

Abstract
Human uracil DNA-glycosylase (UDG) is the prototypic and first identified DNA glycosylase with a vital role in removing deaminated cytosine and incorporated uracil and 5-fluorouracil (5-FU) from DNA. UDG depletion sensitizes cells to high APOBEC3B deaminase and to pemetrexed (PEM) and floxuridine (5-FdU), which are toxic to tumor cells through incorporation of uracil and 5-FU into DNA. To identify small-molecule UDG inhibitors for pre-clinical evaluation, we optimized biochemical screening of a selected diversity collection of >3,000 small-molecules. We found aurintricarboxylic acid (ATA) as an inhibitor of purified UDG at an initial calculated IC50 < 100 nM. Subsequent enzymatic assays confirmed effective ATA inhibition but with an IC50 of 700 nM and showed direct binding to the human UDG with a KD of <700 nM. ATA displays preferential, dose-dependent binding to purified human UDG compared to human 8-oxoguanine DNA glycosylase. ATA did not bind uracil-containing DNA at these concentrations. Yet, combined crystal structure and in silico docking results unveil ATA interactions with the DNA binding channel and uracil-binding pocket in an open, destabilized UDG conformation. Biologically relevant ATA inhibition of UDG was measured in cell lysates from human DLD1 colon cancer cells and in MCF-7 breast cancer cells using a host cell reactivation assay. Collective findings provide proof-of-principle for development of an ATA-based chemotype and “door stopper” strategy targeting inhibitor binding to a destabilized, open pre-catalytic glycosylase conformation that prevents active site closing for functional DNA binding and nucleotide flipping needed to excise altered bases in DNA.
Keywords: DNA repair, UDG, Ligand, Cancer, Structures
1. Introduction
1.1. Base excision repair
Base excision repair (BER) protects DNA from spontaneous base modifications, such as cytosine deamination, endogenous oxygen radicals, extrinsic chemical mutagens, and alkylation as well as base misincorporation during replication due to nucleotide pool imbalance and incorporation of abnormal bases introduced as cancer chemotherapeutic agents (Hitomi et al., 2007; Krokan et al., 1997, 2002). Pyrimidine analogs such as floxuridine (5-FdU) (Longley et al., 2003; McCullough et al., 1999) are metabolized into nucleotides and incorporated into DNA while antifolate drugs such as pemetrexed (PEM) block the thymidylate synthase-mediated conversion of UMP to TMP causing nucleotide pool imbalance and incorporation of uridine during DNA synthesis. These agents kill cells by disrupting DNA replication and they are more toxic in cells that are defective for BER of uracil. As a result, strategies for inhibiting BER represent a promising strategy for sensitizing cancer cells to 5-FdU and PEM.
DNA glycosylases are small monomeric DNA binding proteins that initiate BER by recognizing and then removing altered bases by severing the N-glycosidic bond between base and deoxyribose, forming an apurinic/apyrimidinic (AP) site (Mol et al., 1999). Glycosylases typically do not require co-factors but some do require 4Fe–4S clusters such as MutY and endonuclease III (Guan et al., 1998; Mol et al., 1995c; Thayer et al., 1995), and some can also cleave the AP site (Duncan and Miller, 1980). Eleven known human DNA glycosylases recognize and process a wide range of DNA lesions, including deaminated, alkylated, and oxidized bases (Gossage et al., 2012; Mol et al., 2000a; Tsutakawa et al., 2014). Following damaged base excision, AP endonuclease cleaves the DNA sugar backbone, activating downstream BER proteins from either the small patch or long patch sub-pathway to complete repair (Mol et al., 1995b; Parikh et al., 2000a; Putnam et al., 1999).
1.2. Uracil DNA glycosylase
Uracil-DNA glycosylase (UDG) was the first DNA glycosylase discovered (Krokan et al., 1997, 2000; Wyatt and Wilson, 2009). UDG efficiently removes uracil, as well as the synthetic 5-FU, 5-hydroxyuracil, alloxan, and isodialuric acid, albeit at significantly lower rates. Studies from poxviruses, bacteria, yeast, fish, plants, mammals, and human cells find the different UDGs share a conserved enzymatic activity and active site. In humans, the UNG gene encodes for UDG protein; the gene spans an estimated 13.5 kilobases at chromosome 12q23-q24.1(Krokan et al., 1997; Parikh et al., 2000a; Putnam et al., 1999). Alternative splicing of UNG produces ~35 kDa nuclear and ~33 kDa mitochondrial isoforms. Both share the core enzymatic active site but differ in their N-termini (Krokan et al., 2000).
Crystal structures of human, viral, and E.coli UDG further reveal it is conserved (Kim and Wilson, 2012). Co-crystal structures of UDG protein in complex with DNA support an UDG-DNA interaction mechanism and specificity for uracil-DNA. Ugi, a protein inhibitor, binds and inhibits UDG without contacting its uracil pocket (Mol et al., 1995b; Parikh et al., 2000a; Putnam et al., 1999). Instead it targets the conserved DNA-binding groove in its open conformation (Mol et al., 1995a; Putnam et al., 1999) suggesting potent UDG inhibitors need not bind the uracil pocket.
1.3. Uracil DNA glycosylase and cancer
UDG removes incorporated uracil and 5-FU base lesions from the DNA in cancer cells (Fischer et al., 2007; Pettersen et al., 2011). So blocking UDG with a specific inhibitor could have therapeutic benefits by improving anti-cancer effects of existing chemotherapeutic agents, such as PEM, which promote uracil incorporation through inhibition of thymidylate synthase (TS) (Bulgar et al., 2012; Weeks et al., 2013, 2014). Similarly, 5-FdU also inhibits TS or the drug is metabolized to 5-FdUTP and is incorporated along with uracil due to inhibition of TS (Kim and Wilson, 2012; Longley et al., 2003; Malet-Martino et al., 2002). In fact, shRNA/siRNA-mediated abrogation of UDG expression decreased viability of human cancer cell lines treated with either PEM or 5-FdU (Bulgar et al., 2012; Condie et al., 2015; Weeks et al., 2013, 2014; Yan et al., 2016, 2018).
These data imply that UDG-dependent repair activity can dictate cancer cell sensitivity to chemotherapeutic drugs like PEM and 5-FdU, which are dependent on incorporation of uracil and 5-FU into DNA. So a UDG small-molecule inhibitor could provide an enabling tool for preclinical investigations on the impact of reducing UDG activity on multiple types of cancer cells under therapeutically relevant conditions. More generally, we reasoned that inhibiting the specific DNA glycosylase initiating BER may provide strategic advantages over inhibiting later steps where DNA product binding and direct handoff of intermediates to subsequent pathway enzymes may be more challenging to block with small molecule inhibitors (Mol et al., 2000b; Parikh et al., 1999; Wilson and Kunkel, 2000).
1.4. An effective UDGi tool and novel inhibitor design strategy
The Stivers laboratory described UDG inhibitor screening by a uracil-based tethering approach (Jiang et al., 2005) (Chung et al., 2009; Jiang et al., 2006) and developed uracil-linker compounds complexed with UDG (Chung et al., 2009; Krosky et al., 2006). Here we employed an optimized high-throughput screening (HTS) platform by building upon a robust in vitro biochemical assay (Chung et al., 2009; Krosky et al., 2006) and a select diversity chemical library. We found that the most potent inhibitor was ATA and further characterized its properties as a UDG inhibitor (UDGi) tool compound for cancer research. ATA was previously seen to inhibit UDG (Li et al., 2012), so here starting with an unbiased screen we independently support and extend this finding with biochemical, structural and cellular data. ATA can bind multiple proteins (Cushman et al., 1991; Dixon et al., 2015; Dorjsuren et al., 2011; Ghosh et al., 2009; Gonzalez et al., 1980; Hashem et al., 2009; Hung et al., 2009; Kuban-Jankowska et al., 2017; Obrecht et al., 2019; Tan et al., 2012). Yet, we reasoned that as ATA provides a distinct chemotype from uracil, its ability to bind nucleic acid processing enzymes might prove advantageous.
In fact, identification of the structural mechanism whereby ATA holds UDG in its open destabilized configuration provides a prototypic chemically distinct entity or chemotype for a “door stopper” strategy to guide design of clinical UDG inhibitors. In contrast, binding uracil promotes the closed DNA-binding conformation of UDG, so uracil-like compounds may have larger challenges to avoid being displaced by the larger DNA interface with the entire active site channel compared to compounds that inhibit UDG in a manner similar to that of the ATA complex. As opposed to canonical inhibitors based upon transition state mimicry (Bianchet et al., 2003; Friedman et al., 2009; Jiang et al., 2003, 2004), our results uncover strategic advantages for BER enzymes to instead block the conformational closing needed for damaged DNA substrate recognition and catalysis. By analogy, the identified UDGi acts as a “door stopper” that is binding an inactive conformation like an allosteric inhibitor, but also binds in a manner blocking formation of the active site. ATA holds the active site open such that the closed conformation needed for tight and productive DNA binding plus “lock and key” substrate recognition and catalysis by UDG is precluded.
2. Methodology
2.1. High-throughput small-molecule screening (HTS) biochemical assay
HTS screening assay was used to identify UDG inhibitors. The collection of biologically active molecules was compiled from the LOPAC library (Sigma Aldrich) and Bioactive Compound Library (Selleck Chemicals). A total of 3115 mechanistically annotated compounds were used for screening. All stock solutions were prepared in dimethyl sulfoxide (DMSO) at 3 mM. Upon hits identification, all compounds were purchased from commercial vendors and retested from 10 mM stock solutions in DMSO.
For screening, 384-well assay plates were prepared with final test concentrations of 12.5 μm using a Janus liquid handling platform (Perkin Elmer) equipped with a 50 nL pin transfer tool (V&P Scientific). For concentration-response studies eight concentrations in half-log dilutions, final test plates were prepared from 10 mM DMSO stock solutions using a Janus liquid handling platform (Perkin Elmer) equipped with a standard 96 tip head. A final DMSO concentration of 0.125% was not exceeded in the screening assay and hit validation. The negative controls contained the same percentage of the vehicle. Small molecule solutions were transferred to a 384 well microtiter plate (Corning 3573) where each well contained 20 μL of reaction buffer (2x: 40 mM Tris HCl, 100 mM KCl, 0.4 mM MgCl2, 2 mM DTT, 0.1%Tween), 50 nL of 10 mM compound stock in 100% DMSO, 10 μL of a 40 nM stock of recombinant GST tagged human mitochondrial UDG, expressed in E.coli (Creative BioMart), 10 μL of a double-stranded 14-mer DNA hairpin that contained nine U: A base pairs termed uracil DNA hairpin (100 nM stock, 5′FAM- GCA CUU AAG AAU UGC AAU UCU UAA GUG C-3′Dabcyl, Eurofins Genomics). After 40 min of incubation at room temperature (~23 °C), fluorescence measurements were done using an Enspire plate reader (Perkin Elmer) at an excitation wavelength of 485 nm and emission wavelength of 520 nm. Compound performance was calculated and normalized for each plate with background fluorescence adjustment by the following equation:
Signal over noise ratio (S/N) was determined for each plate, and Z prime was calculated based on the following equation: Z prime = 1 - [(3SD of sample +3SD of control)/(mean of the sample–mean of control)] (Zhang et al., 1999).
Commercial ATA was purchased from Sigma Aldrich (product# A1895). Triammonium ATA salt was purchased from Alfa Asear.
2.2. Multiple uracil-containing DNA hairpin melting
Uracil DNA hairpin-melting analysis was done to study the interaction between ATA and DNA in the absence of UDG protein. DNA was diluted to 1000 nM stock (in water). DNA hairpin was heated to 50 °C for 20 min and allowed to cool for at least 60 min to room temperature. Each reaction (total 40 μL) included: 15.5 μL of ddH20, 4 μL of buffer (2 mM TRIS-HCl, 5 mM KCl, 0.02 MgCl2, 0.1 mM DTT, 0.005% Tween), 0.5 μL of 100% DMSO or ATA. Pre-incubation took place for 20 min at room temperature. Then, 20 μL of uracil DNA hairpin (1000 nM stock, 5′FAM- GCA CUU AAG AAU UGC AAU UCU UAA GUG C-3′Dabcyl) was added to initiate the reaction. A transparent sealant was applied to seal the plate and incubation occurred for 45 min at room temperature. Each plate was spun down gently for 20 sec and was loaded onto an RT-PCR machine (BioRad CFX96, FAM filter). FAM fluorescence intensity was monitored every 1.6 °C/2min from 25°C to 97°C. RFU and negative derivative RFU raw data and plots from the machine were obtained and analyzed. Melting temperature (Tm) value was taken directly from the midpoint on the −dRFU/dt vs. temperature graph.
2.3. UDG-ATA co-crystallization, data collection, and structure refinement
Co-crystallization was done to study the interaction sites between UDG and ATA. UDG was expressed and purified following a published protocol (Parikh et al., 1998). ATA compound was dissolved in water. We grew crystals using the hanging drop method for co-crystallization of the UDG protein construct and ATA at a concentration of 4 mM. Crystallization condition was 2 mM TRIS pH 8.5, 20 mM MgCl2, 21% PEG8000 at 291 K. Diffraction data were collected, processed, and refined. X-ray data for crystallography were collected at SSRL BEAMLINE BL14-1 refined structures have useful statistics and geometry (Table 1). The refined X-ray crystal structure with diffraction data for UDG and bound ATA was deposited in the Protein Data Bank (6VBA).
Table 1.
X-ray diffraction data collection and refinement statistics.
| UDG-ATA | |
|---|---|
| Data collection | |
| Space group | P 21 21 2 |
| Cell dimensions | |
| a, b, c (Å) | 73.65, 54.78, 59.99 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 35.45–1.8 |
| Rsym or Rmerge | 12.5 |
| I/σI | 15 |
| Completeness (%) | 91.91 |
| Redundancy | 11.2 |
| Refinement | |
| Resolution (Å) | 1.8 |
| No. reflections | 21264 |
| Rwork/Rfree | 17.92/23.36 |
| No. atoms | |
| Protein | 1808 |
| Ligand/ion | 62 |
| Water | 259 |
| B-factors | |
| Protein | 23.07 |
| Ligand/ion | 89.20 |
| Water | 31.76 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.0036 |
| Bond angles (°) | 0.8775 |
2.4. Molecular docking
ATA-UDG binding conformations were computationally examined. We selected a series of apoprotein structures of UDG including: PDB 1AKZ which is the apo structure solved at 1.57 Å(Parikh et al., 1998), PDB 1UGH, which is the UDG-Ugi complex solved at 1.9 Å(Mol et al., 1995a), PDB 1SSP, which is a product complex and uracil solved at 1.9 Å(Parikh et al., 1998), and PDB 1EMH, which is the complex of UDG-dsDNA-pseudouracil solved at 1.8 Å (Parikh et al., 2000b). Each structure was prepared for docking by eliminating water and bound ligands. Specifically, for 1UGH we eliminated the Ugi chain, for the 1SSP we eliminated the DNA substrates chains and uracil, and for 1EMH the ds-DNA substrate. The UDG coordinates for each structure were prepared and minimized using Protein Preparation Wizard (Schrödinger Suite 2019-2; Epik, Schrödinger, LLC, New York, NY, 2019; Impact, Schrödinger, LLC, New York, NY, 2019; Prime, Schrödinger, LLC, New York, NY, 2019)(Sastry et al., 2013). We focused molecular docking to the active site region, and selected the residue GLN144 to prepare the docking grid for all structures. We docked ligands with length ≤ 20 Å with cubic box dimensions 15 Å. Ligands used for this study were uracil, salicylic acid, and ATA. The downloaded files contain the atoms and topology of the ligands from PubChem (https://pubchem.ncbi.nlm.nih.gov); we then prepared the ligands for docking experiments using LigPrep (Schrödinger Release 2019-2: LigPrep, Schrödinger, LLC, New York, NY, 2019.). We ran a single docking experiment for each ligand to each of the four prepared UDG structures. We superposed all structures to the UDG Chain of 1SSP to compare the population of docking positions. For the docking experiments, we used Glide Extra Precision (XP) (Schrödinger Release 2019-2: Glide, Schrödinger, LLC, New York, NY, 2019)(Friesner et al., 2004). Each of the ligand conformations generated by LigPrep was automatically docked using flexible docking and ranked based on docking score. The best docking results were selected and compared in different superimposed PyMOL sessions (PyMOL, The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.) to generate figures and 2D ligand-receptor interaction diagram. The computational suite is provided by the SBGrid Consortium (Morin et al., 2013).
2.5. Microscale thermophoresis (MST)
MST (Seidel et al., 2013) was performed to measure the binding affinity between ATA and UDG using the Monolith NT.115 from Nanotemper Technologies. Bacterially expressed and purified full-length human UDG was fluorescently labeled with Atto488 NHS ester according to the manufacturer’s protocol. Briefly, 0.5 mg of protein in 20 mM HEPES, 150 mM NaCl pH 8.0 (final volume 300 μl) was mixed with 10ul Atto488 NHS ester (2 mg/ml in DMSO) and incubated at room temperature for an hour. An equilibrated PD10 desalting column was used to separate labeled proteins from the free dye. Labeling efficiency was determined to be 1:1 (protein to dye) by measuring the absorbance at 280 and 488 nm and the concentration of protein relative to the dye. A solution of ATA was serially diluted from about 25 μM to 12 nM in the presence of fixed concentration of 100 nM Atto488-labeled UDG in 0.01 M HEPES pH 7.4, 0.15 M NaCl, and 0.005% v/v Surfactant P20. The samples were loaded into silica premium capillaries (Polymicro Technologies) and after incubation at room temperature for 15 min measurements were performed at 20 °C, by using 20% LED power and 40% IR-laser power. Measurements were also carried out on 40 and 60% IR-Laser power for comparison. The raw data were exported to GraphPad Prism8 software, and the KD value determined by data fitting with a one-site specific binding model.
2.6. Protein thermal shift
Thermal shifts were done to measure the direct binding between UDG and ATA. Thermal shifts were performed in a 96 well plate format and the individual binding reactions included: 5 μL of protein thermal buffer (Applied Biosystems by Thermo Fisher Scientific #4461335), 10 μL of the purified human UDG (0.8 mg/mL protein stock) from XTAL Biostructures company, recombinant human mitochondrial UDG with a 6xhis tag was expressed in Sf9 insect cells) in 40 mM tris, 150 mM NaCl 1 mM DTT, 10% glycerol, 1uL of either 100% DMSO or compound (working compound stocks: 400μM, 2,000 μM, and 10,000 μM). The molar ratios (MR) of ATA to UDG were as follows: 1.2x, 6.25x, and 31.25x. The reactions were incubated on ice for 20 min in the dark before 2 μL of 10x Sypro orange dye (Applied Biosystems by Thermo Fisher Scientific #4461141) was added to each well. Lastly, 2 μL of ddH20 was added to each reaction to make a total shift reaction volume of 20 μL. Monitoring of fluorescence took place for 2hr (Bioraid CFX96, Hex filter, 25 °C–97 °C, 1.6 °C/2min). RFU and negative derivative RFU raw data and plots from the machine were obtained and analyzed. The melting temperature (Tm) was taken directly from the midpoint on the −dRFU/dt vs. temperature graph.
2.7. Differential scanning fluorimetry assay
Differential scanning fluorimetry (DSF) measurements were performed on a Prometheus NT.48 instrument (NanoTemper Technologies, Munich, Germany). Full-length UDG at 14 μM in 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 pH 7.4 was combined with titrating concentration of ATA (0–125 μM) mixed, incubated at room temperature for 10 min. The capillaries were filled with a 10 μL sample and placed on the sample holder. A temperature gradient of 1 °C·min−1 from 25 to 90 °C was applied and the intrinsic protein fluorescence at 330 and 350 nm was recorded. Additional aggregation onset temperatures were detected via back-reflection light scattering. ATA binding induced an overall decrease in fluorescence intensity in both 330 nm and 350 nm channels and produced a flat ratio curve. Therefore, analysis of single wavelength was used to determine the melting temperature.
2.8. Biochemical DNA cutting assay
DNA cutting activity assay was used to validate the inhibitory effect of ATA on purified human UDG in vitro.
To a 96 well microtiter plate (polypropylene) these reaction components were added: 10 μL of UDG buffer (stock 100 mM Tris-HCl, 250 mM KCl, 1 mM MgCl2, 5 mM DTT, 0.25% Tween), 32 μL of ddH20, 2 μL of DMSO or ATA stock (312 μM, 104 μM, 35 μM or 11.6 μM), and 4 μL of UDG (6,250 pM stock, XTAL Biostructures company recombinant human mitochondrial UDG with a 6xhis tag was expressed in Sf9 insect cells). Following pre-incubation for 20 min at 37 °C, 2 μL of U:A bp DNA duplex 3’TAMsp (500 nM stock, 5’-TCCTGGGTGACAAAGC[deoxyU]AAACACTGTCTCCAAAAAAAATT [TAMsp]-3’, 5’-AATTTTTTTTGGAGACAGTGTTT[A]GCTTTGTCACCCAGGA-3’ ) or T:A bp DNA duplex 3’TAMsp (500nM stock, 5’-TCCTGGGTGACAAAGC[T]AAACACTGTCTCCAAAAAAAATT [TAMsp]-3’, 5’-AATTTTTTTTGGAGACAGTGTTT[A]GCTTTGTCACCCAGGA-3’) was added to initiate the reaction, total reaction volume of 50 μL/well. Each plate was incubated for 120 min at 37 °C. Then, each 96 well plate was sealed and heated at a constant temperature of 95 °C for 55 min using the CFX96 real time system thermocycler. After the heating process, 5 μL of a bromophenol blue denaturing loading dye (20 mg of bromophenol blue, 9.7 ml of formamide and 300 μL of 10M NaOH) was added to each reaction well. DNA products from each reaction were resolved on 20% polyacrylamide nucleic acid denaturing gel (UreaGel 29:1 System EC-829, 7.5M urea +1X TBE, National diagnostics). TBE buffer was prepared by dissolving 54 g of tris base, 28 g of boric acid, and 4.5 g of EDTA in 1.0 L of distilled H2O. Gels were processed at 200 V for 60min in the dark. Gels were imaged based on the fluorescent tags using Typhoon Trio + Variable Mode Imager (Amersham Biosciences) with excitation and emission wavelengths set to 532 nm and 585 nm respectively. Photomultiplier tube voltage (PMT) was set to 400 V, and pixel size resolution set to 100 μm. Gel data were analyzed using ImageJ software. DNA bands were corrected using a background subtraction step. Percent cleavage was calculated by using fluorescence intensity of the cleaved 23-mer DNA strand divided by the sum of the fluorescence bands (23mer+40mer DNA strands)* 100. Relative percent cleavage was generated by using the untreated DMSO condition divided by each treated drug dose *100. Reactions with OGG1 (protein stock from NEBiolabs: humanOGG1) were processed as described above with a different reaction buffer recipe (100 mM TrisHCl, 250 mM KCl, 2 mM MgCl2, 5 mM DTT, 0.25%, 0.5μg/ul of BSA with OGG1 protein stock of 45 nM, 1 μL of OxoG:C bp DNA duplex 3’ Cyanine3 (500 nM stock, 5’-TCCTGGGTGACAAAGC[8oxoG]AAACACTGTCTCCAAAAAAAATT [Cyanine3]-3’, 5’-AATTTTTTTGGAGACAGTGTTT[C]GCTTTGTCACCCAGGA-3’) to a total reaction volume of 50μL/well for an incubation time of 180 min at 37 °C. IC50 was generated using Prism software version 8.4.3, Y=bottom + X* (Top-Bottom/(IC50+X), graphpad.com, non-linear fit, [inhibitor] vs. response (three parameters, R2 = 0.98).
2.9. Cell-free protein extracts from human DLD1 cancer cell lines
DNA cutting activity assay was used to measure ATA inhibitory effect on UDG containing cell extracts in vitro. Total protein lysates were extracted following the manufacturer’s protocol (Millipore CytoBuster protein extraction reagent #71009-3) from human colon DLD1 cancer cell lines: cell lines expressing UDG (wild type and shScramble), a cell line with an sh-knockdown UNG (shKD-UNG), and a cell line with a CRISPR-knockout UDG (KO-UNG). Each 96 well plate contained 10 μM of reaction buffer (final concentration of 20 mM Tris HCl, 50 mM KCl, 0.2 mM MgCl2, 1 mM DTT, 0.05% Tween), distilled H2O, either 2 μL of DMSO or ATA and 20 μg of total whole cell lysate. The reaction was pre-incubated for 20 min at 37 °C before a fluorescently-labeled TAMRA DNA duplex oligo containing a single U:A bp DNA duplex 3’TAMsp (final 30 nM, 5’-TCCTGGGTGACAAAGC[deoxyU]AAACACTGTCTCCAAAAAAAATT [TAMsp]-3’, 5’-AATTTTTTTTGGAGACAGTGTTT[A]GCTTTGTCACCCAGGA-3’) was added to each reaction well with a total reaction volume of 50 μL/well. Plates were further incubated for 20 min at 37 °C. 5 μL of a denaturing loading dye (20 mg of bromophenol blue, 9.7 mL of formamide and 300 μL of 10M NaOH) was added to stop each reaction. DNA products from each reaction were resolved on a 20% denaturing polyacrylamide nucleic acid urea gel (5.3 g of urea was dissolved in 2.3 mL of 5x TBE, 5 mL of 40% bis-acrylamide 29:1 solution). 5x TBE buffer was prepared by dissolving 54 g of tris base, 28 g of boric acid, and 4.5 g of EDTA in a liter of distilled H2O. Gels were processed at 200 V for 60 min at room temperature in the dark. Gels were imaged based on the fluorescent tags using Typhoon Trio + Variable Mode Imager (Amersham Biosciences) with excitation and emission wavelengths set to 532 nm and 585 nm respectively, PMT set to 400, and pixel size resolution set to 100 μm. Gel data were analyzed using ImageJ software. DNA bands were corrected using a constant background band intensity subtraction step. Then, percent cleavage was calculated by using fluorescence intensity of the cleaved 23-mer DNA strand divided by the sum of the fluorescence bands (23mer+40mer DNA strands)* 100 for untreated and treated conditions. Relative percent cleavage was generated by using each treated drug dose divided by the untreated DMSO condition*100. IC50 was estimated using Prism software version 8.4.3, non-linear fit, Y = 100*(X^HillSlope)/(IC50^HillSlope + (X^HillSlope)), graphpad.com, [inhibitor] vs. normalized response (variable slope, R2 = 0.97) averaging points from four gels.
2.9.1. Measurement of uracil removal in reporter plasmids in cells
Removal of uracil in cells was measured by using a reporter plasmid containing a dU:dG base pair (referred to as BFP_U), as described previously (Chaim et al., 2017). In brief, dU is substituted into the transcribed strand at position 191 of the blue fluorescent protein (BFP) gene. When RNA polymerase II incorporates dA opposite dU, transcripts encoding wild type BFP are produced. Upon repair, dU is replaced by dC, leading to production of transcripts encoding a non-fluorescent protein. Hence, fluorescent signal is directly proportional to the dU presence in the reporter plasmid. The reporter plasmid (100 ng per transfection) was mixed with a transfection control plasmid (pmax GFP, 100 ng per transfection) along with a carrier plasmid (800 ng per transfection) that does not produce fluorescent protein to constitute the “uracil repair” cocktail. A corresponding cocktail of damage-free plasmids containing pmax BFP (100 ng per transfection), pmax GFP (100 ng per transfection), and carrier plasmid (800 ng per transfection) constituted the “undamaged” cocktail. After normalizing to expression of the pmax,GFP transfection control, expression of BFP_U in the damaged cocktail was normalized to similarly normalized expression of pmax_BFP in the undamaged cocktail. The reciprocal of BFP_U reporter expression was calculated for each experimental condition. Finally, the reciprocal of BFP_U reporter expression for each inhibitor concentration was normalized to reciprocal of BFP_U reporter expression for cells treated with vehicle alone (DMSO).
Reporter plasmids for measuring removal of hypoxanthine, 8-oxoG opposite dC, and adenine opposite 8-oxoG were as reported previously (Chaim et al., 2017). The reporter plasmid for measuring removal of thymidine opposite dG operates under the same principle as BFP_U and the other glycosylase reporter assays used in this study. Thymidine is inserted into position 444 in the transcribed strand of the pmax mOrange plasmid (such that it lies opposite dG). This creates a premature stop codon, abolishing fluorescence. Removal of thymidine followed by insertion of dC opposite dG restores reporter fluorescence.
2.9.2. Measurement of ATA in vivo using a host-cell reactivation assay
A previously described fluorescent host cell reactivation assay was used to measure initiation of BER at a U:G base pair (Chaim et al., 2017). Human breast cancer MCF-7 cells were seeded at 50,000 cells per well in a 12-well plate, while HAP cells (wild type or UNG knockout) were seeded at 75,000 cells per well in a 6-well plate. Cells were allowed to adhere overnight and then cells in duplicate wells were treated with DMSO or ATA (2.5, 5, or 10 μM) for 24 h such that the DMSO concentration in each well was 0.1%. Following drug treatment, one well was transfected with the undamaged plasmid cocktail and the other well was transfected with the cocktail including the U:G lesion containing reporter plasmid. Transfection was carried out using Lipofectamine 3000 (ThermoFisher) according to the manufacturer’s instructions. In brief, each transfection utilized P3000 reagent (4 μL) and Lipofectamine 3000 reagent (3.75 μL) mixed with the plasmid cocktail in serum-free medium (Opti-MEM, ThermoFisher). For experiments in 12-well plates, 100 μL of medium was used, and 200 μL of medium was used for experiments in 6-well plates. After 24 h, cells were dissociated by trypsinization and analyzed by flow cytometry using an Atune NxT flow cytometer. Gating and compensation were determined by transfection of single-color controls. Reporter expression was calculated for each dose of ATA as previously described and normalized to DMSO treatment (Chaim et al., 2017; Nagel et al., 2014).
2.9.3. Further sigma ATA chemical purity analysis
Purities of final compounds were assessed by analytical reverse-phase HPLC performed with the following method: Dionex UltiMate 3000 series (Thermo Scientific) with a Thermo Scientific Hypersil GOLD C18 (2.1 mm × 50 mm, 3 μm particle size) column with the gradient 10–100% ACN/water (10 min), and 100% ACN/water (5 min) flow = 0.2 mL/min. The mobile phase was buffered with 0.1% formic acid. Mass spectra were obtained on a Thermo Scientific ISQ EC via negative electrospray ionization. Semi-preparative reverse-phase HPLC was performed on an ACCQPrep HP125 system (Teledyne Isco) with a Thermo Scientific Hypersil GOLD C18 (21.2 mm × 250 mm, 5 μm particle size) column. NMR spectra were recorded on an Agilent 400 MR instrument and were calibrated using residual undeuterated solvent as an internal reference (DMSO-d6: 1H NMR δ = 2.50 ppm, 13C NMR δ = 39.52 ppm). The following abbreviations were used to explain NMR peak multiplicities: s = singlet, d = doublet, br = broad. 2-Hydroxybenzoic acid was purchased from TCI (lot: XXHPA-QE). 2,2′-Dihydroxy-5,5′-methylenedibenzoic acid was purchased from Acros (lot: A0387467) and purified by HPLC before use.
5,5′,5”-(hydroxymethanetriyl)tris(2-hydroxybenzoic acid) (1) A powdered mixture consisting of 2,2′-Dihydroxy-5,5′-methylenedibenzoic acid (302 mg, 1.05 mmol) and salicylic acid (145 mg, 1.05 mmol) was added, in portions, to a solution of sodium nitrite (145 mg, 2.10 mmol) in concentrated sulfuric acid (2 mL). The reaction was stirred at room temperature for 24 h. The reaction mixture was added to 100g of ice in a beaker. Crude precipitate was filtered and washed with water. The residue was purified by semi-preparative HPC to afford pure 1 (73 mg, 16%). The preparation of 1 has been previously reported in the literature (Cushman and Kanamathareddy, 1990).
Physical state: red solid; TLC: Rf = 0.14 (DCM/MeOH/TFA 95:5:2, UV active). 1H NMR (400 MHz, DMSO-d6) δ 13.90 (s, 3H), 11.22 (s, 3H), 7.64 (d, J = 2.5 Hz, 3H), 7.29 (dd, J = 8.7, 2.5 Hz, 3H), 6.91 (d, J = 8.7 Hz, 3H), 6.55 (br,1H). 13C NMR(101 MHz, DMSO-d6) δ 171.74, 159.97, 138.25, 135.13, 128.86, 116.61, 111.88, 78.98. MS (ESI-): C22H16O10 [M-H]−: 439.23, [M-H2O-H]−: 421.09, [M + FA-H]−: 485.10, [2M-H]−: 879.33
3. Results
3.1. Identification of the human UDG inhibitor aurintricarboxylic acid
To identify small-molecules that target human UDG activity, we employed high-throughput screening (HTS) on a focused diversity library. We optimized a HTS platform (Chung et al., 2009; Krosky et al., 2006) with an assay measuring removal of multiple uracils from a DNA hairpin (termed uracil DNA hairpin), resulting in loss of hydrogen bonds between U:A base pairs. Consequent melting of the hairpin by UDG at room temperature results in fluorescence when the 5′ end FAM is separated from the 3’ end quencher dabcyl. A small-molecule that blocks the activity of UDG leads to retention of some uracils in the DNA hairpin and reduces the fluorescence level (RFU, relative fluorescence unit). In the absence of inhibitor, monitoring the reaction with UDG protein over 60 min shows on average an increase in the RFU of the DNA substrate (Fig. 1A). Three hundred twenty small-molecules were screened in two independent experiments yielding a correlation coefficient (R2) of 0.711 (Fig. 1B). We successfully screened 3,115 compounds at a concentration of 12.5 μM (Fig. 1C). The final assay conditions have an average signal-to-noise ratio of 3.5, leading to an average Z prime of 0.74. Z prime is a statistical parameter for evaluating the suitability of an assay for HTS, with values above 0.5 required to move forward (Zhang et al., 1999).
Fig. 1.

High-throughput small-molecule screening for human UDG inhibitors.
Panel A: A 60 min reaction time-course experiment was performed to measure human UDG enzymatic activity in the presence of 10 nM UDG (red line) or absence of UDG (black line) uracil DNA hairpin (25 nM) only control. Each data point is the average of 12 reaction replicates from a 384-well plate. Error bars represent standard deviation from the mean.
Panel B: Two independent screens using a plate of 320 compounds were plotted on the same graph with the trend line showing a positive correlation between the two runs with an R2 value of 0.711 using y = mx + b.
Panel C: High-throughput screening with 3115 bioactive compounds. Normalized percent activity of UDG-mediated unquenching of fluorescence DNA hairpin is shown. See methods for further detail regarding the equation that was used to normalize the percent activity of UDG. Control condition denoted as RFU+UDG AVG (DMSO controls without compound); the average RFU value was computed from a set of 32 wells per screening plate. Control condition denotes RFU−UDG AVG (DNA alone, no protein); the average relative fluorescence unit (RFU) value was computed using a set of 32 replicate wells per screening plate.
Panel D: A dose-response curve showing the normalized average % inhibition of UDG-mediated un-quenching of fluorescence from the DNA hairpin over nine different concentrations of ATA, starting with the highest dose of 12.5 μM, then serially diluted to 3.96 μM, 1.25 μM, 0.396 μM, 0.125 μM, 0.0396 μM, 0.01255 μM, 0.003972 μM, 0.001257 μM, and 0.0003978 μM. Each point at each concentration represents a normalized average percent inhibition from three replicate wells from three different 384-well plates. IC50 was generated using Prism software version 8.4.3, non-linear fit, [inhibitor] vs. response (three parameters, R2 = 0.99). Y error bars represent standard deviation from the mean. The average RFU+UDG AVG (UDG + DMSO control without compound) value was computed from a set of 18 wells per screening plate. The average RFU.UDG AVG (uracil DNA hairpin alone) was computed using a set of 9 wells per screening plate.
We eliminated known DNA intercalators and small-molecules that nonspecifically quenched the residual uracil DNA hairpin fluorescence in the absence of UDG protein. We selected and confirmed the activity of aurintricarboxylic acid (ATA) (Sigma Aldrich) as the most potent compound with a reproducible 50% inhibitory constant (IC50) of 40 nM against the purified human UDG (Fig. 1D). ATA provided a chemically distinct entity from uracil, so we proceeded to characterize its activity and mechanism.
3.2. Time course of human UDG activity
To better examine UDG inhibitor activity, we monitored human UDG activity over time. Time course experiments found that ATA at a fixed dose of 500 nM persistently diminished UDG activity over a range of uracil DNA hairpins (Supplementary Fig. 1 A–E). This suggests that ATA blocks the interactions between UDG and uracil DNA hairpin in all tested time points. We did not observe ATA interference with the DNA substrate residual fluorescence signal in the absence of UDG protein over the same time course. We next found that below <250 nM, ATA was unable to significantly reduce UDG-mediated un-quenching of uracil DNA hairpin over a 5-h time course (Supplementary Fig. 1F). In the absence of UDG protein, ATA had no effect on the basal uracil DNA hairpin fluorescence over the same time course (Supplementary Fig. 1G).
3.3. ATA does not directly bind to uracil containing-DNA hairpin substrate
Many chemotherapeutic DNA damaging agents are known to target DNA rather than proteins directly. To test if ATA directly bound DNA, we measured the impact of ATA on the thermal unfolding of the same uracil containing DNA hairpin through FAM fluorescence (directly labeled on the DNA hairpin). We determined the melting temperature (Tm) of the uracil DNA hairpin in the absence of both UDG protein and ATA compound (DNA-only condition). We then titrated in different concentrations of ATA with the uracil DNA hairpin. We found (Supplementary Fig. 1H) that in the absence of UDG, ATA did not shift the melting curve (Tm) of uracil containing DNA at any of the concentrations tested. This observation suggests that ATA does not directly bind uracil containing DNA substrate under these conditions.
3.4. The co-crystal structure of UDG with ATA
To experimentally examine the ATA interaction site with UDG, we co-crystallized UDG with ATA, collected X-ray diffraction data, and refined the crystal structure to 1.8 Å resolution (Table 1). We found ATA bound to the active site of UDG. The ATA omit electron density (Fig. 2A) shows two offset binding positions for ATA. In both positions ATA binds UDG with the oxygens from all three rings, holding three UDG loops in their open conformation thereby preventing the formation of the catalytically competent closed UDG active site. ATA’s binding position blocks the open to closed conformational switch needed for strong DNA binding. The electron density indicated two groups pointed into the active site pocket and bound in two related rotamers. Notably, one moiety stacked with Tyr147 and His148, and hydrogen bonded with Gln152. Refinement and difference map analyses supports an ATA binding mode that both blocks UDG closing and uracil binding.
Fig. 2. The ATA-bound UDG crystal structure is in an open conformation in comparison with UDG-uracil and UDG-DNA bound structures.
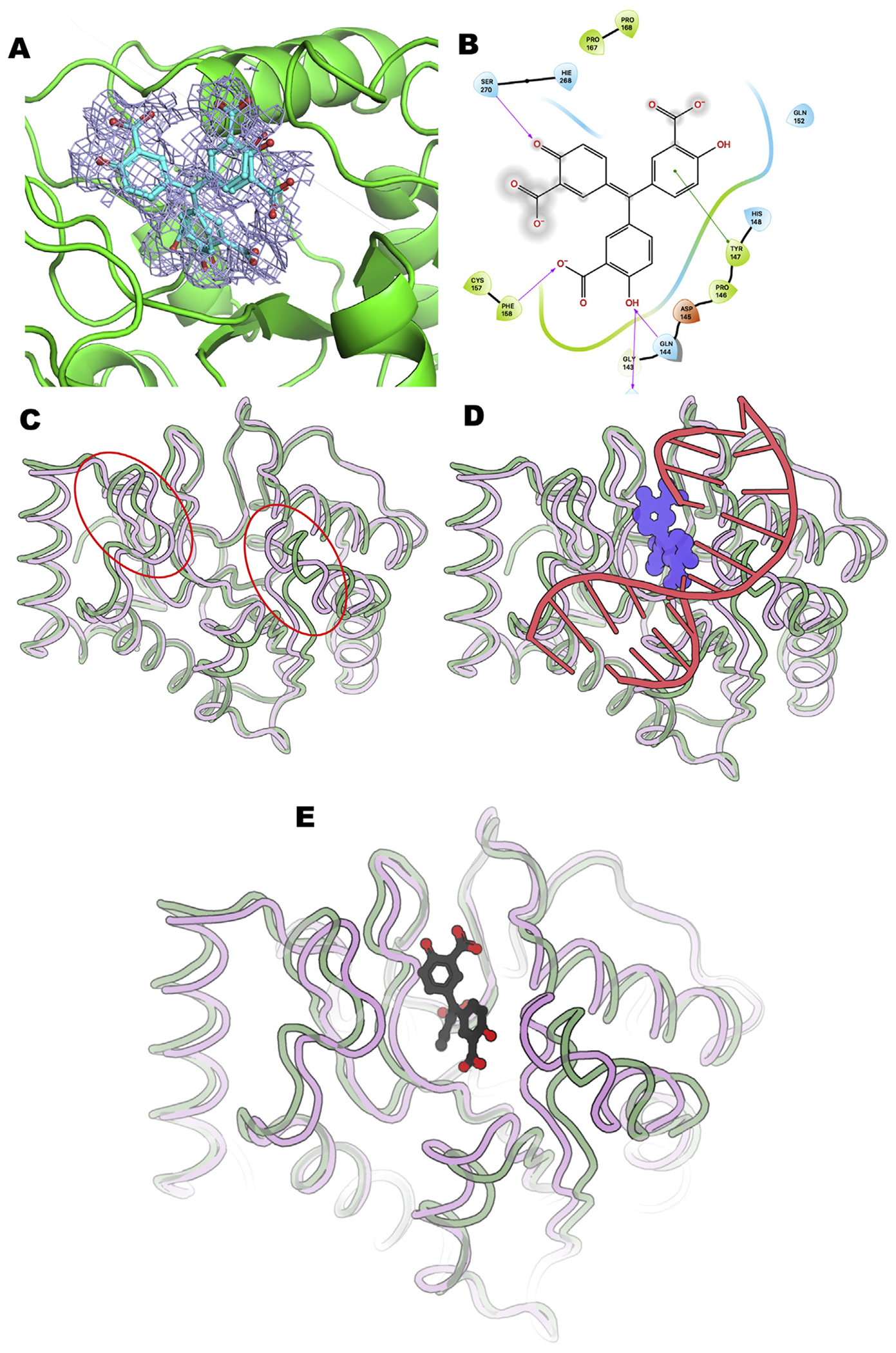
Panel A: UDG electron density omit map contoured at 2 sigma calculated with POLDER (Liebschner et al., 2017) and PHENIX suite (Liebschner et al., 2019) shows ATA binds in the DNA-binding groove in a manner holding UDG in an open conformation catalytically inactive conformation. ATA binds in a dual conformation suggesting ways to improve inhibitor binding to the open conformation. Panel B: 2D interaction map by Maestro molecular viewer shows key loop regions and residues in the UDG-ATA interface. Panel C: Alpha carbon tubes for open UDG (green) superposed onto the closed conformation (pink). Red circles note key differences between open and closed main chain positions. Panel D: UDG binding to dsDNA promotes the closed conformation (pink) as shown for alpha carbon tubes of UDG-dsDNA complex superposed onto open UDG (green) as bound to ATA. Panel E: ATA rings and carboxylates (carbons, black; oxygens, red) bound to the open UDG (green alpha carbon tubes) geometrically block the closed conformation seen for uracil and DNA bound structures (pink).
Importantly, protein electron density showed that both ATA binding modes hold UDG in its open conformation, effectively blocking the open to closed conformational switch by binding to key loop regions and residues shown by the Maestro molecular viewer (Fig. 2B). Panels representing the different UDG conformations are shown in Fig. 2C–E. Alpha carbon tubes represent open UDG (green) superposed onto the closed conformation (pink) (Fig. 2C). This panel shows key differences between open and closed main chain positions (Fig. 2C). UDG binding to dsDNA promotes the closed conformation (pink) as shown for alpha carbon tubes of UDG-dsDNA complex superposed onto open UDG (green) as bound to ATA (Fig. 2D). ATA rings and carboxylates (carbons, black; oxygens, red) bound to the open UDG (green alpha carbon tubes) geometrically block the closed conformation seen for uracil and DNA bound structures (pink) (Fig. 2E).
3.5. Computational analyses of uracil, ATA and salicylic acid binding to UDG
To computationally test ATA binding orientations, we used in silico evaluations of multiple ATA conformations and its composing moieties by docking with Glide (Schrodinger suite for drug discovery). We examined the binding and multiple possible conformations of ATA in complex with UDG for four structures of UDG available in the Protein Data Bank (PDB IDs: 1AKZ, 1UGH, 1EMH, and 1SSP) (Mol et al., 1995a; Parikh et al., 1998, 2000b). As a control, this docking method suitably identifies the uracil base fit to the uracil ring UDG structures. Importantly, ATA docks into poses consistent with the electron density for the DNA-free UDG conformation. We found a similar agreement for the ATA salicylic acid moiety, which also docks into the uracil binding site. We also examined possible ATA binding to UDG conformations bound to uracil in dsDNA, which shows a more closed active site characterized by inward shifts of residues that coordinate DNA binding, bending, uracil nucleotide flipping from duplex DNA and uracil binding. Interestingly, the docking results of ATA to the dsDNA-bound UDG structure show alteration in one of UDG’s β strands, and a shift closing the β strand is part of the open-to-closed transition. These findings are consistent with ATA predominantly binding to UDG in its open conformation and thereby blocking the tight UDG-DNA complex. Collectively, these computational results largely support ATA binding to the open DNA-free UDG (Supplementary Fig. 2A–I).
3.6. ATA binds directly to purified UDG
To experimentally test the binding between the human UDG protein and ligand ATA, we employed microscale thermophoresis (MST). The N-terminal primary amine was labeled with Ato488, and 100 nM labeled-UDG titrated against the increasing concentration of ATA. The MST binding isotherms revealed an affinity binding dissociation constant (KD) for the more soluble triammonium ATA salt to be < 700 nM (Fig. 3A), which was consistent with the DNA hairpin assay results that showed an estimated IC50 of 700 nM. Using an independent protein thermal shift assay with a fluorescence dye that preferentially binds to unfolded hydrophobic regions of the protein, we observed ATA dose-dependent destabilization of the human UDG protein (Fig. 3B).
Fig. 3.
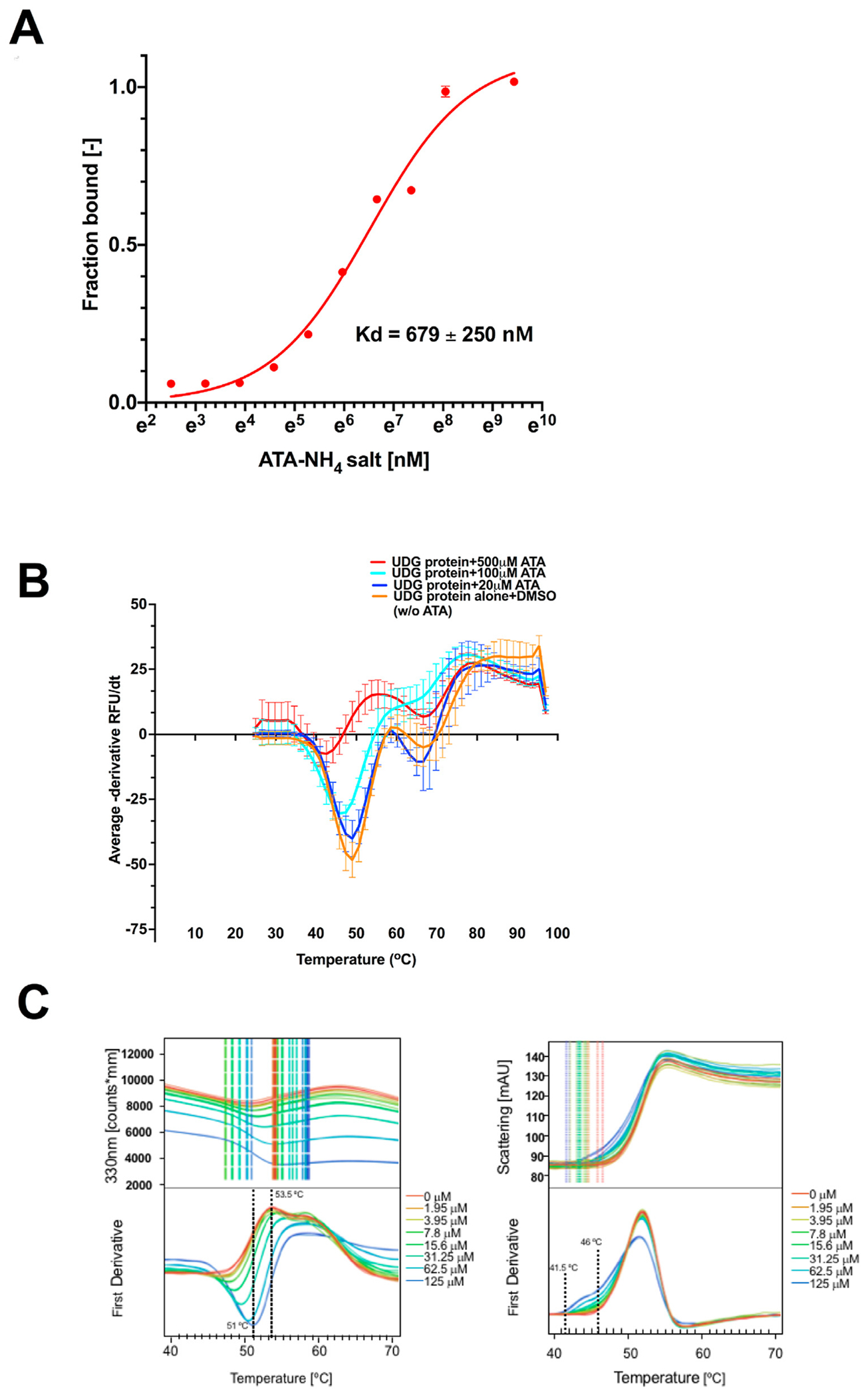
ATA directly binds and destabilizes the human UDG protein.
Panel A: ATA binding to UDG measured by MST. ATA (30 nM–500 μM) was titrated into a fixed concentration of labeled UDG (100 nM). The data for thermophoresis was recorded at 20 °C using the blue LED at 20% and IR-Laser at 40%. The isotherm derived from the raw data and fitted in Prism using one site specific binding model.
Panel B: UDG protein thermal shift with sybro orange dye was performed. Purified human UDG protein was incubated with either DMSO or a dose range of ATA. Fluorescence was monitored for 2hr using Bioraid CFX96, Hex filter, 25–97 °C, 1.6 °C/ 2min. Negative (−) derivative RFU raw data and plots from the machine were obtained and analyzed. The plot shows x = temperature and y = −dRFU/dt. Reaction with UDG (16 μM) protein only + DMSO is shown in orange, UDG + ATA (20 μM) is shown in blue, UDG + ATA (100 μM) is shown in light blue and UDG + ATA (500 μM) is shown in red. Melting temperature (Tm) value was taken directly from the midpoint on the −dRFU/dt vs. temperature graph. Each point on the graph represents an average of three data sets. Y error bars represent standard deviation.
Panel C: Monitoring the thermal stability of UDG in the presence of titrating concentrations of ATA through intrinsic tryptophan fluorescence (left panels). The onset of protein aggregation with increasing concentration of ATA reporting unfolding of proteins (right panels).
To further examine the direct binding of ATA to UDG we used nanoDSF technology to measure the intrinsic tryptophan fluorescence of protein during unfolding (Cimmperman et al., 2008). The thermal stability of UDG in the presence of titrating concentrations of ATA was measured. The results showed a decrease in the mean melting temperature of UDG from 53.5 °C to 51 °C in the presence of ATA, suggesting that ATA destabilizes UDG (Fig. 3C, left panels). Note that the melting temperature for UDG without ATA is 53.5 °C, shown by a dotted line on the first derivative peak corresponding to the red lines on the 330 nm panel. While generally thermal stability or instability is observed by a shift in the peak melting temperature, ATA binding caused protein aggregate formation at lower temperatures. These aggregates associate with surface exposed tryptophans resulting in fluorescence quenching at 330 nm channel. This resulted in a downward shift of the melt curve (first derivative melting temperature peak change from upwards to downward direction) opposed to a peak-shift. This is further confirmed by the scattering data, which marks the beginning of protein denaturation where UDG in the presence of ATA clearly aggregated at lower temperatures than UDG alone (Fig. 3C, right panels). Though we could not derive a binding constant using the protein denaturation assays, our results verified the direct binding of ATA to the human UDG protein. Based on our results, we reasoned ATA binding maintains UDG in an open conformation that loses a β strand hydrogen bond (Putnam et al., 1999).
We further observed that the addition of uracil containing DNA altered the ATA associated decrease in the Tm. of UDG. In the absence of uracil containing DNA, the average maximum Tm change for UDG in the presence of 100 μM ATA is ~3.0 °C (Supplementary Fig. 3A). However, with the addition of 1.0 mM of uracil DNA hairpin, UDG was stabilized, and the addition of 100 μM of ATA did not further change the Tm of UDG protein (Supplementary Fig. 3B). The results suggest that excess uracil containing-DNA could stabilize the UDG protein, consistent with hydrogen-deuterium exchange data that established an extended dsDNA binding region outside of the active site pocket that may complete with the ATA (Roberts et al., 2012). In contrast, excess normal thymidine containing DNA hairpin did not stabilize the UDG protein even at millimolar concentrations (Supplementary Fig. 3C and 3D). Together these results show that UDG stabilization depends upon direct binding to the uracil DNA hairpin and that ATA binds to the DNA-free UDG conformation.
3.7. The efficiency of ATA inhibition of human UDG
To validate ATA inhibition of purified UDG, we employed a dsDNA oligo containing a single U:A base pair and examined heat sensitive DNA cleavage in a gel-based assay. We compared ATA inhibition for UDG acting on uracil-containing dsDNA with inhibition of 8-oxoguanine DNA glycosylase (OGG1) acting on dsDNA containing a single 8-oxoguanine paired with cytosine (8oxG:C bp). ATA demonstrated an average IC50 of 0.70 μM ± 0.41 (700 nM) towards UDG, (R2 = 0.99) (Fig. 4A and B). In contrast, ATA exhibited a 5-fold lower inhibitory effect on human OGG1 protein with an average IC50 of 4.5 μM ± 1.2 μM (4,500 nM, R2 = 0.95) (Fig. 4C and D), despite the recognized cross-reactivity of ATA with nucleotide binding enzymes.
Fig. 4.
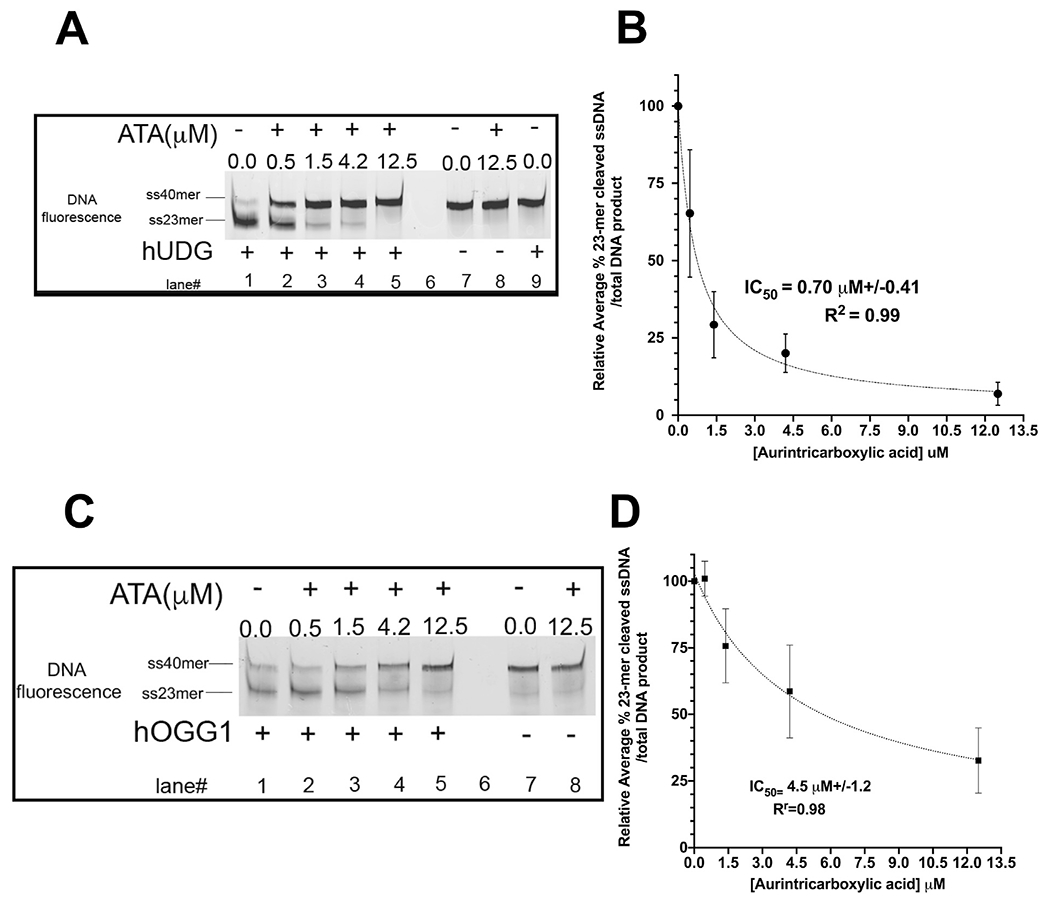
ATA activity on purified human UDG and 8-oxoguanine DNA glycosylases.
Panel A: ATA diminishes UDG-mediated DNA cutting activity by heat in a dose-dependent manner using a single uracil:adenine base-pair DNA cleavage assay. 20% polyacrylamide nucleic acid urea gel electrophoresis was used to resolve the DNA at room temperature, 200 V for 50min. DNA products were visualized by fluorescence using Typhoon Trio + Variable Mode Imager. Analysis and quantification of DNA cleavage were done with a background subtraction step using the 23mer from the DNA only lane. Dose-response plot showing relative average percent 23-mer cleaved single-stranded DNA band divided by the total amount of DNA product bands (23mer+40mer) over different ATA concentrations. The lane with UDG (500pM)+ uracil DNA (20 nM) without ATA (DMSO condition) was set as 100% cleavage and was used as the denominator for calculating subsequent points with inhibitor ATA (12.5 μM, 4.2 μM, 1.3 μM, 0.46 μM). Lane 9 (a control) contained thymidine:adenine bp DNAin the presence of purified UDG did not produce DNA cleavage.
panel B: IC50 was generated using Prism software version 8.4.3, non-linear fit, [inhibitor] vs. response (three parameters, R2 = 0.99) averaging points from four gels. Y error bars represent standard deviation from the mean.
Panel C: ATA displayed a lesser inhibitory effect on purified human OGG1 protein (10 nM) to mediate cleavage of an 8oxG:C bp DNA (10 nM) with a Cy3 fluorescent tag on the 3’ end in vitro.
Panel D: IC50 was generated using Prism software version 8.4.3, non-linear fit, [inhibitor] vs. response (three parameters, R2 = 0.98) averaging points from three gels. Y error bars represent standard deviation from the mean.
3.8. ATA is an effective inhibitor of UDG in human DLD1 colon cancer cell-extracts
To test biological activity, we conducted UDG inhibition assays using whole cell extracts. As a control and to further test the efficiency of UDG over other glycosylases, we generated UDG knockdown (shUNG) and CRISPR-Cas9 knockout (UNG-KO) clones in DLD1 human colon cancer cells (Supplementary Fig. 4) showing shSCR and shUNG knockdown mRNA levels. We performed an in vitro DNA cleavage assay using the uracil containing DNA oligo as described above (Fig. 4). Cell-extracts from the scrambled shRNA and wild type expressing DLD1 cells were used as controls, and UDG-mediated 23mer ssDNA products were readily detectable (Fig. 5A lanes 1 and 5). Cell-extracts derived from UDG knockdown cells, however, showed a drastic reduction in cleavage products (Fig. 5A, lane 3), and the UDG-KO cell extracts showed a complete lack of the ss23mer DNA products (Fig. 5A, lane 7). Treatment with 12.5 μM of ATA, had minimal effect on the 23mer ssDNA generation (Fig. 5A, lanes 2, 4, 6, and 8). However, 50 μM ATA caused near-complete inhibition of 23mer ssDNA appearance in all experimental conditions where UDG was present (scramble shRNA and WT cell extracts, Fig. 5B, lanes 2, 4, and 6). Notably, UNG-KO cell-lysate failed to generate the ss23mer DNA products regardless of the ATA concentrations (Fig. 5A and B, KO UNG lane 8). Thus, our consolidated results revealed that the 23mer ssDNA product could be attributed to UDG activity. Fig. 5B lane 10 (control) shows that in the absence of cell lysate, ATA (50 μM) alone did not dampen the fluorescence signal of the DNA oligo in the reaction and did not generate secondary non-specific cleaved DNA bands. This further suggests that ATA does not bind the DNA substrate.
Fig. 5.

Evaluating the inhibitory effect of ATA on cell-free protein extracts from DLD1 human colon cancer cell line.
Panel A: Human cancer lysates were extracted from DLD1 human colon cancer cell lines: UDG containing DLD1 cells (wild type and sh scramble), DLD1 cell line with a sh-knockdown UDG, and DLD1 cell line with a CRISPR-knockout UDG were treated with ATA. Reactions were done in 96 well plate format using 20 μg of lysate pre-incubated with either vehicle DMSO (gel lanes 1,3,5&7) or 12.5 μM of ATA (gel lanes 2,4,6&8) for 20 min at 37 °C followed by 30 nM of fluorescently-labeled TAMRA DNA duplex oligo (uracil:adenine bp) was added to the reaction for another 20min incubation at 37 °C. Lane 9 contained uracil DNA only (without cell lysate and without the ATA compound). Lane 10 contained uracil DNA +12.5 μM of ATA in the absence of any cell lysate. Reactions were stopped by a denaturing bromophenol blue dye. 20% polyacrylamide nucleic acid urea gels were used and DNA bands were resolved by electrophoresis at room temperature, 200 V for 50 min. DNA products were visualized by fluorescence using Typhoon Trio + Variable Mode Imager.
Panel B: 50.0 μM of ATA was tested in gel lanes 2,4,6,8&10. Reactions were performed and processed same as above.
Panel C: Human UDG containing DLD1 colon cancer lysate was treated with a range of ATA (0–50 μM). Gels were processed and DNA bands were analyzed using ImageJ software. IC50 was estimated using Prism software version 8.4.3, non-linear fit, [inhibitor] vs. normalized response (variable slope, R2 = 0.97). Each data point is an average from four gels.
We next estimated the IC50 in DLD1 UDG containing cell lysate to be 32.5 μM ± 7.03 (Fig. 5C), higher than the 0.7 μM (or 700 nM) IC50 for purified UDG and consistent with ATA having affinity for other macromolecules in the cell lysate. A knockout of UDG or inhibition with ATA completely inhibited the generation of the ss23mer UDG enzymatic products (Fig. 5B), demonstrating ATA inhibitory efficacy towards UDG in cell free extracts.
3.9. ATA inhibition of uracil removal in human cell lines
We used a plasmid-based host cell reactivation assay to measure uracil removal from a U:G base pair in human cell lines (Chaim et al., 2017). Treatment with ATA (2.5 μM) significantly inhibited uracil removal in MCF-7 cells (Fig. 6A). Increasing concentrations of ATA up to 10 μM were not statistically different from the 2.5 μM dose. We next measured the effectiveness of ATA for UDG inhibition by measuring uracil removal in wild type and UDG-knockout HAP cells (Fig. 6B). Knockout of UDG reduced uracil repair by approximately 3-fold. Interestingly, UDG-knockout cells removed uracil from at least 99% of plasmids before fluorescent protein could be expressed suggesting that backup uracil removal pathways operate with high efficiency. ATA (5.0 μM) reduced uracil removal in wild type cells by 1.8-fold, accounting for 60% of the effect of UDG knockout (Fig. 6B). ATA (5 μM) reduced relative uracil removal in UDG-knockout cells from approximately ~0.3 to ~0.1. We cannot quantify the contribution of glycosylases such as SMUG1 to uracil repair in UDG-knockout cells, so the extent to which ATA inhibits these enzymes cannot be determined in this assay. However, the data suggest that inhibition of uracil removal by ATA in wild type cells is predominantly due to UDG inhibition, consistent with its structural interactions with the active site pocket noted above.
Fig. 6.
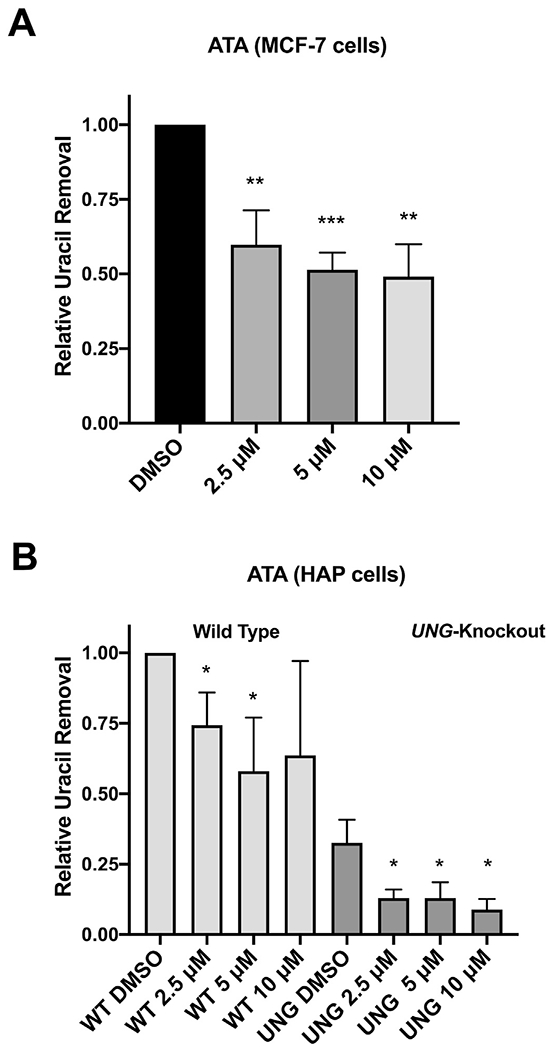
Measurement of ATA in vivo using a host-cell reactivation assay.
Panel A: MCF-7 cells were seeded at 50,000 cells per well in a 12-well plate.
Panel B: HAP cells (wild type or UNG gene knockout) were seeded at 75,000 cells per well in a 6-well plate. Cells were allowed to adhere overnight and then cells in duplicate wells were treated with DMSO or ATA (2.5, 5, or 10 μM) for 24 h (refer to methodology). After 24 h, cells were dissociated by trypsinization and analyzed by flow cytometry using an Atune NxT flow cytometer. Gating and compensation were determined by transfection of single-color controls. Reporter expression was calculated for each dose of ATA as previously described in publications and normalized to DMSO treatment (Chaim et al., 2017; Nagel et al., 2014).
We also compared the inhibitory effects of ATA on the repair of other glycosylase reporter substrates 8oxoG:C (OGG1), A:8oxoG (MPG, MUTYH), hypoxanthine:T (MPG), and T:G base pairs (TDG, MBD4) in MCF-7 cells, (Fig. 7A and B). We found that ATA (2.5 μM) inhibits uracil repair (0.58 reduction in relative repair of uracil, Fig. 7B) significantly more than repair of other transfected reporter plasmids (0.13 reduction in relative repair of 8oxoG:C and 0.35 reduction in relative repair of hypoxanthine:T repair while not inhibiting the repair of a T:G or A:8oxoG base pair, Fig. 7B).
Fig. 7.

Measurement of ATA inhibition on uracil, 8oxoG:C, A:8oxoG:C, Hypoxanthine:T and T:G repairs in MCF-7 cells using a host-cell reactivation assay with doses. Panel A: 1.25 μM and panel B: 2.5 μM. Reporter expression was calculated for each dose of ATA as described in prior publications and normalized to DMSO treatment (Chaim et al., 2017; Nagel et al., 2014).
4. Discussion
Recognition and removal of damaged bases by BER glycosylases is the committed initiating step for BER. Furthermore, subsequent BER steps are common to repair of all base lesions and likely to proceed by DNA product handoffs to avoid the release of toxic and mutagenic intermediates (Huffman et al., 2005; Mol et al., 2000b) that may prove more challenging to completely block by small molecule inhibitors. We focused on prototypic DNA glycosylase UDG since cancer cells expressing high levels of DNA deaminase APOBEC3B and functional p53 can be killed by a UDG protein inhibitor (Serebrenik et al., 2019), supporting the concept that effective small molecule inhibitors of UDG (Grundy and Parsons, 2020) may increase the effectiveness of anticancer therapy.
Here we optimized and applied an assay to screen chemical libraries for UDG inhibitors resulting in identification and characterization of ATA as an exemplary UDGi, (the most potent identified in a screen of more than 3100 bioactive compounds), effective in human cancer cell extracts and in a uracil containing reporter plasmid assay in intact cancer cells. The screen we used is in contrast to a structure-based inhibitor strategy, which would typically screen crystals with chemical fragment libraries and use resulting protein-fragment X-ray crystal structures to design active site inhibitors (Wilson et al., 2020). The ATA results presented herein suggest blocking the active site in the open configuration based on its ability for nucleotide mimicry may be a tool to inform a previously underappreciated strategy to effectively inhibit UDG. Indeed, ATA inhibits other nucleic-acid binding proteins to various degrees (Blumenthal and Landers, 1973; Stewart et al., 1971).
Transition-state approaches allow designing new enzymes from proteins, including catalytic and metal-binding antibodies (Iverson et al., 1990; Kraut, 1988; Lerner et al., 1991) and many enzymes inhibitors are designed as transition-state analogs (Bianchet et al., 2003; Friedman et al., 2009; Jiang et al., 2003, 2004; Parikh et al., 2000b). Alternatively, inhibitor design has also used the concept of anchored plasticity, where binding a stable site is used to push open adjoining regions to gain specificity (Garcin et al., 2008) which can benefit from homologue-based (‘molecular avatar’) templates (Brosey et al., 2021) and pipelines (Houl et al., 2019; Moiani et al., 2018; Shibata et al., 2014).
Yet, UDG recognition of uracil in DNA is cooperative: multiple individual binding interactions of UDG with DNA backbone and uracil nucleotide enable the tight fit of the catalytically competent UDG-DNA substrate complex (Kavli et al., 1996; Slupphaug et al., 1996). These synergistic interactions include conformational closing of the DNA binding channel and uracil pocket. Given the importance of protein motion in enzymatic catalysis, it has been anticipated that disruption of conformational sampling presents an alternative strategy for inhibition (Klinman et al., 2013). By strategically interfering with UDG:DNA interactions and with the conformational closing required for specific uracil recognition and excision, ATA constitutes an effective UDGi despite having multiple bound conformations, not resembling a transition state analog and not pushing open adjoining sites. Instead, ATA binds the open active site, makes interactions with key residues for uracil binding, and resists conformational closing needed for tight DNA binding and for uracil recognition and excision. Strikingly, this is analogous to the strategy employed by a viral Ugi protein inhibitor (Mol et al., 1995a; Putnam et al., 1999) but with extensions into the uracil binding region.
Notably, the open UDG conformation bound by ATA is not in the DNA binding conformation and is less stable than the unbound protein. This may reflect the loss of a beta-strand hydrogen bond as seen in our new structure that matches the prior DNA-free UDG structure (Parikh et al., 1998). This destabilization suggests that an optimal small-molecule might not only inhibit UDG but reduce its stability to effectively reduce its activity in cells by combined inhibition and destabilization. So to further develop ATA-based inhibitors, we plan to make a nonsymmetric derivative and add a thiol to form a covalent connection to Cys157 ~4A from the ATA carboxylate as exposed cysteines are readily modified, which can decrease the protein’s stability (McRee et al., 1990). Although we identified ATA with a fluorescence screen, general approaches to targeting open conformations could employ X-ray scattering to screen directly for binding to a specific conformational state (Brosey and Tainer, 2019).
Taken together, these results teach us fundamental strategies for nucleic acid research and the development of anti-cancer therapeutics. A small molecule inhibitor need not compete with UDG binding to DNA. Compounds that hold UDG in the open (catalytically incompetent) and less stable conformation precluding damaged DNA substrate recognition and base removal may be equally or more effective. This approach may provide a critical enabling strategy for UDG as results from both hydrogen-deuterium exchange and computational docking show that the DNA binding area for UDG (and by implication other BER enzymes) is larger than indicated in crystal structures suggesting it may be quite challenging to block with a small-molecule chemical inhibitor (Roberts et al., 2012). Yet as a “door stopper” ATA does not need to compete with DNA; it only needs to block UDG closing and thus prevent the complementary DNA binding channel and the lock-and-key specific substrate recognition. In this inhibitor design strategy, the ATA chemotype is a critical enabling tool because, unlike uracil and transition state chemotypes that promote formation of the complementary DNA binding channel, ATA blocks this closure and hence blocks strong DNA binding and the functional active site formation. Notably, other DNA base and nucleotide excision enzymes such as MutY, APE1, FEN1, MRE11, WRN and XPG employ an open-to-closed conformational switch to create tight DNA binding and catalytically competent active sites (Manuel et al., 2004; Perry et al., 2006; Syed and Tainer, 2018; Tsutakawa et al., 2014, 2017, 2020; Wang et al., 2017). We therefore propose that the “door stopper” inhibitor strategy identified here merits investigation as a potential general strategy to block excision repair enzymes for understanding their role in cancer biology and identifying novel therapeutics. Currently these findings provide proof-of-principle for development of the ATA chemotype and “door stopper” strategy targeting inhibitor binding to an open pre-catalytic glycosylase conformation preventing functional binding needed to excise damaged bases in DNA.
Indeed, ATA effectively inhibits UDG-mediated cleavage of uracil-containing DNA in human DLD1 colon cancer cell extracts and in human cell lines. These observed reductions of UDG activity in human cancer cell lysates and cells suggests that an ATA-based inhibitor strategy may effectively block intracellular UDG activity, and supports the value of further studies in this regard. The unquenching fluorescence assay from the DNA hairpin with its initial calculated 40 nM IC50 and tests with a single U containing dsDNA oligomer in a DNA cutting assay support an IC50 in the submicromolar range (~0.7uM) consistent with cellular phenotypes and supporting a path for structure-based improvements toward a nonsymmetric and better behaved chemical entity.
ATA contains two structural fragments considered potential liabilities as pan-assay interference compounds (PAINS) (Baell and Holloway, 2010) that exhibit promiscuous biological activity in a variety of assays most commonly due to chemical reactivity. First, ATA contains a quinone-like structural fragment that may be susceptible to conjugate addition of endogenous nucleophiles and potential redox chemistry. Second, the salicylic acid fragment (2-hydroxybenzoic acid moiety) may behave as a potential metal chelator. Yet, with over 85 small molecule drugs that are currently on the market that contain PAINS structural alerts, the use of PAINS filters for removal of hits from further consideration is questionable and likely inappropriate (Capuzzi et al., 2017; Chai and Matyus, 2016; Senger et al., 2016). Instead, we propose that employing the general ATA ability for nucleotide mimicry offers provocative alternative to fragment library screening. We suggest that ATA may prove to be an enabling tool to examine chemical knockdowns of UDG in various types of cancer cells and with combination treatments and to direct chemical and structure-based design of improved functionally-related inhibitors. We hope these UDG-ATA “door stopper” inhibitor findings may prompt new research that opens the door to advance identification of vulnerabilities caused by losses of BER activities and to help pinpoint synthetic lethal relationships potentially useful for developing cancer treatment strategies.
Supplementary Material
Acknowledgments
The authors thank Hung-Ying Kao and William Merrick from the Department of Biochemistry for their helpful advice, Gregory Tochtrop from the Department of Chemistry and Susan Tsutakawa from Lawrence Berkeley Lab for helpful suggestions, and Drew Adams director of the Small Molecule Screening Shared Resource of the Case Comprehensive Cancer Center for expertise regarding high-throughput. We thank former summer students from the laboratory of Dr. Gerson: Eric Stewart (first year medical student from the University of Illinois in Chicago, Case Western School of Medicine Heart, Lung, and Blood summer program 2017). Sergey Kolomiyets (a high school teacher) and Asaan Snipes-Rea (a high school student from the University High School, Cleveland Ohio) in the Case Western Youth Enjoy Science (YES) summer program 2018 and 2019. Mustapha Tauira from Emory University in the Case Western School of Medicine CANSUR summer program 2019.
Funding
This work was supported by in part by grants from the National Institutes of Health [5P30 CA043703 to S.L.G. plus P01 CA092584 and R35 CA220430 to J.A.T.]. J.A.T. efforts are supported by a Robert A. Welch Chemistry Chair and Cancer Prevention and Research Institute of Texas RP180813. Work by D.L. was supported by 5T32HL007118 and Z.D.N. was supported by U01ES029520, P01 CA092584 and P30ES000002.
Footnotes
Declaration of competing interest COI
The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.pbiomolbio.2021.02.004.
References
- Baell JB, Holloway GA, 2010. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Bianchet MA, Seiple LA, Jiang YL, Ichikawa Y, Amzel LM, Stivers JT, 2003. Electrostatic guidance of glycosyl cation migration along the reaction coordinate of uracil DNA glycosylase. Biochemistry 42, 12455–12460. [DOI] [PubMed] [Google Scholar]
- Blumenthal T, Landers TA, 1973. The inhibition of nucleic acid-binding proteins by aurintricarboxylic acid. Biochem. Biophys. Res. Commun 55, 680–688. [DOI] [PubMed] [Google Scholar]
- Brosey CA, Tainer JA, 2019. Evolving SAXS versatility: solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Curr. Opin. Struct. Biol 58, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosey CA, Houl JH, Katsonis P, Balapiti-Modarage LPF, Bommagani S, Moiani D, Bacolla A, Arvai A, Link T, Warden LS, Lichtarge O, Jones DE, Ahmed Z, Tainer JA, 2021. Targeting SARS-CoV-2 Nsp3 macrodomain structure with insights from human poly (ADP-ribose) glycohydrolase (PARG) structures with inhibitors. Prog. Biophys. Mol. Biol 64 (5) 10.1016/j.pbiomolbio.2021.02.002 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgar AD, Weeks LD, Miao Y, Yang S, Xu Y, Guo C, Markowitz S, Oleinick N, Gerson SL, Liu L, 2012. Removal of uracil by uracil DNA glycosylase limits pemetrexed cytotoxicity: overriding the limit with methoxyamine to inhibit base excision repair. Cell Death Dis. 3, e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuzzi SJ, Muratov EN, Tropsha A, 2017. Phantom PAINS: problems with the utility of alerts for pan-assay INterference CompoundS. J. Chem. Inf. Model 57, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai CL, Matyus P, 2016. One size does not fit all: challenging some dogmas and taboos in drug discovery. Future Med. Chem 8, 29–38. [DOI] [PubMed] [Google Scholar]
- Chaim IA, Nagel ZD, Jordan JJ, Mazzucato P, Ngo LP, Samson LD, 2017. In vivo measurements of interindividual differences in DNA glycosylases and APE1 activities. Proc. Natl. Acad. Sci. U. S. A 114, E10379–E10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Parker JB, Bianchet M, Amzel LM, Stivers JT, 2009. Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nat. Chem. Biol 5, 407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmperman P, Baranauskiene L, Jachimoviciute S, Jachno J, Torresan J, Michailoviene V, Matuliene J, Sereikaite J, Bumelis V, Matulis D, 2008. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J 95, 3222–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condie AG, Yan Y, Gerson SL, Wang Y, 2015. A fluorescent probe to measure DNA damage and repair. PLoS One 10, e0131330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman M, Kanamathareddy S, 1990. Synthesis of the covalent hydrate of the incorrectly assumed struture of aurintricarboxylic acid (ATA). Tetrahedron 46, 1491–1498. [Google Scholar]
- Cushman M, Wang PL, Chang SH, Wild C, De Clercq E, Schols D, Goldman ME, Bowen JA, 1991. Preparation and anti-HIV activities of aurin-tricarboxylic acid fractions and analogues: direct correlation of antiviral potency with molecular weight. J. Med. Chem 34, 329–337. [DOI] [PubMed] [Google Scholar]
- Dixon M, Woodrick J, Gupta S, Karmahapatra SK, Devito S, Vasudevan S, Dakshanamurthy S, Adhikari S, Yenugonda VM, Roy R, 2015. Naturally occurring polyphenol, morin hydrate, inhibits enzymatic activity of N-methylpurine DNA glycosylase, a DNA repair enzyme with various roles in human disease. Bioorg. Med. Chem 23, 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorjsuren D, Kim D, Maloney DJ, Wilson DM 3rd, Simeonov A, 2011. Complementary non-radioactive assays for investigation of human flap endonuclease 1 activity. Nucleic Acids Res. 39, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan BK, Miller JH, 1980. Mutagenic deamination of cytosine residues in DNA. Nature 287, 560–561. [DOI] [PubMed] [Google Scholar]
- Fischer F, Baerenfaller K,Jiricny J, 2007.5-Fluorouracil is efficiently removed from DNA by the base excision and mismatch repair systems. Gastroenterology 133, 1858–1868. [DOI] [PubMed] [Google Scholar]
- Friedman JI, Majumdar A, Stivers JT, 2009. Nontarget DNA binding shapes the dynamic landscape for enzymatic recognition of DNA damage. Nucleic Acids Res. 37, 3493–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS, 2004. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED, 2008. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol 4, 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh U, Giri K, Bhattacharyya NP, 2009. Interaction of aurintricarboxylic acid (ATA) with four nucleic acid binding proteins DNase I, RNase A, reverse transcriptase and Taq polymerase. Spectrochim. Acta Mol. Biomol. Spectrosc 74, 1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez RG, Haxo RS, Schleich T, 1980. Mechanism of action of polymeric aurintricarboxylic acid, a potent inhibitor of protein–nucleic acid interactions. Biochemistry 19, 4299–4303. [DOI] [PubMed] [Google Scholar]
- Gossage L, Perry C, Abbotts R, Madhusudan S, 2012. Base excision repair factors are promising prognostic and predictive markers in cancer. Curr. Mol. Pharmacol 5, 115–124. [DOI] [PubMed] [Google Scholar]
- Grundy GJ, Parsons JL, 2020. Base excision repair and its implications to cancer therapy. Essays Biochem. 10.1016/j.pbiomolbio.2021.02.002 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA, 1998. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat. Struct. Biol 5, 1058–1064. [DOI] [PubMed] [Google Scholar]
- Hashem AM, Flaman AS, Farnsworth A, Brown EG, Van Domselaar G, He R, Li X, 2009. Aurintricarboxylic acid is a potent inhibitor of influenza A and B virus neuraminidases. PLoS One 4, e8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi K, Iwai S, Tainer JA, 2007. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair 6, 410–428. [DOI] [PubMed] [Google Scholar]
- Houl JH, Ye Z, Brosey CA, Balapiti-Modarage LPF, Namjoshi S, Bacolla A, Laverty D, Walker BL, Pourfarjam Y, Warden LS, Babu Chinnam N, Moiani D, Stegeman RA, Chen MK, Hung MC, Nagel ZD, Ellenberger T, Kim IK, Jones DE, Ahmed Z, Tainer JA, 2019. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun 10, 5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman JL, Sundheim O, Tainer JA, 2005. DNA base damage recognition and removal: new twists and grooves. Mutat. Res 577, 55–76. [DOI] [PubMed] [Google Scholar]
- Hung HC, Tseng CP, Yang JM, Ju YW, Tseng SN, Chen YF, Chao YS, Hsieh HP, Shih SR, Hsu JT, 2009. Aurintricarboxylic acid inhibits influenza virus neuraminidase. Antivir. Res 81, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson BL, Iverson SA, Roberts VA, Getzoff ED, Tainer JA, Benkovic SJ, Lerner RA, 1990. Metalloantibodies. Science 249, 659–662. [DOI] [PubMed] [Google Scholar]
- Jiang YL, Cao C, Stivers JT, Song F, Ichikawa Y, 2004. The merits of bipartite transition-state mimics for inhibition of uracil DNA glycosylase. Bioorg. Chem 32, 244–262. [DOI] [PubMed] [Google Scholar]
- Jiang YL, Chung S, Krosky DJ, Stivers JT, 2006. Synthesis and high-throughput evaluation of triskelion uracil libraries for inhibition of human dUTPase and UNG2. Bioorg. Med. Chem 14, 5666–5672. [DOI] [PubMed] [Google Scholar]
- Jiang YL, Ichikawa Y, Song F, Stivers JT, 2003. Powering DNA repair through substrate electrostatic interactions. Biochemistry 42, 1922–1929. [DOI] [PubMed] [Google Scholar]
- Jiang YL, Krosky DJ, Seiple L, Stivers JT, 2005. Uracil-directed ligand tethering: an efficient strategy for uracil DNA glycosylase (UNG) inhibitor development. J. Am. Chem. Soc 127, 17412–17420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Slupphaug G, Mol CD, Arvai AS, Peterson SB, Tainer JA, Krokan HE, 1996. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J. 15, 3442–3447. [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Wilson DM 3rd, 2012. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol 5, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman JP, Hammes- Schiffer S, Arora K, 2013. Dynamics in Enzyme Catalysis. Springer, Heidelberg; ; New York. [Google Scholar]
- Kraut J, 1988. How do enzymes work? Science 242, 533–540. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Drablos F, Slupphaug G, 2002. Uracil in DNA–occurrence, consequences and repair. Oncogene 21, 8935–8948. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G, 2000. Base excision repair of DNA in mammalian cells. FEBS Lett. 476, 73–7 . [DOI] [PubMed] [Google Scholar]
- Krokan HE, Standal R, Slupphaug G, 1997. DNA glycosylases in the base excision repair of DNA. Biochem. J 325 (Pt 1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krosky DJ, Bianchet MA, Seiple L, Chung S, Amzel LM, Stivers JT, 2006. Mimicking damaged DNA with a small molecule inhibitor of human UNG2. Nucleic Acids Res. 34, 5872–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuban-Jankowska A, Sahu KK, Gorska-Ponikowska M, Tuszynski JA, Wozniak M, 2017. Inhibitory activity of iron chelators ATA and DFO on MCF-7 breast cancer cells and phosphatases PTP1B and SHP2. Anticancer Res. 37, 4799–4806. [DOI] [PubMed] [Google Scholar]
- Lerner RA, Benkovic SJ, Schultz PG, 1991. At the crossroads of chemistry and immunology: catalytic antibodies. Science 252, 659–667. [DOI] [PubMed] [Google Scholar]
- Li M, Shandilya SM, Carpenter MA, Rathore A, Brown WL, Perkins AL, Harki DA, Solberg J, Hook DJ, Pandey KK, Parniak MA, Johnson JR, Krogan NJ, Somasundaran M, Ali A, Schiffer CA, Harris RS, 2012. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol 7, 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, Moriarty NW, Oeffner RD, Poon BK, Prisant MG, Read RJ, Richardson JS, Richardson DC, Sammito MD, Sobolev OV, Stockwell DH, Terwilliger TC, Urzhumtsev AG, Videau LL, Williams CJ, Adams PD, 2019. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, Adams PD, 2017. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol 73, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG, 2003. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Canc 3, 330–338. [DOI] [PubMed] [Google Scholar]
- Malet-Martino M, Jolimaitre P, Martino R, 2002. The prodrugs of 5-fluorouracil. Curr. Med. Chem. Anticancer Agents 2, 267–310. [DOI] [PubMed] [Google Scholar]
- Manuel RC, Hitomi K, Arvai AS, House PG, Kurtz AJ, Dodson ML, McCullough AK, Tainer JA, Lloyd RS, 2004. Reaction intermediates in the catalytic mechanism of Escherichia coli MutY DNA glycosylase. J. Biol. Chem 279, 46930–46939. [DOI] [PubMed] [Google Scholar]
- McCullough AK, Dodson ML, Lloyd RS, 1999. Initiation of base excision repair: glycosylase mechanisms and structures. Annu. Rev. Biochem 68, 255–285. [DOI] [PubMed] [Google Scholar]
- McRee DE, Redford SM, Getzoff ED, Lepock JR, Hallewell RA, Tainer JA, 1990. Changes in crystallographic structure and thermostability of a Cu,Zn superoxide dismutase mutant resulting from the removal of a buried cysteine. J. Biol. Chem 265, 14234–1424 . [DOI] [PubMed] [Google Scholar]
- Moiani D, Ronato DA, Brosey CA, Arvai AS, Syed A, Masson JY, Petricci E, Tainer JA, 2018. Targeting allostery with avatars to design inhibitors assessed by cell activity: dissecting MRE11 endo- and exonuclease activities. Methods Enzymol. 601, 205–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol CD, Arvai AS, Sanderson RJ, Slupphaug G, Kavli B, Krokan HE, Mosbaugh DW, Tainer JA, 1995a. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell 82, 701–708. [DOI] [PubMed] [Google Scholar]
- Mol CD, Arvai AS, Slupphaug G, Kavli B, Alseth I, Krokan HE, Tainer JA, 1995b. Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell 80, 869–878. [DOI] [PubMed] [Google Scholar]
- Mol CD, Hosfield DJ, Tainer JA, 2000a. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3’ ends justify the means. Mutat. Res 460, 211–229. [DOI] [PubMed] [Google Scholar]
- Mol CD, Izumi T, Mitra S, Tainer JA, 2000b. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected]. Nature 403, 451–456. [DOI] [PubMed] [Google Scholar]
- Mol CD, Kuo CF, Thayer MM, Cunningham RP, Tainer JA, 1995c. Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature 374, 381–386. [DOI] [PubMed] [Google Scholar]
- Mol CD, Parikh SS, Putnam CD, Lo TP, Tainer JA, 1999. DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu. Rev. Biophys. Biomol. Struct 28, 101–128. [DOI] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P, 2013. Collaboration gets the most out of software. eLife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A, Abo RP, Butty VL, Forget AL, Samson LD, 2014. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc. Natl. Acad. Sci. U. S. A 111, E1823–E1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrecht AS, Urban N, Schaefer M, Rose A, Kless A, Meents JE, Lampert A, Abdelrahman A, Muller CE, Schmalzing G, Hausmann R, 2019. Identification of aurintricarboxylic acid as a potent allosteric antagonist of P2X1 and P2X3 receptors. Neuropharmacology 158, 107749. [DOI] [PubMed] [Google Scholar]
- Parikh SS, Mol CD, Hosfield DJ, Tainer JA, 1999. Envisioning the molecular choreography of DNA base excision repair. Curr. Opin. Struct. Biol 9, 37–47. [DOI] [PubMed] [Google Scholar]
- Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA, 1998. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 17, 5214–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SS, Putnam CD, Tainer JA, 2000a. Lessons learned from structural results on uracil-DNA glycosylase. Mutat. Res 460, 183–199. [DOI] [PubMed] [Google Scholar]
- Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA, 2000b. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereo-electronic effects. Proc. Natl. Acad. Sci. U. S. A 97, 5083–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, Cooper PK, Chen DJ, Tainer JA, 2006. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol 13, 414–422. [DOI] [PubMed] [Google Scholar]
- Pettersen HS, Visnes T, Vagbo CB, Svaasand EK, Doseth B, Slupphaug G, Kavli B, Krokan HE, 2011. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 39, 8430–8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Shroyer MJ, Lundquist AJ, Mol CD, Arvai AS, Mosbaugh DW, Tainer JA, 1999. Protein mimicry of DNA from crystal structures of the uracil-DNA glycosylase inhibitor protein and its complex with Escherichia coli uracil-DNA glycosylase. J. Mol. Biol 287, 331–346. [DOI] [PubMed] [Google Scholar]
- Roberts VA, Pique ME, Hsu S, Li S, Slupphaug G, Rambo RP, Jamison JW, Liu T, Lee JH, Tainer JA, Ten Eyck LF, Woods VL Jr., 2012. Combining H/D exchange mass spectroscopy and computational docking reveals extended DNA-binding surface on uracil-DNA glycosylase. Nucleic Acids Res. 40, 6070–6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W, 2013. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Seidel SA, Dijkman PM, Lea WA, van den Bogaart G, Jerabek-Willemsen M, Lazic A, Joseph JS, Srinivasan P, Baaske P, Simeonov A, Katritch I, Melo FA, Ladbury JE, Schreiber G, Watts A, Braun D, Duhr S, 2013. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger MR, Fraga CA, Dantas RF, Silva FP Jr., 2016. Filtering promiscuous compounds in early drug discovery: is it a good idea? Drug Discov. Today 21, 868–872. [DOI] [PubMed] [Google Scholar]
- Serebrenik AA, Starrett GJ, Leenen S, Jarvis MC, Shaban NM, Salamango DJ, Nilsen H, Brown WL, Harris RS, 2019. The deaminase APOBEC3B triggers the death of cells lacking uracil DNA glycosylase. Proc. Natl. Acad. Sci. U. S. A 116, 22158–22163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA, 2014. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA, 1996. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature 384, 87–92. [DOI] [PubMed] [Google Scholar]
- Stewart ML, Grollman AP, Huang MT, 1971. Aurintricarboxylic acid: inhibitor of initiation of protein synthesis. Proc. Natl. Acad. Sci. U. S. A 68, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed A, Tainer JA, 2018. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem 87, 263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan GS, Chiu CH, Garchow BG, Metzler D, Diamond SL, Kiriakidou M, 2012. Small molecule inhibition of RISC loading. ACS Chem. Biol 7, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA, 1995. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 14, 4108–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Lafrance-Vanasse J, Tainer JA, 2014. The cutting edges in DNA repair, licensing, and fidelity: DNA and RNA repair nucleases sculpt DNA to measure twice, cut once. DNA Repair 19, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Sarker AH, Ng C, Arvai AS, Shin DS, Shih B, Jiang S, Thwin AC, Tsai MS, Willcox A, Her MZ, Trego KS, Raetz AG, Rosenberg D, Bacolla A, Hammel M, Griffith JD, Cooper PK, Tainer JA, 2020. Human XPG nuclease structure, assembly, and activities with insights for neurodegeneration and cancer from pathogenic mutations. Proc. Natl. Acad. Sci. U. S. A 117, 14127–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Thompson MJ, Arvai AS, Neil AJ, Shaw SJ, Algasaier SI, Kim JC, Finger LD,Jardine E, Gotham VJB, Sarker AH, Her MZ, Rashid F, Hamdan SM, Mirkin SM, Grasby JA,Tainer JA, 2017. Phosphate steering by Flap Endonuclease 1 promotes 5’-flap specificity and incision to prevent genome instability. Nat. Commun 8, 15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chakravarthy S, Verdine GL, 2017. Structural basis for the lesion-scanning mechanism of the MutY DNA glycosylase. J. Biol. Chem 292, 5007–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks LD, Fu P, Gerson SL, 2013. Uracil-DNA glycosylase expression determines human lung cancer cell sensitivity to pemetrexed. Mol. Canc. Therapeut 12, 2248–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks LD, Zentner GE, Scacheri PC, Gerson SL, 2014. Uracil DNA glycosylase (UNG) loss enhances DNA double strand break formation in human cancer cells exposed to pemetrexed. Cell Death Dis. 5, e1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DM 3rd, Deacon AM, Duncton MAJ, Pellicena P, Georgiadis MM, Yeh AP, Arvai AS, Moiani D, Tainer JA, Das D, 2020. Fragment-and structure-based drug discovery for developing therapeutic agents targeting the DNA Damage Response. Prog. Biophys. Mol. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA, 2000. Passing the baton in base excision repair. Nat. Struct. Biol 7, 176–178. [DOI] [PubMed] [Google Scholar]
- Wyatt MD, Wilson DM 3rd, 2009. Participation of DNA repair in the response to 5-fluorouracil. Cell. Mol. Life Sci 66, 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Han X, Qing Y, Condie AG, Gorityala S, Yang S, Xu Y, Zhang Y, Gerson SL, 2016. Inhibition of uracil DNA glycosylase sensitizes cancer cells to 5-fluorodeoxyuridine through replication fork collapse-induced DNA damage. Oncotarget 7, 59299–59313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Qing Y, Pink JJ, Gerson SL, 2018. Loss of uracil DNA glycosylase selectively resensitizes p53-mutant and -deficient cells to 5-FdU. Mol. Canc. Res 16, 212–221. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR, 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.