

Abstract
Chloride intracellular channels 1 (CLIC1) and 4 (CLIC4) are expressed in endothelial cells and regulate angiogenic behaviors in vitro, and the expression of Clic4 is important for vascular development and function in mice. Here, we found that CLIC1 and CLIC4 in endothelial cells regulate critical G-protein-coupled receptor (GPCR) pathways associated with vascular development and disease. In cultured endothelial cells, we found that CLIC1 and CLIC4 transiently translocated to the plasma membrane in response to sphingosine-1-phosphate (S1P). Both CLIC1 and CLIC4 were essential for mediating S1P-induced activation of the small guanosine triphosphatase (GTPase) Rac1 downstream of the S1P receptor 1 (S1PR1). In contrast, only CLIC1 was essential for S1P-induced activation of the small GTPase RhoA downstream of S1PR2 and S1PR3. Neither were required for other S1P-S1PR signaling outputs. Rescue experiments revealed that CLIC1 and CLIC4 were not functionally interchangeable, suggesting distinct and specific functions for CLICs in transducing GPCR signaling. These CLIC-mediated mechanisms were critical for S1P-induced stimulation of the barrier function in endothelial cell monolayers. Our results define CLICs as previously unknown players in the pathways linking GPCRs to small GTPases and vascular endothelial function.
Introduction
Chloride intracellular channels (CLICs) are considered metamorphic proteins capable of forming a soluble, cytoplasmic structure and a membrane-bound structure (1). The vertebrate CLIC family consists of six members: CLIC1 through CLIC6 (2). CLICs localize to multiple cellular compartments, such as the plasma membrane (3), the mitochondria (4), the nucleus (3), the Golgi (5), and the endoplasmic reticulum (6). CLICs are linked to diverse biological processes, such as apoptosis, differentiation, and migration in various cell types (7, 8).
The mammalian CLIC family is structurally conserved, sharing ~50–60% amino acid sequence homology to each other (9, 10). The primary sequence is consistent with a possible ion channel activity for CLICs, however, the ability of CLICs to form functional ion channels has not been established in cells. Ion channel activity by CLICs has been shown in vitro with artificial planar lipid bilayers (11, 12) but little information about anion or cation selectivity has been obtained (13, 14). The crystal structure of the cytosolic conformations of vertebrate and invertebrate CLICs have been reported and define them as small globular proteins that contain a putative transmembrane domain (TMD) near their N-termini (15, 16). Based on sequence and structural similarity to the omega-class of glutathione-S-transferases (GSTs) (15, 17), CLICs have been proposed to function as GSTs (18). The N-terminus of the cytosolic form is believed to undergo structural reorganization following redox change, leading to membrane insertion (16, 19). The features suggesting GST enzymatic activity for CLICs has been supported by Al Khamici et al. (18) who reported that purified CLIC1, CLIC2, and CLIC4 displayed glutaredoxin-like activity in vitro and identified structural elements required for catalytic activities (18). As with the ion channel hypothesis, it is not clear what physiological processes may depend on the glutaredoxin-like activity of CLICs.
CLICs are known to function in the endothelium during angiogenesis and in physiological functions of the vasculature. Two of the six mammalian CLICs, CLIC1 and CLIC4, are expressed in endothelial cells (ECs) (20, 21). Using knock-down (KD) studies, we previously established that CLIC1 and CLIC4 are necessary for cultured endothelial cell growth and sprouting morphogenesis (20, 21). Reducing CLIC4 expression in human umbilical vein endothelial cells (HUVEC) caused decreased cell proliferation, capillary-like sprouting, and capillary network and lumen formation (20), whereas reducing CLIC1 caused decreased cell proliferation, migration, and lumen formation (21). In mice, genetic deletion of Clic1 or Clic4 individually does not affect viability (22); however, these mice do show various vascular defects. Platelet dysfunction and inhibited clotting phenotypes were observed in Clic1 knockout mice (23). Clic4 knockout mice display a slight reduction in retinal vasculature in neonates (22), smaller kidneys with fewer glomeruli and decreased density in the peritubular capillary network (24), as well as reduced collateral circulation in adult mice (25), and defects in angiogenesis when assessed with a Matrigel plug angiogenesis assay (22). Vascular defects are seen in Clic4 knockout mice under conditions of stress, such as altered oxygen levels in an oxygen toxicity assay (22). Thus, both CLIC1 and CLIC4 contribute to endothelial function and CLIC4 is necessary for vascular development and vascular response to stress.
Despite evidence of vascular function, the molecular mechanisms through which CLICs function in vascular cells, including any role they may have in signal transduction pathways important for EC function, is not clear. Studies in various cell types have suggested potential interactions between CLICs and G-protein-coupled receptors (GPCRs). For example, CLIC4 was found to interact with the histamine H3 receptor, resulting in enhanced H3 receptor surface level expression in brain cells (26), and CLIC6 was shown to physically interact with the C-terminus of dopamine D2 receptors in rat brain cells (27). Further understanding the role of CLICs in GPCR signaling was provided by Ponsioen et al. (28), when they showed that sphingosine-1-phosphate (S1P) induces CLIC4 translocation to the plasma membrane in neuroblastoma cells, establishing a link between CLIC4 and the S1P–S1P receptor (S1PR) signaling pathway. Ponsioen et al. (28) also showed that other GPCR agonists (such as LPA and α-thrombin) and the downstream effector and guanosine triphosphatase (GTPase) RhoA promoted membrane translocation of CLIC4. Similarly, CLIC4 can translocate to the plasma membrane upon stimulation of muscarinic M3 receptors and LPA receptors in HeLa and HEK293 cells (29). Despite these advances, functional roles of CLICs in GPCR signaling are not known.
S1P is a bio-active lipid that binds to and activates the S1PR family of GPCRs, of which there are five in vertebrates (30). Three of these receptors (S1PR1, S1PR2, and S1PR3) are expressed in ECs (31), and S1P–S1PR signaling has been shown to regulate many aspects of EC function and vascular development (30, 32, 33). The aforementioned studies on CLIC function in angiogenesis and CLIC translocation response to S1P led us to hypothesize that the endothelial CLICs (CLIC1 and CLIC4) are previously unknown effectors in S1P signaling in endothelial cells. The S1PRs expressed in ECs link to distinct branches of the canonical GPCR signaling cascade, leading to activation of—among other effectors—members of the membrane localized, small GTPase family: Ras, Rac, Rho and Cdc42 (30, 34). Here, we found that S1P promotes translocation of CLIC1 and CLIC4 from the cytoplasm to the plasma membrane of ECs, suggesting a functional relationship between CLICs and S1PR signaling in these cells. Our data indicate that CLICs are required for S1P-induced Rac1 and RhoA signaling leading to endothelial migration, barrier control, and stress fiber formation. Thus, our findings demonstrate that CLICs are an essential link between GPCRs and Rac1 and RhoA signaling.
Results
S1P promotes CLIC translocation from the cytoplasm to the plasma membrane in endothelial cells
Evaluation of transcript levels by RNA sequencing (RNAseq) analysis demonstrated that CLIC1 and CLIC4 are expressed in multiple EC types, including HUVECs, human microvascular retinal endothelial cells (HRECs), and human dermal lymphatic endothelial cells (HDLECs), whereas the other four family members (CLIC2, 3, 5, or 6) were not substantially expressed in any of the tested ECs (Fig. 1A and fig. S1A). We confirmed the expression of CLIC1 and CLIC4 proteins by Western blot analysis of HUVEC lysates (fig. S1C), further supporting our previously published RT-PCR results (20, 21). We then evaluated the subcellular localization of CLIC1 and CLIC4 in HUVECs, and whether it is regulated by S1P, by immunostaining HUVECs expressing HA epitope-tagged CLIC1 or CLIC4 in HUVECs and treated with S1P. We found that S1P promoted a transient accumulation of HA-CLIC1 and HA-CLIC4 at the plasma membrane (Fig. 1, B and C), with a temporal response of minutes (fig. S1B) that is similar to that reported for CLIC4 in neuroblastoma cells (28). This rapid enrichment of CLIC1 and CLIC4 in the plasma membrane of ECs in response to S1P suggested a functional interaction between CLICs and S1P signaling, which we then explored.
Figure 1. S1P promotes CLIC1 and CLIC4 localization to plasma membrane in endothelial cells.
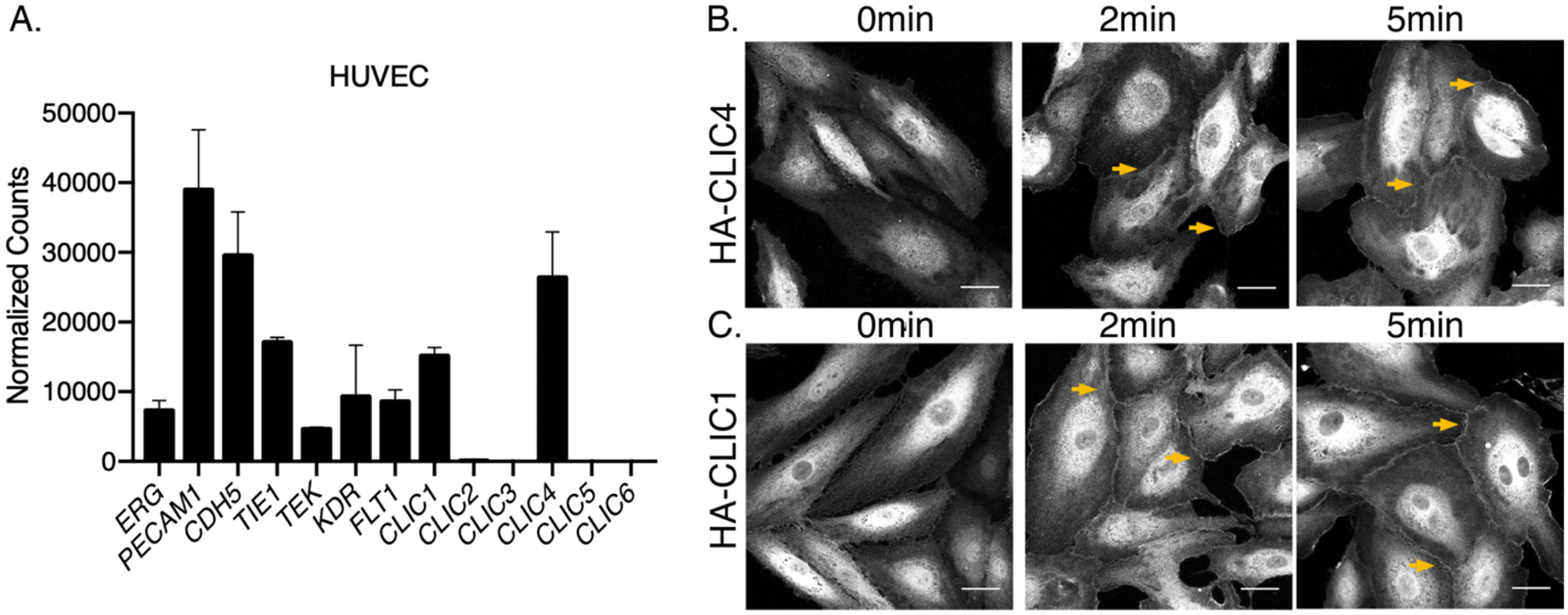
(A) RNA sequencing of human umbilical vein endothelial cells (HUVEC) revealed that only CLIC1 and CLIC4 are expressed. (B and C) HUVECs were infected with HA-CLIC4 (B) and HA-CLIC1 (C), and immunofluorescent studies using HA-antibody were performed after 2min and 5min of 1μM S1P stimulation. Arrows indicate accumulation of HA-CLIC4 and HA-CLIC1 at the plasma membrane. Images are confocal microscopy analysis and representative of three independent experiments. Scale bar = 30 μm
CLICs function redundantly to promote EC viability, and non-redundantly in S1P-induced EC migration, junction formation, and endothelial barrier strengthening
To investigate whether endothelial CLICs are required for S1P-dependent EC functions, we undertook a loss-of-function approach using short-hairpin RNA (shRNA) constructs to knock down the abundance of CLIC1 or CLIC4. HUVECs were infected with lentiviruses encoding control (scrambled) or CLIC1- or CLIC4-specific shRNAs, previously validated (20, 21) and confirmed here (fig. S1C). We used annexin V staining to assess apoptosis in CLIC-KD cells and found that loss of either CLIC alone did not cause significant apoptosis until 48 hours after infection (fig. S1D). In contrast, when we knocked-down CLIC1 and CLIC4 abundance simultaneously (dKD), there was statistically significant cell death observed within 24 hours, which was exacerbated by 48 hours, of lentiviral infection (fig. S1D). The much greater loss of viability in CLIC-dKD cells compared to either single knockdown is also apparent when visually surveying cell morphology (fig. S1E). Single knockdown of CLIC1 or CLIC4 (CLIC1-KD and CLIC4-KD) also showed a reduced cell viability phenotype at later time points (72 and 96 hours), which was confirmed with a quantitative colorimetric MTT cell viability assay (fig. S1F). We conclude that CLIC1 and CLIC4 act redundantly with respect to cell viability, and that CLIC functions are required for EC survival. Therefore, our studies to assess CLIC function in ECs were carried out within 48 hours and incorporated KD of either CLIC1 or CLIC4 by shRNA lentivirus infection.
S1P-S1PR signaling induces several EC responses that facilitate migration and VE-cadherin junction formation, which lead to enhancement of the vascular barrier formed by EC monolayers (35). RNAseq analysis revealed that mRNA encoding the S1PR1 and S1PR3 receptors were the more abundantly expressed than S1PR2 in HUVECs (fig. S1G). We determined the levels of S1PR1 and S1PR3 with Western blot analysis and found S1PR1 and S1PR3 levels are comparable in CLIC-KD cells and control cells (fig. S1H). To assess the roles of CLIC1 and CLIC4 in these S1P-driven responses, we undertook several approaches focusing on measures of endothelial barrier function and regulation of angiogenic responses. To measure endothelial barrier integrity in response to S1P, we used trans-endothelial electrical resistance (TEER) measurements (Fig. 2A and fig. S2A). Baseline resistance when comparing control and CLIC-KD cells was not altered (fig. S2, A and B). In control cells, S1P increased barrier resistance within minutes of S1P challenge; in contrast, CLIC1-KD or CLIC4-KD cells exhibited substantial reduction of the S1P barrier response (Fig. 2A). Immunofluorescent analysis for the VE-cadherin junctions in CLIC1-KD or CLIC4-KD ECs showed that loss of either CLIC resulted in an abnormal zig-zag pattern for these junctions in response to S1P compared to control (fig. S2, C and D), indicating disrupted EC junctions that likely affect barrier function. Finally, to establish whether CLICs play a role in other signaling pathways that regulate barrier function, we examined barrier change in response to VEGF which is known to disrupt EC barrier. We found that the responses of EC monolayers to VEGF was comparable in control, CLIC1-KD, and CLIC4-KD ECs (Fig. 2B and fig. S2B), indicating that CLICs are not involved in VEGF-induced permeability responses.
Figure 2. CLICs regulate S1P-mediated endothelial cell behaviors.
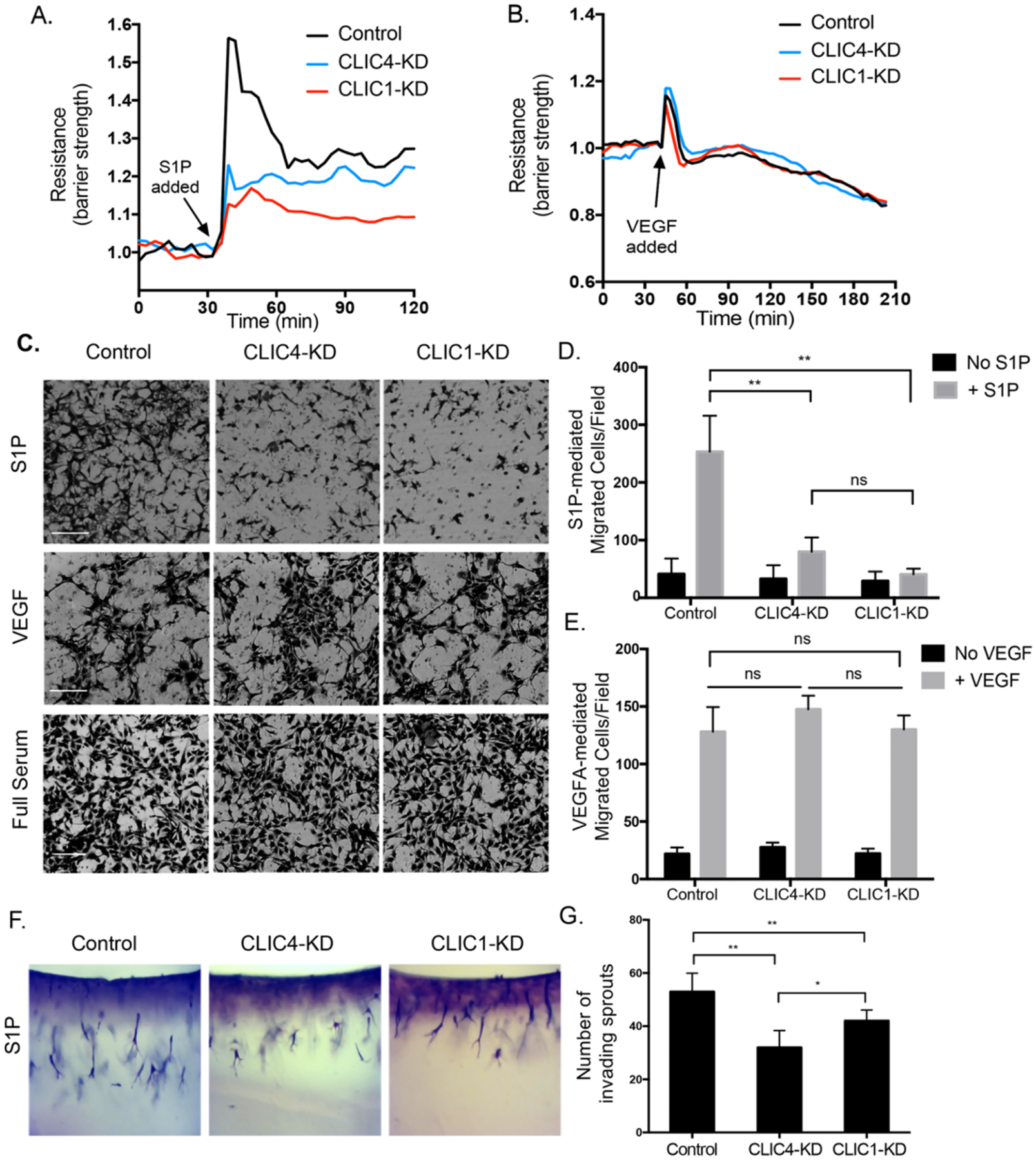
(A and B) HUVECs were serum starved for 2hrs then stimulated with 1μM S1P (A) or 100ng/mL VEGF (B). Trans-endothelial electrical resistance (TEER) was monitored for 2hrs post treatment. Resistance is presented as normalized fold change with respect to time of S1P/VEGF addition. Graphs are representative of four independent experiments. (C-E) HUVECs were seeded in the top chamber of a transwell plate and serum starved for 3hrs. S1P, VEGF, or full serum media was added to the bottom chamber and cells were allowed to migrate for 6hrs. Representative images of three independent experiments. Scale bars = 250μm (C) and quantification of the number of migrated cells per field (D and E). All data (means + SEM) were generated from at least three biological replicates, and significance was determined by ANOVA with Bonferroni correction for multiple comparisons test (** p<0.01, ns: not significant). (F-G) HUVECs were overlaid on collagen matrices laced with 1μm S1P and allowed to invade into the matrix. (F) After 72hrs the collagen matrix was fixed and stained with toluidine blue to allow for visual morphological analysis of sprouts. (G) Quantification of the number of invading sprouts from top to longest sprout. Significance was determined by ANOVA with Bonferroni correction for multiple comparisons test (N=3, *p<0.05, **p<0.01).
To evaluate the role of CLIC1 and CLIC4 in S1P-mediated EC migration, we utilized the Boyden chamber cell migration assay. In this assay, CLIC1-KD or CLIC4-KD ECs exhibited statistically significantly reduced migration towards S1P but not VEGF (Fig. 2, C to E), whereas migration of either cells in response to full serum was comparable to controls. These results indicate the migration defects associated with CLIC1 or CLIC4 depletion are specific to S1P-induced migration. An S1P-induced EC invasion assay (36) revealed a decreased number of invading sprouts when CLIC1 and CLIC4 were knocked down as compared to controls (Fig. 2, F and G), suggesting that CLICs function in the S1P signaling pathway to regulate EC behaviors necessary for S1P-induced sprouting.
CLICs are necessary for endothelial S1P-mediated Rac1 activation
Many endothelial cell types express three of the S1P-receptors; S1PR1, S1PR2 and S1PR3 (31). Our RNAseq analysis (fig. S1G) revealed S1PR1 and S1PR3 are the most abundantly expressed receptors of the three S1PRs in HUVECs. S1P binding to S1PR1 leads to activation of the heterotrimeric G-protein subunit Gαi, which then activates several effectors, including several members of the small GTPase family of proteins (Cdc42, Rac1, and Ras), which signal from the plasma membrane (30, 34). Even though S1PR2 and S1PR3 couple preferentially to Gα12/13 and Gαq, respectively, these receptors can also couple to Gαi (32, 37). The EC behaviors we assayed which require CLIC1 and CLIC4 (migration, junctional VE-cadherin accumulation, and enhancement of barrier function) are all positively regulated by Rac1. We thus sought to directly assess the effect of CLIC1 or CLIC4 loss on S1P-induced Rac1 activity. To this end, we used a Rac1-specific G-LISA; a colorimetric-based assay to quantify activated, GTP-bound Rac1 in cell lysates. In the absence of S1P addition, loss of CLIC1 or CLIC4 did not alter the basal Rac1-GTP levels in a statistically significant manner compared to control cells (fig. S3A). After S1P addition, Rac1-GTP levels were increased in control cells, whereas Rac1-GTP levels were not statistically significantly stimulated in CLIC1-KD or CLIC4-KD cells (Fig. 3A). Thus, the loss of CLIC1 or CLIC4 in ECs abolished S1P-mediated increase of Rac1 activity.
Figure 3. CLICs function in endothelial S1P pathway to regulate Rac1 activity.
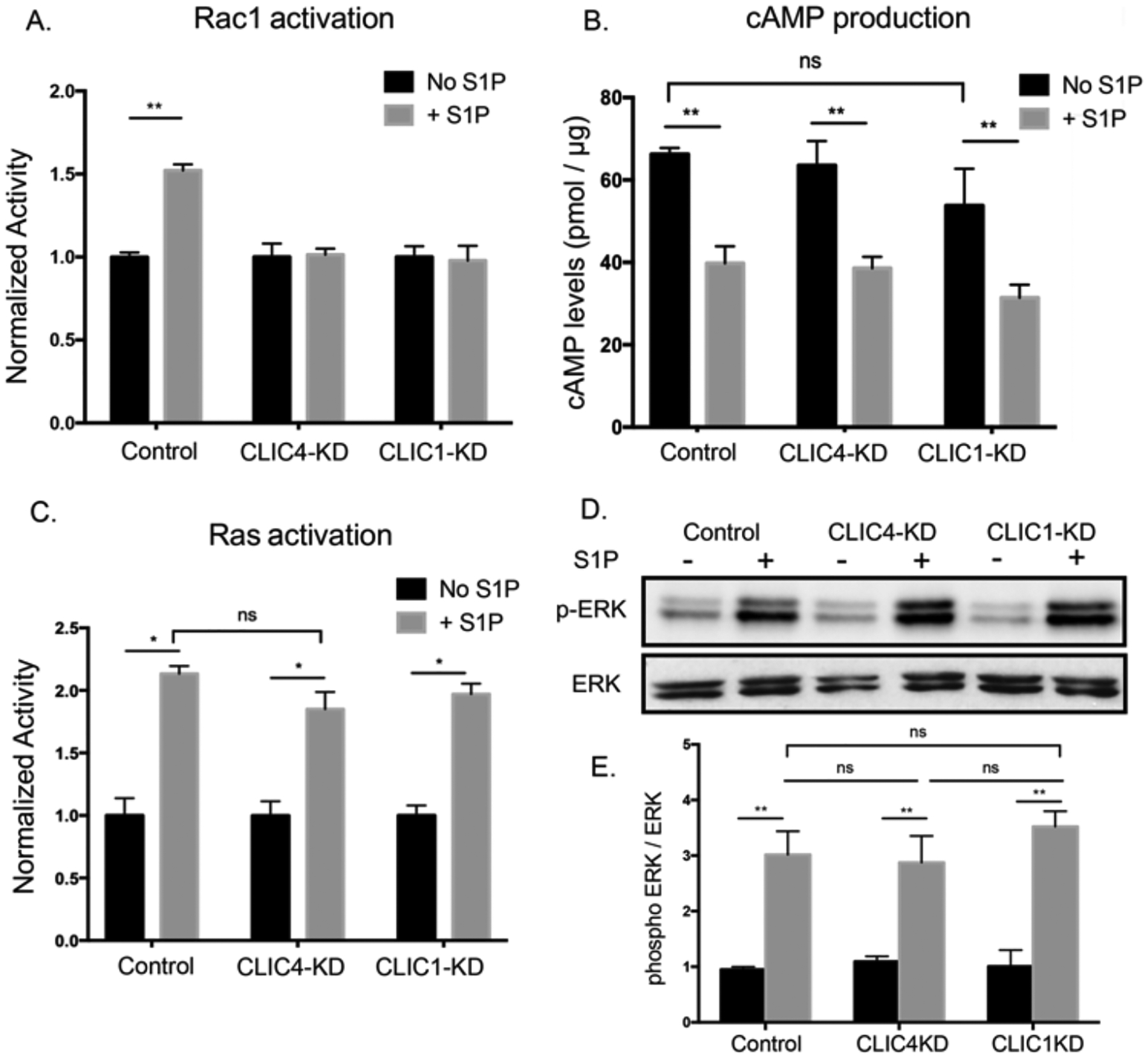
(A) HUVECs were serum starved for 3hrs and lysates were collected following 1μM S1P stimulation for 5min. Rac1 activity was measured by G-LISA assay and normalized to unstimulated cells for each condition (N=3, **p<0.01). (B) HUVECs were treated with IBMX and Forskolin followed by addition of 1μM S1P for 30min. Lysates were collected with 0.1M HCl, and cAMP levels were measured using a competitive immunoassay. (N=3, **p<0.01) (C and D) Following serum starvation and S1P treatment (1μM, 5min), (C) Ras activity was measured by G-LISA assay and normalized to unstimulated cells (N=3, *p<0.05). (D-E) Western blotting for ERK phosphorylation and quantification by densitometry analysis, normalized to the signal of total ERK protein respectively (N=3, **p<0.01). All significance (A-E) was determined by ANOVA with Bonferroni correction for multiple comparisons test.
We then investigated whether CLICs specifically regulate the Rac1 branch of S1PR-Gαi signaling or are more generally involved in the S1PR pathway. For this we considered that another output of S1PR-Gαi is the reduction of intracellular cAMP levels; here, we found that neither CLIC1 nor CLIC4 were required for this output (Fig. 3B). Additionally, we found that loss of neither CLIC1 nor CLIC4 altered S1P-induced Ras activation or ERK phosphorylation in a statistically significant manner (Fig. 3, C to E), which are other outputs of the S1PR-Gαi pathway (30). Further analysis of the potential role of CLIC regulation of Ras in response to epidermal growth factor (EGF) revealed no difference between control and knockdown cells (fig. S3B). It remains to be investigated whether CLICs have a role in the regulation of Cdc42; whereas one study showed Cdc42 was regulated by S1PR-Gαi (34), earlier reports using biochemical approaches had not observed substantial Cdc42 activation in response to S1P (38, 39). Thus, our results demonstrate that CLIC1 and CLIC4 are required for S1P-mediated Rac1 activation but appear not to be required for the other S1PR-Gαi pathways that we tested.
CLIC1 and CLIC4 have non-overlapping functions in regulating S1P-mediated Rac1 activity
The knockdown of an individual CLIC resulted in reduced S1P-induced Rac1 activity (Fig. 3A), suggesting that CLIC1 and CLIC4 are not redundant in their roles in Rac1 activation, as opposed to their role in EC viability (presented above). However, we also note that loss of either CLIC did not cause complete loss of Rac1 activation (fig. S3A) (meaning Rac1 activity is reduced to—but not below—basal levels when CLICs are depleted), raising the question of whether there may be redundancy or compensation between the CLICs. Immunoblotting analysis (fig. S1C) of CLIC-KD ECs showed no evidence for compensation of CLIC expression; that is, reduced levels of CLIC1 did not affect CLIC4 levels, and vice-versa. Unfortunately, we cannot assess the question of redundancy between CLIC1 and CLIC4 in basal Rac1 activity because, as mentioned above, double-knockdown cells were not viable (fig. S1, D and E). Nevertheless, these data suggest that CLIC1 and CLIC4 could cooperate, perhaps in a signaling complex, to couple S1P-induced activation of Rac GTPase.
The finding that depletion of either CLIC individually led to statistically significant loss of S1P-induced Rac1 activation raises the possibility that CLIC1 and CLIC4 may have distinct, non-overlapping, functions in S1PR-induced Rac1 activation. To test this possibility, we tested whether ectopic expression of an individual CLIC could rescue S1P-mediated deficiencies caused by knockdown of the other. To establish such a rescue assay, we first established that ectopic expression of each CLIC could rescue phenotypes caused by shRNA-mediated depletion of the endogenous protein, including restoration of long-term cell viability (>48 hours; fig. S4, A to B), EC barrier function (Fig. 4, A and B), EC migration (Fig. 4, C and D, and fig. S4, C and D), and Rac1 activation (Fig. 4, E and F). Hence, we next tested whether the functions of CLIC1 and CLIC4 in S1P-induced Rac1-mediated signaling are interchangeable or unique. We found that HA-CLIC1 did not rescue barrier function (Fig. 4A), cell migration (Fig. 4C and fig. S4C) or Rac1 activation (Fig. 4E) defects caused by CLIC4 loss. Similarly, we also found that ectopic HA-CLIC4 did not efficiently rescue defects caused by CLIC1 loss (Fig. 4, B, D, and F, and fig. S4D). We therefore conclude that either CLIC1 and CLIC4 have non-overlapping functions mediating S1PR-Gαi-Rac1 signaling responses, or that both CLIC1 and CLIC4 need to be present to facilitate these responses, perhaps in a signaling complex composed of both isotypes.
Figure 4. CLIC1 and CLIC4 function uniquely in regulating Rac1 activity.
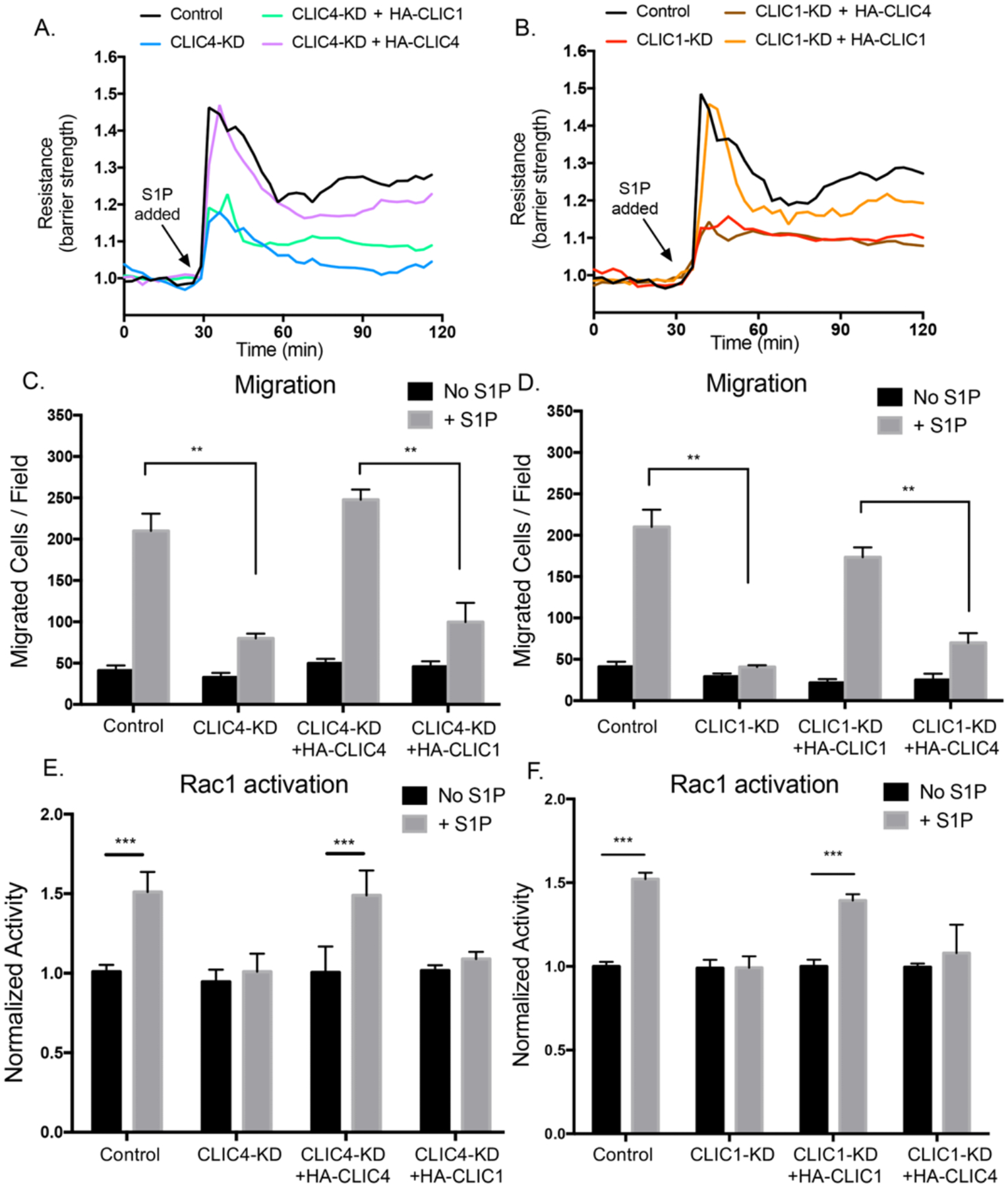
(A and B) HUVECs were serum starved for 2hrs then stimulated with 1μM S1P. Trans-endothelial electrical resistance (TEER) was monitored for 2hrs post treatment. Resistance was normalized with respect to time of S1P addition. Graphs are representative of three independent experiments. (C and D) Boyden chamber migration assay towards 1μM S1P for 6hrs after 3hrs of serum starvation. Quantifications of the number of migrated cells per field are shown. All data (means ± SEM) were generated from at least three biological replicates, and significance was determined by ANOVA with Bonferroni correction for multiple comparisons test (** p<0.01, ns: not significant). (E and F) HUVECs were serum starved for 3hrs then treated with 1μM S1P for 5min. Rac1 activity was measured by G-LISA assay and normalized to unstimulated cells for each condition (ANOVA with Bonferroni correction for multiple comparisons test, n=3, ***p<0.001).
CLIC1 regulates endothelial S1P-driven RhoA activity
In addition to the activation of Gαi, S1P binding to S1PR2 and S1PR3 also activates a Gα12/13-mediated pathway that stimulates the small GTPase RhoA and its downstream effector Rho associated-kinase (ROCK), which leads to actin stress fiber formation and cell contractility (40, 41). To evaluate a potential role for endothelial CLICs in S1PR-Gα12/13-RhoA-ROCK signaling, we used phalloidin to visualize actin structures in the presence or absence of S1P and found that control and CLIC4-KD ECs displayed robust actin stress fiber formation in response to S1P, but that this response was absent in CLIC1-KD ECs (Fig. 5A). This result demonstrates that CLIC1, but not CLIC4, is required for S1P-mediated signaling that leads to actin stress fiber formation.
Figure 5. CLIC1 regulates endothelial S1P-driven RhoA activity.
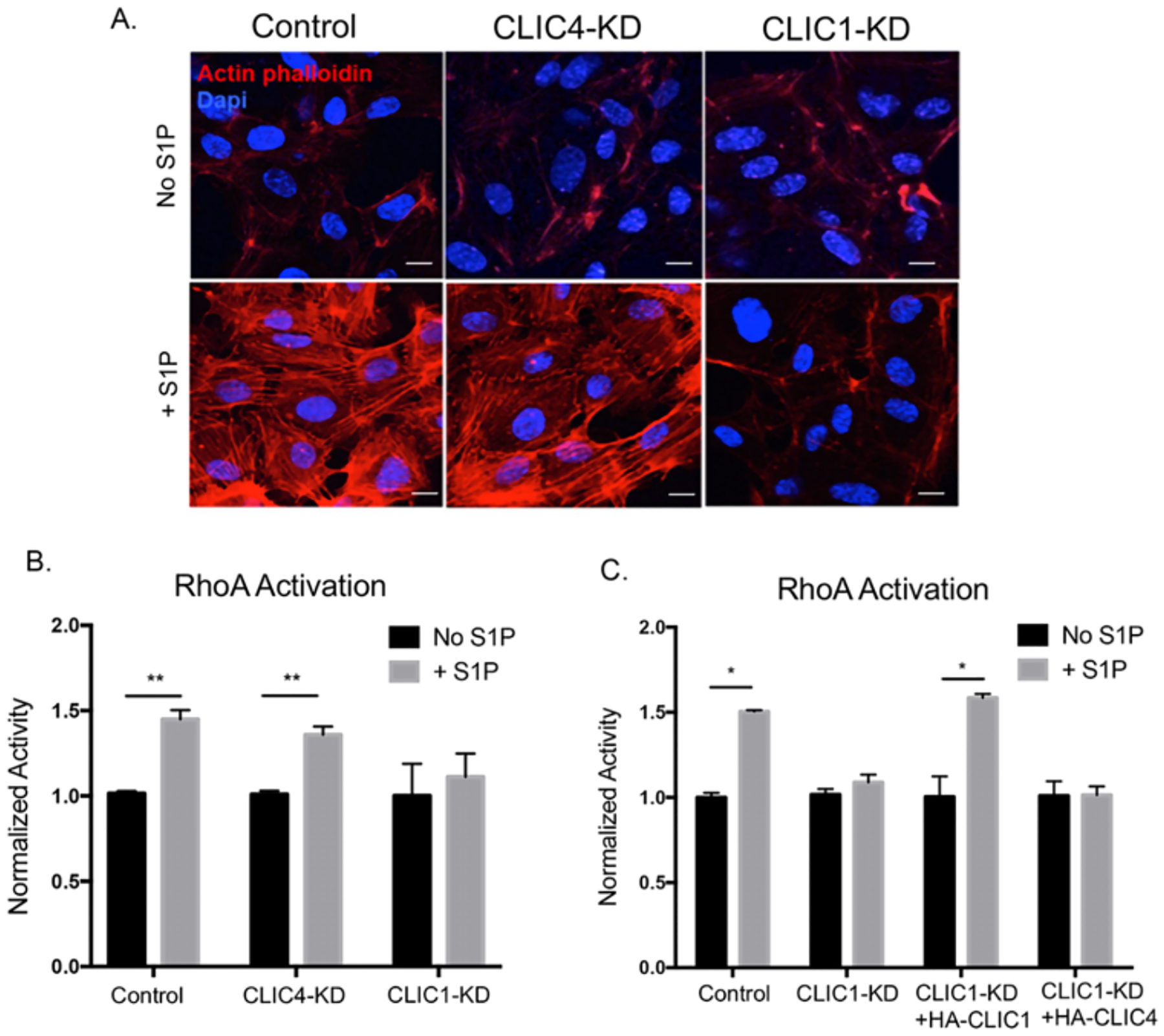
(A) HUVECs were serum starved for 3hrs then stimulated with 1μM S1P for 30min. Cells were fixed and stained with phalloidin to visualize actin stress fibers (red). DAPI was used as a nuclear stain (blue). Confocal images are representative of three independent experiments. Scale bar = 10 μm (B and C) HUVECs were serum starved for 3hrs then treated with 1μM S1P for 5min. RhoA activity was measured by G-LISA assay and normalized to unstimulated cells for each condition (ANOVA with Bonferroni correction for multiple comparisons test, N=3, *p<0.05, **p<0.01).
To investigate whether loss of S1P-induced actin stress fibers in CLIC1-KD cells reflects a reduction in RhoA activity or whether CLIC1 is required elsewhere within the S1PR-Gα12/13-RhoA-ROCK pathway, we directly assessed RhoA activity using a G-LISA assay. Consistent with the CLIC1-KD cell stress fiber phenotype, we found that knockdown of only CLIC1 blocked a S1P-induced RhoA activation (Fig. 5B), suggesting that CLIC1 has a specific role in S1PR-Gα12/13-RhoA signaling. CLIC1 specificity was confirmed by rescue experiments, which showed that ectopic HA-CLIC1, but not HA-CLIC4, rescued S1P-induced RhoA activation in CLIC1-KD cells (Fig. 5C). Thus, we conclude that CLIC1 and CLIC4 have non-overlapping functions in facilitating S1P-S1PR–mediated activation of RhoA.
Discussion
In this study, we found that endothelial expression of CLIC1 and CLIC4 are differentially required for RhoA and Rac1 activation in response to S1P-S1PR signaling, a critical GPCR signal-transduction pathway in vascular development and disease (30, 32). Previous studies implicated CLIC proteins in GPCR signaling inferred from physical interaction (26, 27) or re-localization of CLICs in response to GPCR agonists (28, 29, 42); however, these had not established function for CLICs in GPCR signaling. Our study here demonstrates that S1P promoted the plasma membrane localization of CLIC1 and CLIC4 in ECs, in which S1P signaling is known to play a physiologically relevant role (28). Furthermore, it uncovered both unique and overlapping—but not redundant—functions for CLIC1 and CLIC4 in distinct branches of the S1PR signaling (Fig. 6) that mediate endothelial cell responses to S1P, including migration, barrier regulation, and stress fiber formation.
Figure 6. CLICs function upstream of Rac1 and RhoA in endothelial S1P signaling cascade.
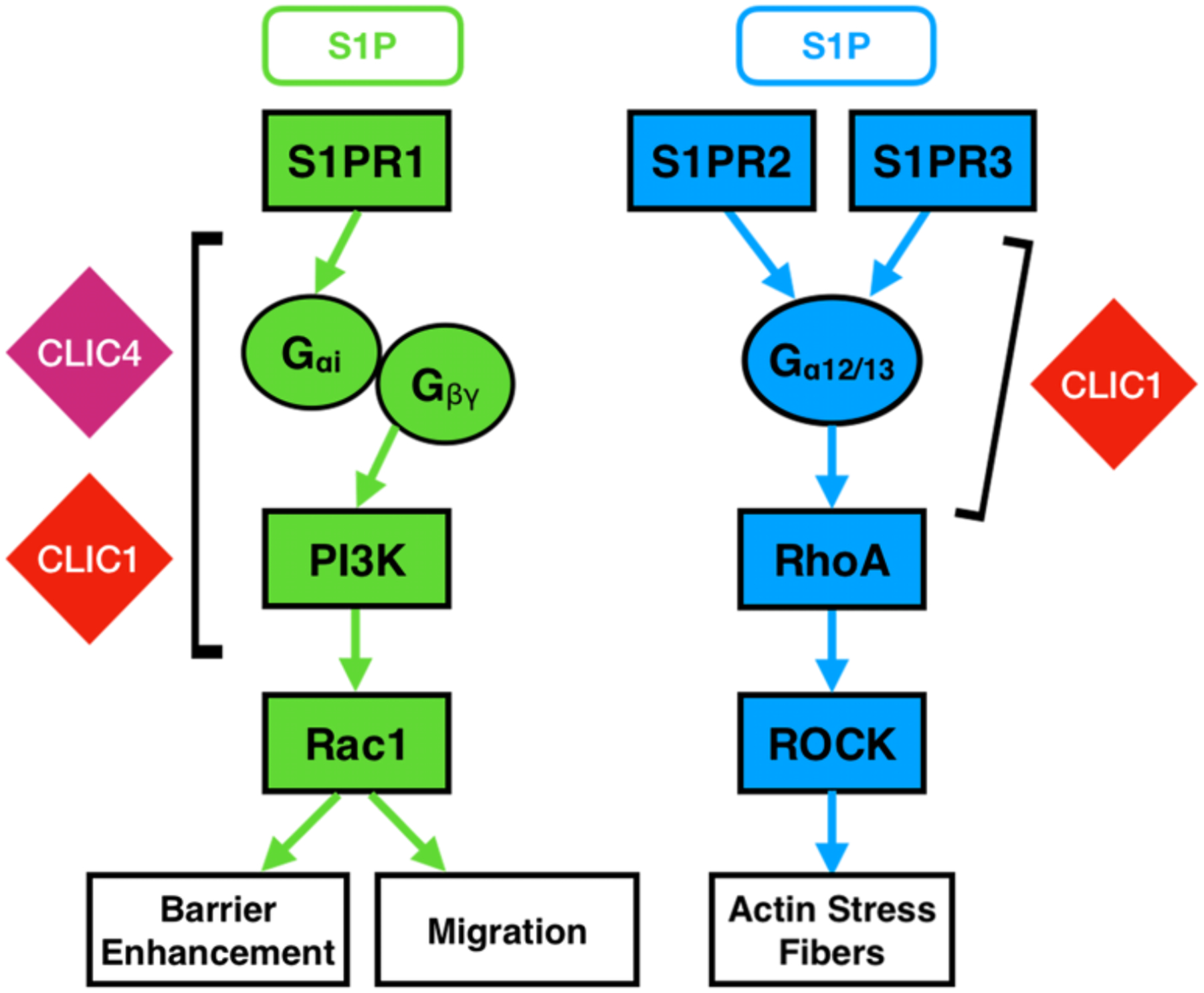
Both CLIC1 and CLIC4 are necessary for S1PR1-mediated endothelial behaviors and function upstream of Rac1. CLIC1, but not CLIC4, is required for S1PR2 and S1PR3 signaling and acts upstream of RhoA.
We speculate that this role for CLIC1 and CLIC4 in RhoA and Rac1 regulation is conserved for other GPCR signaling pathways. In support of a wider role for CLICs as GPCR effectors, previous studies have demonstrated changes in CLIC protein subcellular localization upon treatment of tumor cells [neuroblastoma (28) and HeLa (42)] with the GPCR agonist LPA, and CLIC4 localizes to plasma membrane in response to acetylcholine signaling through muscarinic M3 receptors in HEK293 cells (29). Biochemical interactions of CLIC4 with histamine H3 receptors (26), and CLIC6 with dopamine D2 receptors (27) have also been reported. Although these studies do not demonstrate that CLICs act as effector proteins in these GPCR pathways, they do allow one to hypothesize as to which heterotrimeric G-protein pathways utilize CLICs. Our studies in endothelial cells of S1P receptor signaling implicate GPCRs that couple to Gαi and Gα12/13 subunits as pathways that link to CLICs. These Gα-proteins are also involved in LPA receptor, muscarinic M3 receptor and histamine receptor signaling. To date, our studies do not yet implicate CLICs in GPCRs coupled with Gαs, as none of the S1P/S1PR pathways assessed here signal through this heterotrimeric G-protein subunit (30, 32). We thus speculate that CLICs function downstream of select GPCRs and their associated heterotrimeric G-proteins.
Our results do not indicate that endothelial CLICs function as effectors in VEGF receptor tyrosine kinase pathways in ECs, as we found no evidence that CLIC1 or CLIC4 are critical for VEGF-induced endothelial barrier disruption or EC migration, nor did we find a requirement for CLICs in EGF-mediated Ras activation. Thus, we believe that EC behaviors regulated by receptor tyrosine kinase signaling do not depend on CLIC proteins, suggesting a key role for CLICs in GPCR signaling but not in receptor tyrosine kinase signaling. There is evidence showing that CLIC4 exerts influences upstream of HIF-1α–VEGF signaling by regulating VEGF expression in vivo; however, this occurs by an unknown mechanism (25). Our results here lead us to speculate that the observed regulation of VEGF expression by CLIC4 may be through the modulation of GPCR, RhoA, and/or Rac1 signaling.
Our data support a role for CLICs upstream of Rac and Rho, but published literature describes roles for CLICs downstream of small GTPases. For example, LPA-induced translocation of CLIC4 to the plasma membrane was demonstrated to be driven by RhoA activation in tumor cells (28); consistent with RhoA functioning upstream of CLIC4 to regulate its localization. Do CLICs act both upstream and downstream of Rac1 and RhoA to modulate their activity? CLIC5A appears to stimulate Rac1 in podocytes to protect against hypertension-induced glomerular injury (43), suggesting a role for CLIC5 upstream of this small GTPase. Additionally, in human pulmonary arterial smooth muscle cells (hPASMCs), reduced expression of CLIC4 delayed Rac1 and RhoA activation by S100A4/Mts1 and BMP-2/TGF-ß signaling (44), suggesting that in this context CLIC4 functions upstream of Rac1 and RhoA. Together, these results suggest that CLICs may be involved in the regulation of small Rho-GTPases (Rac1 and RhoA) in complex arrangements that may depend on cellular and signaling contexts. In ECs, loss of CLICs leads to loss of Rac1 and RhoA activation by endothelial S1P signaling, indicating that CLICs act as effectors upstream of Rac1 and RhoA to regulate S1P-induced endothelial behaviors. CLIC1 and CLIC4 re-localize to the plasma membrane upon S1P stimulation, but what drives this process remains unclear. S1PRs undergo internalization upon ligand binding, where the maximum internalization occurs at 15 to 20 min after S1P stimulation (45, 46). We observed CLIC1 and CLIC4 membrane localization was not evident at 15 minutes, which coincides with the time of S1PR internalization. This suggests a possible role of CLICs in S1PR internalization that needs future investigation.
A key issue in the study with CLICs is their potential to function as chloride channels (11, 12). To date, firm evidence of a physiological role for CLIC proteins as ion channels is lacking. It is possible that CLICs utilize channel activity to facilitate the endothelial S1PR-Rac1 or S1PR-RhoA signaling, but we do not believe this to be the case. There is little current evidence that S1P mediates regulation of small GTPases through an ion channel, and CLICs have not been definitively shown to function as ion channels. Another function that has been ascribed to CLICs, based on structural similarities and biochemical analyses in vitro, is that of putative glutathione-S-transferase activity (16, 18). Whether the endothelial CLICs function as GST-like enzymes in vivo, and within S1P signaling, is currently unclear. A critical cysteine residue required for GST-like activity (18) is conserved in the CLIC1 (Cys24) and CLIC4 (Cys35) N-termini, suggesting that both of these proteins could have GST-like activity. Our results indicating unique and non-interchangeable functions for CLIC1 and CLIC4 in S1P-induced Rac1 and RhoA activation suggests that CLIC function in S1PR signaling will not be related to a shared activity, such as the postulated ion channel or GST-like activities, of these proteins. Therefore, we believe that a reasonable model posits that CLICs function in a unique capacity in GPCR signaling.
It remains to be determined whether the functional role for CLICs that we uncovered in endothelial cells is conserved broadly in both vertebrates and invertebrates. Notably, the CLIC homolog in C. elegans, EXC4, is essential in the proper tubulogenic development and maintenance of the excretory canal, and its loss can be rescued by the expression of human CLIC1 fused to the putative transmembrane domain of EXC-4 (47, 48), suggesting a conservation of function across specie. Regardless, CLICs are expressed in many mammalian cell types and have been implicated in multiple cellular functions and diverse signaling pathways (2). We propose that CLICs are a new class of GPCR effectors that link GPCRs to Rho and Rac small GTPases. It is evident that CLICs play important roles in angiogenesis and vascular development (20–22); our future work will determine whether these physiological functions in mammals depend on this role we have discovered for CLICs as GPCR-signaling effectors in ECs.
Materials and Methods
Primary cells and cell culture
Human Umbilical Vein Endothelial Cells (HUVECs) were isolated from human umbilical cords following established protocols (49). The cells used in experimental replicates were isolated from different donors. Cells were grown in EGM™-2 Endothelial Cell Growth Media (Lonza) (including all supplements provided by bullet kits) on culture dishes coated with rat tail type I collagen (Corning). Human Retinal Microvascular Endothelial Cells (HRMECs) were purchased from Cell Systems. Cells were maintained on fibronectin (Sigma) coated plates (Millipore) and in EGM™-2 Endothelial Cell Growth Media (Lonza). 293T cells were acquired from ATCC and maintained in High Glucose DMEM (Gibco) with 10% Heat-Inactivated Fetal Bovine Serum (HI-FBS) and 0.01% penicillin-streptomycin. Unless otherwise noted, cells were cultured under standard conditions in a humidified incubator at 37°C, 5% CO2.
Antibodies and reagents
Sphingosine-1-phosphate (S1P) were obtained from Enzo Life Sciences (BML-SL140). Human VEGF-A and EGF were purchased from R&D system (293-VE) and Sigma (E9644). Antibodies to human CLIC1 (Abcam, ab28722), human CLIC4 (Novus Biologicals, NBP1-85574), tubulin (Sigma, T6074), Phospho-ERK (Cell Signaling Technology, 4370), ERK (Cell Signaling Technology, 4695) were used for immunoblotting. Antibodies to VE-cadherin (Abcam, ab33168), HA (Cell Signaling Technology, C29F4), F-actin (phalloidin, Invitrogen, A12379) were used for immunofluorescence.
RNA isolation and RNAseq analysis
RNA of primary ECs was extracted using the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen, Cat. 74104). RNA quantity and integrity were measured using Bio-analyzer (Agilent TapeStation 4200, UIC Genome Research core) prior to RNA sequencing. Samples were sequenced at ~30 million PE reads with 150 base-paired fragments by Novogene. Raw reads were mapped to the Human database (ENSEMBL/GRCh38) using STAR (version 2.5.0a) and processed with Samtools (version 1.4.1). The counts obtained by FeatureCounts (version 1.5.2) were analyzed by DESeq2 (version 1.18.1) to identify expressed genes.
Gene silencing and overexpression
Human CLIC1 and CLIC4 shRNA-containing constructs were purchased from Sigma-Millipore and screened for significant CLIC1 and CLIC4 knockdown in HUVEC by immunoblotting as described below. Screenings of the shRNAs for both proteins were reported previously (20, 21). Human CLIC4 shRNA targets the 5’-UTR regions with targeting sequence 5′-GCCGTAATGTTGAACAGAATT-3′, and human CLIC1 shRNA targets the coding region with targeting sequences 5’-CCTGTTGCCAAAGTTACACAT-3’ (Sigma). A lentiviral vector expressing scrambled shRNA was used as a control (Sigma-Millipore). Recombinant CLIC1 and CLIC4 DNA inserts were placed into the lentiviral pCCL vector. Wild type CLIC1 and CLIC4 were generated from cDNA of HUVECs. CLIC1 and CLIC4 variants were created by insertion, deletion, or fusion of pCCL-CLIC1 and pCCL-CLIC4 plasmids. DNA sequencing was performed to verify the correct sequence before experiments.
Lentivirus-mediated stable expression of constructs in HUVECs
For lentiviral gene transfer, HEK293T cells were transfected using the calcium phosphate approach with the following combination of plasmids: 3μg of pVSVG, 5μg of pMDLg/pRRE, 2.5μg of pRSV-Rev, and 10μg of pCCL/pLKO vector encoding genes of interest. The lentiviral-containing media was collected 48hrs later, passed through a 0.45μm filter, and then added onto HUVECs. The HUVECs were allowed to express the shRNA or overexpression constructs for at least 48 hours before experiments. pCCL-RFP was performed alongside to ensure infection efficiency was 100%.
MTT viability assay and apoptosis assessment
HUVEC cells were plated 5000 cells/well in triplicate in 96-well plates, and cell viability was determined at 24, 48, 72, and 96-hour time points using the MTT viability assay (Sigma-Millipore manufacturer’s protocol). Cell apoptosis was assessed using Annexin V staining from Biovision (K101) following the manufacturer’s protocol.
Trans-endothelial electrical resistance (TEER) assay
An Electric Cell-substrate Impedance Sensing (ECIS) array plate (Applied Biophysics) containing circular 250μm diameter active electrodes connected in parallel on a common gold pad was coated with rat tail type I collagen (Corning). HUVEC cells were seeded at 50,000 cells per well and allowed to grow overnight. Cells were serum starved (EBM-2, Lonza) for 2 hours, followed by a 30-min baseline resistance stabilization with the ECIS system (Applied Biophysics, model 1600R). 1μM S1P, 100ng/mL VEGF or BSA vehicle control were administered, and trans-endothelial resistance was monitored at a frequency of 4000Hz with measurements taken at 3-min intervals for 2 to 4 hours.
Immunofluorescence assay
HUVECs were plated on 4-well collagen-coated chamber slides (Millipore). Cells were serum starved for 3 hours prior to 1μM S1P treatment for various time points. Cells were then fixed with 4% PFA for 15 mins, washed with PBS, and blocked for 30 mins using a blocking buffer (3% BSA, 1% donkey serum, 0.1% TritonX-100). Primary antibodies for VE-cadherin (1:200, Abcam 33168) and HA (1:1000, Cell Signaling C29F4) were added and incubated overnight at 4°C. Secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 594 (1:1000, Life Technologies) and F-actin phalloidin were added and incubated for 2hrs. Samples were mounted using VectaShield with DAPI (Vector Labs). Slides were imaged using a fluorescent microscope (Zeiss AxioImager Z2), as well as by confocal microscopy using a Zeiss laser scanning microscope (LSM800). Images analysis was done with ZEN software under the same acquisition settings among all cell lines used in each experiment.
Boyden chamber migration assay
HUVECs were serum starved (EBM-2 + 0.5%FBS) overnight. 50,000 cells per well (24-well format, BD-Falcon) were seeded in triplicate on collagen coated inserts of a transwell chamber with 8 μm sized pores (BD Falcon) and incubated in serum free medium (EBM-2, Lonza) for 3 hours. Following starvation, 1.2mL of serum free media supplemented with stimulant (100 ng/ml hVEGF-A or 1μM S1P) was placed in the lower transwell chamber. Cells were allowed to migrate for 6 hours towards the lower chamber stimulant, followed by fixation of the cells with 4% PFA for 15 minutes and incubation with 0.1% crystal violet for 10 minutes to stain the cells. Cells remaining on top of the membrane insert were cleaned off with a cotton swab and the migrated cells at the bottom of the insert were imaged at 10X magnification. Cell migration was measured by counting the number of migrated cells per field, 5 fields for each well, and 3 wells of each condition were averaged for quantification.
3D collagen invasion assay
The 3D collagen invasion assay mediated by S1P was performed as previously described (50). In brief, HUVEC cells were incorporated into collagen matrices. Collagen gels were prepared to a final concentration of 2.5 mg/mL in a transwell system (Corning). Gels were placed into 6mm culture inserts for polymerization and equilibrated at 37°C and 5% CO2. HUVEC cells (50,000/well) were seeded in the upper chamber in M199 containing reduced-serum supplement (Transferrin, BSA, oleic acid and insulin), bFGF (40ng/mL), VEGF (40ng/mL), and ascorbic acid (50 mg/mL). 1μM S1P was added to the collagen matrix. 1μM S1P was added to the bottom chamber. Cultures were allowed to develop for 24 hours followed by changing to fresh medium. Experiments continued for 48 hours before being fixed in 3% glutaraldehyde in PBS and then stained with 0.1% toluidine blue in 30% methanol. Intact cultures were imaged using a Nikon microscope and Olympus camera using Kodachrome 64T color slide film. Three random fields at 10X magnification were selected for quantification and the number of invading cells per high power field (HPF) was counted manually. Intact cultures were cut vertically and photographed from the side to collect representative images of invasion responses.
G-LISA Rac1, RhoA, and Ras activation assays
Assays were performed using Rac1 (Cytoskeleton, BK128), RhoA (Cytoskeleton, BK124), and Ras (Cytoskeleton BK131) G-LISA Activation Assay Kits. HUVECs were serum starved in EBM-2 with 1% serum overnight and with serum-free EBM-2 for an additional 3 hours the following day. The cells were then stimulated with 1μM S1P for Rac1, RhoA, and Ras activation. Cell lysates were harvested and snap frozen in liquid nitrogen. The assay was then performed based on the manufacturer’s protocol.
cAMP measurement
HUVECs were seeded on collagen coated 60mm plates (Corning) and allowed to grow overnight. The next day, cells were serum starved for 3 hours. Cells were then preincubated for 30 minutes with 1mM IBMX (Tocris Bioscience) followed by stimulation for 30 min with various doses of S1P, in the presence of IBMX (1mM) and adenylate cyclase activator forskolin (20μM) (Tocris Bioscience). For Isoproterenol stimulation, 1mM IBMX was present, and forskolin was not added. cAMP levels were then assessed using the Direct cAMP ELISA (enzyme-linked immunosorbent assay) kit (Enzo Life Sciences, ADI-900–066) following the supplier’s protocol.
Immunoblotting
HUVECs were washed with cold PBS, and lysates were collected using TENT buffer (50mM Tris pH 8.0, 2mM EDTA, 150mM NaCl, and 1% Triton X-100) containing protease inhibitor cocktail (EMD Chemicals Inc.). Lysates were prepared for SDS-PAGE using sample buffer containing SDS and β-mercaptoethanol. SDS-PAGE was performed, followed by wet transfer onto a nitrocellulose membrane. The membrane was blocked with 5% non-fat dry milk in TBS-Tween solution. Primary and secondary HRP-antibody incubation was done in 2.5% milk. The membrane was developed using ECL detection reagent (GE Healthcare), and protein bands were imaged.
Statistical analysis
Unless otherwise noted, two-way analysis of variance (ANOVA) with Bonferroni post-hoc analysis was performed on all quantified data to determine significant differences between groups. The statistical tests were analyzed in GraphPad Prism software. P values less than 0.05 were considered statistically significant.
Supplementary Material
Fig. S1. CLIC1 and CLIC4 promote endothelial cell survival and localize to the plasma membrane in response to S1P.
Fig. S2. CLICs regulate S1P-driven barrier integrity and VE-cadherin junction formation.
Fig. S3. Loss of CLICs did not alter basal Rac1 activity or EGF-induced Ras activation.
Fig. S4. Ectopic expression of CLIC1 and CLIC4 rescues endothelial cell viability and S1P-mediated migration.
Acknowledgments:
We would like to thank Anthony Arena and Daniel Shaye (University of Illinois at Chicago) for discussion and critical reading for the manuscript. We are grateful to Zhengjia Chen (University of Illinois at Chicago) for statistical support. The research reported in this publication was partially supported by the University of Illinois Cancer Center Biostatistics Shared Resource (BSR) core.
Funding:
This study was supported by NIH R01 research project grants 5R01HL119043, 5R01HL112626 and 1R01GM134032 to J.K.K. and NIH R35HL135821 grant to T.H.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted for publication in Science Signaling. This version has not undergone final editing. Please refer to the complete version on record at https://www.science.org/journal/signaling. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The raw RNA sequencing data has been deposited in NCBI Gene Expression Omnibus repository (Accession number GSE163568).
References and Notes
- 1.Littler DR, Harrop SJ, Goodchild SC, Phang JM, Mynott AV, Jiang L, Valenzuela SM, Mazzanti M, Brown LJ, Breit SN, Curmi PM, The enigma of the CLIC proteins: Ion channels, redox proteins, enzymes, scaffolding proteins? FEBS Lett 584, 2093–2101 (2010); published online EpubMay 17 ( 10.1016/j.febslet.2010.01.027). [DOI] [PubMed] [Google Scholar]
- 2.Argenzio E, Moolenaar WH, Emerging biological roles of Cl- intracellular channel proteins. J Cell Sci 129, 4165–4174 (2016); published online EpubNov 15 ( 10.1242/jcs.189795). [DOI] [PubMed] [Google Scholar]
- 3.Valenzuela SM, Martin DK, Por SB, Robbins JM, Warton K, Bootcov MR, Schofield PR, Campbell TJ, Breit SN, Molecular cloning and expression of a chloride ion channel of cell nuclei. J Biol Chem 272, 12575–12582 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Fernandez-Salas E, Sagar M, Cheng C, Yuspa SH, Weinberg WC, p53 and tumor necrosis factor alpha regulate the expression of a mitochondrial chloride channel protein. J Biol Chem 274, 36488–36497 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Shanks RA, Larocca MC, Berryman M, Edwards JC, Urushidani T, Navarre J, Goldenring JR, AKAP350 at the Golgi apparatus. II. Association of AKAP350 with a novel chloride intracellular channel (CLIC) family member. J Biol Chem 277, 40973–40980 (2002); published online EpubOct 25 ( 10.1074/jbc.M112277200). [DOI] [PubMed] [Google Scholar]
- 6.Duncan RR, Westwood PK, Boyd A, Ashley RH, Rat brain p64H1, expression of a new member of the p64 chloride channel protein family in endoplasmic reticulum. J Biol Chem 272, 23880–23886 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Suh KS, Mutoh M, Mutoh T, Li L, Ryscavage A, Crutchley JM, Dumont RA, Cheng C, Yuspa SH, CLIC4 mediates and is required for Ca2+-induced keratinocyte differentiation. J Cell Sci 120, 2631–2640 (2007); published online EpubAug 1 ( 10.1242/jcs.002741). [DOI] [PubMed] [Google Scholar]
- 8.Suh KS, Mutoh M, Nagashima K, Fernandez-Salas E, Edwards LE, Hayes DD, Crutchley JM, Marin KG, Dumont RA, Levy JM, Cheng C, Garfield S, Yuspa SH, The organellular chloride channel protein CLIC4/mtCLIC translocates to the nucleus in response to cellular stress and accelerates apoptosis. J Biol Chem 279, 4632–4641 (2004); published online EpubFeb 6 ( 10.1074/jbc.M311632200). [DOI] [PubMed] [Google Scholar]
- 9.Gururaja Rao S, Ponnalagu D, Patel NJ, Singh H, Three Decades of Chloride Intracellular Channel Proteins: From Organelle to Organ Physiology. Curr Protoc Pharmacol 80, 11 21 11–11 21 17 (2018); published online EpubMar ( 10.1002/cpph.36). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gururaja Rao S, Ponnalagu D, Sukur S, Singh H, Sanghvi S, Mei Y, Jin DJ, Singh H, Identification and Characterization of a Bacterial Homolog of Chloride Intracellular Channel (CLIC) Protein. Sci Rep 7, 8500 (2017); published online EpubAug 17 ( 10.1038/s41598-017-08742-z). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh H, Ashley RH, CLIC4 (p64H1) and its putative transmembrane domain form poorly selective, redox-regulated ion channels. Mol Membr Biol 24, 41–52 (2007); published online EpubJan-Feb ( 10.1080/09687860600927907). [DOI] [PubMed] [Google Scholar]
- 12.Singh H, Ashley RH, Redox regulation of CLIC1 by cysteine residues associated with the putative channel pore. Biophys J 90, 1628–1638 (2006); published online EpubMar 1 ( 10.1529/biophysj.105.072678). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Averaimo S, Abeti R, Savalli N, Brown LJ, Curmi PM, Breit SN, Mazzanti M, Point mutations in the transmembrane region of the clic1 ion channel selectively modify its biophysical properties. PLoS One 8, e74523 (2013) 10.1371/journal.pone.0074523). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valenzuela SM, Mazzanti M, Tonini R, Qiu MR, Warton K, Musgrove EA, Campbell TJ, Breit SN, The nuclear chloride ion channel NCC27 is involved in regulation of the cell cycle. J Physiol 529 Pt 3, 541–552 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrop SJ, DeMaere MZ, Fairlie WD, Reztsova T, Valenzuela SM, Mazzanti M, Tonini R, Qiu MR, Jankova L, Warton K, Bauskin AR, Wu WM, Pankhurst S, Campbell TJ, Breit SN, Curmi PM, Crystal structure of a soluble form of the intracellular chloride ion channel CLIC1 (NCC27) at 1.4-A resolution. J Biol Chem 276, 44993–45000 (2001); published online EpubNov 30 ( 10.1074/jbc.M107804200). [DOI] [PubMed] [Google Scholar]
- 16.Littler DR, Assaad NN, Harrop SJ, Brown LJ, Pankhurst GJ, Luciani P, Aguilar MI, Mazzanti M, Berryman MA, Breit SN, Curmi PM, Crystal structure of the soluble form of the redox-regulated chloride ion channel protein CLIC4. FEBS J 272, 4996–5007 (2005); published online EpubOct ( 10.1111/j.1742-4658.2005.04909.x). [DOI] [PubMed] [Google Scholar]
- 17.Dulhunty A, Gage P, Curtis S, Chelvanayagam G, Board P, The glutathione transferase structural family includes a nuclear chloride channel and a ryanodine receptor calcium release channel modulator. J Biol Chem 276, 3319–3323 (2001); published online EpubFeb 2 ( 10.1074/jbc.M007874200). [DOI] [PubMed] [Google Scholar]
- 18.Al Khamici H, Brown LJ, Hossain KR, Hudson AL, Sinclair-Burton AA, Ng JP, Daniel EL, Hare JE, Cornell BA, Curmi PM, Davey MW, Valenzuela SM, Members of the chloride intracellular ion channel protein family demonstrate glutaredoxin-like enzymatic activity. PLoS One 10, e115699 (2015) 10.1371/journal.pone.0115699). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Littler DR, Harrop SJ, Fairlie WD, Brown LJ, Pankhurst GJ, Pankhurst S, DeMaere MZ, Campbell TJ, Bauskin AR, Tonini R, Mazzanti M, Breit SN, Curmi PM, The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J Biol Chem 279, 9298–9305 (2004); published online EpubMar 5 ( 10.1074/jbc.M308444200). [DOI] [PubMed] [Google Scholar]
- 20.Tung JJ, Hobert O, Berryman M, Kitajewski J, Chloride intracellular channel 4 is involved in endothelial proliferation and morphogenesis in vitro. Angiogenesis 12, 209–220 (2009) 10.1007/s10456-009-9139-3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tung JJ, Kitajewski J, Chloride intracellular channel 1 functions in endothelial cell growth and migration. J Angiogenes Res 2, 23 (2010); published online EpubNov 1 ( 10.1186/2040-2384-2-23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ulmasov B, Bruno J, Gordon N, Hartnett ME, Edwards JC, Chloride intracellular channel protein-4 functions in angiogenesis by supporting acidification of vacuoles along the intracellular tubulogenic pathway. Am J Pathol 174, 1084–1096 (2009); published online EpubMar ( 10.2353/ajpath.2009.080625). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu MR, Jiang L, Matthaei KI, Schoenwaelder SM, Kuffner T, Mangin P, Joseph JE, Low J, Connor D, Valenzuela SM, Curmi PM, Brown LJ, Mahaut-Smith M, Jackson SP, Breit SN, Generation and characterization of mice with null mutation of the chloride intracellular channel 1 gene. Genesis 48, 127–136 (2010); published online EpubFeb ( 10.1002/dvg.20590). [DOI] [PubMed] [Google Scholar]
- 24.Edwards JC, Bruno J, Key P, Cheng YW, Absence of chloride intracellular channel 4 (CLIC4) predisposes to acute kidney injury but has minimal impact on recovery. BMC Nephrol 15, 54 (2014) 10.1186/1471-2369-15-54). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chalothorn D, Zhang H, Smith JE, Edwards JC, Faber JE, Chloride intracellular channel-4 is a determinant of native collateral formation in skeletal muscle and brain. Circ Res 105, 89–98 (2009); published online EpubJul 2 ( 10.1161/CIRCRESAHA.109.197145). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda K, Haraguchi M, Kuramasu A, Sato T, Ariake K, Sakagami H, Kondo H, Yanai K, Fukunaga K, Yanagisawa T, Sukegawa J, CLIC4 interacts with histamine H3 receptor and enhances the receptor cell surface expression. Biochem Biophys Res Commun 369, 603–608 (2008); published online EpubMay 2 ( 10.1016/j.bbrc.2008.02.071). [DOI] [PubMed] [Google Scholar]
- 27.Griffon N, Jeanneteau F, Prieur F, Diaz J, Sokoloff P, CLIC6, a member of the intracellular chloride channel family, interacts with dopamine D(2)-like receptors. Brain Res Mol Brain Res 117, 47–57 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Ponsioen B, van Zeijl L, Langeslag M, Berryman M, Littler D, Jalink K, Moolenaar WH, Spatiotemporal regulation of chloride intracellular channel protein CLIC4 by RhoA. Mol Biol Cell 20, 4664–4672 (2009); published online EpubNov ( 10.1091/mbc.E09-06-0529). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lecat S, Matthes HW, Pepperkok R, Simpson JC, Galzi JL, A Fluorescent Live Imaging Screening Assay Based on Translocation Criteria Identifies Novel Cytoplasmic Proteins Implicated in G Protein-coupled Receptor Signaling Pathways. Mol Cell Proteomics 14, 1385–1399 (2015); published online EpubMay ( 10.1074/mcp.M114.046698). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mendelson K, Evans T, Hla T, Sphingosine 1-phosphate signalling. Development 141, 5–9 (2014); published online EpubJan ( 10.1242/dev.094805). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu YP, Yamashita T, Proia RL, The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem 279, 29367–29373 (2004); published online EpubJul 9 ( 10.1074/jbc.M403937200). [DOI] [PubMed] [Google Scholar]
- 32.Blaho VA, Hla T, Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors. Chem Rev 111, 6299–6320 (2011); published online EpubOct 12 ( 10.1021/cr200273u). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yanagida K, Engelbrecht E, Niaudet C, Jung B, Gaengel K, Holton K, Swendeman S, Liu CH, Levesque MV, Kuo A, Fu Z, Smith LEH, Betsholtz C, Hla T, Sphingosine 1-Phosphate Receptor Signaling Establishes AP-1 Gradients to Allow for Retinal Endothelial Cell Specialization. Dev Cell 52, 779–793 e777 (2020); published online EpubMar 23 ( 10.1016/j.devcel.2020.01.016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reinhard NR, Mastop M, Yin T, Wu Y, Bosma EK, Gadella TWJ Jr., Goedhart J, Hordijk PL, The balance between Galphai-Cdc42/Rac and Galpha12/13-RhoA pathways determines endothelial barrier regulation by sphingosine-1-phosphate. Mol Biol Cell 28, 3371–3382 (2017); published online EpubNov 7 ( 10.1091/mbc.E17-03-0136). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D, Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108, 689–701 (2001); published online EpubSep ( 10.1172/JCI12450). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayless KJ, Davis GE, Sphingosine-1-phosphate markedly induces matrix metalloproteinase and integrin-dependent human endothelial cell invasion and lumen formation in three-dimensional collagen and fibrin matrices. Biochem Biophys Res Commun 312, 903–913 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, Ono Y, Shinjo Y, Ishida S, Arang N, Kawakami K, Gutkind JS, Aoki J, Russell RB, Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 177, 1933–1947 e1925 (2019); published online EpubJun 13 ( 10.1016/j.cell.2019.04.044). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Breslin JW, Zhang XE, Worthylake RA, Souza-Smith FM, Involvement of local lamellipodia in endothelial barrier function. PLoS One 10, e0117970 (2015) 10.1371/journal.pone.0117970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vouret-Craviari V, Bourcier C, Boulter E, van Obberghen-Schilling E, Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine-1-phosphate in endothelial cells. J Cell Sci 115, 2475–2484 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, Hla T, Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99, 301–312 (1999); published online EpubOct 29 ( 10.1016/s0092-8674(00)81661-x). [DOI] [PubMed] [Google Scholar]
- 41.Miura Y, Yatomi Y, Rile G, Ohmori T, Satoh K, Ozaki Y, Rho-mediated phosphorylation of focal adhesion kinase and myosin light chain in human endothelial cells stimulated with sphingosine 1-phosphate, a bioactive lysophospholipid released from activated platelets. J Biochem 127, 909–914 (2000); published online EpubMay ( 10.1093/oxfordjournals.jbchem.a022686). [DOI] [PubMed] [Google Scholar]
- 42.Argenzio E, Klarenbeek J, Kedziora KM, Nahidiazar L, Isogai T, Perrakis A, Jalink K, Moolenaar WH, Innocenti M, Profilin binding couples chloride intracellular channel protein CLIC4 to RhoA-mDia2 signaling and filopodium formation. J Biol Chem 293, 19161–19176 (2018); published online EpubDec 14 ( 10.1074/jbc.RA118.002779). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tavasoli M, Li L, Al-Momany A, Zhu LF, Adam BA, Wang Z, Ballermann BJ, The chloride intracellular channel 5A stimulates podocyte Rac1, protecting against hypertension-induced glomerular injury. Kidney Int 89, 833–847 (2016); published online EpubApr ( 10.1016/j.kint.2016.01.001). [DOI] [PubMed] [Google Scholar]
- 44.Spiekerkoetter E, Guignabert C, de Jesus Perez V, Alastalo TP, Powers JM, Wang L, Lawrie A, Ambartsumian N, Schmidt AM, Berryman M, Ashley RH, Rabinovitch M, S100A4 and bone morphogenetic protein-2 codependently induce vascular smooth muscle cell migration via phospho-extracellular signal-regulated kinase and chloride intracellular channel 4. Circ Res 105, 639–647, 613 p following 647 (2009); published online EpubSep 25 ( 10.1161/CIRCRESAHA.109.205120). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu CH, Thangada S, Lee MJ, Van Brocklyn JR, Spiegel S, Hla T, Ligand-induced trafficking of the sphingosine-1-phosphate receptor EDG-1. Mol Biol Cell 10, 1179–1190 (1999); published online EpubApr ( 10.1091/mbc.10.4.1179). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chavez A, Schmidt TT, Yazbeck P, Rajput C, Desai B, Sukriti S, Giantsos-Adams K, Knezevic N, Malik AB, Mehta D, S1PR1 Tyr143 phosphorylation downregulates endothelial cell surface S1PR1 expression and responsiveness. J Cell Sci 128, 878–887 (2015); published online EpubMar 1 ( 10.1242/jcs.154476). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berry KL, Bulow HE, Hall DH, Hobert O, A C. elegans CLIC-like protein required for intracellular tube formation and maintenance. Science 302, 2134–2137 (2003); published online EpubDec 19 ( 10.1126/science.1087667). [DOI] [PubMed] [Google Scholar]
- 48.Berry KL, Hobert O, Mapping functional domains of chloride intracellular channel (CLIC) proteins in vivo. J Mol Biol 359, 1316–1333 (2006); published online EpubJun 23 ( 10.1016/j.jmb.2006.04.046). [DOI] [PubMed] [Google Scholar]
- 49.Jaffe EA, Nachman RL, Becker CG, Minick CR, Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest 52, 2745–2756 (1973); published online EpubNov ( 10.1172/JCI107470). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bayless KJ, Kwak HI, Su SC, Investigating endothelial invasion and sprouting behavior in three-dimensional collagen matrices. Nat Protoc 4, 1888–1898 (2009) 10.1038/nprot.2009.221). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CLIC1 and CLIC4 promote endothelial cell survival and localize to the plasma membrane in response to S1P.
Fig. S2. CLICs regulate S1P-driven barrier integrity and VE-cadherin junction formation.
Fig. S3. Loss of CLICs did not alter basal Rac1 activity or EGF-induced Ras activation.
Fig. S4. Ectopic expression of CLIC1 and CLIC4 rescues endothelial cell viability and S1P-mediated migration.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The raw RNA sequencing data has been deposited in NCBI Gene Expression Omnibus repository (Accession number GSE163568).