

Abstract
Breast cancer metastatic relapse can occur years after therapy, indicating that disseminated breast cancer cells (BCCs) have a prolonged dormant phase before becoming proliferative. A major site of disease dissemination and relapse is bone, although the critical signals that allow circulating BCCs to identify bone microvasculature, enter tissue, and tether to the microenvironment are poorly understood. Using real-time in vivo microscopy of bone marrow (BM) in a breast cancer xenograft model, we show that dormant and proliferating BCCs occupy distinct areas, with dormant BCCs predominantly found in E-selectin– and stromal cell–derived factor 1 (SDF-1)–rich perisinusoidal vascular regions. We use highly specific inhibitors of E-selectin and C-X-C chemokine receptor type 4 (CXCR4) (SDF-1 receptor) to demonstrate that E-selectin and SDF-1 orchestrate opposing roles in BCC trafficking. Whereas E-selectin interactions are critical for allowing BCC entry into the BM, the SDF-1/CXCR4 interaction anchors BCCs to the microenvironment, and its inhibition induces mobilization of dormant micrometastases into circulation. Homing studies with primary BCCs also demonstrate that E-selectin regulates their entry into bone through the sinusoidal niche, and immunohistochemical staining of patient BMs shows dormant micrometastatic disease adjacent to SDF-1+ vasculature. These findings shed light on how BCCs traffic within the host, and suggest that simultaneous blockade of CXCR4 and E-selectin in patients could molecularly excise dormant micrometastases from the protective BM environment, preventing their emergence as relapsed disease.
INTRODUCTION
Patients diagnosed with localized breast cancer, even when treated with adjuvant therapy, remain at risk for late metastatic relapse, most commonly in bone (1). Clinically silent bone marrow (BM) micrometastases can, in fact, be detected in 30% of breast cancer patients with stage I to III disease and predict the likelihood of disease relapse (2). These data suggest that the BM is a particularly attractive location for micrometastases and that characteristics of the BM microenvironment can maintain tumor dormancy for extended periods (3).
Despite our growing understanding of breast cancer cell (BCC)–microenvironment interactions, it is unknown how metastatic BCCs enter the bone. Moreover, circulating tumor cells (CTCs), which may be enriched with cancer stem cells (CSCs), can be detected in the peripheral blood of most metastatic patients and also in early-stage patients even after resection of the primary tumor (4–8). These findings suggest that multidirectional trafficking of BCCs at the metastatic site is a feature of both dormant and clinically apparent metastatic disease. Although the molecular mechanisms that BCCs use to exit the bone are still undefined, CTC frequency does appear to correlate with increasing metastatic disease burden (6).
Understanding the trafficking of dormant BCCs within the bone has been challenging because these events are both rapid and rare. To examine the molecular basis of BCC bone transit, we used high-resolution, real-time fluorescence microscopy to track BCC migration through the calvarial BM in mouse xenograft models of human BCC metastasis. By combining constitutively expressed fluorescent protein labels with cell membrane dyes that are diluted to nondetectable concentrations in proliferating cells, we were able to discriminate the precise anatomic location and movement of dormant versus proliferative metastases (9). In addition, our results were corroborated by genomic, immunohistochemical, and functional analysis of primary patient samples. Together, our findings suggest a molecular framework for metastatic BCC bone migration that is associated with the tumor’s dormancy status.
RESULTS
Genes encoding CXCR4, E-selectin ligands, and their synthetic enzymes are up-regulated in late relapse breast cancer
BCCs express proteins reported to act as E-selectin ligands, as well as the stromal cell–derived factor 1 (SDF-1) receptor, C-X-C chemokine receptor type 4 (CXCR4), and metastatic growth in mouse models can be impaired by inhibition of E-selectin or SDF-1 interactions (10–15). Despite these observations, the specific roles of these molecules in breast cancer progression are undefined. Work from our laboratory has demonstrated that benign and malignant hematopoietic cells enter the BM specifically through a distinct sinusoidal vasculature that has high basal expression of the inflammatory molecules E-selectin and SDF-1 (16). In addition to regulating hematopoietic transit, these sinusoidal and perisinusoidal vascular regions, or niches, are prodormancy microenvironments for self-renewing hematopoietic stem cells (17, 18). We hypothesized that circulating BCCs hijack SDF-1+E-selectin+ hematopoietic vascular gateways to identify and enter the bone and that primary tumor cells that were particularly capable of homing to this prodormancy BM microenvironment would be more likely to produce late relapse. To explore the molecular features of breast tumors associated with metastatic relapse after prolonged dormancy (late recurrence ≥5 years) but not confounded by variations in systemic treatment, we previously compiled breast tumor gene expression data from 4767 patients and established a discovery cohort consisting of 743 lymph node–negative patients who did not receive systemic neoadjuvant or adjuvant chemotherapy or hormonal therapy (Fig. 1A) (19). We defined four subgroups based on gene expression. Of these, group G3 had the fewest recurrences, whereas G4 had an increased risk of late recurrences (≥5 years), although both had a similar distribution of pathologic subtypes (19). We applied these gene expression signatures to analyze a separate data set of 46 samples, where gene expression data from laser capture microdissected tumor epithelium were available (20). We compared the expression of 29 genes representing CXCR4, E-selectin ligands, and enzymes critical for posttranslational processing of E-selectin ligands in these tumor epithelium samples and found that 7 of these genes were elevated in tumors that had late recurrences (table S1 and Fig. 1B). To further determine the correlation of CXCR4 and E-selectin pathway genes with late recurrence in bone, we analyzed the expression of these genes in the estrogen receptor–positive (ER+) subset (697 patients) of our discovery cohort because most ER+ patients experience initial relapse in bone and often at late time points. We found that three of these seven genes were up-regulated in primary tumors with late recurrence compared with tumor samples from recurrence-free patients. In particular, CXCR4 expression correlated with time to recurrence (Fig. 1, C and D).
Fig. 1. BCCs hijack SDF-1+E-selectin+ vascular gateways to enter the bone.

(A) Discovery cohort of untreated, node-negative breast cancer patients revealed four subgroups based on gene expression. Of these, G3 had the highest recurrence-free survival, whereas G4 had an increased risk of late recurrence (≥5 years after diagnosis). In Analysis A, expression of 29 genes representing CXCR4, E-selectin ligands, and enzymes critical for posttranslational processing of E-selectin ligands was analyzed in a data set compiled from 46 microdissected samples (G3, n = 18; G4, n = 10). In Analysis B, these same 29 genes were analyzed in the ER+ subset of our discovery cohort of patients with untreated, node-negative disease [late recurrence (LR) ≥ 5 years, n = 66; recurrence-free (RF) > 15 years, n = 26]. LCM, laser capture microdissection. (B to D) The expression of 7 of 29 genes analyzed was significantly increased in microdissected tumor epithelium of patients with late recurrence (t test). In the ER+ subset of our discovery cohort, 3 of these 29 genes were significantly up-regulated in tumor samples from patients with late recurrence compared to those from patients with recurrence-free survival at ≥15 years (Mann-Whitney U test). (D) CXCR4 expression correlates with time to recurrence in the ER+ subgroup [***P = 0.00097, analysis of variance (ANOVA)] [P values presented in (B); red indicates P < 0.05]. (E) Intravital microscopy was used to image the calvarial BM of mice engrafted with fluorescent membrane dye–labeled BCCs in real time with single-cell resolution. Dextran-FITC (fluorescein isothiocyanate) was used to visualize the blood pool. As shown in the montage image of in vivo micrographs, sinusoidal vasculature, which is known to have a distinct SDF-1+E-selectin+ phenotype, is concentrated in distinct parasagittal locations (montage containing multiple images; scale bar, 200 μm). (F) Fluorescently labeled BCCs were engrafted by intracardiac injection in mice, and the calvarium was imaged in the initial 2 hours after engraftment. BCCs were detected entering the bone specifically in perisinusoidal areas. Cells were not detected in lateral or caudal regions that lack substantial sinusoidal vasculature (montage containing multiple images; scale bar, 100 μm).
The CXCR4/SDF-1 axis does not regulate BCC homing to the BM
To study the precise functions of CXCR4 (SDF-1 receptor) and E-selectin ligands in BCC metastasis, we used video-rate intravital microscopy to track the migration of fluorescently labeled human BCC lines in immunocompromised mice (16). A panel of fluorescently labeled ER+ and ER− BCCs (table S2) were engrafted by intracardiac injection. Video-rate microscopy of BCCs transiting through the calvarium and calvarial BM was then performed, and the vasculature was visualized using high–molecular weight fluorescent dextrans as a blood pool agent.
As shown in a montage of multiple still frame images, the calvarial BM vasculature has a complex anatomy, with dilated sinusoidal capillary beds concentrated medially around a central collecting vein (Fig. 1E). Although BCCs could be identified circulating in the vasculature throughout the calvarium, cells interacted with the endothelial wall only within sinusoidal vascular and perisinusoidal vascular regions, where SDF-1 and E-selectin expression is known to be high (videos S1 and S2) (16). As shown in Fig. 1F, 2 hours after intracardiac injection, BCCs have diapedesed into the BM tissue specifically in these perisinusoidal areas. We noted increased BM homing of ER+ cell lines compared to ER− cells (fig. S1), suggesting that the bone metastatic tropism of ER+ tumors in humans is recapitulated in the xenograft mouse model.
Fig. 2. E-selectin regulates entry of BCCs into bone from peripheral circulation.

(A) Cell surface expression of CXCR4 versus day 1 BCC BM homing (R2 = 0.0013, linear regression; n = 3 per cell line). (B) BCC BM homing in mice treated ± CXCR4 inhibitor AMD3100 (n = 3 each cell line). NS, not significant. (C) BCC BM homing in mice treated ± CXCR4 neutralizing antibodies (MCF-7: n = 3; 1833: n = 4). (D) BCC BM homing in mice treated ± pertussis toxin to broadly inhibit chemokine signaling before engraftment (n = 3 each cell line). (E) E-selectin expression (green) in the sinusoidal regions of calvarial BM was detected by in vivo microscopy using anti–E-selectin–Cy5 antibodies and was colocalized with fluorescently labeled BCCs (cyan) imaged simultaneously on day 1 after intracardiac engraftment (dextran-FITC, red; montage contains multiple images). (F) BCC homing was quantified in mice treated ± E-selectin inhibitor GMI-1271 at both initial (2 hours; MCF-7: **P = 0.0068, n = 3; MDA-MB-231: *P = 0.0356, n = 3; unpaired, two-way t test) and late (20 hours; MCF-7: **P = 0.0052, n = 3; MDA-MB-231: *P = 0.0491, n = 3; unpaired, two-way t test) time points. Representative images shown (BCC, green; dextran-FITC, red; montage contains multiple images). (G) Schematic of MCF-7 stem (CD44+CD24−/low) and non-stem (CD44+CD24−/high) cell isolation by flow sorting and engraftment ± GMI-1271 into female severe combined immunodeficient (SCID) mice. SSC, side scatter; FSC, forward scatter. (H) BM homing of MCF-7 stem and non-stem cell populations ± GMI-1271 at 20 hours after engraftment (**P = 0.0026, n = 3; unpaired, two-way t test). Scale bars, 100 μm.
Given its high expression in organs that are common sites of BCC metastasis, it has been presumed that SDF-1 critically regulates BCC BM homing. We therefore first tested whether SDF-1/CXCR4 interactions mediated BCC entry into BM. Although BCC lines expressed cell surface and intracellular CXCR4 to varying degrees, its expression did not correlate with BM homing potential (Fig. 2A, table S2, and fig. S2). To define the functional role of SDF-1/CXCR4, we used four approaches (fig. S3, A to C). First, we treated mice with the highly specific small-molecule inhibitor of CXCR4, AMD3100 (21). Subcutaneous micro-osmotic pumps were implanted in mice to provide continuous delivery of the molecule, which has a brief therapeutic half-life. Neither ER+ nor ER− BCC homing was affected by AMD3100 inhibition (Fig. 2B). In a second set of experiments, mice were administered anti-CXCR4 neutralizing antibodies to inhibit BCC binding to SDF-1. Again, no effect on homing of either ER+ or ER− cells was observed (Fig. 2C). Next, we pretreated BCCs with pertussis toxin to widely inhibit all chemokine signaling in addition to the CXCR4/SDF-1 chemokine pathway. No effect of pertussis toxin on BCC BM homing was observed compared to phosphate-buffered saline(PBS) control–treated cells (Fig. 2D). Last, we knocked down CXCR4 transcript expression in BCCs with small interfering RNA (siRNA) and found no effect on BM homing potential compared to control siRNA–transduced cells (fig. S4). These data are consistent with previous reports demonstrating that, although CXCR4 inhibition slows metastatic growth in animal models, it does not appear to decrease the frequency of bone metastases (22). Moreover, CXCR4 expression in primary human BCCs correlates with lymph node metastasis but not hematogenous dissemination (23, 24).
Vascular E-selectin mediates BCC homing to the BM
We then turned our attention to the potential role of E-selectin interactions in mediating BCC entry into the bone. In vivo homing studies showed a very high degree of colocalization of BCCs with vascular E-selectin expression in the sinusoidal niche, as detected with fluorescent anti–E-selectin antibodies (Fig. 2E). Using a combination of quantitative polymerase chain reaction, Western blotting, flow cytometry, and immunofluorescence, we then assayed the expression of proteins reported to act as or synthesize E-selectin ligands on leukocytes and/or BCCs (25–29). We found that the BCCs in our studies express multiple potential E-selectin ligands and synthetic enzymes and that all BCCs have functional E-selectin binding activity (figs. S5 to S9). Using an E-selectin–Fc chimera as a functional Western blot probe, we found that ER+ and ER− BCCs express differential E-selectin ligand profiles (figs. S10 and S11). MUC1 emerged as a candidate E-selectin ligand that is more highly expressed by bone-tropic ER+ cells, but further studies will be necessary to confirm the identity of the specific functional E-selectin ligand(s) that regulates BCC bone homing (figs. S12 to S14).
To test the functional role of E-selectin ligands on BCCs, we then treated mice with a highly specific glycomimetic E-selectin binding inhibitor, GMI-1271 (30). In mice treated with inhibitor compared to vehicle control, BCC homing to BM was substantially blocked at both 2 and 20 hours (Fig. 2F and fig. S15). Similar inhibition was observed for both ER+ and ER− cell homing.
Recent data have suggested that the CD44+CD24−/low fraction of both primary and cultured ER+ BCCs is highly enriched for tumor-initiating ability (“stemness”) and that these putative CSCs also have high metastatic potential (31, 32). We therefore wanted to test whether E-selectin inhibition could effectively block this subpopulation from entering the bone. Although many cell lines have a predominantly CD44+CD24+ phenotype, ER+ MCF-7 cells contain a relatively larger CD44+CD24−/low population, facilitating isolation of purified CD44+CD24−/low and CD44+CD24+ cells by fluorescence-activated cell sorting (FACS) (33). We engrafted GMI-1271 versus vehicle control–treated mice with CD44+CD24−/low or CD44+CD24+ FACS-purified MCF-7 BCCs (Fig. 2G; treatment protocol in fig. S15). Whereas homing of non-stem cell populations was not significantly affected by E-selectin inhibition, CSC homing was inhibited more than twofold (Fig. 2H). Consistent with this finding, immunofluorescence staining with E-selectin–Fc fusion protein showed increased binding of E-selectin to CD44+CD24−/low stem cells compared to non-stem cells (fig. S16).
Specific anatomic regions of the BM influence BCC proliferation and dormancy
We next wanted to determine whether the initial sites of BM colonization in the perisinusoidal niche became the foci of metastatic proliferation. To discriminate dormant from proliferative BCCs, we labeled BCCs that constitutively expressed fluorescent tdTomato (tdT) with lipophilic dyes that are stably incorporated into the cell membrane (34). This label is diluted between daughter cells during mitosis and becomes undetectable in proliferative cells after five cell divisions (fig. S17). BCCs were engrafted by intracardiac injection, and the calvarial BM was imaged after 6 weeks. As expected, most engrafted mice did not show substantial BCC proliferation, although dormant BCCs were readily detected in the perisinusoidal niche as DiR+tdT+ nests (Fig. 3A). However, in mice with proliferative BM disease, large DiR−tdT+ cell clusters were identified in physically distinct, lateral, nonsinusoidal regions of the BM, suggesting that perisinusoidal niches maintain a dormant reservoir of BCCs in BM that have the potential to proliferate in alternative BM niches (Fig. 3, B and C).
Fig. 3. SDF-1/CXCR4 interactions tether dormant BCCs to the vascular niche.
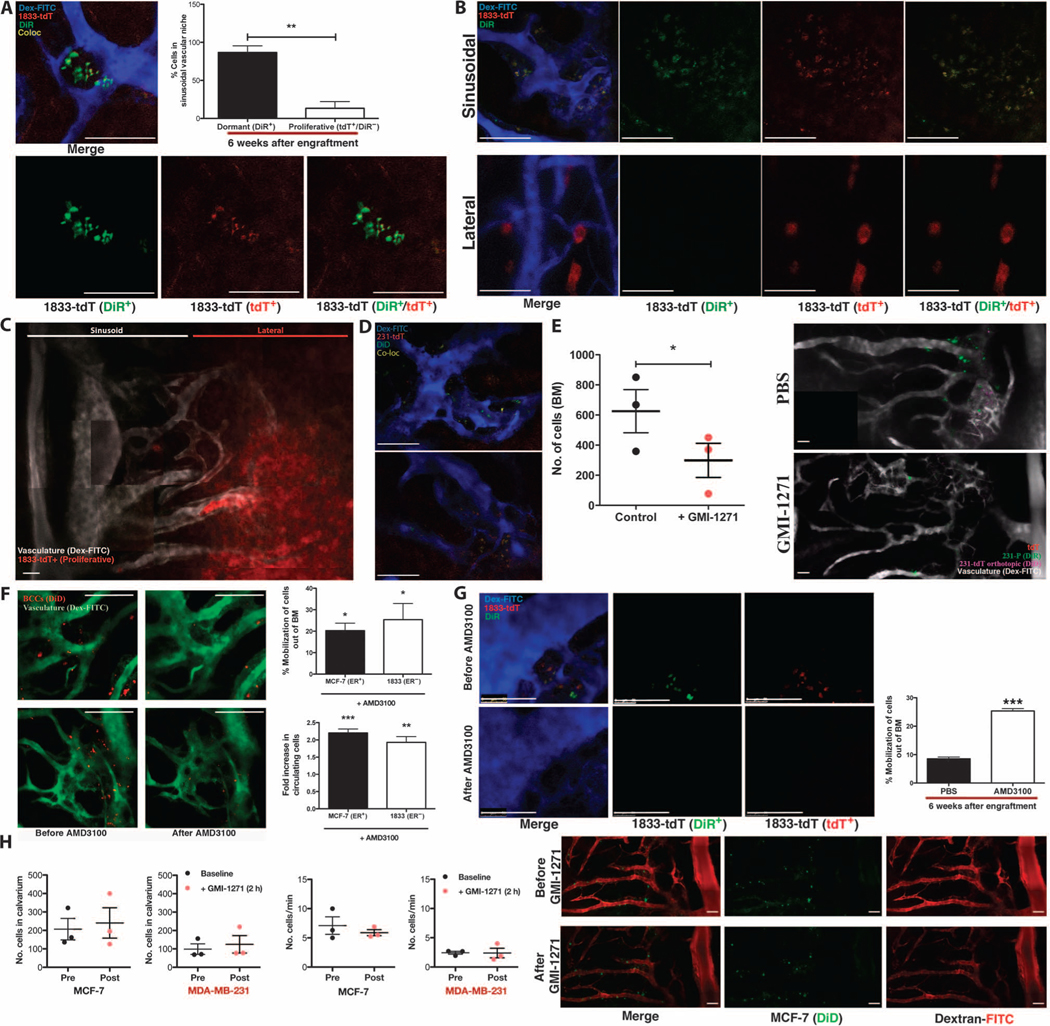
(A) Six weeks after intracardiac engraftment, dormant BCCs (DiR+tdT+) are detected in clusters localized to the sinusoidal vascular region of calvarial BM. Representative images are shown. The percentage of dormant (DiR+tdT+) versus proliferative (DiR−tdT+) BCCs in the sinusoidal niche was calculated (**P = 0.0042, n = 3 mice; unpaired, two-way t test). (B and C) In mice with proliferative bone tumors, rapidly dividing BCCs (DiR−tdT+) were found in distinct lateral, nonsinusoidal regions of BM [representative images of n = 4 experiments; (C) is a montage containing multiple images]. (D) Dormant (DiD+tdT+) BCCs that spontaneously metastasized to bone from orthotopic tumors were detected in the sinusoidal vascular niche at 48 to 79 days. (E) Forty-eight to 79 days after orthotopic engraftment of DiD-labeled tdT+ BCCs, mice were treated ± GMI-1271 and engrafted with DiR-labeled BCCs. DiR-BCCs homed to sinusoids in orthotopic tumor-engrafted mice, with a twofold homing reduction in GMI-1271–treated mice (*P = 0.0126, n = 3; unpaired, two-way t test). Representative images are shown (montage containing multiple images). (F) One day after engraftment, mice were treated ± AMD3100, and images of the calvarium were obtained before and 2 hours after treatment to determine the percentage of BCCs mobilized from BM (MCF-7: *P = 0.0455, n = 3; 1833: *P = 0.0300, n = 4; unpaired, two-way t test). Video-rate microscopy was used to assess the fold increase in peripherally circulating BCCs (MCF-7: ***P = 0.0005, n = 3; 1833: **P = 0.0069, n = 4; unpaired, two-way t test). Representative images are shown. (G) Six weeks after engraftment, mice were treated ± AMD3100. The percentage of dormant DiR+tdT+ BCCs mobilized from BM was calculated (***P = 0.0008, n = 3; unpaired, two-way t test). Representative images are shown. (H) One day after engraftment, mice were treated ± GMI-1271. Images before and 2 hours after treatment demonstrated no significant mobilization of BCCs from BM (n = 3 mice for each cell line; unpaired, two-way t test). Representative images are shown (montage containing multiple images). Scale bars, 100 μm.
Next, we tested whether dormant BCCs that spontaneously metastasized from a primary tumor site were also found in perisinusoidal locations. DiD-labeled BCCs (MDA-MB-231) were engrafted orthotopically in the mammary fat pads, and the calvarial BM was imaged at 48 to 79 days after engraftment. As shown in Fig. 3D, dormant DiD+tdT+ spontaneous metastases were localized in the perisinusoidal niches (fig. S18). When BCCs labeled with a different fluorescent dye (DiR) were then engrafted by intracardiac injection in these same mice, DiR-BCC homing remained perisinusoidal and E-selectin–dependent (Fig. 3E and fig. S19). These data suggest that key bone homing mechanisms remain unchanged in mice that have harbored orthotopic tumors for extended periods of time.
E-selectin and CXCR4/SDF-1 function as opposing vascular gatekeepers regulating BCC entry and exit from the BM
Although our data suggest that E-selectin is the principal mediator of BCC BM homing, previous studies indicate that SDF-1 plays an important role in metastasis growth (15, 35). Given the critical function of SDF-1 in anchoring hematopoietic cells to the BM niche, we hypothesized that SDF-1 similarly tethers BCCs to BM (36, 37). To test this, we engrafted mice with fluorescently labeled BCCs and treated them with a single dose of the CXCR4 antagonist AMD3100 or PBS on D1 (Fig. 3F and fig. S20) or D42 (Fig. 3G and fig. S21) after engraftment. Video-rate microscopy was used to detect BCCs in the calvarium and peripheral blood before and 2 hours after treatment (fig. S22A). A marked number of BCCs were observed to migrate from the BM into peripheral circulation in mice treated with AMD3100 at either time point (Fig. 3, F and G, and videos S3 to S6), indicating that both new and established metastases were anchored to the BM by SDF-1/CXCR4 interactions. Notably, although some proliferative, DiR−tdT+ cells were detected in D42 mice, most mobilized cells were dormant, DiR+tdT+ cells originating from the perisinusoidal niche (fig. S22B). In addition, although CXCR4 blockade had a potent mobilizing effect on BCCs, E-selectin inhibition did not cause BCC migration, emphasizing the specific roles of E-selectin and SDF-1 in controlling access to and persistence within the prodormancy BM niche (Fig. 3H and fig. S23).
E-selectin regulates BM metastasis of primary human BCCs
To further test whether primary human BCCs use E-selectin+ vascular gateways to home to bone, we isolated primary patient BCCs from surgical breast resection tissue and expanded these ex vivo (38). Expanded primary human BCCs were labeled with fluorescent dye, engrafted by intracardiac injection into SCID mice, and imaged using intravital microscopy. As shown in Fig. 4A and fig. S24, primary human BCCs also enter the bone specifically through the sinusoidal vascular niche. In addition, treatment of mice with the E-selectin inhibitor GMI-1271 markedly blocked primary human BCC entry into bone (Fig. 4, B and C).
Fig. 4. Primary human BCCs use E-selectin to enter a dormant sinusoidal perivascular niche in the BM.
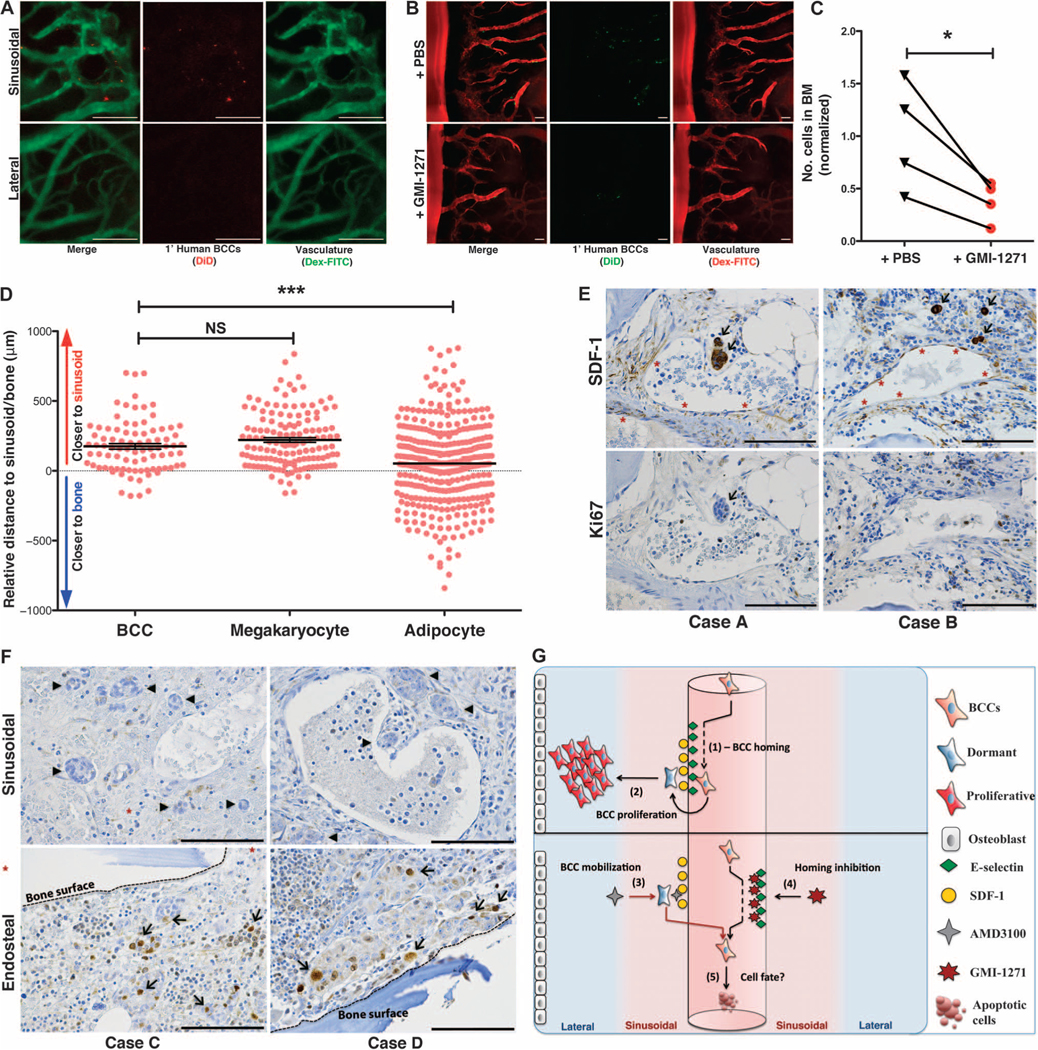
(A) Primary patient BCCs were isolated from surgical resection tissue and engrafted in mice. Intravital calvarial imaging revealed that primary cells home specifically to sinusoidal vasculature (n = 4). Representative images are shown. (B and C) Primary human BCCs were engrafted ± GMI-1271, and BM homing was quantified at 20 hours. Representative images are shown. Homing was decreased almost threefold in GMI-1271–treated mice (*P = 0.0393, n = 4; paired, two-way t test). (D) Histologic sections of BM biopsies from patients with micrometastatic involvement of breast cancer were obtained. The distance of each individual micrometastasis from the nearest sinusoid or bone spicule was measured by hand by a hematopathologist. Distance measurements for megakaryocytes, which are intimately associated with the sinusoidal vasculature, and for adipocytes, which are randomly scattered in bone, were also performed. Similar to megakaryocytes, breast cancer micrometastases were identified near sinusoids, with relative distance measurements that were significantly different from randomly distributed adipocytes [***P = 0.0002, data from three separate patient samples (BCCs, n = 86; megakaryocytes, n = 136; adipocytes, n = 348); unpaired, two-way t test]. (E) Immunohistochemical staining of BM biopsies from micrometastatic disease patients shows that BCCs (arrows) adjacent to SDF-1+ vasculature (asterisks) are Ki67−. (F) BM biopsy samples from patients with macrometastatic disease were stained for Ki67. Perisinusoidal (asterisks) BCCs were Ki67− (arrowheads), but Ki67+ BCCs (arrows) could be identified near bony spicules (dashed lines). (G) Cartoon model of BCC homing (1) and retention and proliferation (2) mechanisms in BM niches. Forced mobilization of dormant cells (3) combined with simultaneous blockade of BM reentry (4) could cause apoptosis or chemosensitize cells deprived of supportive stroma (5). Scale bars, 100 μm.
Perisinusoidal BM niches harbor dormant BCC micrometastases
Having shown that primary human BCCs behave similarly to cell lines in our xenograft studies, we wanted to examine the relationship between BCCs and the BM perisinusoidal niche in human metastatic breast cancer patients. We identified BM biopsies that were performed at our institution as part of staging evaluations before autologous stem cell transplantation. Although most patients had extensive disease involvement, we identified a small series with micrometastatic (≤5% of BM cellularity) disease. Although E-selectin staining could not be performed on the decalcified, paraffin-embedded tissue, we were able to stain for SDF-1 expression by immunohistochemistry. In patients with micrometastatic disease, BCC infiltrates were identified in perisinusoidal, SDF-1+ vascular regions at a rate that was statistically significant compared to chance (***P = 0.0002) (Fig. 4, D and E). Negative Ki67 staining confirmed that these metastases were highly indolent (Fig. 4E). In a separate series of patient cases with macrometastatic BM disease involvement, Ki67+ BCCs could be identified, which are often adjacent to bony surfaces, whereas perisinusoidal BCCs were Ki67− (Fig. 4F). These data corroborate our finding that perisinusoidal niches harbor dormant micrometastases in bone.
DISCUSSION
We used optical imaging technology to track BCC in real time and in vivo with single-cell resolution, and we have identified a portal of entry for BCC metastasis to the bone. This gateway is a morphologically distinct region of sinusoidal vasculature and perisinusoidal venules delineated by localized expression of the inflammatory vascular cell adhesion molecule E-selectin. In addition to controlling the BM entry of the CD44+CD24−/low tumor-initiating subset of BCCs through E-selectin, this BM site functions as a prodormancy niche in which quiescent BCC micrometastases are anchored through SDF-1/CXCR4 interactions.
Breast cancer patients who are diagnosed with early-stage, limited disease can experience metastatic relapse years later, and evidence suggests that this is caused by the proliferation of previously dormant BM micrometastases. Furthermore, clinical studies have shown that early-stage and locally advanced breast cancer patients have detectable CTCs both before and after treatment (4, 39). These observations in patients suggest that dormant CTCs are continuously migrating from micrometastatic sites into the peripheral bloodstream. Exploiting this biologic process through an understanding of the molecular mechanisms involved may present a therapeutic opportunity because epithelial cells that are detached from the stroma typically undergo apoptosis if this separation is prolonged (anoikis). Studies have indeed demonstrated that a large proportion of CTCs are apoptotic, suggesting that loss of cell-matrix adhesion may trigger BCC death in CTCs (40, 41). Forcing dormant BCCs into circulation while blocking their reentry into tissues could therefore gradually clear micrometastases from the bone in a minimally toxic manner.
Current adjuvant chemotherapies and hormonal therapies reduce the risk of relapse by targeting micrometastatic disease, yet these treatments may fail in part because of the nature of the BM microenvironment. Not only is it avid soil for metastasis localization, it may also be a protective haven against chemotherapy. In studies of patients receiving neoadjuvant chemotherapy, many patients with complete responses at the primary site demonstrated persistent micrometastatic disease in the BM (42). Although it is possible that these micrometastases persist after adjuvant chemotherapy solely because of intrinsic chemoresistance, a wealth of in vitro data suggest that BM stromal cells can protect breast cancer from chemotherapy-induced apoptosis (43–45). In addition, stromal interactions may play a role inresistance of BCC stoendocrine therapies (46). A strategy to relocate dormant micrometastases to a more vulnerable environment in peripheral circulation could therefore be used not only to destroy and/or capture intrinsically resistant BCCs but also to leverage the strength of existing adjuvant treatments.
The targets that we have identified in this study are highly “druggable.” The CXCR4 inhibitor AMD3100 is U.S. Food and Drug Administration–approved for hematopoietic stem cell mobilization, and selectin inhibitors are in clinical trials for sickle cell vaso-occlusive crisis, where they have exhibited minimal toxicity (47). Nevertheless, before these or similar agents are used for the treatment of micrometastatic disease, questions regarding the anatomic and biological fate of mobilized BCCs should be taken into careful consideration. For example, will forced mobilization alter the cell cycle status of dormant BCCs? Hematopoietic stem and progenitor cells that enter the peripheral blood after AMD3100 treatment are principally in G0 and, therefore, not susceptible to injury from chemotherapy. Whether dormant BCCs are guided into the cell cycle by forced mobilization remains to be explored. In addition, the duration of treatment and optimal sequencing of chemotherapeutic agents to achieve clinically relevant depletion of micrometastases will need examination. A greater understanding of the mechanisms of BCC homing and mobilization could, however, result in rational use of drugs targeting these processes.
In summary, we have identified a specific vascular “toggle” switch for BCC transit in bone. By blocking E-selectin on the lumen of spe cialized vascular beds in the BM, we can prevent vessel adhesion and subsequent passage of circulating BCCs into the tissue; by inhibiting CXCR4, dormant BCCs inhabiting these same perivascular niches are forced into the bloodstream (Fig. 4G). E-selectin and SDF-1 are thought to influence breast cancer progression, but here, we show that their localized expression and opposing functions in BM niches organize a vascular gateway that also serves as a niche for dormant micrometastases. These data elucidate critical steps in the BCC metastatic cascade by defining a molecular circuit regulating CTC migration. Our findings also present a rationale for therapeutic strategies that could break the foot-hold of dormant metastases in bone to decrease breast cancer relapse.
MATERIALS AND METHODS
Study design
The stromal molecules that regulate BCC homing and retention in the bone were assessed using intravital confocal microscopy analysis in xenograft mouse models. All mouse studies were performed in adherence to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and all institutional guidelines. The migration of fluorescently labeled BCCs in the calvarial BM was assessed using video-rate, high-resolution microscopy after intracardiac or orthotopic injection of fluorescently labeled BCCs. Genomic analysis of available breast cancer patient data sets revealed that CXCR4/SDF-1 and E-selectin expression correlate with late recurrence and, thus, were candidate molecules for mediating BCC entry into the bone. Analysis of the importance of these molecules in BCC bone trafficking included the use of small-molecule inhibitors AMD3100 and GMI-1271 and revealed that E-selectin is a primary regulator of BCC homing, whereas CXCR4/SDF-1 regulates retention of BCCs in the BM. These observations were also confirmed using primary patient BCCs and BM core biopsy samples. Institutional review board (IRB) ethical approval for the use of primary human samples was obtained from Duke University. The n value for each experiment is indicated in the appropriate figure legends.
Materials
General laboratory chemicals and reagents were purchased from Sigma-Aldrich, unless otherwise stated. MDA-MB-231, MCF-7, and T47D cell lines were obtained from the American Type Culture Collection. SUM159 cells were obtained from Asterand. K1 cells were obtained from the Greene Lab (University of Chicago, Chicago, IL). 1833 cells were obtained from the Massagué Lab (Memorial Sloan Kettering Cancer Center, New York, NY). Other reagents were obtained from the following suppliers: AMD3100 (Sigma-Aldrich); GMI-1271 (GlycoMimetics Inc.); fluorescent lipophilic dyes DiD, DiR, and DiI (Invitrogen); high–molecular weight dextran (Invitrogen); AF-647 (Invitrogen); FITC (Life Technologies); Cy5 (GE Healthcare); LIVE/DEAD Near-IR cell viability stain (Invitrogen); Alzet osmotic pump (Durect Co.); anti–CXCR4-PE (phycoerythrin) (clone-1D9, BD Biosciences); rat IgG2a,κ-PE (clone R35-95, BD Biosciences); anti–CD24-PE (clone ML5, BD Biosciences); mouse IgG2a,κ-PE (clone G155-178, BD Biosciences); anti–CD44-APC (allophycocyanin) (clone G44-26, BD Biosciences); mouse IgG2b,κ-APC (clone 27–35, BD Biosciences); anti-hCXCR4 neutralizing antibody (clone 12G5, R&D Systems); mouse IgG2a (clone 20102, R&D Systems); anti-mouse E-selectin (clone 10E9.6, BD Biosciences); rat IgG2a,κ (clone 35–95, BD Biosciences); anti–SDF-1 (clone 79018, R&D Systems); and anti-Ki67 (clone ab15580; Abcam).
Genomic analyses
As we previously described (19), using 743 lymph node–negative breast tumor samples obtained from patients who were treated with local therapy alone, we developed a gene expression classification scheme that distinguishes breast cancer with early or late recurrence using 743 lymph node–negative breast tumor samples obtained from patients who were treated with local therapy alone. We applied this classification to publicly available gene expression data from 46 microdissected breast tumor epithelium samples [Gene Expression Omnibus (GEO) repository accession number GSE5847] and compared gene expressions of 29 genes representing CXCR4, E-selectin ligands, and enzymes critical for posttranslational processing of E-selectin ligands between samples in subgroups with high risk of late recurrence (G4) and with smallest risk of recurrence (G3) using t test. The correlations between selected genes and risk of delayed recurrence were further determined among the 697 ER+ patients. Differences for each pairwise comparison were assessed by Mann-Whitney U test, and the correlation between increasing gene expression and the time of recurrence was measured with ANOVA. Statistical analy6ses were performed with STATISTICA 11 (StatSoft Inc.) and R Project for Statistical Computing.
Cell culture
MDA-MB-231, 1833, MCF-7, and K1 cell lines were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Corning Inc.), supplemented with 10% fetal bovine serum (FBS; Gemini Bio-Products). T47D cells were cultured in RPMI medium (Corning), supplemented with 10% FBS (Gemini). SUM159 cells were cultured in Ham’s F-12 medium (Corning), supplemented with 5% FBS (Gemini). All cultures were maintained at 37°C in a 5% CO2 humidified atmosphere.
Fluorescent dye cell labeling
Cells were fluorescently labeled through incubation with “DiD,” “DiI,” or “DiR” lipophilic dyes (Invitrogen). Cells were isolated at a density of 2.5 × 106 cells/ml, resuspending cells under the following conditions: DiD—12.5 μM DiD in culture medium + 0.3% FBS; DiI—25 μM DiI in culture medium + 0.3% FBS; DiR—25 μM DiR in culture medium + 10% FBS. Cells were protected from the light and rotated at room temperature for 30 min. After incubation, cells were washed twice in PBS, counted, and resuspended at desired concentrations for engraftment. Dye labeling efficiency was tested by flow cytometry.
Mouse engraftment
BCCs were engrafted into female SCID mice by intracardiac injection. Mice were anesthetized with isoflurane and placed into position on their backs. Mouse fur covering the rib cage was removed using Nair hair removal cream. With a 29-gauge needle and 0.3-ml syringe, a 100-μl volume of cells (0.1 × 106 cells in PBS) was injected into the left ventricle. A bright red flash indicated a successful insertion, and this colored flash was monitored about every 25 μl while the injection proceeded. BCCs were also engrafted into the mammary fat pad of SCID mice. Mice were anesthetized, and fur covering the rib cage was removed, as previously described. The mouse was restrained, and using a 29-gauge needle and 0.3-ml syringe, a 100-μl volume of cells (4.3 × 106 cells in PBS) was injected superficially under the skin into the mammary fat pad. Long-term engrafted mice were implanted with estrogen pellets (0.5 mg per pellet, 90-day release; Innovative Research of America) to support ER+ cell proliferation. While mice were anesthetized, fur surrounding the lateral side of the neck was removed. The field was sterilized with alcohol wipes, and an incision was made using sterile surgical instruments. The estrogen pellet was placed under the skin using forceps, and the incision was closed using a suture kit. After engraftment procedures, mice were returned to their cages until ready for imaging. Diagrams illustrating the timing and drug dosing for specific imaging experiments are presented in figs. S3, S15, and S19 to S21.
Intravital confocal microscopy and video-rate imaging
SCID mice were anesthetized with isoflurane, and a rectangular incision was made in the scalp, revealing the intact underlying cortical bone. The region was washed with PBS, and fluorescently labeled dextran (Dex-FITC or Dex-Cy5) was administered by tail vein injection. Mice were placed in a specially designed restrictor, and a coverslip was placed over the exposed calvarial bone. Mice remained anesthetized throughout the procedure. High-resolution images were obtained through the intact mouse skull with a Leica SP5 confocal and multiphoton microscope with a 20×/0.40 NA (numerical aperture) objective lens. The system uses a femtosecond titanium:sapphire laser (Chameleon) for multiphoton or single-photon excitation and multiple Cs lasers (including an argon laser, a HeNe laser, and 561- and 633-nm diode lasers) for single-photon excitation. Images were captured with Leica LAS-AF software using line and frame averaging. The calvarial BM was subdivided into numbered anatomical areas, and overlapping 20× images were captured of the entire region. After the procedure, these images were merged to generate a montage image of the entire calvarium, and cell homing and mobilization counts were obtained. Videos of BCCs in circulation were obtained using the same equipment. Video-rate imaging capture on the central vessel and branching coronal vein of the calvarial vasculature was performed. Videos of a desired vascular region were taken for 5 min, and the fluorescently labeled BCCs observed in circulation or crawling along vascular walls were counted. The number of cells observed in circulation per minute was calculated. The number of circulating cells (number of cells per minute) before and after drug administration (AMD3100 and GMI-1271) was compared.
Flow cytometry and cell sorting
Intracellular and extracellular CXCR4 expression in BCC lines was determined by flow cytometry. Subconfluent cell cultures were trypsinized and counted. For extracellular staining, 1 × 106 cells were washed in PBS and resuspended in 100 μl of wash buffer (autoMACS + 3% bovine serum albumin). Cells were incubated with LIVE/DEAD Near-IR stain (Invitrogen) for 20 min on ice. Cells were washed and subsequently incubated with anti–CXCR4-PE (clone 1D9) or isotype-PE (clone R35-95) for 30 min on ice. Cells were spun at 500g and resuspended in 300 μl of 1% paraformaldehyde (PFA) in PBS. For intracellular CXCR4 staining, after LIVE/DEAD Near-IR incubation, cells were fixed in 1% PFA in PBS on ice for 60 min. Cells were washed and resuspended in 1 ml of 70% EtOH and incubated at −20°C overnight. Cells were subsequently stained for CXCR4 as previously described. Flow cytometry acquisition was performed with a FACSCanto II flow cytometer (BD Biosciences) and analyzed with FlowJo (Tree Star Inc.) software. MCF-7 stem (CD44+/CD24low) and non-stem (CD44+/CD24+) cells were isolated by cell sorting. Subconfluent cultures of MCF-7 cells were trypsinized and stained with LIVE/DEAD Near-IR, as previously described. Cells were subsequently stained with CD24-PE (clone ML5, BD Biosciences) and CD44-APC (clone G44-26, BD Biosciences) on ice for 30 min. Cells were suspended in a buffer of PBS + 1% FBS and collected in a buffer of 50% DMEM/50% FBS. Sorting was performed with a BD FACSAria II cell sorter. After sorting, stem and non-stem cells were counted and resuspended in PBS, and 0.1 × 106 cells in a 100-μl volume were engrafted into mice by intracardiac injection, as described previously.
In vivo immunofluorescence imaging
E-selectin vascular expression in the calvarial BM was imaged in vivo using anti–E-selectin (clone 10E9.6, BD Biosciences) conjugated to the fluorophore AF-647.SCID mice were engrafted with BCCs (0.1 × 106 cells) 20 hours before imaging. Engrafted mice were injected with anti–E-selectin–AF-647 or rat IgG2a,κ–AF-647 isotype control (clone 35–95, BD Biosciences) at a dose of 1 mg/kg 18 hours before imaging. Mice were imaged, and montage images were generated as previously described.
Primary human BCC isolation and ex vivo culture
Isolation and ex vivo culture of primary human mammary epithelial cells were described previously (33). Briefly, tissue specimens were obtained with informed consent from patients. Mammary tissues were minced and digested with a mixture of dispase and collagenase/hyaluronidase (STEMCELL Technologies Inc.), and fat was removed with a 70-μm strainer (BD Biosciences). The ex vivo culture of cells used a fibroblast feeder system. Epithelial cells were cocultured with irradiated (30 Gy) Swiss 3T3 fibroblasts (J2 strain) in F medium [3:1 (v/v) F-12 Nutrient Mixture (Ham)–DMEM (Invitrogen), 5% FBS, hydrocortisone (0.4 μg/ml; Sigma-Aldrich), insulin (5 μg/ml; Sigma-Aldrich), cholera toxin (8.4 ng/ml; Sigma-Aldrich), and epidermal growth factor (10 ng/ml; Invitrogen)] with addition of Y-27632 (5 to 10 μM; Enzo Life Sciences). Cultures were maintained in a 5% CO2 humidified atmosphere at 37°C. Cultured mammary epithelial cells were isolated from feeder cell through differential trypsinization. Feeder/epithelial cocultures were rinsed with PBS and incubated with 0.05% trypsin solution at room temperature for 30 s to 1 min, with close monitoring by phase microscopy. When the feeder cells rounded up and began to detach from the substrate, the cultures were gently tapped, and the detached cells were removed by aspiration. The epithelial colonies remained tightly adherent. The epithelial cells were rinsed again with PBS and then retrypsinized at 37°C for 3 to 5 min. After gentle pipetting to disperse the epithelial cells into a single-cell suspension, the cells were pelleted through PBS containing 10% serum. After centrifugation at 500g, the cell pellets were resuspended in F medium for cell counting and experimental preparation.
Immunohistochemistry
Human BM core biopsy slides (IRB #57532) were obtained from the Duke University Department of Pathology. SDF-1 and Ki67 immunohistochemistry staining was performed under the following conditions: SDF-1 antigen retrieval was performed with Dako EDTA Target Retrieval solution (Dako North America Inc.). Slides were stained with anti–SDF-1 (clone 79018, R&D Systems) at a concentration of 15 μg/ml. Dako Envision System-HRP (DAB), for use with mouse (#K4006, Dako), was used as a secondary antibody. For Ki67, antigen retrieval was performed with Dako EDTA Target Retrieval solution (Dako North America Inc.) Slides were stained with anti-Ki67 (clone ab15580, Abcam) at a concentration of 10 μg/ml. Dako Envision System-HRP (DAB), for use with rabbit (#K4011, Dako), was used as a secondary antibody. Whole slide images of human BM core slides were obtained with an Axio Observer Z1 microscope, Axiocam ICc1 color camera, and Axiovision 4.8 software. The distances of individual human BCCs, megakaryocytes, and adipocytes from sinusoidal vasculature and trabecular bone were measured by a clinical hematopathologist (C.M.M.) using the ImageJ software (version 1.49, NIH) and whole-slide images.
Statistical analysis
Homing cell numbers were determined by manual counting of fluorescent cells detected within the calvarium. The effects of inhibitors versus vehicle controls on BCC homing and mobilization were assessed using unpaired t tests. Statistical differences in the relative distances of BCC metastases (how much closer the metastatic disease was to bone than to the sinusoid or vice versa) were determined by using an unpaired two-way t test. Genomic analysis was performed as described above. All statistical analysis was performed with GraphPad Prism version 5.0, STATISTICA 11, and R Project for Statistical Computing. Significance P values were defined as follows (*P < 0.05; **P < 0.01; ***P < 0.001). Mean ± SEM was graphed.
Supplementary Material
Fig. S1. Bone homing capacity of ER+ and ER− BCC lines.
Fig. S2. Intracellular CXCR4 stores expressed in ER+ and ER− cell lines.
Fig. S3. Schematic for CXCR4-mediated homing inhibition experiments.
Fig. S4. Effect of CXCR4 siRNA knockdown on BCC BM homing.
Fig. S5. E-selectin ligand and synthetic enzyme mRNA transcript expression in ER+ and ER− BCC lines and primary BCCs.
Fig. S6. E-selectin ligand protein expression in ER+ and ER− BCC lines.
Fig. S7. Flow cytometric analysis of E-selectin ligand expression on BCC lines.
Fig. S8. CD44 cell surface expression on ER+ and ER− BCC lines.
Fig. S9. Immunofluorescence analysis of E-selectin ligand expression on BCC lines.
Fig. S10. Western blot analysis of E-selectin ligand expression profiles in BCC lines.
Fig. S11. Western blot analysis of E-selectin ligand expression profiles in BCC lines, with protection of sialic acid residues.
Fig. S12. Immunofluorescence analysis of MUC1 expression on ER+ and ER− BCCs.
Fig. S13. Immunoprecipitation and functional analysis of the E-selectin binding capacity of MUC1 in BCC lines.
Fig. S14. Western blot analysis of E-selectin ligand expression profiles on MCF-7 stem and non-stem cell populations.
Fig. S15. Schematic for E-selectin–mediated homing inhibition experiments.
Fig. S16. Immunofluorescence analysis of E-selectin ligand expression on MCF-7 stem and non-stem cell populations.
Fig. S17. Fluorescent lipophilic membrane dye depletion after successive cell doubling events.
Fig. S18. Spontaneous metastasis of orthotopically engrafted MDA-MB-231 (DiD+tdT+) BCCs to the calvarial BM.
Fig. S19. Schematic for orthotopic engraftment and E-selectin–mediated homing inhibition experiments.
Fig. S20. Schematic for GMI-1271 and AMD3100 mobilization experiments.
Fig. S21. Schematic for AMD3100 mobilization experiments in mice with established disease.
Fig. S22. AMD3100-induced mobilization of dormant BCCs out of the BM and into the circulation.
Fig. S23. Lack of effect of GMI-1271 on BCC mobilization from BM.
Fig. S24. Montage image of primary human BCC homing to sinusoidal regions of the calvarial BM.
Table S1. Gene list.
Table S2. Cell lines.
Circulating BCCs crawling along vascular wall (merge).
Circulating BCCs crawling along vascular wall (DiD channel only).
Circulating BCCs before AMD3100 treatment (merge).
Circulating BCCs before AMD3100 treatment (DiD channel only).
Circulating BCCs after AMD3100 treatment (merge).
Circulating BCCs after AMD3100 treatment (DiD channel only).
Acknowledgments:
We thank Z. Li for assistance with statistical analyses and R. Nanda for helpful discussions. The 1833 and K1 cell lines were provided to us by the laboratories of J. Massagué and G. Greene, respectively.
Funding: This work was supported by the Young Investigator Award program of the American Society of Clinical Oncology (M.L.B.), a Developmental Research Project Award from the University of Chicago Breast Cancer Specialized Program of Research Excellence (D.A.S.), and the Duke Cancer Institute.
Footnotes
Competing interests: J.M. is vice president and chief scientific officer of GlycoMimetics Inc. GlycoMimetics has a patent for GMI-1271.
Data and materials availability: GEO repository accession number GSE5847.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/8/340/340ra73/DC1
Materials and Methods
REFERENCES
- 1.DeVita VT Jr., Lawrence TS, Rosenberg SA, Eds., DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology (Lippincott, Williams & Wilkins, Philadelphia, PA, 2014). [Google Scholar]
- 2.Braun S, Vogl F, Naume B, Janni W, Osborne MP, Coombes RC, Schlimok G, Diel IJ, Gerber B, Gebauer G, Pierga J-Y, Marth C, Oruzio D, Wiedswang G, Solomayer E-F, Kundt G, Strobl B, Fehm T, Wong GYC, Bliss J, Vincent-Salomon A, Pantel K, A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med 353, 793–802 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Weigelt B, Peterse JL, van’t Veer LJ, Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 5, 591–602 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Rack B, Schindlbeck C, Jückstock J, Andergassen U, Hepp P, Zwingers T, Friedl TWP, Lorenz R, Tesch H, Fasching PA, Fehm T, Schneeweiss A, Lichtenegger W, Beckmann MW, Friese K, Pantel K, Janni W; SUCCESS Study Group, Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst 106, dju066 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serrano MJ, Rovira PS, Martinez-Zubiaurre I, Rodriguez MD, Fernández M, Lorente JA, Dynamics of circulating tumor cells in early breast cancer under neoadjuvant therapy. Exp. Ther. Med 4, 43–48 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LWWM, Hayes DF, Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med 351, 781–791 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S, Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 11, R46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lucci A, Hall CS, Lodhi AK, Bhattacharyya A, Anderson AE, Xiao L, Bedrosian I, Kuerer HM, Krishnamurthy S, Circulating tumour cells in non-metastatic breast cancer: A prospective study. Lancet Oncol. 13, 688–695 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA, Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 121, 4821–4831 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myung JH, Gajjar KA, Pearson RM, Launiere CA, Eddington DT, Hong S, Direct measurements on CD24-mediated rolling of human breast cancer MCF-7 cells on E-selectin. Anal. Chem 83, 1078–1083 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shirure VS, Henson KA, Schnaar RL, Nimrichter L, Burdick MM, Gangliosides expressed on breast cancer cells are E-selectin ligands. Biochem. Biophys. Res. Commun 406, 423–429 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A, Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Renkonen J, Paavonen T, Renkonen R, Endothelial and epithelial expression of sialyl Lewisx and sialyl Lewisa in lesions of breast carcinoma. Int. J. Cancer 74, 296–300 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Stübke K, Wicklein D, Herich L, Schumacher U, Nehmann N, Selectin-deficiency reduces the number of spontaneous metastases in a xenograft model of human breast cancer. Cancer Lett. 321, 89–99 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Smith MCP, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD, CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 64, 8604–8612 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, Luster AD, Scadden DT, Lin CP, In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435, 969–973 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugiyama T, Kohara H, Noda M, Nagasawa T, Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Sugiyama T, Nagasawa T, Bone marrow niches for hematopoietic stem cells and immune cells. Inflamm. Allergy Drug Targets 11, 201–206 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Q, Chang JT, Gwin WR, Zhu J, Ambs S, Geradts J, Lyerly HK, A signature of epithelial-mesenchymal plasticity and stromal activation in primary tumor modulates late recurrence in breast cancer independent of disease subtype. Breast Cancer Res. 16, 407–420 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boersma B, Reimers M, Yi M, Ludwig JA, Luke BT, Stephens RM, Yfantis HG, Lee DH, Weinstein JN, Ambs S, A stromal gene signature associated with inflammatory breast cancer. Int. J. Cancer 122, 1324–1332 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Hatse S, Princen K, Bridger G, De Clercq E, Schols D, Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 527, 255–262 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Richert MM, Vaidya KS, Mills CN, Wong D, Korz W, Hurst DR, Welch DR, Inhibition of CCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol. Rep 21, 761–767 (2009). [PubMed] [Google Scholar]
- 23.Kato M, Kitayama J, Kazama S, Nagawa H, Expression pattern of CXC chemokine receptor-4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. 5, R144–R150 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang H, Watkins G, Douglas-Jones A, Mansel RE, Jiang WG, The elevated level of CXCR4 is correlated with nodal metastasis of human breast cancer. Breast 14, 360–367 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Zarbock A, Ley K, McEver RP, Hidalgo A, Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood 118, 6743–6751 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mondal N, Buffone A Jr., Neelamegham S, Distinct glycosyltransferases synthesize E-selectin ligands in human vs. mouse leukocytes. Cell Adh. Migr 7, 288–292 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geng Y, Yeh K, Takatani T, King MR, Three to tango: MUC1 as a ligand for both E-selectin and ICAM-1 in the breast cancer metastatic cascade. Front. Oncol 2, 1–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shirure VS, Reynolds NM, Burdick MM, Mac-2 binding protein is a novel E-selectinlig and expressed by breast cancer cells. PLOS One 7, e44529 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merzaban JS, Burdick MM, Gadhoum SZ, Dagla NM, Chu JT, Fuhlbrigge RC, Sackstein R, Analysis of glycoprotein E–selectin ligands on human and mouse marrow cells enriched for hematopoietic stem/progenitor cell. Blood 118, 1774–1783 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ernst B, Magnani JL, From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov 8, 661–677 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF, Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A 100, 3983–3988 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, Datar RH, Cote RJ, Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 12, 5615–5621 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Murohashi M, Hinohara K, Kuroda M, Isagawa T, Tsuji S, Kobayashi S, Umezawa K, Tojo A, Aburatani H, Gotoh N, Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br. J. Cancer 102, 206–212 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honig MG, Hume RI, DiI and DiO: Versatile fluorescent dyes for neuronal labelling and pathway tracing. Trends Neurosci. 12, 333–341 (1989). [PubMed] [Google Scholar]
- 35.Luker KE, Luker GD, Functions of CXCL12 and CXCR4 in breast cancer. Cancer Lett. 238, 30–41 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, Ponomaryov T, Taichman RS, Arenzana-Seisdedos F, Fujii N, Sandbank J, Zipori D, Lapidot T, G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat. Immunol 3, 687–694 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Nervi B, Link DC, DiPersio JF, Cytokines and hematopoietic stem cell mobilization. J. Cell. Biochem 99, 690–705 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, Haddad BR, Rhim JS, Dritschilo A, Riegel A, McBride A, Schlegel R, ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol 180, 599–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azim HA Jr., Rothé F, Aura CM, Bavington M, Maetens M, Rouas G, Gebhart G, Gamex C, Eidtmann H, Baselga J, Piccart-Gebhart M, Ellis C, Vuylsteke P, Cure H, Domont J, Ferro A, Toral-Peña JC, de Azambuja E, Sotiriou C, Di Cosimo S, Ignatiadis M, Circulating tumor cells and response to neoadjuvant paclitaxel and HER2-targeted therapy: A sub-study from the NeoALTTO phase III trial. Breast 22, 1060–1065 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Méhes G, Witt A, Kubista E, Ambros PF, Circulating breast cancer cells are frequently apoptotic. Am. J. Pathol 159, 17–20 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smerage JB, Budd GT, Doyle GV, Brown M, Paoletti C, Muniz M, Miller MC, Repollet MI, Chianese DA, Connelly MC, Terstappen LWWM, Hayes DF, Monitoring apoptosis and Bcl-2 on circulating tumor cells in patients with metastatic breast cancer. Mol. Oncol 7, 680–692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fehm T, Becker S, Becker-Pergola G, Sotlar K, Gebauer G, Dürr-Störzer S, Neubauer H, Wallwiener D, Solomayer E-F, Presence of apoptotic and nonapoptotic disseminated tumor cells reflects the response to neoadjuvant systemic therapy in breast cancer. Breast Cancer Res. 8, R60 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin FT, Dwyer RM, Kelly J, Khan S, Murphy JM, Curran C, Miller N, Hennessy E, Dockery P, Barry FP, O’Brien T, Kerin MJ, Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: Stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res. Treat 124, 317–326 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Maeda T, Alexander CM, Friedl A, Induction of syndecan-1 expression in stromal fibroblasts promotes proliferation of human breast cancer cells. Cancer Res. 64, 612–621 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, Liu Z.-y., Costes SV, Cho EH, Lockett S, Khanna C, Chambers AF, Green JE, Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 68, 6241–6250 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pontiggia O, Sampayo R, Raffo D, Motter A, Xu R, Bissell MJ, de Kier Joffé EB, Simian M, The tumor microenvironment modulates tamoxifen resistance in breast cancer: A role for soluble stromal factors and fibronectin through b1 integrin. Breast Cancer Res. Treat 133, 459–471 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wun T, Styles L, DeCastro L, Telen MJ, Kuypers FA, Cheung A, Kramer W, Flanner H, Rhee S, Magnani JL, Thackray H, Phase 1 study of the E-selectin inhibitor GMI 1070 in patients with sickle cell anemia. PLOS One 9, e101301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chase SD, Magnani JL, Simon SI, E-selectin ligands as mechanosensitive receptors on neutrophils in health and disease. Ann. Biomed. Eng 40, 849–859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Bone homing capacity of ER+ and ER− BCC lines.
Fig. S2. Intracellular CXCR4 stores expressed in ER+ and ER− cell lines.
Fig. S3. Schematic for CXCR4-mediated homing inhibition experiments.
Fig. S4. Effect of CXCR4 siRNA knockdown on BCC BM homing.
Fig. S5. E-selectin ligand and synthetic enzyme mRNA transcript expression in ER+ and ER− BCC lines and primary BCCs.
Fig. S6. E-selectin ligand protein expression in ER+ and ER− BCC lines.
Fig. S7. Flow cytometric analysis of E-selectin ligand expression on BCC lines.
Fig. S8. CD44 cell surface expression on ER+ and ER− BCC lines.
Fig. S9. Immunofluorescence analysis of E-selectin ligand expression on BCC lines.
Fig. S10. Western blot analysis of E-selectin ligand expression profiles in BCC lines.
Fig. S11. Western blot analysis of E-selectin ligand expression profiles in BCC lines, with protection of sialic acid residues.
Fig. S12. Immunofluorescence analysis of MUC1 expression on ER+ and ER− BCCs.
Fig. S13. Immunoprecipitation and functional analysis of the E-selectin binding capacity of MUC1 in BCC lines.
Fig. S14. Western blot analysis of E-selectin ligand expression profiles on MCF-7 stem and non-stem cell populations.
Fig. S15. Schematic for E-selectin–mediated homing inhibition experiments.
Fig. S16. Immunofluorescence analysis of E-selectin ligand expression on MCF-7 stem and non-stem cell populations.
Fig. S17. Fluorescent lipophilic membrane dye depletion after successive cell doubling events.
Fig. S18. Spontaneous metastasis of orthotopically engrafted MDA-MB-231 (DiD+tdT+) BCCs to the calvarial BM.
Fig. S19. Schematic for orthotopic engraftment and E-selectin–mediated homing inhibition experiments.
Fig. S20. Schematic for GMI-1271 and AMD3100 mobilization experiments.
Fig. S21. Schematic for AMD3100 mobilization experiments in mice with established disease.
Fig. S22. AMD3100-induced mobilization of dormant BCCs out of the BM and into the circulation.
Fig. S23. Lack of effect of GMI-1271 on BCC mobilization from BM.
Fig. S24. Montage image of primary human BCC homing to sinusoidal regions of the calvarial BM.
Table S1. Gene list.
Table S2. Cell lines.
Circulating BCCs crawling along vascular wall (merge).
Circulating BCCs crawling along vascular wall (DiD channel only).
Circulating BCCs before AMD3100 treatment (merge).
Circulating BCCs before AMD3100 treatment (DiD channel only).
Circulating BCCs after AMD3100 treatment (merge).
Circulating BCCs after AMD3100 treatment (DiD channel only).