

Abstract
Since its discovery in 2006, TAR DNA binding protein 43 (TDP-43) has driven rapidly evolving research in neurodegenerative diseases including Amyotrophic Lateral Sclerosis (ALS), Frontotemporal lobar degeneration (FTLD), and limbic predominant age-related TDP-43 encephalopathy (LATE). TDP-43 mislocalization or aggregation is the hallmark of TDP-43 proteinopathy and is associated with cognitive impairment that can be mapped to its regional deposition. Studies in human tissue and model systems demonstrate that TDP-43 may potentiate other proteinopathies such as the amyloid or tau pathology seen in Alzheimer’s Disease (AD) in the combination of AD+LATE. Despite this growing body of literature, there remain gaps in our understanding of whether there is heterogeneity in TDP-43 driven mechanisms across cell types. The growing observations of correlation between TDP-43 proteinopathy and glial pathology suggest a relationship between the two, including pathogenic glial cell-autonomous dysfunction and dysregulated glial immune responses to neuronal TDP-43. Neuroinflammation associated with ALS/FTLD and AD is largely mediated by astrocytic and microglia processes and genetic variants in these diseases regulate innate immune pathways demonstrating the importance of glia. In this review, we discuss the available data on TDP-43 in glia within the context of the neurodegenerative diseases ALS and FTLD and highlight the current lack of information about glial TDP-43 interaction in AD+LATE. TDP-43 has proven to be a significant modulator of cognitive and neuropathological outcomes. A deeper understanding of its role in diverse cell types may provide relevant insights into neurodegenerative syndromes.
Keywords: TDP-43, glia, ALS, FTLD, AD, astrocyte, oligodendrocyte, microglia
INTRODUCTION
Transactive response DNA-binding protein 43 (TDP-43) is an RNA binding protein that is involved in many important cellular functions, including regulation of pre-RNA splicing, mRNA stabilization, DNA repair mechanisms, and stress granule formation (Ratti and Buratti, 2016). In 2006, TDP-43 was discovered as the major pathologic protein comprising the ubiquitinated inclusions in amyotrophic lateral sclerosis (ALS), launching a transformative new direction for research in neurodegenerative diseases (Arai et al., 2006). Shortly after, TDP-43 was also identified as a component of protein inclusions in over half of cases of frontotemporal lobar degeneration (FTLD; Neumann et al., 2006), anchoring FTLD and ALS on opposite sides of a clinical spectrum with shared pathological features. Although there was some debate as to whether TDP-43 was contributing to, or resulting from, the progressive neurodegenerative processes occurring in the brain and spinal cord of these diseases, mutations were ultimately identified in the gene encoding TDP-43, TARDBP, strongly supporting a causative or at least contributory role for TDP-43 in both ALS and FTLD (Gendron et al., 2013). Variants in TARDBP have also been reported in patients initially presenting with parkinsonism and diagnosed with Parkinson’s Disease, suggesting that TDP-43 plays a primary role in neurodegenerative conditions other than ALS and FTLD as well (Quadri et al., 2011; Rayaprolu et al., 2013). As such, aberrant TDP-43 localization and aggregation is emerging as a shared proteinopathy, crossing multiple neurodegenerative diseases most recently to include Alzheimer’s Disease (AD).
It is not clear how disease associated TDP-43 variants drive neuronal injury, but data suggest that the aberrant protein confers both cell autonomous dysfunction in neurons as well as non-cell autonomous injury through glial cells (Gao et al., 2018). Glia function and glia-neuron communication are critical to the maintenance of brain metabolic and network homeostasis (Allen and Barres, 2009; Liddelow and Barres, 2017; Verkhratsky and Nedergaard, 2018; Augusto-Oliveira et al., 2019). TDP-43 expression is ubiquitous in human neural cells and in vitro and in vivo studies suggest possible roles for appropriate TDP-43 function in maintaining normal physiology in oligodendrocytes, astrocyte and microglia (Paolicelli et al., 2017; Wang et al., 2018; LaRocca et al., 2019; Peng et al., 2020). Therefore, glial disruption by dysregulated levels or variant forms of TDP-43 may result in significant downstream detrimental effects to neuronal health and normal brain systems function across the neurodegenerative disease spectrum.
Given the role glia play in the pathophysiology of neurodegenerative diseases, and the presence of TDP-43 in glia themselves, there is value to investigating how TDP-43 proteinopathy impacts glial cells and their interactions with the repertoire of neural cells in brain. Just as our appreciation for glial tau pathology has evolved over the years, the importance of glial TDP-43 pathology is also becoming increasingly apparent (Kovacs, 2018).
In this review, we will discuss the current state of knowledge regarding TDP-43 and its role in glial biology in the context of common neurodegenerative syndromes through evidence borne by clinical and pathological observations. We will explore the glial function of TDP-43 and the glial reaction to TDP-43 in the context of various neurodegenerative disease models focusing on ALS/FTLD and AD. Last, we will suggest that despite the substantial role for glia in AD pathophysiology and growing evidence of comorbid TDP-43 pathology in AD, there remain large gaps in our understanding of how TDP-43 and neuroimmune glia may interact to contribute to AD pathophysiology. The relationship between this multifunctional protein and glia, cells both exquisitely sensitive to the environment and critical in maintaining CNS homeostasis, may reveal novel mechanisms of AD pathophysiology and suggest therapeutic targets aimed at regulating glial responses.
TDP-43
TDP-43 was first identified in 1995 as an HIV-1 TAR regulatory element binding protein found to regulate gene expression (Ou et al., 1995). The 43 kDa protein is a highly conserved nuclear ribonucleoprotein with two RNA recognition motifs through which it binds DNA and RNA as well as a prion-like domain in the C-terminus which contributes to its phase separation properties and aggregation propensity (Gitler and Shorter, 2011). TDP-43 is predominantly localized to the nucleus where it regulates RNA processing. It is shuttled between the nucleus and cytoplasm through active and passive transport in normal physiological states. In the cytoplasm, TDP-43 mediates mRNA stability, RNA transport and granule formation. Numerous studies have identified putative targets of TDP-43 mediated splicing, many of which involve genes implicated in neurological disease (Sephton et al., 2011; Tollervey et al., 2011; Tziortzouda et al., 2021). Preserved nuclear localization of TDP-43 is important for its normal function while aberrant cytoplasmic aggregation of TDP-43 is associated with a toxic gain function in vitro (Barmada et al., 2010; Hergesheimer et al., 2019). TDP-43 mislocalization has motivated attempts to tease apart the relative significance of loss of nuclear TDP-43 localization, gain of toxic TDP-43 function, both mechanisms acting in tandem, or as yet undefined mechanisms responsible for neurodegeneration. Of note, recent work has suggested that formation of TDP-43 aggregates may even be protective (Liu et al., 2013; Bolognesi et al., 2019; de Boer et al., 2020; Suk and Rousseaux, 2020).
TDP-43 also influences cellular responses to stress. Cellular stress promotes phosphorylated TDP-43 separation into liquid droplets, proposed precursors to TDP-43 aggregates. TDP-43 is also recruited to stress granules, cytoplasmic inclusions that regulate mRNA utilization in times of cellular stress (Gasset-Rosa et al., 2019; Nelson et al., 2019b). Typically, after removal of the stressor, stress granules disassemble and TDP-43 will relocalize to the nucleus; however, this process may be dysregulated in the setting of chronic stress (Jo et al., 2020). In this context, stress granules and subsequent recruitment of TDP-43 into insoluble or dissociation resistant aggregates may drive TDP-43 aggregation (Dewey et al., 2012). In turn, TDP-43 can modulate stress granule assembly and disassembly dynamics (Dewey et al., 2012; Khalfallah et al., 2018) and thus improper TDP-43 activity could alter the cellular accommodation to stress. Pathologic TDP-43 can be found in extracellular vesicles isolated from ALS patient samples and has been shown to transmit across cells through microvesicles or independently in in vitro studies (Feiler et al., 2015; Sproviero et al., 2018). Many of these pathogenic mechanisms that have been identified in ALS or FTLD are only now being investigated in AD+LATE now that it is a defined neuropathology. Additionally, how glia contribute to, or protect from, TDP-43 transmission is an outstanding question.
It was not until 2006 that TDP-43 was considered a likely key player in neurodegenerative disease. It was then that phosphorylated TDP-43 was found to be the elusive ubiquitinated protein in ALS/FTLD aggregates observed in the nucleus and cytoplasm of both neurons and glia (Neumann et al., 2006). Subsequently, discovery of pathogenic TDP-43 gene variants in families with ALS confirmed a driving role of TDP-43 in some forms of neurodegeneration (Gitcho et al., 2008; Lagier-Tourenne and Cleveland, 2009). Since that time, TDP-43 function and dysfunction, through its established roles in transcription regulation, splicing, RNA stability, ribonucleoprotein (RNP) granule, and stress granule formation have been linked to neurodegenerative disease pathogenesis (Buratti et al., 2001; Tollervey et al., 2011; Bhardwaj et al., 2013). The list of genes associated with monogenic forms of FTLD and/or ALS TDP-43 proteinopathy is rapidly growing and includes C9orf72, GRN, TBK-1, DCTN1, Ubiquilin 2, and others (Farrer et al., 2009), which provides a starting point for understanding the relationship between TDP-43 and FTLD pathophysiology (Abramzon et al., 2020). TDP-43 deposition is a primary pathological feature of both sporadic and most, but not all, monogenic forms of ALS (Suk and Rousseaux, 2020). However, individuals with SOD1 variant associated ALS do not show TDP-43 pathology (Mackenzie et al., 2007) further supporting the premise that TDP-43 is not solely a downstream consequence of motor neuron degeneration. However, TDP-43 is recognized as a prevalent pathological finding in many neurodegenerative diseases and is independently linked to neurologic syndromes beyond its well-established association with FTLD/ALS (de Boer et al., 2020). The more recently defined neurodegenerative syndrome, Limbic-predominant age-related TDP-43 encephalopathy (LATE) is characterized by TDP-43 deposition in older adults with or without hippocampal sclerosis (HS; discussed below; Nelson et al., 2019a). Common variants at the loci of TMEM106B, ABCC9, and GRN genes are associated with TDP-43 pathology in HS suggesting that TDP-43 is not always a non-specific secondary consequence of a neurodegenerative disease process (Dickson et al., 2010; Van Deerlin et al., 2010; Nelson et al., 2014, 2015; Yu et al., 2015). Work identifying the genetic underpinnings associated with TDP-43 pathology in cognitively impaired individuals more broadly may therefore provide additional mechanistic insight into the degenerative processes leading to dementia.
GLIA IN NEURODEGENERATION
Each of the glial cell types plays a variety of roles in neurodegenerative disease pathogenesis. While extensively reviewed elsewhere (Salter and Stevens, 2017; Gopinath et al., 2020; Escartin et al., 2021), we provide a brief summary of each glial cell type and examples of proposed roles in neurodegeneration here with a focus on evidence from ALS/FTLD and AD.
Astrocytes:
Astrocytes are intimately involved in synaptic signaling, maintenance of the blood brain barrier and CNS homeostasis, neuronal support, and secretion of cytokines and chemokines that can alter the inflammatory profile of adjacent cells (Verkhratsky and Nedergaard, 2018; Linnerbauer and Rothhammer, 2020). These functions are disrupted in human neurodegenerative diseases. “Reactive astrocytes” is the umbrella term for a set of molecular and morphological states that astrocytes assume in response to brain injury and disease (Escartin et al., 2021). They demonstrate cellular hypertrophy, cytoskeleton changes, and elimination of processes, all of which may affect their critical role in synaptic transmission (Verkhratsky and Nedergaard, 2018; Escartin et al., 2021; Smit et al., 2021). While increased levels of GFAP have traditionally been used to assess for the presence of reactive astrocytes, recent work has demonstrated heterogeneity of astrocytes across brain regions, disease states, and homeostasis (Escartin et al., 2019, 2021). Reactive astrocytes may provide both beneficial, or disease-promoting functions (Linnerbauer and Rothhammer, 2020) and a spectrum of astrocytic phenotypes can be documented within the same brain in the disease state. Analyses of patient tissue were invaluable in identifying disease associated astrocytic phenotypes in both ALS and FTLD. Hybrid studies co-culturing patient post-mortem tissue derived astrocytes with stem cell induced motor neurons demonstrate that patient astrocytes generate high levels of proinflammatory cytokines and can confer non-cell autonomous neuronal toxicity in vitro (Haidet-Phillips et al., 2011). Astrocytes are also dynamic contributors to AD pathogenesis. In a murine model of AD, reactive astrocytes are found collocated with amyloid plaques, calcium signaling becomes uncoupled from neuronal signaling, and there is increased release of GABA, glutamate, and other gliotransmitters (Smit et al., 2021). Because of their roles in maintaining synaptic signaling and the blood brain barrier, it is hypothesized that reactive astrocytes with deleterious functions will make good therapeutic targets for neurodegenerative conditions (Escartin et al., 2019; Smit et al., 2021).
Oligodendrocytes:
Oligodendrocytes are the primary myelinating cells of the central nervous system, and are thus particularly important in neuronal signaling (Tognatta and Miller, 2016; Butt et al., 2019). They also secrete neurotrophic factors that maintain neuronal health (Tognatta and Miller, 2016). Oligodendrocytes are the mature form of oligodendrocyte progenitor cells (OPCs), which proliferate throughout the lifespan of the animal (Butt et al., 2019). Proliferation, differentiation, and maturation of OPCs into mature oligodendrocytes were all demonstrated to be altered in neurodegenerative diseases and in aging (Andrea et al., 2016; Tognatta and Miller, 2016; Butt et al., 2019). Like astrocytes, oligodendrocytes and OPCs have only recently been identified as having heterogenous populations between brain regions and grey and white matter, though the functional differences between these subpopulations remain to be discovered (Foerster et al., 2019). In many neurodegenerative conditions as well as aging, remyelination events are common, where OPCs mature into oligodendrocytes and replace lost myelin sheaths (Tognatta and Miller, 2016). However, in most neurodegenerative conditions this remyelination is not sufficient to slow the disease course, which leads to myelin and white matter loss (Tognatta and Miller, 2016; Butt et al., 2019).
Due to their specialized role in maintaining axonal homeostasis, oligodendrocytes require transport of mRNA and translation in the myelin compartment leaving them particularly vulnerable to dysfunction of RNA binding proteins such as TDP-43. Postmortem studies in ALS demonstrate disruption of mRNA delivery and concomitant oligodendroglial pathological protein inclusions (Pons et al., 2020). Knockdown of the oligodendroglial lactate transporter, MCT1, a protein with decreased expression in oligodendrocytes of affected ALS postmortem tissue, results in axon damage and loss of neuronal support underscoring the importance of oligodendroglial metabolic integrity to ALS pathology (Lee et al., 2012). In human AD brain, oligodendrocytes show altered glycolytic and ketolytic processing, indicating that this dysfunction may contribute to observed brain hypometabolism in AD (Saito et al., 2021). OPC senescence may also contribute to neurodegenerative conditions such as AD (Butt et al., 2019). In particular, senolytic treatment in an AD murine model selectively affected OPCs and reduced cognitive deficits as well as reducing AD pathology indicating a role for OPC senescence in modulating the neurodegenerative phenotype (Zhang et al., 2019). Their senescence phenotype as well as significant alteration in neurodegenerative disorders has encouraged researchers to consider OPCs as another potential therapeutic target in neurodegenerative conditions (Tognatta and Miller, 2016; Butt et al., 2019; Zhang et al., 2019).
Microglia:
Microglia, the innate immune cells of the central nervous system, are a major component of neuroinflammation (Salter and Stevens, 2017; Gopinath et al., 2020). It is well known that the role of microglia in neurodegeneration is, to say the least, complicated. In neurodegenerative disease states, microglia have beneficial functions: clearing pathologic peptides and degenerated neurons (Salter and Stevens, 2017; Gopinath et al., 2020). Microglia may also mediate detrimental functions: participating in synaptic stripping and releasing inflammatory cytokines that promote disease progression (Salter and Stevens, 2017; Gopinath et al., 2020).
Genetic studies in familial ALS/FTLD have highlighted innate immune pathways relevant to disease pathogenesis. Progranulin, the protein encoded by the familial FTLD gene, GRN, functions as a chemoattractant in brain to recruit microglia and promote phagocytosis (Pickford et al., 2011). More recently, variants in the TANK-binding kinase 1 (TBK1) gene were found to cause a spectrum of neurodegenerative disease including FTLD, ALS, and parkinsonism (Swift et al., 2021). TBK1 was previously identified as an IKK kinase that can activate NFkB and regulate the inflammatory response in myeloid cells including microglia (Pomerantz and Baltimore, 1999; Yu et al., 2012, p 1). The intersection of ALS/FTLD genes TBK1 and C9orf72 in the stimulator of interferon genes (STING) pathway has led to the suggestion that innate immune interferon pathways may be a potential therapeutic target in ALS (Van Damme and Robberecht, 2021). Both rare and common variants in immune genes expressed by microglia are associated with higher risk of AD (McQuade and Blurton-Jones, 2019) underscoring that primary dysfunction of microglia contributes to AD pathogenesis. The burden of common genetic risk variants in innate immune pathway genes is correlated with impaired cognition and increased amyloid deposition (Femminella et al., 2020). Transcriptomic analyses in several animal models of AD have revealed subpopulations of microglia with unique gene expression during disease (Keren-Shaul et al., 2017; Mathys et al., 2017; Rangaraju et al., 2018), identifying multiple microglia phenotypes (both detrimental and beneficial). Studies in human post-mortem autopsy tissue have attempted to verify these findings, also identifying multiple microglia states in disease (Friedman et al., 2018; Mathys et al., 2019; Nguyen et al., 2020; Srinivasan et al., 2020). In addition to these microglia subtypes, in animal models senescence results in dysregulation of microglia maintenance of the CNS homeostatic environment, suggesting that senescent phenotypes may also play a role in neurodegenerative diseases (Shaerzadeh et al., 2020; Triviño and von Bernhardi, 2021). Similar to astrocytes, this heterogeneity of microglia subtypes has made research to identify specific drivers of disease more complicated.
Overall, glia contribute significantly to neurodegenerative disorders including ALS/FTLD and AD. Each cell type contributes differentially to the neurodegeneration observed. While their heterogeneity is beginning to be investigated to understand disease-relevant mechanisms, ongoing research will help determine which subtypes in what brain regions are important for pathogenic mechanisms in these disorders.
TDP-43 PROTEINOPATHIES
Clinicopathologic correlation of TDP-43 proteinopathies:
TDP-43 proteinopathy refers collectively to neurodegenerative diseases which are characterized by the presence of abnormal accumulations of TDP-43 and associated degenerative changes (de Boer et al., 2020). The various diseases can generally be distinguished clinically by the types of symptoms observed, and pathologically by the pattern of TDP-43 throughout the brain and spinal cord. Classically, the symptoms correlate with the brain regions most affected by the disease. TDP-43 proteinopathies broadly fall into three main categories: motor neuron disease, frontotemporal dementia, and age-related, limbic-predominant disease (neuronal progression summarized in Figure 1). However, it is important to note that TDP-43 pathologic accumulation occurs in less common complex neurodegenerative diseases including Multisystem proteinopathy and Guam Parkinson-dementia complex (de Boer et al., 2020) as well as a number of rare monogenic diseases. Alexander Disease (AxD), caused by pathogenic variants in GFAP, is an autosomal dominant disease causing developmental delay, demyelination, and increased head circumference. Pathological “Rosenthal Filaments” (RF) accumulations in astrocytes is the hallmark of AxD. Phosphorylated TDP-43 colocalizes with RFs in both patient tissue and rodent models of AxD (Walker et al., 2014). A case report of the neurodegenerative autosomal recessive disease, Cockayne syndrome, which causes variable neurological signs including ataxia, retinopathy, and neuropathy is also reported to show both RFs and intraneuronal phosphorylated TDP-43 (Sakurai et al., 2013). These diseases demonstrate the wide variety of TDP-43 proteinopathies. Due to their relative prevalence in the population, we will focus on motor neuron, frontotemporal dementia, and age-related limbic predominant disease for this review.
Figure 1. Clinicopathological Classification of TDP-43 Proteinopathies.
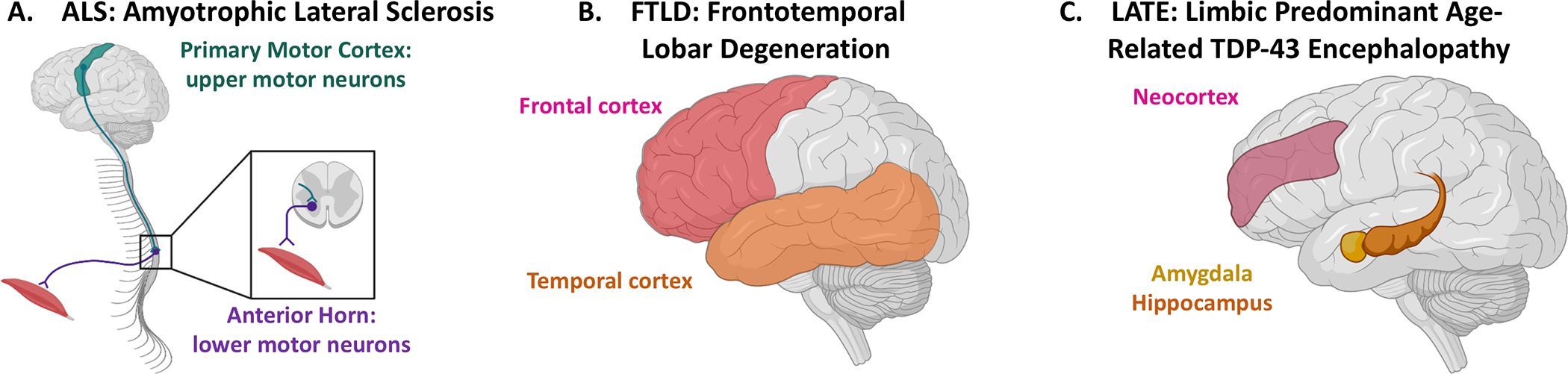
TDP-43 proteinopathies are defined based upon which brain regions contain the greatest abundance of pathology. (A) in ALS, abnormal TDP-43 inclusions are found in both the upper motor neurons of the primary motor cortex (green) as well as lower motor neurons, including those of the anterior horn of the spinal cord (purple). This pattern of pathology correlates with the clinical symptoms of motor neuron dysfunction. (B) FTLD is characterized by abnormal TDP-43 inclusions predominantly affecting cells in the frontal (pink) and temporal (orange) lobes, correlating with the behavioral disturbances classically seen in FTLD. (C) LATE has only recently been described and consensus criteria propose four stages of disease defined by the brain regions involved by abnormal inclusions of TDP-43. While a stage 0 indicates no abnormal TDP-43 aggregates, in stage one the aggregates are limited to the amygdala (yellow); the clinical implications of stage 1 LATE are not well understood. In stage 2 both amygdala and hippocampus (orange) are involved, while by stage 3, neocortex (pink) is also involved. These later stages correlate well with clinical symptoms, including amnestic complaints (hippocampus) and more global cognitive dysfunction (neocortex).
ALS, a prototypical motor neuron disease clinically characterized by progressive upper and lower motor neuron symptoms, was the first neurodegenerative disease in which TDP-43-positive inclusions were identified (Arai et al., 2006). The neuropathology of ALS correlates with the clinical symptoms such that TDP-43-positive inclusions are identified in both primary motor cortex, where upper motor neurons reside, as well as in brainstem motor nuclei and the anterior horns of the spinal cord, thus affecting the lower motor neurons (Figure 1 A). While ALS is defined as a motor neuron disease, a significant proportion of individuals will develop executive cognitive dysfunction or frank frontotemporal dementia (FTD; Liscic et al., 2008; Burrell et al., 2016). FTD represents a heterogeneous spectrum of cognitive disorders with variable phenotypes including marked behavioral changes, executive dysfunction, parkinsonism, and primary progressive aphasia (Seeley, 2008; Olney et al., 2017). The underlying neuropathology of FTD is also highly variable, but approximately 50% of cases are associated with TDP-43 pathology in a frontotemporal lobar pattern (FTLD-TDP; Figure 1B; Ikeda et al., 2004). In these cases, the location of the inclusions also correlates with the clinical symptoms as frontal cortex is particularly important for personality and executive function and the temporal lobe for language.
Although initially thought to be specific for ALS and FTLD-TDP, TDP-43 was soon described in aging and other neurodegenerative diseases, most often in sporadic late-onset AD with up to 57% of individuals reported to have comorbid TDP-43 pathology at autopsy (Matej et al., 2019). Over the last decade, awareness of TDP-43 pathology in aging and AD has increased, culminating in the recent publication of consensus guidelines for neuropathologic evaluation and a common terminology: Limbic Predominant Age-Related TDP-43 Encephalopathy (LATE; Figure 1C; Nelson et al., 2019a). As the name implies, this TDP-43 pathology seen in aging has a characteristic pattern of deposition in the limbic structures of the brain that is distinct from that of ALS and FTLD-TDP. The strong association between LATE and AD also distinguishes LATE from ALS and FTLD-TDP. Clinically, the presence of LATE, with or without AD, is associated with anatomically based cognitive/behavior features related to the regional distribution of TDP-43, such that pathology in the amygdala, hippocampus, and neocortex correlates to emotional lability, amnestic deficits, and global cognitive decline respectively (Nelson et al., 2010, 2019a; Brenowitz et al., 2014; Josephs et al., 2014; Wilson et al., 2016; Bayram et al., 2019; Robinson et al., 2020). The clinical symptoms in the presence of both LATE and AD suggest a possible additive or synergistic effect. In AD, the presence of TDP-43 appears to correlate with the degree of clinical impairment as well as regional pathology and atrophy (Amador-Ortiz et al., 2007; Josephs et al., 2008; Nelson et al., 2010). Individuals found to have LATE and AD neuropathologic change present at autopsy often had greater cognitive deficits, more rapid decline in cognitive function, and more pronounced hippocampal atrophy on ante-mortem imaging than those with AD pathology alone (Josephs et al., 2008, 2017). In addition to evidence that TDP-43 deposition on its own can lead to cognitive decline and potentiate that of AD, there are early suggestions that its absence may be an indicator of cognitive resilience in AD. In a small community-based cohort, individuals who were found to be cognitively intact despite fitting AD neuropathological criteria (resilient) had minimal TDP-43 compared to matched cases with similar AD pathology and dementia (Latimer et al., 2019). These associations suggest that TDP-43 may be a relevant contributor to the disease process in individuals with sporadic late-onset AD, however those mechanisms remain elusive.
Glial TDP-43 pathology in ALS and FTLD-TDP-43:
Neuronal inclusions, intranuclear and cytoplasmic, are the pathognomonic lesions considered necessary and sufficient for the diagnosis of TDP-43 proteinopathies. Particularly in FTLD-TDP and ALS, detailed descriptions and classification schemes were published that distinguish between various subtypes of TDP-43 pathology with a stronger emphasis on neuronal TDP-43. Pathologic TDP-43 inclusions in glial cells are nonetheless well described in ALS and FTLD-TDP (Neumann et al., 2007; Philips et al., 2013), as are the variable glial reactions in brain regions affected by neuronal TDP-43 pathology (Figure 2 A–D). Although some studies employing double labeling for TDP-43 and glial markers have shown TDP-43 inclusions in both astrocytes and oligodendroglia (Neumann et al., 2007; Arai et al., 2010), there is no known correlation between glial TDP-43 and FTLD-TDP subtype. There may be an association between the presence of glial TDP-43 and clinical phenotype however, with a series of more rapidly progressive forms of FTLD-TDP showing marked oligodendroglial TDP-43 pathology (Lee et al., 2017). A recent quantitative analysis of cortex and spinal cord tissue from sporadic and genetic ALS cases mapped the pan-glial expression of TDP-43 as well as the correlation of glial reaction to TDP-43 load (Nolan et al., 2020). Oligodendrocyte accumulation of TDP-43 was a prominent feature, as was previously reported (Philips et al., 2013), while TDP-43 aggregate accumulation within astrocytes themselves was not observed. TDP-43 aggregates were found to colocalize with microglia, although it was not possible to distinguish whether there was aberrant localization of intrinsic microglial TDP-43 or if the microglia were in the process of internalizing external TDP-43. As may be expected, increased expression of a microglia activation marker, CD68, was correlated with higher grey matter TDP-43 immunolabeling in both the sporadic and C9orf72 familial ALS forms (Nolan et al., 2020), highlighting the relationship between TDP-43 and microglia activation though not clarifying the directionality of that interaction.
Figure 2. TDP-43 pathology and glial responses across the TDP-43 proteinopathies.
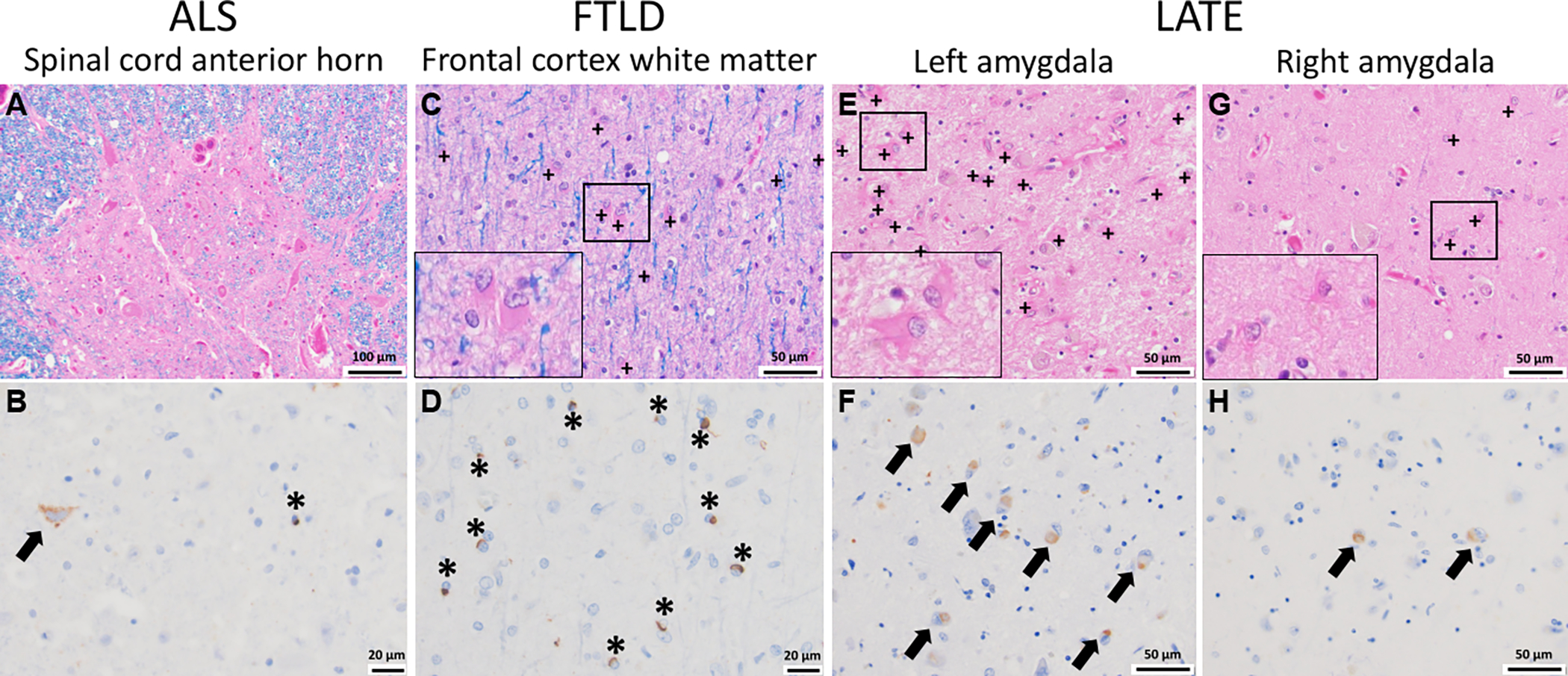
(A, B) In ALS, cells within the anterior horn of the spinal cord are affected (A, H&E/LFB stain) and immunohistochemical staining for phosphorylated TDP-43 highlights inclusions both in neurons (arrow) and glia (asterisk) (B, pTDP-43 immunostain). (C, D) In FTLD, the subcortical white matter is often gliotic as evidenced by increased reactive astroglia (plus signs and inset) (C, H&E/LFB stain) in association with numerous pTD-43 inclusions in oligodendroglia (asterisk, D, pTDP-43 immunostain). (E-H) In LATE, which is often seen comorbid with Alzheimer’s disease, there is a robust glial reaction (E, G, H&E/LFB stain, with reactive astrocytes shown by plus signs and in the insets) in association with the presence of neuronal pTDP-43 inclusions (arrows, F, H, pTDP-43 immunostain). In this case, while pTDP-43 inclusions were found bilaterally in the amygdala, the left side showed significantly more inclusions compared to the right, which correlated with the striking increase in reactive astroglia also seen in the left amygdala (inset). Whether the pTDP-43 inclusions are found in glia as well as neurons (arrows) is not well characterized in the context of AD+LATE. + = reactive astrocyte, arrow = neuronal TDP-43 inclusion, * = glial TDP-43 inclusion
TDP-43 was recently shown to induce inflammation though the STING pathway, a process which activates Interferon type 1 and NFkB signaling and is regulated by the ALS/FTLD-TDP gene TBK-1 (Balka et al., 2020). TDP-43 overexpression induced mitochondrial DNA leakage into the cytosol, activating the DNA sensor cGAS and subsequently STING signaling (Yu et al., 2020). The STING pathway may be one example by which TDP-43 induces activation of microglia, and presumably contributes to observed inflammatory neuropathological change. Recent work suggests that there may be regional differences in the microglia population and response to pathology. A study of familial FTLD-TDP demonstrated that while there was a higher overall burden of microglia in FTLD-TDP white matter, those cells were more dystrophic in contrast to those in the grey matter, which instead appeared more activated (Woollacott et al., 2018). Studies such as these emphasize the importance of further refining our understanding of microglia states and designing studies aimed at targeting particular glial functions in the “right” region of brain.
Glial TDP-43 pathology in LATE:
Unlike ALS and FTLD-TDP, there are only rare mentions in the literature of glial TDP-43 inclusions in the context of LATE and the consensus criteria focuses exclusively on neuronal cytoplasmic and intranuclear inclusions. However, it is unclear whether the paucity of LATE glial TDP-43 descriptions is because LATE, as a recently described pathological entity, was only formally defined in 2019 (Nelson et al., 2019a) or if there is truly a lack of glial TDP-43 pathology in aging. Of note, several studies do mention glial TDP-43 inclusions, usually in summary tables of neuropathologic findings, but do not provide morphologic details or descriptions regarding specific glial cell type or brain regions involved (Uryu et al., 2008; Hunter et al., 2020). It may be that as the neuropathologic evaluation of TDP-43 proteinopathy becomes more routine in the aging population, the full breadth of TDP-43 pathology of LATE will become apparent and lead to more detailed investigation of glial TDP-43 pathology. This progression would be akin to the expansion of the neuropathologic characterization of glial tau pathology in recent years (Kovacs, 2018). This will require a concerted effort to characterize the extent of glial TDP-43 pathology in aged human brains, including double-labeling studies to confirm the cell types involved.
Despite the scarcity of well documented descriptions of TDP-43 mislocalization or aggregation within glia in LATE, the glial response observed in association with the presence of TDP-43 pathology in LATE is well characterized, most notably in the hippocampus. Hippocampal sclerosis of aging, which is defined by marked neuron loss and severe astrogliosis, is often seen in AD and almost always associated with the presence of TDP-43 (Amador-Ortiz et al., 2007; Nelson et al., 2019a).
Astrogliosis is a part of the brain’s response to injury, and reactive astrocytes are a common and non-specific finding in most neurodegenerative diseases. In AD, astrogliosis is often seen in the upper layers of the neocortex, the hippocampus, and the amygdala; but in the presence of comorbid LATE this astrogliosis is markedly increased (Figure 2 E–H). While it is unclear whether TDP-43 plays an active role in the pathophysiologic processes that lead to astrogliosis, recent studies have identified phosphorylated TDP-43 (pTDP-43) in the hippocampus in the absence of hippocampal sclerosis, suggesting that the pTDP-43 pathology precedes, and therefore potentially contributes to, neurodegeneration and reactive gliosis (Power et al., 2018; Nelson et al., 2019a). Currently, it is unknown whether the reactive gliosis is a non-specific response to neuron loss, or a more specific response to the TDP-43 pathology.
Loss of myelin integrity occurs early (Cai and Xiao, 2016; Butt et al., 2019) and is markedly increased in AD, as is the loss of oligodendrocytes (Nasrabady et al., 2018). TDP-43 deposition in oligodendroglia is established in ALS/FTLD-TDP, though similar findings in LATE have not been reported (de Boer et al., 2020). The relationship between pathologic protein aggregates and microglia crosses diseases and is complex, likely changing over the course of disease, though little is known regarding the intersection between microglia and LATE pathology in the context of AD.
TDP-43, GLIA, AND NEURODEGENERATIVE DISEASE MODELS
An abundance of evidence across brain syndromes supports a role for non-cell autonomous mechanisms in neurodegeneration. To better understand the cascade of events resulting in neuronal degeneration and cognitive dysfunction in TDP-43 proteinopathy, it is important to shed light on the relevant pathogenic processes driven by glia.
The discovery of the shared property of RNA/DNA binding by TDP-43 and FUS helped bring RNA processing to the forefront of disease relevant mechanisms (Ling et al., 2013). Glial cells require local RNA translation, RNA transport, and subcellular localization for normal functioning making them vulnerable to acquired or genetic driven RNA processing dysfunction (Barton et al., 2019). In ALS animal models, deletion of TDP-43 in motor neurons alone does not fully recapitulate the human neurodegenerative disease phenotype of motor neuron disease, emphasizing that global CNS dysfunction is related to both neuronal and non-neuronal health and likely the interaction between the two (non-cell autonomous). A murine model of TDP-43 deficiency in which neuronal and glial TDP-43 are depleted develop a severe behavioral phenotype and neurodegeneration reflecting the significant contribution of glia in TDP-43 neurodegeneration (Yang et al., 2014). TDP-43 mediated glial dysfunction is linked to neuronal toxicity (Swarup et al., 2011) and therefore mapping the glial pathways that are dysregulated by TDP-43 or impaired in the presence of pathological TDP-43 is important for a holistic approach to modulating TDP-43 proteinopathy. Model systems are a convenient way to understand the effects of TDP-43 mislocalization on glia. While many studies are directed towards ALS TDP-43 variants, we focus here on studies that may provide insight as to how wildtype TDP-43 may contribute to common neurodegenerative disease.
Astrocytes:
Work in human patient induced pluripotent stem cell derived astrocytes collected from “sporadic” ALS cases demonstrate a relative resilience to cell death by TDP-43 aggregates compared to similarly challenged neurons. Using a seeding paradigm whereby TDP-43 aggregates isolated from patient spinal cord are introduced in culture and assessed for cell to cell spread and cell toxicity, astrocytes remained more unaffected than neurons (Smethurst et al., 2020). Furthermore, the authors report that introducing astrocytes or astrocyte conditioned media to neurons seeded with TDP-43 aggregates promotes clearance of TDP-43 from neurons and decreases in activated caspase 3, a marker of neuronal death (Smethurst et al., 2020). These data indicate that astrocytes may be both resilient to toxicity, and also provide beneficial functions to neurons in the presence of pathological TDP-43. Nevertheless, aggregated TDP-43 can mediate toxic effects on astrocytes contributing to dysfunctional glial physiology and subsequent non-cell autonomous neuronal injury. Astrocytes with TDP-43 inclusions show disrupted aerobic glycolysis and lipid metabolism which in turn may impair metabolic neuronal support (Velebit et al., 2020). In addition, it appears levels of wildtype TDP-43 itself are relevant to intact TDP-43 function. Overexpression of TDP-43 in mice disrupts amino acid homeostasis of astrocytes and neurons (Hebron et al., 2014; Heyburn et al., 2016). Overexpression of TDP-43 via transient transfection in primary murine astrocytes induces expression of PTP1B, an inflammatory mediator also associated with myeloid inflammatory responses (Lee et al., 2020). On the other hand, astrocytes with reduced TDP-43 expression via siRNA mediated knockdown have increased double stranded RNA which stimulates innate immune responses and subsequent cell activation (LaRocca et al., 2019). Localization of TDP-43 in astrocytes, as shown in neurons, may contribute to the astroglial pathologic response seen in neurodegeneration. Expression of the pathologic C-terminal fragment of TDP-43 in vitro in astrocytes leads to cytoplasmic inclusions with subsequent clearing of TDP-43 from the nucleus (Velebit et al., 2020), consistent with previous work demonstrating that restricting TDP-43 to the cytoplasm leads to TDP-43 cytoplasmic aggregation and relocalization of previously nuclear TDP-43 (Winton et al., 2008). Selective glial expression of ALS mutant TDP-43 in rodent models highlights the non-cell autonomous mechanisms of neurodegeneration. Astrocyte specific mutant TDP-43 expression in rats leads to loss of astrocyte glutamate transporters in addition to motor neurons (Tong et al., 2013). However, some human neural studies, which can only be performed in vitro have not yet captured the non-cell autonomous effect of astrocytic mutant TDP-43 expression. An iPSC-based co-culture method demonstrated the cell autonomous effects of pathological TDP-43 cytoplasmic accumulation in astrocytes; however there was no impact on neuronal survival when mutant astrocytes were cultured with motor neurons (Serio et al., 2013).
Oligodendrocytes:
Less is known about TDP-43 in oligodendrocytes outside of the ALS context, though ALS human pathological studies clearly demonstrate oligodendroglial TDP-43 inclusions (Brettschneider et al., 2014). Selective deletion of TDP-43 in astrocytes by conditional excision using cell type specific promoters to drive Cre-mediated recombinase in a floxed TDP-43 murine transgenic model, results in cellular and motor dysfunction as well as acquisition of a reactive astrocytic phenotype, activation of microglia, and loss of mature oligodendrocytes (Peng et al., 2020). Using the same murine model to selectively delete TDP-43 in oligodendrocytes in vivo leads to loss of mature oligodendrocytes and halts maturation of new oligodendrocytes in addition to demonstrating a progressive loss of myelin (Wang et al., 2018). Studies in infectious models of Multiple Sclerosis using Theiler virus link neuroinflammatory processes and oligodendroglial dysfunction. Theiler virus infected mice show a chronic immune mediated demyelinating and behavioral phenotype. Concomitantly, TDP-43 is found to be mislocalized to the cytosol in oligodendrocytes and other glia (Masaki et al., 2019). Of note, human neuropathological studies in sporadic ALS in which TDP-43 aggregates are a hallmark, do not always show a significant loss of oligodendroglial populations. This implies that the complete loss of TDP-43 in animal models may lead to biological effects distinct from sporadic neurodegenerative disease patients who have present, though abnormally localized and aggregated TDP-43.
Microglia:
Work in a murine model engineered to express an inducible pathological TDP-43 which forms neuronal aggregates, a neurodegenerative phenotype, and microglia activation has shed some light on the association between TDP-43 and microglial function (Spiller et al., 2018). Spiller et al. demonstrate the development of a behavioral phenotype and neuronal loss after induction and subsequent accumulation of human TDP-43 (hTDP-43) aggregates. After suppressing expression of the mutant hTDP-43 construct, microglia proliferate, adopting a reactive morphology, and mediate motor neuron functional recovery and TDP-43 clearance (Spiller et al., 2018). These data suggest that once TDP-43 accumulation has led to neuronal dysfunction, microglia can play a restorative role, conferring neuronal protection. Real time imaging of a GFP-tagged wildtype TDP-43 in a zebrafish model of neurodegeneration visualized microglia phagocytosis and localization of neuronal TDP-43 after UV-induced neuronal injury (Svahn et al., 2018). Microglia targeted and phagocytosed UV-injured neurons, internalizing neuronal nuclear TDP-43. In the presence of microglia depletion, however, TDP-43 was mislocalized to the cytoplasm and axons of UV-injured neurons. These findings led the authors to suggest that one mechanism of aberrant TDP-43 accumulation may be defective microglia clearance.
As innate immune cells, microglia respond to microbial or damage associated cues through multiple signaling pathways. These Toll like receptor (TLR), CD-14, and NFkB mediated signaling events drive transcriptional responses to danger signals while the non-canonical NLRP-3 inflammasome complex recognizes intracellular danger molecules and mediates post-translational inflammatory pathways resulting in release of Il-1b, IL-18, and additional cytokines (Heneka et al., 2018). Exposure to wildtype and disease associated genetic variants of TDP-43 in vitro can induce an inflammatory response in microglia by activating NFkB and the NLRP3 inflammasome that is dependent on TDP-43 and CD14 interactions at the cell surface (Zhao et al., 2015). TDP-43 mutant murine models of ALS show induction of NLRP3 expression and inflammasome activation in spinal cord tissue, which occurs in cases of both wildtype and mutant TDP-43 expression. Similarly, microglia isolated from WT or mutant TDP-43 expressing mice release increased IL-1b that is attenuated in the presence of an NLRP3 inhibitor (Deora et al., 2020).
In parallel, inflammatory processes may direct TDP-43 function and localization within glia. In vitro, TDP-43 potentiates the microglia inflammatory response to the canonical TLR4 inflammatory stimulus, lipopolysaccharide (LPS), leading to increased release of inflammatory cytokines (Swarup et al., 2011). TDP-43 and the NFkB p65 subunit have been found to co-immunoprecipitate in ALS patient tissue extracts, though not in tissue of unaffected human subjects (Swarup et al., 2011). This interaction may have functional consequences as suggested by a genetic reporter assay which showed that TDP-43 acts as a co-activator of the NFkB p65 subunit in BV2 cells, a microglia-like cell line. The same group later demonstrated that the inflammatory response induced by LPS in primary microglia and astrocytes in vitro results in relocalization of TDP-43 to the cytosol (Correia et al., 2015). In addition to pathological aggregation of TDP-43 in the cytoplasm, the potentiation of p65 mediated transcription by TDP-43 in the nuclei of affected patients indicates that TDP-43 may mediate a toxic gain of function in the nucleus.
In response to internalization of exogenous aggregated TDP-43, endogenous TDP-43 has been observed to relocate to the cytoplasm from the nucleus in primary in vitro assayed microglia (Leal-Lasarte et al., 2017). Once in the cytoplasm, TDP-43 was again found to activate the NLRP3 inflammasome, inducing IL-1b and IL-18 processing as well as caspase-3 activation. Thus, consistent with findings from TDP-43 transgene expression, TDP-43 exposure can acutely induce the NLRP3 inflammasome and other inflammatory sequences. The question regarding from where microglial aggregated TDP-43 is derived in a patient with a TDP-43 proteinopathy remains to be established. Release from dying neurons is a credible and likely possibility, though uptake either directly from neurons or in extracellular vesicles are also possibilities (Feiler et al., 2015; Sproviero et al., 2018). In total, data in microglia indicate that the aberrant accumulation of TDP-43 in the cytosol may induce intracellular inflammatory signaling by the NLRP3 inflammasome in addition to stimulating cell surface TLR signaling. The loss of nuclear TDP-43 may also result in impaired RNA regulation, though this has not yet been validated experimentally.
The effects of changes in TDP-43 are cell type specific and may be beneficial or deleterious depending on what change in TDP-43 is modeled. Furthermore, the pathogenic effects of TDP-43 may also involve non-neural cells and vasculature in the CNS. For example, neuronal expression of wildtype TDP-43 via intracerebral injection into mouse cortex induces microglial and astrocytic activation (Zamudio et al., 2020) but also leads to increased blood brain barrier permeability. Importantly, the presence of TDP-43 in neurons additionally caused an increased vulnerability to low-dose intra-peritoneal injected LPS induced systemic inflammation (Zamudio et al., 2020). Models such as these begin to interrogate the complex interaction between the CNS and the periphery which undoubtedly is an extremely relevant issue in human patients. While these models provide insight into the pathogenic mechanisms of TDP-43 dysfunction in glial cell types, understanding how these interactions might change in the presence of additional pathology is necessary to better define the pathogenic role of TDP-43 in diseases such as AD.
TDP-43 INTERACTIONS WITH AD PATHOLOGY
TDP-43 pathology is often observed in the presence of AD neuropathologic change. This co-pathology would now be defined as AD + LATE, in other words the combination of AD pathologies Ab plaques and phosphorylated tau neurofibrillary tangles along with neuronal TDP-43 inclusions. When these pathologies occur together, they influence clinical measures, suggesting that the interaction between these different pathological mechanisms may ultimately contribute to cognitive decline.
TDP-43 interaction with Ab:
TDP-43 and amyloid-beta (Ab) often co-occur in AD brain, raising the question whether one can potentiate the deposition of the other. Expressing Ab1–42 via lentivirus in rat cortex led to a concomitant increase in TDP-43 expression, phosphorylation, cytosolic mislocalization, and aggregation, all biochemical features of TDP-43 found in AD brain (Herman et al., 2011, 2012). Using the 3XFAD mouse model, TDP-43 and its 35kDa c-terminal fragment were higher at six months of age which correlated with the accumulated amounts of Ab oligomers in brain (Caccamo et al., 2010). This was further confirmed by reverting the mutant PSEN1 variant to wildtype in the 3XFAD line resulting in a double transgenic (APP/Tau) that does not accumulate Ab oligomers, with a concomitant decrease in TDP-43 expression (Caccamo et al., 2010). Targeted deletion studies demonstrated that loss of forebrain neuronal TDP-43 in the AD model APPswe/PS1ΔE9 mouse enhanced neurodegeneration in the hippocampus and cortex but also reduced both oligomeric and plaque Ab (LaClair et al., 2016). Instead, there was an accumulation of prefibril Ab that correlated with the loss of synaptophysin at eight months of age in APPswe/PS1ΔE9 without TDP-43. In the APP/PS1 murine AD model, TDP-43 oligomers were shown to inhibit the fibril formation of Ab1–40 in vitro, though TDP-43 did not inhibit the seeding of fibrils (Shih et al., 2020). In the same APP/PS1 mouse model, however, the injection of TDP-43 oligomers into brain increased Ab plaques and worsened memory performance, indicating that in vivo conditions for Ab and TDP-43 interaction may differ from the fibril formation in vitro (Shih et al., 2020). Additional data from studies employing lentiviral delivery of TDP-43 into rat cortex indicated that TDP-43 co-localized with BACE1 (a key Ab cleaving enzyme), increased BACE1 expression, and led to increases in APP-c terminal fragment processing, a key mechanism for Ab accumulation (Herman et al., 2012). This direct interaction of TDP-43 and BACE1 may explain some of the previous findings of TDP-43 manipulating the aggregation and accumulation of Ab. Together, these data suggest that TDP-43 is not a by-product of neurodegeneration in AD but may interact with the levels of Ab present, and lead to exacerbated neurodegeneration in these AD models.
TDP-43 interaction with Tau:
Tau is the other primary pathology seen in AD, where in its hyperphosphorylated state, tau forms neurofibrillary tangles and may seed across extracellular space to spread pathology between cells (Goedert et al., 2017). There are several putative mechanisms by which TDP-43 may regulate tau pathology through its mRNA splicing functions. In the healthy brain, 3-repeat (3R) and 4-repeat (4R) tau isoforms are in approximately equal ratio resulting from alternate splicing of exon 10. TDP-43 assists with splicing of tau mRNA, leading to exclusion of exon 10 (Gu et al., 2017) and subsequent decrease of 4R isoforms. Overexpression of TDP-43, or mutations of TDP-43 associated with ALS/FTLD-TDP, both lead to inclusion of exon 10 and increased levels of 3R tau which disrupts the 3R/4R tau ratio in both in vitro and in vivo murine models (Gu et al., 2017). These changes to tau splicing and the 3R/4R tau ratio because of TDP-43 aggregation and depletion from the nucleus are hypothesized to contribute to neurofibrillary tangle formation (Gu et al., 2017). A later study found that de-phosphorylation of TDP-43 at specific serines by protein phosphatases 1α and 1γ subsequently alters the cellular location of TDP-43 and thus its ability to suppress exon 10 inclusion in tau mRNA (Gu et al., 2018). The authors suggest that since protein phosphatase 1 expression is decreased in AD brain (Gong et al., 1993), this reduction could lead to both the hyperphosphorylation of TDP-43 and the inclusion of exon 10 in tau and the subsequent production of neurofibrillary tangles, though this chain of events remains to be demonstrated experimentally. A set of biochemical assays demonstrated that TDP-43 oligomers can serve as a template for oligomeric tau aggregates (Montalbano et al., 2020). Tau oligomers can also alter TDP-43 localization and oligomerization, indicating that tau pathology may induce TDP-43 pathology and vice versa (Montalbano et al., 2020). In vivo, expression of a disease associated TDP-43 variant in cholinergic neurons using an inducible transgenic rat model increases tau pathology in the hippocampus (Moszczynski et al., 2019). This synergism of pathologies even across brain regions is intriguing and fits with hypotheses of both TDP-43 and tau having seeding properties (Jo et al., 2020). Taken together, these studies indicate TDP-43 and tau may influence the pathology of the other through indirect or direct mechanisms.
GLIAL TDP-43 IN AD
While it is clear from other neurodegenerative model systems involving TDP-43 alone that TDP-43 plays a role in glia and in the glial response to neurodegeneration, there is an absence of literature attempting to understand the role of TDP-43 in glia in AD pathogenesis. A few studies have noted additional inflammation as a result of LATE pathology that is increased beyond that seen in AD pathology without TDP-43. However, only one study has directly experimentally investigated the role of TDP-43 in a glial cell type in AD models.
It was repeatedly demonstrated that the presence of TDP-43 pathology induces microglia activation, or other neuroinflammation, beyond that seen in standard AD models. The injection of TDP-43 oligomers into the cortex of APP/PS1 mice caused an increase in Iba-1 expression, often thought to correlate with increased microgliosis (Shih et al., 2020). The increased accumulation of Ab fibrils in the presence of TDP-43 in vivo compared to in vitro was attributed to the contributions of neuroinflammation, as suggested by the enhanced microglial Iba-1 immunoreactivity (Shih et al., 2020). Induced expression of Ab1–42 or TDP-43 alone in rat cortex increases the number of Iba-1 and GFAP positive cells, both glial markers of neuroinflammation (Herman et al., 2012). The combination of Ab and TDP-43 increased the number and protein expression of Iba-1 and GFAP even further, as well as increasing TNFα and IL-6 secretion, indicating that the co-localization of these two pathological proteins may increase neuroinflammation phenotypes (Herman et al., 2012). TDP-43 and mutant tau may synergistically induce inflammatory responses as evidenced by increased Iba-1 immunoreactivity in the setting of co-expression of TDP-43 and mutant tau in rat cortex (Moszczynski et al., 2019). TDP-43 colocalizes with mitochondria in control neurons, and there appears to be significantly higher levels of mitochondrial TDP-43 in human ALS and AD (Wang et al., 2016; Gao et al., 2020). The 5xFAD mouse model brain recapitulates the enhanced TDP-43 levels in mitochondria which is also associated with mitochondrial dysfunction. Inhibiting TDP-43 mitochondrial localization was reported to protect cognition and neuronal loss (Gao et al., 2020). Interestingly, while inhibiting TDP-43 localization to mitochondria does not change the plaque formation or load, it does decrease both neuronal loss and microgliosis (based on number of Iba-1 positive cells) in the 5xFAD mice (Gao et al., 2020). This suggests that augmented TDP-43 localization to mitochondria may change neuronal behavior and neuroinflammatory responses – whether the neuroinflammation is in response to the neuron loss, or a cause of neurodegeneration remains unclear in these experiments. Further studies are needed to accurately and quantitatively assess morphology, cell function, and cytokine and/or gene expression under different AD+TDP-43 pathology conditions to further understand the effects on different glial cell types.
One study directly investigated the effects of manipulating TDP-43 expression in microglia in the presence of AD pathology in murine models. After demonstrating that TDP-43 depletion in the microglia-like cell line, BV2, results in increased phagocytosis and lysosomal biogenesis, the authors studied the in vivo consequence of selectively depleting TDP-43 in microglia on neuroinflammation (Paolicelli et al., 2017). Using an inducible microglia specific TDP-43 knockout line (TDP-43cKO) the authors found enhanced microglia clearance of injected Ab compared to WT mice (Paolicelli et al., 2017). Depletion of TDP-43 in microglia in the context of overexpression of pathological APP (TDP-43cKO/APParc) led to reduced Ab40 and reduced Ab plaques (Paolicelli et al., 2017). However, the TDP-43cKO/APParc mice also had significantly reduced synaptic markers, indicating that synaptic stripping may also occur alongside the Ab clearance. This enhanced phagocytic phenotype in microglia that resulted in reduced synapses was present in TDP-43 cKO mice without the APP transgene suggesting that microglia lacking TDP-43 do not require an inflammatory stimulus to begin the synaptic stripping process. This has additional implications for neurodegenerative disease, where synaptic stripping is hypothesized to be an underlying mechanism of cognitive decline. Similar to what was shown in ALS/FTLD-TDP, the phagocytic marker CD68 is more highly expressed in microglia in individuals with TDP-43 pathology, indicating that the increased phagocytic phenotype seen in animal models may also be present in humans with TDP-43 pathology (Paolicelli et al., 2017). These studies are informative and point to a key role for TDP-43 in microglia in AD, though it is still unknown whether these findings are recapitulated in human brain with AD pathology with or without the co-presence of TDP-43 pathology. Additional studies are needed to determine both whether TDP-43 is depleted from the nucleus in microglia in the setting of pathological TDP-43 aggregates, and whether TDP-43 aggregation in neurons may induce an exaggerated phagocytic phenotype in microglia, potentially leading to synaptic stripping and cognitive decline.
Together, the extant literature, though small, indicates that TDP-43 pathology may further induce activation of microglia and astrocytes and neuroinflammation in AD. There is a suggested bidirectional interaction between TDP-43 and Ab as well as TDP-43 and tau where the two pathological proteins may influence the abnormal accumulation of the other. Further studies are needed to elucidate the interactions of different glia cell types with TDP-43 pathology in AD.
DISCUSSION
Neurodegeneration is a confluence of diverse mechanisms mediated by multiple cell types across a temporal axis (some examples: Lee et al., 2012; Radford et al., 2015; De Strooper and Karran, 2016; Salter and Stevens, 2017; Barton et al., 2019; Butt et al., 2019; Triviño and von Bernhardi, 2021). In this review, we have brought together the research on glia cell types and TDP-43 proteinopathies. Glia clearly interact with TDP-43 in ALS/FTLD-TDP, and we hypothesize that they are similarly important in AD+LATE and other TDP-43 proteinopathy pathophysiology. The mechanisms of TDP-43 mislocalization and aggregation in the TDP-43 proteinopathies ALS, FTLD-TDP are a relatively recent discovery. Although the pathological diagnosis of these disorders focuses on neuronal cytoplasmic inclusions, data from both post-mortem patient tissue as well as model systems demonstrate that glial cells are themselves affected by TDP-43 pathology as well as reactive to TDP-43 pathology in neurons. As a new entity in the neuropathology field, AD+LATE is defined solely by neuronal inclusions and little information exists about glia responses or glial inclusions. Given the significant role glia play in these neurodegenerative disorders it seems crucial to increase our understanding of how TDP-43 and glia interact in these proteinopathies including in AD+LATE.
The cellular responses of glial cells vary by cell type, and it remains to be seen if it varies by brain region as well. For both astrocytes and microglia, model systems suggest that these cells may play beneficial or detrimental roles depending on the type of TDP-43 pathology present. Based on the existing literature, we propose the following overview of TDP-43 and its relationship with glia and neurodegeneration. In the healthy brain, TDP-43 functions normally in splicing, RNA binding, and shuttling between the nucleus and cytoplasm, with the primary concentration in the nucleus. TDP-43 is present ubiquitously in all cell types of the brain (Figure 3A). In ALS/FTLD-TDP, the primary pathology is thought to be neuronal TDP-43 cytoplasmic inclusions (Figure 3B). In both post-mortem patient tissue, and in model systems, glial cytoplasmic inclusions have also been identified. Model systems additionally point to a variety of reparative (clearing TDP-43 from neurons by microglia and astrocytes) and disease-causing (loss of maturation of oligodendrocytes and myelination) mechanisms for TDP-43 in different glial cells (Figure 3B). Additionally, potential mechanisms of transfer of TDP-43 between cells via uptake of endosomes containing TDP-43 aggregates is known (Figure 3B). While the contribution of glia to ALS/FTLD-TDP is somewhat explored, the relationship of TDP-43 and glia in comorbid pathologies such as AD+LATE remains relatively unknown. Like ALS/FTLD-TDP, the primary pathology to confer diagnosis is TDP-43 cytoplasmic inclusions in neurons (Figure 3C). Perhaps because LATE has only recently been defined with a consensus diagnosis, little acknowledgement of the potential contribution of glia to this pathology is described in the literature. We suspect that TDP-43 cytoplasmic inclusions are present in glia as well as in neurons, though this remains to be shown for all glial cell types (Figure 3C). The few studies that exist suggest that TDP-43 mislocalization in glial cells is important for AD pathogenesis. Neuropathology is clear that TDP-43 aggregation in neurons enhances gliosis (Figure 2), suggesting that whether or not TDP-43 dysfunction is present in glia their response may alter cognitive outcomes. Together, these data indicate that much remains to be discovered about whether TDP-43 itself is altered in glial cells in AD+LATE and how that contributes to disease progression.
Figure 3. Glia involvement in TDP-43 proteinopathy is known in ALS/FTLD-TDP while much remains unknown in AD+LATE.
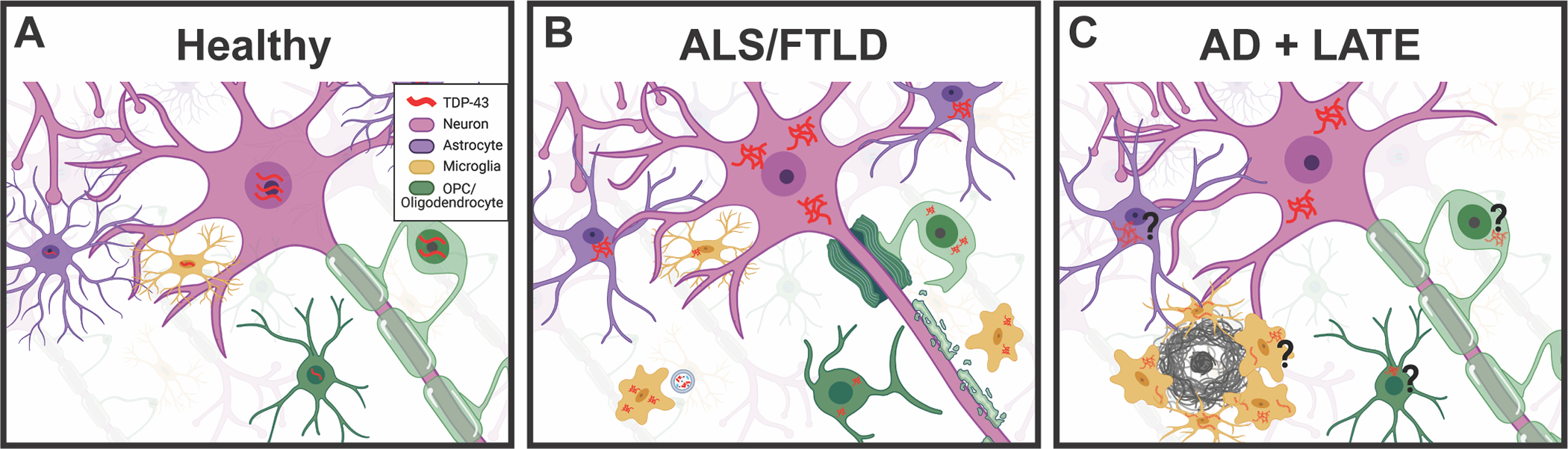
(A) In the healthy brain, TDP-43 is ubiquitously expressed by both neurons and glia. Indicated here in red, TDP-43 shuttles between the nucleus and cytoplasm in normal function, but the majority remains located in the nucleus where it performs its typical RNA binding and splicing functions. (B) In ALS/FTLD TDP-43 aggregates in the cytoplasm of neurons are a hallmark of pathology in about half of cases. In addition, TDP-43 is known to aggregate in the cytoplasm of oligodendrocytes, oligodendrocyte precursor cells (OPCs), astrocytes, and microglia. TDP-43 aggregates may be transferred to microglia through exosomes as depicted. (C) In AD with LATE pathology, TDP-43 aggregates are found in the cytoplasm of neurons shown in solid red. This may lead to abnormal splicing of tau, and increased accumulation of oligomeric amyloid beta. Both of which may further accelerate the AD pathological hallmarks of neurofibrillary tangles and amyloid plaques, though more research is needed. Additionally, it remains unknown whether TDP-43 aggregates in glia such as astrocytes, OPCs, oligodendrocytes, and microglia all of which are activated and responsive to AD pathology. We suspect that TDP-43 pathology exists in these glial cell types as well as in neurons as indicated by the dashed red TDP-43, but further work is needed to confirm this. We hypothesize that mislocalization of TDP-43 is occurring in glia, and that this significantly impacts AD pathological response; however, there is currently little evidence for which cell types are impacted and further research is needed. (Created with Biorender.com)
In addition to understanding the role of TDP-43 in glia, research is needed to further elucidate the interaction of TDP-43 in combination with other pathologies that co-occur in AD+LATE. The synergy between TDP-43 dysfunction and accumulation of Ab as well as tau splicing indicates that TDP-43 may directly enhance primary AD pathology. Additional human tissue-based studies are crucial to this effort to identify the contribution of glia to TDP-43 pathology, particularly longitudinal studies with well characterized participants and an autopsy endpoint to further define the clinical and pathologic associations of TDP-43 and glia. These studies not only need carefully defined tissue sets, but double-labeling to identify TDP-43 inclusions in each of the glial cell types. Such work may help drive better clinical testing approaches to facilitate the identification of individuals with comorbid AD+LATE. AD and ALS/FTLD-TDP are complex diseases whose drug discovery efforts are challenged by clinical, neuropathologic and mechanistic heterogeneity. In the case of AD in particular, which has had a longer run with clinical trials than FTD and ALS, therapeutic efforts have been unsuccessful and there is a pressing need to expand beyond the current approaches. In part, the concern that human clinical trials begin too late in an individual, after disease has already started, is likely true and a contributing factor to repeatedly failed trials. Because glial function is implicated in accelerating disease progression, glial mechanisms are attractive candidates for strategies to halt or slow disease even if the initial insult has begun. Studies to better define the relationship of TDP-43 and specific glial cell types may thus inform strategies which can join the armamentarium of therapies that will be required for successful approaches to these devastating neurodegenerative diseases.
Acknowledgments
Funding Sources:
RF1AG063540 (S.J.K.P.)
P30AG066509 (K.P., C.S.L., S.J.)
K08AG065426 (C.S.L.)
REFERENCES
- Abramzon YA, Fratta P, Traynor BJ, Chia R. 2020. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci 14:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Barres BA. 2009. Neuroscience: Glia - more than just brain glue. Nature 457:675–677. [DOI] [PubMed] [Google Scholar]
- Amador-Ortiz C, Lin W-L, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. 2007. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Annals of Neurology 61:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador-Ortiz C, Lin W-L, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. 2007. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrea R, Ilaria V, José JRA, Arthur B. 2016. Decreased Regenerative Capacity of Oligodendrocyte Progenitor Cells (NG2-Glia) in the Ageing Brain: A Vicious Cycle of Synaptic Dysfunction, Myelin Loss and Neuronal Disruption? Current Alzheimer Research 13:413–418. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochemical and Biophysical Research Communications. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K, Yoshida M, Onaya M, Fujishiro H, Akiyama H. 2010. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology 30:170–181. [DOI] [PubMed] [Google Scholar]
- Augusto-Oliveira M, Arrifano GP, Lopes-Araújo A, Santos-Sacramento L, Takeda PY, Anthony DC, Malva JO, Crespo-Lopez ME. 2019. What Do Microglia Really Do in Healthy Adult Brain? Cells 8:1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D’Silva DB, Moghaddas F, Tailler M, Lawlor KE, Zhan Y, Burns CJ, Wicks IP, Miner JJ, Kile BT, Masters SL, De Nardo D. 2020. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Reports 31:107492. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. 2010. Cytoplasmic Mislocalization of TDP-43 Is Toxic to Neurons and Enhanced by a Mutation Associated with Familial Amyotrophic Lateral Sclerosis. J Neurosci 30:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton SK, Gregory JM, Chandran S, Turner BJ. 2019. Could an Impairment in Local Translation of mRNAs in Glia be Contributing to Pathogenesis in ALS? Front Mol Neurosci [Internet] 12. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6536688/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram E, Shan G, Cummings JL. 2019. Associations between Comorbid TDP-43, Lewy Body Pathology, and Neuropsychiatric Symptoms in Alzheimer’s Disease. J Alzheimers Dis 69:953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj A, Myers MP, Buratti E, Baralle FE. 2013. Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res 41:5062–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer EMJ, Orie VK, Williams T, Baker MR, De Oliveira HM, Polvikoski T, Silsby M, Menon P, van den Bos M, Halliday GM, van den Berg LH, Van Den Bosch L, van Damme P, Kiernan MC, van Es MA, Vucic S. 2020. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry 92:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B, Faure AJ, Seuma M, Schmiedel JM, Tartaglia GG, Lehner B. 2019. The mutational landscape of a prion-like domain. Nat Commun [Internet] 10. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6744496/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz WD, Monsell SE, Schmitt FA, Kukull WA, Nelson PT. 2014. Hippocampal sclerosis of aging is a key Alzheimer’s disease mimic: clinical-pathologic correlations and comparisons with both alzheimer’s disease and non-tauopathic frontotemporal lobar degeneration. J Alzheimers Dis 39:691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Arai K, Del Tredici K, Toledo JB, Robinson JL, Lee EB, Kuwabara S, Shibuya K, Irwin DJ, Fang L, Van Deerlin VM, Elman L, McCluskey L, Ludolph AC, Lee VM-Y, Braak H, Trojanowski JQ. 2014. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol 128:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Dörk T, Zuccato E, Pagani F, Romano M, Baralle FE. 2001. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. The EMBO Journal 20:1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell JR, Halliday GM, Kril JJ, Ittner LM, Götz J, Kiernan MC, Hodges JR. 2016. The frontotemporal dementia-motor neuron disease continuum. Lancet 388:919–931. [DOI] [PubMed] [Google Scholar]
- Butt AM, De La Rocha IC, Rivera A. 2019. Oligodendroglial Cells in Alzheimer’s Disease. Adv Exp Med Biol 1175:325–333. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Magrí A, Oddo S. 2010. Age-dependent changes in TDP-43 levels in a mouse model of Alzheimer disease are linked to Aβ oligomers accumulation. Mol Neurodegener 5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Xiao M. 2016. Oligodendrocytes and Alzheimer’s disease. International Journal of Neuroscience 126:97–104. [DOI] [PubMed] [Google Scholar]
- Correia AS, Patel P, Dutta K, Julien J-P. 2015. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS One [Internet] 10. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4596857/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deora V, Lee JD, Albornoz EA, McAlary L, Jagaraj CJ, Robertson AAB, Atkin JD, Cooper MA, Schroder K, Yerbury JJ, Gordon R, Woodruff TM. 2020. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 68:407–421. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Karran E. 2016. The Cellular Phase of Alzheimer’s Disease. Cell 164:603–615. [DOI] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. 2012. TDP-43 Aggregation In Neurodegeneration: Are Stress Granules The Key? Brain Res 1462:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Baker M, Rademakers R. 2010. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis 7:170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen W-T, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Díaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Götz M, Gutiérrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, Kastanenka KV, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, MacVicar BA, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai KK, Norris CM, Okada S, Oliet SHR, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Pérez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner I-B, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, Sofroniew MV, Verkhratsky A. 2021. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24:312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin C, Guillemaud O, Sauvage M-AC. 2019. Questions and (some) answers on reactive astrocytes. Glia 67:2221–2247. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, Hulihan MM, Kachergus JM, Dächsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier B, Chapon F, Tsuboi Y, Yamada T, Gutmann L, Elibol B, Bhatia KP, Wider C, Vilariño-Güell C, Ross OA, Brown LA, Castanedes-Casey M, Dickson DW, Wszolek ZK. 2009. DCTN1 mutations in Perry syndrome. Nat Genet 41:163–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC, Danzer KM, Weishaupt JH. 2015. TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol 211:897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femminella GD, Harold D, Scott J, Williams J, Edison P, Initiative for the ADN. 2020. The Differential Influence of Immune, Endocytotic, and Lipid Metabolism Genes on Amyloid Deposition and Neurodegeneration in Subjects at Risk of Alzheimer’s Disease. Journal of Alzheimer’s Disease Preprint:1–13. [DOI] [PubMed] [Google Scholar]
- Foerster S, Hill MFE, Franklin RJM. 2019. Diversity in the oligodendrocyte lineage: Plasticity or heterogeneity? Glia 67:1797–1805. [DOI] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee S-H, Haddick PCG, Ngu H, Modrusan Z, Larson JL, Kaminker JS, van der Brug MP, Hansen DV. 2018. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Reports 22:832–847. [DOI] [PubMed] [Google Scholar]
- Gao J, Wang L, Gao C, Arakawa H, Perry G, Wang X. 2020. TDP-43 inhibitory peptide alleviates neurodegeneration and memory loss in an APP transgenic mouse model for Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis 1866:165580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wang L, Huntley ML, Perry G, Wang X. 2018. Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, Cleveland DW. 2019. Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102:339–357.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Rademakers R, Petrucelli L. 2013. TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J Alzheimers Dis 33 Suppl 1:S35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. 2008. TDP-43 A315T Mutation in Familial Motor Neuron Disease. Ann Neurol 63:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Shorter J. 2011. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 5:179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Eisenberg DS, Crowther RA. 2017. Propagation of Tau Aggregates and Neurodegeneration. Annu Rev Neurosci 40:189–210. [DOI] [PubMed] [Google Scholar]
- Gong C-X, Singh TJ, Grundke-Iqbal I, Iqbal K. 1993. Phosphoprotein Phosphatase Activities in Alzheimer Disease Brain. Journal of Neurochemistry 61:921–927. [DOI] [PubMed] [Google Scholar]
- Gopinath A, Collins A, Khoshbouei H, Streit WJ. 2020. Microglia and Other Myeloid Cells in Central Nervous System Health and Disease. J Pharmacol Exp Ther 375:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Chen F, Iqbal K, Gong C-X, Wang X, Liu F. 2017. Transactive response DNA-binding protein 43 (TDP-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. Journal of Biological Chemistry 292:10600–10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wang W, Miao S, Chen F, Wu F, Hu W, Iqbal K, Gong C-X, Liu F. 2018. Protein Phosphatase 1 dephosphorylates TDP-43 and suppresses its function in tau exon 10 inclusion. FEBS Letters 592:402–410. [DOI] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AHM, Kaspar BK. 2011. Astrocytes from Familial and Sporadic ALS Patients are Toxic to Motor Neurons. Nat Biotechnol 29:824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebron M, Chen W, Miessau MJ, Lonskaya I, Moussa CE-H. 2014. Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J Neurochem 129:350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, McManus RM, Latz E. 2018. Inflammasome signalling in brain function and neurodegenerative disease. Nature Reviews Neuroscience 19:610–621. [DOI] [PubMed] [Google Scholar]
- Hergesheimer RC, Chami AA, de Assis DR, Vourc’h P, Andres CR, Corcia P, Lanznaster D, Blasco H. 2019. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: a resolution in sight? Brain 142:1176–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman AM, Khandelwal PJ, Rebeck GW, Moussa CE-H. 2012. Wild Type TDP-43 Induces Neuro-Inflammation and Alters APP Metabolism in Lentiviral Gene Transfer Models. Exp Neurol 235:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman AM, Khandelwal PJ, Stanczyk BB, Rebeck GW, Moussa CE-H. 2011. β-Amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Research 1386:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyburn L, Hebron ML, Smith J, Winston C, Bechara J, Li Z, Lonskaya I, Burns MP, Harris BT, Moussa CE-H. 2016. Tyrosine kinase inhibition reverses TDP-43 effects on synaptic protein expression, astrocytic function and amino acid dis-homeostasis. J Neurochem 139:610–623. [DOI] [PubMed] [Google Scholar]
- Hunter S, Hokkanen SRK, Keage HAD, Fleming J, Minett T, Polvikoski T, Allinson K, Brayne C, the Cambridge City over 75s Cohort collaboration. 2020. TDP-43 Related Neuropathologies and Phosphorylation State: Associations with Age and Clinical Dementia in the Cambridge City over-75s Cohort. Journal of Alzheimer’s Disease 75:337–350. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Ishikawa T, Tanabe H. 2004. Epidemiology of frontotemporal lobar degeneration. Dement Geriatr Cogn Disord 17:265–268. [DOI] [PubMed] [Google Scholar]
- Jo M, Lee S, Jeon Y-M, Kim S, Kwon Y, Kim H-J. 2020. The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp Mol Med 52:1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Dickson DW, Tosakulwong N, Weigand SD, Murray ME, Petrucelli L, Liesinger AM, Senjem ML, Spychalla AJ, Knopman DS, Parisi JE, Petersen RC, Jack CRJ, Whitwell JL. 2017. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: a longitudinal retrospective study. Lancet Neurol 16:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, Rademakers R, Boeve BF, Parisi JE, Smith GE, Ivnik RJ, Petersen RC, Jack CRJ, Dickson DW. 2008. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70:1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, Petrucelli L, Senjem ML, Knopman DS, Boeve BF, Ivnik RJ, Smith GE, Jack CRJ, Parisi JE, Petersen RC, Dickson DW. 2014. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127:811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. 2017. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169:1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Khalfallah Y, Kuta R, Grasmuck C, Prat A, Durham HD, Vande Velde C. 2018. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci Rep [Internet] 8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5953947/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG. 2018. Understanding the Relevance of Aging-Related Tau Astrogliopathy (ARTAG). Neuroglia 1:339–350. [Google Scholar]
- LaClair KD, Donde A, Ling JP, Jeong YH, Chhabra R, Martin LJ, Wong PC. 2016. Depletion of TDP-43 decreases fibril and plaque β-amyloid and exacerbates neurodegeneration in an Alzheimer’s mouse model. Acta Neuropathol 132:859–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. 2009. Rethinking ALS: the FUS about TDP-43. Cell 136:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocca TJ, Mariani A, Watkins LR, Link CD. 2019. TDP-43 knockdown causes innate immune activation via protein kinase R in astrocytes. Neurobiol Dis 132:104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latimer CS, Burke BT, Liachko NF, Currey HN, Kilgore MD, Gibbons LE, Henriksen J, Darvas M, Domoto-Reilly K, Jayadev S, Grabowski TJ, Crane PK, Larson EB, Kraemer BC, Bird TD, Keene CD. 2019. Resistance and resilience to Alzheimer’s disease pathology are associated with reduced cortical pTau and absence of limbic-predominant age-related TDP-43 encephalopathy in a community-based cohort. Acta Neuropathol Commun [Internet] 7. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6556006/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal-Lasarte MM, Franco JM, Labrador-Garrido A, Pozo D, Roodveldt C. 2017. Extracellular TDP-43 aggregates target MAPK/MAK/MRK overlapping kinase (MOK) and trigger caspase-3/IL-18 signaling in microglia. The FASEB Journal 31:2797–2816. [DOI] [PubMed] [Google Scholar]
- Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK, Elman L, Grossman M, Lee VM-Y, Irwin DJ, Van Deerlin VM, Trojanowski JQ. 2017. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 134:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim S, Kang H-Y, Lim HR, Kwon Y, Jo M, Jeon Y-M, Kim SR, Kim K, Ha CM, Lee S, Kim H-J. 2020. The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory response. J Neuroinflammation 17:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang P-W, Pellerin L, Magistretti PJ, Rothstein JD. 2012. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA. 2017. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46:957–967. [DOI] [PubMed] [Google Scholar]
- Ling S-C, Polymenidou M, Cleveland DW. 2013. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 79:416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnerbauer M, Rothhammer V. 2020. Protective Functions of Reactive Astrocytes Following Central Nervous System Insult. Front Immunol 11:573256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. 2008. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol 15:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Yang G, Nonaka T, Arai T, Jia W, Cynader MS. 2013. Reducing TDP-43 aggregation does not prevent its cytotoxicity. Acta Neuropathol Commun 1:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IRA, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM-Y, Trojanowski JQ. 2007. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434. [DOI] [PubMed] [Google Scholar]
- Masaki K, Sonobe Y, Ghadge G, Pytel P, Roos RP. 2019. TDP-43 proteinopathy in Theiler’s murine encephalomyelitis virus infection. PLoS Pathog 15:e1007574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matej R, Tesar A, Rusina R. 2019. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin Biochem 73:26–31. [DOI] [PubMed] [Google Scholar]
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, Tsai L-H. 2017. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep 21:366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H. 2019. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade A, Blurton-Jones M. 2019. Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. Journal of Molecular Biology 431:1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalbano M, McAllen S, Cascio FL, Sengupta U, Garcia S, Bhatt N, Ellsworth A, Heidelman EA, Johnson OD, Doskocil S, Kayed R. 2020. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiology of Disease 146:105130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moszczynski AJ, Harvey M, Fulcher N, de Oliveira C, McCunn P, Donison N, Bartha R, Schmid S, Strong MJ, Volkening K. 2019. Synergistic toxicity in an in vivo model of neurodegeneration through the co-expression of human TDP-43 M337V and tau T175D protein. acta neuropathol commun 7:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. 2018. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun 6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Davis DG, Poduska JW, Patel E, Mendiondo MS, Markesbery WR. 2010. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 20:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C, Coyle-Gilchrist ITS, Chui HC, Fardo DW, Flanagan ME, Halliday G, Hokkanen SRK, Hunter S, Jicha GA, Katsumata Y, Kawas CH, Keene CD, Kovacs GG, Kukull WA, Levey AI, Makkinejad N, Montine TJ, Murayama S, Murray ME, Nag S, Rissman RA, Seeley WW, Sperling RA, White Iii CL, Yu L, Schneider JA. 2019a. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain : a journal of neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Estus S, Abner EL, Parikh I, Malik M, Neltner JH, Ighodaro E, Wang W-X, Wilfred BR, Wang L-S, Kukull WA, Nandakumar K, Farman ML, Poon WW, Corrada MM, Kawas CH, Cribbs DH, Bennett DA, Schneider JA, Larson EB, Crane PK, Valladares O, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Scheff SW, Sonnen JA, Haines JL, Pericak-Vance MA, Mayeux R, Farrer LA, Van Eldik LJ, Horbinski C, Green RC, Gearing M, Poon LW, Kramer PL, Woltjer RL, Montine TJ, Partch AB, Rajic AJ, Richmire K, Monsell SE, Alzheimer’ Disease Genetic Consortium, Schellenberg GD, Fardo DW. 2014. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol 127:825–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Gal Z, Wang W-X, Niedowicz DM, Artiushin SC, Wycoff S, Wei A, Jicha GA, Fardo DW. 2019b. TDP-43 proteinopathy in aging: Associations with risk-associated gene variants and with brain parenchymal thyroid hormone levels. Neurobiol Dis 125:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Wang W-X, Partch AB, Monsell SE, Valladares O, Ellingson SR, Wilfred BR, Naj AC, Wang L-S, Kukull WA, Fardo DW. 2015. Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol 74:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ, Lee VM-Y. 2007. TDP-43-Positive White Matter Pathology in Frontotemporal Lobar Degeneration With Ubiquitin-Positive Inclusions. Journal of Neuropathology & Experimental Neurology 66:177–183. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. [DOI] [PubMed] [Google Scholar]
- Nguyen AT, Wang K, Hu G, Wang X, Miao Z, Azevedo JA, Suh E, Van Deerlin VM, Choi D, Roeder K, Li M, Lee EB. 2020. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol 140:477–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan M, Scott C, Gamarallage MP, Lunn D, Carpenter K, McDonough E, Meyer D, Kaanumalle S, Santamaria-Pang A, Turner MR, Talbot K, Ansorge O. 2020. Quantitative patterns of motor cortex proteinopathy across ALS genotypes. Acta Neuropathol Commun [Internet] 8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7331195/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney NT, Spina S, Miller BL. 2017. Frontotemporal Dementia. Neurol Clin 35:339–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB. 1995. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69:3584–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Jawaid A, Henstridge CM, Valeri A, Merlini M, Robinson JL, Lee EB, Rose J, Appel S, Lee VM-Y, Trojanowski JQ, Spires-Jones T, Schulz PE, Rajendran L. 2017. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 95:297–308.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng AYT, Agrawal I, Ho WY, Yen Y-C, Pinter AJ, Liu J, Phua QXC, Koh KB, Chang J-C, Sanford E, Man JHK, Wong P, Gutmann DH, Tucker-Kellogg G, Ling S-C. 2020. Loss of TDP-43 in astrocytes leads to motor deficits by triggering A1-like reactive phenotype and triglial dysfunction. PNAS 117:29101–29112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Bento-Abreu A, Nonneman A, Haeck W, Staats K, Geelen V, Hersmus N, Küsters B, Van Den Bosch L, Van Damme P, Richardson WD, Robberecht W. 2013. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickford F, Marcus J, Camargo LM, Xiao Q, Graham D, Mo J-R, Burkhardt M, Kulkarni V, Crispino J, Hering H, Hutton M. 2011. Progranulin Is a Chemoattractant for Microglia and Stimulates Their Endocytic Activity. Am J Pathol 178:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz JL, Baltimore D. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J 18:6694–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons AL, Higginbottom A, Cooper-Knock J, Alrafiah A, Alofi E, Kirby J, Shaw PJ, Wood JD, Highley JR. 2020. Oligodendrocyte pathology exceeds axonal pathology in white matter in human amyotrophic lateral sclerosis. The Journal of Pathology 251:262–271. [DOI] [PubMed] [Google Scholar]
- Power MC, Mormino E, Soldan A, James BD, Yu L, Armstrong NM, Bangen KJ, Delano-Wood L, Lamar M, Lim YY, Nudelman K, Zahodne L, Gross AL, Mungas D, Widaman KF, Schneider J. 2018. Combined neuropathological pathways account for age-related risk of dementia. Ann Neurol 84:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri M, Cossu G, Saddi V, Simons EJ, Murgia D, Melis M, Ticca A, Oostra BA, Bonifati V. 2011. Broadening the phenotype of TARDBP mutations: the TARDBP Ala382Thr mutation and Parkinson’s disease in Sardinia. Neurogenetics 12:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford RA, Morsch M, Rayner SL, Cole NJ, Pountney DL, Chung RS. 2015. The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front Cell Neurosci [Internet] 9. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4621294/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, Duong DM, Pennington MW, Lah JJ, Seyfried NT, Levey AI. 2018. Identification and therapeutic modulation of a proinflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener [Internet] 13. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5963076/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratti A, Buratti E. 2016. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. Journal of neurochemistry 138 Suppl:95–111. [DOI] [PubMed] [Google Scholar]
- Rayaprolu S, Fujioka S, Traynor S, Soto-Ortolaza AI, Petrucelli L, Dickson DW, Rademakers R, Boylan KB, Graff-Radford NR, Uitti RJ, Wszolek ZK, Ross OA. 2013. TARDBP mutations in Parkinson’s disease. Parkinsonism Relat Disord 19:312–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JL, Porta S, Garrett FG, Zhang P, Xie SX, Suh E, Van Deerlin VM, Abner EL, Jicha GA, Barber JM, Lee VM-Y, Lee EB, Trojanowski JQ, Nelson PT. 2020. Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain 143:2844–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito ER, Miller JB, Harari O, Cruchaga C, Mihindukulasuriya KA, Kauwe JSK, Bikman BT. 2021. Alzheimer’s disease alters oligodendrocytic glycolytic and ketolytic gene expression. Alzheimer’s & Dementia [Internet] n/a. Available from: 10.1002/alz.12310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Makioka K, Fukuda T, Takatama M, Okamoto K. 2013. Accumulation of phosphorylated TDP-43 in the CNS of a patient with Cockayne syndrome. Neuropathology 33:673–677. [DOI] [PubMed] [Google Scholar]
- Salter MW, Stevens B. 2017. Microglia emerge as central players in brain disease. Nat Med 23:1018–1027. [DOI] [PubMed] [Google Scholar]
- Seeley WW. 2008. Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Curr Opin Neurol 21:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, Moore MJ, Yu G. 2011. Identification of Neuronal RNA Targets of TDP-43-containing Ribonucleoprotein Complexes*♦. Journal of Biological Chemistry 286:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, Burr K, Haghi G, Story D, Nishimura AL, Carrasco MA, Phatnani HP, Shum C, Wilmut I, Maniatis T, Shaw CE, Finkbeiner S, Chandran S. 2013. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci U S A 110:4697–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaerzadeh F, Phan L, Miller D, Dacquel M, Hachmeister W, Hansen C, Bechtle A, Tu D, Martcheva M, Foster TC, Kumar A, Streit WJ, Khoshbouei H. 2020. Microglia senescence occurs in both substantia nigra and ventral tegmental area. Glia 68:2228–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih Y-H, Tu L-H, Chang T-Y, Ganesan K, Chang W-W, Chang P-S, Fang Y-S, Lin Y-T, Jin L-W, Chen Y-R. 2020. TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat Commun [Internet] 11. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7683652/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smethurst P, Risse E, Tyzack GE, Mitchell JS, Taha DM, Chen Y-R, Newcombe J, Collinge J, Sidle K, Patani R. 2020. Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain 143:430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit T, Deshayes NAC, Borchelt DR, Kamphuis W, Middeldorp J, Hol EM. 2021. Reactive astrocytes as treatment targets in Alzheimer’s disease—Systematic review of studies using the APPswePS1dE9 mouse model. Glia [Internet] n/a. Available from: 10.1002/glia.23981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG, Roberts C, Miller KR, Ransohoff RM, Trojanowski JQ, Lee VM-Y. 2018. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci 21:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproviero D, La Salvia S, Giannini M, Crippa V, Gagliardi S, Bernuzzi S, Diamanti L, Ceroni M, Pansarasa O, Poletti A, Cereda C. 2018. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front Neurosci [Internet] 12. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6060258/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan Z, Beach TG, Serrano GE, Hansen DV. 2020. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Reports 31:107843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk TR, Rousseaux MWC. 2020. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener [Internet] 15. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7429473/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svahn AJ, Don EK, Badrock AP, Cole NJ, Graeber MB, Yerbury JJ, Chung R, Morsch M. 2018. Nucleo-cytoplasmic transport of TDP-43 studied in real time: impaired microglia function leads to axonal spreading of TDP-43 in degenerating motor neurons. Acta Neuropathol 136:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup V, Phaneuf D, Dupré N, Petri S, Strong M, Kriz J, Julien J-P. 2011. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB–mediated pathogenic pathways. J Exp Med 208:2429–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift IJ, Bocchetta M, Benotmane H, Woollacott IOC, Shafei R, Rohrer JD. 2021. Variable clinical phenotype in TBK1 mutations: case report of a novel mutation causing primary progressive aphasia and review of the literature. Neurobiology of Aging 99:100.e9–100.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognatta R, Miller RH. 2016. Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology 110:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Hortobágyi T, Nishimura AL, Župunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J. 2011. Characterising the RNA targets and position-dependent splicing regulation by TDP-43; implications for neurodegenerative diseases. Nat Neurosci 14:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X, Li F, Zhou H, Xia X-G. 2013. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J 32:1917–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triviño JJ, von Bernhardi R. 2021. The effect of aged microglia on synaptic impairment and its relevance in neurodegenerative diseases. Neurochem Int 144:104982. [DOI] [PubMed] [Google Scholar]
- Tziortzouda P, Van Den Bosch L, Hirth F. 2021. Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nature Reviews Neuroscience 22:197–208. [DOI] [PubMed] [Google Scholar]
- Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, Miller BL, Kretzschmar HA, Lee VM-Y, Trojanowski JQ, Neumann M. 2008. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67:555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Robberecht W. 2021. STING-Induced Inflammation — A Novel Therapeutic Target in ALS? | NEJM. New England Journal of Medicine [Internet]. Available from: 10.1056/NEJMcibr2031048 [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Sleiman PMA, Martinez-Lage M, Chen-Plotkin A, Wang L-S, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DMA, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell JR, Ghetti B, Spina S, Grafman J, Hodges J, Spillantini MG, Gilman S, Lieberman AP, Kaye JA, Woltjer RL, Bigio EH, Mesulam M, Al-Sarraj S, Troakes C, Rosenberg RN, White CL, Ferrer I, Lladó A, Neumann M, Kretzschmar HA, Hulette CM, Welsh-Bohmer KA, Miller BL, Alzualde A, Lopez de Munain A, McKee AC, Gearing M, Levey AI, Lah JJ, Hardy J, Rohrer JD, Lashley T, Mackenzie IRA, Feldman HH, Hamilton RL, Dekosky ST, van der Zee J, Kumar-Singh S, Van Broeckhoven C, Mayeux R, Vonsattel JPG, Troncoso JC, Kril JJ, Kwok JBJ, Halliday GM, Bird TD, Ince PG, Shaw PJ, Cairns NJ, Morris JC, McLean CA, DeCarli C, Ellis WG, Freeman SH, Frosch MP, Growdon JH, Perl DP, Sano M, Bennett DA, Schneider JA, Beach TG, Reiman EM, Woodruff BK, Cummings J, Vinters HV, Miller CA, Chui HC, Alafuzoff I, Hartikainen P, Seilhean D, Galasko D, Masliah E, Cotman CW, Tuñón MT, Martínez MCC, Munoz DG, Carroll SL, Marson D, Riederer PF, Bogdanovic N, Schellenberg GD, Hakonarson H, et al. 2010. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velebit J, Horvat A, Smolič T, Prpar Mihevc S, Rogelj B, Zorec R, Vardjan N. 2020. Astrocytes with TDP-43 inclusions exhibit reduced noradrenergic cAMP and Ca2+ signaling and dysregulated cell metabolism. Sci Rep [Internet] 10. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7138839/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M. 2018. Physiology of Astroglia. Physiol Rev 98:239–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Daniels CML, Goldman JE, Trojanowski JQ, Lee VM-Y, Messing A. 2014. Astrocytic TDP-43 pathology in Alexander disease. J Neurosci 34:6448–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ho WY, Lim K, Feng J, Tucker-Kellogg G, Nave K-A, Ling S-C. 2018. Cell-autonomous requirement of TDP-43, an ALS/FTD signature protein, for oligodendrocyte survival and myelination. Proc Natl Acad Sci U S A 115:E10941–E10950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, Jiang S, Ma X, Jiang Z, da Rocha EL, Sheng M, Choi H, Lerou PH, Li H, Wang X. 2016. The Inhibition of TDP-43 Mitochondrial Localization Blocks Its Neuronal Toxicity. Nat Med 22:869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Capuano AW, Bennett DA, Schneider JA, Boyle PA. 2016. Temporal course of neurodegenerative effects on cognition in old age. Neuropsychology 30:591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM-Y. 2008. Disturbance of Nuclear and Cytoplasmic TAR DNA-binding Protein (TDP-43) Induces Disease-like Redistribution, Sequestration, and Aggregate Formation. J Biol Chem 283:13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woollacott IOC, Bocchetta M, Sudre CH, Ridha BH, Strand C, Courtney R, Ourselin S, Cardoso MJ, Warren JD, Rossor MN, Revesz T, Fox NC, Holton JL, Lashley T, Rohrer JD. 2018. Pathological correlates of white matter hyperintensities in a case of progranulin mutation associated frontotemporal dementia. Neurocase 24:166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Wang H, Qiao T, Yang B, Aliaga L, Qiu L, Tan W, Salameh J, McKenna-Yasek DM, Smith T, Peng L, Moore MJ, Brown RH, Cai H, Xu Z. 2014. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 111:E1121–E1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C-H, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RRJ, Moecking J, De Nardo D, Balka KR, Calleja DJ, Moghaddas F, Ni E, McLean CA, Samson AL, Tyebji S, Tonkin CJ, Bye CR, Turner BJ, Pepin G, Gantier MP, Rogers KL, McArthur K, Crouch PJ, Masters SL. 2020. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 183:636–649.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA. 2015. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 84:927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Yi Y-S, Yang Y, Oh J, Jeong D, Cho JY. 2012. The Pivotal Role of TBK1 in Inflammatory Responses Mediated by Macrophages. Mediators Inflamm [Internet] 2012. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3523167/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio F, Loon AR, Smeltzer S, Benyamine K, Navalpur Shanmugam NK, Stewart NJF, Lee DC, Nash K, Selenica M-LB. 2020. TDP-43 mediated blood-brain barrier permeability and leukocyte infiltration promote neurodegeneration in a low-grade systemic inflammation mouse model. J Neuroinflammation 17:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Kishimoto Y, Grammatikakis loannis, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Sen JM, Gorospe M, Mattson MP. 2019. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci 22:719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Bell S, Wang J, Wen S, Baloh RH, Appel SH. 2015. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Experimental Neurology 273:24–35. [DOI] [PubMed] [Google Scholar]