

Abstract
Mendelian disorders with cutaneous manifestations comprise a phenotypically heterogeneous group of over 1,000 diseases, and in the majority of them mutant genes have been identified. Mutation detection approaches in these diseases have largely focused on DNA analysis by next-generation sequencing techniques (NGS), including gene-targeted sequencing panels as well as whole-exome and whole-genome sequencing. Genome-wide homozygosity mapping, based on DNA polymorphism, has also assisted in identification of candidate genes in families with consanguinity. However, specific pathogenic variants have not been disclosed in many individual patients when analyzed by NGS, and in particular, DNA-based analysis failed to identify many of the mutations impacting on splicing or gene expression. Whole-transcriptome sequencing by RNA-Seq, with appropriate bioinformatics, provides a robust tool to identify additional mutations to facilitate genetic diagnosis in genodermatoses. RNA-Seq can be used for variant calling and homozygosity mapping similar to DNA-based approaches, but it also allows identification of mutations which result in aberrant transcriptome expression as quantitated by heatmap analysis and altered splicing patterns of RNA as visualized by Sashimi plots. Thus, “clinical RNA-Seq” extends molecular diagnostics of rare genodermatoses, and it could provide a reliable first-tier diagnostic approach to extend mutation databases in patients with heritable skin diseases.
Keywords: gene expression, mutation analysis, RNA-Seq, transcriptome profiling, monogenic genodermatoses, epidermolysis bullosa, ichthyosis
INTRODUCTION
Monogenic heritable diseases comprise a highly heterogeneous group of as many as 10,000 disorders, and some 1,000 of them have cutaneous manifestations (OMIM: http://www.OMIM.org; World Health Organization:https://www.who.int/genomics/public/geneticdiseases/en/index2.html). In some diseases, the manifestations are present only in the skin, thus being non-syndromic, while in some cases the cutaneous manifestations are associated with a number of extracutaneous manifestations being syndromic (Vahidnezhad et al., 2019c). The genetic diagnosis has been established in some of these diseases, many being caused by mutations in several distinct genes. Identification of mutated genes and the specific mutations has greatly advanced our understanding of the pathomechanisms of these, often complex disorders, and such information can be used for confirmation of the diagnosis with subclassification and prognostication. Such rare monogenic disorders can also serve as simplified models for better understanding of common multi-system disorders (Youssefian et al., 2019a). Knowledge of the genetic defect also forms the basis for prenatal testing and preimplantation genetic diagnosis. Furthermore, importantly, the information of the specific mutations is required for potential application of allele-specific treatment approaches that are currently in the developmental pipeline and some of which are already in early clinical trials for heritable skin diseases (Has et al., 2020).
Conventional mutation detection strategies have focused on DNA-based analyses, including next-generation sequencing (NGS) in the form of gene targeted arrays or whole-exome sequencing (WES) and whole-genome sequencing (WGS) (Bamshad et al., 2011; Yang et al., 2013; Adams and Eng, 2018). Information derived from these genomic approaches has also been used for homozygosity mapping (HM) to identify putative candidate genes in consanguineous families (Vahidnezhad et al., 2018a; Vahidnezhad et al., 2019d). However, less than 50% of all cases received genetic diagnosis by DNA-based sequencing techniques as the DNA analysis does not capture many of the disease-causing variants located in the noncoding regions of the genes or because the DNA analysis may overlook the consequences of certain types of mutations (Wright et al., 2018). The latter situation is exemplified by synonymous or silent nucleotide substitutions in exons or in non-canonical splicing sequences both impacting on splicing. In such cases, whole-transcriptome sequencing by RNA-Seq, with appropriate bioinformatics analysis steps, provides a complementary tool to identify additional mutations to facilitate genetic diagnosis of genodermatoses.
RNA-Seq is a multifaceted technique which can be used for variant calling and HM, similar to DNA-based approaches. In addition, RNA-Seq is a powerful tool to identify mutant genes with aberrant expression and perturbed splicing patterns (Figure 1). The importance of this ability of RNA-Seq to identify pathogenic sequence variants is emphasized by the fact that (a) splicing defects are among the major causes of Mendelian disorders and they can be located either deeply in intronic sequences, not captured by WES, or as silent variants inside the exons (Chmel et al., 2015); and (b) pathogenic mutations leading to premature termination codon of translation can cause dramatically reduced gene expression at the mRNA level (Kremer et al., 2017; Fresard et al., 2019). Thus, RNA-Seq can be utilized for interrogation of culprit genes. Furthermore, filtering of the WES results by frequencies is highly efficient for coding sequences of the gene but does not capture intronic or intergenic variants. Some regions in the genome are difficult to sequence, and RNA-Seq can be helpful to find the causative variants in those regions that are not well characterized by DNA-based genome sequencing methods. Collectively, these observations argue that “clinical RNA-Seq” is a powerful tool to facilitate and extend diagnostics of rare sequence variants in genodermatoses. We recognize that the whole-transcriptome sequencing by RNA-Seq with the bioinformatics pipeline is a complex process requiring special expertise and equipment. Therefore, the primary purpose of this summary is to familiarize noncognoscenti to the principles of this technique, and alert the readers of the availability of these contemporary approaches for mutation detection in heritable skin diseases. Also, to enhance readability of this review, a Glossary is enclosed.
Figure 1. RNA-Seq technique workflow, the bioinformatics analysis steps, and potential applications of the information.
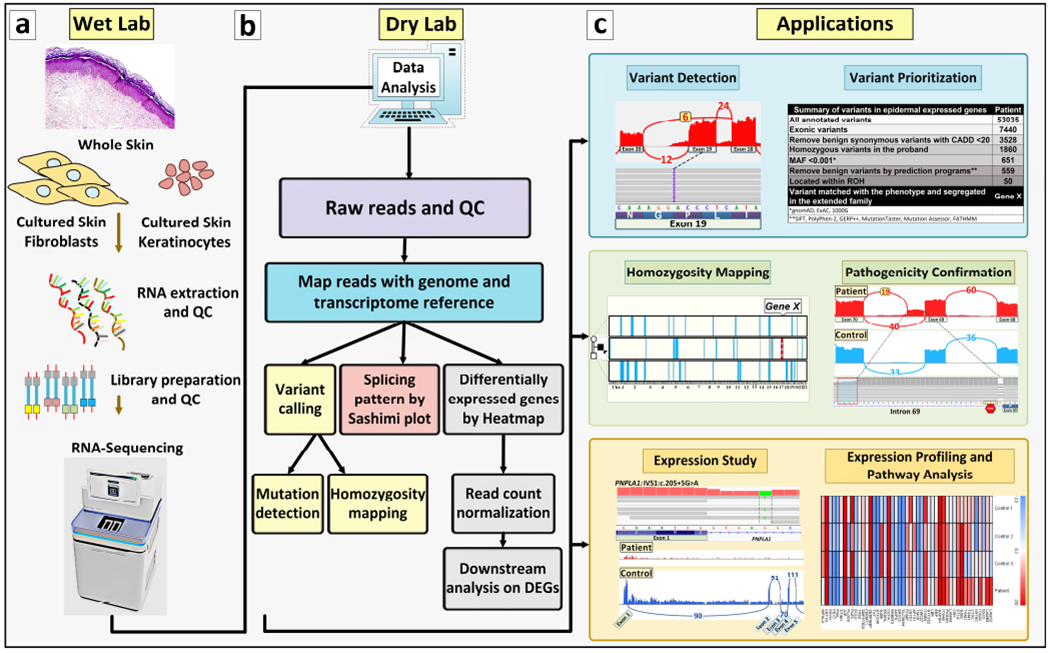
(a, Wet Lab) RNA is extracted from whole skin biopsies, dermal fibroblasts or epidermal keratinocytes and is then sequenced according to protocols and platforms, such as Illumina TruSeq or Takara SMARTer library preparation. The Fastq file will be analyzed by bioinformatics pipelines. (b, Dry Lab) The workflow of bioinformatics analysis includes the steps in the diagram. For details see Figure 2 where the corresponding steps are color coded for clarity. For details of the software packages and databases, see Table 1. (c, Applications) Six potential applications of the RNA-Seq technique are shown; they include variant calling, variant prioritization, homozygosity mapping, validation of variants of unknown significance in the genome, qualitative and quantitative gene expression analysis and differential gene expression/Ingenuity Pathway Analysis (IPA).
RNA-Seq: Work flow and bioinformatics
RNA-Seq is initiated by isolation of RNA from tissues or cells that actively express their genome. In case of skin, RNA can be isolated from a relatively small (3 mm) whole skin biopsy or from cultured cells, such as dermal fibroblasts and epidermal keratinocytes (Figure 1). In this context, the selection of correct cell types is important depending of their gene expression profile. For example, many of the genes involved in cutaneous blistering or cornification disorders are expressed in keratinocytes but not in fibroblasts. Also, it is important to isolate RNA by procedures that preserve the quality of RNA to ensure high quality sequence reads (Vahidnezhad et al., 2020). The biopsies can be transferred to the laboratory in RNA stabilization solution, such as RNAlater™ transport medium, in which RNA is stable at least one week at room temperature, one month at 4°C, and several months at −20°C. Conveniently, there is no need to freeze samples in liquid nitrogen or rush the samples to the laboratory freezer. RNA is then subjected to sequencing initially synthesizing a cDNA library dedicated to RNA-Seq; this approach is different from the conventional cDNA synthesis for Sanger sequencing. RNA-Seq then provides data for bioinformatics analysis, and the different steps of data analysis, as shown in Figure 1, consist of mapping the raw data reads to genome and transcriptome references, alignment of the reads for variant calling and callset refinement, annotation of the variants for HM, as well as counting and normalization of the reads (Figure 1.b). Details of this stepwise process for filtering, including information on bioinformatics softwares and packages and the output files, are shown in Figure 2. (For details of the software packages and databases used for data analysis, variant calling and differentially expressed gene (DEG) analysis, see Table 1). The information on these endpoints, viz. variant detection and prioritization as well as HM, is complementary to the information provided by DNA-based techniques. Filtering of the annotated variants for prioritization assists in identification of candidate genes by first focusing on exonic sequence variants and removal of benign synonymous variants with Combined Annotation Dependent Depletion (CADD) score <20. In case of rare heritable diseases, filtering to include variants with minor allele frequency (MAF) of <0.001, and removal of benign variants by bioinformatics prediction programs, followed by alignment of the candidate genes harboring homozygous sequence variants with runs of homozygosity (ROHs), can significantly reduce the number of variants to be considered as pathogenic. As an example, as shown in Figure 1.c (Variant Prioritization), this approach allowed to reduce the number of variants under consideration from 53,035 to 50. The remaining variants, when matched with the clinical phenotypes, identified a single gene (Gene X) as a candidate gene which was further confirmed by segregation analysis in the family and by modeling of the consequences of the mutation at protein levels, and further corroborated by datasets at mRNA level derived from RNA-seq.
Figure 2. The flowchart of bioinformatics analysis.
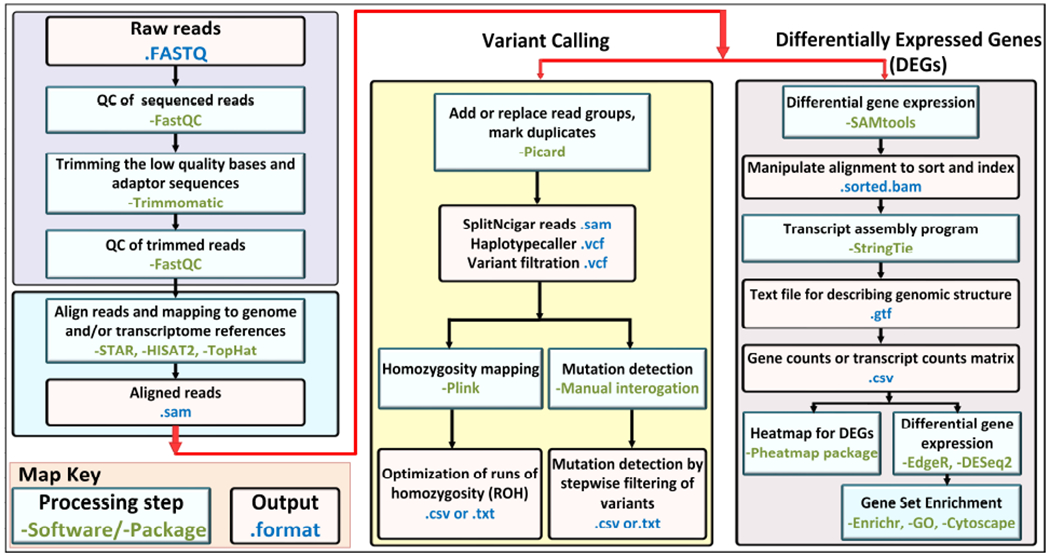
For quality control, the raw FASTQ files are checked by FastQC software, and after trimming the low-quality sequences, FASTQ files are mapped to the genome and/or transcriptome reference sequences by programs such as STAR, HISAT2 and TopHat. The aligned output files are then analyzed by two approaches for variant calling or for differentially expressed genes (DEGs). In variant calling by GATK pipelines (Picard program), the duplicate reads are marked and the variants are called and filtered with HaplotypeCaller. In the next step, the .vcf output file is annotated with ANNOVAR software for mutation detection, and for homozygosity mapping the Plink software is applied. The second approach for differentially expressed genes utilizes SAMtools (Sequence Alignment/Map) for alignment, sorting reads, and indexing, followed by transcript assembly program, such as StringTie, for output file of gene/transcript counts to be used for Heatmap analysis or Differential Gene Expression/Gene Set Enrichment analysis.
Table 1:
Description of computer software and online tools for bioinformatics analyses of NGS data and pathway analysis
| Software | Description and purpose | URL | References |
|---|---|---|---|
| BWA | A package used to map read sequences to a reference genome. | http://bio-bwa.sourceforse.net/ | (Li and Durbin, 2009) |
| GATK | A multi-purpose variant discovery and genotyping tool. | https://software.broadinstitute.org/gatk/ | (McKenna et al., 2010) |
| Picard | Tools used for manipulating sequencing data in different formats such as BAM and VCF files. | http://broadinstitute.github.io/picard | Refer to corresponding URL |
| PLINK | Tools used for whole genome association studies. It can be used for homozygosity mapping and IBD estimation. | http://zzz.bwh.harvard.edu/plink/ | (Purcell et al., 2007) |
| R | A general-purpose software environment for statistical data analysis and visualization. | https://www.r-project.org/ | Refer to corresponding URL |
| SAMtools | Tools can be used for manipulating SAM/BAM formats. | http://samtools.sourceforge.net/ | (Li et al., 2009) |
| FastQC | A quality control tool for high throughput sequence data. | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | (Andrews, 2010) |
| Trimmomatic | A flexible read trimming tool for Illumina NGS data. | http://www.usadellab.org/cms/?page=trimmomatic | (Bolger et al., 2014) |
| STAR | An aligner designed to specifically address many of the challenges of RNA-seq data mapping using a strategy to account for spliced alignments. | https://github.com/alexdobin/STAR/releases | (Dobin et al., 2013) |
| Hisat2 | A fast and sensitive alignment program for mapping next-generation sequencing reads (both DNA and RNA) to a population of human genomes. | http://ccb.jhu.edu/software/hisat2/index.shtml | (Kim et al., 2019) |
| TopHat2 | A fast splice junction mapper for RNA-Seq reads. | https://ccb.jhu.edu/software/tophat/index.shtml | (Kim et al., 2013) |
| StringTie | A fast and highly efficient assembler of RNA-Seq alignments into potential transcripts. | https://ccb.jhu.edu/software/stringtie/ | (Kovaka et al., 2019) |
| Pheatmap package | A useful R package that implement a heatmaps that offers more control over dimensions and appearance. | https://CRAN.R-project.org/package=pheatmap | Refer to corresponding URL |
| EdgeR | A Bioconductor package for differential expression analysis of digital gene expression data. | https://bioconductor.org/packages/edgeR/ | (Robinson et al., 2010) |
| DESeq2 | A Bioconductor package that provides methods to test for differential expression by use of negative binomial generalized linear models | http://bioconductor.org/packages/release/bioc/html/DESeq2.html | (Love et al., 2014) |
| Enrichr | An easy to use intuitive enrichment analysis web-based tool providing various types of visualization summaries of collective functions of gene lists | https://amp.pharm.mssm.edu/Enrichr/ | (Kuleshov et al., 2016) |
| GO | An up-to-date and useful database for enrichment and pathway analysis using RNA-Seq data | http://geneontology.org/ | (Ashburner et al., 2000) |
| Cytoscape | An open source software platform for visualizing complex networks and integrating these with any type of attribute data | https://cytoscape.org/ | (Shannon et al., 2003) |
As noted in the variant prioritization flowchart (Figure 1.c), this variant calling would remove synonymous variants which could result in aberrant splicing, including those residing at the very end of exons at the exon-intron border within a canonical splice site sequence. The confirmation of the pathogenicity resulting in aberrant splicing can be facilitated by RNA-Seq visualized by Sashimi plots demonstrating the pattern of splicing in qualitative and quantitative terms (Figure 1.c). As a consequence of such splice junction mutations, several studies have demonstrated exon skipping, partial or complete intron retention, utilization of alternative splice sequences which are predicted to lead to alterations in translation, with synthesis of defective protein or absence of the protein expression (Kremer et al., 2017). RNA sequencing utilizing Sashimi plots can also demonstrate lack of gene expression at the mRNA level in the case of promoter mutations or large gene deletions. Differentially expressed genes (DEGs) can be quantitated from transcriptome data by heatmap visualization of the expression profiles which can then direct the attention to the most likely pathogenic gene variants among those under consideration (Figure 1.c). It should be noted that gene expression levels in cultured cells can be affected by a number of factors, and analysis of skin biopsies may more accurately reflect the expression profiles in situ.
Examples of the utility of RNA-Seq in facilitating the identification of mutated genes
Over the past decade, our laboratory has focused on mutation detection in large cohorts of heritable skin disorders, including epidermolysis bullosa (EB), Mendelian disorders of cornification (ichthyosis and keratodermas). Pseudoxanthoma elasticum, and more recently epidermodysplasia verruciformis (EV). In each case, DNA and RNA was isolated after obtaining a written, informed consent by the patient, his/her parents or guardians who also consented to the publication of the patient’s images. (These studies were approved by the Institutional Review Boards of the Pasteur Institute of Tehran and Thomas Jefferson University, Philadelphia, PA).
Case 1:
A 1.5-year-old patient with scaly skin, with clinical features consistent with lamellar ichthyosis (Figure 3.a), was analyzed for the underlying genetic mutations by WES, which however was inconclusive (Youssefian et al., 2019b). Subsequently, RNA-Seq of the patient’s skin biopsy revealed a homozygous G>A substitution in the last nucleotide of exon 10 of the TGM1 gene within the canonical splice site sequence which changed the wild-type AG-gt to AA-gt (Figure 3.b). Since this nucleotide substitution (GAG>GAA) did not change the corresponding amino acid, both codons encoding glutamic acid in transglutaminase 1 protein, conventional bioinformatics filtering of DNA data overlooked this variant and did not prioritize it as a pathogenic variant. However, Sashimi plot of the RNA-Seq data revealed that this nucleotide change abolished the canonical splice site at exon 10/intron 10 border, and instead, the patient’s mRNA was processed from two alternate splice sites, the major one residing within intron 10 and the other one within exon 10 resulting in partial retention of intron 10 sequences (Figure 3.b). These altered transcripts were predicted to result in frameshift and abolish the synthesis of functional, full-length transglutaminase 1, explaining the patient’s phenotype. Interestingly, heatmap analysis of the patient’s RNA transcripts, as compared to average transcript levels in three controls, revealed that among the 57 tested genes known to be associated with ichthyosis phenotypes, the level of expression of TGM1 was the lowest (Figure 3.c). This is apparently a reflection of the nonsense-mediated mRNA decay as a result of the splicing mutation disclosed by RNA-Seq.
Figure 3. Examples of the utility of RNA-Seq in mutation detection in challenging cases of heritable skin diseases.
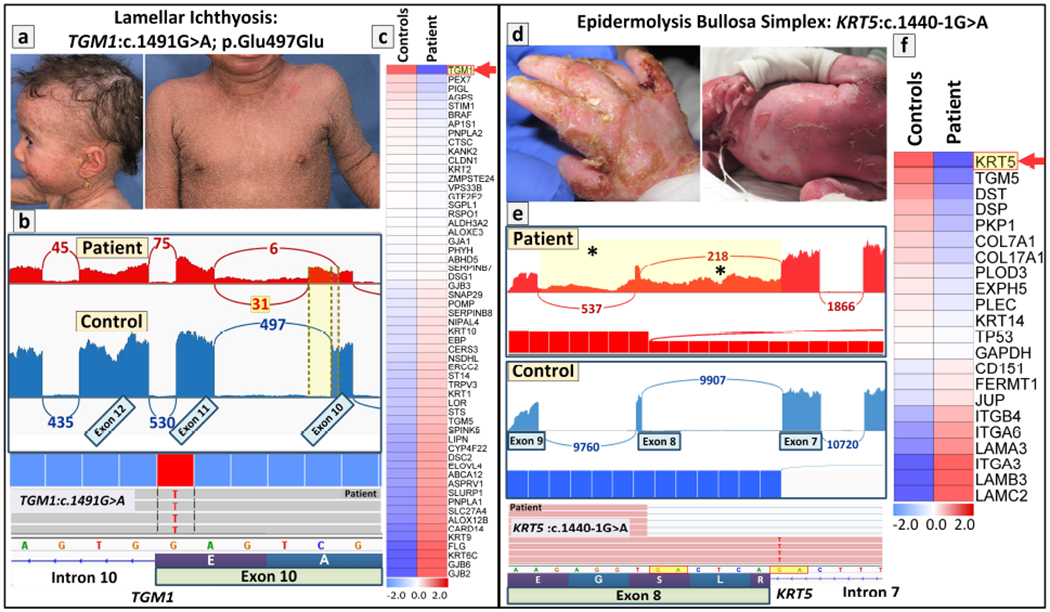
(left panel, a-c) Identification and confirmation of a homozygous synonymous/splice-site mutation in TGM1 in a 1.5-year-old patient with lamellar ichthyosis. (a) The patient manifested with scaly skin, sparse hair and mild erythroderma consistent with diagnosis of lamellar ichthyosis. (b) Sashimi plot of RNA-Seq revealed that a homozygous synonymous mutation results in aberrant splicing and partial intron retention (red), as compared to splicing in control RNA (blue) (upper panel). Screen shot of the genomic sequence visualized by the Integrative Genomics Viewer (IGV) demonstrating a homozygous mutations in TGM1: c.1491G>A at the border of exon10/intron10 (lower panel). (c) Heatmap analysis of the patient’s RNA in comparison to the average quantity of three healthy controls showed that TGM1 had the lowest level of gene expression. (right panel, d-f) Identification and confirmation of a canonical splice-site mutation in KRT5 in a neonate with epidermolysis bullosa simplex (EBS). (d) The neonate with severe generalized EBS presented with fragile skin and mucosa. Milia was present at one week of life. The patient died shortly after birth. (e) Sashimi plot of the transcriptome profile of the mutant KRT5 RNA revealed complex aberrant splicing due to the canonical splice-site mutation of c.1440-1G>A that results in retention of intron 7 and intron 8 sequences in patient (red, asterisks in yellow highlighted area) as compared to splicing in control RNA (blue). Screenshot of the genomic sequence visualized by IGV demonstrating canonical splice-site mutation of KRT5: c.1440-1G>A at the border of exon8/intron7 (lower panel). (f) Differential gene expression of 21 genes associated with blistering phenotype by Heatmap analysis revealed that KRT5 was the most downregulated gene among those associated with EB. Permission to publish the patients’ images was provided by the patient and/or his/her parents (guardians).
Case 2:
The utility of heatmap analysis as a tool to guide in calling of pathogenic variants by RNA-Seq was further illustrated in a neonate with clinical diagnosis consistent with a lethal form of EB (Figure 3.d) (Vahidnezhad et al., 2019a). Analysis of the transcriptome data from RNA-Seq revealed a G>A transition in position −1 of intron 7 just preceding exon 8 in the KRT5 gene (Figure 3.e). Sashimi plot revealed that this homozygous mutation rendered the canonical splice site at intron 7/exon 8 border nonfunctional, and instead an AG sequence six nucleotides upstream within exon 8 was used as an alternate acceptor splice site and lead to retention of intron 7 and 8 sequences in the patient’s cells. Heatmap analysis of 21 genes associated with the blistering phenotype in EB revealed that KRT5 expression was the most downregulated among all these genes (Figure 3.f). Thus, prioritization of the candidate genes was clearly facilitated by quantitative assessment of the transcript levels by heatmap analysis, further supporting the notion of pathogenicity of the sequence variant in KRT5.
Case 3:
The utility of RNA-Seq in confirming the pathogenic consequences of nucleotide changes identified by WES is further illustrated in a 3-year-old patient with EB blistering phenotype (Figure 4, a–c) (Vahidnezhad et al., 2018b). Initial DNA sequencing with an EB-associated gene panel of 21 genes revealed two homozygous mutations in this proband from a consanguineous family. One of the mutations, c.5422C>T, in the EXPH5 gene resulted in a nonsense codon p.Arg1808Ter and was predicted to result in synthesis of truncated exophilin 5 protein and nonsense-mediated RNA decay (Figure 4.a). The second mutation in the COL17A1 gene was a homozygous delA which was predicted to result in a frameshift and termination codon 106 nucleotides downstream of the site of deletion. The mutation in the COL17A1 was indeed shown to result in aberrant splicing, and Sashimi plot revealed partial skipping of exon 4 as well as exon 6, with retention of intron 5 sequences (asterisks in Figure 4.b). Heatmap analysis of these two genes placed them on the top of the three most downregulated genes among the 21 genes known to be associated with skin fragility in the spectrum of EB (Figure 4.c).
Figure 4. Benefits and limitations of RNA-Seq in molecular diagnostic settings in confirmation of the pathogenic consequences of mutations.
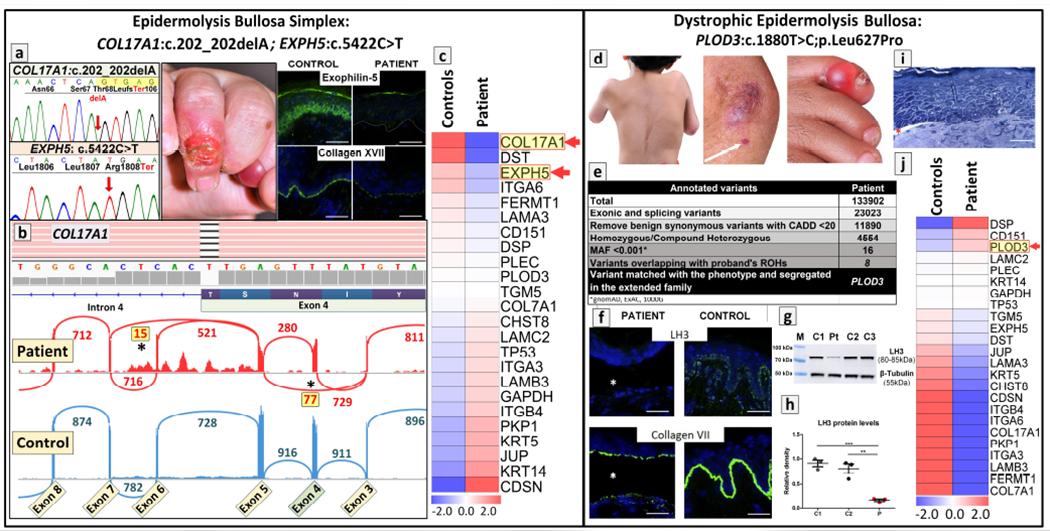
(left panel, a-c) Identification of double homozygous mutations in two distinct genes, EXPH5 and COL17A1, in a patient with epidermolysis bullosa. (a) Next generation sequencing panel of 21 genes identified homozygous mutations in COL17A1 and EXPH5 which were confirmed by Sanger sequencing. Clinical findings in the proband at 11 months of age consisted of blistering and erosions in the fingers. Immunofluorescence staining for exophilin 5 and type XVII collagen demonstrated complete absence (exophilin 5) and/or markedly attenuated and discontinuous pattern (type XVII collagen) in the patient’s skin, as compared to the control skin. (b) Screenshot of the genomic sequence visualized by IGV demonstrating deletion of a nucleotide within exon 4 of COL17A1: c.202_202delA; this frameshift mutation was predicted to result in synthesis of truncated polypeptide (p.Thr68LeufsTer106). Sashimi plot of RNA-Seq revealed complex aberrant splicing due to this frameshift mutation. The donor splice site at the 3′ -end of exon 3 of COL17A1 utilized the acceptor splice site at the intron 3/exon 4 border resulting in partial skipping of exon 4 and 6 (asterisks). (c) Heatmap analysis of the patient’s RNA in comparison to the average quantity of three healthy controls showed that COL17A1 and EXPH5 genes were among the top three most downregulated among the 21 skin fragility-associated genes. (Adopted from Vahidnezhad et al., 2019d, with permission). (right panel, d-j) A missense variant in a novel candidate gene PLOD3 as the cause of syndromic EB identified by WES did not alter transcriptome profiling. (d) The proband manifested with severe scoliosis, joint contractures, and the presence of a tense, hemorrhagic blister on the fifth toe and small erosions on the arm (white arrow) with atrophic scarring at the elbow. (e) Whole exome sequencing identified a total of ~134,000 annotated sequence variants which were filtered by steps indicated to yield 8 variants with minor allele frequency (MAF) of <1:1000 and residing within regions of homozygosity. Matching of the patient’s phenotype identified a mutation in PLOD3, encoding lysyl hydroxylase-3 (LH3). (f) Immunofluorescence reveals complete absence of LH3 in the patient’s skin with a blister (asterisk), while in the control skin, a punctate pattern at the dermal-epidermal junction is noted. Type VII collagen expression is significantly reduced, the remaining protein being primarily in the roof of the blister, as compared to the control skin. (g) Western blotting revealed markedly reduced LH3 protein levels in fibroblasts cultured from the skin of the patient (Pt) as compared to controls (C1–3). Re-probing the filter with an anti-β-tubulin antibody revealed equal protein loading. (h) The LH3 protein levels were quantitated by scanning the bands in Western blots in three separate experiments (mean ± SEM; **p<0.01, ***p<0.001). (i) Histopathology revealed separation at the dermal-epidermal junction (Richardson’s stain). (j) Differential gene expression of 21 genes associated with blistering phenotype by heatmap analysis revealed that PLOD3 gene expression was among the top most expressed genes. Scale bar = 50μm. Permission to publish the patients’ images was provided by the patient and/or his/her parents (guardians) (Adopted from Vahidnezhad et al., 2019c, with permission).
It is of interest to note that EXPH5 and COL17A1 are associated with two different subtypes of EB, viz. simplex and junctional, respectively. In support of pathogenicity of both mutations, the corresponding genes were both located inside of ROHs derived from RNA-Seq data. The combination of these mutations in this consanguineous family explains the blended phenotype which initially made subclassification on the clinical basis difficult. This case also illustrates the importance of genome-wide approaches for mutation detection. Specifically, if methodologies which sequence candidate genes one at a time would have been applied, one of the mutations would probably have been missed. This clearly would have impacted on the ability to provide accurate genetic counseling to the family in terms of risk of inheritance and its use for prenatal testing and preimplantation genetic diagnosis.
As illustrated above, RNA-Seq provides a robust tool to explore pathogenicity of heritable skin diseases by identifying variants at both non-coding and coding regions of the gene. RNA-Seq allows prioritization and interpretation of identified sequence variants and provides information complementary to DNA sequencing by transcriptome analysis at the level of RNA splicing and expression. In this regard, uncovering transcriptional consequences of genetic variants allows prioritization or identifies variants that were missed by the applied filters in the bioinformatics pipeline when analyzing DNA derived data. One of the considerations for RNA-Seq is also its ability to detect genes with mono-allelic expression in search of causal variants, particularly for diseases with recessive mode of inheritance that would not be captured by a single heterozygous variant identified by WES or WGS. Thus, RNA-Seq provides substantial potential to reliably identify pathogenic variants in both known and new disease genes.
Case 4:
This presentation highlights the utility of quantitative transcriptome sequencing as visualized by heatmap analysis in identifying candidate genes with particular emphasis on splice junction variants. It is prudent to point out that these approaches examining the transcriptome levels may not be applicable to missense mutations. As an example, a 4.5-year-old patient with complex connective tissue disorder, including a syndromic form of dystrophic EB, was shown to harbor a missense mutation, c.1880T>C; p.Leu627Pro, in the PLOD3 gene encoding lysyl hydroxylase-3 (LH3), a critical enzyme for post-translational modification of collagens (Figure 4, d–j) (Vahidnezhad et al., 2019b). The study demonstrated that this missense mutation caused absence of LH3 at the protein level but also resulted in reduced collagen VII expression and reduction in anchoring fibrils, apparently due to deficient post-translational modification of collagen VII. While the primary genetic defect was clearly shown to be in PLOD3, the heatmap profile showed that this gene was expressed among the top three highest levels amongst the genes associated with skin fragility in the spectrum of EB. While the reason for upregulation of PLOD3 gene expression is not entirely clear, it may reflect compensatory changes in mRNA levels in response to absent protein (Figure 4.j). Thus, transcript quantitation of DEGs by heatmap in this case of homozygous missense mutations was not helpful in prioritizing or identifying mutant genes.
RNA-Seq reveals an unexpected splicing event in COL7A1
Transcriptome profiling is also able to evaluate tissue and cell-type dependent expression and splicing profiles using the corresponding material as a source of RNA selected at the beginning of the analysis. This consideration highlights one of the limitations of RNA-Seq which requires tissues or cells which express the corresponding genes as starting material. In this context, it is important to note that whole skin biopsy consists not only of epidermal keratinocytes and dermal fibroblasts, but also has a large number of other cell types with unique expression profiles. This issue is highlighted by the demonstration that COL7A1 which encodes type VII collagen was shown to have different splicing pattern in RNA isolated from whole skin as compared to cultured dermal fibroblasts (Figure 5.a). Specifically, Sashimi plot revealed in control skin alternative splicing which was generated by a different exon 18 acceptor site 27 bp upstream from the canonical acceptor site as compared to some, but not all, fibroblast cultures (Figure 5.a). This resulted in insertion of 9 amino acid residues into the fibronectin type III linker domain of the non-collagenous NC-1 region of type VII collagen (Figure 5.b). This splice variant has been previously shown to be differentially expressed in wound edges of patients with epithelizing skin ulcers in patients with EB as compared with normal keratinocytes from steady-state body sites (Figure 5.c) (Sawamura et al., 2003). Our data demonstrating differential expression of this alternative spliced insertion in keratinocytes and fibroblasts provide further evidence of the importance of splice variants in disease processes, and such variants can be visualized by RNA-Seq. This demonstration also emphasized previous observations that cell culture conditions can profoundly alter the gene expression profiles, and for example, the expression levels and transcriptome profiles of fibroblasts in culture may not necessarily reflect those in intact skin (Mahmoudi et al., 2019).
Figure 5. Confirmation of alternative splicing in COL7A1.
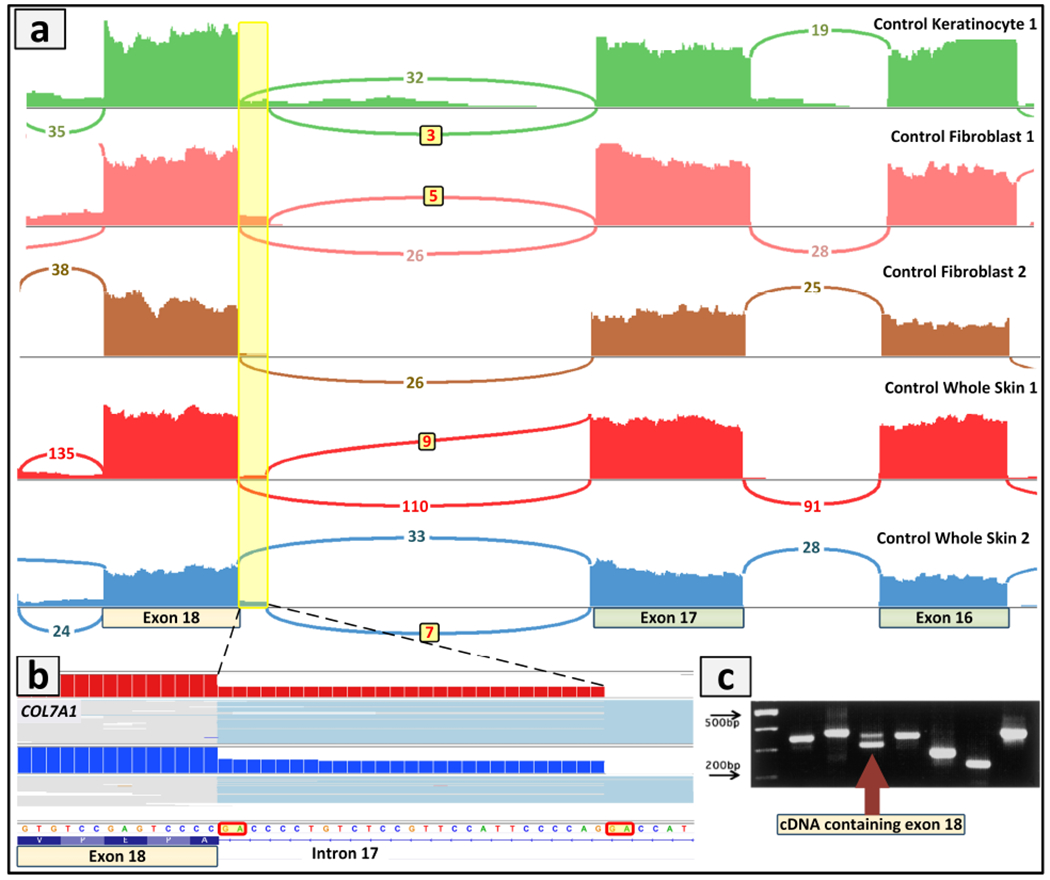
(a) Comparison of COL7A1 gene expression in different sample types, including normal keratinocytes, fibroblasts and whole skin biopsies. The Sashimi plot of RNA-Seq revealed in control skin alternative splicing which was generated by a different exon18 acceptor site 27 bp upstream from the canonical acceptor site as compared to some of the fibroblast cultures. (b) Screenshot of the genomic sequence visualized by IGV demonstrating the 27 bp retention from intron 17 that would result in insertion of 9 amino acid residues into the fibronectin type III linker domain of the non-collagenous NC-1 region of type VII collagen. (c) This alternative splicing has been previously shown to be differentially expressed in wounds of EB patients as compared to normal keratinocytes. The cDNA containing exon 18 and with the 27 bp intron 17 retention is shown by arrow (lower band, exon 18; upper band, exon 18 plus 9 amino acids). (Adopted from Sawamura et al., 2003, with permission).
Clinical utility of transcriptome profiling by RNA-Seq
RNA sequencing is increasingly becoming a complementary way of identifying genes underlying rare heritable diseases bypassing hurdles in interpreting intronic or splice altering variants (Li et al., 2018). It has been indicated that the genetic diagnostic rate for Mendelian diseases by WES is typically only in the range of 20-40% in part due to the fact that WES misses deep intronic, silent or synonymous exonic variants leading to aberrant splicing, and the power of RNA sequencing in solving unrecognized pathogenic variants (Hamanaka et al., 2019) has been demonstrated in studies on a number of heritable skin diseases; some of them were highlighted in this review with the focus on EB and ichthyosis, two heterogeneous disorders which have been associated with 21 and over 67 mutant genes, respectively. Finally, it should be noted that while immunohistochemical/fluorescent staining of the skin samples is often informative and indicates the candidate gene through lack of immunoreactivity for a specific protein, however, this approach does not provide information of the specific mutations (Chmel et al., 2015; He et al., 2016)
Concluding remarks
The feasibility of RNA-Seq applications for heritable skin diseases is emphasized by the ready availability of skin biopsies or the capability to culture epidermal keratinocytes or dermal fibroblasts for RNA isolation. This situation contrasts with many heritable disorders, such as those manifesting with neurologic or internal organ involvement. While transcriptome sequencing has been recognized as complementary to DNA-based next generation sequencing approaches, one could argue that RNA-sequencing, capable of variant calling and HM analogous to DNA-based analysis, with additional capability to reveal aberrant gene splicing and determine the gene expression levels visualized by heatmapping, could be considered as an expedient, reliable, and affordable first-tier diagnostic approach for finding mutations in patients with heritable skin diseases.
Supplementary Material
ACKNOWLEDGEMENTS
Correspondence regarding the technical aspects of this study should be addressed to HV, while those addressing the clinical application should be directed to JU. The authors’ original studies were supported by National Institutes of Health and DEBRA International. Carol Kelly assisted in manuscript preparation. This study is in partial fulfilment of the PhD Thesis of Amir Hossein Saeidian.
Abbreviations:
- NGS
next generation sequencing
- WES
whole-exome sequencing
- WGS
whole-genome sequencing
- HM
homozygosity mapping
- ROH
run of homozygosity
- EB
epidermolysis bullosa
- CADD
Combined Annotation Dependent Depletion
- MAF
minor allele frequency
- DEG
differentially expressed genes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None
CONFLICT OF INTEREST
The authors state no conflict of interest.
REFERENCES
- Adams DR, Eng CM. Next-generation sequencing to diagnose suspected genetic disorders. N Engl J Med 2018;379:1353–62. [DOI] [PubMed] [Google Scholar]
- Andrews S (2010) FastQC: A quality control tool for high throughput sequence data. <http://www.bioinformatics.babraham.ac.uk/projects/fastqc> Accessed.
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011;12:745–55. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmel N, Danescu S, Gruler A, Kiritsi D, Bruckner-Tuderman L, Kreuter A, et al. A deep-intronic FERMT1 mutation causes Kindler syndrome: An explanation for genetically unsolved cases. J Invest Dermatol 2015;135:2876–9. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresard L, Smail C, Ferraro NM, Teran NA, Li X, Smith KS, et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat Med 2019;25:911–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka K, Miyatake S, Koshimizu E, Tsurusaki Y, Mitsuhashi S, Iwama K, et al. RNA sequencing solved the most common but unrecognized NEB pathogenic variant in Japanese nemaline myopathy. Genet Med 2019;21:1629–38. [DOI] [PubMed] [Google Scholar]
- Has C, South AP, Uitto J. Molecular therapeutics in development of epidermolysis bullosa - update 2020. Mol Diagn Ther 2020;(in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Balasubramanian M, Humphreys N, Waruiru C, Brauner M, Kohlhase J, et al. Intronic ITGA3 Mutation Impacts Splicing Regulation and Causes Interstitial Lung Disease, Nephrotic Syndrome, and Epidermolysis Bullosa. J Invest Dermatol 2016;136:1056–9. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, Pertea M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 2019;20:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, Iuso A, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun 2017;8:15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Tian L, Hakonarson H. Increasing diagnostic yield by RNA-Sequencing in rare disease-bypass hurdles of interpreting intronic or splice-altering variants. Ann Transl Med 2018;6:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi S, Mancini E, Xu L, Moore A, Jahanbani F, Hebestreit K, et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 2019;574:553–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamura D, Goto M, Yasukawa K, Kon A, Akiyama M, Shimizu H. Identification of COL7A1 alternative splicing inserting 9 amino acid residues into the fibronectin type III linker domain. J Invest Dermatol 2003;120:942–8. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Jazayeri A, Uitto J. Research techniques made simple: Genome-wide homozygosity/autozygosity mapping is a powerful tool to identify candidate genes in autosomal recessive genetic diseases. J Invest Dermatol 2018a;138:1893–900. [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Touati A, Sotoudeh S, Jazayeri A, et al. Next generation sequencing identifies double homozygous mutations in two distinct genes (EXPH5 and COL17A1) in a patient with concomitant simplex and junctional epidermolysis bullosa. Hum Mutat 2018b;39:1349–54. [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Daneshpazhooh M, Mahmoudi H, Kariminejad A, Fischer J, et al. Biallelic KRT5 mutations in autosomal recessive epidermolysis bullosa simplex, including a complete human keratin 5 “knock-out”. Matrix Biol 2019a;83:48–59. [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Touati A, Pajouhanfar S, Baghdadi T, et al. Mutations in PLOD3, encoding lysyl hydroxylase 3, cause syndromic recessive dystrophic epidermolyis bullosa-like phenotype with abnormal anchoring fibrils and deficiency in type VII collagen. Matrix Biol 2019b;81:91–106. [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Uitto J. Phenotypic spectrum of epidermolysis bullosa: The paradigm of syndromic versus non-syndromic skin fragility disorders. J Invest Dermatol 2019c;139:522–7. [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Zeinali S, Touati A, Abiri M, et al. Genome-wide single nucleotide polymorphism-based autozygosity mapping facilitates identification of mutations in consanguineous families with epidermolysis bullosa. Exp Dermatol 2019d;28:1118–21. [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Sotoudeh S, Liu L, Guy A, Lovell PA, et al. Genomics-based treatment in a patient with two overlapping heritable skin disorders: Epidermolysis bullosa and acrodermatitis enteropathica. Hum Mutat 2020; 10.1002/humu.23980. [DOI] [PubMed] [Google Scholar]
- Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med 2018;20:1216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssefian L, Vahidnezhad H, Saeidian AH, Pajouhanfar S, Sotoudeh S, Mansouri P, et al. Inherited non-alcoholic fatty liver disease and dyslipidemia due to monoallelic ABHD5 mutations. J Hepatol 2019a;71:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssefian L, Vahidnezhad H, Saeidian AH, Touati A, Sotoudeh S, Mahmoudi H, et al. Autosomal recessive congenital ichthyosis: Genomic landscape and phenotypic spectrum in a cohort of 125 consanguineous families. Hum Mutat 2019b;40:288–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.