

Significance Statement
Better understanding of the regulation of auto- and alloantibody production is essential to develop hypothesis-driven therapies for autoimmune kidney diseases and graft rejection. Murine studies demonstrate that erythropoietin (EPO), a kidney-produced hormone, inhibits primary, T cell–dependent humoral immunity. EPO also diminishes autoantibodies and disease severity in murine models of lupus, and significantly reduces secondary humoral immunity in an allogeneic organ transplant model. A direct, STAT5-dependent, inhibitory effect of EPO through its receptor (EPOR) on T follicular helper (TFH) cells that is crucial for B cell maturation mediates these effects. In vitro experiments document that EPO inhibitory effects on TFH formation apply to human cells, providing a rationale for further testing how EPOR activation affects autoimmune kidney diseases and antibody-mediated graft rejection.
Keywords: transplantation, rejection, immunology
Visual Abstract

Abstract
Background
Although high-affinity IgG auto- and alloantibodies are important drivers of kidney inflammation that can result in ESKD, therapeutic approaches that effectively reduce such pathogenic antibodies remain elusive. Erythropoietin (EPO) has immunomodulatory functions, but its effects on antibody production are unknown.
Methods
We assessed the effect and underlying mechanisms of EPO/EPO receptor (EPOR) signaling on primary and secondary, T cell–dependent and T–independent antibody formation using in vitro culture systems, murine models of organ transplantation and lupus nephritis, and mice conditionally deficient for the EPOR expressed on T cells or B cells.
Results
In wild-type mice, recombinant EPO inhibited primary, T cell–dependent humoral immunity to model antigens and strong, polyclonal stimuli, but did not alter T–independent humoral immune responses. EPO also significantly impaired secondary humoral immunity in a potent allogeneic organ transplant model system. The effects required T cell, but not B cell, expression of the EPOR and resulted in diminished frequencies of germinal center (GC) B cells and T follicular helper cells (TFH). In vitro and in vivo experiments showed that EPO directly prevented TFH differentiation and function via a STAT5-dependent mechanism that reduces CD4+ T cell expression of Bcl6. In lupus models, EPO reduced TFH, GC B cells, and autoantibody production, and abrogated autoimmune glomerulonephritis, demonstrating clinical relevance. In vitro studies verified that EPO prevents differentiation of human TFH cells.
Conclusions
Our findings newly demonstrate that EPO inhibits TFH-dependent antibody formation, an observation with potential implications for treating antibody-mediated diseases, including those of the kidney.
An enhanced understanding of the mechanisms regulating antibody formation is important for devising therapeutic strategies for autoimmune kidney diseases and antibody-mediated rejection of transplanted grafts. The development of high-affinity, pathogenic IgG antibodies involves cognate T cell/B cell interactions that cause isotype switch recombination and affinity maturation. Affinity maturation occurs in germinal centers (GCs), specialized compartments within secondary lymphoid organs where antigen-reactive B cells undergo iterative cycles of clonal expansion, immunoglobulin gene somatic hypermutation, and affinity-driven positive selection.1,2 Positive selection of high affinity clones is crucially accomplished via cognate interactions between CD4+CXCR5+PD1+, IL-21–secreting T follicular helper cells (TFH) and GC B cells that have captured, processed, and presented antigen on class II MHC. CD4+CXCR5+PD1+, Foxp3-expressing T follicular regulatory cells (TFR), differentiated from thymic regulatory T cells (Treg), inhibit TFH function and thereby regulate affinity maturation.3 The positively selected B cells can re-enter the GC or differentiate into antibody-producing plasma cells or memory B cells. In the absence of T cells, B cell activation initiated by B cell receptor ligation with nonprotein antigens (e.g., polysaccharides, lipids, nucleic acids) and appropriate costimulatory signals (e.g., toll-like receptor stimulation of C3dg/CD21 ligation) results in low affinity IgM and IgG that can also function as pathogenic mediators of disease.4
Since 2015, our group has been studying the initially unexpected immunoregulatory effects of erythropoietin (EPO), a hypoxia-induced hormone traditionally known for its ability to stimulate erythropoiesis.5 Building on studies performed by others showing that EPO inhibits monocyte activation,6,7 we demonstrated that T cells express the EPO receptor (EPOR) and that EPO inhibits proliferation and differentiation of naïve and memory, CD4+ and CD8+ conventional T cells by uncoupling IL-2 receptor signaling.8,9 We further demonstrated that EPO has protolerogenic properties because it maintains and promotes expansion and function of CD4+ Treg.8 Together, these effects on T cell immunity are clinically significant because EPO therapy inhibits T cell–dependent murine cardiac allograft rejection.8 We further showed that EPO prevents differentiation of IL-17–producing CD4+ T cells and blunts the clinical expression of IL-17–dependent, murine autoimmune kidney disease,10 including murine models of lupus nephritis in which immune complexes deposited in the glomeruli contribute to disease pathogenesis. Whether and how EPO affects the development of humoral immune responses has not been addressed.
Methods
Mice
C57BL/6 (B6, H‐2b), BALB/c (H-2d), (B6 x BALB/c) F1 ([bxd] F1), (B6 x DBA) F1 ([bxd] F1), B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J, MRL/Mp (H-2k), and B6 Cd45.1 mice, 6–8 weeks of age, were purchased from the Jackson Laboratory (Bar Harbor, ME) or bred from Jackson‐derived animals at the Icahn School of Medicine at Mount Sinai. C57BL/6J EPORfl/fl CD4-Cre mice were generated as previously described.10 C57BL/6J EPORfl/fl CD19-Cre were generated crossing EPORfl/fl mice with CD19-CrePOS animals (Jackson Laboratory). T cell receptor–transgenic B6 TEa mice (CD4+ reactive to I‐Ab + peptide I‐Edα53–68) were a gift from A. Rudensky (Memorial Sloan Kettering Cancer Center, New York, NY).
All animals were housed in the Center for Comparative Medicine and Surgery at the Icahn School of Medicine at Mount Sinai under Institutional Animal Care in accordance with guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International. Experiments were performed with age‐ and sex‐matched mice, using animals that were littermates, or were maintained in the same room, and/or were cohoused within the same cages for >2 weeks to limit potential effects of microbiome differences.
Hydroxy-3-nitrophenylacetyl–Keyhole Limpet Hemocyanin Immunization
Groups of animals were immunized with hydroxy-3-nitrophenylacetyl–keyhole limpet hemocyanin (NP-KLH) (Biosearch Technologies, Petaluma, CA). We mixed 100 µg of NP-KLH 1:1 vol/vol in Alum (Immject Alum, ThermoFisher, Waltham, MA) and injected mice intraperitoneally (i.p.).
At 10 days after immunization, serum samples were collected to perform ELISA; spleen cells were collected and analyzed for TFH, TFR, and total and NP+ GC B cell measurements.
2,4,6-Trinitrophenyl-aminoethylcarboxymethyl-Ficoll immunization
For T cell–independent humoral responses, mice were immunized with 50 µg of 2,4,6-trinitrophenyl-aminoethylcarboxymethyl (TNP) Ficoll (Bioresearch Technologies) in 200 μl PBS (i.p.). At 10 days after immunization, spleen cells and serum were collected and analyzed for TNP-specific B cells, IgM, and IgG.
Sheep Red Blood Cell Immunization
Two ml of sheep red blood cells (SRBC; Innovative Research, Novi, MI) were washed twice with 15 ml of PBS and resuspended 1:10 in PBS. Then 200 µl of the SRBC suspension were injected i.p. into mice. At 12 days after immunization, spleen cells were collected and analyzed for TFH, TFR, class switched, and GC B cell measurements.
Heterotopic Heart Transplantation and Allogeneic Spleen Immunization
Murine heterotopic heart transplantation was performed by the microsurgery core in the Icahn School of Medicine at Mount Sinai as previously described.11–13
In selected experiments, recipients were sensitized by allogeneic spleen cell i.p. injections (20 × 106). After 2 weeks, animals with donor-specific antibodies (see below at alloantibody measurement) mean fluorescence intensity (MFI) >1500 at 1:50 dilution received a heart transplant.
TEa TFH Cell Expansion In Vivo
Naïve CD4+ T cells were isolated from B6 TEa mice by magnetic negative selection (StemCell Technologies Inc., Vancouver, BC). Then 5 × 106 B6 TEa naïve CD4+ T cells were injected retro-orbitally into B6 CD45.1 mice. Animals were treated with vehicle or EPO (10,000 IU/kg) for 14 days. Spleen cells were harvested for flow cytometric analyses.
Parent-to-F1 Models
Donor spleen cells were depleted of CD8+ T cells (Dynabeads Flow Comp Mouse CD8 kit, Invitrogen, Carlsbad, CA) and the resultant spleen cells containing 10–15 × 106 CD4+ T cells were injected intravenously (retro-orbitally) into nonirradiated (bxd) F1 recipients.14,15 Flow cytometric analysis verified <1% CD8+ T cells with equal numbers of CD4+ T cell transfers between groups within an experiment.
EPO Treatment
In in vivo experiments, EPO (Retacrit Epoetin Alfa-epbx recombinant, Pfizer, New York, NY, 10,000 units/ml) or PBS was administered i.p. daily at the specified doses.
In Vitro B Cell Stimulation
B cells were enriched from total splenocytes by magnetic negative selection (StemCell Technologies Inc.). Then B cells were stimulated with 1 ug/ml anti-IgM Fab2 plus bacterial LPS (Sigma-Aldrich, St. Louis, MO) at different concentrations (0.1, 1, 10 µg/ml) and incubated in complete RPMI with vehicle or EPO (1000 UI/ml) for 3 days at 37°C in 96-well plates. Cells were collected for flow cytometric analyses.
In Vitro Murine TFH Cell Differentiation
Enriched murine splenic CD4+ T cells (EasySep, StemCell Technologies) were stimulated in RPMI, 10% fetal calf serum, 2 mM Glutamine (Gibco/ThermoFisher), 100 U/ml penicillin/streptomycin (Gibco/ThermoFisher), 50 μM β-mercaptoethanol with anti-CD3/anti-CD28 mAbs (8 μg/ml each, clones 145–2c11/37.51, respectively, from Invitrogen) at 37°C for 5 days with 30 ng/ml murine IL-6 (PeproTech, Rocky Hill, NJ), 20 μg/ml anti–TGF-β (clone 11D.11.15.8, BioXCell, Lebanon, NH), 10 μg/ml anti–IFN-γ, (clone XMG1.2, BioXCell), and 10 μg/ml anti–IL-4 (clone 11B11, BioXCell). Cells were treated with EPO (1000 IU/ml) or vehicle. In selected experiments, 250 μM STAT5 Inhibitor (CAS 285986–31–4, Millipore Sigma, St. Louis, MO) was added to the culture.
For intracellular cytokine staining, cells were cultured for the final 4 hours in the presence of Golgi plug (1 µg/ml; BD Biosciences, San Jose, CA). PMA (50 ng/ml; MilliporeSigma) and ionomycin (500 ng/ml; MilliporeSigma) were added to positive control wells. After surface staining, cells were fixed, permeabilized, and stained for intracellular cytokines. For IL-21 staining, IL-21R subunit/Fc chimera (R&D Systems, Minneapolis, MN) and AffiniPure F(ab′)2 Fragment Goat Anti-human IgG Fcγ (Jackson Immunoresearch, West Grove, PA) were used as the primary and secondary antibody, respectively.
Isolation of Human Peripheral Blood Cell and T Cell Subsets
PBMC were isolated from buffy coats obtained from deidentified healthy donors (New York Blood Bank) through Ficoll–Hypaque (Sigma LifeScience) density gradient centrifugation.
In Vitro Human TFH Cell Differentiation
Naive CD4+ T cells were isolated from buffy coats (EasySep, StemCell Technologies) and activated overnight in RPMI, 10% human serum AB (GeminiBio, West Sacramento, CA), 2 mM glutamine (Gibco/ThermoFisher), 100 U/ml penicillin/streptomycin (Gibco/ThermoFisher), 50 μM β-mercaptoethanol, with anti-huCD3/CD28 dynabeads (10 μl/106 cells, ThermoFisher). Cells were washed and replated in cRPMI with plate-coated anti-huCD3/CD28 mAbs (5 μg/ml each, clone OKT3, Invitrogen, and clone CD28.2, BD Bioscience, respectively) and recombinant human IL-6 (50 ng/ml, PeproTech) with or without EPO (50–100 IU/ml). Cells were incubated at 37°C for 5 days. Intracellular staining for IL-21 was performed using clone A3N2 (eBioscience).
Bcl6 Quantification
Enriched murine splenic CD4+ T cells (EasySep, StemCell Technologies) were stimulated in RPMI, 10% fetal calf serum, 2 mM glutamine (Gibco/ThermoFisher), 100 U/ml penicillin/streptomycin (Gibco/ThermoFisher), 50 μM β-mercaptoethanol with anti-CD3/anti-CD28 mAbs (8 μg/ml each, clones 145–2c11/37.51, respectively, from Invitrogen) at 37°C for 2 days with 30 ng/ml murine IL-6 (PeproTech). Cells were treated with EPO (1000 IU/ml) or vehicle. At day 0 and day 2, cells were stained for PerCP-Cy5.5-anti-Bcl6 (clone 7D1, BioLegend).
Signaling Assays
For intracellular signaling, phospho-flow analyses of murine T cells were distributed into round-bottom 96-well plates (2 × 105 per well) in CTL Free Serum Media (CTL, Shaker Heights, OH), and allowed to rest at 37°C overnight. Cells were kept resting or activated with anti-CD3 and anti-CD28 mAb beads (25 μl/106, ThermoFisher) and IL-6 (30 ng/ml) ±EPO (1000 IU/ml) for 5, 15, 30, 45, and 60 minutes at 37°C. The reaction was stopped by fixation with 4% paraformaldehyde (Affymetrix) for 10 minutes at 37°C. The cells were washed with PBS and permeabilized by resuspension in 90% cold methanol (ThermoFisher Scientific) for 15 minutes on the rocker. Samples were washed three times with PBS 1% FBS (ThermoFisher Scientific, Wilmington, DE) and stained for 2 hours at room temperature for PE–Cy7–anti-CD4 (Clone GK 1.5, BD Pharmigen), Alexa Fluor 647–anti-pSTAT5 (pY69; clone 47/Stat5, BD Phosflow) and PE–anti-pSTAT3 (pY705; clone 4/P-STAT3, BD Phosflow).
In Vitro TFR–TFH–B Cell Coculture Experiments
In vitro T-dependent stimulation assays were performed by culturing 5 × 104 BALB/c enriched B cells (StemCell Technologies Inc.) and 3 × 104 flow-sorted TFH cells from B6, C57BL/6NJ, C57BL/6J EPORfl/fl CD4-CrePOS/NEG or B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J mice preimmunized with BALB/c splenocytes. In selected experiments 1.5 × 104 TFR cells, isolated from preimmunized B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J spleens, were added to the culture. Cells were cultured in R10 medium with 2 μg/ml anti-CD3 (ThermoFisher Scientific), 10 μg/ml anti-IgM (Jackson Immunoresearch), and with vehicle or EPO (1000 UI/ml) for 2 days at 37°C in 96-well plates. Cells were collected for flow cytometric analyses.
Cell Sorting
CD4+ T cells were enriched from total splenocytes by magnetic negative selection (StemCell Technologies Inc.). CD4+-enriched T cells were stained and CD4+TCRβ+ICOS+CXCR5+PD1+ Foxp3-GFP positive (TFR) and Foxp3-GFP negative (TFH) cells were sorted on SH800Z Cell Sorter Sony Biotechnology Inc. (San Jose, CA) (70 μm sorting chip) using optimal purity settings.
ELISA
ELISA was performed on serum samples to assess high affinity and total anti-NP antibodies, using BSA with different degrees of NP conjugation. We used a standard approach as published.16 Briefly, plates were coated overnight at 4°C with 50 µg/ml NP(27)-BSA (Biosearch Technologies) to measure total antibody or 50 µg/ml NP(2)-BSA (Biosearch Technologies) to measure high-affinity antibody. Bound antibodies were detected using anti-mouse IgG1-HRP (Southern Biotech, Birmingham, AL). Anti-TNP (clone 107.3) (BD Pharmingen, Franklin Lakes, NJ) was used to quantify the titers.
Alloantibody Measurement
Serum samples from recipient mice were diluted in PBS as indicated and incubated for 30 minutes at 4°C with syngeneic, donor, or third‐party (MRL/MpJ; H-2k) thymocytes as target cells.17 After a wash step with PBS 1% albumin, the bound antibody was detected by incubation with FITC‐conjugated rat anti‐mouse IgG (eBioscience, San Diego, CA) and quantified by flow cytometry.
Flow Cytometry
We used standard approaches for surface and intracellular staining as published.10 For surface staining, we used APC–anti-CD4 (Biolegend, San Diego, CA), PE–Cy7–anti-CD4 (clone RM4–5), BV510–anti-CD4 (clone GK1.5) (BD Pharmingen), and APC–Cy7–anti-CD4 (clone GK1.5) (Biolegend); biotinylated anti-CXCR5 (BD Pharmingen) followed by Pacific Blue streptavidin (ThermoFisher Scientific), PerCP–Cy5.5–anti-TCRb (clone H57–597) (Biolegend), PE–anti-ICOS (clone 15F9 and clone 7E.17G9) (BD Pharmingen), PE–Cy7–anti-PD1 (clone RMP1–30) (Biolegend), PerCP-Cy5.5 and Pacific Blue–anti-B220 (clone RA 3–6 B2) (BD Pharmingen), APC–Cy7–anti-IgD (clone 11–26c), PE–Cy7–anti-IgM (clone eB121–15F9) (ThermoFisher Scientific), APC and PE–anti-FAS (clone Jo2) (BD Pharmingen), FITC–anti-GL7 (clone Ly-77 and clone GL7) (BD Pharmingen), PE–Cy7-anti-CD38 (clone 90) (eBioscience), PE–Cy7–anti-CD73 (clone TY/11.8) (Biolegend), PE–anti-CD45.1 (clone A20) (BD Pharmingen), APC–anti-TCR Vα2 (clone b20.1) (eBioscience), and TNP–BSA-fluorescein (Biosearch Technologies).
For intracellular staining, we used FITC–anti-Foxp3 (FJK-16S) (ThermoFisher Scientific), PerCP–Cy5.5–anti-IgG (clone Poly45053) (Biolegend), PE–anti-IL17A (clone eBio17B7) (eBioscience), PE–Cy7–anti-IL4 (clone 11B11) (BD Pharmigen), and FITC–anti-INFγ (BD Pharmigen). FITC-anti–EPOR (clone 38407) (R&D Systems) was used to stain EPOR.
Data were acquired (at least 10,000 events, in most cases >100,000 events) on a three-laser Canto II flow cytometer (BD Biosciences) and analyzed using FlowJo (https://www.flowjo.com) software.
Real-time Quantitative RT-PCR
RNA was prepared from mouse CD4+ T cells using Trizol (Invitrogen). cDNA was synthesized using reverse transcription reagent (Applied Biosystems). Real-time PCR assays using the TaqMan universal PCR Master Mix and primer sets for mouse Bcl6 (Mm00477633_m1), Epor (Mm01202755_m1), and Gapdh (Mm99999915_g1) genes were purchased from ThermoFisher. PCR was performed on an Applied Biosystems 7500 Fast system. All experiments were performed at least in triplicate, and Bcl6 and Epor gene expression were normalized to housekeeping gene Gapdh.
Renal Histology
Mice were deeply anesthetized injecting a solution of sterile ketamine (200–300 mg/kg) and xylazine (20–30 mg/kg) in PBS, and transcardially perfused with 4% paraformaldehyde fixate in PBS at a rate of 8–10 ml/min. Kidneys were paraffin embedded (10%) or frozen in Optimal Cutting Temperature compound (Tissue-Tek O.C.T., Sakura) after soaking overnight in 18% sucrose in PBS.
Light Microscopy
Paraffin-embedded kidney sections (3 μm) were stained with periodic acid–Schiff. Images were acquired at random for at least 20 glomeruli per section. Imaging brightfield was performed on the widefield microscope (Zeiss AxioImager Z2M). Histologic scoring was performed in a blinded manner (by A. A.) according to a previously established method.18
Immunofluorescence
Cryosections (5 μm thick) were washed three times with PBS for 5 minutes, then incubated with blocking solution containing 5% BSA for 30 minutes at room temperature and stained for IgG (anti-mouse IgG antibody conjugated with Alexa Fluor 488, 1:500; Jackson Immunoresearch) or C3b (anti-mouse goat FITC-conjugated C3b, 1:100; Cappel, Cincinnati, OH) for 1 hour at room temperature. Nuclei were counterstained with DAPI (Invitrogen). At least 20 glomeruli per section for each animal were randomly acquired using a fluorescence nonconfocal laser scanning microscope (Zeiss AxioImager Z2M with ApoTome.2). IgG and C3b expression were estimated by constructing a contour mask on the monochrome image. Software ImageJ (National Institutes of Health) was used to quantify antibody intensity.
Urinary Albumin and Creatinine Measurement
Urine spot samples were collected from individual mice through gentle restraint and accrual of urine in a collection device. Urine creatinine was quantified using commercial kits from Cayman Chemical (Ann Harbor, MI). Urine albumin was determined using a commercial assay from Bethyl Laboratory Inc. (Montgomery, TX). Albuminuria was expressed as the ratio of urine albumin to creatinine.
BUN Measurement
Blood was collected by submandibular vein or cardiac puncture (terminal) into lithium-heparin–coated microcuvette tubes, 4 μl of plasma collected, and used for BUN measurement in duplicate using a Colorimetric Detection Kit, according to the manufacturer’s instructions (ThermoFisher Scientific, Waltham, MA).
Statistical Analyses
Two group comparisons were analyzed by unpaired Mann–Whitney test or t tests. For histologic score comparisons, we used Wilcoxon test. A two-way repeated-measures ANOVA test was used for multiple comparisons among treatment groups. P values <0.05 were considered significant. All statistical analyses were performed using GraphPad Prism (version 8 for Windows, GraphPad Software, Inc.).
Results
EPO Inhibits T Cell–dependent but Not T-independent Humoral Immunity
We initially tested the effects of administering recombinant EPO on T cell–dependent antibody formation after immunization with the model antigen NP, a hapten, linked to KLH (Figure 1A). We used an NP-ELISA assay16 that detects total (anti NP[27]) or high affinity (anti NP[2]) NP-reactive IgG by varying the degree of NP conjugation. These assays showed that recombinant EPO administration reduced total (Figure 1B) and high-affinity (Figure 1C) NP-reactive IgG by approximately three-fold, the latter indicative of reduced affinity maturation. To next test whether EPO similarly affects a more potent polyclonal antibody response, we transplanted B6 recipients with MHC-disparate BALB/c heart allografts, treated the animals with recombinant EPO (or saline as a control) and quantified serum antidonor class I MHC antibodies 14 days later (Figure 1D). These assays showed that EPO treatment reduced the donor-reactive alloantibody titer by approximately three-fold, with significantly less IgG binding to the allogeneic target cells at all serum concentrations (Figure 1, E–G). As a specificity control, we measured serum from vehicle- and EPO-treated animals for reactivity to third-party target cells (H-2k) and observed none (Supplemental Figure 1).
Figure 1.
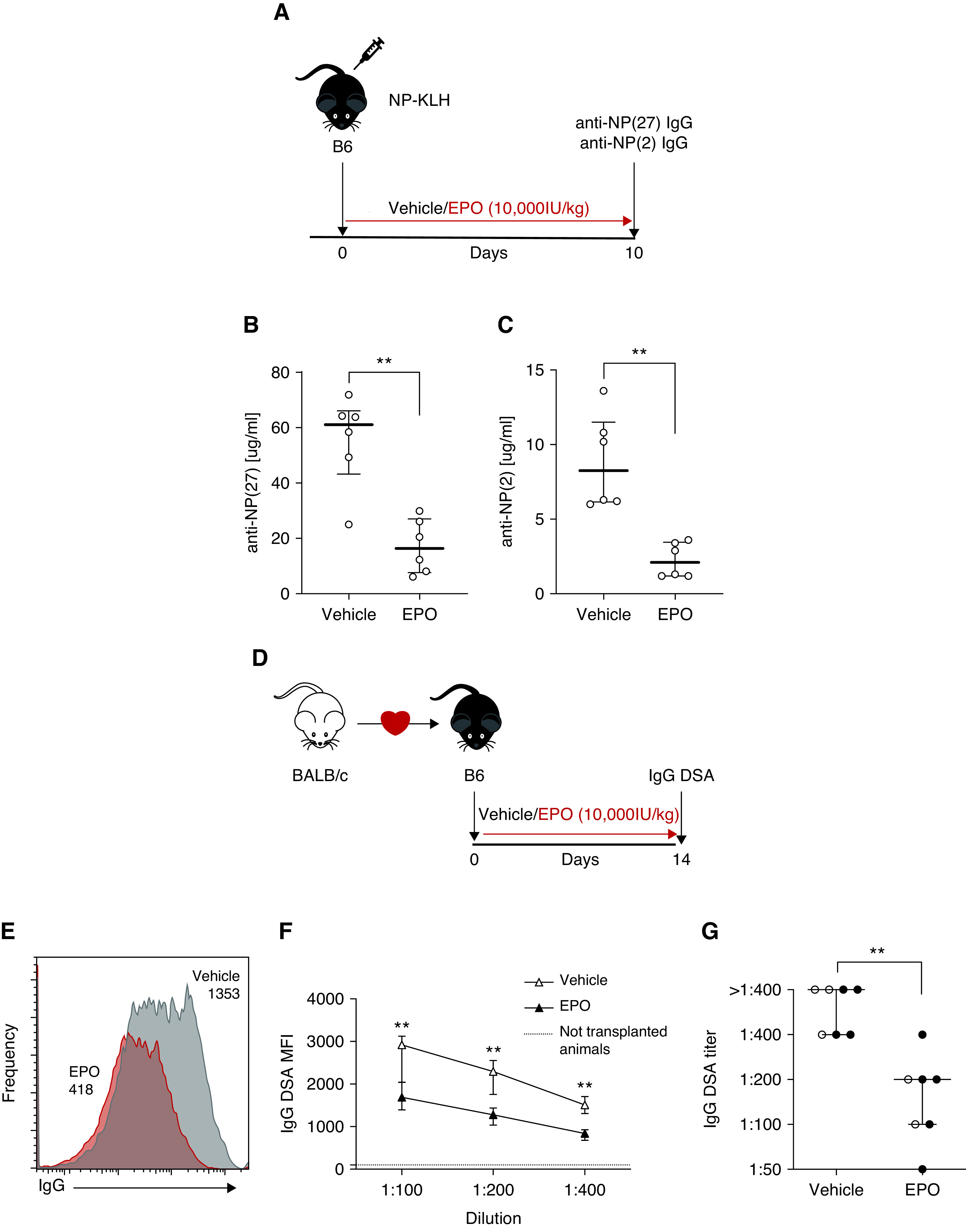
EPO reduces antibody formation after NP-KLH immunization and heart transplantation in mice. (A) Experimental design: B6 mice were immunized with NP-KLH on day 0 and treated with EPO (10,000 IU/kg, i.p.) or vehicle control starting from day 0 to day 10, when the animals were euthanized. (B) Total and (C) high affinity anti-NP antibodies were quantified by ELISA using BSA with different degrees of NP conjugation. (D) B6 recipients of an allogeneic heterotopic transplantation from BALB/c mice were treated with EPO (10,000 IU/kg, i.p.) or vehicle control from the day of transplant up to day 14 post-transplant, when the animals were euthanized. (E)–(G) Representative plots of donor-specific antibodies (DSA) IgG (E), MFI levels (F), and titers (G) in EPO- and vehicle-treated mice. Data represent median±interquartile range (IQR). Empty circles represent male animals, full circles females. **P<0.01 between EPO and vehicle. Data were analyzed using unpaired Mann–Whitney test.
In an effort to explain the above effects of EPO on serum antibodies, we used flow cytometry to quantify splenic B220+Fas+GL7+ GC B cells and B220+IgD-IgM- class-switched B cells on day 10 postimmunization with NP-KLH in groups of EPO-treated and control animals. These assays showed that EPO administration significantly reduced relative and absolute frequencies of total GC B cells (Figure 2, A–C) and NP-specific GC B cells (Figure 2, D–F). We observed similar reduced percentages and absolute numbers of GC B cells and class switched B cells in EPO-treated recipients of allogeneic heart transplants (Figure 2, G–L).
Figure 2.
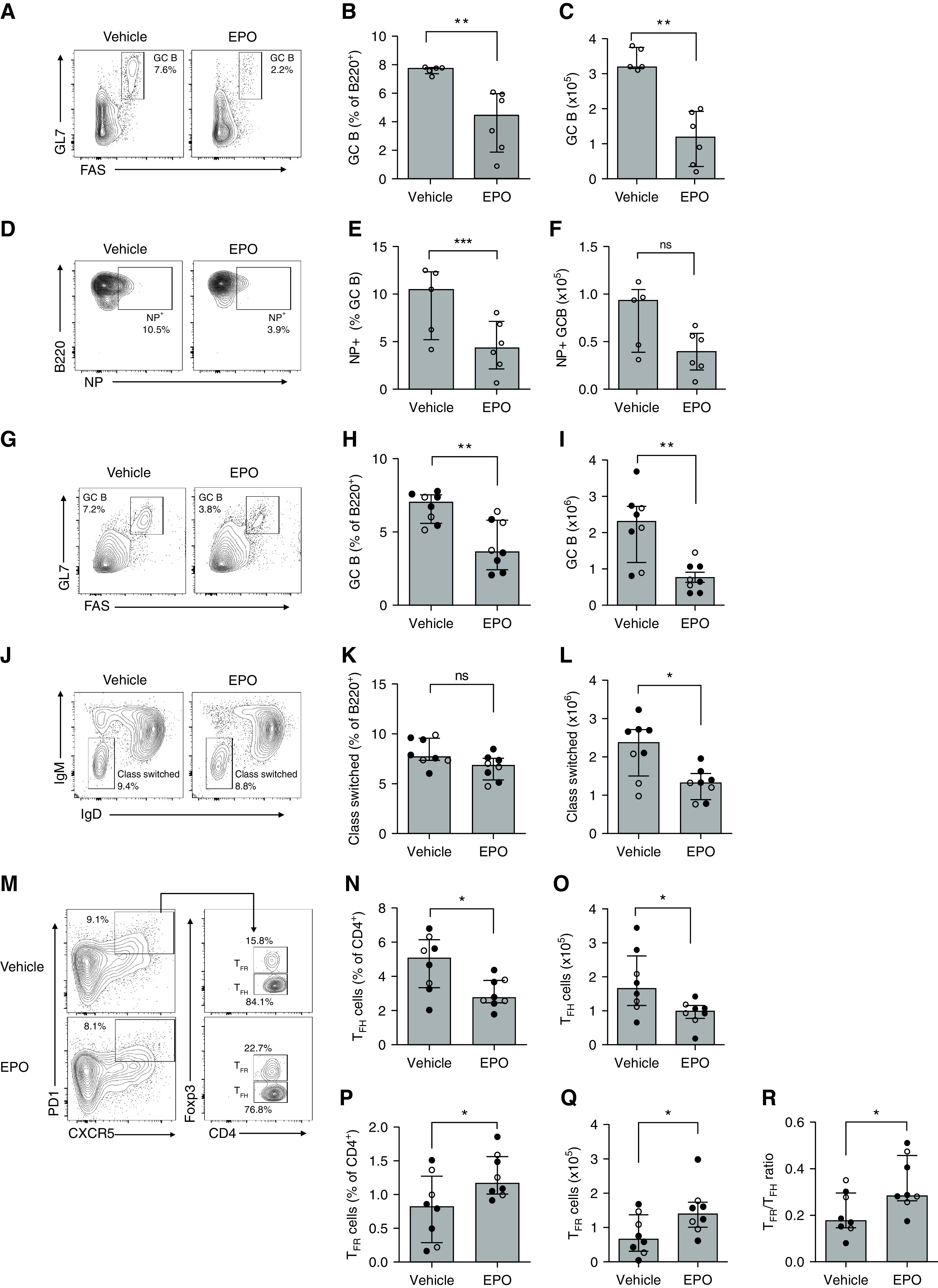
EPO reduces GC and class-switched B cells after NP-KLH immunization and heart transplantation in mice. (A)–(F) Refer to Figure 1A for the experimental design. Representative plots depicting percentages of GC (A) and B220+ NP+ B cells (D) at sacrifice. Quantified percentages (B) and (E) and total numbers (C), (F) of GC and B220+ NP+ GC B cells, respectively. (G)–(L) See Figure 1D for the experimental design. Representative plots depicting percentages of B220+Fas+GL7+ germinal center (GC) (G) and B220+IgM-IgD- class-switched (J) B cells at sacrifice. Quantified percentages (H), (K) and total numbers (I), (L) of GC and class-switched B cells, respectively. Representative plots (M) and data quantification for CD4+PD1+CXCR5+Foxp3- TFH (N), (O), CD4+PD1+CXCR5+Foxp3+ TFR (P), (Q), and TFH/TFR ratio (R) in the same mice at the time of sacrifice. Data represent median±IQR. Empty circles represent male animals, full circles females. *P<0.05; **P<0.01; ***P<0.001; ns: not significant between EPO and vehicle. Data were analyzed using unpaired Mann–Whitney test.
When we quantified total splenic CD4+CXCR5+PD1+ cells (TFH + TFR) in the same transplanted animals, we found lower absolute numbers in the EPO group (Supplemental Figure 2). We next analyzed CD4+CXCR5+PD1+Foxp3- TFH and CD4+CXCR5+PD1+Foxp3+ TFR cells in groups of EPO-treated and control animals immunized with NP-KLH. The assays showed that EPO therapy reduced percentages and absolute numbers of TFH (Figure 2, M–O), and increased percentages and absolute numbers of TFR (Figure 2, M and P–Q), together resulting in increased TFR/TFH ratios (Figure 2R).
To test the effects of EPO administration on T cell–independent antibody production we immunized B6 wild-type (WT) mice with the hapten TNP conjugated to aminoethyl-carboxymethyl-Ficoll, a high molecular-weight polysaccharide that promotes T–independent antibody production,19 and treated them with recombinant EPO or saline control (Figure 3A). Analyses on day 10 postimmunization showed no differences in the percentages of TNP-specific B cells between EPO-treated and control mice (Figure 3, B and C). Nor did we observe an effect of EPO on serum TNP-reactive IgM or IgG (Figure 3, D and E), together indicating that EPO does not directly inhibit B cells in the absence of T cells. In further verification of this notion, when we stimulated purified B cells in culture with anti-IgM Fab2 and varying concentrations of LPS for 48 hours, we observed that LPS increased the IgD- fraction (Figure 3F) and the overall percentages of viable B cells (Figure 3G) in a dose-dependent manner, but addition of EPO had no effect.
Figure 3.
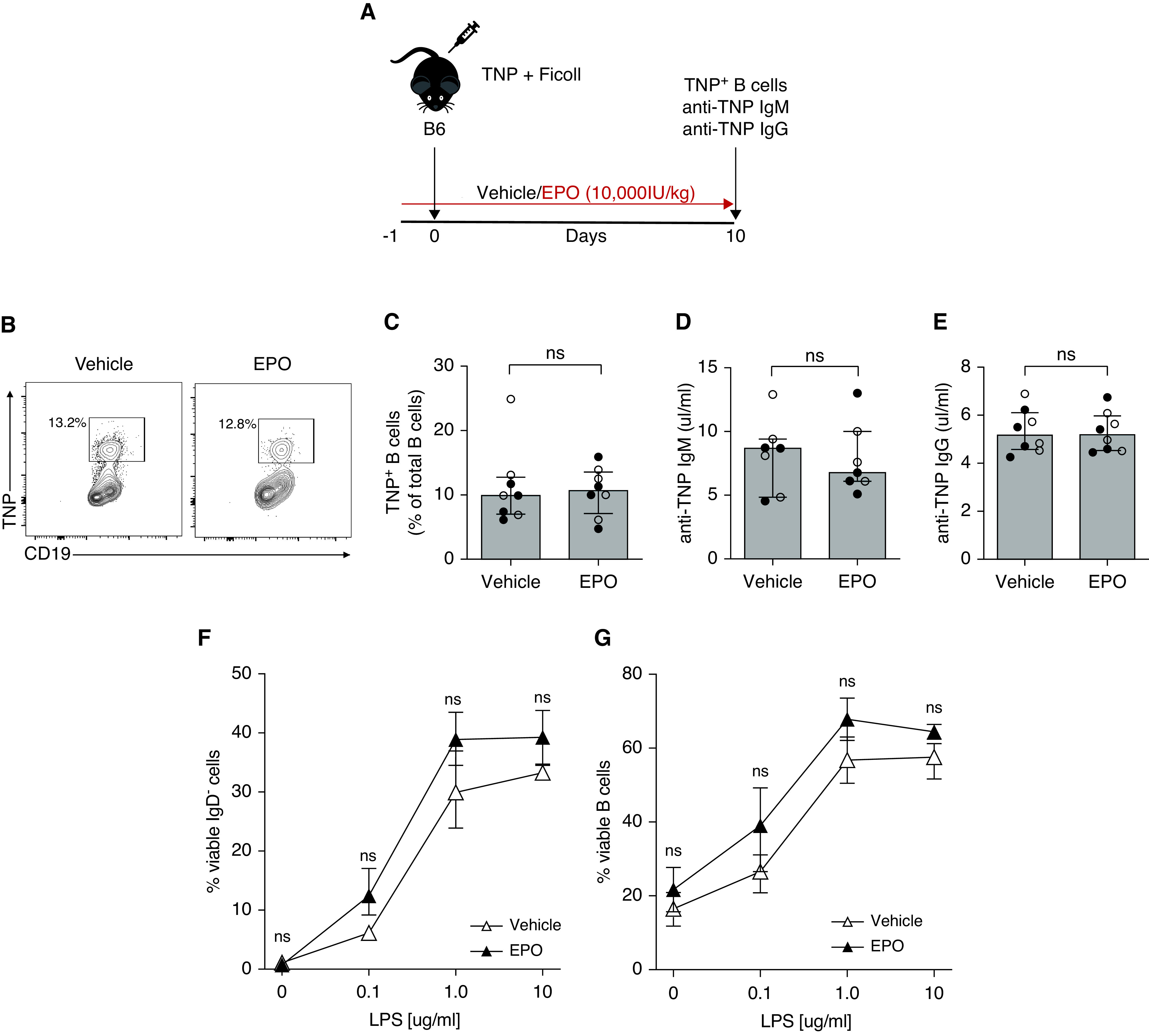
EPO does not directly inhibit B cells. (A) WT B6 mice were immunized with 2,4,6-trinitrophenyl-aminoethylcarboxymethyl-Ficoll and treated with EPO (10,000 IU/kg i.p.) or vehicle control from day -1 to day 10. At day 10, we euthanized the mice to measure TNP-specific B cells and anti-TNP IgM and IgG. Representative plots (B) and data quantification (C) for TNP+CD19+ B cells. Anti-TNP IgM (D) and IgG (E) in sera collected from the same mice at sacrifice. (F), (G) B6 enriched splenic B cells were stimulated with 1 ug/ml anti-IgM Fab2 and increasing LPS concentrations (0.1, 1.0, or 10 µg/ml) and incubated for 2 days at 37°C with EPO (5000 IU/ml) or vehicle control. Quantified percentage of IgD- B cells (F) and total viable B cells (G). Data represent median±IQR. Empty circles represent male animals, full circles represent females. Data were analyzed using unpaired Mann–Whitney test.
EPO Inhibits Humoral Immunity Independent of B Cell-expressed EPOR
Building on our findings that EPO inhibits T cell–dependent but not T–independent antibody formation, we tested whether EPO/EPOR signaling directly on B cells affects humoral immunity using conditional knockout mice. We generated EPORfl/fl CD19-CrePOS mice, in which CD19+ B cells selectively lack EPOR expression (Supplemental Figure 3). We then immunized EPORfl/fl and EPORfl/fl CD19-CrePOS mice with SRBC, a potent polyclonal stimulus that elicits a T cell–dependent GC response, but does not require adjuvant,20 and treated them with EPO or vehicle control (Figure 4A). Then 12 days after immunization, our analyses showed that absence of B cell–expressed EPOR had no effect: EPO administration equivalently reduced GC B cells (Figure 4, B and C) and TFH (Figure 4, D and E), and increased TFR (Figure 4F) and TFR/TFH ratios (Figure 4G) in EPORfl/fl CD19-CrePOS and control EPORfl/fl mice compared with control immunized, vehicle-treated WT animals.
Figure 4.
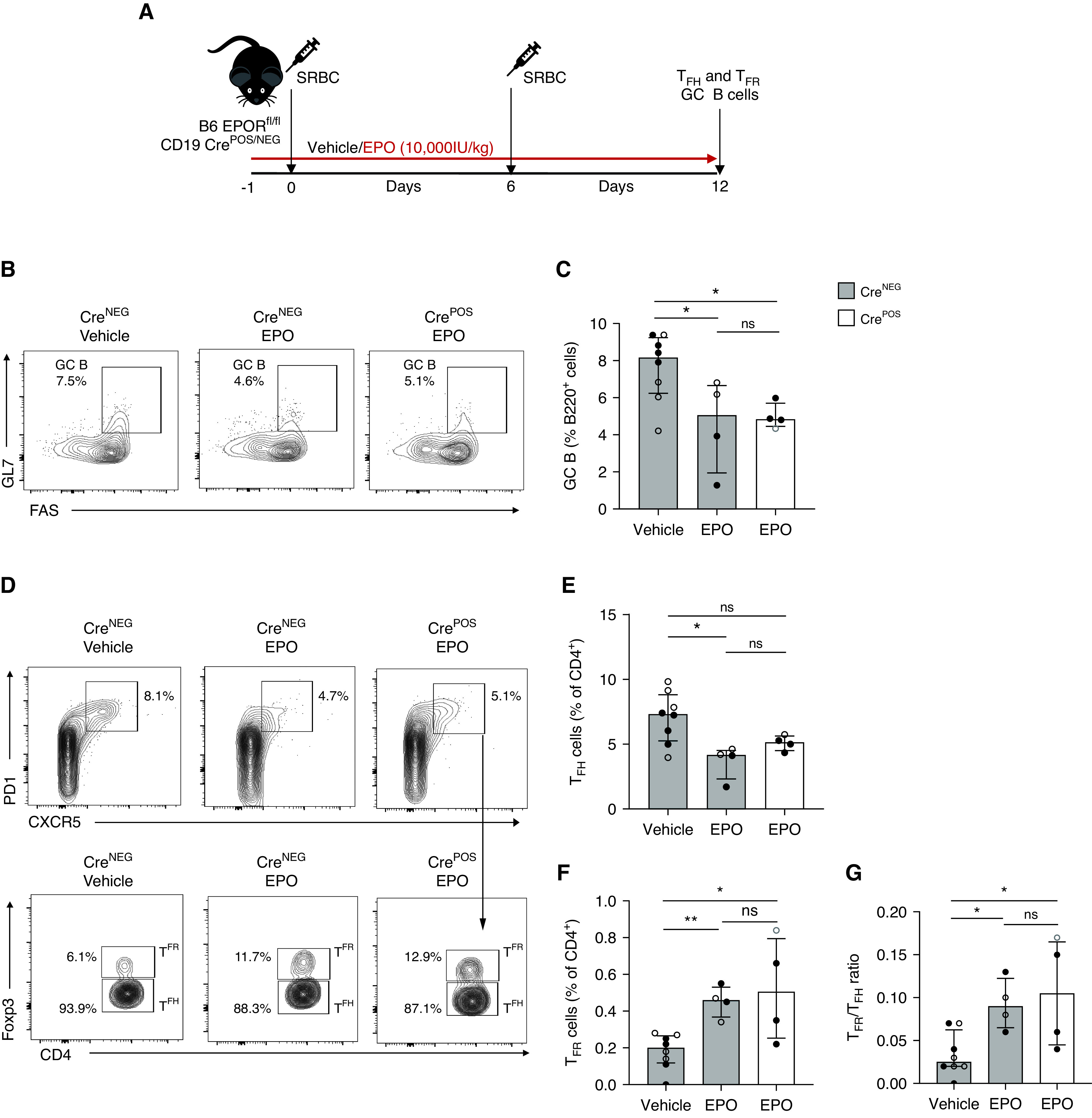
EPO reduces anti-sheep red blood cell (SRBC) antibody production in mice lacking EPOR on B cells. (A) EPORfl/fl CD19-CrePOS and CD19-CreNEG mice were immunized with SRBC on day 0 and 6 and treated with EPO (10,000 IU/kg i.p.) or vehicle control starting from day -1 to day 12. At day 12, we euthanized the mice to measure GC B cells, TFH, and TFR cells. Representative plots (B) and data quantification (C) for B220+Fas+GL7+ GC B cells. Representative plots (D) and data quantification for CD4+PD1+CXCR5+Foxp3- TFH (E), CD4+PD1+CXCR5+Foxp3+ TFR (F), and TFH/TFR ratio (G) in the same mice at the time of sacrifice. Data represent median±IQR. Empty circles represent male animals, full circles represent females. *P<0.05; **P<0.01. Data were analyzed using unpaired Mann–Whitney test.
EPO/EPOR Ligations on T Cells Inhibit TFH Production
We next tested whether EPO limits T cell–dependent B cell responses via preventing TFH differentiation/expansion using EPORfl/fl CD4-CrePOS mice that we generated and verified previously.10 In these mice, EPOR expression is selectively absent in T cells, including CD4+CXCR5+PD1+ TFH cells (Supplemental Figure 3). We immunized EPORfl/fl CD4-CrePOS and control EPORfl/fl mice with SRBC, treated them with EPO or vehicle, and analyzed GC responses by flow cytometry 12 days later (Figure 5A). The absence of T cell–expressed EPOR fully prevented EPO’s inhibitory effects on GC B cells (Figure 5, B–D), class-switched B cells (Figure 5, E–G), and TFH (Figure 5, H–J), and limited the EPO-induced increase in TFR (Figure 5, H and K–M). When we transplanted EPORfl/fl CD4-CrePOS and control EPORfl/fl mice with MHC-disparate BALB/c hearts, we similarly observed that EPO administration had no effect on humoral alloimmunity in the EPORfl/fl CD4-CrePOS recipients (Supplemental Figure 4A). The analyses also showed that EPO reduced donor-reactive alloantibodies (Supplemental Figure 4, B–D), GC B cells (Supplemental Figure 4, E–G), class-switched B cells (Supplemental Figure 4, H–J), and TFH with increased TFR (Supplemental Figure 4, K–P) in EPORfl/fl recipients, but not in EPORfl/fl CD4-CrePOS mice. Together, the data support the conclusion that EPO inhibits humoral immunity via a TFH-dependent mechanism.
Figure 5.
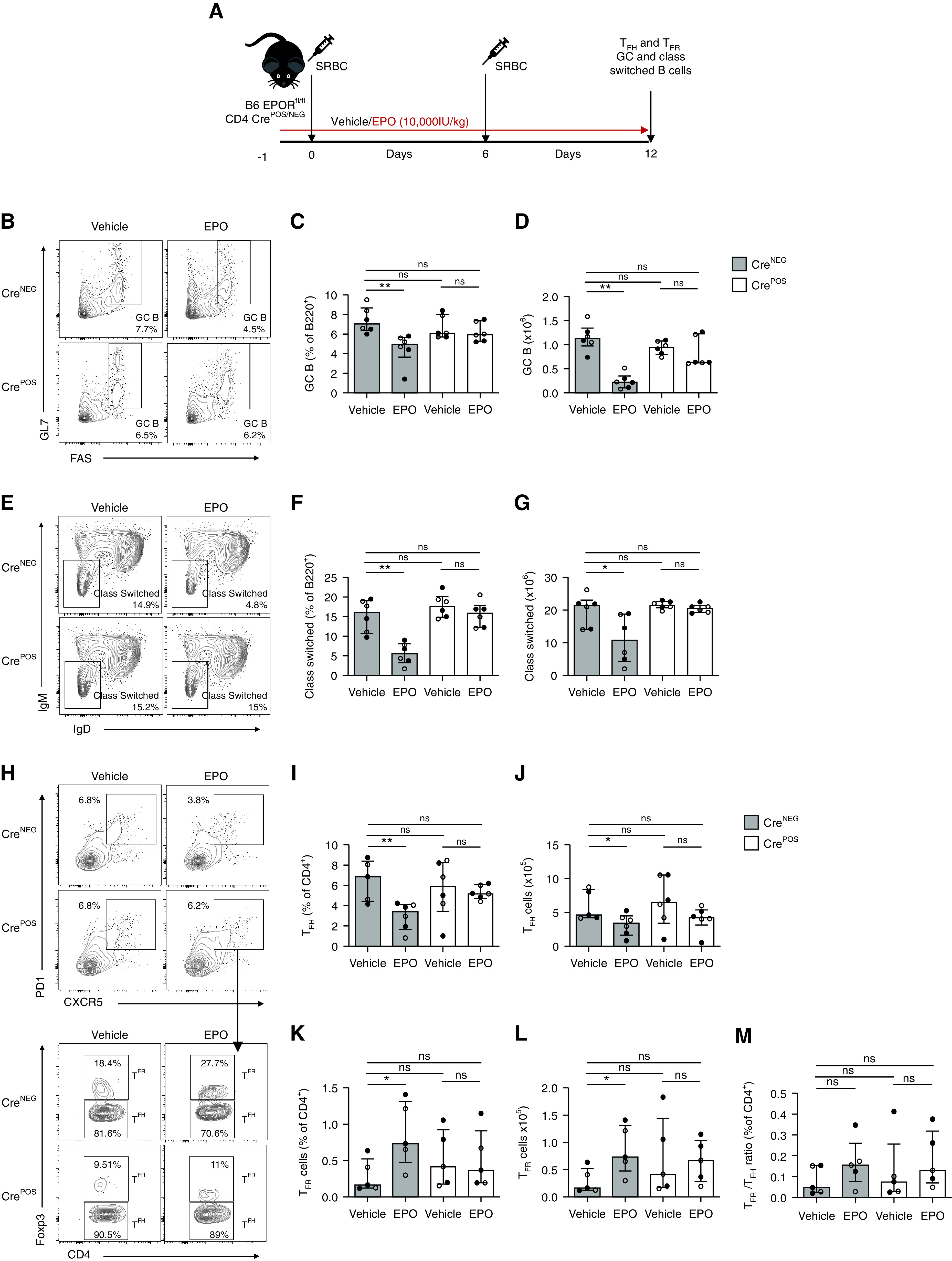
EPO does not affect anti-SRBC antibody production in mice lacking EPOR on T cells. (A) EPORfl/fl CD4-CrePOS and CD4-CreNEG mice were immunized with SRBC on day 0 and 6 and treated with EPO (10,000 IU/kg i.p.) or vehicle control starting from day -1 to day 12. At day 12, we euthanized the mice to measure TFH, TFR, GC, and class switched B cells. (B)–(G) Representative plots (B), (E) and data quantification for GC (C), (D) and class switched (F), (G) B cells. (H)–(M) Representative plots (H) and data quantification for CD4+PD1+CXCR5+Foxp3- TFH (I), (J), CD4+PD1+CXCR5+Foxp3+ TFR (K), (L), and TFH/TFR ratio (M). Data represent median±IQR. Empty circles represent male animals, full circles represent females. *P<0.05; **P<0.01. Data were analyzed using unpaired Mann–Whitney test.
EPO Inhibits TFH Induction and Expansion In Part via Upregulating STAT5
TFH cells differentiate from naïve CD4+ T cells through a multistep process involving IL-6 and IL-21 leading to STAT3 phosphorylation and transcription of transcriptional repressor Bcl6.21 To test the hypothesis that reduced TFH percentages and numbers in EPO-treated animals occur by inhibiting their induction, we stimulated murine naïve CD4+ T cells via their T cell receptor (TCR) in the presence of IL-6, while simultaneously blocking TGF-β, IL-4, and IFN-γ for 5 days, an in vitro system shown by our group and others to induce IL-21–producing CD4+ T cells.11,22 These experiments showed that EPO significantly reduced IL-21 production by murine CD4+ T cells (Figure 6, A and B).
Figure 6.
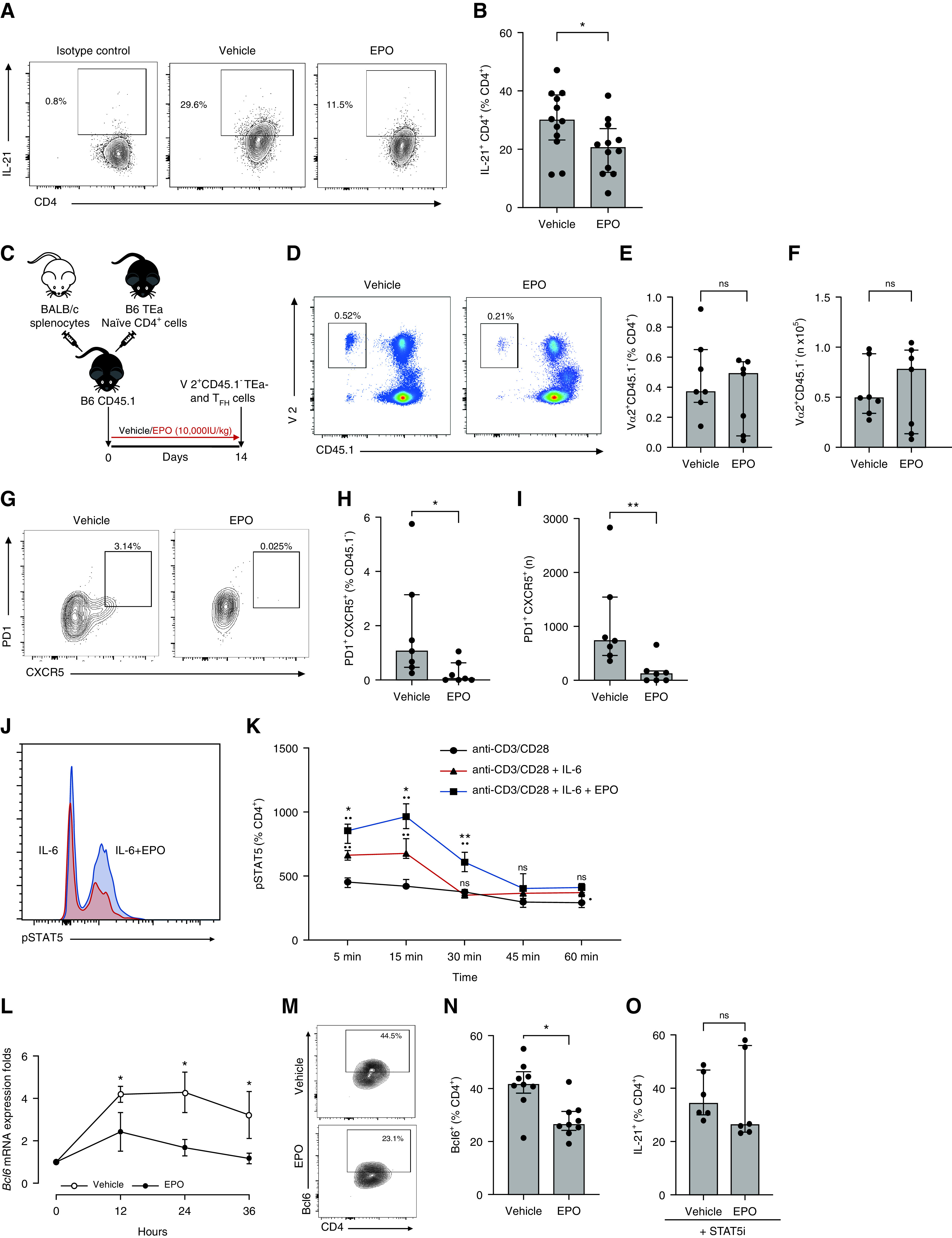
EPO inhibits TFH induction in vitro and in vivo through a STAT5-mediated mechanism. Representative plots (A) for IL-21–producing CD4+ T cells and data quantification (B) of naïve B6 CD62LhiCD44loCD4+ T cells from WT B6 mice cultured under TFH cell–inducing conditions for 5 days in the presence or absence of EPO. Gate was positioned on the basis of isotype control. IL-21+ MFI for vehicle versus EPO: 1213.8±374 versus 901.5±288), *P=0.03; (C) Experimental design: CD4+Vα2+CD45.1- TEa cells were injected into congenic CD45.1 B6 mice, which were immunized with allogeneic BALB/c splenocytes, and treated for 14 days with EPO or vehicle control, then sacrificed. Representative plots (D) and data quantification (E), (F) for total Vα2+CD45.1- at sacrifice. Representative plots (G) and data quantification (H), (I) for CD45.1-PD1+CXCR5+Foxp3- TFH in the same mice. Representative flow plot (J) and data quantification (K) for CD4+pSTAT5+ T cells from WT B6 mice cultured under TFH cell–inducing conditions for 5 days in presence or absence of EPO. Data are from five independent experiments with different donors (median±IQR) *P<0.01 **P<0.0001: the black dot (•) shows comparison versus anti-CD3/CD28 stimulation, asterisk (*) comparison between anti-CD3/CD28 + IL-6 and anti-CD3/CD28 + IL-6 + EPO at the same time point (two-way ANOVA). Time course of Bcl6 gene expression in naïve B6 CD62LhiCD44loCD4+ T cells from WT B6 mice cultured under TFH cell–inducing conditions for 5 days in the presence or absence of EPO (L). Representative plots (M) and data quantification (N) for CD4+BCL6+ T cells from WT B6 mice cultured under TFH cell–inducing conditions for 2 days in presence or absence of EPO. The same quantification at day 0 resulted in absence of Bcl6 (not shown). Data quantification (O) of naïve B6 CD62LhiCD44loCD4+ T cells from WT B6 mice cultured under TFH cell–inducing conditions for 5 days in the presence of a STAT5 inhibitor (STAT5i) and with or without EPO. Each dot represents a different culture well. Data represent median±IQR. *P<0.05, **P<0.01. Data were analyzed using unpaired Mann–Whitney test.
To test the mechanism in vivo, we flow sorted naïve TCR-transgenic CD45.2, TEa CD4+, TCR variable region Vα2+ T cells (H-2b, CD4+ TCR reactive to I-Ab + I-Edα52–68), adoptively transferred them into CD45.1 congenic mice, and injected them with allogeneic BALB/c (H-2d) spleen cells ± recombinant EPO (Figure 6C). Analyses performed 2 weeks later showed that in vivo EPO administration resulted in similar numbers of detectable TCR Vα2+CD45.1- TEa cells (Figure 6, D–F) but with significantly fewer CXCR5+PD1+ TFH within the TEa gate (Figure 6, G–I).
STAT5 is a negative regulator of Bcl6 transcription.23–25 Because EPO/EPOR interaction leads to STAT5 activation by phosphorylation,8,9 we tested whether EPO inhibits Bcl6 transcription via a STAT5 dependent mechanism. We observed that EPO increased STAT5 phosphorylation in murine CD4+ T cells cultured under TFH polarizing conditions (Figure 6, J–K). When we measured the kinetics of Bcl6 gene transcription during differentiation of IL-21–producing CD4+ T cells±EPO, we observed reduced Bcl6 gene expression in the cultures containing EPO (Figure 6L). At 2 days after TFH induction, we documented that EPO reduced Bcl6 protein expression (Figure 6, M–N). To test for a causal link between EPO-induced STAT5 phosphorylation and reduced TFH induction, we induced TFH cells±EPO in the presence of a selective STAT5 inhibitor. These assays revealed EPO had no effect on TFH induction, supporting our working model that EPO inhibits TFH induction by promoting STAT5 phosphorylation, that in turn prevents Bcl6 transcription (Figure 6O).
To test how these mechanisms apply to human cells, we verified that human CD19+ B cells (Supplemental Figure 5A) and CD4+CXCR5+PD1+ TFH (Supplemental Figure 5B) express EPOR. When we tested the effect of EPO on induction of human IL-21+CD4+ T cells in vitro, we observed reduced IL-21 production in the EPO-treated cultures (Supplemental Figure 5, C and D), consistent with the murine findings.
EPO Inhibits The Function of Preformed TFH
To test whether EPO inhibits the function of preformed TFH, we used an in vitro system published by Sage and colleagues, in which in vivo differentiated TFH are tested for their ability to support differentiation of naïve B cells into IgM-IgD-GL7+IgG+ B cells.26 We flow-sorted CD4+CXCR5+PD1+ T cells obtained from groups of B6 EPORfl/fl CD4-CrePOS mice (lacking EPOR on T cells) and control EPORfl/fl B6 mice 1 week after immunization with BALB/c spleen cells (Figure 7A). We cultured them with enriched IgM+IgD+IgG- B cells from naïve mice along with anti-CD3, anti-IgM, and ±EPO or vehicle control. Then 48 hours later, we quantified percentages of B220+IgM-IgD-GL7+IgG+ B cells (Figure 7A).26 Consistent with previous publications,26 control wells containing WT TFH and no EPO showed >10% IgG+ B cells, whereas no IgG+ B cells developed in the absence of TFH (Figure 7, B and C). We observed significantly fewer IgG+ B cells in cultures containing WT TFH and EPO (Figure 7, B and C) but no difference in frequencies of IgG+ B cells in EPO-treated cultures EPOR-deficient TFH, together supporting the conclusion that EPO inhibits TFH function (Figure 7, D and E). Because the isolated CD4+CXCR5+PD1+ T cells used in these assays contain a small percentage of Foxp3+ TFR, we repeated the experiment using flow-sorted CD4+CXCR5+PD1+Foxp3-GFP- TFH isolated from immunized B6 Foxp3-GFP animals (Figure 7F). These assays showed fewer IgG+ B cells in EPO-treated wells (Figure 7, G and H), together confirming that EPO binding to TFH-expressed EPOR directly inhibits the function of preformed TFH.
Figure 7.

EPO directly inhibits inhibitory TFH function independent of TFR. (A) B6 mice received allogeneic immunization with BALB/c splenocytes (20 × 106, i.p.), and after 7 days the animals were euthanized. BALB/c enriched splenic B cells and preimmunized B6 enriched splenic CD4+PD1+CXCR5+ TFH–TFR were stimulated with anti-IgM (10 µg/ml) and anti-CD3 (2 µg/ml). Cells were treated with EPO (1000 IU/ml) or vehicle control, incubated for 2 days at 37°C, and collected to measure IgG+ B cells. Representative plots (B) and quantified percentages (C) of B220+ IgM-IgD- IgG+ B cells from BALB/c B cells cultured alone or with TFH and TFR from preimmunized B6, in the presence of EPO or vehicle. (D)–(E) Same experiment represented in (A), done with EPORfl/flCD4-CrePOS and CD19-CreNEG mice, showing (D) representative plots and (E) quantified percentages of B220+ IgM-IgD- IgG+ B cells. (F) B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J mice received allogeneic immunization with BALB/c splenocytes (20 × 106, i.p.); after 7 days the animals were euthanized, and CD4+PD1+CXCR5+Foxp3- TFH cells were flow sorted. BALB/c enriched splenic B cells and preimmunized B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J TFH were stimulated with anti-IgM (10 µg/ml) and anti-CD3 (2 µg/ml). Cells were treated with EPO (1000 IU/ml) or vehicle control, incubated for two days at 37°C, and collected to measure IgG+ B cells. Representative plots (G) and quantified percentages (H) of B220+ IgM-IgD- IgG+ B cells from BALB/c B cells cultured with preimmunized B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J TFH in the presence of EPO or vehicle control. Dashed line represents the percentage value of B220+ IgM-IgD- IgG+ B cells in a culture with BALB/c B cells and B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J TFH–TFR treated with vehicle control as reference value. Each dot represents a different culture well. Experiments were repeated at least three times. Data represent median ± IQR. *P<0.05; ns: not significant between EPO and vehicle. Data were analyzed using an unpaired t test.
EPO’s Inhibitory Effects Reduce The Pathogenicity of T Cell–dependent Antibody Production In Multiple Disease Models
To assess the clinical significance of the observed effects, we sensitized B6 mice with allogeneic BALB/c splenocytes and quantified serum donor class II–reactive antibodies 2 weeks later. We then allocated mice with similar antibody titers to receive treatment with either EPO or vehicle as a control (Supplemental Figure 6, A–C). The next day we transplanted the sensitized mice with BALB/c hearts and quantified serum alloantibodies 14 days later. These analyses showed that EPO treatment led to significantly lower serum donor-reactive antibody titers versus control-treated animals (Supplemental Figure 6, D and E). EPO-treated animals also showed fewer GC B cells (Supplemental Figure 6, F–H), class-switched B cells (Supplemental Figure 6, I–K), TFH (Supplemental Figure 6, L–N), along with increases in TFR (Supplemental Figure 6, O–Q), together indicating that EPO blunts humoral immunity in sensitized animals. Of note, we observed similar protective effects in male and female recipients.
We next assessed the effects of EPO on the parent-to-F1 model of graft versus host disease (GvHD)27 induced by injecting CD8-depleted B6 spleen cells into nonirradiated semiallogeneic B6xBALB/c (H-2b/d) F1 recipients. In this system, alloreactive donor CD4+ T cells expand and differentiate into TFH cells, with associated induction of GC B cells and production of autoantibodies, including anti-dsDNA antibodies.27 Using this established model, we treated groups of recipients with EPO or vehicle control and quantified frequencies of host-derived TFH. These analyses showed significantly fewer TFH in EPO-treated animals (Supplemental Figure 7, A and B). Further phenotyping showed fewer GC B cells (Supplemental Figure 7, C and D) and lower serum anti-dsDNA antibody levels in EPO-treated animals (Supplemental Figure 7E), together supporting the conclusion that EPO inhibits TFH dependent B cell activation in this GvHD model.
Because the B6 into (bxd)F1 model does not routinely result in a clinical disease phenotype, we performed separate studies in which we administered EPO or vehicle to B6xDBA F1 (H-2b/d) recipients of DBA/2J (DBA) splenocytes (H-2d), a model in which GvHD results in autoantibody formation and lupus-like glomerulonephritis.15 These studies showed that EPO-treated mice contained fewer donor-derived TFH and recipient GC B cells (Figure 8, A and B), lower levels of anti-dsDNA (Figure 8C) and significantly diminished kidney disease severity, the latter documented by diminished glomerular deposition of IgG (Figure 8, D and E) and C3b (Figure 8, F and G) 6 weeks after disease initiation, lower glomerular and tubule-interstitial scores (Figure 8, I and J), and lower BUN and albuminuria (Figure 8, K and L).
Figure 8.

EPO reduces TFH cell induction, GC B cells formation, and anti-dsDNA formation in the B6 into B6 x DBA parent to F1 model of lupus. B6 spleen cells were injected into B6xDBA (bxd) recipients that were treated with EPO (10,000 IU/kg/day, i.p.) or vehicle control until euthanasia on week 10 after injection. Percentages of TFH (A) and GC B cells (B) at sacrifice. Anti-dsDNA autoantibodies in sera collected from the same mice at sacrifice (C). Representative images and quantification of IgG (D), (E) and C3b (F), (G) glomerular deposition in staining of kidney tissues. Original magnification ×20. Differences in IgG (G) and C3b (I) glomerular fluorescent intensity between EPO- and vehicle-treated animals were quantified relative to DAPI using MetaMorph software. At least 20 glomeruli from three animals were included in the analysis. Scale bars: 10 μm. Representative images of periodic acid–Schiff staining of kidney tissue (H). Quantification of glomerular (I) and tubule-interstitial (J) scores, and BUN (K). Albuminuria (L) expressed as the ratio of urine albumin to creatinine. Data represent median±IQR. *P<0.05; **P<0.01. Data were analyzed using unpaired Mann–Whitney test.
Discussion
T cell–dependent, antibody isotype switch recombination and affinity maturation are crucial processes underlying effective antipathogen immunity, but also drive production of autoreactive and alloreactive IgG in many disease states, including autoimmune glomerular diseases and transplant rejection.1,2 Although accumulating data by multiple groups over the past several decades have deciphered the cellular and molecular events involved in these processes, the insights have had a limited effect on therapeutic strategies aimed at reducing pathogenic antibodies to prevent or treat disease.1,2 Herein, we demonstrate the unexpected observation that EPO, a physiologically produced hormone required for red blood cell formation, and commonly used as a therapy for anemia in many clinical scenarios, remarkably blunts development of humoral immunity including pathogenic autoantibodies and alloantibodies. Our cumulative data indicate the mechanism implicates EPO/EPOR ligations on T cells but not on B cells, reducing TFH induction and function required for positive selection of high affinity B cell clones within GC reactions. Although our in vivo data are limited by the fact that our EPOR conditional KO animals had no EPOR expression on all T cells (not selectively on TFH cells), our in vitro experiments strongly support our working model.
We showed herein and in previous publications8,9 that EPOR is expressed by B and T cells, including TFH cells, in humans and mice, consistent with large dataset data.28 Our present data indicate that EPO-induced inhibition of TFH blocks both naïve and memory antibody responses. Long-lived humoral memory immunity is particularly problematic in the context of transplantation (antidonor alloantibodies cause accelerated allograft failure), and in multiple autoimmune diseases. Our findings thus identify previously unrecognized therapeutic target of potential clinical significance, supporting the need for testing EPO/EPOR blockade as treatment strategy for diseases in which ongoing humoral immunity is thought to be pathogenic. Intriguingly, our findings suggest an alternative interpretation of prior data showing that EPO treatment associates with improved graft survival in kidney transplant recipients29 and improved muscular function in patients with rheumatoid arthritis.30 These effects, previously attributed largely to the improved tissue oxygenation induced by EPO, might indeed be at least in part due to the immune modulating properties of this hormone.
Beyond the clinical implications, although others demonstrated that EPO enhances macrophage-mediated clearance of apoptotic bodies and diminishes clinical expression of murine glomerulonephritis,31 our findings suggest that additionally (and perhaps alternatively), the mechanism of EPO’s therapeutic effect is via blockade of TFH-dependent antibody production and affinity maturation.
Bcl6 is a transcription factor required for TFH induction and function, and mice with Bcl6-deficient T cells are not capable of generating TFH cells in response to infections.32 In activated CD4+ T cells, IL-6 and IL-21 promote STAT3 phosphorylation that promotes TFH cell differentiation via upregulation of Bcl6. STAT5, in contrast, represses Bcl6 expression by displacing STAT3 from one of the two regulatory regions to which it binds the Bcl6 gene. Importantly, the repression of Bcl6 mediated by STAT5 is dominant over STAT3-mediated induction.23–25 Because EPO/EPOR interaction leads to STAT5 phosphorylation,8 we tested the hypothesis that EPO inhibitory effects on TFH induction and function are STAT5 dependent. Consistent with this idea, EPO-induced STAT5 phosphorylation was associated with reduced Bcl6 transcription and translation, and an antagonist of STAT5 phosphorylation abolished EPO effects on TFH induction. This is a previously unknown mechanism responsible for the immune modulating effects of EPO, distinct from previously described mechanisms including inhibition of IL-2–induced8 or RAR-related orphan receptor C (RORC)10 signaling in Th1 and Th17 cells, respectively.
The doses of recombinant human EPO used in our murine studies are approximately 20-fold higher than the ones currently used in the clinic to treat anemia. Although there is considerable homology between human and mouse EPO,33 the high doses required to treat mice might be related to the lower affinity of human EPO for murine EPOR. Consistently, the doses of recombinant human EPO required to inhibit TFH differentiation in vitro were significantly lower than the ones required to inhibit differentiation of murine TFH cells. Importantly, we have previously documented that physiologic EPO levels that are produced by the kidney are responsible for Treg induction8 and that even standard therapeutic doses of EPO exert immune modulating effects.8,34 However, we acknowledge that chronic EPO treatment may be associated with increased cardiovascular risk. The use of the nonerythropoietic agonists of heterodimer EPOR35 may represent a more effective strategy to prevent autoantibody or alloantibody formation, while avoiding the side effects of EPO on blood viscosity and platelet function. More studies are needed to assess the immune pharmacodynamic effects of EPOR agonists and how they are affected by the route of administration.
In summary, our results link EPO to TFH–B cell interaction and antibody production. Together with recent evidence that TFH cells likely participate in the pathophysiology of autoantibody and donor-specific antibodies formation in organ transplantation, and in a model of GvHD associated with the production of autoantibodies,36,37 our findings provide a rationale for future studies testing the hypothesis that targeting EPOR signaling38 can prevent and/or treat TFH cell–dependent disease processes, including autoimmune diseases and alloantibody formation in transplant recipients.
Supplementary Material
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Disclosures
E. Fiaccadori reports being a scientific advisor or member via the editorial boards of Blood Purification and the Journal of Nephrology, and reports other interests/relationships as a member of the Italian Society of Nephrology, and Member of the European Society of Parenteral and Enteral Nutrition. P.S. Heeger reports having consultancy agreements with Mallinckrodt Pharmaceuticals. P.T. Sage reports consultancy agreements with Merck, Nuwa, Sanofi, Takeda, and Velox; reports receiving research funding from Jounce, Merck, and Sanofi; and reports being a scientific advisor or member of Immunology Letters. All remaining authors have nothing to disclose.
Funding
This work is supported by the National Institute of Health (National Institute of Allergy and Infectious Diseases [NIAID]) R01 0255A141 (to P. Cravedi), and project 3 of P01 AI123086 ( to P.S. Heeger). A. Cumpelik was supported by a fellowship grant from the American Society of Transplantation.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2021010098/-/DCSupplemental.
Supplemental Figure 1. MFI levels of anti-donor and third-party antibodies in EPO- and vehicle treated B6 recipients of an allogenic transplantation from BALB/c mice .
Supplemental Figure 2. EPO reduces CD4+CXCR5+PD1+ T cells after heart transplantation in mice.
Supplemental Figure 3. EPOR expression in murine T and B cells.
Supplemental Figure 4. EPO does not inhibit formation of alloantibodies in mice lacking EPOR on T cells undergoing heart transplant.
Supplemental Figure 5. Human B and TFH cells express EPOR and EPO inhibits in vitro human TFH induction.
Supplemental Figure 6. EPO prevents DSA increase in presensitized heart transplant recipient mice.
Supplemental Figure 7. EPO prevents immunological changes in the B6 into B6 x BALB/c parent to F1 model.
References
- 1.Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L: Regulation of the germinal center response. Front Immunol 9: 2469, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Silva NS, Klein U: Dynamics of B cells in germinal centres. Nat Rev Immunol 15: 137–148, 2015. PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qi H: T follicular helper cells in space-time. Nat Rev Immunol 16: 612–625, 2016 [DOI] [PubMed] [Google Scholar]
- 4.Cyster JG, Allen CDC: B cell responses: Cell interaction dynamics and decisions. Cell 177: 524–540, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantarelli C, Angeletti A, Cravedi P: Erythropoietin, a multifaceted protein with innate and adaptive immune modulatory activity. Am J Transplant 19: 2407–2414, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spaan M, Groothuismink ZMA, Koning L, Roomer R, Janssen HLA, De Knegt RJ, et al. : Erythropoietin administration suppresses human monocyte function in vitro and during therapy-induced anemia in HCV patients. Antiviral Res 98: 469–475, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Liu B-S, Groothuismink ZMA, Janssen HLA, Boonstra A: Role for IL-10 in inducing functional impairment of monocytes upon TLR4 ligation in patients with chronic HCV infections. J Leukoc Biol 89: 981–988, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Purroy C, Fairchild RL, Tanaka T, Baldwin WM 3rd, Manrique J, Madsen JC, et al. : Erythropoietin receptor-mediated molecular crosstalk promotes T cell immunoregulation and transplant survival. J Am Soc Nephrol 28: 2377–2392, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cravedi P, Manrique J, Hanlon KE, Reid-Adam J, Brody J, Prathuangsuk P, et al. : Immunosuppressive effects of erythropoietin on human alloreactive T cells. J Am Soc Nephrol 25: 2003–2015, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donadei C, Angeletti A, Cantarelli C, D’Agati VD, La Manna G, Fiaccadori E, et al. : Erythropoietin inhibits SGK1-dependent TH17 induction and TH17-dependent kidney disease. JCI Insight 5: 1–19, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chun N, Fairchild RL, Li Y, Liu J, Zhang M, Baldwin WM 3rd, et al. : Complement dependence of murine costimulatory blockade-resistant cellular cardiac allograft rejection. Am J Transplant 17: 2810–2819, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raedler H, Vieyra MB, Leisman S, Lakhani P, Kwan W, Yang M, et al. : Anti-complement component C5 mAb synergizes with CTLA4Ig to inhibit alloreactive T cells and prolong cardiac allograft survival in mice. Am J Transplant 11: 1397–1406, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valujskikh A, Pantenburg B, Heeger PS: Primed allospecific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. Am J Transplant 2: 501–509, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Verghese DA, Chun N, Paz K, Fribourg M, Woodruff TM, Flynn R, et al. : C5aR1 regulates T follicular helper differentiation and chronic graft-versus-host disease bronchiolitis obliterans. JCI Insight 3: e124646, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lang TJ, Nguyen P, Papadimitriou JC, Via CS: Increased severity of murine lupus in female mice is due to enhanced expansion of pathogenic T cells. J Immunol 171: 5795–5801, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Le TV, Kim TH, Chaplin DD: Intraclonal competition inhibits the formation of high-affinity antibody-secreting cells. J Immunol 181: 6027–6037, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cravedi P, Lessman DA, Heeger PS: Eosinophils are not required for the induction and maintenance of an alloantibody response. Am J Transplant 13: 2696–2702, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bajema IM, Wilhelmus S, Alpers CE, Bruijn JA, Colvin RB, Cook HT, et al. : Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: Clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int 93: 789–796, 2018 [DOI] [PubMed] [Google Scholar]
- 19.Hess C, Winkler A, Lorenz AK, Holecska V, Blanchard V, Eiglmeier S, et al. : T cell-independent B cell activation induces immunosuppressive sialylated IgG antibodies. J Clin Invest 123: 3788–3796, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boraschi D, Villa L, Volpini G, Bossù P, Censini S, Ghiara P, et al. : Differential activity of interleukin 1 α and interleukin 1 β in the stimulation of the immune response in vivo. Eur J Immunol 20: 317–321, 1990 [DOI] [PubMed] [Google Scholar]
- 21.Crotty S: T follicular helper cell differentiation, function, and roles in disease. Immunity 41: 529-542, 2014 [DOI] [PMC free article] [PubMed]
- 22.Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. : mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity 45: 540–554, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S: STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med 209: 243–250, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker SR, Nelson EA, Yeh JE, Pinello L, Yuan G-C, Frank DA: STAT5 outcompetes STAT3 to regulate the expression of the oncogenic transcriptional modulator BCL6. Mol Cell Biol 33: 2879–2890, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. : Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol 12: 247–254, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clement RL, Daccache J, Mohammed MT, Diallo A, Blazar BR, Kuchroo VK, et al. : Follicular regulatory T cells control humoral and allergic immunity by restraining early B cell responses. Nat Immunol 20: 1360–1371, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puliaev RA, Puliaeva IA, Ryan AE, Via CS: The patient-into-F1 model of graft-vs-host disease as a model of in vivo T cell function and immunomodulation. Curr Med Chem Immunol Endocr Metab Agents 5: 575–583, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Immunological Genome Project. Available at: https://www.immgen.org/. Accessed 21 May, 2021
- 29.Choukroun G, Kamar N, Dussol B, Etienne I, Cassuto-Viguier E, Toupance O, et al. ; CAPRIT study Investigators: Correction of postkidney transplant anemia reduces progression of allograft nephropathy. J Am Soc Nephrol 23: 360–368, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaltwasser JP, Kessler U, Gottschalk R, Stucki G, Möller B: Effect of recombinant human erythropoietin and intravenous iron on anemia and disease activity in rheumatoid arthritis. J Rheumatol 28: 2430–2436, 2001 [PubMed] [Google Scholar]
- 31.Luo B, Gan W, Liu Z, Shen Z, Wang J, Shi R, et al. : Erythropoeitin signaling in macrophages promotes dying cell clearance and immune tolerance. Immunity 44: 287–302, 2016 [DOI] [PubMed] [Google Scholar]
- 32.Nance JP, Bélanger S, Johnston RJ, Hu JK, Takemori T, Crotty S: Bcl6 middle domain repressor function is required for T follicular helper cell differentiation and utilizes the corepressor MTA3. Proc Natl Acad Sci U S A 112: 13324–13329, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galson DL, Tan CC, Ratcliffe PJ, Bunn HF: Comparison of the human and mouse erythropoietin genes shows extensive homology in the flanking regions. Blood 82: 3321–3326, 1993 [PubMed] [Google Scholar]
- 34.McEachern E, Carroll AM, Fribourg M, Schiano TD, Hartzell S, Bin S, et al. : Erythropoietin administration expands regulatory T cells in patients with autoimmune hepatitis. J Autoimmun 119: 102629, 2021 [DOI] [PubMed] [Google Scholar]
- 35.Brines M, Patel NSA, Villa P, Brines C, Mennini T, De Paola M, et al. : Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc Natl Acad Sci U S A 105: 10925–10930, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forcade E, Kim HT, Cutler C, Wang K, Alho AC, Nikiforow S, et al. : Circulating T follicular helper cells with increased function during chronic graft-versus-host disease. Blood 127: 2489–2497, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knorr DA, Wang H, Aurora M, MacMillan ML, Holtan SG, Bergerson R, et al. : Loss of T follicular helper cells in the peripheral blood of patients with chronic graft-versus-host disease. Biol Blood Marrow Transplant 22: 825–833, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodruff TM, Nandakumar KS, Tedesco F: Inhibiting the C5-C5a receptor axis. Mol Immunol 48: 1631–1642, 2011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.