

Abstract
Triple negative breast cancer is difficult to treat effectively, due to its aggressiveness, drug resistance, and lack of the receptors required for hormonal therapy, particularly at the metastatic stage. Here, we report the development and evaluation of a multifunctional nanoparticle formulation containing an iron oxide core that can deliver doxorubicin, a cytotoxic agent, and polyinosinic:polycytidylic acid (Poly IC), a TLR3 agonist, in a targeted and simultaneous fashion to both breast cancer and dendritic cells. Endoglin-binding peptide (EBP) is used to target both TNBC cells and vasculature epithelia. The nanoparticle demonstrates favorable physicochemical properties and a tumor-specific targeting profile. The nanoparticle induces tumor apoptosis through multiple mechanisms including direct tumor cell killing, dendritic cell-initiated innate and T cell-mediated adaptive immune responses. The nanoparticle markedly inhibits tumor growth and metastasis and substantially extends survival in an aggressive and drug-resistant metastatic mouse model of triple negative breast cancer (TNBC). This study points to a promising platform that may substantially improve the therapeutic efficacy for treating metastatic TNBC.
Keywords: Iron oxide nanoparticles, endoglin binding peptide, doxorubicin, immunotherapy, metastasis, triple negative breast cancer
Graphical Abstract
1. Introduction
Breast cancer is the second leading cause of cancer-related deaths among women in the U.S.1 An estimated 1 in 8 women will develop breast cancer in their lifetime.1 Treating stage IV or metastatic breast cancer is a formidable challenge because of its developed resistance to therapeutics and aggressive proliferation; it can quickly metastasize and spread into multiple organs.2,3 Despite the advances in developing multi-agent treatment approaches, the survival rate for metastatic breast cancer remains low (< 25%).1 The treatment for triple negative breast cancer (TNBC) poses additional challenges as they lack epidermal growth factor receptor 2 (HER-2), estrogen receptors (ER), or progesterone receptors (PR), and thus do not respond to hormonal therapy available in the clinic.4,5
The current treatment options for TNBC commonly starts with surgery to remove the bulk of the tumor mass followed by adjuvant therapy with combinatory therapy such as multi-agent chemotherapy,6 multi-immune agent therapy,7 radio-immunotherapy,8 and chemo-immuno-therapy.9 Recently, the combination of multiple treatments has emerged as a promising approach aimed to circumvent drug resistance and improve outcomes. Among these combinatorial therapies, combined chemo- and immunotherapy has received much attention because of the remarkable progress made in clinical cancer immunotherapy and large accumulation of knowledge in the clinical practice of chemotherapy. However, two major obstacles impede the clinical translation of this combined therapy. First, the two therapeutic agents do not work synchronously or take effect on a cancer cell at the same time even if they are administered together due to the difference in pharmacokinetic profile.10 Second, there is a safety concern of the severe systemic immune responses incurred as a result of off-target cytotoxicity. Nanotechnology has recently shown promise to seamlessly integrate chemo- and immune- therapeutic agents in a single nanoparticle (NP) formulation which allows the multiple agents to reach target cells simultaneously and act synchronously.11 Incorporation of a targeting ligand into such a NP formulation may further improve its safety and therapeutic efficacy by reducing systemic toxicity and required effective dosage of therapeutics. Surface modification can also be employed to optimize the pharmacokinetic profile. Thus a nanotechnology-based approach may substantially improve the efficacy of cancer therapeutics.12,13
Although conceptually viable, the implement of this nanomedicine-based approach has proven to be non-trivial, primarily because of the difficulty in delivering efficacious amounts of therapeutic agents to tumor cells and making drug-loaded NPs sufficiently small. Large-size NPs (>100 nm) can be quickly eliminated by the reticuloendothelial system (RES) before they can reach target sites and yet large-size NPs are less effective in drug delivery than smaller NPs.14 Anticancer drugs used in the clinic for treating breast cancer such as paclitaxel (PTX) and doxorubicin (Dox) are hydrophobic, and it is a challenge to make NPs sufficiently small when the NPs are loaded with hydrophobic drugs that are incompatible with aqueous biological solutions. In the past decade, a number of NP formulations have been developed to deliver hydrophobic chemotherapeutics (e.g. PTX or Dox).15–19 Only Abraxane, a formulation of PTX-loaded NPs, with a size of 130 nm, was approved for the clinic.20 Yet, the use of Abraxane is limited to patients with advanced breast cancer because of its severe systemic toxicity (e.g. hepatic insufficiency, high grade neuropathy and neutropenia)21,22 which is attributable to its large size and non-specificity to targets, uncontrollable dissociation of the non-covalently bound PTX so that that a higher drug dosage is needed to achieve sufficient potency.23 On the other hand, nanomedicine-based immunotherapy can effectively harness the immune system against tumors mostly by targeted delivery of antigen/adjuvant and immune checkpoint modulators or direct cell surface modifications.24 Lipidic, polymeric and protein-based nanoparticles have served as promising carriers for delivering strong immunostimulants such as CpG and ovalbumin25,27 as well as immune checkpoint inhibitors targeting PDL1/PD1 and CTLA-4.28,29 Moreover, liposomes and polystyrene NPs have been conjugated onto the surfaces of T cells, hematopoietic stem cells and antigen-presenting cells to treat antigen-expressing tumor cells.30,31 Some NPs have immunostimulating properties inherently and can activate host immune systems against cancer.32,33 Nevertheless, the majority of these organic NPs have a size typically in the range of 150–300 nm.34–36 Clearly, it would be even more difficult to make small NPs (< 100 nm) that carry both chemo- and immune- therapeutic agents for TNBC. Further, most polymer-based NPs suffer from unpredictable size variation when loaded with therapeutic cargos or when placed in a physiological environment, which results in an inconsistent pharmaceutical profile and therapeutic results.37
Here we report the construction and evaluation of a multifunctional NP formulation that is capable of targeted delivery of chemo- and immuno- therapeutics simultaneously to tumor microenvironment for treating TNBC. We chose the ultra-small (8 nm) iron oxide core coated with a layer of negatively charged silica-polyethylene glycol (PEG) as a robust shape-defining template for subsequent conjugations to conserve size with tumor and vasculature target ligand, an endoglin-binding peptide (EBP), a chemotherapeutic drug doxorubicin (DOX) and an immunomodulating agent polyinosinic:polycytidylic acid (Poly IC). EBP is a small 12-mer histidine-rich peptide with high affinity to endoglin, a transmembrane glycoprotein highly expressed in both vasculature epithelia and TNBC tumors.38–43 DOX, an anti-cancer drug approved by the FDA for clinical use, acts as a cytotoxic agent that induces apoptosis of breast cancer cells.44 Poly IC is a virus-mimic double-stranded RNA (dsRNA) that activates both innate and adoptive immune systems to eliminate cancer cells through activating Toll-like receptor 3 (TLR3).45–49 Poly IC is safe and well-tolerated by patients.50 Layer-by-layer assembly (LBL) has drawn significant attention in the field of drug delivery due to its modular tunability, drug loading versatility and capability of controlled drug release.51 A typical LBL usually requires multiple polyelectrolyte layers for drug protection and assembly stability,52 which could lead to the aforementioned limitations of oversize and unpredictable pharmaceutical profile. The unique configuration of our NP formation rests on the direct deposition of the alternating layers between the positively charged DOX and the negatively charged Poly IC onto the surface of the negatively charged IONPs without the assistance of additional polyelectrolytes. This NP exhibits physicochemical properties tunable by varying the DOX:Poly IC ratio, small size (53 nm) with high-level colloidal stability and a pH responsive drug release profile. In addition, the iron oxide core provides superparamagnetism that enables magnetic resonance imaging (MRI) while the siloxane-crosslinked PEG coating provides negative charges for loading the positively charged DOX and then the negatively charged Poly IC. Systemic injection of our NP formulation demonstrates a safe and favorable pharmaceutical profile, and the NP targeted-delivers chemo- and immuno- therapeutic agents simultaneously to breast cancer cells and enables a combined chemo-immuno-therapy in a synchronous fashion. We show that our NP could inhibit tumor growth and metastasis and extend the survival in both xenografted and orthotopic mouse models of drug-resistant and metastatic breast cancer.
2. Results
2.1. Design of a NP for chemo- and immune-therapy
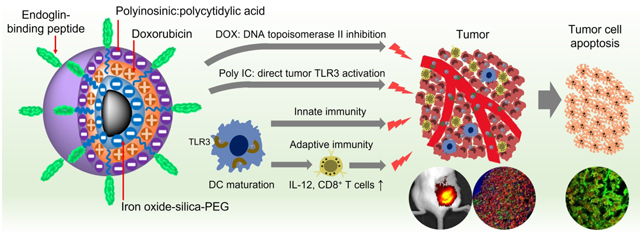
We constructed a multifunctional NP that contains a tumor target peptide, a chemo drug, and an immune therapeutic agent, and can elicit anti-cancer immune responses (Scheme 1). Scheme 1a illustrates the structure and synthesis process of this NP. Iron oxide NPs (IONPs) of 8 nm diameter with siloxane-crosslinked PEG coating53 were conjugated with a tumor-target ligand (EBP, N-terminal cysteine modified) through an NHS-PEG24-maleimide heterobifunctional linker. Negatively charged IONPs are then alternatively loaded with the positively-charged DOX and the negatively-charged Poly IC through a layer-by-layer assembling process. The quantity of DOX loaded is controlled by the thickness of each DOX layer and the number of the layers of DOX and Poly IC.
Scheme 1.

Design of the multifunctional NPs and their possible interaction with tumor and host immune system. a, Schematic illustration of preparation of the EBP-modified and DOX/Poly IC loaded nanocarrier. b, Designed mechanisms of targeted and combined chemo-immuno therapy for TNBC:Tumor vasculature-targeted delivery of DOX (DNA topoisomerase II inhibition) and Poly IC (activation of TLR3 on tumor cell surface) for direct cancer cell killing; activation of host immune system by NPs through dendritic cell (DC) maturation and secretion of cytokines (e.g. IL-12, etc.), and subsequent activation of anti-cancer adaptive (activation of cytotoxic T cells) and innate (activation of NK cells) immune responses. Combined chemotherapeutic and immunological responses gain maximum possible cancer cell killing.
Scheme 1b shows the possible interactions of the NPs with the tumor microenvironment. The EBP mediates the delivery of the NP loaded with DOX and Poly IC to tumor cells and tumor-infiltrating dendritic cells (DCs) by targeting the tumor vasculature. Once the NP gets into tumor cells, the reduced pH in endosomes (pH5–6) or lysosomes (pH4–5) triggers the release of DOX and Poly IC, and consequently induces tumor apoptosis.54–56 On the other hand, Poly IC on NPs may also interact with DCs and enter the cells to activate anti-tumor immune response.56 As antigen-presenting cells, DCs are a key player in the initiation and regulation of immune responses. DCs express TLR3 receptors for Poly IC ligand. Upon binding the TLR3 with Poly IC or NP-Poly IC, the maturation of DCs is stimulated to induce the expression of co-stimulatory molecules (CD80 and CD86), and produces cytokines such as IL-12.57 IL-12 is a potent inducer of anti-tumor immunity through activation of antigen-specific naïve T cells in preclinical models.57,58 Furthermore, mature DCs also activate cytotoxic natural killer (NK) cells for tumor cell destruction.59 Therefore, the designed NP will interact with both tumor and immune system after systemic administration to trigger direct and indirect cancer killing mechanisms in vivo and achieve improved therapeutic outcomes compared to the conventional single-agent treatments.
2.2. Physicochemical properties of NPs.
The EBP-conjugated iron oxide NPs (IONP-EBP) had a negative surface charge (~ −4.3 mV) that facilitated the loading of positively-charged DOX. We tested a series of NP/DOX ratios (Fe/DOX) to optimize DOX loading. Zeta-potential measurement for IONP-EBP at various NP/DOX ratio showed a gradual change of the surface charge from −4.3 mV to +1.26 mV as NP/DOX ratio varied from 10:0 to 10:16 (Fig. 1a). This change in surface charge also indicated the successful DOX loading. The maximum DOX loading was determined to be Fe/DOX = 10:11 (through the ferrozine assay for iron concentration,60 and UV absorbance at 490 nm for DOX concentration) at reaction ratio of 10:16 where the surface charge was near neutral. Evaluation based on the molar density of ~1.64 nmol NP/mg Fe for IONPs yielded the number of DOX molecules per IONP of ~1156. IONP-DOX-EBP remained small and uniform (z-average = 31 nm, PDI = 0.15). After removal of free DOX by size-exclusion chromatography, negatively-charged Poly IC was loaded onto IONP-DOX-EBP at different Fe/Poly IC ratios to produce IONP-DOX-Poly IC-EBP. After loading of Poly IC, the surface charge of NPs changed back to negative and reached a plateau of −17.8 mV when the ratio of Fe/Poly IC reached 10:10 (Fig. 1b). Noticeably, the hydrodynamic size of IONP-DOX-Poly IC-EBP increased with the increase in the ratio of IONP-DOX-EBP to Poly IC (e.g., 216.9 nm with Fe/Poly IC 10:2), which may be attributed to the charged-induced aggregation. On the other hand, the increase in Poly IC amount increased the negative charge, which stabilized the NPs. The hydrodynamic diameter of the NP was reduced to a minimum of 53 nm at the Fe/Poly IC ratio of 10:16 (Fig. 1c).
Fig. 1.

Characterization of DOX and Poly IC loading onto EBP-conjugated IONPs. a, Zeta-potential of IONP-DOX-EBP as a function of Fe/DOX ratio. b, Zeta-potential of IONP-DOX-Poly IC-EBP as a function of Fe/Poly IC ratio (Fe/DOX ratio was 10:11) (HEPES, pH 7.4). c, Hydrodynamic size of IONP-DOX-Poly IC-EBP as a function of Fe/Poly IC ratio (Fe/DOX ratio was 10:11). d, TEM and e, AFM micrographs of IONP-DOX-Poly IC-EBP (Fe/DOX/Poly IC ratio was 10:11:16). Scale bars represent 50 nm. f, Histogram of diameters of IONP-DOX-Poly IC-EBP evaluated from AFM images. g, UV-Vis spectra of DOX, EBP, Poly IC, IONP, IONP-DOX-EBP and IONP-DOX-Poly IC-EBP in PBS buffer (pH 7.4). Arrows indicate the peaks for Poly IC (left) and DOX (right). h, Agarose gel electrophoresis analysis of unbound Poly IC from IONP-DOX-EBP/Poly IC mixtures. Lower panel displays the relative intensities of the gel bands of the upper panel, evaluated with by ImageJ. i, Cumulative release of DOX from IONP-DOX-Poly IC-EBP under different pH conditions at 37°C.
Transmission electron microscopy (TEM) was utilized to characterize the shape and coating of NPs. As shown in the micrographs (Fig. 1d.), IONPs have a core size of ~8 nm. As Poly IC is a dsRNA analog and has high electron density, the coating of Poly IC shows as a light-grey envelope around IONP cores (Fig. 1d). AFM was used to further reveal the coating property of IONP-DOX-Poly IC-EBP. Fig. 1e shows the AFM image of the dehydrated NPs deposited on mica surface. Analysis of these AFM images indicates that IONP-DOX-EBP has an average dehydrated diameter of ~18 nm and the Poly IC loading increases the size to ~22.5 nm (Fig. 1f and Fig. S1 in supplementary information).
Chemical properties of NPs were further characterized by UV-Vis absorbance (Fig. 1g). The IONPs showed strong absorbance in the UV-Vis region but no characteristic peaks. DOX showed absorbance peaks at 260 nm and 500 nm, Poly IC showed absorbance peaks between 240 and 270 nm, corresponding to their excited states. EBP has no unique characteristic peaks. The spectra for IONP-DOX-EBP and IONP-DOX-Poly IC-EBP show a peak at ~500 nm, characteristic of DOX, confirming the successful loading of DOX in these two NP formulations. An additional peak at ~260 nm in the spectrum of IONP-DOX-Poly IC-EBP further confirms the presence of Poly IC in this NP formulation. To determine how much bound and unbound Poly IC from IONP-DOX-EBP/Poly IC mixtures, we analyzed unbounded Poly IC using agarose gel electrophoresis as free Poly IC could move through the gel and be visualized. The band intensities of Poly IC were evaluated with ImageJ software. Approximately 75% of Poly IC were bound to NPs when IONP-DOX-EBP and Poly IC were mixed at the ratio of Fe/Poly IC = 10:16 (Fig. 1h).
2.3. Drug release.
The pH-responsive release of DOX from IONP-DOX-Poly IC-EBP was analyzed using dialysis under three pH conditions, corresponding to those in blood (7.4), cellular endosomes (5.4) and lysosomes (4.5), respectively. The release of DOX under all these conditions were saturated in 10 h (pH 7.4, 4 h; pH 5.4, 8 h; pH 4.5, 10 h). Here a pH-dependent drug release is observed. DOX release is relatively low at high pHs values: only 20% of the loaded DOX was finally released at pH 7.4 and 35% of the loaded DOX at pH 5.4; on the contrary, the final release is high at low pH: ~50% of the loaded DOX was released in the first 4 h and ~100% was released in 10 h at pH 4.5 (Fig. 1i). This suggests that when applied in vivo, DOX will be largely released from NPs after the NPs are internalized by target cells and recruited into lysosomes. Combined with the targeted delivery enabled by the tumor-targeting EBP, high-specificity release of DOX from IONP-DOX-Poly IC-EBP in the lysosomes of tumor cells can be achieved to improve therapeutic efficacy and reduce adverse systemic toxicity. Note that DOX and Poly IC were loaded onto NPs through electrostatic interactions. Release of the positively-charged DOX indicates that the negatively-charged Poly IC was released as well because Poly IC and IONP surface were charge repulsive without DOX. Interaction of Poly IC with its receptor TLR3 occurs in endosomes and lysosomes61 and may not require dissociation of Poly IC from NPs. Therefore, Poly IC can take action as soon as NPs bind to cells and get internalized.
2.4. NP cellular uptake and cancer cell killing.
A murine mammary cancer cell line, 4T1, was used to mimic late stage metastatic breast cancer in human due to its aggressive proliferation and triple negative phenotype.62 The cellular uptake of IONP-DOX-Poly IC-EBP as well as other agents (for comparison) was characterized by confocal laser scanning microscopy (CLSM) and flow cytometry. For both evaluations, all agents (DOX, IONP-DOX, IONP-DOX-Poly IC and IONP-DOX-Poly IC-EBP) carried the same amount of DOX (10 μg/mL) and were incubated with cells for 2 h. The time point was so selected that the time was sufficient for NPs to enter cells while not causing observable cytotoxic effects. As shown in CLSM images (Fig. 2a), all agents were taken up by cells and were mostly localized in nuclei after 2 h of incubation. However, compared to cells treated with free DOX, cells threated with either IONP-DOX or IONP-DOX-Poly IC (IONPs associated with DOX and Poly IC without EBP ligand) showed decreased fluorescence signals, indicating reduced cellular uptake of these two NP formulations. This was expected because the non-targeting NP-bound DOX enters cells through endocytosis which is slower than free DOX’s cell membrane crossing. Cells treated with the targeted NPs (IONP-DOX-EBP or IONP-DOX-Poly IC-EBP), on the other hand, showed similar signal intensities to cells treated with free DOX. EBP targets Endoglin on cell surface and promotes the uptake of NPs. As NPs were not able to freely enter cell nucleus due to their large size for nuclear pore transportation (>5 nm63), the observation also indicates that DOX was successfully released from NPs once the NPs entered cells. The result was confirmed quantitatively by flow cytometry measurements. IONP-DOX (MFI = 315) showed 30% decrease in signal compared to DOX (MFI = 451), while the EBP-conjugated NPs IONP-DOX-EBP (MFI = 474) and IONP-DOX-Poly IC-EBP (MFI = 459) showed compensation of uptake through targeting effect (Fig.2 b, c). Note that such investigation was based on a single time point. A time-dependent uptake of NPs by the cells are worth of further study to gain insight into their pharmacological behaviors in vivo.
Fig. 2.

Cellular uptake and therapeutic effect of various agents on 4T1 cells. For the cellular uptake study, 10 μg/mL DOX or DOX-equivalent NPs ([Fe] ≈ 9 μg/mL) were incubated with cells for 2 h and analyzed. a, CLSM images of cells with membrane and nuclei stained green and blue, respectively. The excitation state fluorescence of DOX (red) was imaged (Ex: 495 nm; Em: 580–654 nm). Scale bar: 100 μm. b, Flow cytometry analysis of DOX uptake into 4T1 cells treated with various agents, performed 2 h after incubation (Ex: 488 nm; Em: 585 nm with 42 nm bandwidth). c, Mean fluorescence intensity (MFI) of DOX from flow cytometry analysis in (c). *** P < 0.005, ****P < 0.0001 by one-way ANOVA with Turkey’s post-hoc test. d, Normalized MFI of DOX at 37°C and 4°C quantified by flow cytometry. **P < 0 0.001, ****P < 0.0001, Student’s unpaired t-test. e, Viability of 4T1 cells incubated with various agents ([Fe] ≈ 0.9 × [DOX]), assessed by Alarma Blue assay. f, Flow cytometry analysis of cell apoptosis induced by i: medium control; ii: IONPs; iii: DOX; iv: IONP-DOX-Poly IC-EBP. DOX concentration was 1 μg/mL equivalent ([Fe] ≈ 0.9 μg/mL) and incubation time was 48 h.
As temperature dependence is a key indicator of endocytosis of NPs, we then performed a test of cell uptake of free DOX and IONP-DOX-Poly IC-EBP at both 4°C and 37°C. Significantly, uptake of DOX into cells were mostly inhibited and only 10% of DOX was taken up by cells at 4°C (Fig. 2d). In contrast, uptake of IONP-DOX-Poly IC-EBP was only partly (~45%) inhibited at 4°C. This is likely due to the phase changes of plasma membrane at low temperature that makes DOX difficult to cross it; this observation agrees with previous findings.64 The result also suggests that, although endocytosis is involved in its uptake, IONP-DOX-Poly IC-EBP may take other pathways to enter cells at low temperature. One possible explanation is that although the NPs had net negative charges, they still had positively charged DOX on the surface. As the molecular structures of coatings on the NP surface could be dynamic rather than rigid, the NP might expose its positively charged regions to the cell membrane during the incubation process, which could facilitate an energy-independent membrane translocation process.
Cellular uptake of these agents determines their therapeutic potency. The viability of cells treated with DOX, Poly IC, IONPs, IONP-DOX, IONP-DOX-Poly IC, and IONP-DOX-Poly IC-EBP, was assessed by the Alamar Blue assay. The results indicate that IONPs were non-toxic to cells. Free Poly IC itself was also non-toxic to cells. The electrostatic repulsion between the negatively charged poly IC and cell membrane significantly limits poly IC’s intracellular access so that poly IC alone shows no cell killing effect. Although all DOX-containing NPs showed similar cell-killing profiles, IONP-DOX had slightly lower potency in this regard (IC50 ≈ 0.64 μg/mL) compared to free DOX, IONP-DOX-Poly IC and IONP-DOX-Poly IC-EBP IC50s ≈ 0.36 μg/mL) (Fig. 2e). This result is consistent with cellular uptake study (Fig. 2c), in which the IONP-DOX incurred the least cellular uptake among these agents. Tumor apoptosis assay by Annexin V and PI staining confirmed that the cellular apoptosis induced by IONP-DOX-Poly IC-EBP is comparable to that induced by free DOX (Fig. 2f).
2.5. In vitro immune response of bone marrow-derived dendritic cells (BMDCs) to NPs.
Poly IC exerts anti-cancer immunity through activation of dendritic cells (DCs) and secretion of cytokines.55,65 The direct immune response of IONP-DOX-Poly IC-EBP in vitro was examined using bone marrow-derived dendritic cells (BMDCs). All agents were labeled with Cy5 for fluorescence analysis. 10 μg/mL Poly IC or an agent (either IONP-DOX-EBP or IONP-DOX-Poly IC-EBP) carrying equivalent Poly IC was incubated with BMDC cells for 1 h at 37°C. It was seen that although both free DOX and IONP-DOX-EBP can enter BMDCs, DOX from IONP-DOX-Poly IC-EBP remained in cytoplasm without entering nuclei. Although 1 h is not long enough to exclude the likelihood of these NPs to enter the nucleus, the reduced uptake into the nucleus during initial contact suggests the stimulation of BMDCs with reduced cytotoxicity to these cells Fig. 3a).66 In fact, DOX is reported to have low toxicity to BMDCs, and does not upregulate their CD80 or CD86 expressions.67 To examine the maturation of BMDCs, free Poly IC, IONP-DOX-EBP and IONP-DOX-Poly IC-EBP were incubated with cells for 24 h. The expression levels of CD80 and CD86, the BMDC maturation surface markers, were found elevated in cells treated with Poly IC-containing samples (2~2.5× higher than untreated controls). IONP-DOX-Poly IC-EBP showed the strongest therapeutic effect among all the agents as shown by mean fluorescence intensities of anti-CD86 and anti-CD86 quantified by flow cytometry (Fig. 3, b–e). The enhanced BMDC stimulation by IONP-DOX-Poly IC-EBP is likely due to the NP-mediated Poly IC delivery into endosomes or cytoplasm where TLR3 resides.68 Further, the production of IL-12 in cellular supernatants was quantified by ELISA. As a product of matured DCs, IL-12 assist in differentiation of naïve T cells into T helper 1 (TH1) effector cells.57 Fig. 3f indicates that Poly IC and IONP-DOX-Poly IC-EBP induced equivalent production of IL 12. While we expect that these agents would have a therapeutic effect on target tumor cells, their potential cytotoxicity is undesirable.69 We thus tested the cytotoxicity of these agents against BMDCs. IONP-DOX-EBP showed a ~25% of cell killing with a high DOX dose,67 while the IONP-DOX-Poly IC-EBP had negligible effect to cell viability (Fig. 3g). The low cytotoxicity of IONP-DOX-Poly IC-EBP to DCs is likely due to the combinatory effect of insensitivity of DCs to DOX and reduced NP uptake by the nucleus. These results indicate that IONP-DOX-Poly IC-EBP can both kill cancer cells by Dox and activate immune cells against cancer cells without causing significant toxicity to DCs.
Fig. 3.

Cellular responses of BMDCs to various agents. For all assays, 10 μg/mL Poly IC or an agent carrying equivalent Poly IC was incubated with cells. a, Confocal fluorescence microscopy imaging of cellular uptake of free DOX, IONP-DOX-EBP and IONP-DOX-Poly IC-EBP into BMDCs. Cell nuclei were stained with DAPI. DOX was imaged at Ex: 495 nm and Em: 580–654 nm. NPs were labeled with Cy5 (red) and imaged at Ex: 652 nm and Em: 665–745 nm. b-c, Flow cytometry study of DC maturation. BMDCs were incubated with Poly IC, IONP-DOX-EBP or IONP-DOX-Poly IC-EBP for 24 h and the expression of (b) CD86 and (c) CD80 were evaluated. d-e, Mean fluorescence intensities of anti-CD86 and anti-CD80 antibodies, respectively, derived from (b) and (c). f, Production of IL-12 by BMDCs in cellular supernatants 24 h after incubation with Poly IC, IONP-DOX-EBP or IONP-DOX-Poly IC-EBP, quantified by ELISA. g, Toxicity of Poly IC, IONP-DOX-EBP and IONP-DOX-Poly IC-EBP on BMDCs after 24 h incubation, assessed by the Alamar Blue viability assay. **P < 0.01, ***P < 0.005, ****P < 0.0001 by one-way ANOVA with Turkey’s post-hoc test.
2.6. In vivo innate and antigen-specific T cell immune response
Activation of DCs in mice by Poly IC (i.e., upregulation of CD80 and CD86 as shown in Fig. 3) would lead to production of IL-12 in blood (Fig. 4a). To confirm if IONP-DOX-Poly IC-EBP can induce IL-12 in vivo, we quantified plasma IL-12 levels in mice after IONP-DOX-Poly IC-EBP, DOX, Poly IC were injected intravenously. 24-h post-injection, blood was drawn from mice and plasma was separated. IL-12 contents in plasma were determined by ELISA assay.65 The result showed that DOX did not increase the IL-12 level, suggesting that DCs were not activated. In contrast, rapid elevation of the IL-12 level was observed in blood drawn from mice treated with either free Poly IC or IONP-DOX-Poly IC-EBP. The peak of the IL-12 level was reached at 1.5 h post injection and the level then gradually decreased over time. Notably, IONP-DOX-Poly IC-EBP induced a level of IL-12 ~2.6 times higher than free Poly IC (Fig. 4b).
Fig. 4.

In vivo assessment of immune response induced by IONP-DOX-Poly IC-EBP. a, Schematic illustration of activation of immune response by IONP-DOX-Poly IC-EBP. b, Production of IL-12 in mouse serum by intravenous injection of DOX (5 mg/kg), Poly IC (18 mg/kg), or IONP-DOX-Poly IC-EBP (DOX 10 mg/kg, Poly IC 18 mg/kg), assessed by ELISA. Serum was collected before injection and 1.5, 6.5, and 24 h post-injection. *P < 0.05, **P < 0.01, ****P < 0.0001 by two-way ANOVA with Turkey’s post-hoc test. c, Flow cytometry analysis of single cell suspensions processed from tumor and spleen of mice treated with IONP-DOX, Poly IC, or IONP-DOX-Poly IC-EBP (DOX 10 mg/kg and Poly IC 18 mg/kg equivalent). Mice were euthanized three days after treatments. Single cell suspensions of tumors and spleens were stained with anti- CD8, CD25, CD69 antibodies for analysis.
Poly IC has also been shown to directly or indirectly activate T cells and trigger adapted anti-tumor immune response.70,71 This process includes the proliferation of cytotoxic T cells (CD8+) and activation of T cells (i.e. upregulation of CD25 and CD69, Fig. 4a). We next tested if Poly IC containing NPs can activate T cells and kill tumors in tumor-bearing mice. Three days after treatments, tumor bearing mice were euthanized. Tumor and spleen cells were harvested and stained with anti-CD8, anti-CD25, and anti-CD69 antibodies and analyzed by flow cytometry (Fig. 4c). tumor cells of untreated mice showed low levels of CD8 (3.94%), CD25 (1.76%) and CD69 (1.87%). In contrast, the administration of free Poly IC and IONP-DOX-Poly IC-EBP resulted in a considerable increase of CD8 (8.63% and 15.3%), CD25 (7.91% and 18.5%), and CD69 (3.57% and 12.1%), respectively. Notably, IONP-DOX-Poly IC-EBP was seen to cause more activation of T cells than free Poly IC. In spleen samples, only IONP-DOX-Poly IC-EBP was found to cause a significant increase of activated T cells (CD8: 17.3%, CD25: 5.08%, CD69: 2.08%), which may be attributable to the spleen accumulation of NPs. Free Poly IC is not able to accumulate in spleens with substantial amount. Compared to free Poly IC, IONP-DOX-Poly ICEBP NPs induced systemic immune response which can produce persistent anti-tumor effect.
2.7. In vivo MR and NIR fluorescence imaging of tumor-bearing mice treated with IONP-DOX-Poly IC-EBP-Cy5.5.
To further exploit the MRI capability of our multifunctional NP, we evaluated the magnetic properties of IONP-DOX-Poly IC-EBP in solution by MR imaging and then investigated its biodistribution in mice by in vivo MR imaging. As the surface coating may affect the T2 relaxivity of IONPs, we tested the in vitro T2 properties of PEGylated, EBP-conjugated, and DOX/Poly IC co-loaded IONPs. Results showed the r2 value of IONP-DOX-Poly IC-EBP-Cy5.5 is comparable to those reported NPs (Fig. S2, supplementary information).
Mice bearing 4T1 tumors were treated with IONP-DOX-Poly IC-EBP-Cy5.5 and MRI images of mice were acquired prior to and one hour post i.v. injection. Fig. 5a shows the images of a representative mouse. One hour after i.v. injection of IONP-DOX-Poly IC-EBP-Cy5.5, darkened areas appeared in tumor, indicating the intratumoral location of the injected NPs. Darkened areas were also found in kidneys and spleen, indicating the accumulation of NPs in these organs. One day after NP administration, more dark areas (increased T2) were observed in tumors, indicating increased NP accumulation. Increased T2 was also observed in kidney, which is apparently due to the renal clearance of NPs. Conversely, the T2 signal intensity in spleen was reduced (Fig. 5b). This observation can be explained as follows. Shortly after injection, NPs were circulating in blood and gradually accumulated in tumor, kidney, and spleen. One day after injection, NPs relocated from spleen to tumors and continuously excreted by the urinary system. The in vivo MRI not only proved the usefulness of these NPs as an effective T2 contrast agent, but also provided key information about biodistribution and relocation of NPs in mice.
Fig. 5.

MR and NIR epifluorescence imaging of mice bearing 4T1 tumors and treated using IONP-DOX-Poly IC-EBP-Cy5.5. a, In vivo MRI of two cross sections (upper and lower panels, respectively) of abdomen of the same mouse before, and 1 and 24 h after administration of IONP-DOX-Poly IC-EBP. Red arrows and dashed circles indicate tumor, yellow and green dashed circles indicate kidney or spleen, respectively. b, Relative MR intensity in tumor, kidney and spleen in mice, acquired pre-injection, 1 h and 1 d post-injection. c, Live IVIS images of mice bearing 4T1-luc tumors and treated with IONP-DOX-Poly IC-EBP-Cy5.5. The images were acquired before i.v. administration and 1 h to 6 days post-administration.
Bioluminescence and fluorescence imaging of live mice was also carried out to validate the in vivo targeting efficacy. IONP-DOX-Poly IC-EBP-Cy5.5 was injected intravenously into 4T1-luc tumor-bearing mice one week after tumor inoculation. The images were taken from 1 h to 6 d after injection. The result showed that the NPs co-localized with tumor at 1 h and continued to accumulate in tumor at 1 d and 2 d, and the accumulation lasted for nearly a week (Fig. 5c). This observation is consistent with cellular uptake studies and in vivo MRI, and confirms targeting of 4T1 cancer cells by EBP both in vitro and in vivo.
2.8. Biodistribution and pharmacokinetics of NPs in a flank-tumor mouse model.
The accumulation of NPs in various organs in tumor-bearing mice was analyzed by quantifying Cy5.5 intensities of Cy5.5-conjugated NPs. Mice were euthanized at 1, 24 and 48 h after NPs injections and organs were harvested. Fig. 6a shows radiant efficiencies (Dox fluence intensity) for various tissues. The results showed that at 1 h, NPs mostly accumulated in liver, spleen, kidneys and started to accumulate in tumor. At 24 h, NPs continued to accumulate in tumor, liver and kidneys, but were being eliminated from spleen. Some NPs were also found in the lungs at 24 h. At 48 h, NP accumulation virtually was unchanged in liver, spleen and kidney remained, but slightly increased in tumor (Fig. 6a). This result agrees well with those obtained by MR imaging (Fig. 5a and 5b). It should be noted that it is expected that NPs accumulate in the liver in a large quantity as a result of the clearance by the reticuloendothelial system (RES).14 It’s also known that the accumulation of NPs is size-dependent,72,73 and we have designed our NPs with a proper size to minimize the liver accumulation.
Fig. 6.

In vivo biodistribution and pharmacokinetics of IONP-DOX-Poly IC-EBP. a, Biodistribution of NPs (labeled with Cy5.5) quantified by measuring Cy5.5 fluorescence intensity in various organs. **P < 0.01, ****P < 0.0001 by two-way ANOVA with Turkey’s post-hoc test. b, Pharmacokinetics of IONP-DOX-Poly IC-EBP assessed by quantifying DOX fluorescence intensities. DOX: Ex, 500 nm; Em, 600 nm. Fluorescence intensity of DOX at 1 h was normalized to 1.
The blood circulation half-time of IONP-DOX-PolyIC-EBP was determined by quantifying DOX fluorescence signals in blood of BALB/c mice over time. The DOX signal intensity versus time relationship is shown in Fig. 6b from which it was determined that the half-lives of IONP-DOX-Poly IC-EBP is 4.8 h. The long blood half-life was primarily attributed to the small and uniform size of NPs, and to the hydrophilic coating of PEG molecules.
2.9. Tissue uptake of DOX and Histopathology in NP and free DOX treated tumor-bearing mice.
To investigate the targeted DOX delivery of IONP-DOX-Poly IC-EBP, we evaluated the uptake of DOX into various organs of mice administered intravascularly with free DOX or IONP-DOX-Poly IC-EBP (10 mg/kg DOX per mouse). Images of tissue sections of various organs from mice in Fig. 7a shows the deposition of DOX in various organs 48 h post-injection. Significant differences in DOX uptake by liver and tumors were observed. For mice treated with DOX, the liver displayed a strong and evenly distributed fluorescence signal. Previous investigations have suggested that the fluorescence signal might result from original DOX and its fluorescent metabolites by hepatic enzymes.74 In contrast, the livers from mice treated with IONP-DOX-Poly IC-EBP showed a scattered spot distribution of DOX. This could result from the internalized, agglomerated NPs taken up by liver macrophages, which is common to all types of NPs.75 No observable DOX signal was found in tumors of mice treated with free DOX. In contrast, strong DOX signals were observed in tumors of mice treated with IONP-DOX-Poly IC-EBP. The DOX was mainly located near blood vessels, likely due to uptake of NPs mediated by overexpressed Endoglin on these vessels. A plot of DOX uptake vs various tissues derived by quantifying fluorescence DOX intensity based on images (Fig. 7a) is shown in Fig. S3 (Supplementary Information). IONP-DOX-Poly IC-EBP has much higher accumulation in tumor than DOX, which explains why IONP-DOX-Poly IC-EBP is much more effective in tumor cell killing than DOX. It is also noted that the distributions of both DOX and IONP-DOX-Poly IC-EBP were heterogenous, presumably due to the heterogenic nature of this type of tumors.
Fig. 7.

Uptake of DOX into tumor and tissue histology of various organs in mice treated with free DOX or IONP-DOX-Poly IC-EBP. BALB/c mice were treated with saline (negative control), DOX (positive control) or IONP-DOX-Poly IC-EBP (therapeutic agent). DOX dose was 10 mg/kg and Poly IC dose was 18 mg/kg, and tissues were collected 48 h after a single i.v. injection. a, Confocal fluorescence microscopy imagines of tissues sections of various organs/tumors. Red: cell membrane (WGA-AF647). Blue: cell nucleus (DAPI). Green: DOX (Ex: 495 nm; Em: 600–650 nm). Scale bar: 75 μm. b, H&E stained tissue sections of heart, liver, spleen, lung, kidney and tumor from mice treated with the same conditions in a.
It is worthwhile noting that the 4T1 flank tumor model was used by design for studying the in vivo immune response, tumor targeting, NP biodistribution as well as pathological analysis. The flank tumor model of 4T1 is localized and has little chance to metastasize into other organs compared to the orthotopic model,76 which facilitates the investigation and interpretation of in vivo targeting and biodistribution (e.g., no tumor metastasizing into lung/liver/kidney to complicate biodistribution and pathological analysis). The orthotopic tumor model, on the other hand, is more clinically relevant and tends to metastasize; we thus used it for studying the metastasis inhibition and survival analysis.
To assess systemic toxicity and DOX uptake in various organs and tumors, mice were treated with DOX or IONP-DOX-Poly IC-EBP. One week after mice were inoculated with cancer cells, a single intravenous injection of DOX or IONP-DOX-Poly IC-EBP (10 mg/kg DOX equivalent per mouse) was given to each mouse. Forty-eight hours post-injection, mice were sacrificed, and organs/tumors were collected and processed for H&E analysis. The H&E stained images of heart tissue sections indicate that a severe heart damage was incurred in mice treated with DOX as evidenced by appearance of stripped spaces in myocardium, whereas minimal or no apparent heart damage was found in mice treated with IONP-DOX-Poly IC-EBP (Fig. 7b, first column). No pathological changes in organs other than heart were found in mice treated with either DOX or IONP-DOX-Poly IC-EBP. The cardiac toxicity of DOX has been well known, which is due to complex mechanisms.77 Our results indicate that by loading DOX into our NP formulation, the cardiotoxicity of DOX may be reduced or eliminated.
2.10. Therapeutic efficacy in tumor growth inhibition and inducing apoptosis in flank tumor model.
The luciferase-transfected 4T1 cells (4T1-luc) were inoculated into flanks of female wild-type BALB/c mice. This mouse model is appropriate for targeted chemo-immuno combinatorial therapy because it has an aggressive tumor proliferation profile and triple negative phenotypes. More importantly, the syngeneic mice have uncompromised immune systems which is ideal to test immunotherapeutics. To evaluate the treatment efficacy, tumor growth in mice was monitored over time before and after treatment. Each group of mice were administered with one of following agents via i.v. injection: PBS, drug-free IONPs, DOX, Poly IC, IONP-DOX-EBP and IONP-DOX-Poly IC-EBP. The treatment started 7 days after tumor inoculation and three dosages were given in every three days: DOX, 10 mg/kg in NP formulations and free form, 3d interval; Poly IC, 18 mg/kg, 3d interval (Fig. 8a). It should be noted that we also used a lower dosage (5 mg/kg) for free DOX than the dosage for our NP formulations (10 mg/kg) because the severe toxicity incurred by free DOX at 10 mg/kg caused mouse death in less than 2 weeks (Fig. S4a, Supplementary Information). In contrast, mice administered with NPs at a dose of 10 mg/kg DOX survived with some weight loss, indicating an increased maximum tolerable dose (MTD) when DOX is incorporated into our NP formulation (Fig. S4a, Supplementary Information). Fig. 8b shows the average tumor volume measured over time for each mouse group with the treatment started at day 7 and completed at day 13 (3 doses). The tumor growth inhibition by a therapeutic agent is defined as the reduction in tumor volume when compared to the average tumor volume of the mouse group treated with PBS. Treating mice with bare IONPs, free DOX or free Poly IC showed no (e.g., IONPs) or small (e.g., free DOX, free Poly IC) inhibition to tumor growth as compared to PBS-treated mice (Fig. 8b). Comparatively, IONP-DOX-EBP and IONP-DOX-Poly IC showed substantial tumor inhibition. Significantly, IONP-DOX-Poly IC-EBP showed strongest tumor growth inhibition among all the treatment options (~85%). Fig. 8c shows images of mice bearing 4T1-luc tumors 48 h after intravascular administration of various agents with IVIS. The bioluminescence generated from luciferase in cancer cells in mice was weakest in IONP-DOX-Poly IC-EBP treated ones among all treatments (Fig. 8c). This result showed its robust efficacy in a preclinical model due to the effects from targeted and combined chemo-immuno therapeutics.
Fig. 8.

In vivo evaluation of NPs in a 4T1-luc flank tumor model of wild-type BALB/c mice. a, Schedule of tumor inoculation, treatment regimen, and monitoring. b, Tumor size as a function of time over the 28-day period shown in a., starting from tumor inoculation (day 0), for mice treated with eight agents (n = 4/per agent). **P < 0.01, ***P < 0.005 by two-way ANOVA with Turkey’s post-hoc test. Each mouse was administered three times by i.v. injection and the tumor volume was measured every three days, both following the schedule shown in a. The agents administered include PBS, IONPs, DOX (5 and 10 mg/kg), Poly IC (18 mg/kg), IONP-DOX-EBP (DOX 10 mg/kg), IONP-DOX-Poly IC (DOX 10 mg/kg, Poly IC 18 mg/kg), and IONP-DOX-Poly IC-EBP (DOX 10 mg/kg, Poly IC 18 mg/kg). Black dashed arrow indicates tumor growth curve (also black dashed) with free 10 mg/kg DOX treatment. c, Live IVIS images of mice bearing 4T1-luc tumors 48 h after intravascular administration of various agents: (i) PBS, (ii) IONPs, (iii) DOX 5 mg/kg, (iv) DOX 10 mg/kg, (v) Poly IC (18 mg/kg), (vi) IONP-DOX-EBP (DOX 10 mg/kg), (vii) IONP-DOX-Poly IC (DOX 10 mg/kg, Poly IC 18 mg/kg), and (viii) IONP-DOX-Poly IC-EBP (DOX 10 mg/kg, Poly IC 18 mg/kg). d, Confocal fluorescence microscopic images of tumor tissue sections harvested from mice treated with various agents shown in a. Tumor tissue sections were stained with Annexin V (green) for apoptotic cells and propidium iodide (red) for nuclei. Scale bar = 100 μm.
To investigate whether tumor cells underwent apoptosis in mice treated with various agents, each mouse was administered via iv injection with one of the following agents (i) PBS, (ii) IONPs, (iii) DOX 5 mg/kg, (iv) DOX 10 mg/kg, (v) Poly IC (18 mg/kg), (vi) IONP-DOX-EBP (DOX 10 mg/kg), (vii) IONP-DOX-Poly IC (DOX 10 mg/kg, Poly IC 18 mg/kg), and (viii) IONP-DOX-Poly IC-EBP (DOX 10 mg/kg, Poly IC 18 mg/kg). Tumor tissues were collected 48 h post injection and stained with Annexin V-Alexa Fluor 647 for apoptotic cells and propidium iodide (PI) for nucleic acids. Confocal microscopic images of these tumor sections showed that treatment of IONP-DOX-Poly IC-EBP induced massive apoptosis in tumor (Fig. 8d, viii). Tumors in mice that underwent other treatment options showed only minor apoptosis of various degrees (Fig. 8d i–vii; Fig. S4b, Supplementary Information.). DOX induces DNA damage and apoptosis through inhibiting DNA topoisomerase II.44 Direct activation of TLR3 on breast cancer cells by Poly IC can also trigger apoptosis.78 Furthermore, the anti-tumor immunity induced by interaction between Poly IC and DCs can cause tumor death through apoptosis.56 Together, the multifunctional NP-mediated multiple attack to cancer cells, combined with targeted delivery, achieved the maximum tumor inhibition compared to any of the single-agent treatment options.
2.11. Inhibition of tumor growth and metastasis in orthotropic tumor model.
We further evaluated the therapeutic efficacy of IONP-DOX-Poly IC-EBP in a second mouse model: a 4T1-luc orthotopic tumor model. Unlike in the flank tumor model where tumor cells were inoculated in the flank, 4T1-luc cells were directly implanted subcutaneously at mammary gland, the same anatomical site of breast cancer in human. Cancer cells grown in mammary gland have the tendency to metastasize to various organs (Fig. 9a). Thus, this model allows the examination of the capability of our NPs in inhibiting tumor metastasis in addition to tumor growth. Based on the investigation conducted in the flank tumor model above, two treatment options were selected for study in the second mouse model: free DOX 5 mg/kg and IONP-DOX-Poly IC-EBP (DOX 10 mg/kg, Poly IC 18 mg/kg), with each treatment including 5 instead of 3 sequential i.v. injections 3 days apart. Tumor volumes were measured over time using a caliper, and mice survival and tumor metastasis were monitored throughout the experiment (Fig. 9b). Treatment with IONP-DOX-Poly IC-EBP resulted in greater and more sustainable inhibition to tumor growth as shown by a flat growth curve lasting till 19 days (in contrast to 16 days with only 3 injections) (Fig. 9c). Tumor growth was monitored for 25 days. No treatment caused weight loss in mice during this period (Fig. 9d). Overall survival was continuously monitored till all mice were euthanized (Fig. 9e). The median survival times were 28.5, 32.5 and 37 d for PBS, free DOX, and IONP-DOX-Poly IC-EPB treated mice, respectively. Twenty-two days after tumor inoculation, metastasis was monitored by IVIS bioluminescence imaging. Un-treated mice showed metastasis as early as day 22 after tumor inoculation (Fig. 9f). Metastasis was found in liver (day 22), kidneys (day 22) and lungs (day 34) as shown by bioluminescence in PBS treated mice (Supplementary Information, Fig. S5). Free DOX treatment delayed the metastasis to day 31. Kidney metastasis was observed and marked with red arrow c. IONP-DOX-Poly IC-EBP showed the potent treatment and delayed the metastasis to day 45 till lung metastasis was observed (Supplementary Information, Fig. S5).
Fig. 9.

Tumor growth and metastasis in a 4T1-luc primary tumor model. a, Cartoon illustration of primary breast tumor and potential metastatic sites in a mouse. b, Schedule of tumor inoculation, treatment regimen, and monitoring. c, Sizes of 4T1-luc tumors of mice under various treatments: PBS, DOX (5 mg/kg each injection) and IONP-DOX-Poly IC-EBP (DOX 10 mg/kg, Poly IC 18 mg/kg each injection) injected intravenously. Five injections were given following the schedule shown in b. *p < 0.05 between IONP-DOX-Poly IC-EBP and untreated groups, by two-way ANOVA with Turkey’s post-hoc test. d, Body weights of mice from all groups (n = 6) as a function of time. e, Kaplan-Meier survival curves of mice treated with various agents. f, Representative fluorescence images of mice from treatment groups: PBS, DOX, and IONP-DOX-Poly IC-EBP. Arrows indicate primary tumor and possible metastatic sites. a: primary tumor, b: liver, c: kidney(s).
3. Discussion
Chemotherapy has been shown to play an important role in positive tumor immune response modulation by enhancing tumor antigenicity and adjuvanticity.79 Chemo-immuno-therapy could be a promising option in treating TNBC as shown by the recent approval of the combination of Atezolizumab (anti-PD-L1) with paclitaxel.80 However, those combinatorial therapies show improved efficacy only in a small subset patients. The promise of combined chemo- and immune- therapy for TNBC has been shadowed by the lack of an effective means to circumvent a number of obstacles including physiological and cellular barriers to the delivery of sufficient amounts of chemo- and immune- therapeutic agents to tumors. Here, we present an EBP-conjugated and DOX/Poly IC co-loaded IONP for targeted delivery of chemo-immuno therapy for treating TNBC. The IONP-DOX-Poly IC-EBP is designed to circumvent common limitations of therapeutic agent delivery and endowed with many favorable attributes beyond those provided by existing carriers. The design of our technique has the following unique features: 1) a new assembly method to make a small and stable NPs by electrolyte-free layer-by-layer deposition. Such method can be applied not only to DOX and Poly IC, but also to other charged therapeutic molecules in general; 2) a targeted, chemo-immuno combinatory therapy for metastatic TNBC. The targeting ligand enhances the delivery of both DOX and Poly IC on IONP into tumor microenvironments including tumor cells and immature DCs; 3) superparamagnetic properties of IONPs render them detectable by MRI, thus enabling non-invasive tumor diagnosis and treatment response monitoring.
The targeting of triple negative breast cancer has been challenging due to the lack of cell surface receptors.81 Several cancer-specific cell surface targets were investigated such as PD-L1, CD44, CXCR4, uPAR, αvβ3 integrin, LIV-1, etc.82–88 Targeting these receptors has shown several limitations including (a) high dependence of cancer cell expression, (b) conjugation of antibodies which are bulky, showing side effects,89 and expensive, and (c) high systemic background of targets.90 Among several potential TNBC targets, endoglin is a co-receptor of transforming growth factor-beta and plays a crucial role in vasculature development and angiogenesis of breast cancer.91 Endoglin is involved in the proliferation and invasion of breast cancer cells, and high endoglin expression is correlated with a high risk for metastasis in patients.92 Monoclonal antibodies against endoglin have been used to target TNBC in mice models using NPs as a carrier and have shown a specific targeting effect.43,93 The present study utilizes an endoglin-specific small peptide which is much less bulky and allows for increased loading of therapeutic molecules. Our NPs are able to enter cancer cells effectively both in vitro (Fig.3) and in vivo (Fig. 5).
Co-loading of both chemotherapeutics and immunotherapeutics on a single NP had been technically challenging because of their diametrically different physicochemical properties that precluded co-loading. In previous studies, NPs have been employed to simultaneously deliver both chemo- and immuno- therapeutic antigens and/or adjuvants to tumors through active targeting or through enhanced permeability and retention effects.94 Most of these NPs were large in size (much greater than 100 nm), and as a result, experienced a short half-life in blood, which led to low drug accumulation in tumors.95,96 Although the alternative of loading immuno- and chemo-therapeutics on different NPs for separate delivery can avoid oversized NP,97 the fact that immuno- and chemo-drugs cannot reach the same cell at the same time could drastically impair their therapeutic synergy. On the contrary, our NP’s LBL design directly deposits positively charged DOX and negatively charged Poly IC on the surface of negatively charged ultra- small and stable PEG-coated IONP, eliminating the need for additional polyelectrolytes which could otherwise cause confounding immune responses for encapsulation and stabilization.98 Through modular surface engineering, our NP exhibits efficient Dox/poly IC co-loading and tunable physicochemical properties including size and surface charge (Fig. 1). The final NP bearing targeting peptide is spherical in shape and ultrasmall in size with a hydrodynamic diameter of ~53 nm. The NP demonstrates a long circulation time in blood (4.8 h) (Fig. 6b) and releases drug payload in an acidic environment (Fig. 2) and induces, concurrently, dendritic cell-mediated innate and T cell-mediated adaptive immune responses. The NP triggers DC maturation as confirmed by upregulation of BMDC surface markers (CD80 and CD86) (Fig. 3d and e) without inflicting significant cytotoxicity on BMDCs, likely due to the insensitivity of DCs to DOX and the reduced drug uptake by the nucleus. It is reported that DCs in the 4T1 tumor microenvironment are functionally defective and exhibit a characteristic of immature phenotype, which is responsible for immunosuppression of 4T1.99 As a result of DC activation, both tumor and spleen generated antigen-specific immune response (Fig. 4c), and a higher level of production of IL-12 was observed by systemic injection of IONP-DOX-Poly IC-EBP as compared to injection of free Poly IC (Fig. 4b). Although several approaches had been introduced to deliver IL-12 gene to tumors, the delivery and transfection efficiency were limited and the gene carriers (e.g., PEI) showed severe toxicity to biological system.100,101 In tumor-bearing mice, our NP effectively activates antigen-specific T cell response (CD8+, CD25+, CD69+) more than free Poly IC through systemic injections. This is likely due to the targeted delivery enabled by EBP (Fig. 3 and Fig. 5) and the small size of our NP (Fig. 1). Systemic injection of IONP-DOX-Poly IC-EBP resulted in the greatest tumor growth inhibition in the flank tumor model among all the treatment options (Fig. 8). Further, our NP is non-toxic to liver, spleen, lung and kidney (Fig. 7), and markedly reduced the cardiotoxicity of DOX and enabled the use of higher DOX dosing through targeted delivery to tumor sites (Fig. 8b).
The clinical potential of IONP-DOX-Poly IC-EBP was evaluated in the aggressive 4T1 breast cancer mouse model by systemic injection. 4T1 breast adenocarcinoma cells were used to mimic stage IV metastatic human breast cancer. 4T1 cells are highly aggressive and can spontaneously metastasize to various organs when injected into BALB/c mice.102 The 4T1 cells also have a “triple negative” phenotype (lack of ER, PR and HER2 receptors on cell surface) and are resistant to DOX treatment by expression of P-glycoprotein (efflux transporter of DOX).103 Various combinations of treatment options have been investigated to treat 4T1 tumors, including radio-immunotherapy,8 multi-agent chemotherapy,6 multi-antibody therapy,7 etc. But no effective treatment has been identified for the 4T1 induced breast metastasis. Our NP also demonstrated tumor targeting capability (Fig. 5), and markedly increased the therapeutic efficacy against tumor growth (Fig. 8) and metastasis as compared to the treatment with DOX alone (Fig. 9 and Fig. S5). In addition to the therapeutic function, our NP provides MRI and optical imaging capability enabled by IONP core and near-IR fluorophore, respectively, allowing examination of NP localization in vivo (Fig. 5).
In summary, we have developed a multifunctional NP formulation that can deliver chemo- and immuno- therapeutic agents simultaneously targeting breast cancer cells in a synchronous fashion. The NP demonstrates physicochemical properties favorable for in vivo application. The NP induces tumor apoptosis by multiple mechanisms including direct tumor cell killing, dendritic cell-mediated innate response and T cell-mediated adaptive immune response. Our NP demonstrated a potent ability to inhibit tumor growth and metastasis, and to extend survival in an aggressive and metastatic mouse model of triple negative breast cancer (TNBC). The non-invasive image-guided approaches could be quite useful in clinic to enable monitoring of therapeutic response and improve the success rate of cancer treatment. This study points to a new strategy that may substantially improve the outcome of treating metastatic TNBC.
4. Experimental Section
4.1. Materials.
3-(triethoxysilyl)propyl succinic anhydride (SATES) was purchased from Gelest (Arlington, VA). 2000 MW mono-amine functionalized poly(ethylene) glycol (mPEG2K-NH2) was purchased from Laysan Bio (Arab, AL). EBP was customized from GenScript (Piscataway, NJ). The 2-iminothiolane (Traut’s reagent) was purchased from Molecular Biosciences (Boulder, CO). NHS-PEG24-maleimide, Annexin V-Alexa Fluor 647 and Wheat germ agglutinin-Alexa Fluor 647 were purchased from Thermo Fisher Scientific (Rockford, IL). DOX was purchased from LC Laboratories (Woburn, MA). Annexin V-FITC was purchased from BD Biosciences (San Diego, CA). NHS-Cy5 was purchased from Lumiprobe Corp. (Hallandale Beach, FL). Anti-mouse antibodies CD86-Alexa Fluor 647 (GL-1) and CD80-PE (16–10A1) were purchased from BioLegend (San Diego, CA). The mouse IL-12 P40/70 ELISA kit was purchased from Raybiotech Inc. (Norcross, GA). D-Luciferin was purchased from PerkinElmer Inc. (Waltham, MA). All other chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO).
4.2. NP synthesis and surface PEGylation.
Oleic acid-stabilized iron oxide NPs with an 8-nm core were synthesized following a published method.104 Coating these NPs with silane-PEG2000-NH2 was adapted from our previous published method.105 For a typical batch, 50 mg of iron oxide NPs was suspended in 43 mL of anhydrous toluene followed by addition of 70 μl of triethylamine in a 3-neck round-bottom flask fitted with a Graham condenser. The flask was sealed with a rubber septum and purged with nitrogen. The solution was heated to 100°C and 0.15 mL of SATES was added to the flask. 281.25 mg of mPEG2K-NH2 was dissolved in 7 mL of anhydrous toluene and the resultant solution was added to the flask 15 minutes later. An additional 75 μL of SATES was injected 1 h after the mPEG2K-NH2 injection, and the solution was allowed to react for an additional 6.75 h. The solution was transferred to a single-neck round-bottom flask and NPs were precipitated with hexane. The NP precipitate was dispersed in tetrahydrofuran (THF), sonicated for 10 min, and precipitated with hexane. The resulting NP pellet was suspended in 10 mL anhydrous THF and sonicated for 10 min. 93.75 mg of mPEG2K-NH2 and 281.25 mg of mPEG2K-NH2 were dissolved in 12 mL anhydrous THF and added to the NP solution. The flask was then sealed with a septum and purged with nitrogen. 18.75 mg of N, N’-dicyclohexylcarbodiimide (DCC) was dissolved in 2 mL anhydrous THF and added to the flask, and the reaction solution was placed in a sonication bath at 25°C and allowed to react for 16 h. Fully PEGylated NPs were precipitated with hexane, redispersed in 20 mL of ethanol, sonicated for 10 min, and precipitated again with hexane. The pellet was fully dried and dispersed in deionized water under sonication for 10 min. The NPs were purified through size exclusion gel chromatography (Sephacryl S-200).
4.3. NP conjugations.
EBP (CAHKHVHHVPVRL) was conjugated onto IONPs by NHS-PEG-maleimide crosslinking chemistry. The EBP has a cysteine modification at N-terminal to introduce a thiol group which is maleimide reactive. For conjugation of IONP with PEG, 1 mL of 3 mg of IONPs (the amount of NPs was determined by [Fe] concentration) in PBS buffer was first incubated with 2.13 μL of SM(PEG)24 (250 mM in DMSO) for 30 min on a rocker. Free SM(PEG)24 was removed by purification through a PD-10 desalting column (GE Healthcare, Piscataway, NJ) equilibrated with PBS. PEG−maleimide modified IONPs were then mixed with the EBP and allowed to react for 30 min, and unreacted EBP was removed using S-200 sephacryl resin equilibrated with PBS to obtain IONP-EBP. Fe concentration was determined by a ferrozine assay. To optimize DOX loading onto IONP-EBP, different Fe/DOX ratios (10:2 to 10:16, w/w) were tested. DOX (5 mg/mL in deionized water) and IONP-EBP were mixed in PBS and incubated overnight on a rocker. Unbound DOX was removed by passing samples through sephacryl S-200 columns to get IONP-DOX-EBP. Fe concentration was quantified by a ferrozine assay,60,106 and DOX concentrations were quantified by UV absorbance at 500 nm. Poly IC (10 mg/mL in deionized water) was then mixed with IONP-DOX-EBP with various ratios of Fe to Poly IC (10:2~10:16, w/w) and incubated for 20 min to make IONP-DOX-Poly IC-EBP. IONP-DOX-Poly IC-EBP was freshly made for all assays.
To make Cy5.5- labeled NPs, IONP-DOX-EBP (3 mg) was incubated for 1 h with NHS- Cy5.5 (7.2 μL, 5 mg/mL in DMSO) in PBS before purification by an S-200 sephacryl column in PBS. For NPs used in animal studies, the sample preparations were scaled up proportionally.
4.4. NP characterizations.
To prepare NP samples for TEM imaging, NP solution (4 μL) was transferred onto a TEM grid (copper grid, 300-mesh, coated with carbon and Formvar film). After drying the solution in air using a filter paper, TEM images were acquired on a Tecnai G2 F20 electron microscope (FEI, Hillsboro, OR) operating at a voltage of 200 kV.
The hydrodynamic size and ζ-potential of NPs were characterized using a Zetasizer Nano-ZS (Malvern Instruments, Worcestershire, UK). The analyses were performed at the room temperature. The pH value of all NP solutions for ζ-potential measurements was 7.4 (20 mM HEPES buffer).
Samples for AFM were prepared by dropping and drying a low concentration of NP solution on freshly cleaved mica. The samples were then imaged using a Bruker Dimension Icon AFM (Madison, WI, USA) in tapping mode in air, with an antimony-doped silicon cantilever (FESP, Bruker, Madison, WI, USA). This cantilever has a nominal spring constant of 2.8 N/m, a resonant frequency of 75 kHz, a length of 225 μm, and a tip radius of 8 nm. Resulting images were processed with Gwyddion software. The absorbance of different solutions was recorded by a UV–Vis spectrometer (Agilent Technologies, Santa Clara, CA). For agarose gel electrophoresis, free Poly IC and different NP-Poly IC mixtures were loaded onto a 0.8% agarose gel (premixed with ethidium bromide) and ran for 30 min at a voltage of 100 V. Gel was imaged by a Gel Doc XR imaging system (Bio-Rad).
pH-responsive DOX release was assessed by a dialysis method. IONP-DOX-Poly IC-EBP solution (PBS, pH 7.4) was loaded into three dialysis tubings (1 mL each, 14 kDa MW cutoff). Sealed dialysis tubings were then immersed in 30 mL of different buffers (PBS, pH 7.4; Sodium acetate buffer, pH 5.4; MES buffer, pH 4.5) in a 37°C water bath and stirred for 72 h. The DOX released was sampled (0.5 mL each) at different time points (1, 2, 4, 6, 8, 10, 24, 48 and 72 h) and quantified by fluorescence reading (SpectraMax i3 multimode microplate reader, Molecular Devices). Cumulative release was calculated through DOX concentration and converted to total percentage of drug released over time.
4.5. Cellular studies in 4T1 breast cancer cells.
4.5.1. Cell culture.
4T1 and 4T1-luc cells were provided by Stanley Riddell laboratory in Fred Hutchinson Cancer Research Center. Cells were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic (Thermo Fisher Scientific, Rockford, IL). Cells were cultured in an incubator maintained at 37°C, 5% CO2 and 95% humidity.
4.5.2. Characterization of cellular uptake by confocal laser scanning microscopy.
4T1 cells were seeded onto glass-bottom petri dishes (Mattech). After overnight incubation, cells were incubated with different agents (10 μg/mL DOX or DOX equivalent NPs) for 2 h at either 37°C or 4°C. Cells were washed 3 times with cold PBS, fixed with 4% paraformaldehyde for 15 min at 37°C and stained with 5 μg mL WGA-Alexa Fluor 647 for 5 min at 37°C, followed by 3 times of PBS washing (5 min each). Cells were then incubated with DAPI for 5 min at 37°C, followed by PBS washing. Cells were then mounted with VECTASHIELD mounting medium (Vector Laboratories, Inc. Burlingame, CA). The images of cells were acquired using a Leica SP8X confocal laser scanning microscope (Leica, Germany).
4.5.3. Cell uptake assay by flow cytometry.
4T1 cells were incubated with different agents for 2 h (10 μg/mL DOX or DOX equivalent NPs) at either 37°C or 4°C, followed by washing with cold PBS for 3 times. Cells were then trypsinized and resuspended in cold PBS and analyzed by flow cytometry (FACSCanto II, BD Biosciences). Each condition was triplicated.
4.5.4. Alamar Blue cell viability assay.
Cells were seeded in a 96-well plate and incubated overnight. In the following day, the medium was replaced with medium containing various agents. The cells were then incubated for 72 h. Cell viability was assessed using the Alamar Blue assay. Briefly, the medium was replaced with cell culture medium containing the reagent and allowed to incubate for 2 h. Following the incubation, a microplate reader (SpectraMax i3 multimode microplate reader, Molecular Devices) was used to determine the fluorescence intensity of the dye (550ex/590em). The fluorescence intensity from NP or free drug treated cells was compared to those from untreated control cells to determine percent viability. Each condition was triplicated.
4.5.5. Cell apoptosis assay by flow cytometry.
4T1 cells were seeded into 6-well plates and incubated overnight. DOX or DOX-equivalent NPs were added into cells (1 μg/mL DOX final concentration). Cells were incubated with NPs for 48 h. Cells were then trypsinized, aspirated, and washed once with PBS. Cells were then counted and suspended in 0.1 mL Annexin V binding buffer containing 50 μg/mL propidium iodide and 5 μL FITC-Annexin V reagent. Cells were further incubated for 15 min at room temperature in dark. 0.4 mL Annexin V binding buffer was added prior to analysis by flow cytometry. Data acquisition was performed on FACSCanto II and analyzed by FlowJo software (Treestar, Inc., San Carlos, CA). Each condition was triplicated.
4.6. Cellular studies of bone marrow-derived dendritic cells (BMDCs).
4.6.1. BMDCs preparation.
BMDC were generated from female BALB/c mice of 6–8 weeks old. The isolation of bone marrow cells was carried out following a previous report.107 Cells were resuspended in RPMI-1640 supplemented with 10% FBS, 20 ng/mL recombinant murine GM-CSF (Shenandoah Biotechnology, Inc, Warwick, PA), and antibiotics (Pen Strep). Cells were allowed to differentiate for 7 d with one addition of fresh media at day 3. At day 7, cells were aspirated and counted for assays. Medium containing 10 ng/mL GM-CSF was used in assays.
4.6.2. Confocal microscopic imaging of cellular uptake.
Cells were seeded onto a chamber slide (Nunc™ Lab-Tek™ II Chamber Slide™ System, Thermo Fisher Scientific Rockford, IL) with a density of 50,000 per well and incubated overnight. DOX, IONP-DOX-EBP-Cy5, or IONP-DOX-Poly IC-EBP-Cy5 was added into cells at a concentration of 10 μg/mL Poly IC-equivalency and incubated for 1 h. Cells were then washed and imaged with a Leica SP8X confocal laser scanning microscope.
4.6.3. BMDCs maturation.
Cells were seeded onto a 6-well plate at a density of 300,000 per well and incubated overnight. Poly IC, IONP-DOX-EBP or IONP-DOX-Poly IC-EBP was added into cells at a concentration of 10 μg/mL Poly IC-equivalency and incubated for 24 h. Cells only in medium were used as control. Cells were then trypsinized, aspirated and co-stained with CD80-PE and CD86-AF647 according to manufacturer’s instruction. Cells were then analyzed by flow cytometry on FACSCanto II. All experiments were performed in triplication.
4.6.4. BMDC viability.
Cells were seeded onto a 96-well plate at a density of 30,000 per well and incubated overnight. Poly IC, IONP-DOX-EBP or IONP-DOX-Poly IC-EBP was incubated with cells at a concentration of 10 μg/mL Poly IC-equivalency for 24 h. Cells only in medium were used as control. Cell viability was then assessed by aforementioned Alamar blue assay. Each condition was triplicated.
4.6.5. IL-12 production by ELISA assay.
All conditions used were same as those for viability assay. Twenty-four hours after addition of agents into cell medium, supernatants were collected and IL-12 levels in medium were assessed by an ELISA assay kit following the manufacturer’s protocol. Each condition was triplicated.
4.7. Animal Studies.
All animal studies were conducted in accordance with University of Washington Institute of Animal Care and Use Committee (IACUC) approved protocols as well as with federal guidelines. Five-week-old female BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, Maine) and housed in the animal research facility.
4.7.1. Pharmacokinetics of IONP-DOX-Poly IC-EBP-Cy5.
Six-week-old mice were administered with IONP-DOX-Poly IC-EBP-Cy5 through i.v. injection (dosage: 10 mg/kg DOX or equivalent). At 1, 2, 4, 8, 24 and 72 h post-injection, blood (5–25 μL) was collected from tail vein. The total amount of blood withdrawn from each mouse never exceeded one percent of the total body weight of the animal during the experiment. Whole blood was diluted with PBS and spun using a benchtop centrifuge for 2 min at 5000 g to separate the plasma. The diluted plasma was added to a 96-well black plate. The plate was scanned on a SpectraMax i3 plate reader (fluorescence mode) to measure Cy5 (ex, 646 nm; em, 676 nm) and DOX (ex, 500 nm; em, 600 nm) fluorescence signals. Three mice were used for each group.
4.7.2. Serum IL-12 determination.
The same conditions in pharmacokinetics study were used here except that blood was collected before agent administration and 2, 6.5 and 24 h after administration. IL-12 in plasma was assessed using an ELISA kit following the manufacturer’s instruction and quantified with a SpectraMax i3 plate reader.
4.7.3. Antigen-specific T cell response by flow cytometry
Twelve 6-week-old female BALB/c mice were used in this study. 4T1 cells (105 cells per mouse) were inoculated subcutaneously into #9 mammary glands. The tumors were allowed to grow into palpable masses. Ten days after tumor inoculation, the 12 mice were separated into 4 treatment groups. Four treatments, including IONP-DOX-EBP-Poly IC (10 mg/kg DOX and Poly IC), IONP-DOX-EBP (10 mg/kg DOX), free Poly IC (10 mg/kg Poly IC) and PBS, were administered intravenously into four groups of mice bearing tumors (200 μL per mouse), respectively. Three days after administration, mice were euthanized, and tumors and spleens were harvested.
Harvested tumors and spleens were then sectioned, squeezed through 70 μm cell strainers and washed with PBS w/1% v/v ratio of FBS to obtain single cell suspensions. Cells were spun down with a centrifuge at 1500 rpm for 4 min and resuspended in 750 μL PBS w/1% v/v FBS. Anti-CD8-PE, antiCD25-APC, and anti-CD69-FITC (Biolegend Inc.) solutions (3 μL each) were then mixed with 241 μL of PBS w/1% v/v FBS (total antibody solution volume for each single cell suspension was 250 μL). The cell/antibody suspension was then incubated in dark at room temperature for 30 mins. After incubation, 11 mL of PBS w/1% v/v FBS was added to all cell/antibody suspensions followed by centrifuging at 1500 rpm for 4 mins. Finally, all cell pellets were resuspended in 500 μL PBS w/1% v/v FBS and analyzed by flow cytometry (FACSCanto II).
4.7.4. Near-IR fluorescence and bioluminescence imaging.
Accumulation of IONP-DOX-Poly IC-EBP-Cy5.5 in tumor was assessed by near-IR fluorescence imaging. One week after tumor inoculation, IONP-DOX-Poly IC-EBP-Cy5.5 was administered into mice through i.v. injection. At 0.5 h, 6 h, 1 d, 2 d, 4 d and 7 d post-injection, fluorescence and/or bioluminescence images, as well as optical photographs, were taken by a XENOGEN IVIS 200 imaging system (PerkinElmer Inc.) with imaging parameters: excitation wavelength: 710 nm; emission filter: ICG; exposure time: 1 second; binning factor: 2; f/stop: 4. For bioluminescence imaging of tumors, mice were injected with 150 mg kg–1 luciferin intraperitoneally at day 24 after first administrations and imaged with an IVIS system. Imaging parameters are emission filter: open; exposure time: 30 seconds; binning factor: 2; f/stop: 4.
4.7.5. Tumor growth inhibition study in 4T1-luc flank tumor model.
6-week-old female BALB/c mice were used in this study. 4T1-luc cells were transfected to stably express luciferase so that bioluminescence imaging could be used to monitor tumor growth. 4T1-luc cells were trypsinized and suspended in PBS (107 cells/mL) and injected subcutaneously into the right flanks of mice (0.1 mL per mouse). Seven days after tumor inoculation, twenty eight mice were randomly divided into 7 groups and each group was administered through intravenous injection with three doses of one of the following agents: PBS, IONPs, DOX, Poly IC, IONP-DOX-EBP, IONP-DOX-Poly IC, and IONP-DOX-Poly IC-EBP. For free DOX injection, the dose was 5 mg/kg. For all NP agents, doses of DOX and Poly IC were 10 mg/kg and 18 mg/kg, respectively. Each mouse received an injection at day 7, 10 and 13 after tumor inoculation. The tumor size was measured by a caliper and the tumor volume was calculated by equation: V = width2 × length/2. The tumor size was measured every 3 days starting at day 4 after tumor cell inoculation. The body weight was also monitored after first agent administration.
4.7.6. Tumor growth inhibition, survival and metastasis study in 4T1-luc primary tumor model.
6-week-old female BALB/c mice were used in this study. 4T1-luc cells were injected subcutaneously into the #9 mammary glands (106 cells in 0.05 mL PBS per mouse). Seven days after tumor inoculation, eighteen mice were randomly divided into 3 groups and each group was administered through intravenous injection with five doses of one of the following agents: PBS, DOX, and IONP-DOX-Poly IC-EBP. For free DOX, dose was 5 mg/kg per injection. For IONP-DOX-Poly IC-EBP, doses of DOX and Poly IC were 10 mg/kg and 18 mg/kg, respectively. Each mouse received an injection at day 7, 10, 13, 16 and 19 after tumor inoculation. The tumor size and body weight were monitored after first administration. Metastasis was monitored by IVIS imaging starting at day 22 after tumor inoculation. Mice were imaged at day 22, 25, 28, 31, 34, 37, 41, 45 and 50 Mice were euthanized following the approved animal protocol.
4.8. Processing and imaging of sectioned organs and tumors.
One week after inoculated with 4T1-luc cells, mice were injected with various agents intravenously. Forty-eight hours later, mice were euthanized and organs (heart, kidney, liver, lung, and spleen) and tumors were harvested and pre-served in 10% formalin for 48 h. Formalin-fixed tissue samples were first transferred from PBS to 70% ethanol and maintained for 2 hours, and then were transferred into 95% ethanol/5% methanol and maintained for an additional 2 hours. The samples were transferred in absolute ethanol and maintained for 1 hour, and then transferred again into 3 consecutive absolute xylene solutions, and maintained in each for 1 hour. The samples were dehydrated and then placed in 2 consecutive melted paraffin baths and maintained in each for 2 hours. The tissue samples were then embedded in paraffin blocks and sectioned at 10 um thickness and loaded onto microscope slides. The loaded slides were heated to remove excess paraffin and adhere the samples to the slides. The samples were then deparaffinized by a series of xylene, ethanol and PBS baths.
4.8.1. Tumor apoptosis test by Annexin-V and confocal microscopic imaging.
The deparaffinized tissue samples from the above process were stained with PI and Annexin V-Alexa Fluor 647, mounted with Prolong Gold mounting medium, and imaged with a Leica SP8X confocal microscope. Annexin V stained cells are apoptotic.
4.8.2. H&E staining and imaging for histopathology.
The deparaffinized samples were stained with haemotoxylin and eosin, and mounted with Prolong Gold mounting medium. Microscopic images of tissues were acquired using a Nikon ECLIPSE TE 2000-S microscope.
4.8.3. DOX uptake in organs and tumors by confocal microscopic imaging.
The deparaffinized tissue samples were stained with DAPI and WGA-AF647, and mounted with Prolong Gold mounting medium. Samples were imaged with a Leica SP8X confocal microscope.
4.9. MR Imaging.
4.9.1. In vitro MR imaging.
Magnetic properties of IONP-DOX-Poly IC-EBP were assessed using MR Imaging. Quantitative T2 and T2-weighted scan sequences were used to determine R2 relaxivity values and T2-weighted signal changes as a function of Fe concentration, respectively. MR imaging was performed using a Bruker Avance III 600 MHz 14 T vertical-bore spectrometer. NPs in phosphate buffered saline were pipetted into glass vials (3.25 mm I.D., 5 mm O.D., 200 μL volume). The vials were fixed in place inside a water reservoir; the water served as a homogeneous background signal to minimize magnetic susceptibility variations near the samples. The secured vials were placed in a 25-mm single-channel 1H radiofrequency receive coil (PB Micro 2.5). Relaxation properties of NPs were evaluated with a quantitative T2 multi-spin multi-echo (MSME) pulse sequence with TR = 2500 ms, TE = 6.7 + 6n ms (n = 0–16), and 78 × 156 μm2 in-plane resolution with 0.5 mm slice thickness for 14 slices. T2-weighted images were acquired with a rapid acquisition with refocused echoes (RARE) pulse sequence with TR = 4000 ms, TE = 6.78 ms, and 78 × 52 μm2 in-plane resolution with 0.5 mm slice thickness for 14 slices. Analysis of MRI data was accomplished with the FMRIB software library (FSL), Paravision 5.1 analysis package (Bruker), and ImageJ (NIH). T2 values were determined within a circular, 100-voxel region of interest.
4.9.2. In vivo MR imaging.
In vivo MR imaging was performed using the same imaging system used for in vitro MR imaging. A T2-weighted scan sequence was used to acquire MR images prior to and following tail-vein injection of IONP-DOX-Poly IC-EBP. A mouse was anesthetized with isoflurane (Piramal Healthcare) and attached to a coil-integrated respiratory monitoring system (SA Instruments; MR-compatible small animal monitoring and gating system) with nose-cone for oxygen/anesthetic, ear-bar head holder, circulating temperature control bath, and residual gas extraction. Abdominal scans were acquired using a RARE T2-weighted scan sequence (TR = 4000 ms, TE = 27 ms, in-plane resolution = 62 × 94 μm2, matrix = 384 × 256) with slices oriented in the transverse plane with 0.5 mm slice thickness and 0.75 mm interslice. Analysis of in vivo MR images was performed using the Paravsion 5.1 analysis package (Bruker) and ImageJ (NIH).
4.10. Statistical analysis.
Student’s unpaired t-tests were performed for comparison between treatment groups. One-way and two-way analyses of variance (ANOVA) followed by Turkey’s post-hoc multiple comparison tests were used for comparison of multiple groups. Statistical analyses were performed in Microsoft Excel or GraphPad Prism software.
Supplementary Material
Acknowledgements
The work is supported by NIH grants R01EB026890 and R01CA161953. Q. M. and R. R. acknowledges support from an NIH Ruth L. Kirschstein T32 Fellowship (T32CA138312). We also acknowledge the support from the NIH to the UW W. M. Keck Microscopy Center (S10 OD016240). We acknowledge UW Shared IVIS Core and the NIH Shared Instrumentation Grant 1S10OD010652-01. This work was also supported by Biomedical Research Support Shared Instrumentation Grant (S10RR029021 to 14 T HRIM Facility). We also acknowledge Mackenzie Halbert for assisting with AFM image acquisitions. We acknowledge the use of the equipment on NP characterization in Nanoengineering & Science Institute and Molecular Engineering & Science Institute supported by NSF (grant NNCI-1542101).
Footnotes
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information. Raw data generated for this study are available from the corresponding author on reasonable request.
Author Credits
Q. M. and M. Z. designed the research. Q.M., G.L. performed the experiments and analyses. M.K. made oxide nanoparticles. R.A.R. performed the measurement of magnetic nanoparticles and acquired MR images of cells and mice. F.C.C, H.W. assisted in vivo experiments and data analysis. J. Y. advised on experiments on evaluation of immune treatment efficacy in vitro and in vivo. Q.M., G.L, and M. Z. wrote the manuscript. M.Z supervised the study. All authors discussed the results and commented on the manuscript.
Competing interests
The authors declare no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at xxxxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, et al. , CA: A Cancer Journal for Clinicians (2020) 70 (1), 7. [DOI] [PubMed] [Google Scholar]
- 2.Jones SE, Clinical Breast Cancer (2008) 8 (3), 224. [DOI] [PubMed] [Google Scholar]
- 3.Lu J, et al. , Cancer Research (2009) 69 (12), 4951. [DOI] [PubMed] [Google Scholar]
- 4.Foulkes WD, et al. , New England Journal of Medicine (2010) 363 (20), 1938. [DOI] [PubMed] [Google Scholar]
- 5.Yao H, et al. , Oncotarget (2017) 8 (1), 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bandyopadhyay A, et al. , PLOS ONE (2010) 5 (4), e10365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uno T, et al. , Nat Med (2006) 12 (6), 693. [DOI] [PubMed] [Google Scholar]
- 8.Filatenkov A, et al. , Radiation research (2014) 182 (2), 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oner G, et al. , Breast J (2020) 26 (5), 995. [DOI] [PubMed] [Google Scholar]
- 10.Lopez JS, and Banerji U, Nature reviews. Clinical oncology (2017) 14 (1), 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang J, et al. , Science Advances (2020) 6 (35), eabc3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mu Q, et al. , Expert Opinion on Drug Delivery (2016), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell MJ, et al. , Nat Rev Cancer (2017) 17 (11), 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilhelm S, et al. , Nature Reviews Materials (2016) 1 (5), 16014 [Google Scholar]
- 15.Irby D, et al. , Mol Pharm (2017) 14 (5), 1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larrañeta E, et al. , J Funct Biomater (2018) 9 (1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mo ZC, et al. , Adv Drug Deliv Rev (2016) 106 (Pt A), 132. [DOI] [PubMed] [Google Scholar]
- 18.Xue P, et al. , Colloids Surf B Biointerfaces (2019) 180, 202. [DOI] [PubMed] [Google Scholar]
- 19.Yang G, et al. , Angew Chem Int Ed Engl (2019) 58 (40), 14357. [DOI] [PubMed] [Google Scholar]
- 20.Gradishar WJ, Expert Opinion on Pharmacotherapy (2006) 7 (8), 1041. [DOI] [PubMed] [Google Scholar]
- 21.Villano JL, et al. , Invest New Drugs (2006) 24 (5), 455. [DOI] [PubMed] [Google Scholar]
- 22.Lee H, et al. , Scientific reports (2020) 10 (1), 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao M, et al. , PloS one (2015) 10 (7), e0131429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoon HY, et al. , Biomaterials (2018) 178, 597. [DOI] [PubMed] [Google Scholar]
- 25.Kuai R, et al. , Nat Mater (2017) 16 (4), 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu G, et al. , Nat Commun (2017) 8 (1), 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oberli MA, et al. , Nano Lett (2017) 17 (3), 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, et al. , Nano Lett (2016) 16 (9), 5503. [DOI] [PubMed] [Google Scholar]
- 29.Li SY, et al. , J Control Release (2016) 231, 17. [DOI] [PubMed] [Google Scholar]
- 30.Stephan MT, et al. , Nature Medicine (2010) 16 (9), 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan HF, et al. , Nature Nanotechnology (2017) 12 (8), 763. [DOI] [PubMed] [Google Scholar]
- 32.Yazdi MH, et al. , Arzneimittelforschung (2012) 62 (11), 525. [DOI] [PubMed] [Google Scholar]
- 33.Lizotte PH, et al. , Nature nanotechnology (2016) 11 (3), 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Koker S, et al. , Angew Chem Int Ed Engl (2016) 55 (4), 1334. [DOI] [PubMed] [Google Scholar]
- 35.Schmid D, et al. , Nat Commun (2017) 8 (1), 1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan Y, et al. , Nano Letters (2017) 17 (12), 7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin G, et al. , Advanced Functional Materials n/a (n/a), 2007096 [Google Scholar]
- 38.Bi, X., et al., 582
- 39.Chen F, et al. , Acs Nano (2013) 7 (10), 9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong H, et al. , European Journal of Nuclear Medicine and Molecular Imaging (2011) 38 (7), 1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi SX, et al. , Biomaterials (2013) 34 (12), 3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsujie M, et al. , International Journal of Oncology (2006) 29 (5), 1087. [PubMed] [Google Scholar]
- 43.Uneda S, et al. , International Journal of Cancer (2009) 125 (6), 1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tewey K, et al. , Science (1984) 226 (4673), 466. [DOI] [PubMed] [Google Scholar]
- 45.Dennis JW, Cancer Research (1986) 46 (10), 5131. [PubMed] [Google Scholar]
- 46.Schmidt KN, et al. , Journal of Immunology (2004) 172 (1), 138. [DOI] [PubMed] [Google Scholar]
- 47.Wesch D, et al. , Journal of Immunology (2006) 176 (3), 1348. [DOI] [PubMed] [Google Scholar]
- 48.Hirabayashi K, et al. , Cancer Research (1999) 59 (17), 4325. [PubMed] [Google Scholar]
- 49.Salazar AM, et al. , Neurosurgery (1996) 38 (6), 1096. [PubMed] [Google Scholar]
- 50.Okada H, et al. , J. Clin. Oncol. (2011) 29 (3), 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramasamy T, et al. , Acta Biomaterialia (2014) 10 (12), 5116. [DOI] [PubMed] [Google Scholar]
- 52.Correa S, et al. , J Control Release (2016) 240, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fang C, et al. , Small (2009) 5 (14), 1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, et al. , Scientific reports (2016) 6, 21225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hernandez-Gil J, et al. , Advanced healthcare materials (2015) 4 (7), 1034. [DOI] [PubMed] [Google Scholar]
- 56.Cheng YS, and Xu F, Cancer biology & therapy (2010) 10 (12), 1219. [DOI] [PubMed] [Google Scholar]
- 57.Medzhitov R, Nat Rev Immunol (2001) 1 (2), 135. [DOI] [PubMed] [Google Scholar]
- 58.Tugues S, et al. , Cell Death and Differentiation (2015) 22 (2), 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandez NC, et al. , Nat Med (1999) 5 (4), 405. [DOI] [PubMed] [Google Scholar]
- 60.Lin G, et al. , Biomaterials Science (2021) 9 (2), 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsumoto M, et al. , Journal of immunology (Baltimore, Md. : 1950) (2003) 171 (6), 3154. [DOI] [PubMed] [Google Scholar]
- 62.Jeon H, et al. , Oncotarget (2016) [Google Scholar]
- 63.Knockenhauer KE, and Schwartz TU, Cell (2016) 164 (6), 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lane P, et al. , Cancer Res (1987) 47 (15), 4038. [PubMed] [Google Scholar]
- 65.Cobaleda-Siles M, et al. , Small (Weinheim an der Bergstrasse, Germany) (2014) 10 (24), 5054. [DOI] [PubMed] [Google Scholar]
- 66.Momparler RL, et al. , Cancer Res (1976) 36 (8), 2891. [PubMed] [Google Scholar]
- 67.Tanaka H, et al. , Cancer Res (2009) 69 (17), 6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chaturvedi A, and Pierce SK, Traffic (2009) 10 (6), 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Villiers CL, et al. , Journal of Nanoparticle Research (2010) 12 (1), 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salem ML, et al. , Vaccine (2009) 27 (4), 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lövgren T, et al. , Cancer Immunology, Immunotherapy (2017) 66 (10), 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang L, et al. , Nanoscale (2015) 7 (2), 625. [DOI] [PubMed] [Google Scholar]
- 73.Blanco E, et al. , Nat Biotechnol (2015) 33 (9), 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sturgill MG, et al. , Cancer investigation (2000) 18 (3), 197. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Y-N, et al. , Journal of Controlled Release (2016) 240, 332. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Y, et al. , Oncol Lett (2018) 15 (5), 6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carvalho FS, et al. , Med. Res. Rev. (2014) 34 (1), 106. [DOI] [PubMed] [Google Scholar]
- 78.Salaun B, et al. , The Journal of Immunology (2006) 176 (8), 4894. [DOI] [PubMed] [Google Scholar]
- 79.Galon J, and Bruni D, Nat Rev Drug Discov (2019) 18 (3), 197. [DOI] [PubMed] [Google Scholar]
- 80.Schmid P, et al. , The New England journal of medicine (2018) 379 (22), 2108. [DOI] [PubMed] [Google Scholar]
- 81.Kalimutho M, et al. , Trends in Pharmacological Sciences (2015) 36 (12), 822. [DOI] [PubMed] [Google Scholar]
- 82.Li C-W, et al. , Cancer Cell (2018) 33 (2), 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mittapalli RK, et al. , Molecular Cancer Therapeutics (2013) 12 (11), 2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang S, et al. , Nanomedicine: Nanotechnology, Biology and Medicine (2016) 12 (2), 411. [DOI] [PubMed] [Google Scholar]
- 85.Deng X, et al. , Biomaterials (2014) 35 (14), 4333. [DOI] [PubMed] [Google Scholar]
- 86.Misra AC, et al. , Biomacromolecules (2015) 16 (8), 2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruan SB, et al. , RSC Advances (2015) 5 (79), 64303 [Google Scholar]
- 88.Sussman D, et al. , Mol Cancer Ther (2014) 13 (12), 2991. [DOI] [PubMed] [Google Scholar]
- 89.Hansel TT, et al. , Nat Rev Drug Discov (2010) 9 (4), 325. [DOI] [PubMed] [Google Scholar]
- 90.Kennel SJ, et al. , Journal of Cell Science (1993) 104 (2), 373. [DOI] [PubMed] [Google Scholar]
- 91.Bodey B, et al. , Anticancer research (1998) 18 (5A), 3621. [PubMed] [Google Scholar]
- 92.Dales JP, et al. , American journal of clinical pathology (2003) 119 (3), 374. [DOI] [PubMed] [Google Scholar]
- 93.Chen F, et al. , ACS Nano (2013) 7 (10), 9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shao K, et al. , ACS Nano (2015) 9 (1), 16. [DOI] [PubMed] [Google Scholar]
- 95.Bauleth-Ramos T, et al. , Advanced Functional Materials (2017) 27 (42), 1703303 [Google Scholar]
- 96.Liu Y, et al. , Acta Biomater (2018) 66, 310. [DOI] [PubMed] [Google Scholar]
- 97.Wang T, et al. , Nanoscale (2019) 11 (29), 13934. [DOI] [PubMed] [Google Scholar]
- 98.Zhang P, et al. , ACS Nano (2015) 9 (6), 6465. [DOI] [PubMed] [Google Scholar]
- 99.Trad M, et al. , Biomed Res Int (2015) 2015, 891236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dehshahri A, et al. , Applied Biochemistry and Biotechnology (2016) 179 (2), 251. [DOI] [PubMed] [Google Scholar]
- 101.Sheikhsaran F, et al. , Colloids and Surfaces B: Biointerfaces [DOI] [PubMed] [Google Scholar]
- 102.Tao K, et al. , BMC Cancer (2008) 8 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bao L, et al. , The American Journal of Pathology (2011) 178 (2), 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Park J, et al. , Nat Mater (2004) 3 (12), 891. [DOI] [PubMed] [Google Scholar]
- 105.Mu Q, et al. , Journal of materials chemistry. B, Materials for biology and medicine (2016) 4 (1), 3226835125 [Google Scholar]
- 106.Riemer J, et al. , Anal Biochem (2004) 331 (2), 370. [DOI] [PubMed] [Google Scholar]
- 107.Madaan A, et al. , Journal of Biological Methods (2014) 1 (1) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.