

Abstract
The majority of adult patients with acute lymphoblastic leukemia (ALL) suffer relapse, and in patients with central nervous system (CNS) metastasis, prognosis is particularly poor. We recently demonstrated a novel route of ALL CNS metastasis dependent on PI3Kδ regulation of the laminin receptor integrin α6. B-ALL cells did not, however, rely on PI3Kδ signaling for growth. Here we show that broad targeting of PI3K isoforms can induce growth arrest in B-ALL, reducing systemic disease burden in mice treated with a single agent pan-PI3Ki, copanlisib. Moreover, we show that cellular stress activates PI3K/Akt-dependent survival pathways in B-ALL, exposing their vulnerability to PI3Kδ and pan-PI3Ki. The addition of a brief course of copanlisib to chemotherapy delivered the combined benefits of increased survival, decreased systemic disease, and reduced CNS metastasis. These data suggest the promising, multifaceted potential of pan-PI3Ki for B-ALL CNS prophylaxis, systemic disease control, and chemosensitization.
Keywords: CNS relapse, PI3K inhibition, acute lymphoblastic leukemia, chemosensitization, microenvironment
Introduction
Adult B-cell acute lymphoblastic leukemia (B-ALL) is an aggressive malignancy in which the majority of patients relapse despite high rates of initial remission [1]. ALL is also particularly neurotropic [2]. Despite intensive cytotoxic chemotherapy or craniospinal irradiation administered as central nervous system (CNS) prophylaxis with induction regimens, 5–10% of patients relapse in the CNS [3]. Once relapsed, prognosis is grim and patients die within months [3]. A limited understanding of the mechanisms of CNS relapse has prevented effective, targeted drug development and remains a major area of unmet clinical need [3].
Our lab recently discovered a novel mechanism of B-ALL invasion into the CNS that is regulated by phosphatidylinositol 3-kinase (PI3K), a druggable kinase in the B-cell receptor signaling pathway [4]. Using multiple mouse models, we demonstrated the proof of principle that PI3Kδ inhibition (PI3Kδi) decreases B-ALL metastatic spread to the CNS [4]. We discovered that B-ALL cells migrate into the CNS along the outer surface of vessels passaging directly between vertebral or calvarial BM, through fenestrations in the cortical bone, and into the adjacent leptomeninges (LM) [4]. This process is dependent on interactions between the vascular basement membrane laminin and α6 integrin, a laminin receptor whose expression by B-ALL blasts we found was downregulated by PI3Kδi [4]. B-ALL expression of α6 integrin has also been associated with the persistence of residual disease after treatment, suggesting a potential role in chemo-resistance [5–7.
In our prior study of single agent PI3Kδi in B-ALL mice, we observed minimal effects of single agent PI3Kδ isoform-selective inhibition on systemic disease progression. This finding could reflect a lack of dependency of B-ALL cells on PI3K signaling for in vivo growth. Alternatively, additional PI3K isoforms may play distinct or compensatory roles in cellular processes that promote ALL disease progression in the periphery [8,9]. To our knowledge, detailed analyses of the effects of PI3K inhibition (PI3Ki) on B-ALL in vivo have not, however, been undertaken.
In the current study, we examine the differential effects of δ isoform-specific vs. pan-PI3Ki in B-ALL in vitro and in vivo. Our data suggest important potential therapeutic benefits of pan-class I PI3Ki with the FDA approved agent, copanlisib, including decreasing CNS invasion, enhancing systemic responses to chemotherapy and arresting proliferating blasts in quiescent G0. These findings have substantial therapeutic implications for the use of PI3Ki as a targeted therapeutic in B-ALL.
Methods
Flow cytometry
For integrin α6 staining, cells were treated with vehicle or copanlisib for 72 h in RPMI + 0.3% FBS. Flow cytometry analysis was performed + as previously described [4].
Adhesion assay
Recombinant human laminin 511 (Biolamina) was plated overnight on 96-well tissue culture plates (10 μg/mL). Cells (25 × 103) were plated in 100 μL of media and allowed to adhere for 20–30 min before a 45 min vehicle or copanlisib treatment.
Three-dimensional invasion assay
Prior to the invasion assay, cells were treated with vehicle or copanlisib for 72 h in RPMI + 0.3% FBS. Invasion assays were performed as previously described [4].
Mouse engraftment
Pathogen-free 6- to 8-week old male and female SCID mice were intravenously engrafted with 5 × 106 cells. All experimental procedures were approved by the Animal Care and Use Committee of Duke University and performed in accordance with the ethical guidelines.
Drug administration
Copanlisib was formulated in PEG400/H20 (20/80, v/v) acidified water solution (2% trifluoroacetic acid). For single agent studies copanlisib (14 mg/kg) was intravenously administered twice daily, twice weekly from day 1 post-engraftment. For chemotherapeutic/PI3Ki combination studies Ara-C (Hospira Inc.) was administered by intraperitoneal injection at 500 mg/m2 every day for 5 days from day 20 post-engraftment. Copanlisib (14 mg/kg) or vehicle was administered twice a day on days 21 and 25. GS-649443 (2 mg/kg) or vehicle was administered twice daily for 7 days from day 20 by oral gavage. In Nalm-6 β-catenin reporter GFP engrafted mice, Ara-C was administered as above on day 20 only and GS-649443 was administered from day 20 twice daily for 3 days by oral gavage. Paired-animal analysis and animal monitoring was performed as previously described [4].
Cell culture, immunofluorescence, Western blot, BM and CSF cell collection, histology, statistical analysis
See Supplemental Table 1 and supplemental methods for a detailed description.
Results
PI3Kδ inhibition sensitizes ALL cells to chemotherapy in vitro
We previously showed that the PI3Kδ inhibitor GS-649443, a tool compound of idelalisib optimized for bioavailability in mice, significantly improves survival in B-ALL xenograft and PDX leukemic mice by blocking integrin α6-laminin mediated CNS metastasis. Despite this striking effect on CNS migration, PI3Kδi had no effect on BM disease burden or ALL apoptosis (Supplemental Figure 1) in xenograft mice, suggesting that ALL cells may not depend on PI3Kδ for survival under normal conditions in a supportive microenvironment [4]. Indeed, we observed that Akt signaling activity downstream of PI3K was low in ALL cell lines cultured in complete media, while pAkt increased in response to serum starvation (Supplemental Figure 2). Based on these findings, we hypothesized that cytotoxic stress induced by cytarabine (Ara-C) chemotherapy would similarly induce PI3K-dependent survival responses in ALL cells, rendering them vulnerable to PI3Ki in vitro and in vivo.
Akt and GSK3β phosphorylation occur downstream of PI3K activation to regulate key anti-apoptotic signaling pathways [10]. We measured pAkt and pGSK3β in multiple ALL cell lines by Western blotting (WB) at 24, 48, and 72 h following a three hour 100 nM Ara-C pulse. We observed a time-dependent increase in both pAkt and pGSK3β (Figure 1(A) and Supplemental Figure 3). A profound increase in pAkt and pGSK3β was seen in Nalm-6 cells at 48 h, while a more modest increase in phosphorylation in RCH-ACV cells was detected at 24 h.
Figure 1.
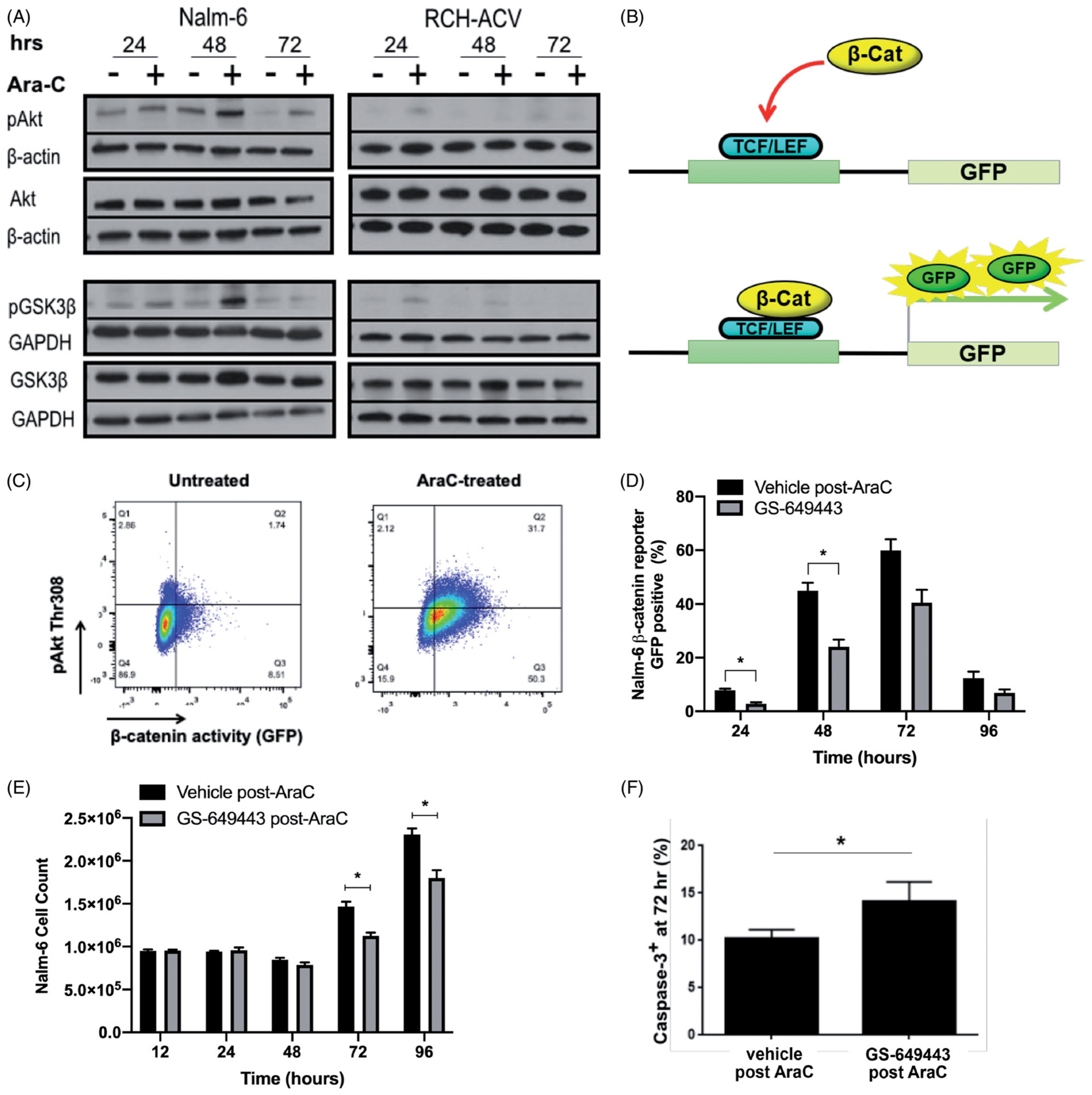
PI3Kδ inhibition chemosensitizes through blockade of Ara-C mediated Akt-dependent survival signaling. (A) Western blot analysis of Akt and GSK3β phosphorylation in Nalm-6 and RCH-ACV cell lines over time following three hour exposure to Ara-C. Images are representative of 3 biologically independent replicates. (B) Schematic illustrating β-catenin reporter GFP transcription. (C) Representative image of flow cytometry gating strategy used to detect pAkt and β-catenin reporter GFP positivity following three hour exposure of cells to Ara-C. (D) β-catenin reporter GFP positivity detected over time by flow cytometry in Nalm-6 cells treated with vehicle or GS-649443 after three hour Ara-C exposure. Data are mean ± s.e.m.; unpaired two-sided Student’s t-test, n = 3 biologically independent experiments; p < 0.05, p < 0.01. (E) Proliferation over time of Nalm-6 cells treated with vehicle or GS-649443 after three hour exposure to post-Ara-C. Data are mean ± s.e.m.; unpaired two-sided Student’s t-test, n = 3 biologically independent experiments; p < 0.05. (F) Caspase-3 positivity in Nalm-6 cells as detected by flow cytome try following 72 h treatment with vehicle or GS-649443 after three hour exposure to Ara-C. Data are mean ± s.e.m.; unpaired two-sided Student’s t-test, n = 3 biologically independent experiments; p < 0.05.
GSK3β plays an important role in initiating anti-apoptotic signaling through the Wnt/β-catenin pathway. We therefore treated Nalm-6 β-catenin reporter GFP cells with Ara-C for 3 h, and then analyzed for pAkt expression and GFP signal 24 h later using flow cytometry (Figure 1(B,C)). Consistent with the observed increase in Akt and GSK3β phosphorylation by WB, we found that the Nalm-6 cell line up-regulates β-catenin signaling in response to Ara-C treatment, and that Akt phosphorylation frequently co-occurs with β-catenin activity (Supplemental Figure 4(A)).
We next tested whether PI3Ki could prevent anti-apoptotic responses to cell stress and sensitize ALL to Ara-C chemotherapy. We treated Nalm-6 β-catenin GFP cells with a three hour Ara-C pulsation, followed by vehicle or GS-649443 treatment. Cells were analyzed at 12, 24, 48, 72 and 96 h by flow cytometry. We found that PI3Kδi decreased both β-catenin signaling activity and pAkt expression in Nalm-6 cells 24- and 48 h post-Ara-C (Figure 1(D) and Supplemental Figure 4). This correlated with a decrease in ALL proliferation and an increase in caspase-3 positive cells at 72 h, suggesting that combination treatment enhances cell kill from chemotherapy through suppression of PI3K-dependent anti-apoptotic pathways. (Figure 1(E,F)).
PI3Kδ inhibition sensitizes ALL cells to chemotherapy in vivo
To determine whether these chemosensitizing effects occurred in vivo, SCID mice were engrafted with Nalm-6 cells and treated with a 5-day regimen of Ara-C combined with 7 days of GS-649443 or vehicle beginning 20 days post-engraftment (Figure 2(A)). We found that the combination of Ara-C and GS-649443 significantly increased survival compared to Ara-C and vehicle (Figure 2(B)). We also found that while all Ara-C/vehicle treated mice developed hindlimb paralysis (HLP), a typical symptom of CNS metastasis, only 1/6 of Ara-C/GS-649443 treated mice developed HLP at clinical endpoint (Figure 2(C)).
Figure 2.
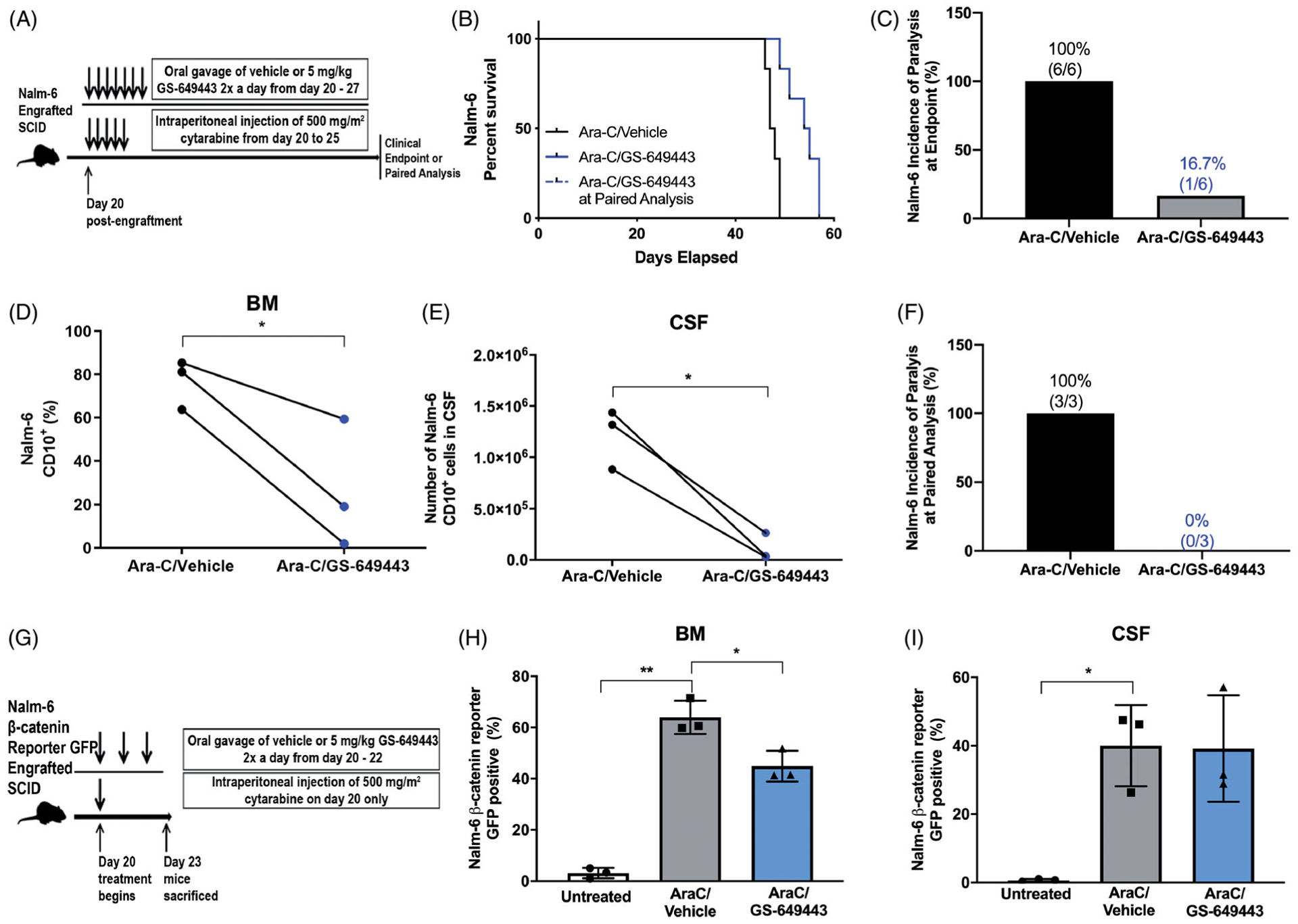
PI3Kδ inhibition potentiates ALL to cytotoxic chemotherapy in vivo, resulting in increased survival as well as decreased BM and CNS disease. (A) Schematic of Ara-C/GS-649443 treatment of leukemic mice. (B) Kaplan-Meier survival curve of Nalm-6 engrafted SCID mice treated with Ara-C/vehicle or Ara-C/GS-649443. Two-sided log rank Mantel-Cox test, n = 6 mice per group. (C) Incidence of hind-limb paralysis at clinical endpoint in Nalm-6 mice; p = 0.0023. (D and E) Disease burden in the BM (percentage of CD10+ ALL cells) and in the CNS (number of CD10+ ALL cells) of Ara-C/vehicle- and Ara-C/GS-649443-treated Nalm-6 mice euthanized at matched time points. Paired two-sided Student’s t-test, n = 3 mice per treatment group; p < 0.05. (F) Incidence of hind-limb paralysis in Ara-C/vehicle- and Ara-C/GS-649443-treated Nalm-6 engrafted mice at paired analysis. (G) Schematic of Ara-C/GS-649443 treatment of mice engrafted with Nalm-6 β-catenin reporter GFP cells. (H and I) Percentage of Nalm-6 cells reporting β-catenin activity in the BM and CNS of mice treated with Ara-C, Ara-C/vehicle or Ara-C/GS-649443. Paired two-sided Student’s t-test, n = 3 mice per treatment group; p < 0.05, p < 0.01.
Paired analysis of disease burden in mice sacrificed at matched time points revealed that compared to Ara-C alone, the combination of Ara-C and GS-649443 significantly decreased ALL blasts in the BM (Figure 2(D)). Given the minimal effect of single agent GS-649443 on B-ALL BM disease [4], these data suggest that PI3Kδi in combination with Ara-C is chemosensitizing. Moreover, although GS-649433 does not penetrate the blood-brain barrier in healthy or leukemic mice [4], the effect of Ara-C and GS-649433 combined treatment on systemic disease also served to dramatically reduce CNS metastasis. CSF blasts were significantly decreased at paired analysis, and the incidence of HLP in Ara-C/vehicle treated mice was 100% compared to 0% in the Ara-C/GS-649443 treated mice (Figure 2(E,F)).
The Wnt/β-catenin pathway is among the key anti-apoptotic signaling responses regulated by PI3K activity. To show that PI3Ki blocks Wnt/β-catenin signaling in vivo as well as in vitro, Nalm-6 β-catenin reporter GFP-engrafted SCID mice were treated with a single dose of Ara-C or vehicle 20 days post-engraftment ± 3-days of GS-649443 (Figure 2(G)). Mice were euthanized on day 4 post-treatment and CD10 + ALL cells in the BM and CSF were analyzed by flow cytometry for β-catenin reporter GFP expression (Figure 2(H,I)). We found that β-catenin activity was increased in the BM and CSF of all Ara-C treated mice. Consistent with our in vitro findings, we demonstrated that PI3Kδi significantly downregulated ALL β-catenin responses in the BM. Predictably, PI3Kδi did not change β-catenin activity in CSF blasts, as GS-649443 does not penetrate this site [4].
PI3Ki effects on α6 expression and laminin-mediated migration in B-ALL cells is mediated predominantly through the PI3Kδ isoform
Although the PI3K class I isoforms α, β, γ and δ are structurally similar, their unique roles in regulating diverse cell processes are an area of active investigation [11]. We therefore sought to determine whether PI3Kδ is the primary isoform regulating ALL integrin α6 expression and function in CNS invasion, and whether targeting additional isoforms would better control ALL proliferation and survival in the BM. We first assayed numerous B-ALL cell lines as well as primary patient blasts and found that while the δ isoform showed the highest relative expression, α, β and γ isoforms were detectable in all samples (Figure 3(A)).
Figure 3.
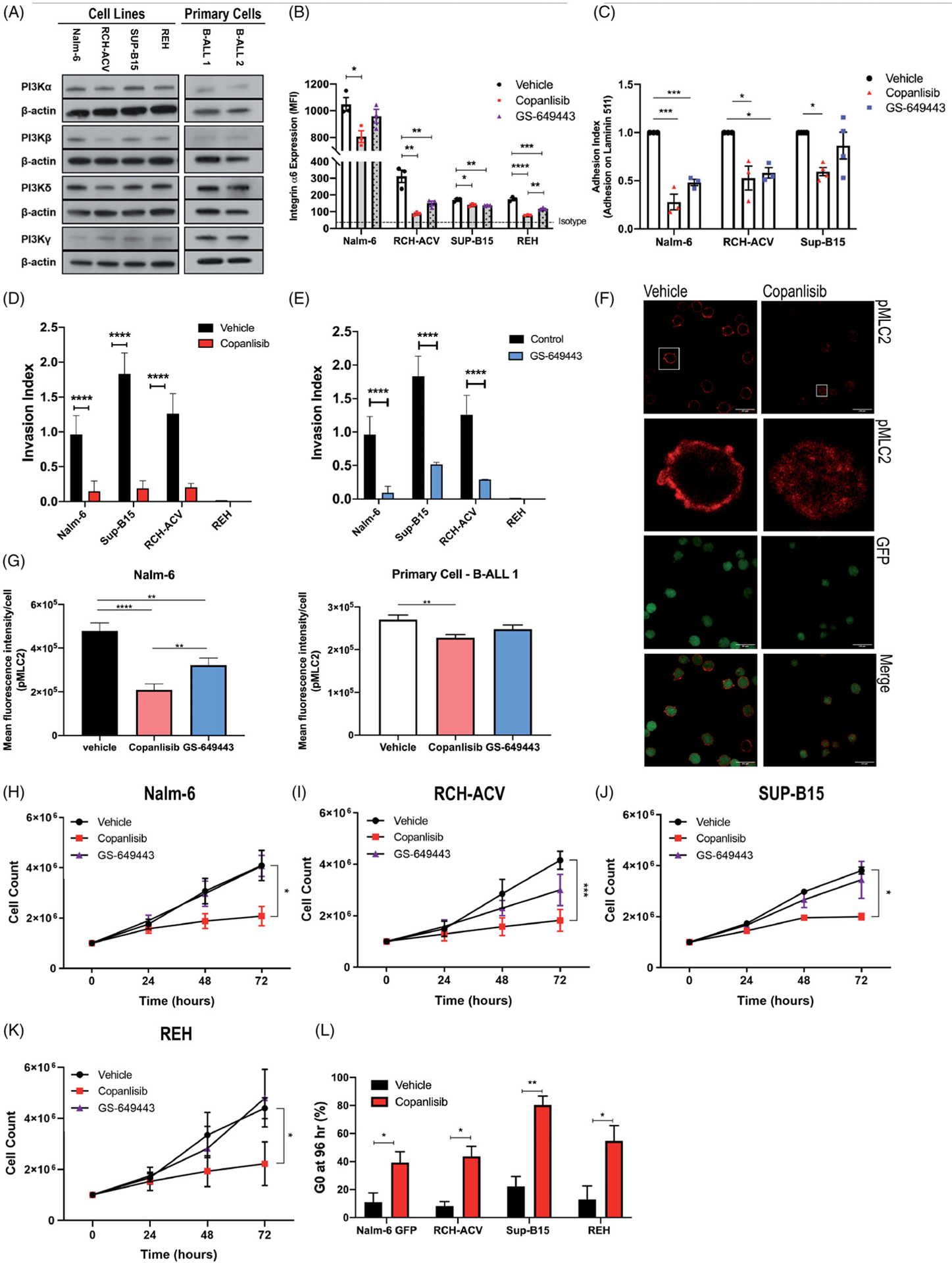
Effects of PI3Kδ vs. pan-PI3K inhibition on B-ALL cells in vitro. (A) Western blot analysis of PI3Kα, PI3βK, PI3Kδ and PI3Kc expression in B-ALL cell lines (Nalm-6, RCH-ACV, Sup-B15, and REH) and two primary patient B-ALL samples. Image is a representative of 3 biologically independent experiments. (B) Flow cytometry of integrin α6 surface expression in B-ALL cell lines after 72 h treatment with vehicle, copanlisib or GS-649443. Data are mean ± s.e.m.; ANOVA with Tukey test, n = 3 biologically independent experiments; *p<0.05, **p<0.01, ***p<0.001, ***p<0.0001. (C) Quantification of adherent B-ALL cells on laminin 511-coated plates and stained with anti-pMLC2 (red) following vehicle or copanlisib treatment. (G) Mean pixel density quantification of pMLC2 staining in Nalm-6 or primary patient B-ALL cells bound to laminin 511-coated plates (from F). Data are mean ± s.e.m.; unpaired two-sided Student’s t-test; p < 0.0001. Nalm-6 cells, n = 100; primary patient B-ALL cells, n 200. (H – K), B-ALL cell proliferation over 72 hrs of treatment with vehicle, GS-649443, or=copanlisib. Data are mean ± s.e.m.;=unpaired two-sided Student’s t-test at 72 h, n = 3 biologically independent experiments; p < 0.05, p < 0.001. (L) Flow cytometry analysis using Hoechst and Ki67 staining shows a G0 cell cycle arrest in B-ALL cell lines following 72 h of treatment with copanlisib. Data are mean ± s.e.m.; unpaired two-sided Student’s t-test, n = 3 biologically independent experiments; p < 0.05, p < 0.01.
We treated the B-ALL cell lines with copanlisib or GS-649443 and analyzed α6 integrin cell surface expression levels by flow cytometry (Figure 3(B)). Both copanlisib and GS-649443 treatment decreased B-ALL α6 integrin expression compared to vehicle. In the REH cell line, copanlisib treatment elicited a statistically significant, though small absolute greater reduction in α6 integrin expression compared to GS-649443. In all other B-ALL cell lines, however, no significant difference between pan- vs. delta-PI3Ki was observed, suggesting that α6 expression is principally regulated through the PI3Kδ isoform (Figure 3(B)).
We next compared the effect of pan- vs. PI3Kδi on functional responses to laminin in vitro. Pan-PI3Ki with copanlisib significantly decreased B-ALL adhesion to laminin as well as invasion along laminin-containing matrices, though results were similar to those obtained with PI3Kδi. (Figure 3(C–E)). We previously showed that PI3Kδi of ALL cell migration is mediated through reduction in pMLC2, which regulates actinomysin contractility downstream of extra-cellular matrix engagement [4,12–14]. Consistent with this, we found that pan-PI3Ki more potently decreased MLC2 phosphorylation in Nalm-6 and primary ALL cells bound to laminin compared to PI3Kδi alone (Figure 3(F,G)).
Pan-PI3Ki, but not PI3Kδi, potently blocks ALL cell proliferation through induction of G0 cell cycle arrest
PI3K signaling is known to regulate cell cycle machinery [15,16], yet our prior in vivo study of single agent GS-649443 showed no effect on BM burden in xenograft mice [4]. Furthermore, analysis of BM blasts from vehicle vs. GS-649443 treated mice revealed only modest effects on the proportion of cells in G1 vs. S phase (Supplemental Figure 5). Previous in vitro work has shown that idelalisib can slow B-ALL proliferation, though these studies were performed at micromolar concentrations of drug that are well above its IC50 of 0.7 nM and are unattainable in vivo [17,18]. While single agent PI3Kδi thus appears ineffective to control BALL proliferation, we sought to determine whether pan-PI3Ki could impact B-ALL growth at clinically achievable nM concentrations [19]. We found that 100 nM copanlisib substantially arrested B-ALL growth, yet PI3Kδi with 100 nM 10 μM GS-649443 showed modest or no effects on B-ALL proliferation in vitro (Figure 3(H–K) and Supplemental Figure 6). These data suggest that PI3Kδi alone does not significantly alter ALL cell proliferation, due either to compensatory upregulation of other isoform activity, or because PI3K-mediated growth effects are predominantly mediated through α, β or γ downstream pathways.
We next measured cell cycle and apoptotic indices of cultured cells by flow cytometry. Hoechst and Ki67 staining revealed a significant increase in the proportion of cells in quiescent G0 phase (Figure 3(L)) and a decrease in S and G2/M phases for most B-ALL cell lines treated with copanlisib compared to vehicle (Supplemental Figure 7). Annexin V and PI/7-AAD staining did not demonstrate an increase in apoptosis (Supplemental Figure 8), suggesting that the principal cause of decreased proliferation was G0 arrest.
Pan-PI3Ki augments stress-induced B-ALL cell death in vitro
In addition to the growth inhibitory and CNS prophylaxis potential for pan-PI3Ki we next sought to confirm that copanlisib could also serve as a potent chemo-sensitizing agent in vitro and in vivo. In proliferation assays, the combination of Ara-C and copanlisib more effectively inhibited growth of Nalm-6 and RCH-ACV cells compared to Ara-C alone (Figure 4(A,B)). To determine whether copanlisib was acting to block anti-apoptotic responses to chemotherapy, we measured cell death by Annexin V and PI/7-AAD staining. We found that copanlisib treatment following Ara-C pulsation enhanced apoptosis of Nalm-6 cells compared to Ara-C alone (Figure 4(C)). Nalm-6 β-catenin reporter activity also decreased when cells were treated with copanlisib following Ara-C pulsation compared to Ara-C alone, suggesting that pan-PI3Ki can attenuate chemotherapy-induced stress response (Figure 4(D)).
Figure 4.
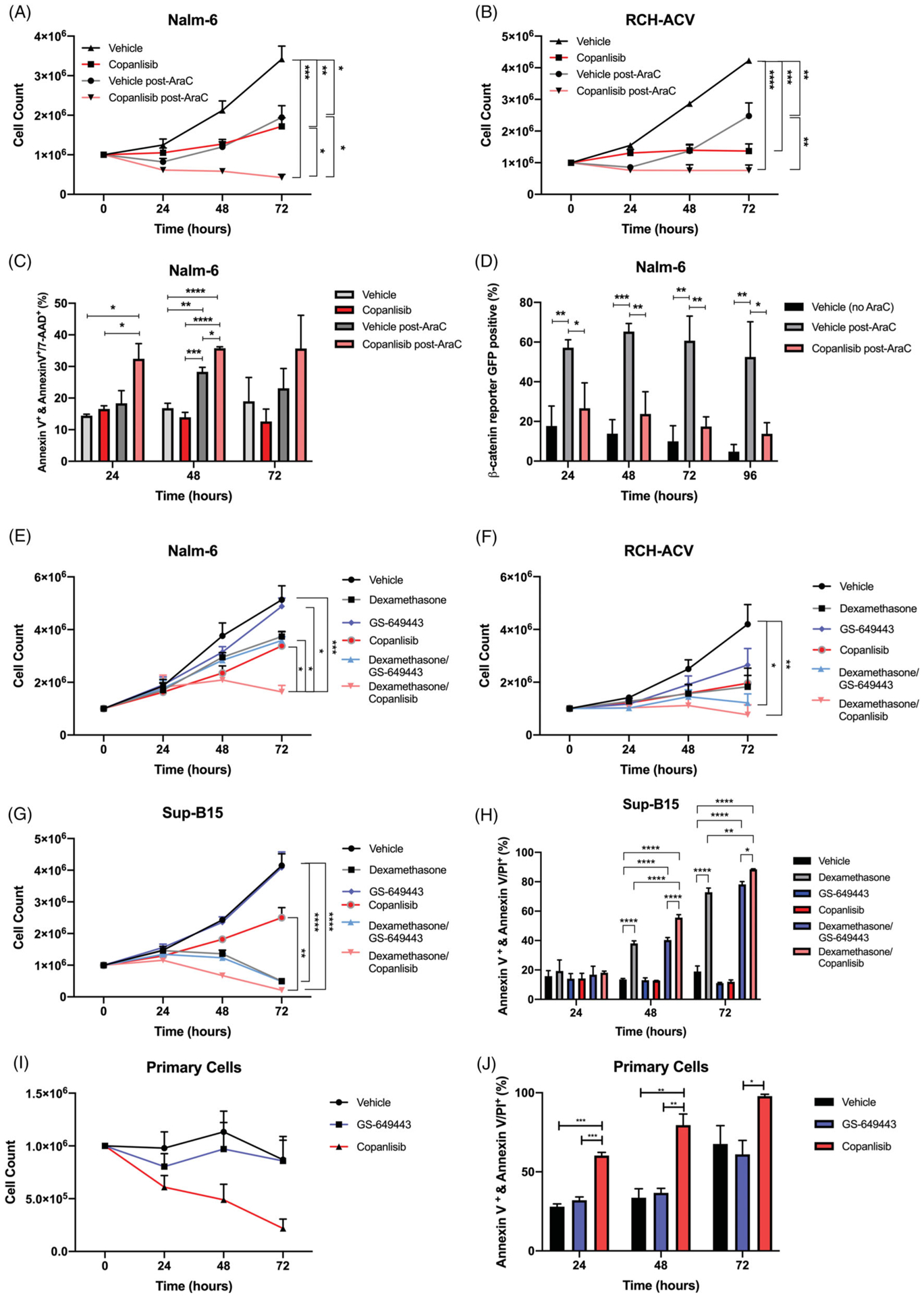
Pan-PI3Ki augments chemotherapy and steroid-induced B-ALL cell death in vitro. (A and B) Proliferation of Nalm-6 and RCH-ACV cells treated with vehicle or copanlisib after three hour exposure to Ara-C or PBS. Data are mean ± s.e.m.; ANOVA with Tukey test at 72 h, n = 3 biologically independent experiments; p < 0.05, p < 0.01, p < 0.001, p < 0.0001. (C) Apoptosis in Nalm-6 cells as detected by Annexin V+Annexin V/PI or Annexin V/7-AAD. Cells were treated with vehicle or copanlisib alone or vehicle or copanlisib after three hour exposure to Ara-C. Data are mean ± s.e.m.; ANOVA with Tukey test, n 3 biologically independent experiments; p < 0.05, p < 0.01, p < 0.001, p < 0.0001. (D) β-catenin reporter GFP positivity detected over time by flow cytometry in Nalm-6 cells treated with vehicle or copanlisib after three hour exposure to Ara-C. Data are mean ± s.e.m.; unpaired two-sided Student’s t-test, n = 3 biologically independent experiments; p < 0.05, p < 0.01. (E–G) Proliferation of Nalm-6, RCH-ACV, or Sup-B15 cells treated with vehicle, GS-649443, or copanlisib alone or in combination with dexamethasone. Data are mean ± s.e.m.; ANOVA with Tukey test at 72 h, n = 3 biologically independent experiments; p <0.05, **p < 0.01, p < 0.001, p < 0.0001. (H) Apoptosis in Sup-B15 cells as detected by Annexin V+Annexin V/PI or Annexin V/7-AAD. Cells were treated with vehicle, GS-649443, or copanlisib alone or in combination with dexamethasone. Data are mean ± s.e.m.; ANOVA with Tukey test, n = 3 biologically independent experiments; p < 0.05, p < 0.01, p < 0.0001. (I) Proliferation of primary patient B-ALL cells treated with vehicle, GS-649443, or copanlisib. Data are mean ± s.e.m. (J) Apoptosis in primary patient B-ALL cells as detected by Annexin V+Annexin V/PI. Cells were treated vehicle, GS-649443, or copanlisib. Data are mean ± s.e.m.
In addition to cytotoxic chemotherapies, glucocorticoids such as dexamethasone are an important component of ALL treatment regimens known to induce apoptotic cell death in lymphoblasts [20]. Though not well understood, some evidence suggests that glucocorticoid resistance is mediated by Akt activation and that PI3K/mTOR inhibiton may sensitize ALL cells to steroids [21–24]. To determine if pan-PI3K or PI3Kδ inhibitors could enhance B-ALL steroid-induced death at clinically relevant concentrations, we treated multiple B-ALL lines with dexamethasone ± 100 nM copanlisib or GS-649443. We found that PI3Kδi had minimal effects on steroid-induced apoptosis, while pan-PI3Ki effectively sensitized cells to dexamethasone (Figure 4(E–H)).
Although ALL cell lines are selected for microenvironment-independent growth, most primary ALL cells suffer significant cytotoxic stress ex vivo. We therefore predicted that, even when cultured in optimal conditions, primary B-ALL cells would demonstrate sensitivity to PI3Ki. We observed that primary B-ALL cultured with the pan-PI3Ki copanlisib rapidly underwent apoptosis (Figure 4(I,J) and Supplemental Figure 9). Interestingly, selective PI3Kδi did not induce apoptosis, suggesting that, primary ALL cells in culture, similar to B-ALL cell lines exposed to steroids, depend on non-δ isoform anti-apoptotic PI3K activity.
Pan-PI3Ki decreases CNS invasion, inhibits BM disease progression, and improves response to chemotherapy in leukemic mice
We next tested the effects of copanlisib on disease progression in leukemic mice. In contrast to a half-life of 39.1 h in humans, copanlisib is rapidly metabolized in mice (t1/2~0.7 h) [25]. We treated RCH-ACV or Nalm-6 engrafted SCID mice twice daily, twice weekly with 14 mg/kg copanlisib, a previously described [26] intermittent dosing regimen (Figure 5(A)). In RCH-ACV engrafted mice, copanlisib treatment significantly improved survival (Figure 5(B)). The difference in survival suggests a benefit compared to our previously published survival data using GS-649443, though this was not statistically significant (data not shown) [4]. We also observed a decreased incidence of HLP at clinical endpoint in the copanlisib treated mice (50% of mice) compared to the vehicle (100% of mice), indicating that copanlisib attenuates CNS metastasis (Figure 5(C)).
Figure 5.
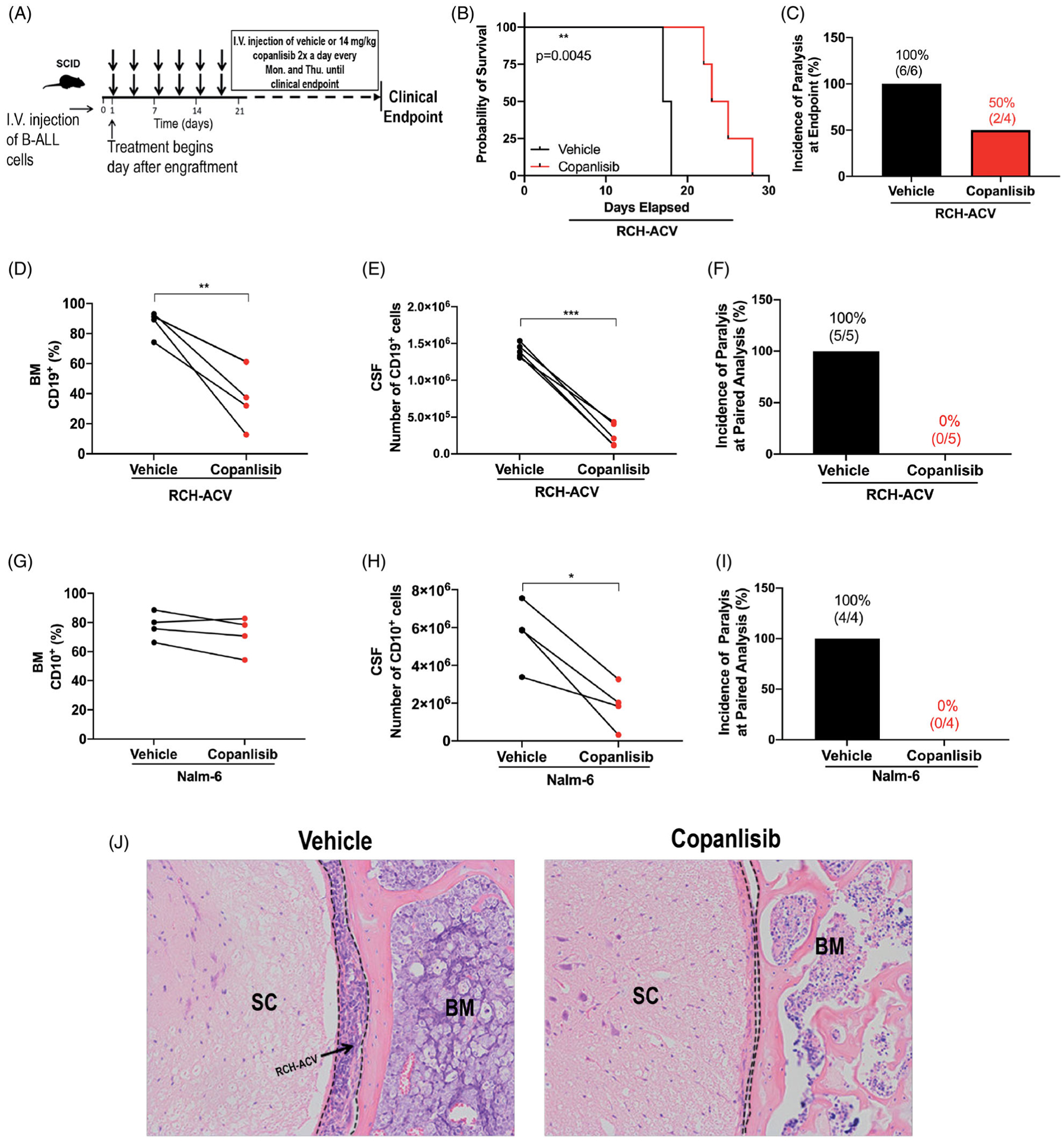
Copanlisib decreases CNS invasion, inhibits BM disease progression, and improves response to chemotherapy in leukemic mice. (A) Schematic of copanlisib treatment of leukemic mice. (B) Kaplan-Meier survival curve of RCH-ACV engrafted SCID mice treated with copanlisib vs. vehicle. Two-sided log rank Mantel-Cox test, vehicle: n = 6 mice, copanlisib: n = 4 mice; p = 0.0045. (C) Incidence of hind-limb paralysis at clinical endpoint in RCH-ACV engrafted mice. (D and E) Disease burden in the BM (percentage of CD19+ ALL cells) and in the CNS (number of CD19+ ALL cells) of vehicle- and copanlisib-treated RCH-ACV engrafted mice euthanized at matched time points. Paired two-sided Student’s t-test, n 5 mice per treatment group; p < 0.01, p < 0.001. (F) Incidence of hind-limb paralysis in RCH-ACV engrafted mice at paired=analysis. (G and H) Disease burden in the BM (percentage of CD10+ ALL cells) and in the CNS (number of CD10+ ALL cells) of vehicle- and copanlisib-treated Nalm-6 engrafted mice euthanized at matched time points. Paired two-sided Student’s t-test, n = 4 mice per treatment group; p < 0.05. (I) Incidence of hind-limb paralysis in Nalm-6 engrafted mice at paired analysis. (J) RCH-ACV ALL cells in subarachnoid space (dashed line) of the spinal cord (SC) at paired analysis. Representative histologic sections of the vertebrae are shown. H&E, hematoxylin and eosin. n = 5 mice per treatment group.
To quantify the effect of pan-PI3Ki on systemic and CNS disease burden, we measured lymphoblast involvement in the BM and CSF of copanlisib or vehicle-treated mice at matched timepoints. In contrast to our previously published results showing a minimal effect of single agent PI3Kδi on systemic disease [4], we found that copanlisib significantly decreased disease in the BM of RCH-ACV engrafted mice at matched timepoints (Figure 5(D)). CNS involvement was even more markedly decreased (Figure 5(E)). This was reflected by a 0% incidence of paralysis at paired analysis in the copanlisib treated mice compared to 100% in the vehicle arm (Figure 5(F)). In the Nalm-6 model, there was a trend toward decreased BM disease burden in the copanlisib treatment arm, (Figure 5(G)) in contrast again to prior data showing no effect of PI3Kδi on BM blasts (Supplemental Figure 10) [4]. There was, however, a significant decrease in CNS involvement and correspondingly, no copanlisib treated mice developed HLP (Figure 5(H–J)).
To model the effect of combining pan-PI3Ki with induction chemotherapy, we tested whether a brief pulse of copanlisib in combination with Ara-C could decrease BM MRD compared to chemotherapy alone. We treated Nalm-6 engrafted SCID mice with Ara-C for 5 days beginning 20 days post-engraftment (Figure 6(A)). The mice were treated with 2 doses of copanlisib overlapping with chemotherapy. Paired analysis showed that treatment with Ara-C in combination with pan-PI3Ki significantly decreased BM disease burden and reduced CNS metastasis (Figure 6(B,C)). The incidence of HLP at paired analysis was 0% in the Ara-C/copanlisib treated mice compared to 100% in the Ara-C/vehicle treated mice (Figure 6(D)). Finally, endpoint analysis revealed that a brief pulse of copanlisib in combination with Ara-C significantly prolonged survival (Figure 6(E)).
Figure 6.
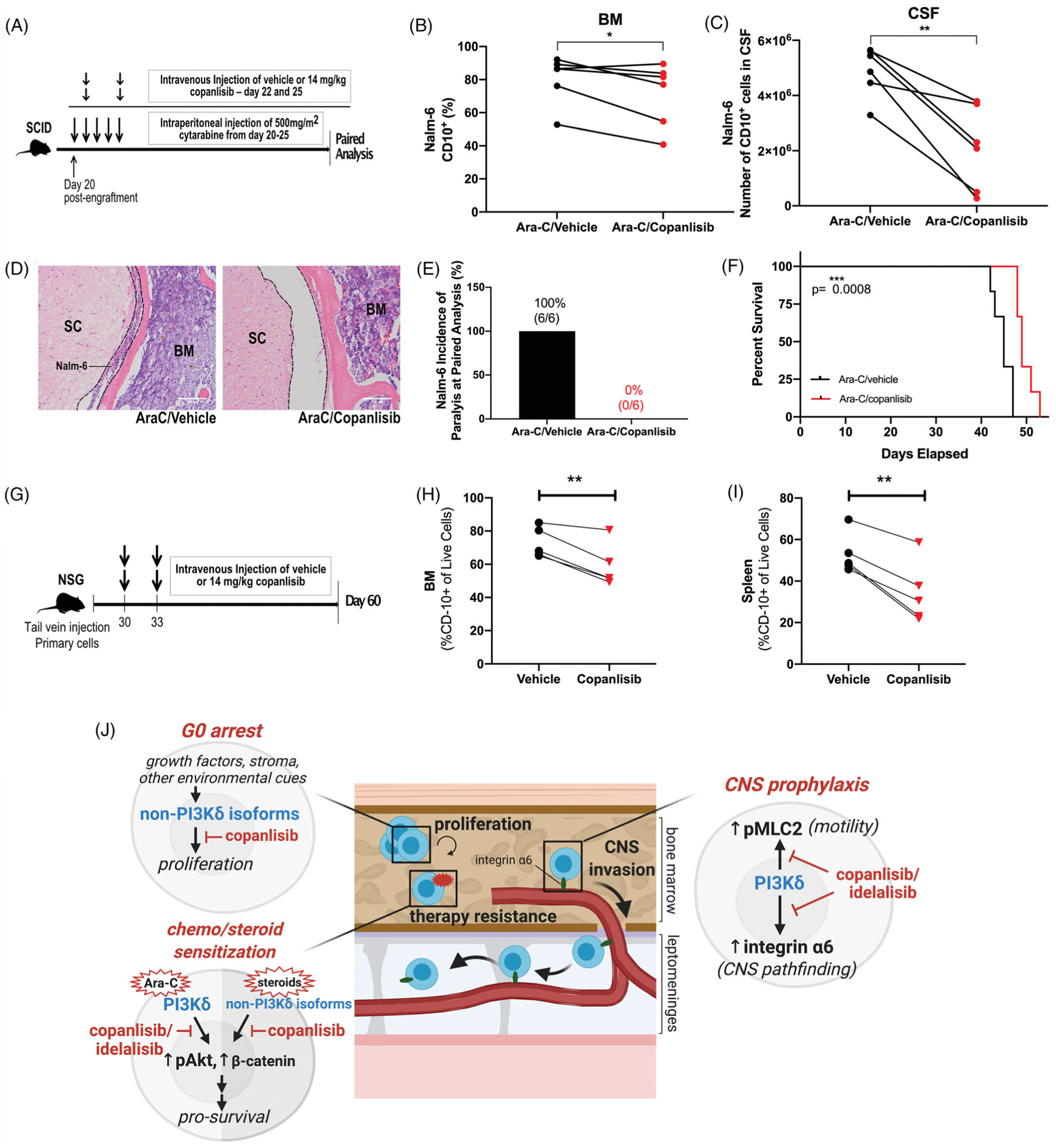
Copanlisib potentiates ALL to cytotoxic chemotherapy in vivo, resulting in increased survival as well as decreased BM and CNS disease. (A) Schematic of Ara-C/copanlisib treatment of leukemic mice. (B and C) Disease burden in the BM (percentage of CD10+ ALL cells) and in the CNS (number of CD10+ ALL cells) of Ara-C/vehicle and Ara-C/copanlisib-treated Nalm-6 engrafted mice euthanized at matched time points. Paired two-sided Student’s t-test, n = 6 mice per treatment group; p < 0.05, p < 0.01. (D) Nalm-6 ALL cells in subarachnoid space (dashed line) of the spinal cord (SC) at paired analysis following treatment with Ara-C/ vehicle and Ara-C/copanlisib. Representative sections of the vertebrae are shown. H&E, hematoxylin and eosin. n = 6 mice per treatment group. (E) Incidence of hind-limb paralysis in Ara-C/vehicle- and Ara-C/copanlisib-treated Nalm-6 engrafted mice at paired analysis. (F) Kaplan-Meier survival curve of Nalm-6 (GFP) engrafted SCID mice treated with Ara-C/vehicle or Ara-C/copanlisib. Two-sided log rank Mantel-Cox test, n = 6 mice per group, p < 0.001. (G) Schematic of copanlisib treatment of PDX leukemic mice. (H–I) Disease burden in the BM and spleen (percentage of CD10+ ALL cells) of vehicle and copanlisib treated primary B-ALL engrafted NSG mice at day 60. Paired two-sided Student’s t-test, n = 5 mice per treatment group; p < 0.01 (J) Model of PI3Ki action in B-ALL: PI3K-dependent ALL cell proliferation in the BM is predominantly mediated by PI3Kα/β/γ isoforms, with pan-PI3K inhibition via copanlisib leading to a G0 cell cycle arrest. PI3Kδ and PI3Kα/β/γ signaling activate Akt-dependent pro-survival pathways conferring resistance to chemotherapy or steroids respectfully. Lastly, PI3Kδ signaling leads to the upregulation of integrin α6 and the activation of MLC2-dependent motility, allowing for invasion along laminin+emissary vessels that passage from the calvarial and vertebral BM into the leptomeninges.
Primary ALL cells typically proliferate slower in mice than ALL cell lines, suggesting they are less adapted to the host microenvironment. Cellular stress in this setting likely created dependencies on anti-apoptotic signaling pathways, including those downstream of PI3K. We therefore hypothesized that primary B-ALL in PDX mice could be sensitive to a short pulse of PI3Ki, even in the absence of cytotoxic chemotherapy. NSG mice at 30 days post-engraftment with primary B-ALL cells were thus given 2 days of copanlisib 3 days apart and sacrificed on day 60. Quantification of CD10+ blasts demon strated significantly decreased tumor burden in the BM and spleen one month after this brief exposure to copanlisib (Figure 6(H,I)).
Discussion
Current management of ALL relies on intensive drug regimens that combine multiple lymphotoxic agents including steroids and cytotoxic chemotherapeutics [1,27,28]. Despite high response rates, the majority of adult patients will suffer fatal disease relapse arising from minimal residual disease (MRD) unresponsive to the initial regimen [28–30]. While some of these patients will be successfully treated with salvage regimens, those that experience disease relapse in the CNS are almost uniformly incurable [3,31]. For this reason, new, molecularly targeted approaches to eradicate MRD and prevent CNS metastasis are critical areas of unmet need.
PI3Ki has shown substantial efficacy in indolent B cell malignancies but has yet to occupy a role in ALL treatment [19,25,32]. PI3K signaling is, however, a highly attractive therapeutic target in B-ALL given its important role at the crossroads of microenvironment signals that have the potential to protect MRD and fuel tumor invasiveness. PI3K lies downstream of an array of integrin, cytokine, and chemokine receptors that are critical mediators of stromal and other host tissue signals. These signals are capable of inducing ALL motility, proliferation, and anti-apoptotic survival programs [9,11,15,33], and their disruption may enhance current therapies without introducing overlapping toxicities.
While prior in vitro studies have shown the potential benefit of PI3Ki in B-ALL[17,18,34–36], this study demonstrates for the first time in vivo, in B-ALL mouse models that PI3Ki can serve a multifunctional role counteracting migration, proliferation, and chemotherapy-induced survival responses. We show that both PI3Kδi and pan-PI3Ki block Akt-independent motility pathways that lead to CNS invasion, as well as Akt-dependent anti-apoptotic signaling. While PI3Kδ appears to play a minor role in cell cycle progression, we found that pan-PI3Ki in contrast slows B-ALL growth by inducing a G0 cell cycle arrest. Compared to our previously published findings in B-ALL mice treated with single agent PI3Kδi GS-6494443 [4], our current data suggest that pan-PI3Ki with copanlisib produces improved BM responses and a deeper CNS prophylactic benefit. This was observed despite the relatively poor pharmacokinetic profile of copanlisib in mice relative to GS-649443, a PI3Kδ inhibitor optimized for murine use. We therefore surmise that the sustained levels of PI3Ki achieved in humans after copanlisib dosing [37] could lead to substantial therapeutic responses in patients. Our findings also suggest that biomarkers of CNS metastasis potential (integrin α6 expression), chemosensitization (Akt/GSK3β phosphorylation), and proliferative response (Ki67) could predict clinical responses and guide the use of PI3Ki in specific patients. We therefore propose that pan-PI3Ki is a promising tool that can be incorporated in current regimens to target B-ALL cells in multiple ways - as a CNS prophylaxis agent, as a maintenance therapeutic to slow the growth of systemic disease, and alongside standard chemotherapy to enhance tumor cell death (Figure 6(J)).
Lastly, an unexpected finding was that a single week of PI3Ki in combination with Ara-C substantially decreased CNS disease progression in mice. Future research on the effect of brief or intermittent versus continuous treatment with PI3Ki on ALL CNS invasion will be helpful in limiting adverse effects and maximizing patient outcomes. These encouraging data also warrant further investigation into the potential of PI3Ki to sensitize ALL cells to other standard of care agents. Ongoing studies to understand differential effects of PI3Kδ versus pan-PI3Ki on host immune cell populations, including T regulatory cells, will also guide our use of these agents in the clinic and potential combination with immune modulating therapies. In summary, our data show that pan-PI3Ki may represent a feasible, multidimensional, and synergistic approach to the significant hurdles of B-ALL MRD eradication and CNS prophylaxis.
Supplementary Material
Ackowledgements
We thank Zhiguo Li and Kevin Brennan for consultation on statistical analysis. Figure 6(J) schematic created with BioRender.
Funding
This work was supported by the Error! Hyperlink reference not valid. and by NIH/NCI R01-CA234580. GS-649443 was provided by Stacey Tannheimer at Gilead Sciences, Inc.
Footnotes
Disclosure statement
We are reporting that we have received the drug, GS-649443, from Gilead Sciences, Inc.
Data availability statement
Please contact the corresponding author for original data and protocols.
References
- [1].Oriol A, Vives S, Hernández-Rivas J-M, et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA study group. Haematologica. 2010;95(4):589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Surapaneni UR, Cortes JE, Thomas D, et al. Central nervous system relapse in adults with acute lymphoblastic leukemia. Cancer. 2002;94(3):773–779. [DOI] [PubMed] [Google Scholar]
- [3].Larson RA. Managing CNS disease in adults with acute lymphoblastic leukemia. Leuk Lymphoma. 2018; 59(1):3–13. January [DOI] [PubMed] [Google Scholar]
- [4].Yao H, Price TT, Cantelli G, et al. Leukaemia hijacks a neural mechanism to invade the central nervous system. Nature. 2018;560(7716):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Coustan-Smith E, Song G, Clark C, et al. New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2011;117(2): 6267–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].DiGiuseppe JA, Fuller SG, Borowitz MJ. Overexpression of CD49f in precursor B-cell acute lymphoblastic leukemia: potential usefulness in minimal residual disease detection. Cytometry B Clin Cytom. 2009;76(2):150–155. [DOI] [PubMed] [Google Scholar]
- [7].Gang EJ, Kim HN, Hsieh YT, et al. Integrin α6 mediates drug resistance of acute lymphoblastic B-cell leukemia. Blood. 2020;136:210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Reif K, Okkenhaug K, Sasaki T, et al. Cutting edge: differential roles for phosphoinositide 3-kinases, p110γ and p110δ, in lymphocyte chemotaxis and homing. J Immunol. 2004;173(4):2236. [DOI] [PubMed] [Google Scholar]
- [9].Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15(1): 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Franke TF, Hornik CP, Segev L, et al. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22(56): 8983–8998. [DOI] [PubMed] [Google Scholar]
- [11].Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, et al. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010. May;11(5): 329–341. [DOI] [PubMed] [Google Scholar]
- [12].Hanna S, El-Sibai M. Signaling networks of Rho GTPases in cell motility. Cell Signalling. 2013;25(10): 1955–1961. [DOI] [PubMed] [Google Scholar]
- [13].Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Reiske HR, Kao S-C, Cary LA, et al. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J Biol Chem. 1999; 274(18):12361–12366. [DOI] [PubMed] [Google Scholar]
- [15].Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. [DOI] [PubMed] [Google Scholar]
- [16].Katso R, Okkenhaug K, Ahmadi K, et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. [DOI] [PubMed] [Google Scholar]
- [17].Evangelisti C, Cappellini A, Oliveira M, et al. Phosphatidylinositol 3-kinase inhibition potentiates glucocorticoid response in B-cell acute lymphoblastic leukemia. J Cell Physiol. 2018;233(3):1796–1811. [DOI] [PubMed] [Google Scholar]
- [18].Lannutti BJ, Meadows SA, Herman SEM, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Patnaik A, Appleman LJ, Tolcher AW, et al. First-inhuman phase I study of copanlisib (BAY 80–6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol. 2016;27(10): 1928–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Smith LK, Cidlowski JA. Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Prog Brain Res. 2010;182:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hall CP, Reynolds CP, Kang MH. Modulation of glucocorticoid resistance in pediatric T-cell acute lymphoblastic leukemia by increasing BIM expression with the PI3K/mTOR inhibitor BEZ235. Clin Cancer Res. 2016;22(3):621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Safaroghli-Azar A, Bashash D, Sadreazami P, et al. PI3K-δ inhibition using CAL-101 exerts apoptotic effects and increases doxorubicin-induced cell death in pre-B-acute lymphoblastic leukemia cells. Anti Cancer Drugs. 2017;28(4):436–445. [DOI] [PubMed] [Google Scholar]
- [23].Silveira AB, Laranjeira ABA, Rodrigues GOL, et al. PI3K inhibition synergizes with glucocorticoids but antagonizes with methotrexate in T-cell acute lymphoblastic leukemia. Oncotarget. 2015;6(15):13105–13118. Vol [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xie M, Yang A, Ma J, et al. Akt2 mediates glucocorticoid resistance in lymphoid malignancies through FoxO3a/Bim axis and serves as a direct target for resistance reversal. Cell Death Dis. 2019;9(10): 1013–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Krause G, Hassenrück F, Hallek M. Copanlisib for treatment of B-cell malignancies: the development of a PI3K inhibitor with considerable differences to idelalisib. Drug Des Devel Ther. 2018;12:2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu N, Rowley BR, Bull CO, et al. BAY 80–6946 is a highly selective intravenous PI3K inhibitor with potent p110 alpha and p110delta activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12(11): 2319–2330. [DOI] [PubMed] [Google Scholar]
- [27].Gökbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120(9):1868–1876. [DOI] [PubMed] [Google Scholar]
- [28].Jabbour E, Short NJ, Jorgensen JL, et al. Differential impact of minimal residual disease negativity according to the salvage status in patients with relapsed/ refractory B-cell acute lymphoblastic leukemia. Cancer. 2017;123(2):294–302. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jabbour E, Gökbuget N, Advani A, et al. Impact of minimal residual disease status in patients with relapsed/refractory acute lymphoblastic leukemia treated with inotuzumab ozogamicin in the phase III INO-VATE trial. Leuk Res. 2020;88:106283–105835. [DOI] [PubMed] [Google Scholar]
- [30].Short NJ, Jabbour E. Minimal residual disease in acute lymphoblastic leukemia: how to recognize and treat it. Curr Oncol Rep. 2017;19(1):1534–6269. ( [DOI] [PubMed] [Google Scholar]
- [31].Pui C-H, Howard SC. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol. 2008;9(3):257–268. [DOI] [PubMed] [Google Scholar]
- [32].Miller BW, Przepiorka D, de Claro RA, et al. FDA approval: idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin Cancer Res. 2015;21(7): 1525–1529. April 1 [DOI] [PubMed] [Google Scholar]
- [33].Liu P, Cheng H, Roberts TM, et al. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lonetti A, Cappellini A, Spart a AM, et al. PI3K pan-inhibition impairs more efficiently proliferation and survival of T-cell acute lymphoblastic leukemia cell lines when compared to isoform-selective PI3K inhibitors. Oncotarget. 2015;6(12):10399–10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sklarz LM, Gladbach YS, Ernst M, et al. Combination of the PI3K inhibitor Idelalisib with the conventional cytostatics cytarabine and dexamethasone leads to changes in pathway activation that induce anti-proliferative effects in B lymphoblastic leukaemia cell lines. Cancer Cell Int. 2020;20(1):390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bashash D, Safaroghli-Azar A, Delshad M, et al. Inhibitor of pan class-I PI3K induces differentially apoptotic pathways in acute leukemia cells: shedding new light on NVP-BKM120 mechanism of action. Int J Biochem Cell Biol. 2016;79:308–317. [DOI] [PubMed] [Google Scholar]
- [37].Dreyling M, Santoro A, Mollica L, et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol. 2017;35(35):3898–3905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Please contact the corresponding author for original data and protocols.