

Abstract
Autosomal dominant tubulointerstitial kidney disease (ADTKD) refers to a group of disorders with a bland urinary sediment, slowly progressive chronic kidney disease (CKD), and autosomal dominant inheritance. Due to advances in genetic diagnosis, ADTKD is becoming increasingly recognized as a cause of CKD in both children and adults. ADTKD-REN presents in childhood with mild hypotension, CKD, hyperkalemia, acidosis, and anemia. ADTKD-UMOD is associated with gout and CKD that may present in adolescence and slowly progresses to end-stage kidney disease. HNF1β mutations often presents in childhood with anatomic abnormalities such as multicystic or dysplastic kidneys, in addition to CKD and a number of other extra-kidney manifestations. ADTKD-MUC1 is less common in childhood, and progressive CKD is its sole clinical manifestation, usually beginning in the late teenage years. This review describes the pathophysiology, genetics, clinical characteristics, diagnosis, and treatment of the different forms of ADTKD, with an emphasis on diagnosis. We also present data on kidney function in children with ADTKD from the Wake Forest Rare Inherited Kidney Disease Registry.
Keywords: uromodulin, mucin-1, renin, autosomal dominant, pediatric, chronic kidney disease, inherited, review
Introduction:
Autosomal dominant tubulointerstitial kidney disease (ADTKD) has three cardinal features: autosomal dominant inheritance, slowly progressive chronic kidney disease (CKD), and a bland urinary sediment[1, 2]. The most common causes of ADTKD are mutations in UMOD [3], MUC1 [4], HNF1β[5], and REN [6, 7]. In this review, we will describe these conditions, with an emphasis on their clinical presentation and management in childhood.
The most important and distinguishing characteristic of these genetic conditions is autosomal dominant inheritance. While many family members may be affected, the presence of CKD of any type that affects a parent and a child is strongly suggestive of an autosomal dominant disorder. CKD is slowly progressive in all forms of ADTKD, but there is a wide spectrum of disease, with some individuals reaching end-stage kidney disease (ESKD) in their teens (rarely), while other individuals in the same family may not develop ESKD until past age 80 years[8]. While ESKD from ADTKD rarely occurs in childhood, CKD is often present. Thus, autosomal dominant inheritance, slowly progressive CKD, and a bland urinary sediment are the most important findings and are present in the vast majority of children with this disorder.
An important distinction in the clinical diagnosis of children with a bland urinary sediment and CKD is whether they may have nephronophthisis or ADTKD. Nephronophthisis is an autosomal recessive disorder caused by homozygous or compound heterozygous mutations in one of over 20 genes expressed in kidney tubular ciliary proteins. [9]. [9–11]. The infantile form of nephronophthisis may present at birth, with ESKD often developing within the first years of life[12]. In the juvenile form of nephronophthisis, patients may present with polydipsia, polyuria, growth retardation, and CKD[12], with a median age of ESKD of approximately 13 years[13], though ESKD has occurred as late as age 56 years [14]. Thus, in children with a bland urinary sediment, CKD, and no evidence of kidney disease in either parent, one should first consider nephronophthisis. In contrast, ADTKD is an autosomal dominant disorder, with a parent usually being affected. While ADTKD-REN can present in childhood, the median age of ESKD of approximately 54 years in ADTKD-UMOD and 46 years in ADTKD-MUC1[15] (see Fig. 1). As with nephronophthisis, there is a spectrum with regards to the age of ESKD in ADTKD, with patients as young as 6 years having developed ESKD[16–19]. Thus, in children with CKD and a bland urinary sediment, one may first consider ADTKD if a parent also has CKD.
Fig. 1.
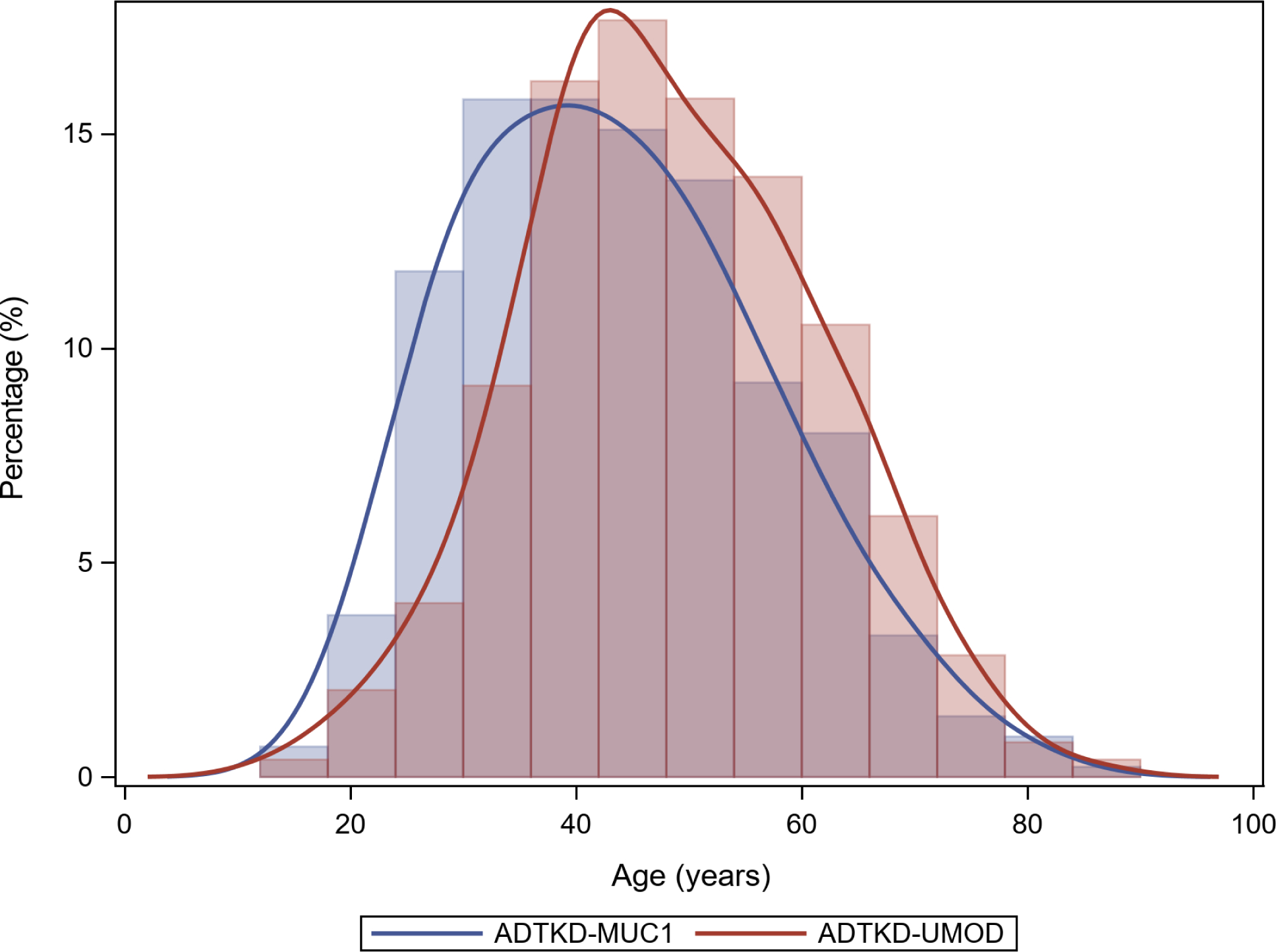
Distribution of the age of end-stage kidney disease in ADTKD-UMOD and ADTKD-MUC1 from the Wake Forest Rare Inherited Kidney Disease Registry, including 493 individuals with ADTKD-UMOD and 424 individuals with ADTKD-MUC1
Congenital anomalies of the kidney and urinary tract (CAKUT) may also present in childhood with CKD and structural abnormalities of the kidney, including multicystic kidneys, kidney dysplasia or agenesis, vesico-ureteral reflux and other urinary tract abnormalities. Kidney dysplasia refers to abnormal kidney architecture with immature nephrons and undifferentiated stroma. Kidney dysplasia may be identified on ultrasound as echogenic kidneys, with kidneys being small or of normal size, often with accompanying cysts[20]. Many patients will have CKD as a result of these anatomic changes, and family members may have CKD with no known CAKUT findings[21]. HNF1β mutations are a frequent cause of CAKUT and are most commonly associated with bilateral multicystic kidneys or kidney dysplasia [22]. In addition, the findings of echogenic kidneys on ultrasound with CKD and a positive family history are similar to changes that may be seen in nephronophthisis, ADTKD-REN, MUC1, and UMOD.
Given the overlap in clinical presentations between nephronophthisis, CAKUT, and ADTKD, and the possibility of de novo mutations in ADTKD, gene panels encompassing a large number of genetic disorders of the kidney or whole exome sequencing are the most efficient method to identify genetic causes of CKD in childhood. A collaborative approach will provide the highest probability of making a correct diagnosis. Adult and pediatric nephrologists should work together to compare manifestations of disease in the family across the entire age spectrum. Urologists and other specialists may also provide input when other clinical manifestations exist. Importantly, a genetics counselor will be most helpful in establishing inheritance, identifying appropriate diagnostic options, and obtaining consent from the family regarding benefits and risks of diagnosis. The genetics counselor will also be helpful in educating the family when a diagnosis is made and providing information and screening to other family members. Finally, specialists in these rare disorders may be called upon to provide their expertise.
Methods:
In addition to a review of the literature, data is included from an ongoing cohort study of patients with ADTKD at Wake Forest School of Medicine[23]. The registry was started in 1996, and all patients with ADTKD are invited to participate. At present, there are 469 individuals and 163 families with ADTKD-UMOD, 307 individuals and 99 families with ADTKD-MUC1, and 23 individuals and 13 families with ADTKD-REN. The protocol has been approved by the Institutional Review Board of Wake Forest School of Medicine. Estimated glomerular filtration rate (eGFR) was measured with the Pottel equation to facilitate comparison of measurements between childhood and early adulthood[24].
ADTKD-UMOD:
Pathophysiology and genetics:
ADTKD-UMOD is due to mutations in the UMOD gene encoding the glycoprotein uromodulin (also known as Tamm Horsfall glycoprotein)[3]. The mature uromodulin protein is a membrane-anchored glycoprotein containing 616 amino acids, with 48 cysteine residues contributing to 24 disulfide bridges that are crucial for proper protein folding. Uromodulin undergoes extensive cross-linking in the endoplasmic reticulum[25]. Uromodulin is predominantly expressed in the thick ascending limb of Henle and to a lesser degree in the distal convoluted tubule[26]. UMOD mutations affect the amino acid sequence of the uromodulin protein, resulting in improper folding of uromodulin and deposition in the endoplasmic reticulum[27]. Mutant uromodulin deposition leads to endoplasmic reticulum stress and accelerated apoptosis, tubular cell dropout and nephron loss leading to CKD. Wild-type uromodulin enhances the delivery of the furosemide-sensitive NKCC2 transporter and the thiazide-sensitive sodium chloride cotransporter NCC to the apical surface of tubular cells, leading to sodium retention [26, 28, 29]. Decreased uromodulin production in ADTKD-UMOD leads to a decreased presence of the NCC and NKCC2 transporters on the apical surface, resulting in decreased sodium reabsorption and a mild natriuresis. The mild natriuresis leads to a secondary increase in proximal tubular uptake of sodium and secondarily uric acid. Thus, affected individuals develop hypouricosuric hyperuricemia, a cardinal feature of ADTKD-UMOD.
Clinical characteristics:
Hyperuricemia is present early in life, and the most common pediatric presentation of ADTKD-UMOD is adolescent gout (see Fig. 2). In the Wake Forest cohort of 459 individuals with ADTKD-UMOD, there was a 50% lifetime risk of developing gout (vs. 4% prevalence in the general population[30]), and 8% developed gout in childhood at a mean age of 15.2±2.8 years [15]. A 5 year-old was the youngest individual to develop gout. Due to the common occurrence of hyperuricemia in ADTKD-UMOD, the condition was initially termed familial juvenile hyperuricemic nephropathy[31, 32]. Patients often present with classic symptoms of podagra or knee inflammation. As gout can be triggered by excess physical activity [33], teenagers may develop a gout attack after engaging in sports. The condition is frequently misdiagnosed by physicians due to the rarity of pediatric gout. However, parents in affected families will often offer the diagnosis, as they are familiar with the clinical manifestations of gout and look to it as a harbinger of ADTKD-UMOD.
Fig. 2.
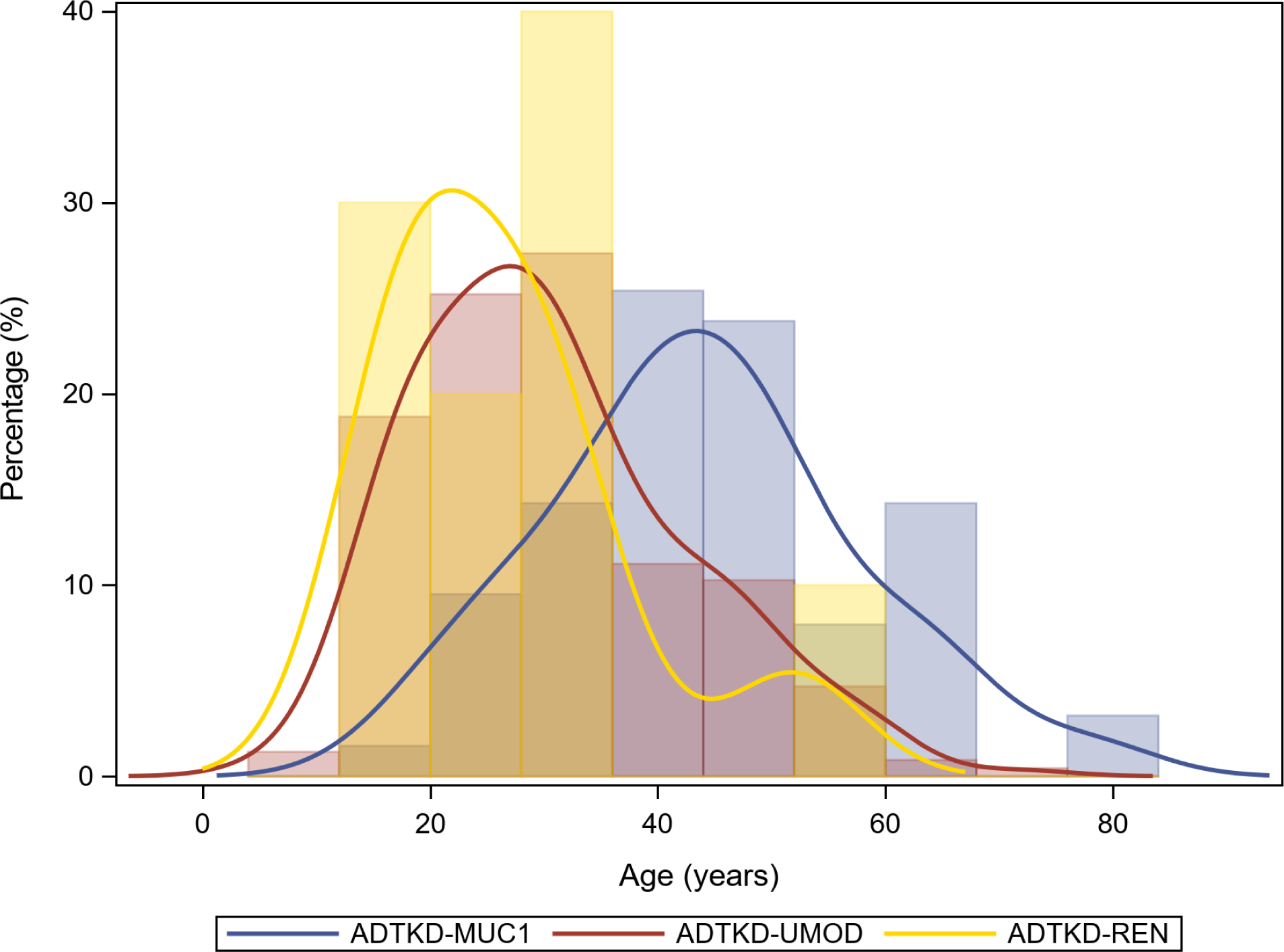
Distribution curve of gout by age, including 63 individuals with ADTKD-MUC1, 234 individuals with ADTKD-UMOD, and 10 individuals with ADTKD-REN
CKD often occurs in children with ADTKD-UMOD (see Fig. 3). The Wake Forest cohort included data on 34 children <18 years with ADTKD-UMOD. At the time of first measurement, the mean eGFR was 72.3±20.1 ml/min/1.73m2 at a mean age of 12.5±4.0 years. There were 11(32%) children with stage 3 CKD who presented at a mean age of 15.1±2.8 years. Fifteen (44%) individuals had stage 2 CKD, with a mean age at measurement of 11.7±4.1 years. There were 7 (21%) individuals with an eGFR > 90 ml/min/1.73m2 with a mean age of presentation of 10.4±3.9 years. Thus, the majority of children with ADTKD-UMOD have CKD, becoming more prevalent as these individuals age during childhood.
Fig. 3.
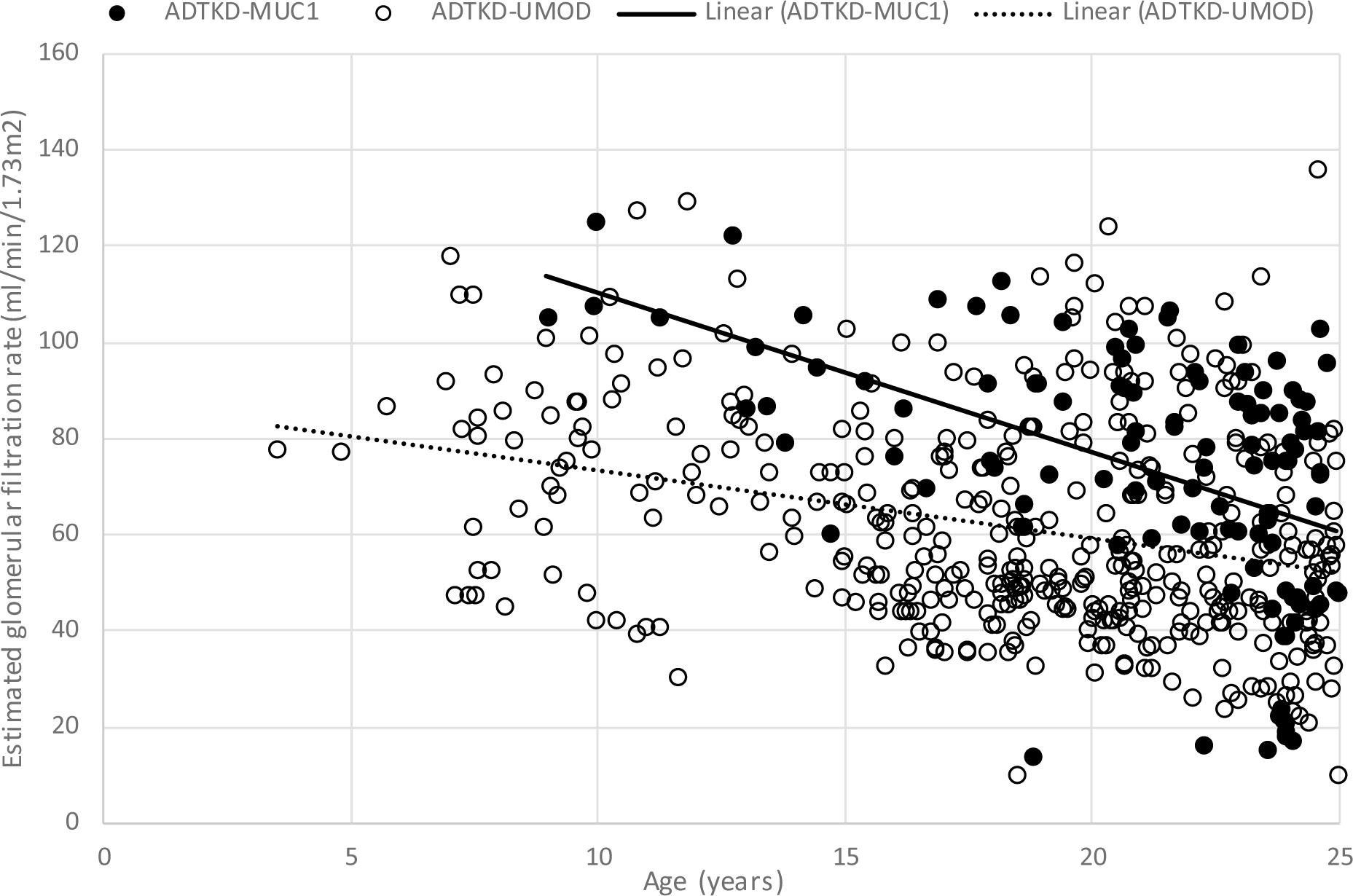
Estimated glomerular filtration rate (eGFR) values prior to age 24 for 47 individuals with ADTKD-MUC1, 81 with ADTKD-UMOD. The eGFR was calculated using the Pottel equation[24]. An eGFR of 10 ml/min/1.73m2 was assigned when a patient reached end-stage kidney disease
While CKD is common in childhood, progression to ESKD is rare prior to adulthood [34, 35]. The earliest age of ESKD reported is in a child with a Cys248Trp mutation who presented at age 3 with decreased eGFR, urinary concentration defect, hypertension, and mild proteinuria[36]. The patient proceeded to ESKD at age 6. Other family members proceeded to dialysis between ages 18 and 52, illustrating the wide variation of age of ESKD in families with ADTKD-UMOD that remains unexplained. In another family with an in-frame deletion of nucleotides 668–767 a child developed ESKD at age 12 years [19]. We have also identified a family with a p.C52S mutation, whose family members developed ESKD at 12, 14, 18 and 26 years. Other families with this mutation were found to have a mean age of ESKD of 43 and 30 years. In these families presenting at an early age, gout is usually present, and there is often a family history of ADTKD with similar early presentation of family members. Other characteristics of the disease remain similar to patients with older presentation.
Diagnosis:
The key factor leading to a diagnosis is the presence of CKD in both the patient and an affected parent and possibly other family members. Hypouricosuric hyperuricemia is present in almost all children with this disorder, and the presence of gout strongly suggests this diagnosis. When evaluating pediatric patients for hyperuricemia, it is important to use age-related norms for serum urate levels [37–39]. Fractional excretion of urate (FEurate ) is usually less than 0.05 (5%) in children with ADTKD-UMOD[40]. In the Wake Forest cohort, the mean FEurate was 0.044±0.025 in affected children with a range of 0.016–0.101. However, in a large cohort study of normal children the FEurate was <0.05 in 42% of children between 5 and 10 years of age[37], making FEurate an unreliable screening test. While hyperuricemia is seen in most cases of ADTKD-UMOD, it can be seen in CKD of any cause. Children with ADTKD-UMOD will have a bland urinary sediment and a kidney ultrasound with normal or smaller kidney size and kidney cysts in about 20% of cases.
The differential diagnosis for ADTKD-UMOD includes nephronophthisis. Patients with nephronophthisis also have a bland urinary sediment and normal or small kidneys on ultrasound. The key differentiating factor is that nephronophthisis is autosomal recessive, with only other siblings affected, while in ADTKD-UMOD, there is usually at least a parent affected. Clinical findings associated with nephronophthisis can include retinitis pigmentosa, hepatic fibrosis, and situs inversus, as well as other neurological, skeletal, and ocular manifestations[11]. Hyperuricemia and gout are more common in ADTKD-UMOD. Hyperuricemia and gout in childhood may also lead to consideration of urate overproduction syndromes such as PRS1 overactivity or HPRT deficiency. Both of these conditions are X-linked and associated with overproduction of urate rather than under-excretion. Gout is usually present much earlier in life in these disorders, and untreated patients often have urate nephrolithiasis.
Genetic testing:
Mutational analysis of the UMOD gene is the definitive diagnostic test. Kidney biopsies are not recommended for diagnosis, as biopsies can be associated with significant complications, and pathologic findings are nonspecific and non-diagnostic for ADTKD-UMOD. Similar pathologic changes may be seen in nephronophthisis. While directed genetic analysis of the UMOD gene can be performed, genetic panels will rule out other possible causes (such as nephronophthisis and ADTKD-REN).
Treatment:
At present, there is no specific treatment for ADTKD-UMOD. Given the mild natriuresis that patients experience, sodium restriction is not indicated and may exacerbate hyperuricemia. Young patients who present with gout should be treated with allopurinol[41], as, due to the genetic nature of this condition, patients are highly likely to have many subsequent gout attacks, and tophi may develop over time. Several past publications suggested that allopurinol might slow progression[42]. A weakness of these studies was the lack of a genetic diagnosis and short-term follow-up. Given these considerations, we recommend treatment with allopurinol if patients have pediatric gout but not to prevent CKD progression. If patients have a significantly elevated serum urate level, one might consider using allopurinol preventively if, for example, the patient is a competitive athlete who wishes to avoid the possibility of a gout attack unexpectedly before an important sporting event. Allopurinol prevents gout by lowering serum uric acid levels and is not a treatment for acute gout. When allopurinol is initiated, there may be an increased incidence of gout for the first several months. Allopurinol should not be stopped in the setting of an acute gout flare. If a skin rash develops, allopurinol should be stopped immediately and the patient evaluated, as allopurinol has rarely been associated with Stevens Johnson syndrome. Low-purine diets are not recommended, as they are less likely to be effective and have substantial impact on quality of life[41].
Genetic screening:
Parents in families with ADTKD-UMOD may wish to have their children tested for UMOD mutations. Genetic societies discourage the testing of children for genetic disorders unless a specific therapy is available. If genetic testing is considered, the child should be informed of the benefits and risks of diagnosis and should give assent. Risks include a lifelong diagnosis that could affect the acquisition of health and life insurance in some countries. In adolescent patients who are active in athletics, one might consider genetic screening or measurement of uric acid levels, as gout prevention would prevent inopportune gout attacks. For patients in whom the mean familial age of ESKD is >45 years and gout is infrequent, one should discourage genetic testing because the condition is unlikely to affect the child before age 18 years and is very unlikely to affect the patient for many years. It is also important to consider providing information and screening of other family members. Many affected adult family members may be undiagnosed, as well as their children. Such evaluation will also be helpful in the identification of unaffected potential kidney donors. Referral to a genetic counsellor will be helpful in screening, education, and also in making a genetic diagnosis.
Summary:
ADTKD-UMOD can present in childhood with hyperuricemia, gout, CKD, and a family history of CKD. Only in rare instances does ADTKD-UMOD result in ESKD in children. Patients have a bland urinary sediment and often have many affected family members. Genetic diagnosis can be performed in childhood, but only with the assent of the child and only if avoidance of gout is a high priority. Gout is well controlled with allopurinol.
ADTKD-MUC1:
Pathophysiology and genetics:
ADTKD-MUC1 is due to mutations in the MUC1 gene that encodes mucin-1, one of 20 different mucins found in humans. A combination of different mucins lines epithelial surfaces, including the respiratory tract, gastrointestinal tract, mammary ducts, and sebaceous ducts[43]. Mucin-1 is a transmembrane protein with a cytoplasmic tail that is responsible for intracellular signaling. Mucin-1 contains a repetitive sequence of 20 amino acids (known as a variable number of tandem repeats (VNTR)). There are between 20 and 120 repeats in the mucin-1 VNTR as determined by the paternal and maternal VNTR lengths. Sugar residues bind to the VNTR, contributing to strong O- and N-glycosylation and giving the mucin-1 molecule its mucinous properties[44]. The most common mutation in ADTKD-MUC1 is the addition of a cytosine residue to a seven cytosine tract in the VNTR, resulting in a unique frameshift protein within the VNTR. Other mutations may also cause ADTKD-MUC1, including the addition of a guanosine residue or loss of two cytosine residues; all mutations result in the creation of the same frameshift protein[45]. The mutated MUC1 protein deposits in the endoplasmic reticulum Golgi intermediate compartment (ERGIC), where secretory proteins are transported in cargo vesicles between the ER exit sites and the Golgi apparatus[46]. The deposition of this abnormal protein results in accelerated tubular cell death, nephron dropout, and progressive CKD. Unfortunately, due to the high cytosine/guanosine content and the repetitive nature of the VNTR, genetic mutational analysis cannot be carried out with standard genetic sequencing[4, 45, 47].
Clinical characteristics:
While mucin-1 is expressed in many tissues and the mutated protein can be identified in many epithelial tissues of affected individuals[45], the only clinical characteristic of ADTKD-MUC1 is CKD, and all secondary clinical manifestations (e.g. hypertension and gout) are due to CKD and occur with the same frequency as in other forms of CKD. The isolated kidney phenotype, despite the broad expression of mucin-1 in multiple tissues, remains unexplained. Affected individuals will have a bland urinary sediment with minimal proteinuria, and the kidney ultrasound will be unremarkable in childhood. While children with ADTKD-MUC1 may have a mildly decreased eGFR, this condition is unlikely to present in childhood, and there is no benefit of screening in children.
In the Wake Forest cohort, 18 individuals with ADTKD-MUC1 underwent eGFR measurement prior to age 18. The mean age of these individuals was 14.1±2.8 years and the mean eGFR was 94.3±18.1 ml/min/1.73m2. There were no individuals with Stage 3 CKD, and 8(44%) had Stage 2 CKD, presenting at a mean age of 15.2±1.7 years. Thus, most children with ADTKD-MUC1 will have a normal or mildly decreased eGFR. Fig. 3 shows eGFR values in individuals with ADTKD-MUC1. The median age of ESKD in this condition is 46 years[48], with the youngest age of ESKD being 16 years[8].
Diagnosis:
Given the lack of symptoms, diagnosis rests on the presence of CKD and a bland urinary sediment in a patient who has a parent who also suffers from CKD.
Genetic testing:
Due to the high guanosine/cytosine content of the VNTR, disease causing mutations in MUC1 are missed by Sanger sequencing. For this reason, gene panels and whole exome sequencing will NOT find disease-causing mutations in this gene, even if they clam to do so. Genetic testing is available free of charge through the Broad Institute for children with CKD and evidence of autosomal dominant transmission. Contact ableyer@wakehealth.edu to arrange. Other options that may be available for genetic sequencing include SNaPshot minisequencing, and single molecule real-time sequencing. These techniques will identify the cytosine duplication that is present in approximately 95% of individuals with this disorder[45] but will miss other mutations. Immunohistochemical examination of urinary cells for the frameshift mucin-1 protein and other research techniques to identify mutations other than the cytosine duplication are being developed[45, 47, 49].
Treatment:
There is no currently approved treatment for this condition. The Broad Institute of Harvard and MIT is developing a compound that removes the mutated protein from the ERGIC and has been found to be effective an a murine model [50].
Genetic screening:
Genetic screening of children is highly discouraged, as this condition will rarely require treatment in childhood. Prenatal genetic testing is currently unavailable in the US.
Summary:
ADTKD-MUC1 is caused by frameshift mutations in the VNTR of the MUC1 gene. The only clinical manifestation of this condition is CKD, and the disease rarely presents in childhood.
ADTKD-REN:
Pathophysiology and genetics:
ADTKD-REN is caused by mutations in the REN gene encoding renin. Preprorenin is synthesized in the juxtaglomerular cells of the kidney. Preprorenin includes the signal peptide, which directs the immature renin to the endoplasmic reticulum, and the prosegment, which ensures proper folding of the renin molecule. The mature renin molecule is enzymatically active and is responsible for blood pressure control, potassium and bicarbonate homeostasis, and augmenting erythropoiesis. In addition, renin is important in kidney tubulogenesis. Recessive or compound heterozygous mutations in almost all genes of the renin-angiotensin system can cause kidney tubular dysgenesis, which is characterized by a high mortality and abnormal kidney development[51]. In contrast, heterozygous mutations in the REN gene result in the production of wild-type renin from a normal renin allele and mutated renin from the mutated renin allele. Clinical manifestations of this disorder result from a decrease in normal renin activity as well as from the deposition of the mutant renin within tubular cells[6]. Mutations that cause ADTKD-REN may occur in the signal peptide, prosegment, or mature renin molecule[7]. Mutations in the signal peptide and the prosegment often present in childhood. Mutations in the region encoding mature renin present in a similar manner and at a similar age to patients with ADTKD-UMOD. All mutations can be identified by standard genetic testing.
Clinical characteristics:
The clinical characteristics of this condition were recently described for a large international cohort of families with this disorder[7]. There are several characteristic presentations in childhood. Individuals with signal peptide mutations in the international cohort presented with acute kidney injury (10%), anemia, acidosis and kidney failure (13%), anemia alone (31%), CKD (22%) and gout (25%). Individuals with prosegment mutations presented with anemia, CKD, or gout. In addition, as patients have decreased renin and angiotensin production, they are prone to acute kidney injury, similar to patients receiving angiotensin converting enzyme inhibitors. Thus, patients may present in childhood during a viral illness with acute kidney injury, hyperkalemia, and acidemia. Patients presenting with acute kidney injury respond well to volume repletion, but a decreased eGFR often remains. In all pediatric presentations, patients will have anemia, which is due to low erythropoietin levels.
Chronic kidney disease:
Patients have a low eGFR that is present at earliest measurement and may be due to abnormalities in embryonic kidney development. Fig. 4 shows the distribution of eGFR values vs. age for individuals with REN mutations. The eGFR values were almost all less than 70 ml/min/1.73m2 from an early age. Despite the initial low eGFR, only one child developed ESKD before age 20 years. Fig. 4 shows the slow progression that occurred through age 40 years, with the median age of ESKD being 53 years in the signal peptide group and 51 years in the prosegment group.
Fig. 4.
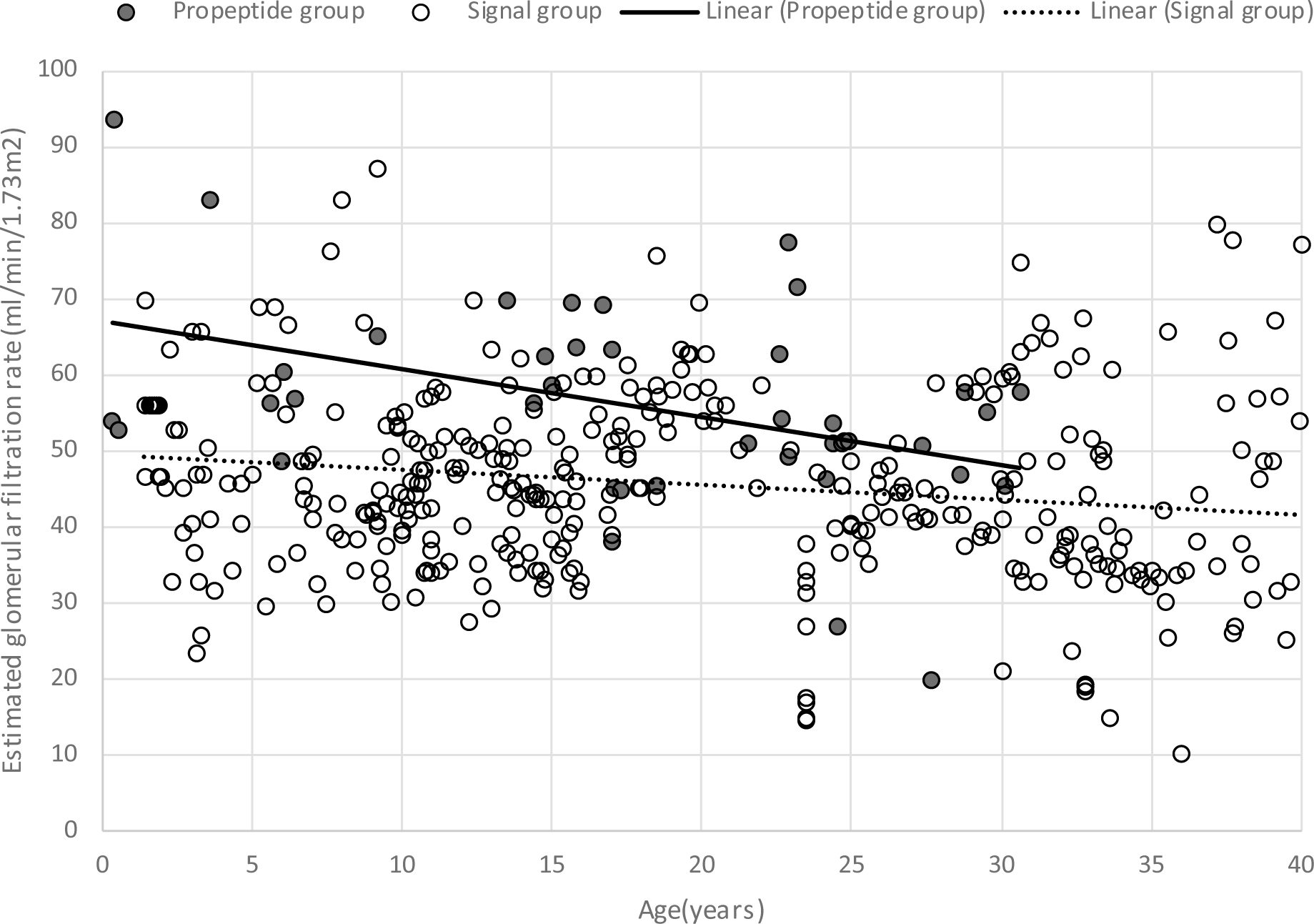
Estimated glomerular filtration rate (eGFR) values prior to age 40 years in patients with ADTKD-REN. There were multiple eGFR measurements from 7 patients in the prosegment group and 24 in the signal peptide group
Electrolytes:
Serum potassium levels usually vary between 4 mEq/L and 6 mEq/l and remain relatively stable over time. Some patients require dietary potassium restriction. Acidosis is a common feature of ADTKD-REN, with patients often having serum bicarbonate levels between 15 and 24 mEq/l, with a mean bicarbonate level of 22 mEq/L.
Anemia:
The mean hemoglobin level for children with ADTKD-REN is approximately 10 g/dl, with the lowest hemoglobin level of 7.5 g/dl in the international cohort study[7]. Patients with hemoglobin levels > 9 g/dl may not require erythropoietin treatment. Hemoglobin levels will usually remain stable in childhood; in males, hemoglobin levels tend to rise after age 20 due to increased testosterone secretion.
Hyperuricemia is present from childhood and more severe in males, with gout developing in approximately 50% of individuals with ADTKD-REN, though this usually occurs in adulthood.
Diagnosis:
ADTKD-REN should be considered in children with mildly low blood pressure, hyperkalemia, acidemia, and anemia out of proportion to CKD, especially if a parent is affected with CKD. Plasma renin levels are often low but may be in the low end of the normal range, complicating diagnosis. CKD is present from childhood, with most children having an eGFR of approximately 50 ml/min/1.73m2. The presence of low eGFR, mild hyperkalemia, and anemia helps to differentiate this condition from renal tubular acidosis.
Genetic testing:
Mutations are easily identified by Sanger sequencing of the REN gene, and testing is available through gene panels and whole exome sequencing.
Treatment:
Fludrocortisone can be used to treat the renin/aldosterone deficiency in ADTKD-REN[7, 52, 53]. Treatment with fludrocortisone will improve serum potassium and serum bicarbonate levels. While serum bicarbonate levels will also respond to oral alkali administration, the latter requires a larger pill or liquid burden, and most patients do not achieve optimal serum bicarbonate levels with this regimen[7]. In addition, fludrocortisone will result in a small increase in blood pressure and a secondary increase in eGFR. Anemia is easily treated with erythropoietin but may not be required. Many children remain asymptomatic with hemoglobin levels in the 10 g/dl range without erythropoietin.
Genetic screening:
Genetic screening of siblings and children of affected individuals should be considered due to the numerous therapeutic interventions that are possible and the early onset of clinical manifestations.
Summary:
ADTKD-REN is associated with early-onset CKD and manifestations of renin deficiency, including mild hypotension, hyperkalemia, anemia, and acidemia. The condition is easily diagnosed with genetic testing. Patients respond well to treatment with fludrocortisone. Despite most individuals presenting with an eGFR < 50 ml/min/1.73m2 before age 5, GFR remains preserved through childhood and adolescence.
ADTKD-HNF1β
Pathophysiology and genetics:
Hepatocyte nuclear factor 1β is a transcription factor that is important in the embryonic development of the kidney, pancreas, liver, and the urogenital tract. HNF1β regulates PKHD1 (mutations causing autosomal recessive polycystic kidney disease), PKD2, and GLIS2(mutations associated with nephronophthisis), playing an important role in maintenance of the kidney tubular structure[54]. As HNF1β is important in the development of many organs, heterozygous mutations may result in pancreatic atrophy and diabetes mellitus, unexplained liver function test abnormalities, kidney malformations, and CKD[55]. Loss of preservation of tubular structure results in the frequent occurrence of kidney cysts. Loss of regulation of FXDY2 (which encodes the gamma subunit of the Na+ K+ ATPase regulating epithelial ion transport) results in hypomagnesemia[56].
Approximately 50% of HNF1β mutations are de novo, and approximately 50% involve whole gene deletions[5, 22], with no correlation between mutation type and phenotypic expression. Incomplete penetrance is an important characteristic of this disorder, which makes diagnosis elusive. HNF1β deletion is included in the 17q12 microdeletion syndrome; characteristics of this syndrome include gynecologic abnormalities, epilepsy, lipodystrophy, autism, and esophageal malformations[55].
Clinical findings:
In childhood, structurally abnormal kidneys are the most common presenting symptom[22], with approximately 20% of CAKUT cases being caused by HNF1β mutations[5]. In one study, HNF1β mutations were found in 10% of CAKUT cases in adults and children, with 65% having cysts. Consistent with other studies, all patients were found to have bilateral kidney anomalies, with multicystic kidneys and kidney dysplasia as the most common findings[57].
A cohort study from the German Multicenter HNF1β Childhood Registry[22] provides the most comprehensive information about HNF1β mutations in childhood. In this study, prenatal kidney dysplasia (hyperechogenic kidneys) was seen in 32/62 (52%) patients. Bilateral kidney cysts were noted in 74% of patients in follow up, with 16% having unilateral cysts, and 10% with no cysts. Overall, these children had slow CKD progression. Cystic disease was non-progressive in 72%, with a mean eGFR loss of −0.33 ml/min/1.73m2/year. Patients who had increasing cysts over time had a more accelerated decline in eGFR of approximately −3 ml/min/1.3m2/year. Other findings included hypomagnesemia (24%), hyperuricemia (37%) and elevated liver enzymes (21%). Hyperglycemia occurred in only 8%[22].
The mnemonic MAGIC LUCID includes the many varied clinical findings in ADTKD- HNF1β (see Table 2).
Table 2.
Clinical manifestations of HNF1β Mutations (MAGIC LUCID)
| Magnesium. Hypomagnesemia related to kidney magnesium losses |
| Autosomal dominant |
| Genital tract abnormalities, including bicornuate uterus, absent uterus, vaginal hypoplasia |
| Incomplete penetrance |
| Cysts of the kidney and other structural abnormalities, including multicystic kidneys, fetal bilateral hyperechogenic kidneys, kidney agenesis, and hypoplastic kidneys. Less commonly, vesicoureteral reflux and hydronephrosis |
| Liver test abnormalities. Liver biopsies are normal. |
| Uric acid elevation. Hypouricosuric hyperuricemia. Gout may occur. |
| Chronic kidney disease |
| Inherited |
| Diabetes and pancreatic anomalies. Maturity onset diabetes of youth 5. Atrophy of the pancreatic tail |
Diagnosis:
Diagnosis can be elusive due to incomplete penetrance and the fact that 50% of mutations are de novo. Affected family members will be affected by a varied combination or none of these conditions. For example, a child may have multicystic kidneys, while the patient’s affected mother is entirely asymptomatic, and a maternal uncle may have early-onset diabetes mellitus, gout, and CKD. Targeted questioning about conditions associated with this disorder in family members should be used in children found to have kidney structural abnormalities or CKD. The presence of bilateral kidney anomalies[57], hypomagnesemia and/or pancreas anomalies are more specific indicators of ADTKD- HNF1β[58]. Clinicians should use age-appropriate norms when evaluating serum magnesium levels[59]. Patients with HNF1β mutations can also present in a similar fashion as autosomal recessive polycystic kidney disease, with Potter’s sequence, oligohydramnios, and enlarged polycystic kidneys due to the role of HNF1β as a master regulator of PKHD1 and PKD2 genes[60]. When taking a family history, it is important to recognize the incomplete penetrance that occurs in this disorder and the varied clinical effects. Even if both parents are asymptomatic, it is important to obtain an extended family history, with emphasis on early onset diabetes, gout, and chronic kidney disease. The patient’s parents may not realize that family members suffer from other conditions such as hypomagnesemia and unexplained liver function abnormalities.
Genetic diagnosis:
Diagnosis of HNF1β mutations can be difficult, as there is a high incidence of gene deletions that will be missed by performing only gene sequencing. For patients presenting with findings restricted to CAKUT, one might initially consider a chromosomal microarray, as at least 10% of CAKUT cases are associated with copy-number variants[61, 62]. One might also consider a gene panel including HNF1β or whole exome sequencing to identify monogenic causes of CAKUT[57, 63]. When evaluating patients with a syndrome complex consistent with ADTKD-HNF1β, one might consider multiplex ligation –dependent probe amplification (MLPA), which will identify copy number variations in HNF1β as well as a gene panel or whole exome sequencing to identify mutations in HNF1β and other genes.
Treatment:
All affected individuals should have a kidney ultrasound and screening for liver function test abnormalities, hypomagnesemia, hyperglycemia, and hyperuricemia. Based on baseline kidney ultrasound findings and eGFR, patients may need annual ultrasounds and should undergo periodic measurements of serum creatinine. Gynecologic examination for uterine abnormalities may be considered when age-appropriate. Affected individuals should be monitored for the development of diabetes mellitus. Diabetes treatment usually requires insulin therapy due to pancreatic hypoplasia and hepatic insulin resistance in this disorder[64]. There have been no formal studies of the treatment of hypomagnesemia in this disorder. If treatment is required, frequent administration of magnesium supplementation will be required, because administered magnesium will be quickly excreted in the urine. Patients who develop gout should be treated with allopurinol to prevent recurrent gout and development of tophi over time. There are no specific treatments for CKD in this disorder.
Genetic screening:
Genetic screening should only be performed in childhood if actionable conditions may be identified. Screening of children would be helpful in identifying hypomagnesemia CKD, and hyperglycemia. For this reason, genetic screening may be considered. Given difficulties in genetic testing, incomplete penetrance, and the variation in clinical findings, a genetics counsellor is extremely helpful in guiding diagnosis, information patients of risks and benefits of diagnosis, and in providing knowledge and information about screening to family members.
Summary:
ADTKD-HNF1β is a multisystemic disease that often presents in childhood with bilateral kidney anomalies, often multicystic kidneys. Though cysts are often present early in life, most children have a slowly progressive increase in cysts with very slow loss of kidney function. Incomplete penetrance and a varied clinical presentation makes diagnosis difficult.
Other conditions:
There are a number of other autosomal dominant conditions in which tubulointerstitial kidney disease occurs. SEC61A1 encodes the protein transport protein Sec61 subunit alpha isoform 1 (SEC61α). It is a component of the mammalian endoplasmic reticulum (ER) translocon, a structure facilitating biosynthesis and transport of secretory proteins as well as intracellular calcium ions (Ca2+) homeostasis. Mutations in SEC61A1 are rare, with six families reported[65–68]. Individuals with missense mutations affecting the translocon pore present with intrauterine growth retardation, dysplastic kidneys, congenital anemia, neutropenia gout, and CKD [65, 66]. Individuals with mutations in transmembrane helices presented with common variable immunodeficiency [68]. Genetic diagnosis is easily performed by conventional testing and is part of standard kidney genetic panels. Alagille syndrome [69, 70], Townes-Brocks syndrome[71, 72], and HDR (hypoparathyroidism, deafness, and kidney abnormalities) syndrome [73] may all have CKD and genitourinary tract abnormalities as clinical findings. Renal coloboma syndrome caused by PAX2 mutations is an autosomal dominant condition usually associated with proteinuria and glomerular changes. However, patients may have a broad spectrum of kidney presentations[74], including tubulointerstitial kidney disease. Mutations in mitochondrial DNA are not autosomal dominant disorders but may present with CKD in childhood that could appear autosomal dominant [75].
Summary:
ADTKD is becoming increasingly recognized as a cause of CKD and ESKD in adults and CKD in children. There are now more than 300 families identified with these disorders in the United States. Most forms of ADTKD do not result in progression to ESKD in childhood. Patients may be identified due to presentation with CKD in childhood or for screening due to family history. In some forms hyperuricemia is common. Diagnosis can usually be made by gene panels, except for ADTKD-MUC1, which requires dedicated genetic testing. Genetic diagnosis of HNF1β mutations may also require chromosomal microarray. Readers are encouraged to contact the authors (ableyer@wakehealth.edu) to discuss potential cases and to obtain genetic testing for ADTKD-MUC1 and other causes of ADTKD.
Table 1.
Causes of autosomal dominant tubulointerstitial kidney disease
| Clinical condition | Mutated genes | Kidney manifestations | Extra-kidney manifestations | Diagnostic considerations in addition to family history of kidney disease |
| ADTKD-UMOD | UMOD | Hypouricosuric hyperuricemia, CKD | Gout may occur in adolescence | Gout and CKD |
| ADTKD-MUC1 | MUC1 | CKD | None | Specialized genetic testing. Mutations not identified by gene panels or whole exome sequencing |
| ADTKD-REN | REN | Hyperkalemia, acidosis, hypouricosuric hyperuricemia, CKD | Gout, anemia, mild hypotension | Patients are prone to AKI during viral illnesses |
| ADTKD-HNF1β | HNF1β | Fetal hyperechogenic kidneys, multicystic and dysplastic kidneys, kidney agenesis, hypomagnesemia, hyperuricemia | MAGIC LUCID (see Table 2) | Targeted questioning for family history of associated conditions in patients with kidney cysts |
| ADTKD-SEC61A1 | SEC61A1 | CKD | Neutropenia, intrauterine growth retardation | Extremely rare |
| Alagille syndrome | JAG1 or NOTCH2 | Vesico-ureteral reflux, CKD | Deep-set eyes, broad forehead, triangular chin, butterfly vertebrae, stenosis of pulmonary arteries, cholestasis | Deep-set eyes, broad forehead and triangular chin as well as other extra-kidney manifestations. |
| Townes-Brocks syndrome | SALL1 | Solitary kidney, dysplastic kidneys, vesico-ureteral reflux, CKD | External ear malformations, syndactyly and triphalangeal thumb, imperforate anus | Findings of hand and ear abnormalities, imperforate anus |
| HDR syndrome | GATA3 | CKD | H(hypoparathyroidism), D(deafness) | Consider this diagnosis in patients with hypocalcemia and deafness |
Acknowledgements:
SK and colleagues were supported by the Ministry of Health of the Czech Republic (grant NV17-29786A, NU21-07-00033), the Ministry of Education of the Czech Republic (grant LTAUSA19068) and by institutional programs of Charles University in Prague (UNCE/MED/007 and PROGRES-Q26/LF1). The National Center for Medical Genomics (LM2018132) kindly provided sequencing and genotyping. AJB was funded by NIH-NIDDK R21 DK106584, CKD Biomarkers Consortium, the Slim Health Foundation, the Black-Brogan Foundation, Soli Deo Gloria. MTW is supported by Children’s Clinical Research Advisory Committee (CCRAC), Department of Defense (W81XWH1910205), and the NIH (P30 DK079328-11, R01DK119631). The authors thank Emily Johnson,M.S. for her help in preparation of figures.
Footnotes
Conflict of Interest: The authors have no conflicts of interest to report.
References:
- 1.Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, Bleyer AJ (2019) Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers 5:60.DOI 10.1038/s41572-019-0109-9 [DOI] [PubMed] [Google Scholar]
- 2.Bleyer AJ, Kidd K, Zivna M, Kmoch S (2017) Autosomal Dominant Tubulointerstitial Kidney Disease. Adv Chronic Kidney Dis 24:86–93.DOI S1548–5595(16)30140–9 [pii]; 10.1053/j.ackd.2016.11.012 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, Shirts B, Xu L, Zhu H, Barmada MM, Bleyer AJ (2002) Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet 39:882–892.DOI 10.1136/jmg.39.12.882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirby A, Gnirke A, Jaffe DB, Baresova V, Pochet N, Blumenstiel B, Ye C, Aird D, Stevens C, Robinson JT, Cabili MN, Gat-Viks I, Kelliher E, Daza R, DeFelice M, Hulkova H, Sovova J, Vylet’al P, Antignac C, Guttman M, Handsaker RE, Perrin D, Steelman S, Sigurdsson S, Scheinman SJ, Sougnez C, Cibulskis K, Parkin M, Green T, Rossin E, Zody MC, Xavier RJ, Pollak MR, Alper SL, Lindblad-Toh K, Gabriel S, Hart PS, Regev A, Nusbaum C, Kmoch S, Bleyer AJ, Lander ES, Daly MJ (2013) Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 45:299–303.DOI 10.1038/ng.2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heidet L, Decramer S, Pawtowski A, Moriniere V, Bandin F, Knebelmann B, Lebre AS, Faguer S, Guigonis V, Antignac C, Salomon R (2010) Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol 5:1079–1090.DOI 10.2215/CJN.06810909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zivna M, Hulkova H, Matignon M, Hodanova K, Vylet’al P, Kalbacova M, Baresova V, Sikora J, Blazkova H, Zivny J, Ivanek R, Stranecky V, Sovova J, Claes K, Lerut E, Fryns JP, Hart PS, Hart TC, Adams JN, Pawtowski A, Clemessy M, Gasc JM, Gubler MC, Antignac C, Elleder M, Kapp K, Grimbert P, Bleyer AJ, Kmoch S (2009) Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, and chronic kidney failure. Am J Hum Genet 85:204–213.DOI 10.1016/j.ajhg.2009.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zivna M, Kidd K, Zaidan M, Vyletal P, Baresova V, Hodanova K, Sovova J, Hartmannova H, Votruba M, Treslova H, Jedlickova I, Sikora J, Hulkova H, Robins V, Hnizda A, Zivny J, Papagregoriou G, Mesnard L, Beck BB, Wenzel A, Tory K, Haeffner K, Wolf MTF, Bleyer ME, Sayer JA, Ong ACM, Balogh L, Jakubowska A, Laszkiewicz A, Clissold R, Shaw-Smith C, Munshi R, Haws RM, Izzi C, Capelli I, Santostefano M, Graziano C, Scolari F, Sussman A, Trachtman H, Decramer S, Matignon M, Grimbert P, Shoemaker LR, Stavrou C, Abdelwahed M, Belghith N, Sinclair M, Claes K, Kopel T, Moe S, Deltas C, Knebelmann B, Rampoldi L, Kmoch S, Bleyer AJ (2020) An International Cohort Study of Autosomal Dominant Tubulointerstitial Kidney Disease due to REN Mutations Identifies Distinct Clinical Subtypes. Kidney Int.DOI 10.1016/j.kint.2020.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bleyer AJ, Kmoch S, Antignac C, Robins V, Kidd K, Kelsoe JR, Hladik G, Klemmer P, Knohl SJ, Scheinman SJ, Vo N, Santi A, Harris A, Canaday O, Weller N, Hulick PJ, Vogel K, Rahbari-Oskoui FF, Tuazon J, Deltas C, Somers D, Megarbane A, Kimmel PL, Sperati CJ, Orr-Urtreger A, Ben-Shachar S, Waugh DA, McGinn S, Bleyer AJ Jr., Hodanova K, Vylet’al P, Zivna M, Hart TC, Hart PS (2014) Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin J Am Soc Nephrol 9:527–535.DOI CJN.06380613 [pii]; 10.2215/CJN.06380613 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolf MT, Hildebrandt F (2011) Nephronophthisis. Pediatr Nephrol 26:181–194.DOI 10.1007/s00467-010-1585-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braun DA, Hildebrandt F (2017) Ciliopathies. Cold Spring Harb Perspect Biol 9.DOI 10.1101/cshperspect.a028191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf MT (2015) Nephronophthisis and related syndromes. Curr Opin Pediatr 27:201–211.DOI 10.1097/MOP.0000000000000194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stokman M, Lilien M, Knoers N (1993) Nephronophthisis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A (eds) GeneReviews((R)), Seattle (WA). [PubMed] [Google Scholar]
- 13.Stokman MF, van der Zwaag B, van de Kar N, van Haelst MM, van Eerde AM, van der Heijden JW, Kroes HY, Ippel E, Schulp AJA, van Gassen KL, van Rooij I, Giles RH, Beales PL, Roepman R, Arts HH, Bongers E, Renkema KY, Knoers N, van Reeuwijk J, Lilien MR (2018) Clinical and genetic analyses of a Dutch cohort of 40 patients with a nephronophthisis-related ciliopathy. Pediatr Nephrol 33:1701–1712.DOI 10.1007/s00467-018-3958-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Georges B, Cosyns JP, Dahan K, Snyers B, Carlier B, Loute G, Pirson Y (2000) Late-onset renal failure in Senior-Loken syndrome. Am J Kidney Dis 36:1271–1275.DOI 10.1053/ajkd.2000.19845 [DOI] [PubMed] [Google Scholar]
- 15.Kidd K, Vylet’al P, Schaeffer C, Olinger E, Zivna M, Hodanova K, Robins V, Johnson E, Taylor A, Martin L, Izzi C, Jorge SC, Calado J, Torres RJ, Lhotta K, Steubl D, Gale DP, Gast C, Gombos E, Ainsworth HC, Chen YM, Almeida JR, de Souza CF, Silveira C, Raposeiro R, Weller N, Conlon PJ, Murray SL, Benson KA, Cavalleri GL, Votruba M, Vrbacka A, Amoroso A, Gianchino D, Caridi G, Ghiggeri GM, Divers J, Scolari F, Devuyst O, Rampoldi L, Kmoch S, Bleyer AJ (2020) Genetic and Clinical Predictors of Age of ESKD in Individuals With Autosomal Dominant Tubulointerstitial Kidney Disease Due to UMOD Mutations. Kidney Int Rep 5:1472–1485.DOI 10.1016/j.ekir.2020.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moskowitz JL, Piret SE, Lhotta K, Kitzler TM, Tashman AP, Velez E, Thakker RV, Kotanko P (2013) Association between genotype and phenotype in uromodulin-associated kidney disease. Clin J Am Soc Nephrol 8:1349–1357.DOI CJN.11151012 [pii]; 10.2215/CJN.11151012 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf MT, Beck BB, Zaucke F, Kunze A, Misselwitz J, Ruley J, Ronda T, Fischer A, Eifinger F, Licht C, Otto E, Hoppe B, Hildebrandt F (2007) The Uromodulin C744G mutation causes MCKD2 and FJHN in children and adults and may be due to a possible founder effect. Kidney Int 71:574–581.DOI 10.1038/sj.ki.5002089 [DOI] [PubMed] [Google Scholar]
- 18.Schaffer P, Gombos E, Meichelbeck K, Kiss A, Hart PS, Bleyer AJ (2010) Childhood course of renal insufficiency in a family with a uromodulin gene mutation. Pediatr Nephrol 25:1355–1360.DOI 10.1007/s00467-009-1436-y [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tramma D, Samourkasidou D (2020) Hyperuricemia and Early-onset Chronic Kidney Disease in a 7-year-old Child. Indian Pediatr 57:765. [PubMed] [Google Scholar]
- 20.Jain S, Chen F (2019) Developmental pathology of congenital kidney and urinary tract anomalies. Clin Kidney J 12:382–399.DOI 10.1093/ckj/sfy112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Connaughton DM, Kennedy C, Shril S, Mann N, Murray SL, Williams PA, Conlon E, Nakayama M, van der Ven AT, Ityel H, Kause F, Kolvenbach CM, Dai R, Vivante A, Braun DA, Schneider R, Kitzler TM, Moloney B, Moran CP, Smyth JS, Kennedy A, Benson K, Stapleton C, Denton M, Magee C, O’Seaghdha CM, Plant WD, Griffin MD, Awan A, Sweeney C, Mane SM, Lifton RP, Griffin B, Leavey S, Casserly L, de Freitas DG, Holian J, Dorman A, Doyle B, Lavin PJ, Little MA, Conlon PJ, Hildebrandt F (2019) Monogenic causes of chronic kidney disease in adults. Kidney Int 95:914–928.DOI 10.1016/j.kint.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okorn C, Goertz A, Vester U, Beck BB, Bergmann C, Habbig S, Konig J, Konrad M, Muller D, Oh J, Ortiz-Bruchle N, Patzer L, Schild R, Seeman T, Staude H, Thumfart J, Tonshoff B, Walden U, Weber L, Zaniew M, Zappel H, Hoyer PF, Weber S (2019) HNF1B nephropathy has a slow-progressive phenotype in childhood-with the exception of very early onset cases: results of the German Multicenter HNF1B Childhood Registry. Pediatr Nephrol 34:1065–1075.DOI 10.1007/s00467-018-4188-8 [DOI] [PubMed] [Google Scholar]
- 23.Bleyer AJ, Kidd K, Robins V, Martin L, Taylor A, Santi A, Tsoumas G, Hunt A, Swain E, Abbas M, Akinbola E, Vidya S, Moossavi S, Bleyer AJ Jr., Zivna M, Hartmannova H, Hodanova K, Vyletal P, Votruba M, Harden M, Blumenstiel B, Greka A, Kmoch S (2019) Outcomes of patient self-referral for the diagnosis of several rare inherited kidney diseases. Genet Med.DOI 10.1038/s41436-019-0617-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pottel H, Hoste L, Dubourg L, Ebert N, Schaeffner E, Eriksen BO, Melsom T, Lamb EJ, Rule AD, Turner ST, Glassock RJ, De Souza V, Selistre L, Mariat C, Martens F, Delanaye P (2016) An estimated glomerular filtration rate equation for the full age spectrum. Nephrol Dial Transplant 31:798–806.DOI 10.1093/ndt/gfv454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O (2011) The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int 80:338–347.DOI ki2011134 [pii]; 10.1038/ki.2011.134 [doi] [DOI] [PubMed] [Google Scholar]
- 26.Tokonami N, Takata T, Beyeler J, Ehrbar I, Yoshifuji A, Christensen EI, Loffing J, Devuyst O, Olinger EG (2018) Uromodulin is expressed in the distal convoluted tubule, where it is critical for regulation of the sodium chloride cotransporter NCC. Kidney Int 94:701–715.DOI 10.1016/j.kint.2018.04.021 [DOI] [PubMed] [Google Scholar]
- 27.Vylet’al P, Kublova M, Kalbacova M, Hodanova K, Baresova V, Stiburkova B, Sikora J, Hulkova H, Zivny J, Majewski J, Simmonds A, Fryns JP, Venkat-Raman G, Elleder M, Kmoch S (2006) Alterations of uromodulin biology: a common denominator of the genetically heterogeneous FJHN/MCKD syndrome. Kidney Int 70:1155–1169.DOI 5001728 [pii]; 10.1038/sj.ki.5001728 [doi] [DOI] [PubMed] [Google Scholar]
- 28.Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, Raffi H, Rampoldi L, Uchida S, Hille C, Dosche C, Kumar S, Castaneda-Bueno M, Gamba G, Bachmann S (2011) Activation of the bumetanide-sensitive Na+,K+,2Cl− cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J Biol Chem 286:30200–30210.DOI M111.222968 [pii]; 10.1074/jbc.M111.222968 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trudu M, Janas S, Lanzani C, Debaix H, Schaeffer C, Ikehata M, Citterio L, Demaretz S, Trevisani F, Ristagno G, Glaudemans B, Laghmani K, Dell’Antonio G, team S, Loffing J, Rastaldi MP, Manunta P, Devuyst O, Rampoldi L (2013) Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med 19:1655–1660.DOI 10.1038/nm.3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh G, Lingala B, Mithal A (2019) Gout and hyperuricaemia in the USA: prevalence and trends. Rheumatology (Oxford) 58:2177–2180.DOI 10.1093/rheumatology/kez196 [DOI] [PubMed] [Google Scholar]
- 31.Moro F, Ogg CS, Simmonds HA, Cameron JS, Chantler C, McBride MB, Duley JA, Davies PM (1991) Familial juvenile gouty nephropathy with renal urate hypoexcretion preceding renal disease. Clin Nephrol 9:263–269 [PubMed] [Google Scholar]
- 32.Pavelka K Jr., Sebesta I, Blovska J, Maly J, Chadimova M (1996) [Familial juvenile gouty nephropathy]. Cas Lek Cesk 135:668–671 [PubMed] [Google Scholar]
- 33.Abhishek A, Valdes AM, Jenkins W, Zhang W, Doherty M (2017) Triggers of acute attacks of gout, does age of gout onset matter? A primary care based cross-sectional study. PLoS One 12:e0186096.DOI 10.1371/journal.pone.0186096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaffer P, Gombos E, Meichelbeck K, Kiss A, Hart PS, Bleyer AJ (2010) Childhood course of renal insufficiency in a family with a uromodulin gene mutation. Pediatr Nephrol 25:1355–1360.DOI 10.1007/s00467-009-1436-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carucci NS, Caridi G, Lugani F, Barone C, Conti G (2019) A novel UMOD gene mutation associated with chronic kidney failure at a young age. Clin Nephrol 92:151–155.DOI 10.5414/CN109128 [DOI] [PubMed] [Google Scholar]
- 36.Wolf MT, Beck BB, Zaucke F, Kunze A, Misselwitz J, Ruley J, Ronda T, Fischer A, Eifinger F, Licht C, Otto E, Hoppe B, Hildebrandt F (2007) The Uromodulin C744G mutation causes MCKD2 and FJHN in children and adults and may be due to a possible founder effect. Kidney Int 71:574–581.DOI 5002089 [pii]; 10.1038/sj.ki.5002089 [doi] [DOI] [PubMed] [Google Scholar]
- 37.Stiburkova B, Bleyer AJ (2012) Changes in serum urate and urate excretion with age. Adv Chronic Kidney Dis 19:372–376.DOI S1548–5595(12)00147–4 [pii]; 10.1053/j.ackd.2012.07.010 [doi] [DOI] [PubMed] [Google Scholar]
- 38.Colantonio DA, Kyriakopoulou L, Chan MK, Daly CH, Brinc D, Venner AA, Pasic MD, Armbruster D, Adeli K (2012) Closing the gaps in pediatric laboratory reference intervals: a CALIPER database of 40 biochemical markers in a healthy and multiethnic population of children. Clin Chem 58:854–868.DOI 10.1373/clinchem.2011.177741 [DOI] [PubMed] [Google Scholar]
- 39.Ridefelt P, Hilsted L, Juul A, Hellberg D, Rustad P (2018) Pediatric reference intervals for general clinical chemistry components - merging of studies from Denmark and Sweden. Scand J Clin Lab Invest 78:365–372.DOI 10.1080/00365513.2018.1474493 [DOI] [PubMed] [Google Scholar]
- 40.Bleyer AJ, Woodard AS, Shihabi Z, Sandhu J, Zhu H, Satko SG, Weller N, Deterding E, McBride D, Gorry MC, Xu L, Ganier D, Hart TC (2003) Clinical characterization of a family with a mutation in the uromodulin (Tamm-Horsfall glycoprotein) gene. Kidney Int 64:36–42.DOI 10.1046/j.1523-1755.2003.00081.x [DOI] [PubMed] [Google Scholar]
- 41.Eckardt KU, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, Deltas C, Hosking A, Kmoch S, Rampoldi L, Wiesener M, Wolf MT, Devuyst O (2015) Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management-A KDIGO consensus report. Kidney Int.DOI ki201528 [pii]; 10.1038/ki.2015.28 [doi] [DOI] [PubMed] [Google Scholar]
- 42.Moro F, Simmonds HA, Cameron JS, Ogg CS, Williams GD, McBride MB, Davis PM (1991) Does Allopurinol affect the progression of familial juvenile gouty nephropathy? In: Harkness RA, al e (eds) Purine and Pyrimidine Metabolism in Man VII. Plenum Press, New York, pp 199–202. [DOI] [PubMed] [Google Scholar]
- 43.Apostolopoulos V, Stojanovska L, Gargosky SE (2015) MUC1 (CD227): a multi-tasked molecule. Cell Mol Life Sci 72:4475–4500.DOI 10.1007/s00018-015-2014-z [doi];10.1007/s00018–015-2014-z [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanisch FG, Muller S (2000) MUC1: the polymorphic appearance of a human mucin. Glycobiology 10:439–449.DOI 10.1093/glycob/10.5.439 [DOI] [PubMed] [Google Scholar]
- 45.Zivna M, Kidd K, Pristoupilova A, Baresova V, Defelice M, Blumenstiel B, Harden M, Conlon P, Lavin P, Connaughton DM, Hartmannova H, Hodanova K, Stranecky V, Vrbacka A, Vyletal P, Zivny J, Votruba M, Sovova J, Hulkova H, Robins V, Perry R, Wenzel A, Beck BB, Seeman T, Viklicky O, Rajnochova-Bloudickova S, Papagregoriou G, Deltas CC, Alper SL, Greka A, Bleyer AJ, Kmoch S (2018) Noninvasive Immunohistochemical Diagnosis and Novel MUC1 Mutations Causing Autosomal Dominant Tubulointerstitial Kidney Disease. J Am Soc Nephrol.DOI ASN.2018020180 [pii]; 10.1681/ASN.2018020180 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dvela-Levitt M, Kost-Alimova M, Emani M, Kohnert E, Thompson R, Sidhom EH, Rivadeneira A, Sahakian N, Roignot J, Papagregoriou G, Montesinos MS, Clark AR, McKinney D, Gutierrez J, Roth M, Ronco L, Elonga E, Carter TA, Gnirke A, Melanson M, Hartland K, Wieder N, Hsu JC, Deltas C, Hughey R, Bleyer AJ, Kmoch S, Živná M, Barešova V, Kota S, Schlondorff J, Heiman M, Alper SL, Wagner F, Weins A, Golub TR, Lander ES, Greka A (2019) Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 178:521–535.e523.DOI 10.1016/j.cell.2019.07.002 [DOI] [PubMed] [Google Scholar]
- 47.Wenzel A, Altmueller J, Ekici AB, Popp B, Stueber K, Thiele H, Pannes A, Staubach S, Salido E, Nuernberg P, Reinhardt R, Reis A, Rump P, Hanisch FG, Wolf MTF, Wiesener M, Huettel B, Beck BB (2018) Single molecule real time sequencing in ADTKD-MUC1 allows complete assembly of the VNTR and exact positioning of causative mutations. Sci Rep 8:4170.DOI 10.1038/s41598-018-22428-0 [doi];10.1038/s41598–018-22428–0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olinger E, Hofmann P, Kidd K, Dufour I, Belge H, Schaeffer C, Kipp A, Bonny O, Deltas C, Demoulin N, Fehr T, Fuster DG, Gale DP, Goffin E, Hodanova K, Huynh-Do U, Kistler A, Morelle J, Papagregoriou G, Pirson Y, Sandford R, Sayer JA, Torra R, Venzin C, Venzin R, Vogt B, Zivna M, Greka A, Dahan K, Rampoldi L, Kmoch S, Bleyer AJ, Devuyst O (2020) Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1. Kidney Int 98:717–731.DOI 10.1016/j.kint.2020.04.038 [DOI] [PubMed] [Google Scholar]
- 49.Knaup KX, Hackenbeck T, Popp B, Stoeckert J, Wenzel A, Buttner-Herold M, Pfister F, Schueler M, Seven D, May AM, Halbritter J, Grone HJ, Reis A, Beck BB, Amann K, Ekici AB, Wiesener MS (2018) Biallelic Expression of Mucin-1 in Autosomal Dominant Tubulointerstitial Kidney Disease: Implications for Nongenetic Disease Recognition. J Am Soc Nephrol 29:2298–2309.DOI 10.1681/ASN.2018030245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dvela-Levitt M, Kost-Alimova M, Emani M, Kohnert E, Thompson R, Sidhom EH, Rivadeneira A, Sahakian N, Roignot J, Papagregoriou G, Montesinos MS, Clark AR, McKinney D, Gutierrez J, Roth M, Ronco L, Elonga E, Carter TA, Gnirke A, Melanson M, Hartland K, Wieder N, Hsu JC, Deltas C, Hughey R, Bleyer AJ, Kmoch S, Zivna M, Baresova V, Kota S, Schlondorff J, Heiman M, Alper SL, Wagner F, Weins A, Golub TR, Lander ES, Greka A (2019) Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 178:521–535 e523.DOI 10.1016/j.cell.2019.07.002 [DOI] [PubMed] [Google Scholar]
- 51.Gribouval O, Gonzales M, Neuhaus T, Aziza J, Bieth E, Laurent N, Bouton JM, Feuillet F, Makni S, Ben Amar H, Laube G, Delezoide AL, Bouvier R, Dijoud F, Ollagnon-Roman E, Roume J, Joubert M, Antignac C, Gubler MC (2005) Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet 37:964–968.DOI 10.1038/ng1623 [DOI] [PubMed] [Google Scholar]
- 52.Bleyer AJ, Zivná M, Hulková H, Hodanová K, Vyletal P, Sikora J, Zivný J, Sovová J, Hart TC, Adams JN, Elleder M, Kapp K, Haws R, Cornell LD, Kmoch S, Hart PS (2010) Clinical and molecular characterization of a family with a dominant renin gene mutation and response to treatment with fludrocortisone. Clin Nephrol 74:411–422.DOI 10.5414/cnp74411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kmoch S, Zivna M, Bleyer AJ (1993) Autosomal Dominant Tubulointerstitial Kidney Disease, REN-Related.DOI NBK53700 [bookaccession] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferre S, Igarashi P (2019) New insights into the role of HNF-1beta in kidney (patho)physiology. Pediatr Nephrol 34:1325–1335.DOI 10.1007/s00467-018-3990-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bockenhauer D, Jaureguiberry G (2016) HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol 31:707–714.DOI 10.1007/s00467-015-3142-2 [DOI] [PubMed] [Google Scholar]
- 56.Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, van’t Hoff W, Marks SD, Trompeter RS, Tullus K, Winyard PJ, Cansick J, Mushtaq I, Dhillon HK, Bingham C, Edghill EL, Shroff R, Stanescu H, Ryffel GU, Ellard S, Bockenhauer D (2009) HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 20:1123–1131.DOI 10.1681/ASN.2008060633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raaijmakers A, Corveleyn A, Devriendt K, van Tienoven TP, Allegaert K, Van Dyck M, van den Heuvel L, Kuypers D, Claes K, Mekahli D, Levtchenko E (2015) Criteria for HNF1B analysis in patients with congenital abnormalities of kidney and urinary tract. Nephrol Dial Transplant 30:835–842.DOI 10.1093/ndt/gfu370 [DOI] [PubMed] [Google Scholar]
- 58.Faguer S, Chassaing N, Bandin F, Prouheze C, Garnier A, Casemayou A, Huart A, Schanstra JP, Calvas P, Decramer S, Chauveau D (2014) The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int 86:1007–1015.DOI S0085–2538(15)30417–8 [pii]; 10.1038/ki.2014.202 [doi] [DOI] [PubMed] [Google Scholar]
- 59.Kolbuc M, Lessmeier L, Salamon-Slowinska D, Malecka I, Pawlaczyk K, Walkowiak J, Wysocki J, Beck BB, Zaniew M (2020) Hypomagnesemia is underestimated in children with HNF1B mutations. Pediatr Nephrol 35:1877–1886.DOI 10.1007/s00467-020-04576-6 [DOI] [PubMed] [Google Scholar]
- 60.Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, Hiesberger T, Fiette L, Igarashi P, Yaniv M, Pontoglio M (2004) A transcriptional network in polycystic kidney disease. EMBO J 23:1657–1668.DOI 10.1038/sj.emboj.7600160 [doi];7600160 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verbitsky M, Westland R, Perez A, Kiryluk K, Liu Q, Krithivasan P, Mitrotti A, Fasel DA, Batourina E, Sampson MG, Bodria M, Werth M, Kao C, Martino J, Capone VP, Vivante A, Shril S, Kil BH, Marasa M, Zhang JY, Na YJ, Lim TY, Ahram D, Weng PL, Heinzen EL, Carrea A, Piaggio G, Gesualdo L, Manca V, Masnata G, Gigante M, Cusi D, Izzi C, Scolari F, van Wijk JAE, Saraga M, Santoro D, Conti G, Zamboli P, White H, Drozdz D, Zachwieja K, Miklaszewska M, Tkaczyk M, Tomczyk D, Krakowska A, Sikora P, Jarmolinski T, Borszewska-Kornacka MK, Pawluch R, Szczepanska M, Adamczyk P, Mizerska-Wasiak M, Krzemien G, Szmigielska A, Zaniew M, Dobson MG, Darlow JM, Puri P, Barton DE, Furth SL, Warady BA, Gucev Z, Lozanovski VJ, Tasic V, Pisani I, Allegri L, Rodas LM, Campistol JM, Jeanpierre C, Alam S, Casale P, Wong CS, Lin F, Miranda DM, Oliveira EA, Simoes ESAC, Barasch JM, Levy B, Wu N, Hildebrandt F, Ghiggeri GM, Latos-Bielenska A, Materna-Kiryluk A, Zhang F, Hakonarson H, Papaioannou VE, Mendelsohn CL, Gharavi AG, Sanna-Cherchi S (2019) The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet 51:117–127.DOI 10.1038/s41588-018-0281-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, Nees SN, Verbitsky M, Perry BJ, Sterken R, Lozanovski VJ, Materna-Kiryluk A, Barlassina C, Kini A, Corbani V, Carrea A, Somenzi D, Murtas C, Ristoska-Bojkovska N, Izzi C, Bianco B, Zaniew M, Flogelova H, Weng PL, Kacak N, Giberti S, Gigante M, Arapovic A, Drnasin K, Caridi G, Curioni S, Allegri F, Ammenti A, Ferretti S, Goj V, Bernardo L, Jobanputra V, Chung WK, Lifton RP, Sanders S, State M, Clark LN, Saraga M, Padmanabhan S, Dominiczak AF, Foroud T, Gesualdo L, Gucev Z, Allegri L, Latos-Bielenska A, Cusi D, Scolari F, Tasic V, Hakonarson H, Ghiggeri GM, Gharavi AG (2012) Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 91:987–997.DOI 10.1016/j.ajhg.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van der Ven AT, Connaughton DM, Ityel H, Mann N, Nakayama M, Chen J, Vivante A, Hwang DY, Schulz J, Braun DA, Schmidt JM, Schapiro D, Schneider R, Warejko JK, Daga A, Majmundar AJ, Tan W, Jobst-Schwan T, Hermle T, Widmeier E, Ashraf S, Amar A, Hoogstraaten CA, Hugo H, Kitzler TM, Kause F, Kolvenbach CM, Dai R, Spaneas L, Amann K, Stein DR, Baum MA, Somers MJG, Rodig NM, Ferguson MA, Traum AZ, Daouk GH, Bogdanovic R, Stajic N, Soliman NA, Kari JA, El Desoky S, Fathy HM, Milosevic D, Al-Saffar M, Awad HS, Eid LA, Selvin A, Senguttuvan P, Sanna-Cherchi S, Rehm HL, MacArthur DG, Lek M, Laricchia KM, Wilson MW, Mane SM, Lifton RP, Lee RS, Bauer SB, Lu W, Reutter HM, Tasic V, Shril S, Hildebrandt F (2018) Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 29:2348–2361.DOI 10.1681/ASN.2017121265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Urakami T (2019) Maturity-onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes Metab Syndr Obes 12:1047–1056.DOI 10.2147/DMSO.S179793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bolar NA, Golzio C, Zivna M, Hayot G, Van HC, Schepers D, Vandeweyer G, Hoischen A, Huyghe JR, Raes A, Matthys E, Sys E, Azou M, Gubler MC, Praet M, Van CG, McFadden K, Pediaditakis I, Pristoupilova A, Hodanova K, Vyletal P, Hartmannova H, Stranecky V, Hulkova H, Baresova V, Jedlickova I, Sovova J, Hnizda A, Kidd K, Bleyer AJ, Spong RS, Vande WJ, Mortier G, Brunner H, Van LL, Kmoch S, Katsanis N, Loeys BL (2016) Heterozygous Loss-of-Function SEC61A1 Mutations Cause Autosomal-Dominant Tubulo-Interstitial and Glomerulocystic Kidney Disease with Anemia. Am J Hum Genet 99:174–187.DOI S0002–9297(16)30199–9 [pii]; 10.1016/j.ajhg.2016.05.028 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Espino-Hernandez M, Palma Milla C, Vara-Martin J, Gonzalez-Granado LI (2020) De novo SEC61A1 mutation in autosomal dominant tubulo-interstitial kidney disease: Phenotype expansion and review of literature. J Paediatr Child Health.DOI 10.1111/jpc.15148 [DOI] [PubMed] [Google Scholar]
- 67.Van Nieuwenhove E, Barber JS, Neumann J, Smeets E, Willemsen M, Pasciuto E, Prezzemolo T, Lagou V, Seldeslachts L, Malengier-Devlies B, Metzemaekers M, Hassdenteufel S, Kerstens A, van der Kant R, Rousseau F, Schymkowitz J, Di Marino D, Lang S, Zimmermann R, Schlenner S, Munck S, Proost P, Matthys P, Devalck C, Boeckx N, Claessens F, Wouters C, Humblet-Baron S, Meyts I, Liston A (2020) Defective Sec61alpha1 underlies a novel cause of autosomal dominant severe congenital neutropenia. J Allergy Clin Immunol 146:1180–1193.DOI 10.1016/j.jaci.2020.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schubert D, Klein MC, Hassdenteufel S, Caballero-Oteyza A, Yang L, Proietti M, Bulashevska A, Kemming J, Kuhn J, Winzer S, Rusch S, Fliegauf M, Schaffer AA, Pfeffer S, Geiger R, Cavalie A, Cao H, Yang F, Li Y, Rizzi M, Eibel H, Kobbe R, Marks AL, Peppers BP, Hostoffer RW, Puck JM, Zimmermann R, Grimbacher B (2018) Plasma cell deficiency in human subjects with heterozygous mutations in Sec61 translocon alpha 1 subunit (SEC61A1). J Allergy Clin Immunol 141:1427–1438.DOI 10.1016/j.jaci.2017.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N (2018) Systematic Review: The Epidemiology, Natural History, and Burden of Alagille Syndrome. J Pediatr Gastroenterol Nutr 67:148–156.DOI 10.1097/MPG.0000000000001958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kamath BM, Podkameni G, Hutchinson AL, Leonard LD, Gerfen J, Krantz ID, Piccoli DA, Spinner NB, Loomes KM, Meyers K (2012) Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A 158A:85–89.DOI 10.1002/ajmg.a.34369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reardon W, Casserly LF, Birkenhager R, Kohlhase J (2007) Kidney failure in Townes-Brocks syndrome: an under recognized phenomenon? Am J Med Genet A 143A:2588–2591.DOI 10.1002/ajmg.a.31699 [DOI] [PubMed] [Google Scholar]
- 72.Bleyer AJ, Hart PS, Kmoch S (2010) Hereditary interstitial kidney disease. Semin Nephrol 30:366–373.DOI 10.1016/j.semnephrol.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lemos MC, Thakker RV (2020) Hypoparathyroidism, deafness, and renal dysplasia syndrome: 20 Years after the identification of the first GATA3 mutations. Hum Mutat 41:1341–1350.DOI 10.1002/humu.24052 [DOI] [PubMed] [Google Scholar]
- 74.Rasmussen M, Nielsen ML, Manak JR, Mogensen H, Lildballe DL (2021) PAX2 variant associated with bilateral kidney agenesis and broad intrafamilial disease variability. Clin Kidney J 14:704–706.DOI 10.1093/ckj/sfaa013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Connor TM, Hoer S, Mallett A, Gale DP, Gomez-Duran A, Posse V, Antrobus R, Moreno P, Sciacovelli M, Frezza C, Duff J, Sheerin NS, Sayer JA, Ashcroft M, Wiesener MS, Hudson G, Gustafsson CM, Chinnery PF, Maxwell PH (2017) Mutations in mitochondrial DNA causing tubulointerstitial kidney disease. PLoS Genet 13:e1006620.DOI 10.1371/journal.pgen.1006620 [DOI] [PMC free article] [PubMed] [Google Scholar]