

Abstract
Germline loss-of-function mutations in BRIP1 are associated with ovarian carcinoma and may also contribute to breast cancer risk, particularly among patients who develop disease at an early age. Normal BRIP1 activity is required for DNA interstrand crosslink repair, and is thus central to the maintenance of genome stability. Although pathogenic mutations have been identified in BRIP1, genetic testing more often reveals missense variants for which the impact on molecular function and subsequent roles in cancer risk are uncertain. Next generation sequencing of germline DNA in 2,160 early-onset breast cancer and 1,199 ovarian cancer patients revealed nearly 2% of patients carry a very rare missense variant (MAF<0.0001) in BRIP1. This is 3 fold higher than the frequency of all rare BRIP1 missense alleles reported in more than 60,000 individuals of the general population (p<0.0001, Chi-square). Using CRISPR-Cas9 gene editing technology and rescue assays, we functionally characterized 20 of these missense variants, focusing on the altered protein’s ability to repair interstrand crosslink damage. 75% of the characterized variants rendered the protein hypomorph or null. In a clinical cohort of >117,000 breast and ovarian cancer patients who underwent panel testing, the combined odds ratio associated with BRIP1 hypomorph or null missense carriers compared to the general population was 2.30 (95%CI=1.60-3.30, p<0.0001). These findings suggest that novel missense variants within the helicase domain of BRIP1 may confer risk for both breast and ovarian cancer and highlight the importance of functional testing for additional variants.
Keywords: Hereditary breast cancer, hereditary ovarian cancer, BRIP1, FANCJ, functional characterization
Introduction
BRCA1 interacting protein C-terminal helicase 1 (BRIP1) is a member of the Fanconi anemia (FA) pathway of proteins with an established role in DNA interstrand crosslink (ICL) repair (1, 2). The N-terminal domain is comprised of seven, highly conserved DEAH helicase motifs that function in the unwinding of DNA, preferentially at forked duplex substrates and D-loops(3, 4), and in the resolution of guanine quadruplexes (G4-DNA)(5–7). The N-terminal region also includes an iron-sulfur (Fe-S) binding domain that distinguishes BRIP1 from other DEAH helicase family members. A single missense mutation in this domain (A349P) abrogates BRIP1’s ability to unwind DNA strands and displace proteins bound to DNA (8, 9). The C-terminal domain contains an essential phosphorylation site S990 required for the binding of BRIP1 to the tumor suppressor, BRCA1 (10). Cells lacking BRIP1 are unable to repair ICL damage, resulting in cell death(2, 11) and homozygosity or compound heterozygosity for missense, nonsense or frameshift mutations leads to Fanconi anemia (12, 13).
BRIP1 was reported to be the third most common ovarian cancer susceptibility gene with nearly 0.9-2.5% of all ovarian cancer patients carrying a splice, stop or frameshift defect (14–16). Rare missense variants are also associated with an increased risk for ovarian cancer, but the function of these variants is unknown (14). Mutations in BRCA1 and BRCA2, also members of the FA pathway, confer high risk for the development of ovarian and breast cancers (17–19). Given that BRIP1 is a member of the FA pathway it was logically assumed that BRIP1 mutations might similarly confer risk for both ovarian and breast cancers. Family studies initially pointed to truncating mutations in BRIP1 (including the hotspot FA allele, R798X) as low-penetrance breast cancer susceptibility alleles (20, 21). Recent studies, however, have brought into question the role that pathogenic BRIP1 mutations play in risk for breast cancer (16, 22, 23).
Next-generation sequencing (NGS) for cancer susceptibility alleles frequently includes analysis of BRIP1. BRIP1 missense variants are identified more frequently than nonsense or frameshift mutations. The vast majority of these missense alleles are reported as variants of uncertain significance (VUS). As of May 2019, 950 missense variants in BRIP1 have been reported to ClinVar with 933 clinically classified as a VUS. Most of the BRIP1 variants in ClinVar are linked to the conditions “familial cancer of the breast” and “neoplasm of the ovary” (24). To better understand how BRIP1 VUSs contribute to cancer risk, we functionally characterized rare and novel missense variants identified in ovarian and early-onset breast cancer patients by assessing interstrand crosslink DNA repair activity. The missense variants that we classified as deleterious (nulls and hypomorphs) were significantly enriched in a cohort of >117,000 breast and ovarian cancer patients who underwent clinical testing. Our findings highlight the importance of functional testing for BRIP1 VUSs and the need for further studies to determine the role that missense variants play in cancer risk.
Materials and Methods
Cohorts and Patient Materials
Early-onset breast cancer cohort -
Breast cancer subjects were recruited to The Young Women’s Breast Cancer Program (YWBCP) at Washington University, St. Louis during the period 2005-2012 (IRB-approved protocols 04-1009 and 2012C0097). The YWBCP enrolled women from all 50 states. The majority of subjects were Caucasian. African Americans made up 3.7% of the cohort, and there were smaller numbers of Native American and Asians. The 2,160 subjects investigated in this study all had invasive breast cancer diagnosed at age ≤40 years. For those subjects for whom BRCA1 and/or BRCA2 testing data were available, 15.7% carried mutations. Germline DNA was prepared form white blood cells for the vast majority of subjects. All coding exons in BRIP1 (GenBank: NM_032043) were sequenced to an average of >500 reads using the TruSeq Custom Amplicon Kit v1.5 and a MiSeq instrument with Reagent Kit v2 (Illumina). Variants were identified using Miseq Reporter software (v2.5.1).
Ovarian cancer cohort –
1,199 ovarian cancer cases were identified from the University of Washington (UW) Gynecologic Oncology Tissue Bank, the Mayo Clinic Ovarian Spore Tissue Bank or from the ARIEL2 part 1 clinical trial. Sequencing of germline DNA was completed using BROCA as previously described (25). Clearly damaging mutations for the UW and ARIEL2 cases have been previously reported (15).
Clinical diagnostic laboratory breast and ovarian cancer cohorts -
Data in the clinical laboratory cohort included 117,346 women referred for clinical genetic testing with a multi-gene panel inclusive of BRIP1 in the period (July 2012-Sept 2018). Multi-gene panel testing on genomic DNA isolated from blood or saliva was carried out by NGS with deletion/duplication analysis. Targeted coding exons and adjacent intronic nucleotides of the BRIP1 gene were enriched for sequencing. Additional follow-up Sanger sequencing was performed for any regions missing or with insufficient read depth coverage for reliable heterozygous variant detection (26). ICD10 codes were used to filter the de-identified data for applicable cases.
Exome Aggregation Consortium (ExAC) and FLOSSIES -
Two publically available, online sequencing datasets were utilized to estimate population frequencies of BRIP1 variants. Germline variants from 60,327 unrelated individuals of European, African, Latino, South Asian, East Asian and Middle Eastern descent were analyzed from ExAC (27). Additionally, germline variants from 9,884 cancer-free women, over the age of 70, of European and African descent from the Fabulous Ladies Over 70 project (FLOSSIES, https//whi.color.com) were analyzed. We chose to include all the FLOSSIES controls to better sample BRIP1 genetic diversity among women of African descent.
CRISPR Cas9
pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138). CRISPR-Cas9 target sites near candidate missense variants were identified using CRISPOR (28). Guide sequences with in silico specificity scores >50 and zero exonic, off-target effects were selected for exons 3, 8, 15 and 16. Guide RNAs were cloned into the pSpCas9(BB)-2A-GFP construct as described (29). HeLa and HEK293TN cells were co-transfected with CRISPR constructs and DNA template oligos containing missense variants using Lipofectamine™ 2000 (Thermo Fisher Scientific). After 48 hours, live GFP positive cells were single-cell plated using a FACSAria™ III (BD Biosciences) flow cytometer. DNA ligase IV inhibitor SCR7 [1.0uM] (APExBIO) was added to the media and colonies were maintained in 96-well plates. DNA was prepared from individual HeLa and HEK293TN clones using Quick-DNA Miniprep Kit (Zymo Research) and incorporation of missense variants was detected using PCR and Sanger sequencing (primers list in Supplemental Methods). Clones that failed to incorporate missense variants but contained homozygous or compound heterozygous frameshift mutations were further screened by western blot for loss of protein expression and chosen as null controls.
Cell Culture
HeLa (obtained from J. Parvin laboratory, Columbus, OH) and HEK293TN (System Bio, LV900A-1) cells were cultured in DMEM (Sigma Aldrich) with 10% FBS at 37°C in a 5% CO2-containing atmosphere. Cell lines were confirmed mycoplasma negative using the MycoAlert assay (Lonza). The estimated number of passages between authentication of cancer cell lines and completion of experiments is 20. Additional details regarding authentication are provided in the Supplemental Methods.
Karyotypes
Cells were incubated for 14 hours at 37°C in media containing MMC [60nM]. KaryoMAX™ Colcemid™ Solution (Thermo Fisher Scientific) was added dropwise and cells were incubated for an additional 45 mins at 37°C. Cells were collected, re-suspended in 0.56% KCl and placed in a 37°C water bath for 10 mins to bloat. After incubation, cells were immediately spun down, removed from KCl solution and fixed in 3:1 methanol to acetic acid. Cells were dropped onto slides in 55°C humid chamber and stained with KaryoMAX™ Giemsa Stain Solution (Thermo Fisher Scientific). Metaphase spreads were imaged on BIOREVO BZ-9000 (Keyence) at 100X magnification and images were captured and processed with BZ-II Viewer.
Clonogenic Growth Assays
Five thousand cells were plated in each well of a six-well dish and allowed to adhere. Cells were incubated for 24 hours at 37°C in media containing MMC (Sigma Aldrich), cisplatin (Sigma Aldrich), hydroxyurea (Sigma Aldrich) or TMPyP4 (Abcam) then washed 2X with PBS before fresh media was added. After seven days, cells were washed with PBS, fixed in 7:1 methanol to acetic acid for 10 mins and incubated with crystal violet (0.5% in 25% methanol) for 30 mins at room temperature with gentle shaking. Crystal violet was removed and cells were washed 3X with water and allowed to dry. Cells were de-stained with methanol for 30 mins at room temperature with gentle shaking. Equal volumes of solution were removed from the de-stained cells and plated in optically clear 96-well plates. Optical density of crystal violet was measured at 580nm using the Synergy™ 2 Microplate Reader with Gen5™ Software v1 (BioTek). Colony survival values plotted are relative to an untreated control.
Cell Cycle Analysis
Cells were incubated for 16 hours at 37°C in normal media or media containing MMC (Sigma Aldrich). Cells were trypsinized, collected as a single-cell suspension and fixed in ice-cold 70% ethanol for at least 10 mins on ice. Fixed cells were RNaseA treated for 30 mins at room temperature. Propidium iodide was added and cells were incubated for an additional 15 mins before analysis using a FACSCalibur flow cytometer (BD Biosciences).
Cycloheximide Chase Assay
HeLa BRIP1−/− clone D2 was transfected with pCDH-BRIP1 WT and mutant constructs using Lipofectamine™ 2000 (Thermo Fisher Scientific). At 48 hours post-transfection, equal number of transfected cells were plated into 12 well plates and allowed to adhere. Cycloheximide [30ug/mL] was added at time 0 and cells were collected at 0, 1, 2, 4, 6, and 8 hours. Protein was quantified by western blot and values were normalized to time 0. Analysis of protein degradation was performed from three independent experiments for each construct.
Statistical Analysis
Statistical tests were performed using GraphPad Prism 5 for Windows (GraphPad Software, Inc). Analysis of G2/M accumulation was performed from three independent experiments. P-values were calculated using one-way ANOVA with Tukey’s post-hoc test for pairwise comparisons. Western blot quantification of CRISPR clones was performed from three independent experiments. P-values were calculated using one-way ANOVA with Dunnett’s multiple comparison test to BRIP1+/+ clone. Protein half-life was determined using a nonlinear regression one-phase decay. P-values by functional grouping were calculated using one-way ANOVA with Tukey’s multiple comparison test. Results were considered statistically significant when p<0.05.
Results
Increased number of BRIP1 missense allele carriers in ovarian and early onset breast cancer cohorts
Sequencing germline DNA from 1,199 ovarian and 2,160 early-onset breast cancer patients revealed 6 ovarian (0.50%) and 8 breast cancer (0.37%) patients carried a frameshift or nonsense mutation in BRIP1 (Supplemental Figure S1). The hotspot R798X mutation was seen in 1 ovarian and 3 breast cancer patients along with 4 additional nonsense and 4 frameshifts. The frequency of truncating mutations in both cohorts was higher than reported by ExAC (0.19%) and FLOSSIES (0.19%). Consistent with previous reports (16), the enrichment of truncating mutations reached statistical significance in the ovarian cancer cohort (p<0.05 Fisher exact test) but not the breast cancer cohort.
Rare missense alleles (MAF≤0.0005) were more common than nonsense and frameshift mutations. Thirty-six ovarian (3.0%) and 43 breast cancer (2.0%) patients carried a rare or novel missense allele (Figure 1, (30, 31)). The frequency of rare alleles was significantly higher (p<0.001 both cohorts, Pearson Chi-square test) than all rare alleles reported by the ExAC general population (1.6%) and FLOSSIES (1.4%), with the greatest enrichment observed for very rare variants (MAF<0.0001, p<0.0001). The majority of rare alleles (54-of-64) clustered within the highly conserved helicase domain (1-888 amino acids) and are predicted in silico to be deleterious to protein function (Supplemental Table S1) (32). The P47A mutation, known to have reduced ATPase activity and helicase deficiency (3) was observed in both the breast and ovarian cancer cohorts. Four additional rare alleles in the C-terminal region of the helicase domain were seen in both cohorts (Q540L, I691L, Q740H and A745T).
Figure 1. Graphical summary of BRIP1 rare (MAF<0.0005) and novel missense variants identified in ovarian and early-onset breast cancer patients.
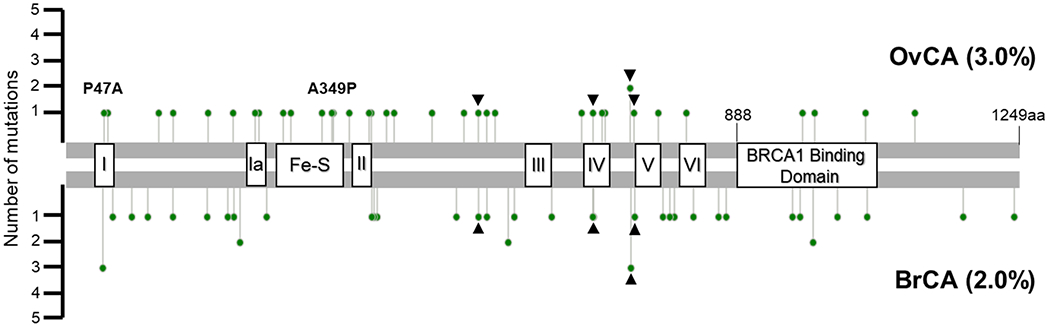
The ATPase reduced, helicase deficient P47A (helicase domain I) was seen in both cohorts. Helicase deficient, dominant negative mutant A349P (Fe-S domain, 276-362aa) was identified in a single ovarian cancer patient. Rare alleles in the C-terminal region of the helicase domain (Q540L, I691L, Q740H and A745T) identified in both cohorts are indicated with black arrowheads. Mutation plot generated using cBioPortal MutationMapper tool. Helicase domain motifs: I (39-57); Ia (245-258); II (385-398); III (610-624); IV (689-710); V (748-775); VI (819-836). BRCA1 binding domain: 888-1063aa.
Several of the missense alleles we identified have been described as mutations. The A349P and R707C variants associated with FA (12, 33) were each seen once in the ovarian cohort. Substitution of proline for alanine at amino acid 349 results in helicase deficiency and the mutant protein acts as a dominant-negative in vitro (9). The novel K52R allele was identified in one individual with ovarian cancer. This mutation, like A349P perturbs WT helicase function in a dominant negative manner (1). A subset of the missense variants identified fall within the seven highly conserved motifs of a DEAH helicase, but most lie outside of these domains (Supplemental Table S1). None of the missense alleles involve the amino acids known to be essential for BRCA1 binding (S990, P991 and F993) (10).
Of the 64 different rare variants identified in breast or ovarian cancer patients, 57 appear in ClinVar. Most are classified as VUSs (n=46) or having conflicting interpretations of pathogenicity (n=10) in association with familial cancer of breast, neoplasm of ovary, Fanconi anemia complementation group J (FA-J) and/or hereditary cancer-predisposing syndrome (review dates 2016-2018). Only A349P is described as likely pathogenic (Supplemental Table S2).
Of the 36 ovarian cancer patients and 43 breast cancer patients carrying a rare BRIP1 missense variant, six (7.6%) also carried a pathogenic mutation in BRCA1 or BRCA2 (Supplemental Table S1). The co-occurrence of BRCA1/2 mutations was not observed in any of the BRIP1 frameshift or nonsense mutations carriers.
Missense alleles in BRIP1 confer sensitivity to ICL damage
To assess the function of missense variants in the C-terminal helicase region, we utilized CRISPR-Cas9 gene editing to create isogenic HeLa cell lines lacking BRIP1 protein and/or expressing candidate missense variants identified in our cohorts. The helicase deficient P47A and A349P mutations were generated to serve as positive controls for loss-of-function missense mutations (Supplemental Figure S2). We generated HeLa cell lines expressing two representative candidate VUSs identified in our cohorts, A745T and D791V. A745T was identified in both cohorts and D791V was seen in an early-onset breast cancer patient with a strong family history of breast and other cancers. HeLa clones expressing missense mutations carry the mutation of interest on one homologue and a frameshift mutation, predicted to undergo nonsense-mediated decay, on the other. RT-PCR sequencing confirmed that the missense allele was expressed with trace or no expression of the frameshift transcript consistent with compound heterozygosity (Supplemental Figure S2A). Quantitative RT-PCR demonstrated that BRIP1 transcript levels were similar in clones expressing the missense alleles to those in BRIP1+/+ and BRIP1+/− clones (a positive control for hemizygosity). As expected, qPCR showed significantly reduced BRIP1 transcript levels in −/− clones (Supplemental Figure S2B).
Cells lacking BRIP1 protein are sensitive to ICL damage and exhibit characteristic chromosomal aberrations when treated with ICL agents. These clastogenic effects are seen in patients with FA-J (34). Mitomycin C (MMC) treatment of BRIP1−/− and BRIP1 A349P/- HeLa clones resulted in characteristic chromosomal breaks, gaps and radial formations (Figure 2A). Karyotypes of BRIP1 P47A/- cells treated with MMC showed modest numbers of chromosomal breaks and gaps but were characterized by many acentric fragments that were seen much more frequently than in the wildtype and null clones (Figure 2A). BRIP1 D791V/- clones had karyotypes similar to BRIP1 P47A/- cells with increased acentric fragments and chromosomal breaks. BRIP1 A745T/- clones, on the other hand, had normal karyotypes, similar to wildtype HeLa cells (Figure 2A).
Figure 2. Missense alleles in BRIP1 increase sensitivity to ICL damage.
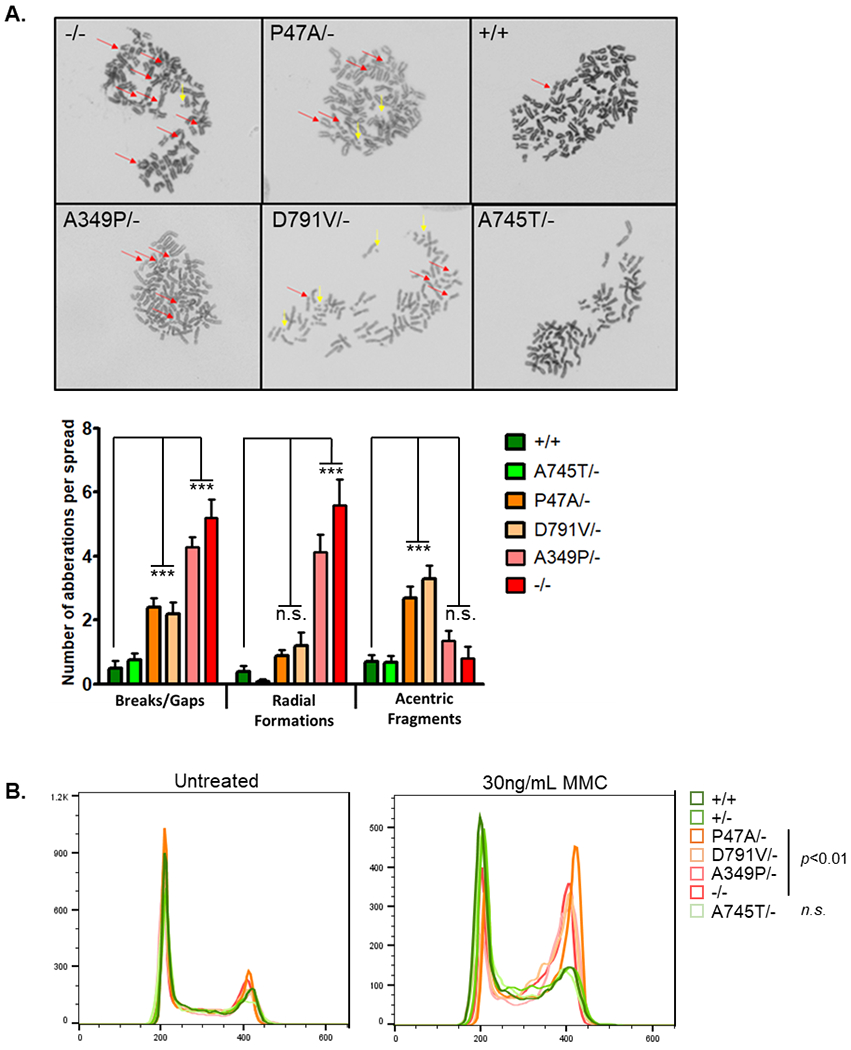
A) Representative karyotypes of independent HeLa clones after exposure to MMC [60nM]. Chromosomal breaks, gaps, radials (red arrows) and acentric fragments (yellow arrows) were observed in −/−, A349P/-, P47A/- and D791V/−. The average number of breaks/gaps, radial formations and acentric fragments per metaphase spread were calculated for each genotype (n=15 spreads per cell line). Statistical significance determined by two way ANOVA with a Bonferroni posttest to BRIP1+/+ (*** p<0.0001). B) Overlay of representative cell cycles from independent HeLa clones with and without MMC treatment. Statistical significance calculated by averaging percentage of cells in G2/M from 3 replicates (one-way ANOVA with Tukey’s multiple comparison test).
There were no differences in proliferation between the clones (ATP cell viability assay) and no differences in cell cycle when cells were grown in normal media (Figure 2B). Treatment with MMC, however, was associated with a statistically significant increase in G2/M for the clones expressing P47A, D791V, A349P and null alleles compared to BRIP1+/+,+/− and A745T/- clones (Figure 2B). Accumulation of cells in G2/M and increased rates of apoptosis are phenotypes observed in FA-J patient cells (2).
To further assess the sensitivity of mutants to ICL, we performed clonogenic growth assays with clones treated with increasing concentrations of MMC. As expected, null and A349P mutant expressing cells were extremely sensitive, showing reduced cell viability at concentrations as low as 2.5ng/mL. The estimated IC50s were between 4 and 7ng/mL for null and A349P cells and strikingly different than the IC50 for wildtype cells (>30ng/mL) (Figure 3). Interestingly, clones expressing the helicase deficient P47A allele and D791V allele showed intermediate sensitivity to MMC (Figure 3). They were largely resistant to MMC at low doses but more much more sensitive than wildtype cells at high doses (IC50 between 5 and 15ng/mL), consistent with a hypomorphic phenotype. In contrast, clones expressing the A745T allele were resistant to MMC damage, much like wildtype clones.
Figure 3. Missense alleles classified as null, hypomorphic or wildtype based on ICL damage induced cell killing.
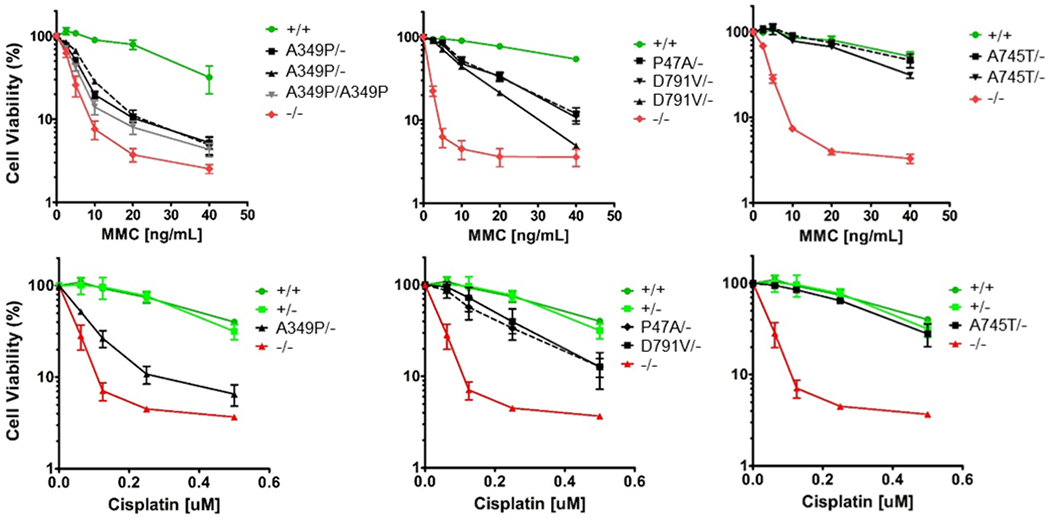
Clonogenic growth assays of independent HeLa clones after exposure to increasing concentrations of MMC and cisplatin. Data presented are the average of three replicates with error bars indicating one standard deviation. Cisplatin experiments were performed simultaneously with a single BRIP1+/+. +/− and −/− clone for control.
Treatment with a second ICL agent, cisplatin, paralleled the findings with MMC (Figure 3). BRIP1−/− and BRIP1 A349P/- clones were much more sensitive to the cytotoxic effects (IC50 ~0.1uM) than clones expressing hypomorphic alleles, P47A and D791V (IC50 ~0.2uM) and homozygous or hemizygous wildtype alleles (IC50 ~0.4uM).
BRIP1 localizes to sites of stalled replication forks and may be necessary for replication fork restart and the timely progression through cell cycle (35, 36). To test whether BRIP1 activity was necessary for release of stalled replication forks in our cell lines, we performed the same clonogenic growth assays for cells treated with hydroxyurea, an antimetabolite that stalls replication forks. Although there was some variability in the sensitivity among the HeLa clones investigated, it was not associated with BRIP1 genotype (Supplemental Figure S3).
Several studies have shown that BRIP1 preferentially binds and can unwind G4-DNA in vitro and thus BRIP1 may be essential for the resolution of G4 structures in vivo (5, 6, 9, 37, 38). We performed clonogenic growth assays using the G4 stabilizing agent, TMPyP4 and did not observe any differences in cell viability between clones (Supplemental Figure S3). However, we noticed that even wildtype HeLa cell lines were exquisitely sensitive to this particular drug, which is known to have many off-target effects, and thus may not reflect the effects of G4 stabilization alone.
CRISPR-Cas9 gene editing of HEK293TN cells validated the findings with HeLa clones. As was seen with HeLa cells, no differences in cell viability were observed in HEK293TN treated with hydroxyurea (Supplemental Figure S4A). Genetically modified HEK293TN cells confirmed BRIP1 D791V/- and BRIP1−/− clones were sensitive to MMC and cisplatin, and showed G2/M phase accumulation similar to that observed in the genetically modified HeLa clones (Supplemental Figure S4B).
Unstable proteins linked to hypomorphic variants
Hypomorphic alleles can reflect deleterious changes in transcription, RNA processing or stability, and/or changes in the protein that affect its function and/or stability. It has been shown that the substitution of a proline for an alanine at position 47 results in an unstable protein (1). Western blot analysis of BRIP1 in our HeLa and HEK293TN CRISPR clones showed that BRIP1 P47A/- and BRIP1 D791V/- clones had significantly lower levels of endogenous BRIP1 than wildtype (+/+), +/−, A745T/-, and A349P/- clones (Figure 4A). Using a cycloheximide chase assay, we determined that endogenous A349P and WT proteins had similar BRIP1 half-lives at 5-6 hours (Figure 4B). Because BRIP1 levels in the P47A and D791V clones were so low at baseline, it was not possible to accurately determine the half-lives of the endogenous proteins.
Figure 4. BRIP1 hypomorphs exhibit protein instability.
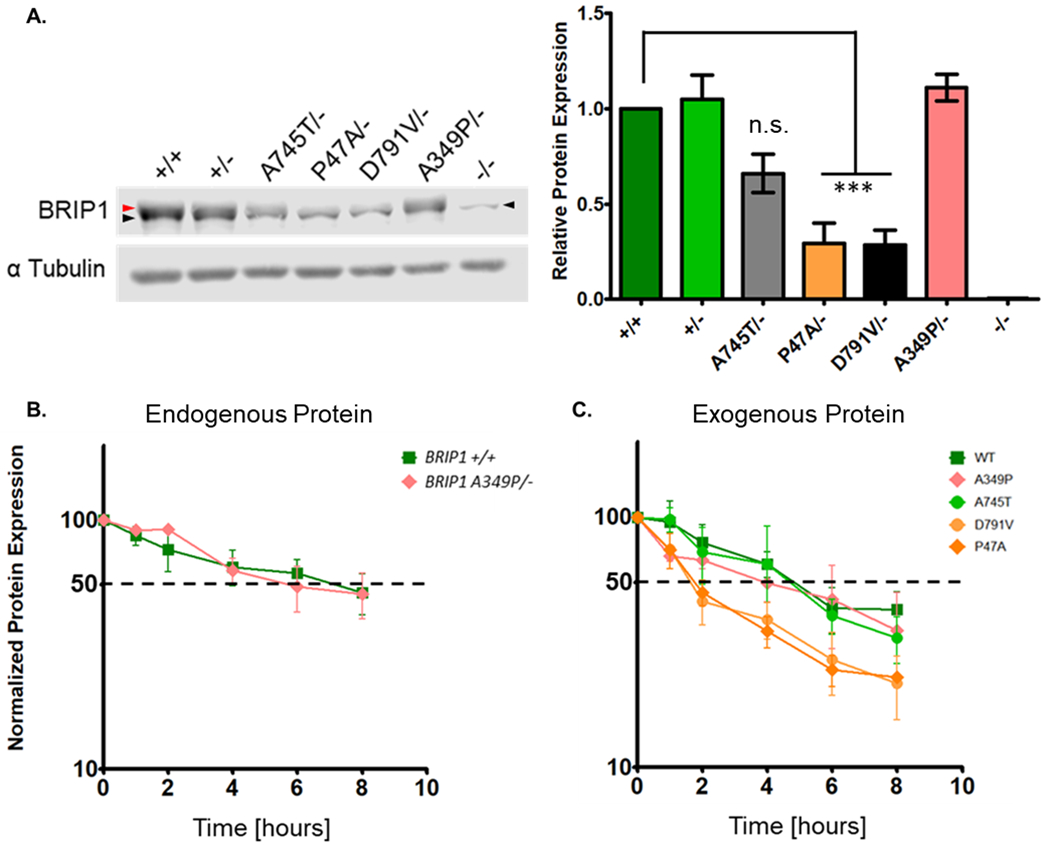
A) Representative western blot showing variable protein expression in HeLa clones (red arrowhead denotes BRIP1 protein; black arrow head denotes non-specific band of lower mass). Right panel: Quantification of relative BRIP1 levels. Data presented are the average of three replicates with error bars indicating one standard deviation. Statistical significance determined by one way ANOVA with a Dunnett’s multiple comparison test to BRIP1+/+ (*** p<0.0001). B) Cycloheximide chase analysis of endogenous wildtype and A349P protein. Fifty percent protein expression denoted by dotted black line. Data presented are the average of three replicates with error bars indicating one standard deviation. C) Cycloheximdie chase analysis of exogenously expressed wildtype, A349P, A745T, D791V and P47A protein. Half-life denoted by dotted black line. Data presented are the average of three replicates with error bars indicating one standard deviation.
Overexpression of the mutant proteins in a HeLa BRIP1−/− cell line combined with cycloheximide chase assays showed that exogenous wildtype (+/+), A349P and A745T proteins have half-lives of 4-5 hours. The P47A and D791V hypomorphs showed decreased protein stability with a half-life less than 2 hours (Figure 4C), confirming that P47A is an unstable protein and pointing to protein stability as a factor contributing to ICL sensitivity in D791V cells.
Rescue experiments classify additional missense alleles as wildtype, hypomorphic or null
In an effort to functionally characterize additional rare and novel alleles identified in patients with breast and ovarian cancer we undertook rescue experiments using our BRIP1−/− HeLa cell lines. Expression constructs for 20 missense variants were evaluated. These included the previously characterized P47A and A349P as controls plus 18 variants of unknown function, located in the C-terminal helicase region (15 from our discovery cohorts and 3 very rare variants previously reported in ovarian cases (14)). HeLa clones stably expressing Y822H, E636K, E511G, G690E and, the hypomorph D791V were readily established. Clonogenic growth assays and cell cycle analysis revealed that none of the alleles fully rescued the ICL effects despite having BRIP1 protein levels comparable to wildtype clones (Supplemental Figure S5A–B). Y822H, E636K, E511G and D791V all showed partial rescue in cell viability assays, similar to the endogenous hypomorph P47A and D791V alleles (CRISPR-Cas9 clones, Figure 3). Cell cycle analysis showed that when treated with MMC, the HeLa cells stably expressing those four missense alleles were all characterized by accumulation of cells in G2/M.
Of the five variants investigated, G690E was the only example in which there was no evidence for rescue of MMC cytotoxicity in the clonogenic growth assay. The dose response curve for the G690E rescue was nearly identical to that seen with the null clone and similar to the endogenous A349P allele (CRISPR-Cas9 clone, Figure 3). The substitution of a glutamic acid for a glutamine at position 690 was thus considered to result in a null protein. Given that A349P protein can increase sensitivity of cells to ICL damage in the presence of wildtype protein and is thus classified as a dominant negative(9), we created a stable clone expressing G690E in BRIP1+/+ HeLa cells and tested cell viability after exposure to MMC. The presence of G690E exogenous protein did not increase the sensitivity of wildtype cells to ICL damage (Supplemental Figure S5C) indicating that it is unlikely to act as a dominant negative. Analysis of protein stability showed that the G690E protein has a reduced half-life (~2 hours), comparable to D791V. The G690E variant, although null, was thus different than the A349P dominant negative (a stable protein). Reduced protein stability might explain in part why G690E it is a loss-of-function mutation (Supplemental Figure S5D) but other biochemical defects cannot be ruled out.
Despite multiple attempts (5 independent transfections), we were unable to establish stable clones expressing wildtype or the wildtype-like A745T protein in HeLa BRIP1−/− cells. The inability to stably express other members of the FA pathway has been reported (39). We determined that stable transfection of BRIP1−/− HeLa cells to express exogenous BRIP1 variants was possible only with hypomorphic or null alleles.
We devised a transient rescue assay that allowed us to characterize all missense variants (wildtype, hypomorph and null alleles). BRIP1−/− HeLa cells were transfected with pCDH-Puro-BRIP1 constructs, grown 48 hours and then put under puromycin selection and challenged with MMC. Cells were allowed to grow 10-15 days and subjected to a second MMC challenge. Fifteen-20 days later total numbers of clones were determined (Supplemental Figure S6A–B). Survival after two rounds of MMC treatment was dependent on exogenous protein expression (Supplemental Figure S6C). The MMC doses used for these transient rescue assays were determined on the basis of effects observed with known positive and negative controls. The high dose (30ng/mL) kills or severely impedes growth of all BRIP1−/− cells and cells expressing null or hypomorph alleles of BRIP1. The lower dose (15ng/mL) kills or severely impedes BRIP1−/− cells and cells expressing null alleles, but has a minor or no effect on cells expressing hypomorphic alleles (detailed methods describing the transient rescue assay can be found in Supplemental Materials and Methods with representative images shown in Supplemental Figure S6B).
Growth of clones that survived two rounds of MMC selection was used to classify variants. If greater than 50% of the transfected, twice challenged cells grew out at both low and high doses of MMC (compared to the unchallenged puromycin only selected population), then the re-expressed allele was deemed wildtype in function. If less than 50% of challenged cells grew out at either low or high dose of MMC, re-expressed alleles were classified as hypomorphs, having partial rescue ability but not to wildtype levels. If none of the challenged cells grew out at either dose of MMC, alleles were deemed null in function. All transient rescue experiments were replicated (2-4 replicates). Alleles with vagaries in protein expression and/or phenotype across replicates were excluded. In addition to testing alleles for ICL damage sensitivity, we assessed protein stability of each allele by transient exogenous overexpression of mutant proteins in HeLa null cell lines (Supplemental Figure S7).
Twenty missense alleles, located in the helicase domain, were successfully screened (Table 1). Of these, five alleles (25%) were classified as wildtype, including A745T and Q740H alleles that were identified in both of our cohorts. Nine variants (45%) fell into the category of hypomorphs, including both the P47A and D791V alleles characterized in our CRISPR model, as well as the three additional hypomorph alleles characterized by stable rescue. The remaining 6 alleles (30%) were classified as null, including the A349P and G690E allele. Most of the alleles classified as hypomorph and null created proteins with significantly shorter half-lives than wildtype protein, with null proteins trending to be more unstable than hypomorphs (Table 1, Supplemental Figure S7). The exception to this observation is the stable, dominant-negative, null A349P protein. We noted that some of the wildtype like alleles created proteins with much greater half-lives than WT protein and may in fact result in an increased stability of BRIP1 protein.
Table 1.
Key features of functionally characterized missense alleles
| Functional class | Expression construct | Conserved motif | Clone growth | Protein half-life [95% CI] | CONDEL | ||
|---|---|---|---|---|---|---|---|
| Puro only | MMC 15 | ng/mL 30 | |||||
| Wildtype | Wildtype | +++ | ++ | ++ | 5.7 [3.9, 8.5] | NA | |
| S407G | +++ | ++ | ++ | 8.2 [4.2, 10.8] | Damaging | ||
| H587L | +++ | ++ | ++ | 4.0 [3.0, 6.3] | Neutral | ||
| Q740H | +++ | ++ | ++ | 8.1 [5.5, 11.2] | Damaging | ||
| A745T | +++ | ++ | ++ | 4.9 [3.2, 6.11] | Damaging | ||
| L844I | +++ | ++ | ++ | ND | Neutral | ||
|
| |||||||
| Hypomorph | P47Aa | H-I | +++ | ++ | + | 2.9 [2.3, 4.0] | Damaging |
| E511G | +++ | ++ | + | 4.4 [3.5, 6.0] | Damaging | ||
| E636K | +++ | + | -- | 3.4 [2.6, 4.6] | Damaging | ||
| I691L | H-IV | +++ | ++ | + | 2.7 [2.0, 4.1] | Damaging | |
| G649S | +++ | ++ | + | 3.6 [2.3, 4.8] | Damaging | ||
| I782V | +++ | ++ | + | ND | Neutral | ||
| D791V | +++ | + | -- | 3.0 [2.3, 4.2] | Damaging | ||
| K797R | +++ | + | -- | 2.5 [1.7, 3.7] | Damaging | ||
| Y822H | H-VI | +++ | ++ | + | 3.8 [3.3, 4.5] | Damaging | |
|
| |||||||
| Null | A349Pa | Fe-S | +++ | -- | -- | 5.6 [3.8, 8.6] | Damaging |
| V676E | +++ | -- | -- | 2.6 [2.1, 3.3] | Damaging | ||
| G690E | H-IV | +++ | -- | -- | 2.3 [1.5, 3.3] | Damaging | |
| R777C | +++ | -- | -- | 2.2 [1.5, 3.7] | Damaging | ||
| C832Y | H-VI | +++ | -- | -- | 2.9 [2.3, 3.9] | Damaging | |
| R865W | +++ | -- | -- | 1.8 [1.4, 2.4] | Damaging | ||
| EV | +++ | -- | -- | NA | NA | ||
ND – not determined; NA – not applicable
previously characterized as deleterious missense variant
BRIP1 null and hypomorphs are seen in women with breast and ovarian cancer who have undergone clinical cancer genetic testing
BRIP1 is included in many cancer susceptibility NGS multi-gene panel tests (MGPT) and is frequently evaluated in patients with breast and ovarian cancer. Clinical testing data for 101,759 breast cancer patients and 15,587 ovarian cancer patients who had NGS MGPT in the period 2012-2018 identified 14 of the 20 missense alleles that we functionally classified as wildtype, hypomorphic or null. Of the six alleles not detected (E636K, G649S, V676E, D791V, C832Y, L844I), two were reported in ClinVar as having been identified in patients with familial cancer of the breast.
Of the 14 alleles identified in the clinical cohort and evaluated in this functional study, 323 breast (0.32%) and 56 ovarian (0.36%) cancer patients carried one of the alleles tested. The wildtype Q740H and A745T were the most frequently observed rare variants (Figure 5, Table 2). However, 161 patients (0.14%) carried an allele deemed null or hypomorphic in our functional assays. Strikingly, the I691L hypomorph allele was identified in 15 additional breast and ovarian cancer patients but was not observed in FLOSSIES or ExAC controls. Clinical testing confirms P47A as a recurrent BRIP1 risk allele and the frequency of our newly characterized hypomorph and null alleles is comparable to the frequency observed for A349P.
Figure 5. Occurrence of functionally characterized variants in clinical testing.
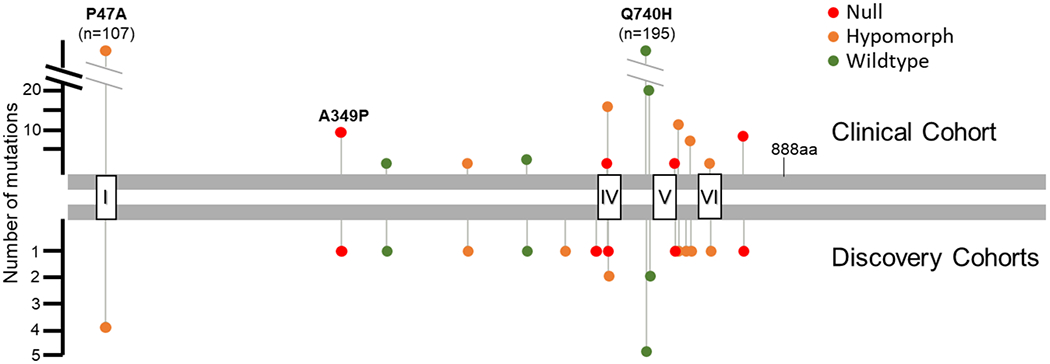
Q740H and A745T account for a majority of the wildtype alleles (green) identified. P47A was the most common hypomorph allele (orange) detected. Null alleles (red) were seen at lower frequencies. Helicase domain motifs: III (610-624); IV (689-710); V (748-775); VI (819-836).
Table 2.
Occurrence of functionally characterized missense alleles in patient and control populations
| Discovery Cohorts (n=3,359) | Clinical Cohort | EXAC (n=60,327) | FLOSSIES (n=9,884) | |||
|---|---|---|---|---|---|---|
| Breast Cancer (n=101,759) | Ovarian Cancer (n=15,587) | |||||
| Wildtypes | S407G | 1 | 1 | -- | -- | -- |
| H587L | 1 | 2 | -- | -- | -- | |
| Q740H | 5 | 163 | 32 | 56 | 8 | |
| A745T | 2 | 18 | 2 | 8 | 1 | |
| L844Ib | -- | -- | -- | -- | -- | |
| Hypomorphs | P47Aa | 4 | 96 | 11 | 29 | 5 |
| E511G | 1 | 1 | -- | -- | -- | |
| E636K | 1 | -- | -- | -- | -- | |
| I691L | 2 | 13 | 2 | -- | -- | |
| G649Sb | -- | -- | -- | -- | -- | |
| I782V | 1 | 10 | 1 | 2 | 1 | |
| D791V | 1 | -- | -- | -- | -- | |
| K797R | 1 | 3 | 4 | -- | 1 | |
| Y822H | 1 | 1 | -- | 1 | -- | |
| Nulls | A349Pa | 1 | 7 | 2 | 3 | -- |
| V676E | 1 | -- | -- | -- | -- | |
| G690E | 1 | 1 | -- | -- | -- | |
| R777C | 1 | 1 | -- | -- | -- | |
| C832Yb | -- | -- | -- | -- | -- | |
| R865W | 1 | 6 | 2 | 1 | 2 | |
previously characterized as deleterious missense variant
rare missense variants previously reported in ovarian cancer cases(14)
Combining the discovery and clinical cohorts, 178 ovarian and breast cancer patients carried a hypomorph or null missense variant (0.15%) which is significantly greater than the expected frequency in the general population (ExAC 0.06%; odds ratio = 2.47, 95%CI=1.73-3.54, p<0.0001).
Discussion
Our study shows that greater than 2% of patients with ovarian and early-onset breast cancer carry a rare missense variant in BRIP1 DNA helicase. At present, the effect of these missense variants on protein function and the cancer risk they confer is largely unknown. Our functional characterization of 20 rare missense variants in the BRIP1 helicase domain (18 of which had not previously been studied) revealed that most (15-of-20, 75%) significantly impaired ICL repair and are thus loss-of-function defects. A majority were hypomorphs (9-of-15) and six were considered to be nulls.
Breast and ovarian cancer patients will frequently undergo genetic testing for mutations in cancer susceptibility genes. Testing has been widely adopted because of the recognized benefit to patients and their family members. Positive or negative findings help guide cancer surveillance for the patient and her family members. Identification of a germline mutation in BRCA1 or BRCA2 is central to both primary prevention (surgical prophylaxis) and for those women who have breast or ovarian cancers, to decisions regarding best approaches for adjuvant and/or second-line treatment. Risk reducing oophorectomy and mastectomy in patients with BRCA1/2 mutations is associated with lower rates of ovarian and breast cancer with a decrease in all-cause mortality(40). Currently, germline testing is most often for a large number of cancer susceptibly genes. The role that mutations play in some of the genes tested is not fully understood and frequently they are low or moderate penetrance mutations. A complexity of panel testing is that rare and/or previously uncharacterized VUSs will be identified more frequently than pathogenic mutations. Rare variants, particularly those identified in moderate to low penetrance susceptibility genes, pose a challenge to health care providers. The risk associated with these variants is usually unknown and family-based studies (penetrance and expressivity analyses as well as tumor studies) to classify variants are not feasible. Ambiguity surrounding the potential function of rare variants can create unwanted anxiety in patients and family members and may lead to unnecessary medical interventions (41–43). Functional studies are needed to better interpret cancer risk and guide disease management strategies.
Reduced protein stability is a frequent feature of hypomorphic alleles. We found that most of the hypomorph and null alleles we evaluated have reduced half-lives and low steady state levels of BRIP1 protein. Although lower protein levels could explain the reduction in ICL repair activity we observed, it is likely that some of the missense changes directly affect the helicase activity of the mutant protein. We showed that BRIP1+/− HeLa and BRIP1+/− HEK293TN cells do not show an increase in sensitivity to MMC or cisplatin (Figure 2B and 3, Supplemental Figure S4A–B), and from that we conclude haploinsufficiency does not confer increase sensitivity to ICL damage. One of the controls we investigated, the unstable P47A protein, is ATPase deficient and lacks helicase activity (3). Biochemical characterization of helicase activity of the unstable alleles could further determine the functional effects of missense alleles that are associated with reduced ICL repair. A second control allele we investigated (A349P) is a null based on assays that measure effects of MMC on chromosome fragmentation, cell viability and cell cycle, but has normal protein stability (Figures 2, 3, and 4B–C). Other BRIP1 functions that could be affected by single amino acid substitutions include DNA binding, nuclear localization, processivity of unwinding, displacement of proteins, binding to other proteins, dimerization, and recruitment to sites of damage (4, 9, 44–47). Ideally, all of these functions could be considered as part of missense variant analysis. The currently available reagents present challenges to fully characterize the function of BRIP1 protein variants. The lack of specific of antibodies for G4 DNA made it impossible for us to assess the effects of variants on G4 DNA resolution. Additional reagents will be critical for in vivo functional assays.
Creating BRIP1 null cell lines in HeLa and HEK293 was relatively simple as both cell lines seem to thrive without BRIP1 protein. In contrast, rescuing the null phenotype with re-expression of wildtype proteins proved to be unexpectedly difficult. Stable clones rescued with wildtype BRIP1 protein could only be created after challenging the cells with ICL damaging agents to force survival dependency on the exogenously expressed protein. A doxycycline inducible, controlled expression system could provide a way to overcome this obstacle and should be explored in future efforts to characterize additional variants.
In our initial discovery efforts, we noticed that three very rare alleles (R579C, K979E and Q227E) were seen twice in our breast cancer cohort but were not observed in the ovarian cancer cohort. In addition, a cluster of five different missense alleles, located in the highly conserved Fe-S domain, were identified in our ovarian cancer cohort but none were observed in this domain in the breast cancer cohort (Figure 1). These idiosyncrasies may allude to functional regions of the protein that are cancer type specific and could explain the difference in occurrence of BRIP1 mutations between ovarian and breast cancer.
A745T and Q740H account for a majority of the wildtype alleles identified in ovarian and breast cancer patients who underwent clinical genetic testing and carry one of the rare missense alleles functionally tested in our study. Of the remaining individuals who carry one of the other 18 alleles tested, most harbor a putative hypomorph or null variant. Remarkably, 15 patients carry the hypomorph I691L allele, which was not observed in either ExAC or FLOSSIES control populations. This suggests that some rare alleles in BRIP1 are more penetrant than others.
Although BRIP1 mutations have been described as the third or fourth most frequent causes of inherited ovarian cancer (14–16, 48), the analyses have been largely restricted to nonsense or frameshift alleles. Family-based studies to analyze penetrance of very rare alleles are impractical, making it difficult to assess cancer risk associated with rare missense alleles identified in BRIP1. The clinical utility for understanding the functional impact of missense variants in susceptibility genes has been made clear by the extensive research on BRIP1 binding partner, BRCA1. Classification of VUSs identified in ovarian and breast cancer patients can contribute to earlier detection and screening of disease and improve treatment options by personalizing care with targeted therapies. This study highlights the importance of uncovering the functions of BRIP1 in tumor development and the role of BRIP1 missense variants in protein function.
Supplementary Material
Significance:
Functional characterization of rare variants of uncertain significance in BRIP1 revealed that 75% demonstrate loss-of-function activity, suggesting rare missense alleles in BRIP1 confer risk for both breast and ovarian cancer.
Acknowledgments
We wish to thank the dedicated staff at the Siteman Cancer Center and all of the patients who participated The Young Women’s Breast Cancer Program (YWBCP). Specifically, we acknowledge Kay Coker and Diane Struckhoff for their key role in recruitment subject to the YWBCP. We wish to thank Thomas Ludwig and Dongju Park for their Karyotyping expertise. We also thank the Genomics Shared Resource and the Analytical Cytometry Shared Resource at the Ohio State University Comprehensive Cancer Center for support with Sanger sequencing and flow cytometry. And, we wish to thank Virginia Speare and Patrick Reineke at Ambry for their contributions to clinical data curation and manuscript editing.
The work was supported in part by Celebrate Fitness and the Siteman Cancer Center, Washington University, St. Louis and Marsha Rivkin Center for Ovarian Cancer Research (Marsha Rivkin Center GRT00041690) Pilot Study Award [PJG]. Funding for sequencing the ovarian cancer cases came from an Ovarian Cancer Research Foundation Program Project Development Award [EMS and SHK], Department of Defense Ovarian Cancer Research Program OC120506 [EMS and SHK] and Mayo Clinic Ovarian Cancer Spore 5P50CA136393 [SHK]. This work was also supported in part by Pelotonia [CLM].
Funding:
Department of Defense Ovarian Cancer Research Program OC120506 [E.M.S. and S.H.K.] and Mayo Clinic Ovarian Cancer Spore 5P50CA136393 [S.H.K.]
Footnotes
Disclosure of Potential Conflicts of Interest: M.E.R. is an employee of Ambry Genetics. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, et al. 2001. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105:149–160. [DOI] [PubMed] [Google Scholar]
- 2.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, and Cantor SB 2005. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8:255–265. [DOI] [PubMed] [Google Scholar]
- 3.Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, and Livingston DM 2004. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci U S A 101:2357–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta R, Sharma S, Sommers JA, Jin Z, Cantor SB, and Brosh RM Jr. 2005. Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J Biol Chem 280:25450–25460. [DOI] [PubMed] [Google Scholar]
- 5.London TB, Barber LJ, Mosedale G, Kelly GP, Balasubramanian S, Hickson ID, Boulton SJ, and Hiom K 2008. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem 283:36132–36139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Y, Shin-ya K, and Brosh RM Jr. 2008. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol 28:4116–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bharti SK, Sommers JA, George F, Kuper J, Hamon F, Shin-ya K, Teulade-Fichou MP, Kisker C, and Brosh RM Jr. 2013. Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J Biol Chem 288:28217–28229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, and White MF 2006. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol Cell 23:801–808. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y, Sommers JA, Suhasini AN, Leonard T, Deakyne JS, Mazin AV, Shin-Ya K, Kitao H, and Brosh RM Jr. 2010. Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood 116:3780–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X, Chini CC, He M, Mer G, and Chen J 2003. The BRCT domain is a phospho-protein binding domain. Science 302:639–642. [DOI] [PubMed] [Google Scholar]
- 11.Bridge WL, Vandenberg CJ, Franklin RJ, and Hiom K 2005. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet 37:953–957. [DOI] [PubMed] [Google Scholar]
- 12.Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, et al. 2005. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet 37:931–933. [DOI] [PubMed] [Google Scholar]
- 13.Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, et al. 2005. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet 37:934–935. [DOI] [PubMed] [Google Scholar]
- 14.Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN, Intermaggio MP, Fraser L, Gentry-Maharaj A, Hayward J, Philpott S, et al. 2015. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J Natl Cancer Inst 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, Bernards SS, Casadei S, Yi Q, Burger RA, et al. 2016. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol 2:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber-Lassalle N, Hauke J, Ramser J, Richters L, Gross E, Blumcke B, Gehrig A, Kahlert AK, Muller CR, Hackmann K, et al. 2018. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res 20:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, et al. 1998. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 62:676–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, et al. 2017. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 317:2402–2416. [DOI] [PubMed] [Google Scholar]
- 19.Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, and Weber BL 2002. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst 94:1365–1372. [DOI] [PubMed] [Google Scholar]
- 20.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, et al. 2006. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 38:1239–1241. [DOI] [PubMed] [Google Scholar]
- 21.De Nicolo A, Tancredi M, Lombardi G, Flemma CC, Barbuti S, Di Cristofano C, Sobhian B, Bevilacqua G, Drapkin R, and Caligo MA 2008. A novel breast cancer-associated BRIP1 (FANCJ/BACH1) germ-line mutation impairs protein stability and function. Clin Cancer Res 14:4672–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Easton DF, Lesueur F, Decker B, Michailidou K, Li J, Allen J, Luccarini C, Pooley KA, Shah M, Bolla MK, et al. 2016. No evidence that protein truncating variants in BRIP1 are associated with breast cancer risk: implications for gene panel testing. J Med Genet 53:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, Hallberg E, Moore R, Thomas A, Lilyquist J, et al. 2017. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. 2016. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44:D862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, and King MC 2010. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A 107:12629–12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mu W, Lu HM, Chen J, Li S, and Elliott AM 2016. Sanger Confirmation Is Required to Achieve Optimal Sensitivity and Specificity in Next-Generation Sequencing Panel Testing. J Mol Diagn 18:923–932. [DOI] [PubMed] [Google Scholar]
- 27.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. 2016. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez-Perez A, and Lopez-Bigas N 2011. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bharti SK, Sommers JA, Awate S, Bellani MA, Khan I, Bradley L, King GA, Seol Y, Vidhyasagar V, Wu Y, et al. 2018. A minimal threshold of FANCJ helicase activity is required for its response to replication stress or double-strand break repair. Nucleic Acids Res 46:6238–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sasaki MS, and Tonomura A 1973. A high susceptibility of Fanconi’s anemia to chromosome breakage by DNA cross-linking agents. Cancer Research 33:1829–1836. [PubMed] [Google Scholar]
- 35.Kumaraswamy E, and Shiekhattar R 2007. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol 27:6733–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwab RA, Nieminuszczy J, Shin-ya K, and Niedzwiedz W 2013. FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. The Journal of cell biology 201:33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarkies P, Murat P, Phillips LG, Patel KJ, Balasubramanian S, and Sale JE 2012. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res 40:1485–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castillo Bosch P, Segura-Bayona S, Koole W, van Heteren JT, Dewar JM, Tijsterman M, and Knipscheer P 2014. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J 33:2521–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue Y, Li Y, Guo R, Ling C, and Wang W 2008. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum Mol Genet 17:1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domchek SM, Friebel TM, Singer CF, Evans DG, Lynch HT, Isaacs C, Garber JE, Neuhausen SL, Matloff E, Eeles R, et al. 2010. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 304:967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson ER, Rowley SM, Li N, McInerny S, Devereux L, Wong-Brown MW, Trainer AH, Mitchell G, Scott RJ, James PA, et al. 2016. Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J Clin Oncol 34:1455–1459. [DOI] [PubMed] [Google Scholar]
- 42.Kurian AW, Li Y, Hamilton AS, Ward KC, Hawley ST, Morrow M, McLeod MC, Jagsi R, and Katz SJ 2017. Gaps in Incorporating Germline Genetic Testing Into Treatment Decision-Making for Early-Stage Breast Cancer. J Clin Oncol 35:2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffman-Andrews L 2017. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci 4:648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lei H, and Vorechovsky I 2003. BACH1 517C-->T transition impairs protein translocation to nucleus: a role in breast cancer susceptibility? Int J Cancer 104:389–391. [DOI] [PubMed] [Google Scholar]
- 45.Gupta R, Sharma S, Sommers JA, Kenny MK, Cantor SB, and Brosh RM Jr. 2007. FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood 110:2390–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sommers JA, Rawtani N, Gupta R, Bugreev DV, Mazin AV, Cantor SB, and Brosh RM Jr. 2009. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J Biol Chem 284:7505–7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Y, Sommers JA, Loiland JA, Kitao H, Kuper J, Kisker C, and Brosh RM Jr. 2012. The Q motif of Fanconi anemia group J protein (FANCJ) DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J Biol Chem 287:21699–21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurian AW, Ward KC, Howlader N, Deapen D, Hamilton AS, Mariotto A, Miller D, Penberthy LS, and Katz SJ 2019. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J Clin Oncol 37:1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.