

Abstract
Summary
The Interface Contact definition with Adaptable Atom Types (INTERCAAT) was developed to determine the atomic interactions between molecules that form a known three dimensional structure. First, INTERCAAT creates a Voronoi tessellation where each atom acts as a seed. Interactions are defined by atoms that share a hyperplane and whose distance is less than the sum of each atoms’ Van der Waals radii plus the diameter of a solvent molecule. Interacting atoms are then classified and interactions are filtered based on compatibility. INTERCAAT implements an adaptive atom classification method; therefore, it can explore interfaces between a variety macromolecules.
Availability and implementation
Source code is freely available at: https://gitlab.com/fiserlab.org/intercaat.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Exploring interfaces of macromolecular interactions from Protein Data Bank (PDB) coordinate files (Berman et al., 2000) is an essential everyday task in bioinformatics. Several software tools have been developed to utilize PDB coordinates to visualize and analyze inter and intra molecular interactions (Sobolev et al., 1999). Determining residues that form the interface between proteins is surprisingly complicated. A recent study demonstrated that on average only about 80% of residue overlap between any two alternative interface prediction methods (Gil and Fiser, 2019). This is due to the subjective definitions guiding these methods, some of which focus on changes in solvent accessibility, while others focus on variable distance thresholds requiring specific contacts between interacting residues. Our current effort focused on establishing a generic method to accurately assess interfaces using an advanced geometrical approach, considering the compatibility of interactions, and providing adjustable options that the user can modify to explore alternative definitions. Another advantage of INTERCAAT is that it uses an adaptive atom classification function and therefore can explore interactions between a variety of molecules beyond proteins e.g. interactions with nucleic acids or lipids.
First, INTERCAAT parses a PDB file and creates a Voronoi tessellation between atoms. A Voronoi diagram is computed via Delaunay triangulation. An atomic interaction is established between two atoms if they share a bounded hyperplane and are within a distance less than the sum of the atoms Van der Waals radii plus the diameter of a solvent molecule. We should point out that our methodology treats the atoms as points in the Voronoi tessellation and then as spheres for the distance cutoff. This is a small conflict considering the Van der Waals radii among heavy atoms are not drastically different. Atomic interactions can be further filtered to show only ‘legitimate’ interactions. Legitimacy depends on the hydrophobic/hydrophilic properties of the interacting atoms (Sobolev et al., 1999). Atoms can belong to one of eight classes and if each atom class is compatible, their interaction is considered ‘legitimate’. For a residue on the query chain to be considered as part of the interface, it is required to have a minimum number of interactions with the interacting chain(s) to prevent accidental classifications. Voronoi tessellations were first used in a protein context in 1974 (Richards, 1974) but have since been used to investigate a series of protein related issues including residue volumes, packing, folding and binding (Poupon, 2004).
2 Software design
INTERCAAT was developed in a Linux environment. It requires three inputs while six additional switches are optional. The required inputs include the name of a PDB file, the chain ID of the query chain whose interface needs to be determined, and the chain ID(s) of the chain(s) interacting with the query chain. The optional switches include setting the minimum required number of interactions of the query chain required, whether to display an interaction matrix, whether to consider class compatibility of interactions, setting the solvent molecule radius, whether to include chains other than the query and interacting chains in the Voronoi calculation, and finally, an optional file path of the PDB file. Each optional switch has default values; which, along with an input example, can be displayed with the programs help function. The output displays every atomic interaction between the query chain and the interacting chain(s), the distance between the interacting atoms and the assigned atom classes. The compatibility matrix, if displayed, shows each interface residue in the query chain and the corresponding number of atomic interactions.
The atomic class of an atom is not predetermined based on the residue it belongs to. Instead, the program determines its class-based solely on its coordinates and particular atom type. Therefore, the program is able to classify atoms from most molecules including proteins, DNA, RNA, etc. INTERCAAT currently recognizes common biological atoms: C, N, O, P, S, Cl, F and Br. If an unknown atom is input into INTERCAAT it will assign it an arbitrary Van der Waal radius equal to 1.8 angstroms and assign its class as ‘?’. Any atom with class ‘?’ will be considered universally compatible. It is up to the user to determine if the interaction makes sense or update the script to handle new atom types.
INTERCAAT consists of two programs written in python version 3.8.6, an .ini configuration file and the qhull package. The two python files contain the main script and the functions. The configuration file must be changed to specify the path to call qhull. Qhull software calculates the Voronoi tessellation between atoms (Barber, 1996). If the user does not have the qhull program or prefers not to use it, it can be specified in the configuration file to run the Voronoi calculation using python instead. The downside to this is that the program will run much slower. For convenience, both python scripts are well commented.
3 Implementation
Benchmarking is not really possible in the sense that there is no gold standard of interface definitions available. However, three different databases were utilized to compare results of INTERCAAT. These include 320, 105 and 125 interfaces defined by the BioLiP database (Yang et al., 2013), the Nox database (Zhu et al., 2006) and the Dockb database (Vreven et al., 2015), respectively. Although all of these comparisons focused on protein–protein interactions, we should point out that INTERCAAT was developed with adaptive atom classification capabilities and it is not restricted to protein–protein interactions (Supplementary Material).
The goal of the comparisons was to evaluate the performance of INTERCAAT as well as to determine the optimal input for the minimum atomic interactions necessary for a residue to be considered part of the interface. This was done by comparing the common interface residues predicted by both INTERCAAT and the other databases, the unique residues INTERCAAT predicted and the unique residues predicted by the other databases. In addition to plotting box and whisker plots, where the whiskers have a cutoff at the 5th and 95th percentiles, F1 scores were calculated to quantify these results into a single score. F1 scores represent a tests accuracy by measuring the harmonic mean utilizing a test’s precision and recall (Fig. 1).
Fig. 1.
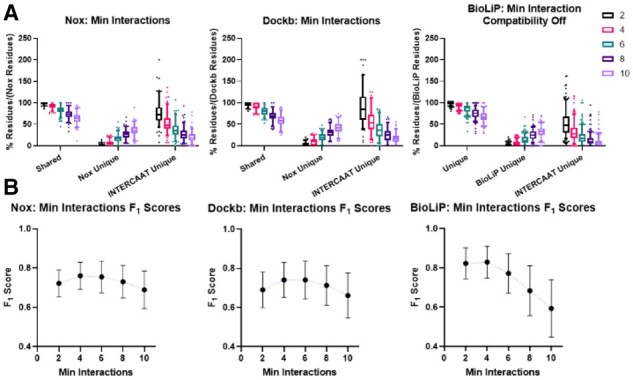
Comparison of INTERCAAT against three other databases for protein–protein interfaces while modulating minimum interactions. (a) The percentage of common interface residues identified by both INTERCAAT and the other database divided by the total amount of residues identified by the other database for different minimum interaction cutoffs. (b) F1 scores
The F1 scores assumed that the true positives were the shared predicted residues, the false positives were the unique residues predicted by INTERCAAT and the false negatives were the unique residues predicted by another database. As the minimum interactions requirement was increased, we consistently observed that the recall of INTERCAAT decreased and the precision increased. A minimum interaction requirement of four emerged as the optimal balance between precision and recall. The resulting F1 scores of INTERCAAT against the other databases was approximately 0.8, as expected (Gil and Fiser, 2019).
Funding
This work was supported by NIH grants GM136357 and AI141816.
Conflict of Interest: The authors declare no conflict of interest.
Supplementary Material
Contributor Information
Steven Grudman, Department of Systems and Computational Biology, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
J Eduardo Fajardo, Department of Systems and Computational Biology, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Andras Fiser, Department of Systems and Computational Biology, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
References
- Barber C.B. et al. (1996) The quickhull algorithm for convex hulls. ACM Trans. Math. Softw., 22, 469–483. [Google Scholar]
- Berman H.M. et al. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil N., Fiser A. (2019) The choice of sequence homologs included in multiple sequence alignments has a dramatic impact on evolutionary conservation analysis. Bioinformatics, 35, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poupon A. (2004) Voronoi and Voronoi-related tessellations in studies of protein structure and interaction. Curr. Opin. Struct. Biol., 14, 233–241. [DOI] [PubMed] [Google Scholar]
- Richards F.M. (1974) The interpretation of protein structures: total volume, group volume distributions and packing density. J. Mol. Biol., 82, 1–14. [DOI] [PubMed] [Google Scholar]
- Sobolev V. et al. (1999) Automated analysis of interatomic contacts in proteins. Bioinformatics, 15, 327–332. [DOI] [PubMed] [Google Scholar]
- Vreven T. et al. (2015) Updates to the integrated protein–protein interaction benchmarks: docking benchmark version 5 and affinity benchmark version 2. J. Mol. Biol., 427, 3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. (2013) BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res., 41, D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. et al. (2006) NOXclass: prediction of protein–protein interaction types. BMC Bioinformatics, 7, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.