

Significance Statement
Transcription factors (TFs) play crucial roles in kidney development and diseases by recognizing specific DNA sequences to control gene expression programs. The kidney’s cellular heterogeneity poses substantial challenges to identifying the genomic binding sites and direct target genes of TFs in vivo. We apply the cleavage under targets and release using nuclease (CUT&RUN) technique, together with transcriptomic analysis, to identify cAMP response element-binding protein (CREB) target genes in cystic epithelial cells of autosomal dominant polycystic kidney disease (ADPKD) in a mouse model. CREB binds to and activates ribosomal biogenesis genes, and inhibition of CREB retards cyst growth in the ADPKD models. CUT&RUN is a powerful method for genome-scale profiling and identifying direct targets of TFs from small numbers of specific kidney cells.
Keywords: ADPKD, CREB, transcription factors, cAMP, kidney diseases, CUT&RUN
Visual Abstract

Abstract
Background
Genome-wide mapping of transcription factor (TF) binding sites is essential to identify a TF’s direct target genes in kidney development and diseases. However, due to the cellular complexity of the kidney and limited numbers of a given cell type, it has been challenging to determine the binding sites of a TF in vivo. cAMP response element-binding protein (CREB) is phosphorylated and hyperactive in autosomal dominant polycystic kidney disease (ADPKD). We focus on CREB as an example to profile genomic loci bound by a TF and to identify its target genes using low numbers of specific kidney cells.
Methods
Cleavage under targets and release using nuclease (CUT&RUN) assays were performed with Dolichos biflorus agglutinin (DBA)–positive tubular epithelial cells from normal and ADPKD mouse kidneys. Pharmacologic inhibition of CREB with 666-15 and genetic inhibition with A-CREB were undertaken using ADPKD mouse models.
Results
CUT&RUN to profile genome-wide distribution of phosphorylated CREB (p-CREB) indicated correlation of p-CREB binding with active histone modifications (H3K4me3 and H3K27ac) in cystic epithelial cells. Integrative analysis with CUT&RUN and RNA-sequencing revealed CREB direct targets, including genes involved in ribosome biogenesis and protein synthesis. Pharmacologic and genetic inhibition of CREB suppressed cyst growth in ADPKD mouse models.
Conclusions
CREB promotes cystogenesis by activating ribosome biogenesis genes. CUT&RUN, coupled with transcriptomic analysis, enables interrogation of TF binding and identification of direct TF targets from a low number of specific kidney cells.
Gene expression programs that establish and maintain cellular homeostasis of kidney are largely controlled by transcription factors (TFs). TFs bind to sequence-specific cis-regulatory DNA elements and exert cell type– and context-specific transcriptional responses.1 Disturbance of TF activities is closely associated with the progression of kidney diseases.2,3 Therefore, identification of direct target genes regulated by disease-related TFs is crucial to dissect the mechanisms of disease progression. The most widely used cistromic analysis for identifying TF targets genome wide is chromatin immunoprecipitation (ChIP) coupled with sequencing (ChIP-seq), which typically requires large numbers of cells. It is challenging to adopt this technique to analyze TF targets in specific diseased renal cells in vivo due to the difficulty of obtaining sufficient diseased cells from the complex renal tissue. Many studies used whole kidney tissue or cell line models, but such approaches have numerous limitations. Using the entire kidney tissue, which is a mixture of >40 cell types,4–6 impedes characterization of the function and regulation of particular diseased cells. The use of renal cell lines is also not ideal because they lack cell-cell interaction and cell-environmental communication, and because their immortalization processes result in altered responses to signals. Recently, a new method called cleavage under targets and release using nuclease (CUT&RUN) was developed for mapping TF targets,7–9 which has two major advantages over ChIP: first, it requires only a small number of cells; and, second, in CUT&RUN, unlike in ChIP, the chromatin does not need to be fragmented first, which helps to maintain protein-DNA interactions in their natural state. This new method can more accurately identify where TFs bind to DNA, from yeast to human cells. Thus, CUT&RUN may be ideal for analyzing the unique TF targets in specific renal cells.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disease and is characterized by numerous renal cysts filled with large volumes of fluid, which leads to renal failure in over half of patients by the time they are 60.10,11 Although it has long been known that mutation of polycystic kidney disease 1 (PKD1) or PKD2 in renal epithelial cells turns them into cystic cells, causing the formation of numerous renal cysts, the underlying mechanisms are still elusive, which limits the development of effective treatments.12,13 Identifying disease-perturbed TFs and their targets that drive the cell-fate change in ADPKD, from normal renal epithelial cells to cystic cells, should help to uncover the pathogenic mechanisms of this disease and, thus, help to identify potential therapeutic targets. Studies over the past two decades have revealed that the dysregulation of cAMP signaling plays a central role in ADPKD.14–16 The cAMP response element (CRE)–binding protein (CREB) family of TFs—including CREB, CRE modulator (CREM), and activating transcription factor 1 (ATF1)—are the main nuclear effectors of cAMP signaling.17,18 As previously reported, activation of cAMP–protein kinase A signaling promotes the phosphorylation at serine 133 (Ser133) of the kinase-inducible domain of CREB, which triggers CREB’s association with the histone acetyltransferase coactivators CREB-binding protein/p300 to enhance target gene expression.19 CREB governs a wide range of cellular processes, including metabolism, cell survival and proliferation, differentiation, apoptosis, long-term memory, and immune responses.20–23 However, whether and how CREB is involved in the pathogenesis of ADPKD is unknown. As a TF, CREB influences various biologic processes by regulating distinct targets in different contexts.20 Thus, identifying ADPKD-specific CREB targets is essential to define the role of CREB in ADPKD.
In this study, we identified CREB targets in cystic cells by coupling the CUT&RUN technique and transcriptomic analysis. We found that the CREB targets in cystic cells included genes involved in ribosome biogenesis and protein synthesis. Functional experiments in ADPKD mouse models further demonstrated that abnormally activated CREB promotes cyst growth. This work demonstrates that CUT&RUN is a powerful method for genome-scale profiling and identifying direct targets of TFs from small numbers of specific kidney cells.
Methods
Study Approval
All mouse experiments were approved by the ethics committee of the Tianjin Medical University. Patient specimens were obtained from cyst reduction surgery or nephrectomy. All patients involved provided informed consent. Human studies were approved by the ethics committee of Shandong Provincial Hospital.
Mouse Models
Pkd1fl/fl; Cre/Esr1+ mice were described previously.24 To induce Pkd1 deletion, a single intraperitoneal injection of 10 mg/kg tamoxifen (T5648; Sigma) was given at postnatal day 10 (P10) for the early-onset mouse model. Tamoxifen was dissolved in corn oil (C8267; Sigma). To generate the late-onset mouse model, 250 mg/kg tamoxifen was given at P25 and P28 by intraperitoneal injection. 666-15 (HY101120; MedChem Express) was dissolved in DMSO and diluted with 5% Tween-80 before injection. For the early-onset mouse model, mice were euthanized at P29. 666-15 (5 mg/kg) was given once daily for five consecutive days, followed by two days of rest. 666-15 treatment was started on P11 and continued for 3 weeks. For the late-onset mouse model, mice were euthanized at P115. 666-15 treatment was initiated from P55 and continued for 2 months. Both sexes were used in the early-onset model, and only male mice were used in the late-onset model.
A-CREB transgenic mice in the C57BL/6 background were generated by Shanghai Biomodel Organism Science and Technology Development Co., Ltd. The PiggyBac transposon system was used to randomly insert the A-CREB sequence into the mouse genome by injection into fertilized eggs. Pkd1fl/fl; Cdh16-Cre mice were generated by crossing Pkd1fl/fl mice with Cdh16-Cre mice. Pkd1fl/fl; Cdh16-Cre mice were then crossed with A-CREB transgenic mice. Both sexes were used in the kidney-specific mouse model, and mice were euthanized at P7. Five pairs of kidneys were collected for tissue section analysis and the rest were used for RNA or protein preparation.
Cell Culture
Primary Dolichos biflorus agglutinin (DBA)–positive cells were isolated as described previously.25 Briefly, kidneys from early-onset mice at P29 were finely minced and digested using a gentle collagenase/hyaluronidase solution (07919; STEMCELL Technologies), which was diluted with EpiCult-B Mouse Medium (05610; STEMCELL Technologies). A single-cell suspension was then generated by further digestion using 0.05% trypsin-EDTA, phenol red (25200056; Gibco) and 1 U/mL dispase (07923; STEMCELL Technologies). Cell clumps were excluded using a 40-μm cell strainer (352340; Corning). DBA-positive cells from collecting/distal tubules were then enriched using biotinylated DBA antibody (B-1035; Vector Laboratories) and a Collection Biotin Binder kit (11533D; Invitrogen). Isolated cells were used for CUT&RUN analysis and RNA or protein preparation.
WT 9-12 cells (DBA-positive and immortalized with wild-type [WT] simian virus 40) were cultured in DMEM/F-12 medium supplemented with 10% FBS.26 Human embryonic kidney 293T cells were purchased from the American Type Culture Collection. Cells were cultured in DMEM supplemented with 10% FBS.
ChIP-Seq
ChIP was performed as described before.27 Briefly, WT 9-12 cells were crosslinked using 1% formaldehyde (F8775; Sigma Aldrich) for 10 minutes. Chromatin was sheared to a size range of 200–500 bp. Immunoprecipitation was performed using 5 μl of the phosphorylated CREB (p-CREB; at Ser133) antibody (9198; Cell Signaling Technology). Purified DNA was sequenced on the Illumina NovaSeq platform.
CUT&RUN
CUT&RUN was performed as previously described, with several modifications.8 Briefly, isolated DBA-positive cells were divided into 100,000 per tube and washed once with 1 ml wash buffer (20 mM HEPES at pH 7.5, 150 mM sodium chloride, 0.5 mM spermidine, 1% BSA, cOmplete Protease Inhibitor Cocktail, and PhosSTOP inhibitor). ConA beads (BP531; BangsLab) were prewashed with binding buffer (20 mM HEPES–potassium hydroxide at pH 7.9, 10 mM potassium chloride, 1 mM calcium chloride, and 1 mM manganese chloride). Washed cells were resuspended with 100 μl wash buffer and incubated with 10 μl prewashed ConA beads for 10 minutes at room temperature. ConA bead–binding cells were incubated with 50 μl of antibody buffer (wash buffer containing 2 mM EDTA) containing 0.5 μg of anti-H3K4me3 (05-745R; Millipore) or anti-H3K27ac (ab4729; Abcam) or anti–p-CREB (Ser133) antibody for 10 minutes at room temperature. Cells were washed twice with 800 μl dig-wash buffer (wash buffer containing 0.05% digitonin) and then incubated with 100 μl dig-wash buffer containing 700 ng/ml Protein A-Micrococcal Nuclease (pA-MN) fusion protein (homemade) for 10 minutes at room temperature. Samples were washed twice with 800 μl dig-wash buffer, and then placed on wet ice for 5 minutes to chill to 0°C. To initiate MNase digestion, 2 μL of 100 mM calcium chloride was added to the above samples, followed by incubation for 30 minutes on ice. Digestion was stopped by adding 100 μl 2× stop buffer (200 mM sodium chloride, 20 mM EDTA, 4 mM EGTA, 0.05 mg/ml RNaseA, 0.04 mg/ml DNase-free glycogen, and 10 pg/ml spike-in DNA purified from Escherichia coli), followed by incubation for 30 minutes at 37°C. Two microliters of 10% SDS and 2.5 μl of proteinase K were added to samples and incubated for 1 hour at 50°C. DNA was then purified using phenol/chloroform/isoamyl alcohol (25:24:1). Library construction was performed with a VAHTS Universal DNA Library Prep Kit from Illumina V3 (ND607; Vazyme). Sequencing was performed on the Illumina NovaSeq platform. Genomic data were deposited in the Gene Expression Omnibus (GSE165416).
RNA Sequencing
Total RNA was isolated from DBA-positive cells using TRIzol (15596018; Invitrogen) according to the manufacturer’s instructions. Sequencing was performed on the Illumina NovaSeq platform. Genomic data were deposited in the Gene Expression Omnibus (GSE165417).
Bioinformatic Analysis
ChIP-seq reads were aligned to the human reference genome (hg38) by bowtie2 (version 2.3.4.1) with default parameters. CUT&RUN reads were aligned to the human (hg38) or mouse (mm10) reference genome by bowtie2 (version 2.3.4.1) with the following parameters: –local, –very-sensitive-local, –no-unal, –no-mixed, –no-discordant, –phred33, -I 10, -X 700. Unmapped and duplicated reads were removed using SAMtools (version 1.7). To visualize data on Integrative Genomics Viewer, BAM files were converted to bigWig files using deepTools (version 3.4.3). Peak calling was performed using HOMER (version 4.11). Annotation of peaks was performed using HOMER and GREAT (version 4.0.4). RNA-sequencing (RNA-seq) reads were aligned to the mouse reference genome (mm10) by HISAT2 (version 2.1.0) with default parameters. Reads were counted by featureCounts (version 1.6.0) and differentially expressed genes were calculated by DESeq2. Fold change of ≤−1.5 or ≥1.5 and adjusted P<0.05 were considered as significant differential expression. Comparison of WT and ADPKD under DMSO treatment was used to define ADPKD differentially expressed genes. 666-15–rescued genes were defined as ADPKD-upregulated genes whose expression was downregulated after 666-15 treatment. Function enrichment analysis was performed using KOBAS (version 3.0).
Histology and Immunohistochemistry Analyses
Paraffin-embedded human and mouse kidneys were cut into 6- to 8-μm sections and stained with hematoxylin and eosin for histologic analysis. For immunochemistry analysis, human tissue sections were treated with 3% hydrogen peroxide for 15 minutes and then blocked with 5% BSA for 1 hour at room temperature. After incubation with primary antibodies, sections were incubated with anti-rabbit secondary antibody conjugated with horseradish peroxidase (PV-6000; Zsgb Bio). For H score calculation, staining intensity (0, 1+, 2+, or 3+) was determined for each cell in a fixed field. The percentage of cells at each staining intensity level was then calculated.28,29 H score=(1×[% cells 1+]+2×[% cells 2+]+3×[% cells 3+]).
Immunofluorescence
The mouse kidneys were embedded in 100% Optimal Cutting Temperature compound. Kidney sections were blocked with 5% BSA and incubated with primary antibodies overnight at 4°C. Sections were then incubated with goat anti-rabbit secondary antibody labeled with Alexa Fluor 488 (Thermo Fisher Scientific). Sections were imaged by fluorescence microscope (DMi8; Leica).
Statistical Analysis
The unpaired, two-tailed t test was performed using GraphPad Prism (version 8.0.2). Results were presented as means±SEMs. P<0.05 was considered statistically significant.
Results
Activation of CREB in ADPKD Kidneys
To assess the activation of CREB in ADPKD, we first analyzed the state of CREB in Pkd1fl/fl; Cre/Esr1+ mice, an ADPKD mouse model. Consistent with previous studies,30–32 we found that, although total CREB protein showed little change, the phosphorylation of CREB at Ser133 increased markedly in kidneys from Pkd1−/− mice compared with that in WT mice (Figure 1A). Notably, p-CREB staining was most prominent in cyst-lining epithelial cells (Figure 1B). The collecting duct (CD) is the most affected renal tubular segment in ADPKD, especially in the late stage of the disease, when more than half of the renal cysts are of CD origin.33 We isolated cells positive for DBA, a CD/tubule marker, from mouse kidneys. As shown in Figure 1C, p-CREB increased markedly in DBA-positive cells isolated from ADPKD kidneys, confirming activation of the CREB pathway in the diseased cell population. To determine when CREB is activated in the ADPKD models, we examined p-CREB levels in the cystic tubular cells and observed positive staining of p-CREB, on P16 in the early-onset model and P55 in the late-onset model, which gradually increased during disease progression (Supplemental Figure 1, A and B). We further performed a quantitative analysis of p-CREB in kidneys from patients with ADPKD (Supplemental Table 1). The p-CREB in ADPKD kidneys was remarkably higher than in normal kidneys, showing intense nuclear staining in cells lining the renal cysts (Figure 1, D and E, Supplemental Figure 1C). Altogether, these results suggest that p-CREB is overactivated in ADPKD kidneys.
Figure 1.
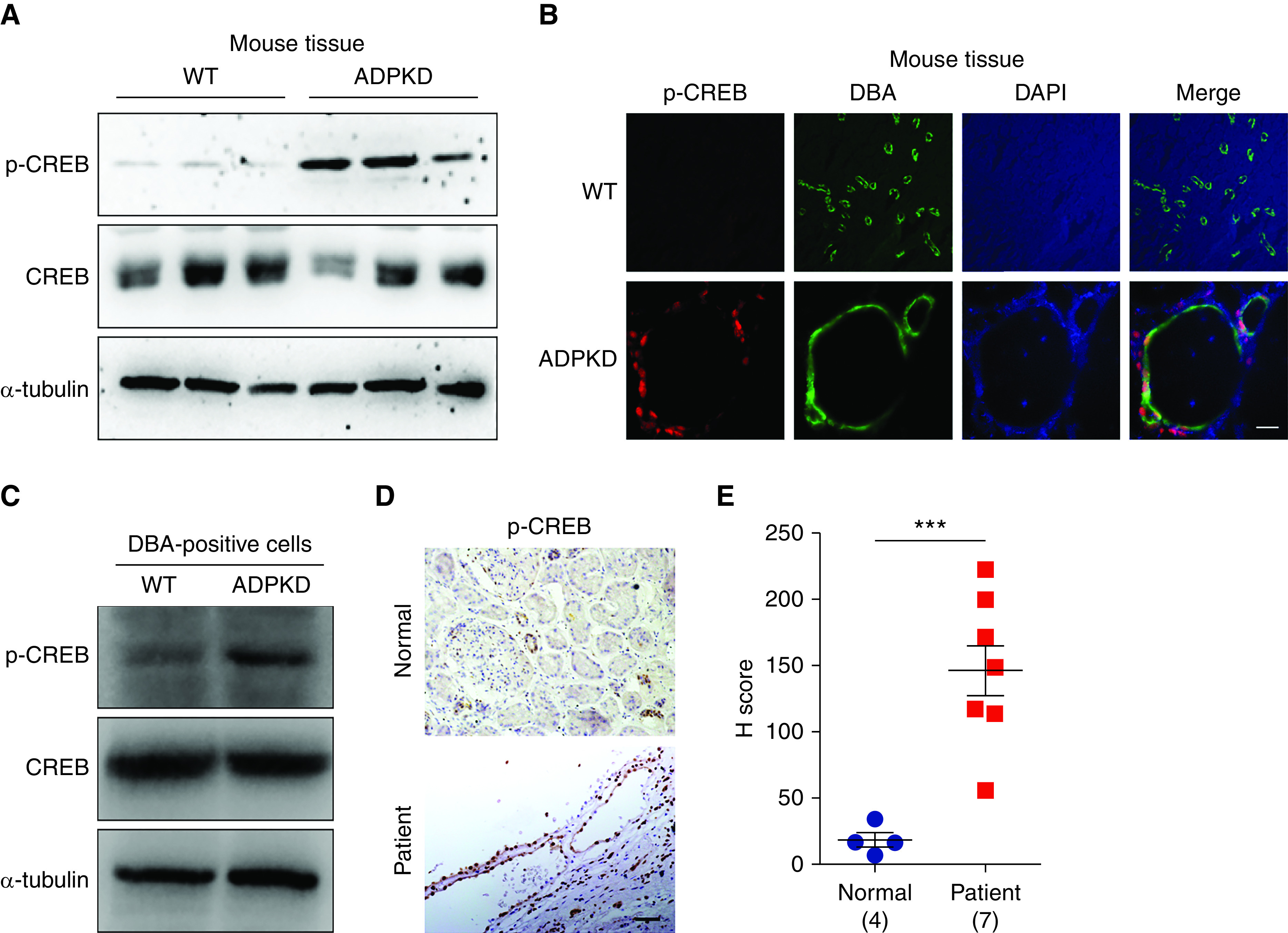
Phosphorylation of CREB is increased in ADPKD kidneys. (A) Western blot analysis of p-CREB (Ser133) in kidneys from WT and early-onset ADPKD mice. (B) Immunofluorescence analysis of p-CREB level in kidneys from WT and ADPKD mice. Scale bar, 25 μm. Original magnification, 650×. (C) Western blot analysis of p-CREB in DBA-positive cells from WT and ADPKD mice. (D) Immunohistochemistry analysis of p-CREB level in normal kidney and kidney from a patient with ADPKD. Scale bar, 50 μm. Original magnification, 200×. (E) The signal density of p-CREB was determined by the H score. Data are represented as means±SEMs, and were analyzed using the unpaired, two-sided t test. ***P<0.001. DAPI, 4′,6-diamidino-2-phenylindole.
Genomic Characterization of p-CREB Distribution in Cystic Cells
Because the CUT&RUN technique can identify the genomic binding sites of a TF in a small number of cells, it is a strong candidate approach for identifying targets bound by p-CREB in cystic cells in ADPKD kidneys. To determine whether the results obtained with the CUT&RUN method in a small number of kidney cells were comparable with those obtained with the traditional ChIP-seq technique in a large number of cells, we selected a patient-derived CD-origin cell line (WT 9-12) and used CUT&RUN and ChIP-seq to identify the p-CREB binding peaks, with the cell numbers being 1 × 105 and 1 × 107, respectively. As shown in Supplemental Figure 2A, although the cell number is 100-fold lower, the signals obtained from CUT&RUN are comparable with those from ChIP-seq. The p-CREB peaks, obtained by both techniques, show a similar genome-wide distribution, with more than half of the peaks located in gene promoters (Supplemental Figure 2B). The signal obtained from p-CREB CUT&RUN was correlated with that from ChIP-seq (Supplemental Figure 2, C and D). As shown in Supplemental Figure 2E, CUT&RUN and ChIP-seq profiles also showed similar distribution. A comparison of CUT&RUN and ChIP-seq track graphs of p-CREB for several representative genes is shown in Supplemental Figure 2F.
Next, we analyzed the genomic distribution of H3K4me3 in the same way as p-CREB, and found that the CUT&RUN method could also detect comparable H3K4me3 signals to ChIP-seq, despite using 100 times fewer cells (Supplemental Figure 3A). The signal obtained from H3K4me3 CUT&RUN and ChIP-seq also showed high correlation (Supplemental Figure 3, B and C) and a similar distribution profile (Supplemental Figure 3D). A comparison of CUT&RUN and ChIP-seq track graphs of H3K4me3 for several representative genes is shown in Supplemental Figure 3E. Together, these results suggest that the CUT&RUN method is ideal for reliably identifying binding targets of TFs and chromatin modifications in a small number of renal cells.
To precisely analyze the role of activated CREB in the cystic cells, DBA-positive cells from kidneys of WT and ADPKD mice were enriched, and the genomic distribution of p-CREB in these cells was profiled by CUT&RUN (Figure 2A). We identified 844 unique p-CREB binding peaks in ADPKD cells (Figure 2B, Supplemental Table 2). Metagene analysis showed that the signals of these peaks are much higher in ADPKD cells than in WT cells (Figure 2C). Motif analysis of these p-CREB peaks also revealed the enrichment of CRE (Figure 2D), consistent with the fact that CREB is a CRE binding protein. We found that over half of these peaks were located in gene promoters (Figure 2E), positioned within 5 kb upstream and downstream of the transcription start site of the neighboring gene (Figure 2F). As shown in Supplemental Figure 4, p-CREB binding genes were prominently enriched in the cell cycle, RNA metabolism, cellular responses to stress, the immune system, and the ribosome. We noticed that the p-CREB signal strength was different in the p-CREB binding regions and wondered whether different p-CREB binding strengths were associated with varying states of chromatin. To address this, we profiled the genome-wide distribution of two transcriptionally permissive histone modifications (H3K4me3 and H3K27ac) by CUT&RUN and compared the chromatin permissiveness across different groups of p-CREB binding peaks: low (reads <30), medium (reads 30–50), and high (reads >50). As shown in Figure 2G, genomic regions with higher p-CREB occupancy are decorated with more H3K4me3 and H3K27ac modifications, suggesting stronger p-CREB binding is associated with more active chromatin status. CUT&RUN track graphs of p-CREB, H3K4me3, and H3K27ac for representative gene regions from different p-CREB binding groups are shown in Figure 2H.
Figure 2.
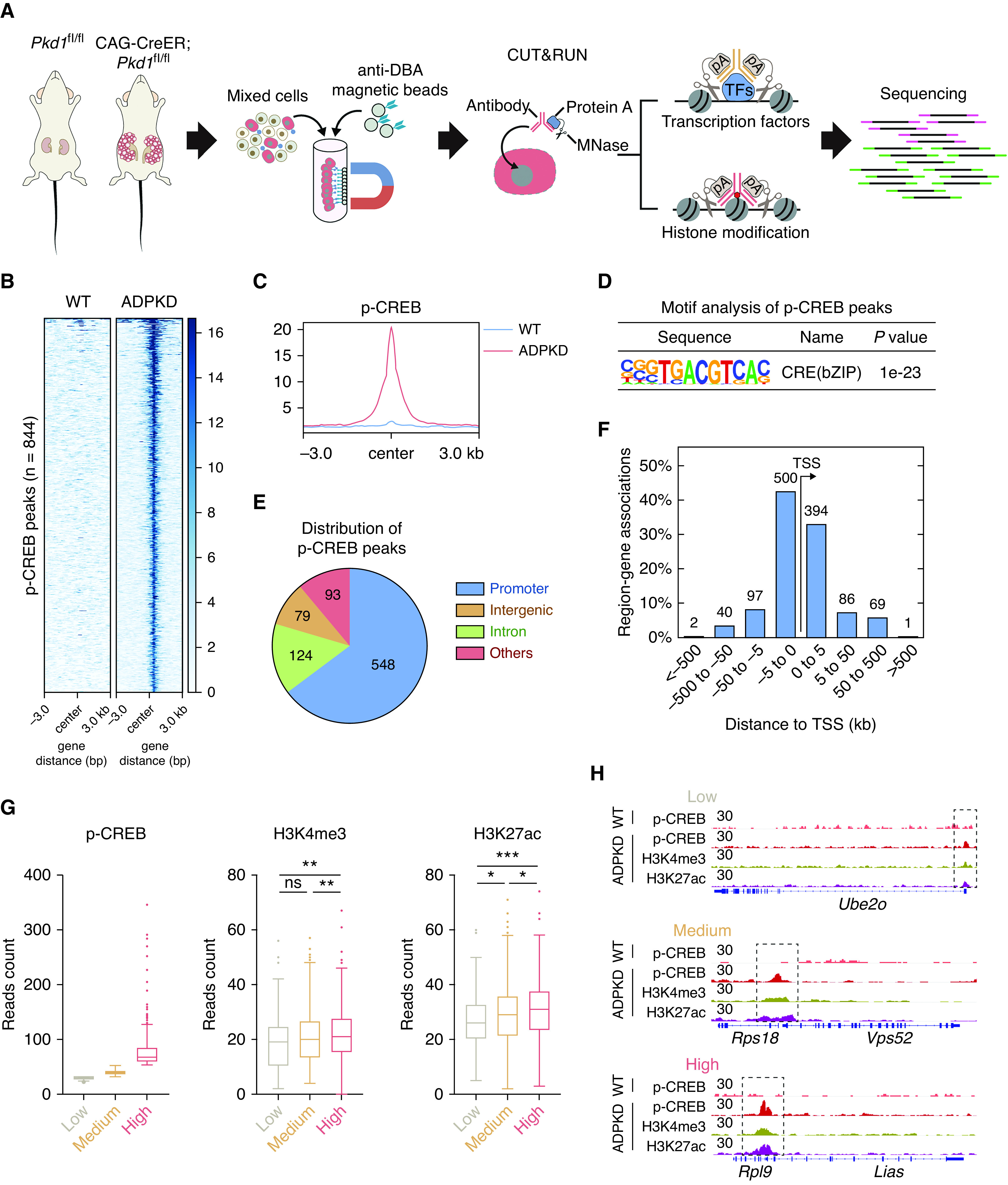
Genomic profiling reveals that stronger p-CREB binding is associated with more active chromatin. (A) Schematic overview of genomic profiling of p-CREB and histone modifications by CUT&RUN followed by sequencing pA, protein A. An early-onset mouse (n=1) was used for the following analysis. (B) Heat map of normalized p-CREB signals in DBA-positive cells from WT and ADPKD mice. (C) Composite plots of normalized p-CREB signals in DBA-positive cells from WT and ADPKD mice. (D) Motif analysis of p-CREB peaks in DBA-positive cells from ADPKD mice. (E) Genomic distribution of p-CREB peaks in DBA-positive cells from ADPKD mice. (F) Number of genes around p-CREB peaks at different distances from adjacent transcription start site (TSS). (G) Box plots of p-CREB, H3K4me3, and H3K27ac reads in the indicated groups. Low, reads of p-CREB were <30; medium, reads of p-CREB were between 30 and 50; high, reads of p-CREB were >50. (H) Representative CUT&RUN tracks of p-CREB, H3K4me3, and H3K27ac in the indicated regions. Data were analyzed using the unpaired, two-sided t test. *P<0.05, **P<0.01, ***P<0.001. pA, protein A.
Activation of CREB Increases the Expression of Cystogenesis-Associated Genes
Notably, the p-CREB antibody can recognize other phosphorylated–kinase-inducible domain proteins, including phosphorylated-ATF1 and phosphorylated-CREM. Therefore, p-CREB binding peaks may represent CREB/ATF1/CREM binding sites. To identify direct CREB target genes, we examined the changes of gene expression after CREB inhibition with 666-15, a cell-permeable naphthol derivative that acts as a highly potent and selective CREB inhibitor.34 As shown in Supplemental Figure 5, 666-15 dose dependently repressed the expression of CRE-luciferase reporter genes. 666-15 has previously been shown to readily achieve pharmacologically relevant concentrations for CREB inhibition and to be well tolerated in vivo.35 Therefore, we treated WT and ADPKD mice with 666-15, and harvested DBA-positive cells for RNA-seq analysis (Figure 3A). We found that gene expression was dysregulated for many genes in ADPKD cells, and 666-15 treatment markedly rescued the expression of many of these ADPKD-disturbed genes (Figure 3B, Supplemental Table 3), including ADPKD-upregulated genes and ADPKD-downregulated genes (Figure 3C). Because p-CREB induced by cAMP is generally associated with gene activation,36 we, therefore, chose genes that were upregulated in ADPKD and reversed by 666-15 treatment for further investigation, and we defined them as 666-15–rescued genes.
Figure 3.
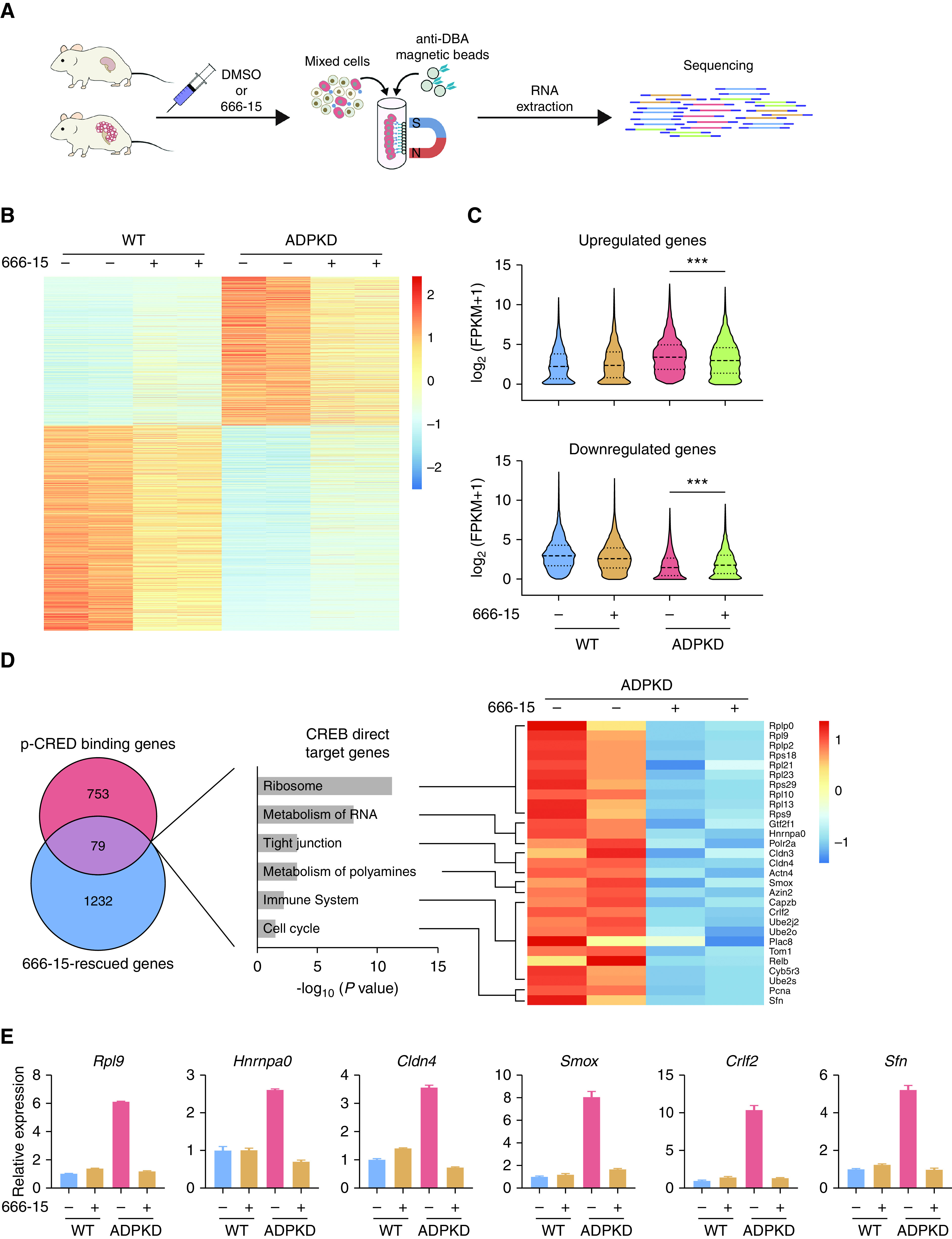
p-CREB regulates ADPKD-associated genes. (A) Schematic overview of transcriptomic analysis in DBA-positive cells from early-onset mice at P29. (B) Heatmap of differentially expressed genes in DBA-positive cells from the indicated groups. (C) Violin plots showing the relative fragments per kilobase of transcript per million mapped reads (FPKM) values of upregulated genes and downregulated genes in DBA-positive cells from the indicated groups. (D) Identification and function enrichment analysis of CREB direct target genes. (E) Quantitative PCR validation of representative genes in DBA-positive cells from the indicated groups. ***P<0.001.
To discover CREB direct target genes, we performed an integrative analysis of p-CREB binding genes with 666-15–rescued genes. As shown in Figure 3D, 79 genes were identified as CREB direct targets. Function enrichment analysis of CREB direct targets revealed prominent enrichment of genes related to cell proliferation and inflammation-related pathways, including ribosome biogenesis,37,38 metabolism of RNA,39,40 the tight junction,41,42 metabolism of polyamines,43–45 the cell cycle, and the immune response46 (Figure 3D), supporting the connection between CREB activation and cystic cell proliferation and inflammation. We validated several CREB direct targets from each enriched pathway by quantitative RT-PCR analysis and confirmed that 666-15 treatment rescued their expression in cystic cells (Figure 3E). We further overlapped CREB target genes with other previously published PKD transcriptomic datasets. PKD-upregulated genes were defined as genes whose expression was upregulated in at least two independent studies, as described previously.47 By comparing our CREB direct targets (n=79) and the PKD-upregulated genes (n=775), we identified nine common genes (Supplemental Figure 6A). As shown in Supplemental Figure 6B, 666-15 treatment rescued the expression of five representative genes in cystic cells. Collectively, these integrative analyses identified CREB direct target genes in cystic kidney epithelial cells.
Pharmacological Inhibition of CREB Suppresses Renal Cyst Growth in an Early-Onset Mouse Model
The above results suggest that activated CREB is responsible for the dysregulation of cell proliferation and inflammation during cystogenesis, two major phenotypic manifestations of ADPKD cells.10,11 A previous report showed that CREB inhibition reduced cyst expansion using the Madin–Darby canine kidney in vitro cyst model.48 However, whether CREB inhibition could suppress cyst growth in vivo is still unknown. To address this, we first examined the effect of 666-15 treatment in an early-onset ADPKD mouse model. To generate the early-onset mouse model, we induced Pkd1 gene deletion by a single injection of tamoxifen (10 mg/kg) at P10. 666-15 (5 mg/kg) treatment was initiated at P11 and continued for 3 weeks. Treatment was applied once daily for five successive days, followed by two days of rest (Figure 4A). Compared with vehicle treatment, administration of 666-15 decreased the kidney size (Figure 4B), kidney weight/body weight (KW/body wt) ratio (Figure 4C), cystic index (Figure 4, D and E), and cell proliferation (Supplemental Figure 7). Furthermore, we also observed that 666-15 treatment improved kidney function, as assessed by BUN (Figure 4F). Together, these results demonstrate that pharmacologically inhibiting CREB activity delays ADPKD progression in the early-onset mouse model.
Figure 4.
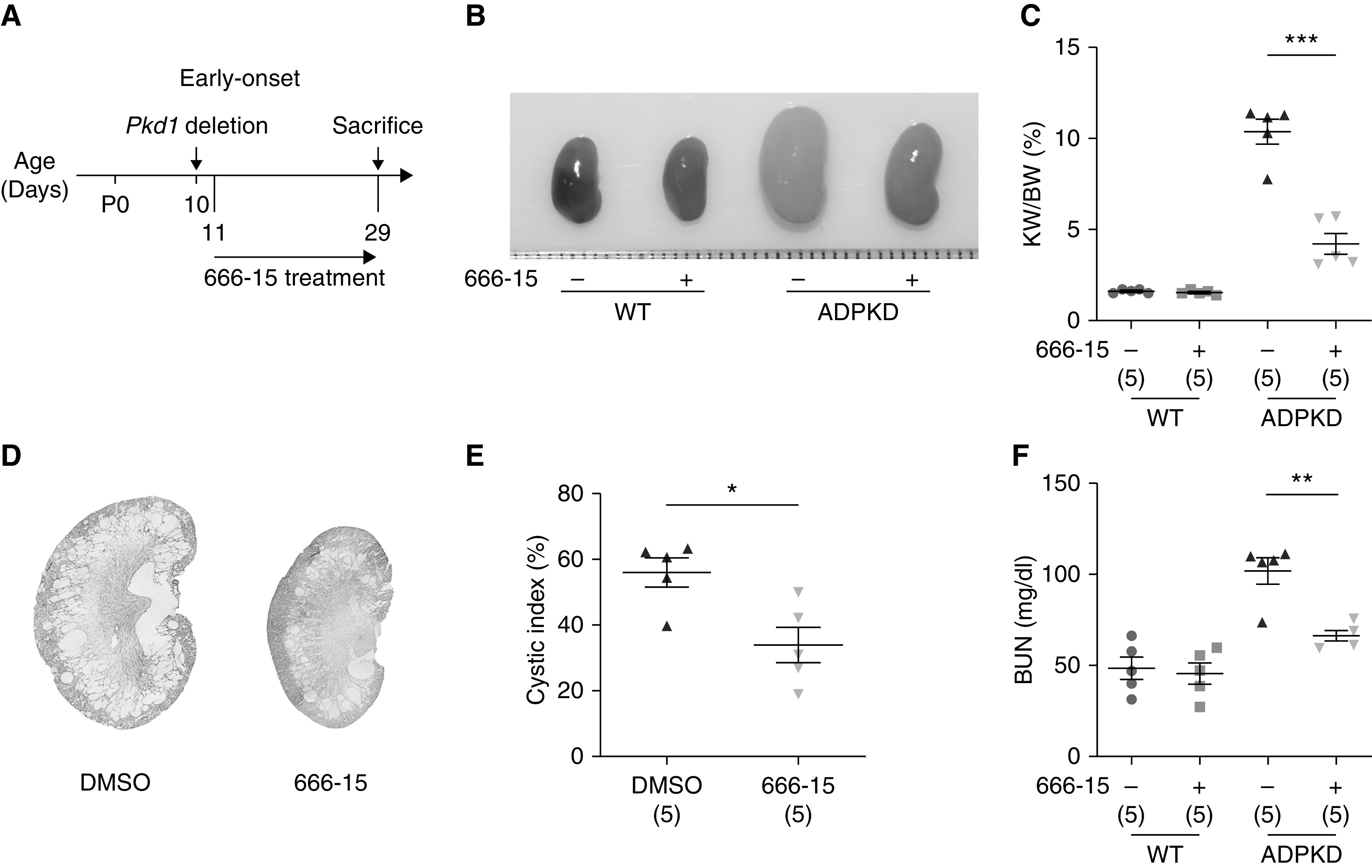
666-15 treatment inhibits renal cyst growth in the early-onset ADPKD mouse model. (A) Schematic view of experimental design in the early-onset ADPKD model. (B) Representative images of P29 kidneys from WT and ADPKD mice treated with DMSO or 666-15. (C) KW/body wt (BW) ratios in the indicated groups of mice. (D) Hematoxylin and eosin staining of kidney sections from ADPKD mice treated with DMSO or 666-15. (E) Cystic index of hematoxylin and eosin–stained kidney sections from ADPKD mice treated with DMSO or 666-15. (F) Plasma BUN levels of P29 mice from the indicated groups. Data are represented as means±SEMs, and were analyzed using the unpaired, two-sided t test in (C), (E), and (F). *P<0.05, **P<0.01, ***P<0.001.
Pharmacological Inhibition of CREB Suppresses Renal Cyst Growth in a Late-Onset Mouse Model
To more closely mimic human ADPKD progression and to evaluate the effects of long-term 666-15 treatment, we used a late-onset ADPKD mouse model and addressed whether 666-15 could reduce cyst growth in animals with established ADPKD. To this end, we induced Pkd1 gene deletion by two injections of tamoxifen (250 mg/kg) at P25 and P28. 666-15 (5 mg/kg) treatment was initiated at P55 and continued for 2 months (Figure 5A), during which 666-15 was administered once daily for 5 days, followed by two rest days. We found that, compared with vehicle treatment, 666-15 treatment delayed cyst growth, as indicated by decreased kidney size (Figure 5B), KW/body wt ratio (Figure 5C), cyst index (Figure 5, D and E), and BUN level (Figure 5F). We observed no systemic toxicity, such as loss of body weight or behavioral changes, in 666-15–treated mice. Together, these results demonstrate that pharmacologically inhibiting CREB activity effectively delays disease progression in the mouse model of late-onset ADPKD.
Figure 5.
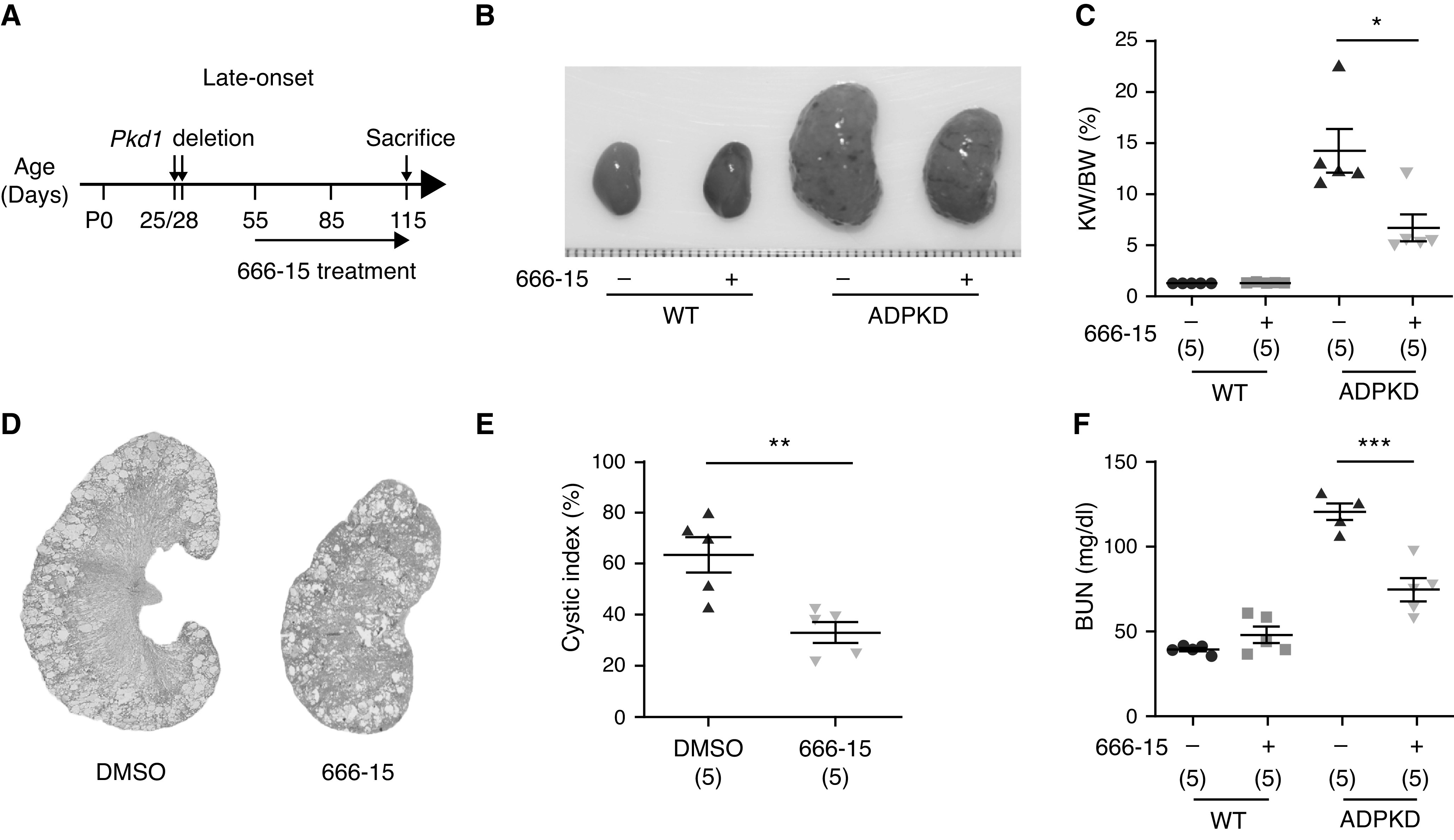
666-15 treatment inhibits renal cyst growth in the late-onset ADPKD mouse model. (A) Schematic view of experimental design in the late-onset ADPKD model. (B) Representative images of P115 kidneys from WT and ADPKD mice treated with DMSO or 666-15. (C) KW/body wt (BW) ratios in the indicated groups of mice. (D) Hematoxylin and eosin staining of kidney sections from ADPKD mice treated with DMSO or 666-15. (E) Cystic index of hematoxylin and eosin–stained kidney sections from ADPKD mice treated with DMSO or 666-15. (F) Plasma BUN levels of P115 mice from the indicated groups. Data are represented as means±SEMs, and were analyzed using the unpaired, two-sided t test in (C), (E), and (F). *P<0.05, **P<0.01, ***P<0.001.
Genetic Inhibition of CREB Delays Cyst Formation In Vivo
Because knockout of CREB is embryonic lethal,49 we adopted A-CREB, a dominant-negative inhibitor of CREB.50 Overexpression of A-CREB repressed the expression of CRE-luciferase reporter genes (Supplemental Figure 8, A and B). To further validate the effect of genetically inhibiting CREB in ADPKD progression in vivo, we generated A-CREB transgenic mice and crossed them with Pkd1fl/fl; Cdh16-Cre mice. The expression of A-CREB was confirmed by Western blotting analysis (Supplemental Figure 8C). As shown in Figure 6, A–E, and Supplemental Figure 9, overexpression of A-CREB decreased kidney size, KW/body wt ratio, cystic index, and cell proliferation, compared with control. Analysis of BUN revealed that A-CREB treatment improved kidney function. Together, these data demonstrate that genetic inhibition of CREB delays cyst formation in vivo.
Figure 6.
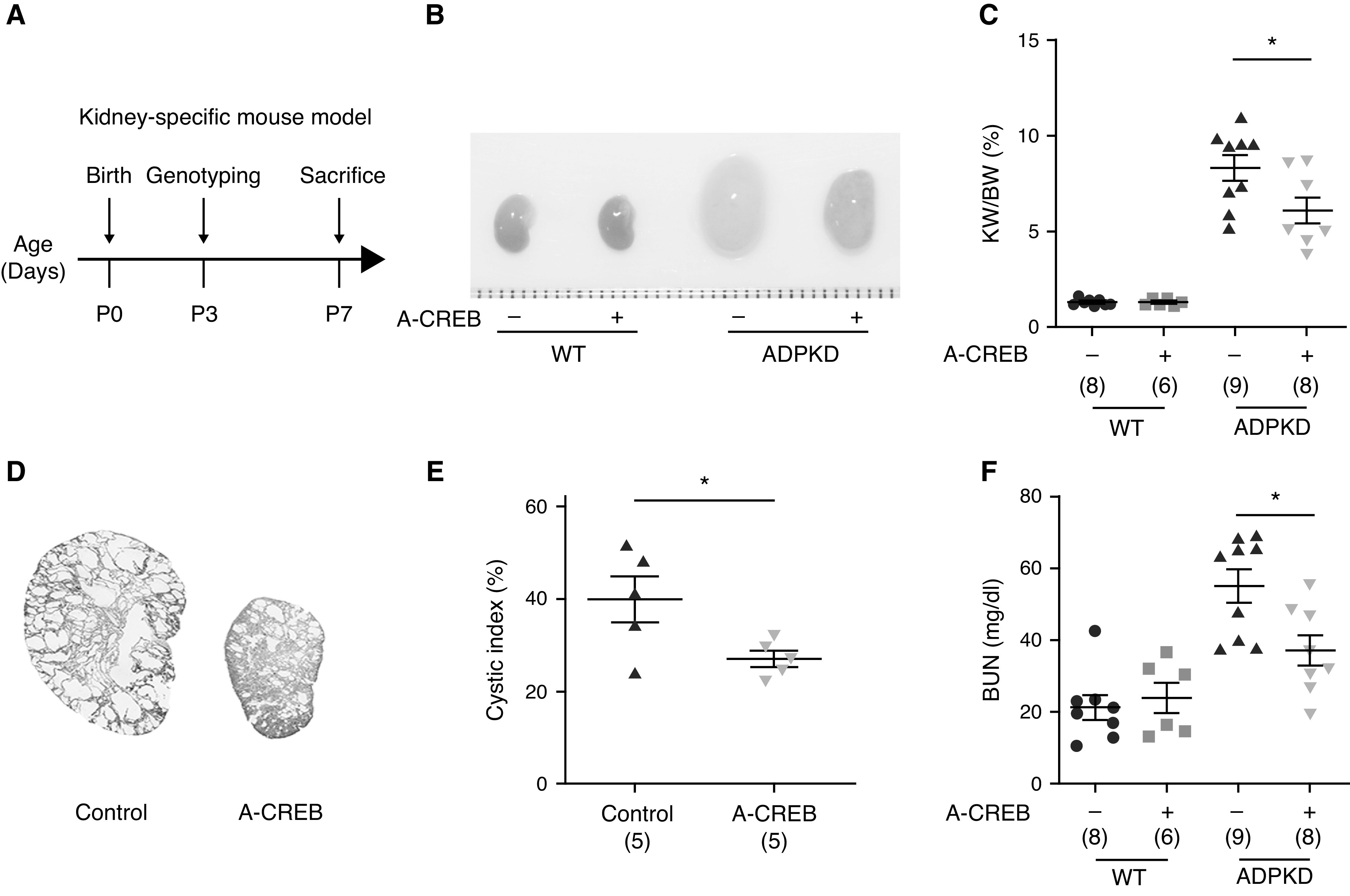
Genetic inhibition of p-CREB ameliorates ADPKD progression. (A) Schematic view of experimental design in the kidney-specific mouse model. (B) Representative images of P7 kidneys from WT and ADPKD mice with or without A-CREB expression. (C) KW/body wt (BW) ratios in the indicated groups of mice. (D) Hematoxylin and eosin staining of kidney sections from ADPKD mice with or without A-CREB expression. (E) Cystic index of hematoxylin and eosin–stained kidney sections in (D). (F) Plasma BUN levels of P7 mice from the indicated groups. Data are represented as means±SEMs, and were analyzed using the unpaired, two-sided t test in (C), (E), and (F). *P<0.05.
Discussion
TFs bind to specific DNA sequences and control gene expression programs that are crucial for establishing and maintaining kidney homeostasis. Due to the kidney’s cellular heterogeneity, substantial challenges are posed to identifying genomic binding sites and direct target genes of TFs in vivo. Here, we showed that CUT&RUN is a powerful method for mapping genomic locations of TFs and profiling histone modifications from low cell numbers. We also showed that CUT&RUN combined with transcriptomic analysis is highly effective for identifying direct targets of TFs in specific renal cells. This study provides a model for research in renal diseases, where obtaining large numbers of purified diseased cells from animal models or patients is quite challenging. Moreover, because kidney development is initiated from small populations of cells within the embryo, the strategy used in this study should also be applicable to characterize the target genes of key developmental TFs from a limited number of embryonic cells, which is essential for the understanding of kidney development.
Improved understanding of the dysregulated signaling and pathologic features of ADPKD has revealed marked similarities to such features in cancers.51 These broad similarities between ADPKD and cancer suggest that many agents developed for cancer should also be applicable to ADPKD treatment, for which very few therapeutic options to delay disease development are currently available. Several recent preclinical and clinical studies of ADPKD have shown the therapeutic benefits of such drug repurposing.52–54 Overexpression or hyperactivation of CREB has been reported in many hematopoietic and solid tumors.55 Novel CREB inhibitors with higher solubility and bioavailability have been actively under development and shown promise in preclinical studies.36 Our integrative analysis of CUT&RUN and RNA-seq revealed CREB direct targets, many of which are involved in pathways related to cystogenesis, including cell proliferation and inflammation. Interestingly, the most significantly enriched pathway is associated with ribosome biogenesis and protein synthesis, supporting a high bioenergetic demand for sustained cell proliferation during cystogenesis. Hyperactivation of ribosome biogenesis has been implicated in cancer initiation and progression by increasing protein synthesis to support cell proliferation.37 Thus, our current work, showing the increased abundance of many ribosome proteins in ADPKD cells and identifying these ribosome genes as CREB direct targets, adds a new layer of similarity between ADPKD and cancer cells and provides evidence supporting a key role of CREB in ADPKD cell proliferation. Although many of the CREB direct target genes support the connection between CREB activation and cystic expansion and inflammation, their direct roles in cystogenesis are currently unknown, which warrants further investigation.
In addition to responding to cAMP/protein kinase A signaling, CREB is also a substrate for various cellular kinases, including protein kinase C, AKT, and calcium/calmodulin-dependent kinases.17 Importantly, these pathways are also frequently disturbed in ADPKD.12,13 Thus, it is reasonable to speculate that CREB lies at the hub, orchestrating the expression of a cystogenesis-associated transcriptome and integrating multiple disease-disturbed signaling pathways in ADPKD cells. Supporting this idea, the specific CREB targets we identified in cystic cells include many genes that are involved in disease-related pathways of ADPKD. Functional experiments show that targeted pharmacologic and genetic inhibition of CREB activity can substantially delay disease progression. Activation of CREB, through its phosphorylation, occurs mainly in cystic cells, and not in normal cells surrounding the cysts. Therefore, targeted inhibition of CREB may selectively inhibit cyst growth while sparing normal parenchyma, providing a further rationale for developing CREB inhibitors as potential therapeutic agents for ADPKD.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by National Natural Science Foundation of China grants 81770658 and 82070689 (to Y. Chen), and Tianjin Municipal Science and Technology Commission grant 19JCJQJC63800 (to Y. Chen).
Supplementary Material
Acknowledgments
We thank Lijia Li and Fangnong Lai for their technical assistance in the CUT&RUN experiments.
Z. Liu performed mouse and biochemistry studies and bioinformatics analysis; Y. Liu performed human specimen analysis and mouse studies; L. Dang assisted with the biochemical studies; M. Geng performed mouse studies; Y. Sun and Y. Lu provided expertise on mouse studies; Z. Fang analyzed data and edited the manuscript; H. Xiong provided human specimens; and Y. Chen conceived of, designed, and supervised the project; analyzed data; and wrote the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2021010101/-/DCSupplemental.
Supplemental Table 1. ADPKD patient information.
Supplemental Table 2. Annotation of p-CREB binding peaks in DBA+ cells from ADPKD kidney.
Supplemental Table 3. Comparison of differentially expressed genes in DBA+ cells from WT and ADPKD mice treated with or without 666-15.
Supplemental Figure 1. Phosphorylation of CREB increases steadily in ADPKD kidneys.
Supplemental Figure 2. Comparison of p-CREB CUT&RUN and ChIP-seq.
Supplemental Figure 3. Comparison of H3K4me3 CUT&RUN and ChIP-seq.
Supplemental Figure 4. Function enrichment analysis of p-CREB binding genes.
Supplemental Figure 5. Inhibition of CREB transactivity by 666-15 treatment.
Supplemental Figure 6. p-CREB target genes previously reported as cystogenesis-associated genes.
Supplemental Figure 7. 666-15 suppresses cystic cell proliferation in the early-onset model.
Supplemental Figure 8. Inhibition of CREB by overexpressing A-CREB.
Supplemental Figure 9. 666-15 suppresses cystic cell proliferation in the kidney-specific mouse model.
References
- 1.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. : The human transcription factors. Cell 172: 650–665, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Formica C, Malas T, Balog J, Verburg L, ’t Hoen PAC, Peters DJM: Characterisation of transcription factor profiles in polycystic kidney disease (PKD): Identification and validation of STAT3 and RUNX1 in the injury/repair response and PKD progression. J Mol Med (Berl) 97: 1643–1656, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR: Targeting the progression of chronic kidney disease. Nat Rev Nephrol 16: 269–288, 2020 [DOI] [PubMed] [Google Scholar]
- 4.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al. : Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindström NO, McMahon JA, Guo J, Tran T, Guo Q, Rutledge E, et al. : Conserved and divergent features of human and mouse kidney organogenesis. J Am Soc Nephrol 29: 785–805, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, et al. : A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun 10: 2832, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skene PJ, Henikoff S: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6: e21856, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skene PJ, Henikoff JG, Henikoff S: Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13: 1006–1019, 2018 [DOI] [PubMed] [Google Scholar]
- 9.Xia W, Xu J, Yu G, Yao G, Xu K, Ma X, et al. : Resetting histone modifications during human parental-to-zygotic transition. Science 365: 353–360, 2019 [DOI] [PubMed] [Google Scholar]
- 10.Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE: Polycystic kidney disease. Nat Rev Dis Primers 4: 50, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornec-Le Gall E, Alam A, Perrone RD: Autosomal dominant polycystic kidney disease. Lancet 393: 919–935, 2019 [DOI] [PubMed] [Google Scholar]
- 12.Chapin HC, Caplan MJ: The cell biology of polycystic kidney disease. J Cell Biol 191: 701–710, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris PC, Torres VE: Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 124: 2315–2324, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH 2nd: Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med 10: 363–364, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Chebib FT, Sussman CR, Wang X, Harris PC, Torres VE: Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat Rev Nephrol 11: 451–464, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Perrone RD, Koch G, et al. ; REPRISE Trial Investigators: Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med 377: 1930–1942, 2017 [DOI] [PubMed] [Google Scholar]
- 17.Mayr B, Montminy M: Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg D, Groussin L, Jullian E, Perlemoine K, Bertagna X, Bertherat J: Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Ann N Y Acad Sci 968: 65–74, 2002 [DOI] [PubMed] [Google Scholar]
- 19.Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, et al. : Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature 370: 226–229, 1994 [DOI] [PubMed] [Google Scholar]
- 20.Altarejos JY, Montminy M: CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 12: 141–151, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakhina V, Arey RN, Kaletsky R, Kauffman A, Stein G, Keyes W, et al. : Genome-wide functional analysis of CREB/long-term memory-dependent transcription reveals distinct basal and memory gene expression programs. Neuron 85: 330–345, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han J, Li E, Chen L, Zhang Y, Wei F, Liu J, et al. : The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 524: 243–246, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, et al. : Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A 102: 4459–4464, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y, Liu Z, Cao X, Lu Y, Mi Z, He C, et al. : Activation of P-TEFb by cAMP-PKA signaling in autosomal dominant polycystic kidney disease. Sci Adv 5: eaaw3593, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menezes LF, Lin CC, Zhou F, Germino GG: Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 5: 183–192, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loghman-Adham M, Nauli SM, Soto CE, Kariuki B, Zhou J: Immortalized epithelial cells from human autosomal dominant polycystic kidney cysts. Am J Physiol Renal Physiol 285: F397–F412, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Lu Y, Sun Y, Liu Z, Lu Y, Zhu X, Lan B, et al. : Activation of NRF2 ameliorates oxidative stress and cystogenesis in autosomal dominant polycystic kidney disease. Sci Transl Med 12: eaba3613, 2020 [DOI] [PubMed] [Google Scholar]
- 28.Thike AA, Chng MJ, Fook-Chong S, Tan PH: Immunohistochemical expression of hormone receptors in invasive breast carcinoma: correlation of results of H-score with pathological parameters. Pathology 33: 21–25, 2001 [PubMed] [Google Scholar]
- 29.Mi Z, Song Y, Cao X, Lu Y, Liu Z, Zhu X, et al. : Super-enhancer-driven metabolic reprogramming promotes cystogenesis in autosomal dominant polycystic kidney disease. Nat Metab 2: 717–731, 2020 [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Ward CJ, Harris PC, Torres VE: Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int 77: 129–140, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puri P, Schaefer CM, Bushnell D, Taglienti ME, Kreidberg JA, Yoder BK, et al. : Ectopic phosphorylated creb marks dedifferentiated proximal tubules in cystic kidney disease. Am J Pathol 188: 84–94, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye H, Wang X, Constans MM, Sussman CR, Chebib FT, Irazabal MV, et al. : The regulatory 1α subunit of protein kinase A modulates renal cystogenesis. Am J Physiol Renal Physiol 313: F677–F686, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, et al. : Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 122: 4257–4273, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie F, Li BX, Kassenbrock A, Xue C, Wang X, Qian DZ, et al. : Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J Med Chem 58: 5075–5087, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li BX, Gardner R, Xue C, Qian DZ, Xie F, Thomas G, et al. : Systemic inhibition of CREB is well-tolerated in vivo. Sci Rep 6: 34513, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steven A, Friedrich M, Jank P, Heimer N, Budczies J, Denkert C, et al. : What turns CREB on? And off? And why does it matter? Cell Mol Life Sci 77: 4049–4067, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pelletier J, Thomas G, Volarević S: Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat Rev Cancer 18: 51–63, 2018 [DOI] [PubMed] [Google Scholar]
- 38.Bursać S, Prodan Y, Pullen N, Bartek J, Volarević S: Dysregulated ribosome biogenesis reveals therapeutic liabilities in cancer. Trends Cancer 7: 57–76, 2021 [DOI] [PubMed] [Google Scholar]
- 39.Young DJ, Stoddart A, Nakitandwe J, Chen SC, Qian Z, Downing JR, et al. : Knockdown of Hnrnpa0, a del(5q) gene, alters myeloid cell fate in murine cells through regulation of AU-rich transcripts. Haematologica 99: 1032–1040, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Zhang X, Han C, Wan G, Huang X, Ivan C, et al. : TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 520: 697–701, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Díaz-Coránguez M, Liu X, Antonetti DA: Tight junctions in cell proliferation. Int J Mol Sci 20: 5972, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu AS, Kanzawa SA, Usorov A, Lantinga-van Leeuwen IS, Peters DJ: Tight junction composition is altered in the epithelium of polycystic kidneys. J Pathol 216: 120–128, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arruabarrena-Aristorena A, Zabala-Letona A, Carracedo A: Oil for the cancer engine: The cross-talk between oncogenic signaling and polyamine metabolism. Sci Adv 4: eaar2606, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Mao Y, Zhao L, Li L, Wu J, Zhao M, et al. : p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 567: 253–256, 2019 [DOI] [PubMed] [Google Scholar]
- 45.Zabala-Letona A, Arruabarrena-Aristorena A, Martín-Martín N, Fernandez-Ruiz S, Sutherland JD, Clasquin M, et al. : mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 547: 109–113, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y, Chen M, Zhou J, Lv J, Song S, Fu L, et al. : Interactions between macrophages and cyst-lining epithelial cells promote kidney cyst growth in Pkd1-deficient mice. J Am Soc Nephrol 29: 2310–2325, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malas TB, Formica C, Leonhard WN, Rao P, Granchi Z, Roos M, et al. : Meta-analysis of polycystic kidney disease expression profiles defines strong involvement of injury repair processes. Am J Physiol Renal Physiol 312: F806–F817, 2017 [DOI] [PubMed] [Google Scholar]
- 48.Kakade VR, Tao S, Rajagopal M, Zhou X, Li X, Yu AS, et al. : A cAMP and CREB-mediated feed-forward mechanism regulates GSK3β in polycystic kidney disease. J Mol Cell Biol 8: 464–476, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bleckmann SC, Blendy JA, Rudolph D, Monaghan AP, Schmid W, Schütz G: Activating transcription factor 1 and CREB are important for cell survival during early mouse development. Mol Cell Biol 22: 1919–1925, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C: A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol 18: 967–977, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seeger-Nukpezah T, Geynisman DM, Nikonova AS, Benzing T, Golemis EA: The hallmarks of cancer: Relevance to the pathogenesis of polycystic kidney disease. Nat Rev Nephrol 11: 515–534, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asawa RR, Danchik C, Zahkarov A, Chen Y, Voss T, Jadhav A, et al. : A high-throughput screening platform for polycystic kidney disease (PKD) drug repurposing utilizing murine and human ADPKD cells. Sci Rep 10: 4203, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kiseleva AA, Golemis EA: Informatics-guided drug repurposing for autosomal dominant polycystic kidney disease (ADPKD). EBioMedicine 52: 102628, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malas TB, Leonhard WN, Bange H, Granchi Z, Hettne KM, Van Westen GJP, et al. : Prioritization of novel ADPKD drug candidates from disease-stage specific gene expression profiles. EBioMedicine 51: 102585, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sapio L, Salzillo A, Ragone A, Illiano M, Spina A, Naviglio S: Targeting CREB in cancer therapy: A key candidate or one of many? An update. Cancers (Basel) 12: 3166, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.