

Abstract
Mitochondrial reactive oxygen species (ROS) have emerged as an important mechanism of disease and redox signaling in the cellular system. Under basal or pathological conditions, electron leakage for ROS production is primarily mediated by complexes I and III of the electron transport chain (ETC) and by the proton motive force (PMF), consisting of a membrane potential (ΔΨ) and a proton gradient (ΔpH). Several factors control redox status in mitochondria, including ROS, the PMF, oxidative posttranslational modifications (OPTM) of the ETC subunits, SOD2, and cytochrome c heme lyase (HCCS). In the mitochondrial PMF, increased ΔpH-supported backpressure due to diminishing electron transport and chemiosmosis promotes a more reductive mitochondrial physiological setting. OPTM by protein cysteine sulfonation in complex I and complex III has been shown to affect enzymatic catalysis, the proton gradient, redox status, and enzyme-mediated ROS production. Pathological conditions associated with oxidative or nitrosative stress, such as myocardial ischemia and reperfusion (I/R), increase mitochondrial ROS production and redox dysfunction via oxidative injury to complexes I and III, intensely enhancing protein cysteine sulfonation and impairing heme integrity. The physiological conditions of reductive stress induced by gains in SOD2 function normalize I/R-mediated ROS overproduction and redox dysfunction. Further insight into the cellular mechanisms by which HCCS, biogenesis of c-type cytochrome, and OPTM regulate PMF and ROS production in mitochondria will enrich our understanding of redox signal transduction and identify new therapeutic targets for cardiovascular diseases in which oxidative stress perturbs normal redox signaling.
Keywords: complex I, complex III, ischemia and reperfusion, oxidative posttranslational modification, proton motive force
INTRODUCTION
Mitochondria are the powerhouses of the living cell, producing most of the cell’s energy by oxidative phosphorylation. The process of energy transduction requires the coordinated action of four major respiratory enzyme complexes (complexes I–IV) and ATP synthase (F0F1−ATPase). For mammalian complexes I−IV, complete high-resolution structures are now available (1–4), enabling to follow electron flow in the electron transport chain (ETC). Mitochondrial energy transduction for ATP synthesis is driven by the oxidation of NADH and FADH2, which is carried out by the ETC located within the inner membrane. The ETC mediates a stepwise electron flow from NADH or succinate to molecular oxygen through a series of electron carriers including complex I, complex II, ubiquinone, complex III, cytochrome c, and complex IV (Fig. 1). Electron flow mediated by the ETC can drive proton translocation from the matrix side to the cytoplasmic side. ATP synthesis is then catalyzed by F0F1−ATPase and driven by chemiosmosis, enabling the flow of protons back across the membrane (Fig. 1). This process is called oxidative phosphorylation.
Figure 1.

Schematic picture explaining the mechanism of oxygen free radical(s) generation mediated by electron transport chain, the electrochemical gradient (Δp), or the proton motive force (PMF) in mitochondria. In the presence of ADP, PMF provides the driving force for reentry of H+ to matrix by chemiosmosis, resulting in ATP synthesis. Blue arrows indicate the path of forward electron transport from NADH or FADH2 to O2, and red arrow specifies reverse electron flow from FADH2-linked succinate to complex I. Brown arrows indicate the sites mediating •O2− generation in mitochondria. As electrons pass through the chain, protons are pumped from the mitochondrial matrix to the intermembrane space (IMS), thereby establishing an electrochemical potential gradient, also called proton motive force, across the inner membrane. The positive and negative charges on the membrane denote the membrane potential (ΔΨ). The proton gradient, denoted by ΔpH for the difference of pH across the membrane. Δp- or ΔpH-supported backpressure can contribute to •O2− generation when electron transport is slowed down under the physiological conditions of low ADP or low Po2. The common inhibitors used for studying the ETC components, ΔpH, and ΔΨ are indicated in brick red. ETC, mitochondrial electron transport chain.
In addition, mitochondria play a central role in the regulation of programmed cell death via redox dysfunction. They trigger apoptosis by impairing electron transport and energy metabolism, by forming mitochondrial permeability transition pore (MPTP), releasing cytochrome c and activating caspase that mediates apoptosis, and by altering the cellular redox potential via overproduction of reactive oxygen species (ROS) (5, 6). The aforementioned mechanisms can help to explain a variety of cardiovascular diseases caused by mitochondrial dysfunction. In this review article, we focus on the mechanism of redox regulation by mitochondrial complex I and complex III of the ETC, oxidative posttranslational modification (OPTM) of complex I and complex III, the proton motive force (PMF), and/or electrochemical gradient (Δp), manganese superoxide dismutase (SOD2), and cytochrome c heme lyase (HCCS). We examine the related physiological implications and the pathways in which these mechanisms control the disease process of myocardial ischemia and reperfusion (I/R).
HOW MITOCHONDRIA GENERATE ROS AND REDOX HOMEOSTASIS
Oxidative Phosphorylation, Endogenous ROS, Proton Motive Force, and Redox Homeostasis
Three major enzymatic systems were directly identified as important sources of oxygen-free radical generation in the cardiovascular system: 1) xanthine oxidase, mainly within endothelial cells; 2) the mitochondrial electron transport chain, mainly from the cardiomyocytes; and 3) NADPH oxidase, primarily within leukocytes (7). In cardiomyocytes, mitochondria comprise 30%–40% of the volume, and generate ∼90% of the ATP (8). Mitochondria are also one of the major sources of ROS generation in the cardiomyocytes. Under most physiologic conditions, NADH- or FADH2-linked electron transport to O2 is tightly coupled to oxidative phosphorylation for ATP synthesis. However, oxidative phosphorylation is also the major endogenous source of ROS produced by mitochondria (5, 6) because the mitochondrial •O2− is primarily generated by electron leakage from the ETC. Under the physiological conditions of low cellular ADP-Pi, the oxygen tension (Po2) in mitochondria is low; O2 consumption by the ETC does not meet the needs of oxidative phosphorylation. A decrease in the rate of mitochondrial phosphorylation increases the electron leakage from the ETC and subsequent production of •O2− . The •O2− is converted to H2O2 by mitochondrial SOD (SOD2), and H2O2 is further converted to H2O by glutathione peroxidase (GPx2) in the presence of glutathione (GSH). Oxidized glutathione (GSSG) is reduced back to free GSH by mitochondrial glutathione reductase (GR2) in the presence of NADPH in a redox cycle.
The proton motive force (PMF; also termed electrochemical gradient, Δp) across the inner mitochondrial membrane is generated from proton pumping of complex I, complex III, and complex IV and represents the potential energy driving the protons to reenter the matrix for ATP synthesis via chemiosmosis. The PMF consists of a proton gradient (ΔpH) and a membrane potential (ΔΨ). The electron flux mediated by ETC is inversely dependent on ΔpH and ΔΨ, and the electron slip is inversely dependent on electron flux. Physiological states of low Po2 and low ADP-Pi/ATP ratio for a given reductive pressure (high NADH/NAD+ and FADH2/FAD ratios) can slow electron flux and increase membrane potential. This will limit proton pumping from matrix to inner membrane space, limiting electron flux and promoting electron slip to O2 at complex I and complex III to produce •O2−. Therefore, the ΔpH-supported backpressure (9) with high ΔΨ inhibits chemiosmosis for coupling and functions as a driving force of endogenous mitochondrial ROS production.
This rationale has been demonstrated by electron paramagnetic resonance (EPR) assay (10, 11). The •O2− generated by isolated mitochondria can be induced by glutamate plus malate (NADH-linked) and measured using the EPR spin-trapping technique with 5',5'-dimethyl pyrroline N-oxide (DMPO). Supplementation of NADH-energized mitochondria with ADP enables chemiosmosis for coupling and initiates state 3 respiration for ATP synthesis and dissipates ΔpH, which results in minimal •O2− generation by mitochondria, which can be detected by EPR spin-trapping assay or measuring downstream H2O2 generation by Amplex Red (10). The experimental result indicates that a decrease in ΔpH is correlated with decreased •O2− generation by mitochondria under the coupling conditions. Further addition of oligomycin A to NADH-energized mitochondria blocks the proton channel of F0F1−ATPase and initiates the respiratory condition independently of ADP, which gradually increases and restores ΔpH and related backpressure. As a result, the limited efficiency of electron flux increases the •O2− generation induced by glutamate plus malate, which can be determined by EPR spin-trapping or measuring downstream H2O2 generation. The concept of electron slip for •O2− generation driven by ΔpH or ΔΨ can be tested by specific mitochondrial uncouplers nigericin and valinomycin (10). Nigericin, an ionophore functioning as a H+/K+ antiporter, can abolish ΔpH, thus allowing ΔΨ to increase to the full magnitude of the PMF (Fig. 1), and valinomycin functions as a K+ mobile carrier ionophore to collapse ΔΨ. Whereas an uncoupler (e.g., carbonyl cyanide-p-trifluoromethoxyphenylhydrazone, FCCP) functions as both a proton carrier and a charge carrier that collapses both ΔpH and ΔΨ. The addition of nigericin or valinomycin to mitochondria uncouples the mitochondria, dissipating ΔpH or ΔΨ, respectively, and reduces NADH-linked •O2− production. Analysis of the downstream H2O2 produced by NADH-energized mitochondria indicates that the rate of •O2− generation by the ETC is decreased by the uncoupler FCCP or an inhibitor of Δp (12, 13). These results support increased ΔΨ- and ΔpH-supported backpressure as the source of endogenous •O2− generation in mitochondria (Fig. 1).
Under the physiological conditions of low ADP-Pi, the increased backpressure generated by ΔpH and ΔΨ can inhibit the electron transport activity of the ETC, which subsequently predisposes the mitochondria or myocytes to be more reductive (or increases reductive pressure). As demonstrated by EPR redox analysis using the spin probe of CMH (1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine.HCl) (10) or PCA [3-(carboxy)-2,2,5,5-tetramethylpyrrolidin-1-oxyl] (14, 15), dissipation of ΔpH and ΔΨ with nigericin and valinomycin prompts the redox status of mitochondria or myocytes to become more oxidized. This evidence supports the roles of ΔpH, ΔΨ, and endogenous ROS as the physiological effectors of redox homeostasis in mitochondria.
SOD2, Nitric Oxide, Catalase, and Redox Regulation of Mitochondria
Overexpressing SOD2 in the myocyte of the murine heart (cardiac-specific SOD2-tg mouse model) induces a cardiac phenotype of supernormal function via enhancing mitochondrial bioenergetics and H2O2-mediated metabolic dilation (16). The SOD2-tg mouse myocardium and cardiac mitochondria exhibit a more reductive physiological setting, despite the increased rate of H2O2 generation by energetic mitochondria. A more reductive milieu in the myocardium of SOD2-tg mice has been proven to alleviate systolic dysfunction induced by the antineoplastic agent carmustine (BCNU) via neutralizing oxidative stress in the heart (11). A highly reductive physiological setting in the cardiac mitochondria of SOD2-tg mice is also reported to correct the I/R-induced redox alteration and reverse the impaired ETC activities of complex I and complex III in the postischemic heart (17, 18). However, overexpressing SOD2 in the myocytes also imposes reductive stress (19), which induces a phenotypic switch to hypertrophy with marked misfolding of SOD2 and associated protein aggregates (16, 20).
The NO produced by eNOS is known to function as one of the paracrine factors for endothelial cell-to-myocyte communication. The mitochondria of the cardiovascular system are an important target for the NO generated by eNOS. Under the physiological conditions of low Po2, NO competes with O2 in reversibly binding to the heme a3 of complex IV, decreasing the rate of ADP-independent oxygen consumption and alleviating •O2− production (21). This concept has been further demonstrated in the eNOS−/− murine heart (22). Genetic deletion of eNOS induces a more oxidized physiological redox setting in the myocardium and cardiac mitochondria, leading to marked dysfunction of the mitochondria (22). Overexpression of SOD2 in the eNOS−/− murine heart normalizes the redox alteration and rescues its mitochondrial function via partial restoration of the state 3 respiration rate and respiratory control index (Supplemental Fig. S1, https://doi.org/10.6084/m9.figshare.16691935).
Overexpressing catalase in the mitochondria (mouse model of mCAT) is reported to attenuate the phenotype of cardiac aging (23). Like the mouse model of SOD2-tg, the mCAT murine heart displays a more reductive physiological setting, which normalizes the redox imbalance of the mitochondria and myocardium during the aging process.
FACTORS INVOLVED IN •O2− GENERATION AND REDOX STATUS IN THE MITOCHONDRIA OF THE POSTISCHEMIC HEART
Electron transport mediated by both complex I and complex III generates PMF for energy transduction and ATP synthesis. Complex I and complex III are also two main segments of the ETC widely recognized to be responsible for •O2− generation. One, on complex I, generates •O2− largely via electron leakage from the reduced flavin mononucleotide (FMN) (24, 25) and the quinone-binding site or domain (Fig. 1). The other, on complex III, mediates •O2− production mainly through the Q cycle mechanism, in which electron leakage for •O2− generation results from ubisemiquinone (•Qo− in Figs. 1 and 3) at the Qo site (29). The •O2− production mediated by complex III exhibits bidirectionality with a distinct membrane sidedness or topology. Experimental evidence supports that •O2− is released to both sides of the inner mitochondrial membrane by the •Q (30, 31).
Mediation of •O2− overproduction and redox dysfunction by complex I or complex III is highly relevant in the disease conditions of myocardial ischemia and reperfusion (I/R) (17, 18). Mitochondrial dysfunction is the disease hallmark of I/R injury (10, 17, 32, 33). Impairment of mitochondrial function during myocardial I/R is caused by oxidative stress. In the ischemic heart, oxygen delivery to the myocardium is not sufficient to meet the need for substrate oxidation in mitochondria during the physiological conditions of hypoxia, leaving the mitochondria in a highly reductive state. Because of the significant depletion of ADP and low electron transport activity of ETC (34), this results in increased ΔpH-supported backpressure. Reperfusion is a “double-edged sword.” Reestablishment of blood flow is mandatory to salvage the ischemic myocardium from infarction, but reperfusion per se can contribute to injury and ultimate infarct size. Reintroduction of O2 with reperfusion greatly increases electron leakage along with a decrease in scavenging capacity, leading to marked overproduction of •O2− by complex I and complex III in the mitochondria (17, 18). This overproduction of ROS converts mitochondrial thiol redox to a more oxidative physiological setting, leading to redox dysfunction primarily caused by oxidation of free thiols and protein thiols. Oxidative posttranslational modification (OPTM) with protein cysteine oxidation of complex I and complex III leading to consequent impairment of the PMF is emerged as an important mechanism in inducing a more oxidative physiological setting and redox dysfunction in the postischemic heart (17, 18).
Mediation of •O2− Generation and Redox Dysfunction by Complex I in the Postischemic Heart
Mitochondrial complex I [EC 1.6.5.3. NADH: ubiquinone reductase (NQR)] is the first energy-conserving segment of the ETC. Purified bovine heart complex I contains 45 different polypeptides with a total molecular mass approaching 980 kDa (35). Conformational changes of reversible A/D (active/deactive state) transition are important to the catalytic mechanism of complex I (36, 37). The A/D transition has been proposed as a mechanism that provides a physiological response of mitochondrial respiratory chain to low oxygen tension or anoxia conditions. Ischemic condition is shown to induce progressive accumulation of D-form complex I ex vivo, and slow reactivation of D-form enzyme takes place during early phase of reoxygenation ex vivo (38, 39). D-form of the enzyme can be specifically labeled with sulfhydryl reagent whereas the A-form is relatively insensitive to sulfhydryl reagent.
Mediation of •O2− generation by complex I has been reported to be involved in electron leakage from the FMN moiety (40, 41), the quinone-binding domain (42–44), and the iron-sulfur cluster (43). Further evidence shows that the •O2− generation by complex I during reverse/forward electron transport can be inhibited by an uncoupler or nigericin, suggesting that •O2− production by complex I is more dependent on ΔpH than ΔΨ under normal physiological conditions (10, 13, 44).
Complex I is rich in cysteine residues and hosts associated redox protein thiols that are highly susceptible to oxidative stress, leading to reversible and irreversible OPTM, including protein cysteine S-glutathionylation (45–49), protein tyrosine nitration (50), and protein cysteine sulfonation (17). The evidence for I/R-mediated in vivo S-glutathionylation and tyrosine nitration of complex I is limited to experimental results based on immunoblotting using antibodies against glutathione (GSH) and 3-nitrotyrosine, presumably due to the low abundance of glutathionylation and nitration induced by I/R in vivo. Conversely, I/R-mediated irreversible cysteine sulfonation of mitochondrial complex I in vivo has been reported recently based on the experimental evidence of using unbiased and robust proteomics with MS (mass spectrometry) analysis (17). Label-free quantitation (LFQ) indicated that cysteine sulfonation of complex I in cardiac mitochondria is nearly undetectable due to the highly reductive milieu under the physiological conditions of ischemia in vivo. Reperfusion following 30 min of ischemia induces a significantly high abundance of cysteine sulfonation in complex I of the postischemic heart. Proteomic analysis with MS mapping of sulfonated cysteines from the complex I from the mitochondria of the postischemic heart has provided deep insights into the molecular mechanisms of how I/R decreases the catalytic function of complex I and how I/R mediates electron leakage from complex I as well as how I/R impairs redox homeostasis and ΔpH.
With a chaotropic agent, purified complex I can be resolved into three subcomplexes: a flavoprotein fraction (Fp), an ion sulfur protein fraction (Ip), and a hydrophobic fraction (Hp). Proteomic analysis has revealed that I/R in vivo predisposes complex I to a more oxidized status via enhancing protein cysteine sulfonation across the Fp, Ip, and Hp (Table 1) (17). Intense cysteine sulfonation of the 51 kDa subunit (ndufv1) supports the FMN moiety as a major site of •O2− generation mediated by complex I during postischemic reperfusion. The cysteine ligands are sulfonated for the iron-sulfur clusters at five centers: N3 (C425 of the 51 kDa subunit), N1a (C134/C139 of the 24 kDa subunit), N1b (C92 of the 75 kDa subunit), N4 (C226 of the 75 kDa subunit), and N2 (C158/C188 of the PSST subunit). This result indicates that I/R impairs electron transport activity but increases electron leakage for •O2− locally to induce sulfonation of the cysteine ligands for the metal centers of the 51 kDa, 24 kDa (ndufv2), 75 kDa (ndus1), and PSST (ndufs7) subunits (Table 1). Proteomic analysis further supports the domain of the ubiquinone-binding site as the other major site of I/R-induced •O2− generation, as indicated by cysteine sulfonation of the N2 cluster (C158/C188 of the PSST subunit) and the PSST (C175) and 49 kDa (ndufs2, C146) subunits, which consequently impairs the enzymatic function of ubiquinone reduction and increases •O2− generation from the N2 cluster or semiquinone (Table 1).
Table 1.
Summary of functional effects of I/R-mediated site-specific protein sulfonation of the mitochondrial complex I in the postischemic heart∗
| Functional Impact on Complex I Activity and Redox Status | Subunits and Cysteine Residues of I/R-Mediated Sulfonation of Complex I | Mechanisms of Functional Impact Resulted from Specific Cysteine Sulfonation | Reference |
|---|---|---|---|
| Enzymatic activity and ROS generation involved in FMN moiety and iron-sulfur clusters | 51 kDa N3 (C425) | Impairing main electron transfer pathway and decreasing electron transport activity of complex I Increasing ROS produced from FMNH• and iron-sulfur clusters |
17, 40 |
| 75 kDa N1b (C92) | 17 | ||
| 75 kDa N4 (C226) | 17, 49 | ||
| PSST N2 (C158/C188) | 17 | ||
| 24 kDa N1a (C134/C139) | Impairing antioxidant function of N1a cluster, increasing accumulation of FMNH• for •O2− generation, thus decreasing electron transport activity | 17 | |
| Enzymatic activity and ROS generation involved in ubiquinone-binding site and semiquinone | PSST N2 (C158/C188) | Impairing main electron transfer pathway, decreasing the efficiency of ubiquinone reduction and ubiquinone-binding, increasing semiquinone for •O2− generation | 17 |
| PSST (C175) | 17 | ||
| 49 kDa (C146/C326/C347) | 17 | ||
| Transition of deactive form to active form | ND3 (C39) | Reversible S-nitrosylation of C39 delays reactivation of D-from and respiratory chain. Irreversible sulfonation of C39 prevents the D-from of complex I from undergoing reactivation | 51, this work |
| Pumping of protons, pH, and PMF | ND4L (C98) | Affecting E channel (ND4L) and channel of distal antiport-like subunit (ND5), decreasing efficiency of proton pumping, ΔpH, and PMF | This work |
| ND5 (C371/C341/C329) | This work | ||
| Redox alterations | 51 kDa (C286/C125/C187/C206/C238) | Affecting redox regulation and antioxidant defense | 17, 40, 47 |
| 75 kDa (C354/C554/C564/C727) | 17, 46, 49 | ||
| 24 kDa N1a (C134/C139) | Impairing antioxidant function of N1a cluster via altering redox potential of N1a center | 17 | |
| Structural integrity | 42 kDa (C112/C183/C253) | Affecting the structural integrity of complex I via weakening docking to ND2 | 17 |
| 13 kDa (C79) | Affecting the structural integrity of complex I structure via impairing Zn2+ binding | 17 | |
| 15 kDa (C43) | Breaking intramolecular disulfide bond and impacting structural integrity | 17 |
FMN, flavin mononucleotide; I/R, ischemia and reperfusion; PMF, proton motive force; ROS, reactive oxygen species. ∗Reference 17.
Proteomics with MS analysis also indicated significantly enhancing cysteine oxidation of the Hp subcomplex at the ND3 (C39), ND4L (C98), and ND5 (C329/C341/C371) subunits under stress conditions of I/R (Fig. 2, Supplemental Fig. S2 is available at https://doi.org/10.6084/m9.figshare.17125595, and Table 1). The ND4L subunit is a structural part of the E channel (hosting ND1, ND6, and ND4L) that functions to translocate protons by a coupling mechanism of complex I (1, 52, 53). The ND5 is one of three distal antiporter-like subunits hosting channels required for the pumping of protons (1, 52, 53). Together with increased sulfonation of the ubiquinone-binding domain, I/R-mediated sulfonation of ND4L and ND5 may decrease the efficiency of proton pumping, which reduces backpressure and PMF generated by complex I (Table 1).
Figure 2.
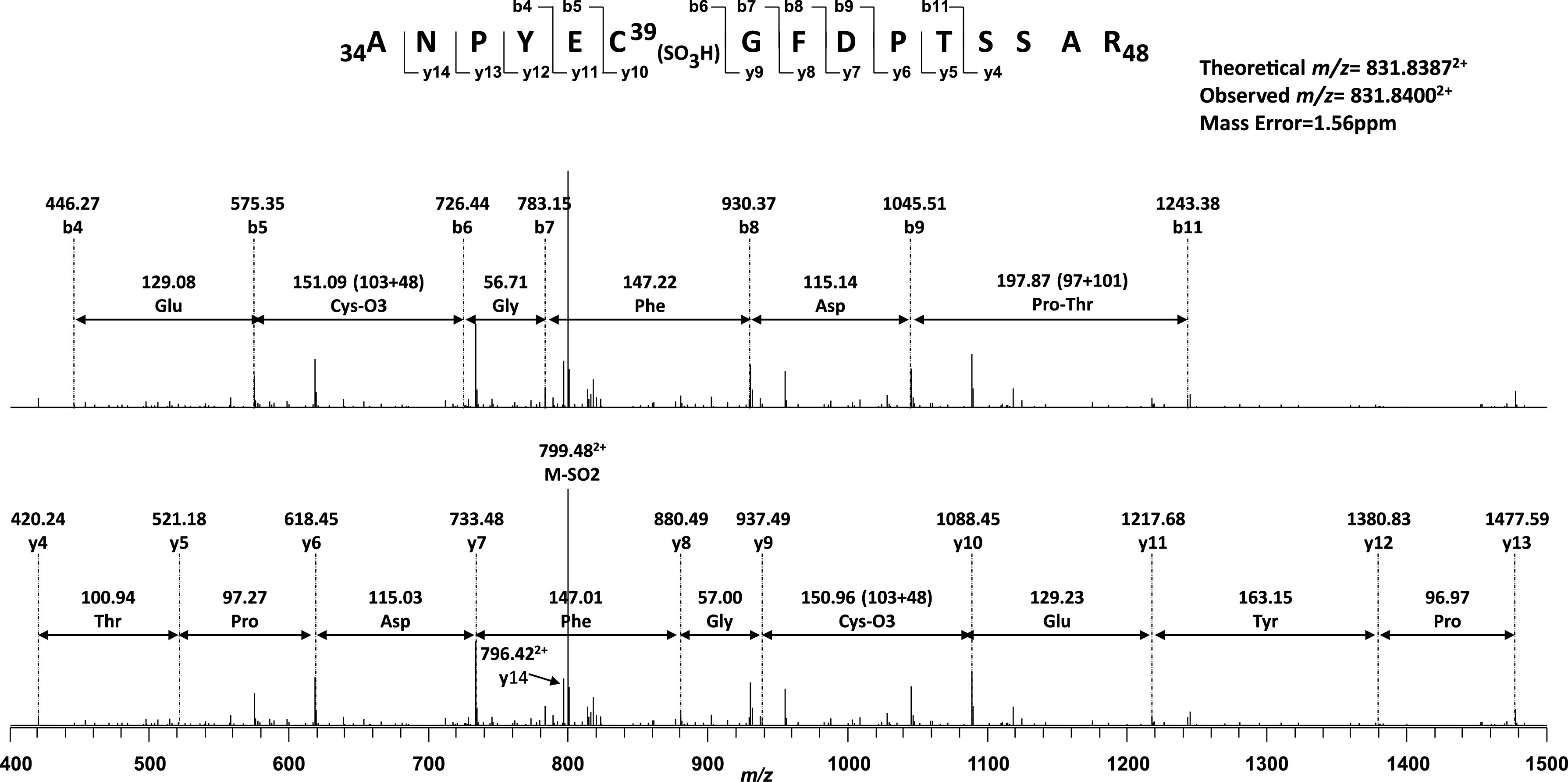
MS/MS spectrum of the doubly protonated molecular ion of the sulfonated peptide [34ANPYEC39(SO3H)GFDPTSSAR48] of the ND3 subunit from the complex I of postischemic rat heart. The underline indicates the cysteine-39 residue of ND3 subunit (Mt-ND3). The sequence-specific ions are labeled as y and b ions on the spectrum. The amino acid involved in S-sulfonation is denoted by SO3H in parentheses. A mass difference between y10 and y9 fragment ions (150.96 Da) corresponds to cysteine-39 sulfonation (151 Da).
Sulfhydryl group at the C39 of ND3 subunit has been reported important to mediate the mechanism of A/D transition via conformational change of complex I ex vivo (51). Irreversible covalent modification of the ND3 at C39 residue prevents the D-form of complex I from undergoing reactivation and exacerbates irreversible impairment of the enzymatic function. The stress condition of I/R mediates an irreversible protein oxidation at the C39 of the ND3 supports the above concept in vivo (Fig. 2). Enhanced sulfonation of C39 of the ND3 subunit indicates progression of reperfusion after ischemia damages the intrinsic property of reversible A/D transition in vivo, predisposing the complex I inactive state and aggravating mitochondrial dysfunction. However, reversible S-nitrosylation of C39 can delay reactivation of the enzyme and associated respiratory chain (51). Thus rapid reactivation of D-form is hypothetically featured as main contributor of ROS production, and slowing rapid reactivation of complex I may represent a therapeutic strategy of postischemic injury (36).
Redox alterations mediated by I/R are clearly supported by detection of enhancing sulfonation of both 51 kDa (ndufv1) and 75 kDa (ndus1) subunits at C286, C125, C187, C206, C238 and C354, C554, C564, C727 that are not involved in the ligands of iron-sulfur clusters (Table 1). The in vivo results strongly support the additional function role of 51 and 75 kDa subunits in redox regulation and antioxidant defense against oxidant attack, which has been established by ample evidence of in vitro studies (46–49, 54).
Proteomic analysis further disclosed that I/R mediates increased sulfonation of 42 kDa (ndufa10), 15 kDa (ndufs5), and 13 kDa (ndufs6) subunits important for stabilizing the intact complex I during enzyme turnover and assembly of the complex I (Table 1). Increased site-specific cysteine oxidation of the 42, 15, and 13 kDa subunits may negatively impact the structural integrity of mammalian complex I via impairing docking to ND2 (1, 55), intramolecular disulfide (1), and Zn2+-binding (56) synergistically promoting I/R-mediated complex I injury and consequent mitochondrial dysfunction in the postischemic heart.
I/R-mediated cysteine sulfonation of the complex I in vivo can be alleviated in the mouse model of cardiac-specific overexpression of SOD2 in the mitochondria of myocytes (SOD2-tg murine model) (16, 17). Therefore, •O2− could traverse within intramolecular complex I under the conditions of I/R (57). Protein sulfonation of nonmetal binding cysteines or nonquinone-binding cysteines in the complex I was likely mediated by intramolecular movement of •O2−. The conditions of myocardial I/R facilitate protonation of •O2− (pKa 4.9). Protonated form of •O2− released from N3, N1b, N4, N2, N1a clusters, FMN, and semiquinone could traverse intrasubunit or intersubunits within the complex I, inducing sulfonation of 51 kDa, 75 kDa, 13 kDa, PSST, and 49 kDa subunits. Protonated •O2− released from N2 center and associated ubiquinone-binding pocket could further freely diffuse across membrane to sulfonate the subunits of ND3, ND4L, ND5, and 15 kDa. The 42 kDa subunit is loosely bound to complex I, residing on the matrix face of the membrane and the peripheral arm without contacting any hydrophilic core subunits. Intense sulfonation of the 42 kDa during I/R was likely mediated by the protonated •O2− released in matrix and traversed from the FMN.
Mediation of •O2− Generation and Redox Dysfunction by Complex III in the Postischemic Heart
Mitochondrial complex III catalyzes the electron transfer from ubiquinol (QH2) to ferricytochrome c, which is coupled to proton translocation for ATP synthesis. The redox centers of complex III consist of QH2, hemes bL (low potential b or b566), bH (high potential b or b562), and c1, and the Rieske iron-sulfur cluster. The electron transfer from QH2 to cytochrome c follows the Q cycle mechanism. The Q cycle, first proposed by Peter Mitchell, is now accepted as the mechanism describing how complex III moves the protons for the biochemical generation of ΔpH, which is used for ATP generation by complex III (Fig. 3). The Q cycle pathway further serves as an important mechanism for generating the ΔpH and ΔΨ and as an endogenous source of •O2− by complex III as shown in the Fig. 3 (26, 58–60).
Figure 3.

Electron transport, PMF, and superoxide generation mediated by the Q cycle mechanism in complex III. The diagram is adapted from Ref. (26) with modification. Two electrons of ubiquinol (QH2) go down two different paths. As the first electron is transferred from ubiquinol (QH2) to cyt c, semiquinone (•Qo−) is formed at the membrane of cytoplasmic side and 2H+ are released to IMS (H+/e− = 2). The second electron goes through heme bL and heme bH of cytochrome b and is directed toward matrix side. The second electron and 2H+ are accepted by stable semiquinone (•Qi−) at the matrix side and result in generation of ΔpH (solid bar arrow in dark gray). Unstable semiquinone, •Qo−, is endogenous source of •O2− production. The electron flow through low potential heme b (bL) is proposed as the other source of •O2− production as backpressure is increased (26). Gray areas symbolize the reactions involved in •O2− production. P, O, and C represent positive, outside, and cytoplasmic side, respectively. N, I, and M stand for negative, inside, and matrix side, respectively. Solid bars in light gray arrow show the opposition of electron transfer from the bL to bH by the membrane potential (ΔΨ). Inhibition of bH reoxidation by antimycin A is indicated by black square block. Myxothiazol and stigmatellin are two •Qo− inhibitors. Myxothiazol binds at the cytochrome bL Qo site of the complex III in the “heme bL proximal” position, blocking electron transfer to the Rieske iron-sulfur cluster (27). Stigmatellin binds at the cytochrome b Qo site in the heme bL distal position and associated with RISP in hydrogen-binding contact (28). IMS, intermembrane space; ΔpH, proton gradient; PMF, proton motive force.
In the Q cycle mechanism, there are two semiquinones formed in different parts of the cycle (Fig. 3). An unstable semiquinone (•Qo−) is formed near the cytoplasmic site (outer site or positive site). The other semiquinone (•Qi−), formed near the matrix side (inner site or negative site), is a stable semiquinone and EPR detectable. During the enzyme turnover of complex III, one electron from QH2 is sequentially transferred to the Rieske iron-sulfur protein (RISP), then to cytochrome c1 (cyc1), and then to ferricytochrome c (cyt c). QH2 contains two electrons, but cytochrome c only accepts one electron. This leaves an unstable semiquinone (•Qo−) formed at the cytoplasmic site and releases two H+ into the intermembrane space (IMS). One electron from the unstable semiquinone is transferred to a low potential heme b (bL), and then transferred to a high potential heme b (bH). One electron from bH is then transferred to ubiquinone (Q) to form a stable semiquinone at the matrix side. The stable semiquinone can accept one electron and two H+ from the matrix from a second single turnover to complete the cycle, thus pumping two protons, forming QH2, and generating ΔpH and PMF (Fig. 3). In addition to •Qo− as an endogenous source of •O2−, the electron flow from heme bL to heme bH can result in leakage for •O2− generation driven by ΔpH or ΔΨ when backpressure is high (Fig. 3) (26).
Oxidative injury of complex III is further marked in the mitochondria of the postischemic heart. Proteomic analysis indicates that I/R conditions induce impairment of heme c1 via inhibiting thioether bond formation between two vinyl groups of protoporphyrin IX and two cysteines of cytochrome c1. I/R conditions also induce destruction of the Rieske iron-sulfur cluster via enhancing sulfonation of the cysteine ligand at the C236 residue of RISP (Table 2) (18). The evidence of proteomic studies thus supports the disease mechanism of I/R injury to complex III in cardiac mitochondria, carried out via impairing the proton motive Q cycle mechanism, decreasing electron transport activity, and diminishing ΔpH. Furthermore, I/R-mediated thioether defect of cyt c1 and C236 sulfonation of RISP impairs the PMF driven by the Q cycle mechanism of the complex III in the postischemic heart. Cardiac-specific transgenic expression of SOD2 in mitochondria nearly fully rescues the I/R injury of the complex III (18). Therefore, overproducing ROS mediates damage to RISP via weakening the metal binding as a major mechanism of reperfusion injury of complex III.
Table 2.
Summary of the functional effects of I/R-mediated heme destruction, thioether defect of heme c1, and site-specific protein sulfonation of mitochondrial complex III in the postischemic heart∗
| Functional Impact on Complex III Activity and Redox Status | I/R-Mediated Enhancing Heme Impairment and Site-Specific Cysteine Sulfonation of Complex III | Mechanisms of Functional Impact Resulted from Heme Impairment and Specific Cysteine Sulfonation |
|---|---|---|
| Q-cycle mediating electron transport activity, ROS production, and PMF | Alkylated C122/C125 of cytochrome c1 via carbamidomethylation | Impairing heme integrity of cytochrome c1, decreasing electron transport activity driven by Q-cycle and increasing ROS from the heme c1 moiety |
| C236 sulfonation of Rieske iron-sulfur protein (uqcrfs1) | Impairing 2Fe-2S integrity of RISP, decreasing electron transport activity driven by Q-cycle and increasing ROS from the 2Fe-2S cluster | |
| Electron transfer between cytochrome c1 (cyc1) and cyt c | C140 sulfonation of cytochrome c1 (cyc1) | Fingerprint of •O2− at Qo site and released to IMS |
| C220 sulfonation of cytochrome c1 | Weakening binding interaction of cyc1 with hinge protein and impacting proper complex formation of cyc1 and cyt c Fingerprint of •O2− at Qo site and released to IMS |
|
| C65 sulfonation of hinge protein (uqcrh) | ||
| Redox alterations | C69 sulfonation of core 1 (uqcrc1) | Affecting redox regulation and antioxidant defense Fingerprint of •O2− released to or produced in matrix Little impact on electron transport activity of complex III |
| C268 sulfonation of core 1 | ||
| C380 sulfonation of core 1 | ||
| C410 sulfonation of core 1 | ||
| C445 sulfonation of core 1 | ||
| C453 sulfonation of core 1 | ||
| C191 sulfonation of core 2 (uqcrc2) |
I/R, ischemia and reperfusion; PMF, proton motive force; RISP, Rieske iron-sulfur protein; ROS, reactive oxygen species.
∗Reference 18.
Lesnefsky and Hoppel have identified ischemia-mediated damage to the Rieske iron-sulfur cluster of complex III in the isolated aging rat heart using low-temperature EPR (loss of characteristic of g = 1.79 in the trough of EPR spectrum at cryogenic temperature of 15 K) (61), which was not observed in the mitochondria of adult rat heart (18). However, the mechanism remains unclear how ischemic condition ex vivo induces damage to the 2Fe-2S cluster of the complex III.
The c-type cytochromes (cytochrome c and cytochrome c1) are characterized by the covalent attachment of heme to protein via two thioether bonds. These cysteines are always observed in a CxxCH heme-binding motif, the first cysteine to 2-vinyl and the second cysteine to 4-vinyl. These covalent heme attachments provide stability to c-type cytochromes, increasing electron transfer efficiency and minimizing electron leakage in the form of •O2−. The mechanism of heme attachment to protein by thioether formation is catalyzed by cytochrome c heme lyase (or holocytochrome c synthetase, encoded by the HCCS gene) localized in the IMS. Therefore, HCCS mediates c-type cytochrome biogenesis and its heme integrity in mitochondria, indirectly minimizes the electron leakage from complex III, and exploits the activity of cyt c to scavenge •O2−.
In mice and humans, the HCCS gene is located on the X chromosome and is subject to X chromosome inactivation. Analysis of human tissue-specific expression via genome-wide integration of transcriptomics and proteomics indicates heart as the tissue with highest expression level of HCCS (62). A cardiac-specific conditional knockout to inactivate the X-linked HCCS gene in mice results in a different phenotype between males and females during embryonic development. In contrast to the midgestational lethality of HCCS knockout in males, heterogeneous knockout females appear normal after birth, displaying a remarkable regenerative capacity of the post-midgestational heart (63).
The stress conditions of I/R significantly suppress the level of HCCS in the mitochondria (18), thus contributing to the mechanism of complex III injury in the postischemic heart via inducing defects in the thioether bonds of c-type heme (Table 2). Therefore, I/R-mediated complex III injury via damaging the c-type heme and Rieske iron-sulfur cluster further amplifies the mitochondrial oxidative stress by increasing electron leakage for ROS production and decreasing the O2− scavenging ability of cyt c. Proteomic analysis further detected I/R-enhancing sulfonation of C140 and C220 from the cytochrome c1 subunit of complex III (Table 2). Together with the evidence of elevated C65 sulfonation of hinge protein (uqcrh), stress conditions of I/R weaken the interaction of cytochrome c1 and hinge protein, impact proper complex formation of cytochrome c1 and cyt c, and decrease electron transport efficiency from ubiquinol to cyt c.
In addition to impairing the Q cycle mechanism, the pathological conditions of I/R also mediate enhanced cysteine sulfonation of the core 1 (uqcrc1) and core 2 (uqcrc2) subunits, which has been proven to have no significant effect on the QCR activity and O2− generation activity mediated by complex III in vitro (Table 2) (18). Since cysteine sulfonation of complex III in cardiac mitochondria is virtually undetectable under ischemic conditions in vivo, any increase in cysteine sulfonation of core 1 and core 2 subunits as mediated by reperfusion thus contributes to a more oxidative physiological setting in the postischemic heart.
I/R mediates extensive cysteine oxidation of the core 1 subunit (C69, C268, C380, C410, C445, C453) and the unique C191 sulfonation of the core 2 subunit, which are localized in the matrix compartment. They are presumably induced by a portion of the ROS generated by complex III and the FMN moiety of the complex I. Protonated •O2− is released to the matrix side and traversed to surface-exposed cysteines of the core 1 and core 2 subunits, mediating sulfonation of core 1 and core 2 subunits. However, I/R-induced cysteine sulfonation detected in the RISP (C236), cytochrome c1 (C140 and C220), and hinge protein (C65) is caused by ROS overproduced by the Qo site of complex III via intramolecular traverse, because these residues are localized within the IMS. Thus, strong evidence is provided for the topological bidirectionality of ROS generation by complex III during myocardial I/R (30, 31, 64).
Reversible Cysteine Sulfoxidation of Complex I and Complex III in the Ischemic Heart
It appears that the proteomic method does not distinguish the level of oxidation that impacts potential reversibility. Basal and enhanced levels of protein cysteine sulfinaton and sufonation have been consistently detected in the complex I and complex III in the mitochondria of normal and postischemic myocardium (17, 18). The physiological conditions of ischemia thus reverse the basal level of reversible cysteine sulfoxidation to free sulfhydryl of complex I and complex III in vivo (17, 18), which is consistent with the concept of reversible cysteine sulfoxidation as an oxygen sensor (65). However, direct evidence based on MS analysis for protein cysteine sulfenation (converting Cys-SH to Cys-SOH) is not available yet, presumably due to nature of reversibility with short half-life. A novel biotinylated dimedone has been developed by Eaton’s laboratory to label protein sulfenic acids in the systems of perfused heart and myocytes (65). Isolated rat heart subjected to hypoxia resulted in a striking loss of labeling, which returned when oxygen was resupplied, concluding protein sulfenic acid as oxygen sensor.
The Relative Contributions of Sulfhydryl Nitrosylation versus Sulfoxidation in the Mitochondrial Complex I and Complex III
Cysteine sulfhydryl nitrosylation of mitochondrial respiratory chain features as an important cardioprotective mechanism via the pathway of slowing reactivation of respiratory chain or shielding the cysteine residues from ROS-induced sulfoxidaiton. Perfusion of mitochondria-targeted S-nitrosothiol (MitoSNO) during early reperfusion after ischemia ex vivo nitrosates the C39 residue of complex I ND3 subunit and quench the ROS overproduction via deaccelerating reactivation of D-form enzyme (51). Ischemic precondition (IPC) has shown to induce protein nitrosothiol (SNO) that provides cardioprotection by protecting cysteine residues from sulfoxidation during I/R injury (66, 67). Proteomic studies of isolated murine heart have indicated significant increase in cysteine nitrosylation with IPC suppressed I/R-induced sulfoxidation at the C67 site of 42 kDa subunit in the complex I and C268/C380 sites of core 1 subunit in the complex III (67). IPC-mediated sulfhydryl nitrosylation of other ETC complexes to protect protein reactive thiols from sulfoxidation also includes C568 of the 70 kDa subunit (SdhA) of the complex II and C115 of the subunit Vb of the complex IV (67).
Redox Regulation with Protein S-Glutathionylation in the Postischemic Heart
The mitochondrial redox pool is enriched in GSH with a high physiological concentration (68, 69); overproduction of ROS increases the ratio of GSSG to GSH in the postischemic heart (45). The 51 and 75 kDa subunits of the complex I are modified by reversible S-glutathionylation in vitro and in vivo (45–48, 54). Intrinsic S-glutathionylated complex I can be detected in the rat myocardium and enhanced in the postischemic heart (45). In vitro studies using isolated mitochondria also indicate that increasing S-glutathionylation of complex I is favored by the conditions of oxidative stress such as exposure to organic peroxide, excess NO, or the thiol oxidant diamide. In vitro oxidant exposure of mitochondria also resulted in impaired complex I activity, but reversal of S-glutathionylation failed to restore complex I activity, suggesting a nonessential role for S-glutathionylation in oxidant-induced damage to complex I activity (46). In vitro S-glutathionylation of complex I with a low dosage of GSSG leads to marginal enhancement of the electron transfer efficiency and a decrease in the electron leakage (47). Therefore, an increase in S-glutathionylation of complex I in vivo is likely important in preventing oxidative damage to the enzyme. In contrast to complex I, complex II exhibits the opposite glutathionylation pattern following myocardial ischemia and reperfusion [i.e., deglutathionylation, see Chen et al. (33)]. In vivo myocardial I/R, as well as Langendorff isolated perfused rat heart, resulted in decreased intrinsic S-glutathionylation of the Fp (70 kDa) subunit of complex II (33). Studies of isolated complex II also indicated that S-glutathionylation increased electron transfer activity somewhat and decreased electron leakage for •O2− generation, suggesting that I/R-mediated deglutathionylation contributes to the decrease in complex II activity observed in the postischemic myocardium (33).
Redox Modification with Protein Methionine Oxidation in the Postischemic Heart
Methionine serves a unique role as the 6th heme ligand of the c-type cytochrome. ROS-induced protein methionine oxidation of cyt c in vitro enhances it peroxidase activity and peroxide-mediated protein-centered tyrosyl radical (70). The formation of cyt c-derived tyrosyl radical and tyrosine ortho-semiquinone is further reported to mediate cellular apoptosis (71, 72). Two distinct classes of methionine sulfoxide reductase, MsrA and MsrB, function as antioxidant enzymes to catalyze reduction of methionine sulfoxide to methionine. However, transgenic overexpressing MsrA targeting to mitochondria fails to protect the murine heart from I/R injury ex vivo presumably due to lacking myristoylation of MsrA in mitochondria (73). Experimental investigation of the proteomics for protein methionine oxidation in the postischemic heart has proven challenging because of the lack of methodology capable of effectively blocking methionine from autooxidation.
FUTURE DIRECTIONS AND PERSPECTIVE
Ongoing further evaluation of the role of SOD2 and HCCS in the normal and postischemic hearts will enable increased understanding of how mitochondrial ROS mediates cellular signaling in coordinating reductive stress and biogenesis of c-type cytochrome to cardiovascular physiology and disease. Further elucidation of the SOD2 or catalase-induced reductive stress and c-type cytochrome biogenesis that regulate PMF, mitochondrial ROS generation, OPTM, and related redox status in myocytes under physiological or pathological conditions is a major frontier in the fields of redox regulation and signaling.
Currently, specific cysteine-sulfonated domains of complex I and complex III have been identified in the postischemic heart, but it is currently unknown whether any identified sulfonated domain is regulated by the nitric oxide signaling, I/R-induced neutrophil infiltration and activation, or whether c-type cytochrome biogenesis mediated by HCCS can normalize redox dysfunction conferred by the OPTM induced by I/R, or how reductive pressure imposed by SOD2 and catalase differentially normalizes the redox dysfunction of I/R, or reversible protein S-sulfhydration (persulfidation) and associated endogenous H2S signaling pathways rescues mitochondrial ETC from irreversible cysteine oxidation induced by I/R (74).
Future studies are likely to reveal new layers of complexity: characterization of the PMF and Δp-supported backpressure as regulated by more diverse mechanisms of mitochondrial ROS production and regulation, including how SOD2, catalase, and HCCS regulate the PMF in basal conditions, how they restore the PMF during postischemic reperfusion, and how activation of neutrophil, H2S signaling, and NO metabolism in mitochondria can impact the PMF and redox setting during the I/R-induced inflammatory progress. As this frontier of knowledge advances, continued attention to the fundamental process of mitochondrial ROS production and associated redox regulation will be needed to understand disease pathogenesis and develop improved treatments to ameliorate disease.
Conclusions
Mitochondrial ROS play critical roles in disease pathogenesis and redox signal transduction. Complex I and complex III are the most extensively characterized ETC complexes mediating ROS generation in mitochondria, and they appear to be responsible for the majority of mitochondrial ROS in cellular physiology and disease. In the cardiovascular system, substantial evidence supports the primary physiological function of mitochondrial ROS (i.e., H2O2) as a metabolic dilator to mediate intracellular signaling of myocardial blood flow to support cardiac work (16, 75–77). Overexpressing SOD2 in the myocytes imposes a reductive stress, which alleviates the conditions of elevated oxidative stress. In the disease of myocardial ischemia and reperfusion, oxidative injury to complex I and complex III with increased cysteine sulfonation and heme damage can weaken its catalytic function and the function driving PMF and enhance ROS production in a vicious cycle (Fig. 4). Overexpressing SOD2 in the myocytes protects complex I and complex III from I/R-mediated oxidative injury as well as normalizes I/R-induced redox dysfunction (17, 18). Downregulation of HCCS mediates c-type heme defects in the disease progression of acute myocardial I/R (18). As proposed in the Fig. 4, the HCCS of the IMS thus serves as an upstream mediator in conserving the integrity of the c-type cytochrome and minimizing electron leakage, whereas the SOD2 of the matrix is a downstream scavenger of •O2− that maintains redox homeostasis in mitochondria.
Figure 4.

Redox pathway in the mitochondria of postischemic heart. The pathway shows that acute I/R mediates overproducing •O2− by complex I and complex III in mitochondria, which enhances protein cysteine sulfonation (PrSO3H) of complex I and complex III. Under I/R conditions, PrSO3H of complex I and complex III is further enhanced by impairment of GR2, GPx, and SOD2 of mitochondrial antioxidant enzyme pathway. Acute I/R also mediates mitochondrial injury via downregulation of HCCS and related c-type heme damage of complex III and cyt c. HCCS, cytochrome c heme lyase or holocytochrome c synthase; I/R, ischemia and reperfusion.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16691935.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.17125595.
GRANTS
This work was supported in part by NIH grants (HL83237).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C-L.C. and Y.-R.C. conceived and designed research; C-L.C., L.Z., and Y.-R.C. performed experiments; C-L.C., L.Z., Z.J., T.K., and Y.-R.C. analyzed data; T.K. and Y.-R.C. interpreted results of experiments; Y.-R.C. prepared figures; Y.-R.C. drafted manuscript; T.K. and Y.-R.C. edited and revised manuscript; C-L.C., L.Z., Z.J., T.K., and Y.-R.C. approved final version of manuscript.
REFERENCES
- 1.Fiedorczuk K, Letts JA, Degliesposti G, Kaszuba K, Skehel M, Sazanov LA. Atomic structure of the entire mammalian mitochondrial complex I. Nature 538: 406–410, 2016. doi: 10.1038/nature19794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121: 1043–1057, 2005. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 3.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 281: 64–71, 1998. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 4.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 272: 1136–1144, 1996. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 5.Wallace DC. Mitochondrial diseases in man and mouse. Science 283: 1482–1488, 1999. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 6.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359–407, 2005. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res 70: 181–190, 2006. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 8.Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol 69: 51–67, 2007. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 9.Davis EJ, Davis-van Thienen WI. Force-flow and back-pressure relationships in mitochondrial energy transduction: an examination of extended state 3-state 4 transitions. Arch Biochem Biophys 275: 449–458, 1989. doi: 10.1016/0003-9861(89)90391-3. [DOI] [PubMed] [Google Scholar]
- 10.Kang PT, Chen C-L, Lin P, Chilian WM, Chen Y-R. Impairment of pH gradient and membrane potential mediates redox dysfunction in the mitochondria of the post-ischemic heart. Basic Res Cardiol 112: 36, 2017. [Erratum in Basic Res Cardiol 112: 49, 2017] doi: 10.1007/s00395-017-0626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang PT, Chen CL, Ren P, Guarini G, Chen YR. BCNU-induced gR2 DEFECT mediates S-glutathionylation of complex I and respiratory uncoupling in myocardium. Biochem Pharmacol 89: 490–502, 2014. doi: 10.1016/j.bcp.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Votyakova TV, Reynolds IJ. DeltaPsi(m)-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem 79: 266–277, 2001. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 13.Lambert AJ, Brand MD. Superoxide production by NADH: ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J 382: 511–517, 2004. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu X, Liu B, Zhou S, Chen YR, Deng Y, Zweier JL, He G. Ischemic preconditioning prevents in vivo hyperoxygenation in postischemic myocardium with preservation of mitochondrial oxygen consumption. Am J Physiol Heart Circ Physiol 293: H1442–H1450, 2007. doi: 10.1152/ajpheart.00256.2007. [DOI] [PubMed] [Google Scholar]
- 15.Zhu X, Zuo L, Cardounel AJ, Zweier JL, He G. Characterization of in vivo tissue redox status, oxygenation, and formation of reactive oxygen species in postischemic myocardium. Antioxid Redox Signal 9: 447–455, 2007. doi: 10.1089/ars.2006.1389. [DOI] [PubMed] [Google Scholar]
- 16.Kang PT, Chen CL, Ohanyan V, Luther DJ, Meszaros JG, Chilian WM, Chen YR. Overexpressing superoxide dismutase 2 induces a supernormal cardiac function by enhancing redox-dependent mitochondrial function and metabolic dilation. J Mol Cell Cardiol 88: 14–28, 2015. doi: 10.1016/j.yjmcc.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang PT, Chen C-L, Lin P, Zhang L, Zweier JL, Chen Y-R. Mitochondrial complex I in the post-ischemic heart: reperfusion-mediated oxidative injury and protein cysteine sulfonation. J Mol Cell Cardiol 121: 190–204, 2018. doi: 10.1016/j.yjmcc.2018.07.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CL, Kang PT, Zhang L, Xiao K, Zweier JL, Chilian WM, Chen YR. Reperfusion mediates heme impairment with increased protein cysteine sulfonation of mitochondrial complex III in the post-ischemic heart. J Mol Cell Cardiol 161: 23–38, 2021. doi: 10.1016/j.yjmcc.2021.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajasekaran NS, Connell P, Christians ES, Yan L-J, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ, Benjamin IJ. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 130: 427–439, 2007. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Chen CL, Kang PT, Jin Z, Chen YR. Differential protein acetylation assists import of excess SOD2 into mitochondria and mediates SOD2 aggregation associated with cardiac hypertrophy in the murine SOD2-tg heart. Free Radic Biol Med 108: 595–609, 2017. doi: 10.1016/j.freeradbiomed.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown GC, Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res 75: 283–290, 2007. doi: 10.1016/j.cardiores.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 22.Kang PT, Chen CL, Chen YR. Increased mitochondrial prooxidant activity mediates up-regulation of Complex I S-glutathionylation via protein thiyl radical in the murine heart of eNOS−/−. Free Radic Biol Med 79: 56–68, 2015. doi: 10.1016/j.freeradbiomed.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 119: 2789–2797, 2009. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cadenas E, Boveris A, Ragan CI, Stoppani AO. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys 180: 248–257, 1977. doi: 10.1016/0003-9861(77)90035-2. [DOI] [PubMed] [Google Scholar]
- 25.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 191: 421–427, 1980. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YR, Chen CL, Yeh A, Liu X, Zweier JL. Direct and indirect roles of cytochrome b in the mediation of superoxide generation and NO catabolism by mitochondrial succinate-cytochrome c reductase. J Biol Chem 281: 13159–13168, 2006. doi: 10.1074/jbc.M513627200. [DOI] [PubMed] [Google Scholar]
- 27.Kim H, Xia D, Yu CA, Xia JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Inhibitor binding changes domain mobility in the iron-sulfur protein of the mitochondrial bc1 complex from bovine heart. Proc Natl Acad Sci USA 95: 8026–8033, 1998. doi: 10.1073/pnas.95.14.8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH. Electron transfer by domain movement in cytochrome bc1. Nature 392: 677–684, 1998. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 29.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237: 408–414, 1985. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 30.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279: 49064–49073, 2004. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 31.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277: 44784–44790, 2002. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 32.Chen CL, Chen J, Rawale S, Varadharaj S, Kaumaya PPT, Zweier JL, Chen YR. Protein tyrosine nitration of the flavin subunit is associated with oxidative modification of mitochondrial complex II in the post-ischemic myocardium. J Biol Chem 283: 27991–28003, 2008. doi: 10.1074/jbc.M802691200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen YR, Chen CL, Pfeiffer DR, Zweier JL. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem 282: 32640–32654, 2007. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 34.Lasley RD, Ely SW, Berne RM, Mentzer RM Jr.. Allopurinol enhanced adenine nucleotide repletion after myocardial ischemia in the isolated rat heart. J Clin Invest 81: 16–20, 1988. doi: 10.1172/JCI113288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem 281: 32724–32727, 2006. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 36.Babot M, Birch A, Labarbuta P, Galkin A. Characterisation of the active/de-active transition of mitochondrial complex I. Biochim Biophys Acta 1837: 1083–1092, 2014. doi: 10.1016/j.bbabio.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinogradov AD. Catalytic properties of the mitochondrial NADH–ubiquinone oxidoreductase (complex I) and the pseudo-reversible active/inactive enzyme transition. Biochim Biophys Acta 1364: 169–185, 1998. doi: 10.1016/S0005-2728(98)00026-7. [DOI] [PubMed] [Google Scholar]
- 38.Maklashina E, Kotlyar AB, Karliner JS, Cecchini G. Effect of oxygen on activation state of complex I and lack of oxaloacetate inhibition of complex II in Langendorff perfused rat heart. FEBS Lett 556: 64–68, 2004. doi: 10.1016/S0014-5793(03)01369-3. [DOI] [PubMed] [Google Scholar]
- 39.Gorenkova N, Robinson E, Grieve DJ, Galkin A. Conformational change of mitochondrial complex I increases ROS sensitivity during ischemia. Antioxid Redox Signal 19: 1459–1468, 2013. doi: 10.1089/ars.2012.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen YR, Chen CL, Zhang L, Green-Church KB, Zweier JL. Superoxide generation from mitochondrial NADH dehydrogenase induces self-inactivation with specific protein radical formation. J Biol Chem 280: 37339–37348, 2005. doi: 10.1074/jbc.M503936200. [DOI] [PubMed] [Google Scholar]
- 41.Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem 279: 4127–4135, 2004. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- 42.Ohnishi ST, Shinzawa-Itoh K, Ohta K, Yoshikawa S, Ohnishi T. New insights into the superoxide generation sites in bovine heart NADH-ubiquinone oxidoreductase (complex I): the significance of protein-associated ubiquinone and the dynamic shifting of generation sites between semiflavin and semiquinone radicals. Biochim Biophys Acta 1797: 1901–1909, 2010. doi: 10.1016/j.bbabio.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 43.Ohnishi ST, Ohnishi T, Muranaka S, Fujita H, Kimura H, Uemura K, Yoshida K, Utsumi K. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J Bioenerg Biomembr 37: 1–15, 2005. doi: 10.1007/s10863-005-4117-y. [DOI] [PubMed] [Google Scholar]
- 44.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH: ubiquinone oxidoreductase (complex I). J Biol Chem 279: 39414–39420, 2004. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 45.Chen J, Chen CL, Rawale S, Chen CA, Zweier JL, Kaumaya PT, Chen YR. Peptide-based antibodies against glutathione-binding domains suppress superoxide production mediated by mitochondrial complex I. J Biol Chem 285: 3168–3180, 2010. doi: 10.1074/jbc.M109.056846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hurd TR, Requejo R, Filipovska A, Brown S, Prime TA, Robinson AJ, Fearnley IM, Murphy MP. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem 283: 24801–24815, 2008. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen CL, Zhang L, Yeh A, Chen CA, Green-Church KB, Zweier JL, Chen YR. Site-specific S-glutathiolation of mitochondrial NADH ubiquinone reductase. Biochemistry 46: 5754–5765, 2007. doi: 10.1021/bi602580c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor ER, Hurrell F, Shannon RJ, Lin TK, Hirst J, Murphy MP. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J Biol Chem 278: 19603–19610, 2003. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 49.Kang PT, Zhang L, Chen CL, Chen J, Green KB, Chen YR. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic Biol Med 53: 962–973, 2012. doi: 10.1016/j.freeradbiomed.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, Chen YR, He G. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochim Biophys Acta 1794: 476–485, 2009. doi: 10.1016/j.bbapap.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cochemé HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RAJ, Krieg T, Brookes PS, Murphy MP. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19: 753–759, 2013. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berrisford JM, Baradaran R, Sazanov LA. Structure of bacterial respiratory complex I. Biochim Biophys Acta 1857: 892–901, 2016. doi: 10.1016/j.bbabio.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 53.Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature 494: 443–448, 2013. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem 279: 47939–47951, 2004. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 55.Zhu J, Vinothkumar KR, Hirst J. Structure of mammalian respiratory complex I. Nature 536: 354–358, 2016. doi: 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta 1817: 851–862, 2012. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 57.Ganesana M, Erlichman JS, Andreescu S. Real-time monitoring of superoxide accumulation and antioxidant activity in a brain slice model using an electrochemical cytochrome c biosensor. Free Radic Biol Med 53: 2240–2249, 2012. doi: 10.1016/j.freeradbiomed.2012.10.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cape JL, Bowman MK, Kramer DM. A semiquinone intermediate generated at the Qo site of the cytochrome bc1 complex: importance for the Q-cycle and superoxide production. Proc Natl Acad Sci USA 104: 7887–7892, 2007. doi: 10.1073/pnas.0702621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry 42: 6493–6499, 2003. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 60.Zhang L, Yu L, Yu CA. Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J Biol Chem 273: 33972–33976, 1998. doi: 10.1074/jbc.273.51.33972. [DOI] [PubMed] [Google Scholar]
- 61.Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ, Hoppel CL. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys 385: 117–128, 2001. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
- 62.Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjöstedt E, Lundberg E, Szigyarto CA, Skogs M, Takanen JO, Berling H, Tegel H, Mulder J, Nilsson P, Schwenk JM, Lindskog C, Danielsson F, Mardinoglu A, Sivertsson A, von Feilitzen K, Forsberg M, Zwahlen M, Olsson I, Navani S, Huss M, Nielsen J, Ponten F, Uhlén M. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13: 397–406, 2014. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drenckhahn J-D, Schwarz QP, Gray S, Laskowski A, Kiriazis H, Ming Z, Harvey RP, Du X-J, Thorburn DR, Cox TC. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev Cell 15: 521–533, 2008. doi: 10.1016/j.devcel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 64.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res 114: 524–537, 2014. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Charles RL, Schröder E, May G, Free P, Gaffney PR, Wait R, Begum S, Heads RJ, Eaton P. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics 6: 1473–1484, 2007. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 66.Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, Steenbergen C. Characterization of potential S-nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol 300: H1327–H1335, 2011. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res 108: 418–426, 2011. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol 46: 215–234, 2006. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 69.Costa NJ, Dahm CC, Hurrell F, Taylor ER, Murphy MP. Interactions of mitochondrial thiols with nitric oxide. Antioxid Redox Signal 5: 291–305, 2003. doi: 10.1089/152308603322110878. [DOI] [PubMed] [Google Scholar]
- 70.Chen YR, Deterding LJ, Sturgeon BE, Tomer KB, Mason RP. Protein oxidation of cytochrome C by reactive halogen species enhances its peroxidase activity. J Biol Chem 277: 29781–29791, 2002. doi: 10.1074/jbc.M200709200. [DOI] [PubMed] [Google Scholar]
- 71.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1: 223–232, 2005. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 72.Chen YR, Chen CL, Chen W, Zweier JL, Augusto O, Radi R, Mason RP. Formation of protein tyrosine ortho-semiquinone radical and nitrotyrosine from cytochrome c-derived tyrosyl radical. J Biol Chem 279: 18054–18062, 2004. doi: 10.1074/jbc.M307706200. [DOI] [PubMed] [Google Scholar]
- 73.Zhao H, Sun J, Deschamps AM, Kim G, Liu C, Murphy E, Levine RL. Myristoylated methionine sulfoxide reductase A protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 301: H1513–H1518, 2011. doi: 10.1152/ajpheart.00441.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zivanovic J, Kouroussis E, Kohl JB, Adhikari B, Bursac B, Schott-Roux S, Petrovic D, Miljkovic JL, Thomas-Lopez D, Jung Y, Miler M, Mitchell S, Milosevic V, Gomes JE, Benhar M, Gonzalez-Zorn B, Ivanovic-Burmazovic I, Torregrossa R, Mitchell JR, Whiteman M, Schwarz G, Snyder SH, Paul BD, Carroll KS, Filipovic MR. Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-sulfhydration. Cell Metab 30: 1152–1170, 2019. doi: 10.1016/j.cmet.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saitoh S, Zhang C, Tune JD, Potter B, Kiyooka T, Rogers PA, Knudson JD, Dick GM, Swafford A, Chilian WM. Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol 26: 2614–2621, 2006. doi: 10.1161/01.ATV.0000249408.55796.da. [DOI] [PubMed] [Google Scholar]
- 76.Rogers PA, Chilian WM, Bratz IN, Bryan RM Jr, Dick GM. H2O2 activates redox- and 4-aminopyridine-sensitive Kv channels in coronary vascular smooth muscle. Am J Physiol Heart Circ Physiol 292: H1404–H1411, 2007. doi: 10.1152/ajpheart.00696.2006. [DOI] [PubMed] [Google Scholar]
- 77.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res 110: 471–480, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16691935.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.17125595.