similar in these disorders. Likewise, postsynaptic dopaminergic terminal imaging has lower diagnostic accuracy when compared with [18F]FDG‐PET studies in the prediction of a clinical diagnosis of PD or APDs at 2 years of follow‐up. 47 , 48
Imaging Techniques Available in Research in Parkinsonisms
Neuromelanin Imaging
Neuromelanin (NM) is a dark granular intracellular pigment found in catecholaminergic neurons of the SNc and locus coeruleus (LC), which provides protection for endogenous dopamine, which is highly susceptible to oxidative stress. 49 NM‐containing regions present high signal intensity on T1‐weighted fast spin‐echo images at 3 T.
In the past decade, several studies have used NM‐sensitive MRI to evaluate patients with PD, showing high accuracy in the differential diagnosis from HCs. 11
In a study of patients with early PD, the lateral part of the SN showed the most significant signal attenuation. Remarkably, the change in LC signal was similar to that of the SN, and the signal attenuation in LC presented higher sensitivity and specificity than that of the SN in differentiating PD from HCs (82% sensitivity and 90% specificity for LC and 73% sensitivity and 87% specificity for SN), suggesting early neuronal depletion in this structure as well. 50
The role of NM imaging in distinguishing PD from APDs is less clear. Some studies suggested lower reductions in NM signal in the SN in PSP versus MSA and PD, 51 whereas others have found that the NM‐sensitive substantia nigra pars compacta (SNpc) area and midbrain volume were significantly smaller in patients with PSP compared with patients with PD and HCs. 52
The investigations of the relationship of NM‐MRI and DAT‐SPECT have found a positive correlation between the loss of NM signal and the corresponding striatal 123I‐FP‐CIT binding of striatal dopamine presynaptic terminals. 53
This is a promising technique to improve PD diagnosis, and although there are some studies that show that the visual analysis of NM imaging offers high diagnostic values and comparable results with quantitative analyses, 50 there are some steps that still need to be taken in terms of imaging protocols and analysis before this technique can be used in clinical practice.
Iron Imaging
Quantitative measurements of iron deposition are mostly used in research protocols using iron‐sensitive techniques such as relaxation rate R2* and quantitative susceptibility mapping (QSM). Compared with relaxation rates, SWI and QSM generated through magnitude and phase images from gradient‐echo MRI sequences reflect susceptibility of local tissues being less influenced by changes in water content, local water diffusion rates and macroscopic magnetic field inhomogeneities. Therefore, these techniques are potentially superior methods of measuring iron in vivo, and QSM is a promising biomarker of disease‐related iron accumulation in PD, more reproducible than R2* and more suited to multicenter studies. 54
In addition, QSM allows to quantify the magnetic susceptibility value of brain tissue from gradient‐echo MRI and provide excellent contrast between gray matter nuclei and surrounding tissues and is more precise than SWI. 20
According to a recent meta‐analysis of iron imaging studies, iron overload in the SN was detected in patients with PD compared with HCs, even in the early stages of the disease, confirming that R2* and QSM provide reliable markers of iron content in PD, although QSM seemed to be a more robust biomarker than R2*. Nonetheless, imaging processing guidelines for QSM are not yet fully standardized. 55
Diffusion Tensor Imaging
The DWI and DTI techniques allow quantitative analysis of microstructural changes in parkinsonisms, and therefore several studies have analyzed DTI of the SN as a diagnostic marker for PD.
Some studies found highly significant FA reduction in SN, 56 whereas others concluded that there was no sufficient evidence for nigral DTI parameters to serve as a biomarker for PD. 57
A further study applying a multitarget DTI approach using different DTI metrics (FA, radial diffusivity, longitudinal diffusivity, and MD) focused on the BG and cerebellum in 15 patients with PD and 12 patients with ET found that DTI measurements from the caudate and SN separated PD from ET with a sensitivity of 92% and a specificity of 87%. 58
Recently, advanced postprocessing methods including free‐water (FW) imaging and neurite orientation dispersion and density imaging have been introduced.
FW imaging is an advanced dMRI method that allows for the separation of the fluid (ie, FW) and tissue (FW‐corrected FA) components. Recent work suggests that FW imaging might be helpful for the diagnosis of PD based on regional changes across the BG and cerebellum and their tracts. 59 Moreover, it was observed that FW values increase in the posterior SN of patients with PD compared with HCs, correlate with motor symptom severity, and increase longitudinally for 1 year, suggesting that it may be a suitable marker of disease progression. 60
A recent multicenter study provided an objective, validated, and generalizable dMRI approach to distinguish different forms of parkinsonian syndromes using disease‐specific machine‐learning comparisons of FW and FW‐corrected FA with 60 template regions and tracts of interest. 61
Postsynaptic Dopaminergic Imaging
Dopamine receptors are found on postsynaptic dopaminergic neurons. The output responses to dopaminergic stimulation can be imaged measuring D2/D3 receptor activity using different radioligands such as [11C]Raclopride, carbon‐labelled ((+)‐4‐propyl‐3,4,4a,5,6,10b‐hexahydro‐2H‐naphtho[1,2‐b][1,4]oxazin‐9‐ol hydrochloride) [11C]PHNO, [18F]Fallypride, or carbon‐labelled ((S)‐N‐((1‐ethyl‐2‐pyrrolidinyl)methyl)‐5‐bromo‐2,3‐dimethoxybenzamide) [11C]FLB‐457. 62
PET studies in patients with PD show that the striatal binding to the postsynaptic dopaminergic receptors is either normal or increased. The reduction of striatal dopamine may lead to D2 receptor upregulation as a compensatory reaction, which presents an increased activity contralateral to the clinically most affected side. In contrast, patients with APDs present reduced uptake attributed to the loss of postsynaptic receptors. 63
In the clinical setting, the role of postsynaptic D2 receptors for differential diagnosis is poor because of the low negative predictive value. 64 Therefore, this technique is mostly used in research centers for investigational purposes.
Tau Pet
Molecular imaging of pathologically misfolded and hyperphosphorylated tau proteins is of immense interest in PSP and CBD; however, its use is within the scope of research studies.
The variance in the appearance of tau pathology (3R vs. 4R, paired helical vs. straight filaments and different aggregation states) complicates the development of tau‐imaging tracers, none of the current tracers have surpassed exploratory stages, and their specificity is still questionable, especially the off‐target binding of the first generation of tracer molecules to the monoamino oxidase (MAO) enzyme family. 65 , 66
In PSP, studies with first‐generation tracers mostly using [18F]‐AV1451 found elevated retention in the pallidum, midbrain, dentate nucleus of the cerebellum, TH, caudate nucleus, and frontal cortical regions, whereas Coakeley et al did not report such elevated binding. 67 However, evidence of unspecificity regarding an elevated signal in the posterior putamen in patients with MSA put this utility into question. 68
Second‐generation tracers ([18F]‐PI2620, [18F]‐MK‐6240, [18F]‐RO‐948, and [18F]‐PM‐PBB3) hold the promise of greater specificity. [18F]‐PI‐2620 currently is the most validated tracer with relatively high in vitro binding affinity for aggregated 4‐repeat isoforms and no substantial off‐target binding to β‐amyloid or MAO A or B. 69 This tracer was used in a recently published German multicenter study that compared patients with PSP with α‐synucleinopathies, AD, and HCs and found that patients with PSP‐RS and non‐RS showed significant elevation of tracer binding in PSP target regions (Fig. 4) When using classification by at least 1 positive target region, the sensitivity and specificity for detection of PSP‐RS versus any control group were 85% and 77%, respectively.
Tau‐PET may evolve as an in vivo modality supportive of the pathological PSP diagnosis at the individual patient level. However, the currently available evidence regarding its sensitivity and specificity, as assessed against the neuropathological gold standard, is too limited to draw diagnostic conclusions. 22
Practical Perspectives of Imaging in Parkinsonisms
Can Imaging Differentiate PD from Nondegenerative Conditions?
Although PD diagnosis still remains a clinical diagnosis, the new MDS clinical diagnostic criteria include imaging as “supportive criteria” (cardiac sympathetic denervation using (123)I‐meta‐iodobenzylguanidine ‐MIBG‐ scintigraphy) and “absolute exclusion criteria” (normal presynaptic dopaminergic terminal scan).
In this regard, the use of DAT‐SPECT was approved by the FDA for routine clinical use in the differential diagnosis of PD and ET. Moreover, it may also help to differentiate other causes of parkinsonisms such as those attributed to neuroleptic treatment or vascular or functional parkinsonism. Other PET and SPECT markers of presynaptic dopaminergic terminal function are used in clinical practice, which contribute to the differential diagnosis.
However, they are unable to differentiate PD from other neurodegenerative parkinsonisms such as MSA and PSP and should not be used isolated for this purpose.
Approximately 10% of patients with a diagnosis of PD may present normal functional imaging (Scans without Evidence of Dopamine Deficit, SWEDDs). 70 This situation reinforces the importance of integrating imaging with clinical diagnostic criteria and a careful neurological examination.
Newer imaging techniques such as DNH and NM‐MR are emerging and may probably enter the clinical practice to help distinguish PD from nondegenerative conditions.
Can Imaging Differentiate PD from APDs?
Recent advances in imaging using different markers of neurodegeneration such as MRI and iron imaging or DWI showed that the diagnostic accuracy in the differential diagnosis of PD and APDs increased by combining these methodologies, analyzing patterns of atrophy in the MCP, SCP, putamen, midbrain, the MRPI index, the distribution of iron deposition and DWI maps.(Figs. 2 and 3).
Similarly, glucose metabolism imaging with [18F]FDG‐PET can reliably help in the differential diagnosis of PD from APDs, and they have been demonstrated to be superior to D2/D3 SPECT (Figs. 4 and 6). 48
Imaging Work‐Up of Parkinsonisms in Clinical Practice
The role of imaging biomarkers has progressed from merely excluding secondary causes of parkinsonisms to aiding in the differential diagnosis of PD from APDs based on changes in brainstem structures and the BG.
Here we propose 2 imaging‐based algorithms, 1 for MRI (Fig. 1) and other for PET/SPECT imaging (Fig. 6), to guide the clinician in the use of imaging methods for differential diagnosis between PD and APDs.
Briefly, both imaging algorithms reflect the basic steps that may not be missed in clinical practice when assessing a parkinsonian disorder: the first step is to rule out a secondary cause of the disorder, the second step is to evaluate if the etiology of the disorder is clearly degenerative (typical features, absence of red flags, good response to dopaminergic trial), and the third step refers to unclear degenerative parkinsonism cases or atypical parkinsonism (atypical features, presence of red flags, poor response to dopaminergic medication, no evidence of dopaminergic degeneration in imaging). In this situation, other imaging methods can be integrated with clinical diagnostic criteria and a detailed clinical and neurological evaluation to aid in the differential diagnostic process.
Conclusion
We delineated a pragmatic imaging‐based diagnostic approach based on current experience and published literature to guide the use of imaging methods in movement disorders clinics for the differential diagnosis of parkinsonisms. However, there are still great challenges to face regarding priorities in the use of imaging technologies in PD and APDs, depending on whether it is in clinical practice and/or in research protocols. The challenges may include decision making on various aspects such as standardization of the imaging interpretation and normal/pathological thresholds, the quality performance in detecting early disease and disease progression, and the application of multimodal imaging.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
C.P.: 1A, 1B, 1C, 2A
A.P.S.: 1A, 1B, 1C, 2A, 2B
T.v.E.: 1A, 1B, 1C, 2A, 2B
R.C.: 1A, 1B, 1C, 2B
K.S.: 1A, 1B, 1C, 2A, 2B
V.K.: 1A. 1B, 1C, 2A, 2B
J.E.A.:1 A, 1B, 1C, 2A, 2B
S.L.: 1 A, 1B, 1C, 2A, 2B
Disclosures
Ethical Compliance Statement
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The authors confirm that the approval of an institutional review board was not required for this work. The authors confirm that patient consent was not required for this work.
Funding Sources and Conflicts of Interest
Antonio P. Strafella is supported by the Canada Research Chair program and Canadian Institutes of Health Research (PJ8–1699695). Stephane Lehericy is supported by the Investissements d'Avenir (IAIHU‐06 Paris Institute of Neurosciences–Instituts hospitalo‐universitaires‐IHU and ANR‐11‐INBS‐0006) and Biogen Inc.
Financial Disclosures for Previous 12 Months
Antonio P. Strafella is a consultant for Hoffman La Roche and received honoraria from GE Health Care Canada LTD and Hoffman La Roche. Valtteri Kaasinen serves as an advisory board member of Abbvie; has received speaker's honoraria from Orion Pharma, Teva, GE Healthcare, Abbvie, and Nordic Infucare AB; travel expenses from Nordic Infucare AB; and research funding from the Finnish Alcohol Research Foundation, the Päivikki and Sakari Sohlberg Foundation, the International Parkinson and Movement Disorder Society, and Finnish governmental research funding (VTR). Klaus Seppi reports research grants from the Fonds zur Förderung der wissenschaftlichen Forschung (FWF) Austrian Science Fund, The Michael J. Fox Foundation, and AOP Orphan Pharmaceuticals AG, outside the submitted work and personal fees from Teva, Union chimique belge (UCB), Lundbeck, AOP Orphan Pharmaceuticals AG, Roche, Grünenthal, Stada, Licher Pharma, Abbvie, and the International Parkinson and Movement Disorders Society outside the submitted work. Cecilia Peralta is a consultant for Eurofarma and has received honoraria.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Tondo G, Esposito M, Dervenoulas G, Wilson H, Politis M, Pagano G. Hybrid PET‐MRI applications in movement disorders. Int Rev Neurobiol 2019;144:211–257. [DOI] [PubMed] [Google Scholar]
- 2. Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G. Accuracy of clinical diagnosis of Parkinson disease: a systematic review and meta‐analysis. Neurology 2016;86(6):566–576. [DOI] [PubMed] [Google Scholar]
- 3. Adler CH, Beach T, Hentz J, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: a clinicopathologic study. Neurology 2014;83(5):406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fiford CM, Ridgway GR, Cash DM, et al. Patterns of progressive atrophy vary with age in Alzheimer's disease patients. Neurobiol Aging 2018;63:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30(12):1591–1560. [DOI] [PubMed] [Google Scholar]
- 6. Parkinson's Disease, National Clinical Guideline for Diagnosis and Management in Primary and Secondary Care NICE Clinical Guidelines, N°35 National Collaborating Centre for Chronic Conditions. London: Royal College of Physicians; 2006 [PubMed]
- 7. Meijer FJA, Goraj B, Bloem BR, Esselink RAJ. Clinical application of brain MRI in the diagnostic work‐up of parkinsonism. J Parkinsons Dis 2017;7(2):211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mahlknecht P, Hotter A, Hussl A, Esterhammer R, Schocke M, Seppi K. Significance of MRI in diagnosis and differential diagnosis of Parkinson's disease. Neurodegener Dis 2010;7(5):300–318. [DOI] [PubMed] [Google Scholar]
- 9. Tinaz S, Courtney MG, Stern CE. Focal cortical and subcortical atrophy in early Parkinson's disease. Mov Disord 2011;26:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine‐containing neurons in Parkinson's disease. Brain 1999;122(Pt 8):1437–1448. [DOI] [PubMed] [Google Scholar]
- 11. Pavese N, Tai Y. Nigrosome imaging and Neuromelanin sensitive MRI in diagnostic evaluation of parkinsonism. Mov Disord Clin Pract 2018;5(2):131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lehericy S, Vaillancourt D, Seppi K, et al. The role of high‐field magnetic resonance imaging in Parkinsonian disorders: pushing the boundaries forward. Mov Disord 2017;32(4):510–525. [DOI] [PubMed] [Google Scholar]
- 13. Mahlknecht P, Krismer F, Poewe W, Seppi K. Meta‐analysis of dorsolateral Nigral Hyperintensity on magnetic resonance imaging as a marker for Parkinson's disease. Mov Disord 2017;32(4):619–623. [DOI] [PubMed] [Google Scholar]
- 14. Schwarz ST, Xing Y, Naidu S, et al. Protocol of a single group prospective observational study on the diagnostic value of 3T susceptibility weighted MRI of nigrosome‐1 in patients with parkinsonian symptoms: the N3iPD study (nigrosomal iron imaging in Parkinson's disease). BMJ Open 2017;7(12):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim YE, Sung HY, Lee J. Nigrosome 1 imaging: technical considerations and clinical applications. Br J Radiol 2019;92(1101):20180842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zwanenburg J, van der Kolk A, Luijten P. Ultra‐high‐field MR imaging: Research Tool or Clinical Need?. PET Clin 2013;8(3):311–328. [DOI] [PubMed] [Google Scholar]
- 17. Ceravolo R, Antonini A, Frosini D, et al. Nigral anatomy and striatal denervation in genetic parkinsonism: a family report. Mov Disord 2015;30(8):1148–1149. [DOI] [PubMed] [Google Scholar]
- 18. Dexter D, Wells F, Lees AJ, et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J Neurochem 1989;52(6):1830–1836. [DOI] [PubMed] [Google Scholar]
- 19. Dexter D, Carayon A, Javoy‐Agid A, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991;114(Pt 4):1953–1975. [DOI] [PubMed] [Google Scholar]
- 20. Heim B, Krismer F, De Marzi R, Seppi K. Magnetic resonance imaging for the diagnosis of Parkinson's disease. J Neural Transm (Vienna) 2017;124(8):915–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shoeibi A, Olfati N, Litvan I. Frontrunner in translation: progressive supranuclear palsy. Front Neurol 2019;10:1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Höglinger G, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology 2008;246(1):214–221. [DOI] [PubMed] [Google Scholar]
- 24. Lee W. Conventional magnetic resonance imaging in the diagnosis of parkinsonian disorders: a meta‐analysis. Mov Disord Clin Pract 2020;8(2):217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71(9):670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fanciulli A, Stankovic I, Krismer F, Seppi K, Levin J, Wenning K. Multiple system atrophy. Int Rev Neurobiol 2019;149:137–192. [DOI] [PubMed] [Google Scholar]
- 27. Heim B, Krismer F, Seppi K. Structural imaging in atypical parkinsonism. Int Rev Neurobiol 2018;142:67–148. [DOI] [PubMed] [Google Scholar]
- 28. Mestre T, Gupta A, Lang AE. MRI signs of multiple system atrophy preceding the clinical diagnosis: the case for an imaging‐supported probable MSA diagnostic category. J Neurol Neurosurg Psychiatry 2016;87(4):443–444. [DOI] [PubMed] [Google Scholar]
- 29. Nicoletti G, Fera F, Condino F, et al. MR imaging of middle cerebellar peduncle width: differentiation of multiple system atrophy from Parkinson disease. Radiology 2006;239(3):825–830. [DOI] [PubMed] [Google Scholar]
- 30. Reimao S, Guerreiro C, Seppi K, Ferrerira J, Poewe W. A standardized MR imaging protocol for parkinsonism. Mov Disord 2020;35(10):1745–1750. [DOI] [PubMed] [Google Scholar]
- 31. Drake‐Perez M, Boto J, Fitsiori A, Lovblad K, Vargas MI. Clinical applications of diffusion weighted imaging in neuroradiology. Insights Imaging 2018;9(4):535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bajaj S, Krismer F, Palma JA, et al. Diffusion‐weighted MRI distinguishes Parkinson disease from the parkinsonian variant of multiple system atrophy: a systematic review and meta‐analysis. PLoS One 2017;12(12).1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mangesius S, Hussl A, Krismer F, et al. MR planimetry in neurodegenerative parkinsonism yields high diagnostic accuracy for PSP. Parkinsonism Relat Disord 2018;46:47–55. [DOI] [PubMed] [Google Scholar]
- 34. Nicoletti G, Tonon C, Raffaele L, et al. Apparent diffusion coefficient of the superior cerebellar peduncle differentiates progressive supranuclear palsy from Parkinson's disease. Mov Disord 2008;23(16):2370–2376. [DOI] [PubMed] [Google Scholar]
- 35. Arena JE, Stoessl AJ. Optimizing diagnosis in Parkinson's disease: Radionuclide imaging. Parkinsonism Relat Disord 2016;22((suppl 1)):S47–S51. [DOI] [PubMed] [Google Scholar]
- 36. Poston KL, Eidelberg D. FDG PET in the evaluation of Parkinson's disease. PET Clin 2010;5(1):55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaasinen V, Vahlberg T. Striatal dopamine in Parkinson disease: a meta‐analysis of imaging studies. Ann Neurol 2017;82:873–882. [DOI] [PubMed] [Google Scholar]
- 38. Lin SC, Lin KJ, Hsiao IT, et al. In vivo detection of monoaminergic degeneration in early Parkinson disease by (18)F‐9‐fluoropropyl‐(+)‐dihydrotetrabenzazine PET. J Nucl Med 2014;55(1):73–79. [DOI] [PubMed] [Google Scholar]
- 39. De la Fuente FR, Sossi V, McCormick S, Schulzer M, Ruth TJ, Stoessl AJ. Visualizing vesicular dopamine dynamics in Parkinson's disease. Synapse 2009;63(8):713–716. [DOI] [PubMed] [Google Scholar]
- 40. Saari L, Kivinen K, Gardberg M, Joutsa J, Noponen T, Kaasinen V. Dopamine transporter imaging does not predict the number of nigral neurons in Parkinson disease. Neurology 2017;88(15):1461–1467. [DOI] [PubMed] [Google Scholar]
- 41. Honkanen EA, Saari L, Orte K, Gardberg M, Noponen T, Joutsa J, Kaasinen V. No link between striatal dopaminergic axons and dopamine transporter imaging in Parkinson's disease. Mov Disord 2019;34(10):1562–1566. [DOI] [PubMed] [Google Scholar]
- 42. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 2013;36(10):587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Granert O, Drzezga AE, Boecker H, et al. Metabolic topology of neurodegenerative disorders: influence of cognitive and motor deficits. J Nucl Med 2015;56(12):1916–1921. [DOI] [PubMed] [Google Scholar]
- 44. Holtbernd F, Ma Y, Peng S, et al. Dopaminergic correlates of metabolic network activity in parkinson's disease. Hum Brain Mapp 2015;36:3575–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meyer PT, Frings L, Rücker G, Hellwig S. (18)F‐FDG PET in parkinsonism: differential diagnosis and evaluation of cognitive impairment. J Nucl Med 2017;58(12):1888–1898. [DOI] [PubMed] [Google Scholar]
- 46. Eckert T, Tang C, Ma Y, et al. Abnormal metabolic networks in atypical parkinsonism. Mov Disord 2008;23(5):727–733. [DOI] [PubMed] [Google Scholar]
- 47. Pirker W, Djamshidian S, Asenbaum S, et al. Progression of dopaminergic degeneration in Parkinson's disease and atypical parkinsonism: a longitudinal beta‐CIT SPECT study. Mov Disord 2002;17:45–53. [DOI] [PubMed] [Google Scholar]
- 48. Hellwig S, Amtage F, Kreft A, et al. [18F]FDG‐PET is superior to [123I]IBZM‐SPECT for the differential diagnosis of parkinsonism. Neurology 2012;79(13):1314–1322. [DOI] [PubMed] [Google Scholar]
- 49. Zecca L, Bellei C, Costi P, et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc Natl Acad Sci U S A 2008;105(45):17567–17572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohtsuka C, Sasaki M, Konno K, et al. Changes in substantia nigra and locus coeruleus in patients with early‐stage Parkinson's disease using neuromelanin‐sensitive MR imaging. Neurosci Lett 2013;541:93–98. [DOI] [PubMed] [Google Scholar]
- 51. Ohtsuka C, Sasaki M, Konno K, Kato K, Takahashi J, Yamashita F, Terayama Y. Differentiation of early‐stage parkinsonisms using Neuromelanin‐sensitive magnetic resonance imaging. Parkinsonism Relat Disord 2014;20(7):755–760. [DOI] [PubMed] [Google Scholar]
- 52. Taniguchi D, Hatano T, Kamagata K, et al. Neuromelanin imaging and midbrain volumetry in progressive supranuclear palsy and Parkinson's disease. Mov Disord 2018;33(9):1488–1492. [DOI] [PubMed] [Google Scholar]
- 53. Isaias IU, Trujillo P, Summers P, et al. Neuromelanin imaging and dopaminergic loss in Parkinson's disease. Front Aging Neurosci 2016;8:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Du G, Liu T, Lewis M, et al. Quantitative susceptibility mapping of the midbrain in Parkinson's disease. Mov Disord 2016;31(3):317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pyatigorskaya N, Sanz‐Morère CB, Rahul Gaurav R, et al. Iron imaging as a diagnostic tool for Parkinson's disease: a systematic review and meta‐analysis. Front Neurol 2020;11:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vaillancourt D, Spraker M, Prodoehl J, et al. High‐resolution diffusion tensor imaging in the substantia nigra of de novo Parkinson disease. Neurology 2009;72(16):1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schwarz S, Abaei M, Gontu V, Morgan S, Bajaj N, Auer D. Diffusion tensor imaging of nigral degeneration in Parkinson's disease: a region‐of‐interest and voxel‐based study at 3 T and systematic review with meta‐analysis. Neuroimage Clin 2013;14(3):481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Prodoehl J, Li H, Planetta P, et al. Diffusion tensor imaging of Parkinson's disease, atypical parkinsonism, and essential tremor. Mov Disord 2013;28(13):1816–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mitchell T, Archer D, Chu W, et al. Neurite orientation dispersion and density imaging (NODDI) and free‐water imaging in parkinsonism. Hum Brain Mapp 2019;40(17):5094–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ofori E, Pasternak O, Planetta P, et al. Longitudinal changes in free‐water within the Substantia Nigra of Parkinson's disease. Brain 2015;138(Pt 8):2322–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Archer DB, Bricker JT, Winston TC, et al. Development and validation of the automated imaging differentiation in parkinsonism (AID‐P): a multicentre machine learning study. Lancet Digit Health 2019;1(5):e222–e231. [DOI] [PubMed] [Google Scholar]
- 62. Strafella AP, Bohnen NI, Perlmutter JS, et al. Molecular imaging to track Parkinson's disease and atypical parkinsonisms: new imaging frontiers. Mov Disord 2017;32(2):181–192. [DOI] [PubMed] [Google Scholar]
- 63. Antonini A, Leenders KL, Vontobel P, et al. Complementary PET studies of striatal neuronal function in the differential diagnosis between multiple system atrophy and Parkinson's disease. Brain 1997;120(Pt 12):2187–2195. [DOI] [PubMed] [Google Scholar]
- 64. Vlaar AM, van Kroonenburgh MJ, Kessels AG, Weber WE. Meta‐analysis of the literature on diagnostic accuracy of SPECT in Parkinsonian syndromes. BMC Neurol 2007;7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Eimeren T, Bischof G, Drzezga A. Is tau imaging more than just upside‐down 18 F‐FDG imaging? J Nucl Med 2017;58(9):1357–1359. [DOI] [PubMed] [Google Scholar]
- 66. Villemagne V, Doré V, Burnham S, Masters C, Rowe C. Imaging tau and amyloid‐β Proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol 2018;14(4):225–236. [DOI] [PubMed] [Google Scholar]
- 67. Coakeley S, Cho SC, Koshimori Y, et al. [18 F] AV‐1451 binding to neuromelanin in the substantia nigra in PD and PSP. Brain Struct Funct 2018;223(2):589–595. [DOI] [PubMed] [Google Scholar]
- 68. Cho H, Choi JY, Lee SH, Ryu YH, Lee MS, Lyoo CH. 18 F‐AV‐1451 binds to putamen in multiple system atrophy. Mov Disord 2017;32(1):171–173. [DOI] [PubMed] [Google Scholar]
- 69. Bullich S, Barret O, Constantinescu C, et al. Evaluation of Dosimetry, quantitative methods, and test‐retest variability of 18 F‐PI‐2620 PET for the assessment of tau deposits in the human brain. J Nucl Med 2020;61(6):920–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schwingenschuh P, Ruge D, Edwards M, et al. Distinguishing SWEDDs patients with asymmetric resting tremor from Parkinson's disease: a clinical and electrophysiological study. Mov Disord 2010;25(5):560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brendel M, Barthel H, van Eimeren T. et al. Assessment of 18F‐PI‐2620 as a Biomarker in Progressive Supranuclear Palsy. JAMA Neurol. 2020;77(11):1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]