

Abstract
Translation initiation factor 2 (eIF2) is a heterotrimeric protein that transfers methionyl-initiator tRNAMet to the small ribosomal subunit in a ternary complex with GTP. The eIF2 phosphorylated on serine 51 of its α subunit [eIF2(αP)] acts as competitive inhibitor of its guanine nucleotide exchange factor, eIF2B, impairing formation of the ternary complex and thereby inhibiting translation initiation. eIF2B is comprised of catalytic and regulatory subcomplexes harboring independent eIF2 binding sites; however, it was unknown whether the α subunit of eIF2 directly contacts any eIF2B subunits or whether this interaction is modulated by phosphorylation. We found that recombinant eIF2α (glutathione S-transferase [GST]–SUI2) bound to the eIF2B regulatory subcomplex in vitro, in a manner stimulated by Ser-51 phosphorylation. Genetic data suggest that this direct interaction also occurred in vivo, allowing overexpressed SUI2 to compete with eIF2(αP) holoprotein for binding to the eIF2B regulatory subcomplex. Mutations in SUI2 and in the eIF2B regulatory subunit GCD7 that eliminated inhibition of eIF2B by eIF2(αP) also impaired binding of phosphorylated GST-SUI2 to the eIF2B regulatory subunits. These findings provide strong evidence that tight binding of phosphorylated SUI2 to the eIF2B regulatory subcomplex is crucial for the inhibition of eIF2B and attendant downregulation of protein synthesis exerted by eIF2(αP). We propose that this regulatory interaction prevents association of the eIF2B catalytic subcomplex with the β and γ subunits of eIF2 in the manner required for GDP-GTP exchange.
In the process of translation initiation as it occurs in eukaryotes, the methionyl-initiator tRNA (Met-tRNA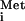 ) is transferred to the 40S ribosomal subunit in a ternary complex consisting of Met-tRNA
) is transferred to the 40S ribosomal subunit in a ternary complex consisting of Met-tRNA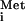 , the heterotrimeric initiation factor 2 (eIF2), and GTP. The resulting 43S preinitiation complex binds to the mRNA, scans for the AUG start codon, and triggers hydrolysis of the GTP bound to eIF2 upon base pairing between Met-tRNA
, the heterotrimeric initiation factor 2 (eIF2), and GTP. The resulting 43S preinitiation complex binds to the mRNA, scans for the AUG start codon, and triggers hydrolysis of the GTP bound to eIF2 upon base pairing between Met-tRNA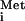 and the AUG. After release of eIF2-GDP, the 60S ribosomal subunit joins to form the 80S initiation complex. The eIF2-GDP is inactive for binding Met-tRNA
and the AUG. After release of eIF2-GDP, the 60S ribosomal subunit joins to form the 80S initiation complex. The eIF2-GDP is inactive for binding Met-tRNA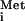 and must be converted to eIF2-GTP to regenerate the ternary complex. This recycling reaction is stimulated by the guanine nucleotide exchange factor (GEF) eIF2B and is a major target of translational control by a conserved mechanism involving phosphorylation of eIF2. The eIF2 phosphorylated on serine 51 of its α subunit [eIF2(αP)] is a competitive inhibitor of eIF2B. As eIF2 generally occurs in excess of eIF2B, and phosphorylation of eIF2-GDP increases its affinity for eIF2B, the recycling of eIF2 can be inhibited by phosphorylation of only a fraction of eIF2 (12).
and must be converted to eIF2-GTP to regenerate the ternary complex. This recycling reaction is stimulated by the guanine nucleotide exchange factor (GEF) eIF2B and is a major target of translational control by a conserved mechanism involving phosphorylation of eIF2. The eIF2 phosphorylated on serine 51 of its α subunit [eIF2(αP)] is a competitive inhibitor of eIF2B. As eIF2 generally occurs in excess of eIF2B, and phosphorylation of eIF2-GDP increases its affinity for eIF2B, the recycling of eIF2 can be inhibited by phosphorylation of only a fraction of eIF2 (12).
Four different eIF2α kinases that are activated by different starvation or stress conditions (shown in parentheses) have been identified in mammalian cells: HRI (heme deprivation), PKR (double-stranded RNA produced in virus-infected cells), PERK (unfolded proteins in the endoplasmic reticulum), and GCN2 (amino acid starvation) (3, 12, 14, 25). Activation of the mammalian kinases PKR and HRI leads to a high level of eIF2α phosphorylation sufficient to inhibit general translation initiation as an adaptive response to virus infection or heme starvation, respectively. GCN2 is the sole eIF2α kinase in budding yeast. When activated in amino acid-starved cells, GCN2 induces the translation of GCN4 mRNA, encoding a transcriptional activator of amino acid biosynthetic genes. Translational control of GCN4 involves four short open reading frames (uORFs) in the leader of the mRNA. In nonstarvation conditions, ribosomes translate uORF1, reinitiate at uORF2 to 4, and fail to reach the GCN4 start codon. In starved cells, phosphorylation of eIF2 by GCN2 reduces eIF2B function and lowers the concentration of ternary complexes. Consequently, many ribosomes that resume scanning after translating uORF1 fail to rebind the ternary complex until scanning past uORF4 and thus reinitiate at the GCN4 start site instead. General translation and cell growth are inhibited when eIF2 is phosphorylated at higher levels than occurs in amino acid-starved wild-type yeast, as in GCN2c mutants bearing constitutively activated forms of the kinase (12).
eIF2B contains five subunits (Table 1) and is found in a 1:1 complex with its substrate, eIF2 (6, 23). The ɛ, δ, γ, and β subunits of yeast eIF2B (encoded by GCD6, GCD2, GCD1, and GCD7, respectively) are essential, and nonlethal mutations in these genes lead to temperature-sensitive growth (Ts− phenotype) and derepression of GCN4 translation (Gcd− phenotype), indicative of reduced ternary complex levels. In contrast, deletion of GCN3 (encoding eIF2Bα) has no effect on cell growth and confers a Gcn− phenotype (failure to induce GCN4) (11), suggesting that GCN3 is required primarily for inhibition of eIF2B by eIF2(αP). GCD2 and GCD7 have sequence similarity to GCN3, and when all three proteins were overexpressed in yeast, they formed a stable subcomplex that reduced the inhibitory effect of eIF2(αP) on translation initiation (29). This subcomplex had no GEF activity in vitro but could bind to purified eIF2 holoprotein in a manner stimulated by phosphorylation of Ser-51 on the α subunit (21). Hence, it was proposed that the overexpressed GCD2-GCD7-GCN3 subcomplex sequestered the inhibitor eIF2(αP)-GDP and allowed native eIF2B to recycle the unphosphorylated eIF2-GDP (21).
TABLE 1.
Yeast genes encoding the subunits of eIF2 and its GEF, eIF2B
| Factor | Subunit | Gene |
|---|---|---|
| eIF2 | eIF2α | SUI2 |
| eIF2β | SUI3 | |
| eIF2γ | GCD11 | |
| eIF2B | eIF2Bα | GCN3a |
| eIF2Bβ | GCD7a | |
| eIF2Bδ | GCD2a | |
| eIF2Bγ | GCD1b | |
| eIF2Bɛ | GCD6b |
Part of the regulatory subcomplex.
Part of the catalytic subcomplex.
Additional evidence implicating GCD2 and GCD7 as regulatory subunits in eIF2B was provided by the isolation of point mutations in these proteins that eliminate the effects of eIF2(αP) on translation in yeast, conferring a Gcn− phenotype and suppressing the growth inhibition of GCN2c alleles (22, 28). These mutations could decrease the affinity of eIF2B for eIF2(αP)-GDP or allow eIF2B to accept eIF2(αP)-GDP as a substrate. Evidence for the latter mechanism came from in vitro GDP-GTP exchange assays using purified eIF2(αP)-[3H]GDP and cell extracts containing overexpressed eIF2B subunits. Unlike wild-type eIF2B, the mutant complexes containing Gcn− substitution GCD7-S119P (22) or GCD7-I118T,D178Y (28) catalyzed nucleotide exchange at nearly identical rates on phosphorylated or unphosphorylated eIF2-[3H]GDP. Thus, these GCD7 mutations allowed eIF2B to utilize the competitive inhibitor eIF2(αP)-GDP as a substrate (21).
Remarkably, the two-subunit complex comprised of eIF2B subunits GCD6 and GCD1 has GEF activity greater than that of five-subunit eIF2B and can accept phosphorylated and unphosphorylated eIF2-GDP as equivalent substrates. Thus, GCD6 and GCD1 comprise an unregulated catalytic subcomplex in eIF2B. Accordingly, we proposed that the GCD2-GCD7-GCN3 regulatory subcomplex is required to inhibit the GCD6-GCD1 catalytic subcomplex when the substrate is phosphorylated (21). We envisioned that binding of phosphorylated eIF2-GDP to the eIF2B regulatory subcomplex would preclude its interaction with the active site in the GCD1-GCD6 catalytic subcomplex. The Gcn− mutations in GCD7 would overcome this nonproductive interaction and allow binding of eIF2(αP)-GDP to eIF2B in the manner required for nucleotide exchange (21).
Previously, we suggested that the homologous regulatory segments in GCN3, GCD2, and GCD7 are juxtaposed to form a binding site for the phosphorylated N-terminal portion of eIF2α (22). Consistent with this idea, Gcn− mutations were obtained in yeast eIF2α (encoded by SUI2) in residues surrounding Ser-51 that reduce the inhibitory effect of eIF2(αP)-GDP on eIF2B activity in vivo. These mutations alter residues Ile-58, Leu-84, Arg-88, and Val-89 (27). Alanine substitution of Ser-48 has a similar effect in mammalian cells (4, 7, 15, 18). Moreover, addition of recombinant human eIF2α-S48A to rabbit reticulocyte lysates reduced the abundance of 15S complexes containing eIF2, thought to represent inactive eIF2B-eIF2(αP)-GDP complexes stabilized by Ser-51 phosphorylation (26). This last finding suggests that mutation of Ser-48 to Ala reduces the affinity of eIF2(αP)-GDP for eIF2B as a means of overcoming the inhibition by Ser-51 phosphorylation.
At odds with our proposal that eIF2B regulatory subunits interact directly with eIF2α (22, 27), no binding was detected between eIF2B holoprotein and phosphorylated recombinant rat eIF2α. By contrast, recombinant eIF2β showed significant binding to eIF2B holoprotein, and it also bound to the isolated δ and ɛ subunits of eIF2B (GCD2 and GCD6, respectively, in yeast) (17). Based on these findings, it was suggested that eIF2α does not contact eIF2B and that Ser-51 phosphorylation elicits a conformational change in eIF2 that enhances interaction between eIF2β and the δ and ɛ subunits of eIF2B (17).
In this study, we show for the first time that the GCD2-GCD7-GCN3 regulatory subcomplex of yeast eIF2B can form a stable complex in vitro with a recombinant form of the α subunit of eIF2. This glutathione S-transferase (GST)–SUI2 fusion protein formed a stable complex with the regulatory subcomplex, but not with the catalytic subcomplex of eIF2B, in a manner stimulated by phosphorylation of Ser-51 in the recombinant protein. None of the individual eIF2B subunits was capable of this stable interaction, consistent with the idea that the binding domain for phosphorylated SUI2 [SUI2(P)] in eIF2B requires contributions from all three regulatory subunits. We present genetic data that SUI2(P) competes effectively with eIF2(αP) for association with the GCD2-GCD7-GCN3 subcomplex in vivo, providing evidence that binding of SUI2(P) to the eIF2B regulatory subunits is a physiological interaction. Furthermore, in vitro binding of GST-SUI2(P) to the eIF2B regulatory subunits was impaired by Gcn− mutations in SUI2 and also by those in GCD7 shown previously to permit eIF2(αP)-GDP to be accepted as a substrate by eIF2B in vitro. These last findings provide compelling evidence that tight binding of SUI2(P) to the eIF2B regulatory subunits is crucial for the negative regulation of eIF2B function when the substrate eIF2-GDP is phosphorylated.
MATERIALS AND METHODS
Yeast strains and plasmids.
Yeast strains and plasmids used in this study are shown in Tables 2 and 3, respectively; details of their construction will be made available on request.
TABLE 2.
Yeast strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| BJ1995 | MATα leu2 trp1 ura3-52 gal2 pep4-3 prb1-1122 | 13 |
| H2767 | MATα leu2 trp1 ura3-52 gal2 pep4-3 prb1-1122 gcd6Δ′ gcd7Δ p1871 (GCD2 GCD7 GCN3 URA3) pTK1.11 (GCD1-2xFlag-His GCD6 LEU2) | 20 and this study |
| GP3299 | MATa leu2-3 leu-112 ura3-52 trp1-Δ63 gcn2Δ gcd2Δ::hisG pAV1003 (GCD2-K627T TRP1) pTK4 (PKR-Flag-His URA3) | 22 and this study |
| GP3511 | MATα leu2-3 leu2-112 ura3-52 inol gcn2Δ pep4::LEU2 sui2ΔHIS4-lacZ pAV1089 (SUI2 SUI3 GCD11-His) | 21 |
| H1402 | MATα leu2-3,112 ura3-52 inol | 21 |
| H1608 | MATα leu2-3,112 ura3-52 inol GCN2c-M719V-E1537G HIS4-lacZ | 24 |
TABLE 3.
Plasmids used in this study
| Plasmid | Yeast gene | Marker(s) | Parent vector | Reference or source |
|---|---|---|---|---|
| pRS425 | Empty vector | LEU2, 2μm | 5 | |
| pRS426 | Empty vector | URA3, 2μm | 5 | |
| p1873 | GCD1 GCD6 | LEU2, 2μm | pRS425 | 9 |
| p1871 | GCD2 GCD7 GCN3 | URA3, 2μm | pRS426 | 9 |
| p2297 | GCD2 | URA3, 2μm | pRS426 | 29 |
| p2302 | GCD1 GCD6 | URA3, 2μm | pRS426 | 29 |
| p2304 | GCN3 | URA3, 2μm | pRS426 | 29 |
| p2305 | GCD7 | URA3, 2μm | pRS426 | 29 |
| pAV1139 | GCD2 GCD7-S119P GCN3 | URA3, 2μm | pRS426 | 21 |
| pAV1140 | GCD2 GCD7-I118T D178Y GCN3 | URA3, 2μm | pRS426 | 21 |
| pAV1089 | SUI2 SUI3 GCD11-His | URA3, 2μm | pRS426 | 21 |
| p1861 | GST | Ampr | pGEX-5X-3 | Pharmacia |
| p2565 | GST-SUI2 | Ampr | pGEX-5X-3 | W. Yang |
| pTK29 | SUI2 | LEU2, 2μm | pRS426 | This study |
| pTK1.11 | GCD1-2xFlag-His GCD6 | URA3, 2μm | pRS425 | This study |
| pTK4 | PKR-Flag-His under GAL1-CYC1 promoter | URA3, leu2-d, 2μm | pEMBLyex4 | This study |
| pTK6 | GST-SUI2-S51A | Ampr | pGEX-5X-3 | This study |
| pTK17 | GST-SUI2-His (aaa 1–245) | Ampr | pGEX-6p-2 | This study |
| pTK18 | GST-SUI2-His (aa 1–197) | Ampr | pGEX-6p-2 | This study |
| pTK19 | GST-SUI2-His (aa 1–140) | Ampr | pGEX-6p-2 | This study |
| pTK20 | GST-SUI2-His (aa 1–100) | Ampr | pGEX-6p-2 | This study |
| pTK22 | GST-SUI2-K79A | Ampr | pGEX-5X-3 | This study |
| pTK23 | GST-SUI2-G80A | Ampr | pGEX-5X-3 | This study |
| pTK24 | GST-SUI2-D83A | Ampr | pGEX-5X-3 | This study |
| pTK26 | GST-SUI2-R88T | Ampr | pGEX-5X-3 | This study |
| pTK27 | GST-SUI2-E49N | Ampr | pGEX-5X-3 | This study |
| pTK28 | GST-SUI2-E49Q | Ampr | pGEX-5X-3 | This study |
aa, amino acids.
Purification.
His6-eIF2B was overexpressed from plasmids pTK1.11 and p1871 in yeast strain H2767. Cells were grown at 30°C in YPD to an optical density at 600 nm (OD600) of 8 to 10, harvested, washed with ice-cold distilled water, resuspended in breaking buffer (75 mM Tris-HCl [pH 7.5], 100 mM KCl, 1 mM Na2EDTA, 14 mM 2-mercaptoethanol, 10 μM GDP, 50 mM NaF) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg each of pepstatin A, leupeptin, and aprotinin per ml), and broken using the French press or a bead beater. For the French press, 1 ml of lysis buffer was used per g cells, whereas twice the volume was used for the bead beater. The whole-cell extract (WCE) was centrifuged at 4°C for 15 min at 8,000 × g to remove cellular debris, and the supernatant was subjected to ultracentrifugation at 200,000 × g for 2 h to pellet the ribosomes. Ribosomes were resuspended gently in buffer A (20 mM Tris-HCl [pH 7.5], 0.1 mM MgCl2, 10% glycerol, 10 μM GDP, 2 mM 2-mercaptoethanol, 50 mM NaF, protease inhibitors) containing 500 mM KCl (buffer A-500) and incubated for 1 h at 4°C with slow stirring. The ribosomal salt wash (RSW) was obtained by ultracentrifugation of the ribosomal suspension at 200,000 × g for 2 h. Ni-silica resin (Qiagen) was washed twice in buffer A-500 containing 5 mM imidazole, and 1.2 ml of a 50% slurry in the same buffer was added to the RSW and incubated overnight at 4°C. The mixture was centrifuged at 1,000 rpm for 2 min, the supernatant was discarded, and the resin was washed five times, each with 10 volumes of buffer A-500 containing 5 mM imidazole. The resin was washed three more times with 10 volumes of buffer A containing 1 M KCl and 5 mM imidazole and twice with buffer B (Tris-buffered saline, 10% glycerol, 10 mM imidazole, 0.5 mM PMSF, complete protease inhibitor tablets without EDTA [Boehringer Mannheim]). eIF2B was eluted three times, each with 1 ml of buffer B containing 250 mM imidazole. Protein concentrations of the eluates were determined using the Bradford assay (Bio-rad), and the peak fractions were pooled.
Plasmid pTK4 was used to overexpress Flag- and His6-tagged PKR from a galactose-inducible promoter in yeast strain GP3299. The strain was grown to saturation in 100 ml of SD medium with minimal supplements and diluted to 400 ml of the same medium but with 10% galactose and 2% raffinose as carbon sources (SGAL), grown overnight, and used to inoculate 10 liters of SGAL medium at a starting OD600 of 0.1. The cells were grown in a fermenter to an OD600 of ∼6 and harvested by zonal centrifugation using a Sorvall centrifuge at 15,000 rpm in a TZ-28 rotor. The cells were washed in ice-cold distilled water, resuspended in Flag binding buffer (FBB) (20 mM sodium phosphate [pH 7.0], 500 mM NaCl, 0.1% Triton X-100, complete protease inhibitor tablets without EDTA, 4 μg of leupeptin per ml, 1 mM PMSF, 10 mM NaF, 50 mM β-glycerophosphate, 125 μM Na3VO4), and broken in the bead beater. The WCE was clarified by centrifugation at 15,000 × g for 1 h. Two milliliters of anti-Flag-M2 affinity gel (50%) (Sigma) was washed five times in 10 ml of FBB, resuspended in 1 ml of FBB, and added to the WCE. The mixture was incubated overnight on a Labquake shaker (Barnstead-Thermolyne) at 4°C, and the resin was collected by centrifugation at 3,000 rpm for 5 min in an open-bucket rotor using a Beckman J-6B centrifuge. The resin was washed five times, each with 10 volumes of FBB, and the PKR was eluted four times, each with 1 ml of FBB containing Flag peptide (400 μg/ml; Sigma) by incubation on a nutator for 10 min at 4°C and collecting the resin as described above. The eluates were pooled and concentrated in a Centricon 30 concentrator (Amicon). The eluate was adjusted to 1 mM dithrothreitol (DTT)–10 mM MgCl2–0.1 mM EDTA–10% glycerol and stored at 70°C after rapid freezing using liquid nitrogen.
His6-tagged eIF2 was purified from GP3511 as described previously (21) except that Na3VO4 was omitted. The partially purified eIF2 from the heparin-Sepharose column was applied to a 1-ml column of Hi Trap Q Sepharose using the syringe mode (Amersham Pharmacia Biotech) (G. Pavitt and A. G. Hinnebusch, unpublished observations) and washed sequentially with 5 ml each of heparin-100 and heparin-200 before eluting the eIF2 with 5 ml of heparin-300 (21). The eluate was concentrated in a Centricon 30 spin concentrator and stored in liquid nitrogen after rapid freezing.
Yeast WCE preparation.
WCEs used for GST pull-down assays were prepared from the transformants of yeast strain BJ1995 overexpressing the appropriate eIF2B subunits as described previously (21), except that the cells were broken by vortexing with acid-washed glass beads five times for 1 min each, with 1-min intervals on ice, and including 75 mM Tris-HCl (pH 7.5), 1 mM EDTA, and complete protease inhibitor tablets in the breaking buffer. To prepare WCEs for Ni+2-silica pull-down assays, 0.1 mM EDTA and 5 mM 2-mercaptoethanol were used instead of 1 mM EDTA and 1 mM DTT in the breaking buffer.
GST pull-down assays.
GST-SUI2 fusions were expressed in Escherichia coli BL21(DE3) (Novagen), using 0.4 mM isopropyl-B-d-thiogalactopyranoside to induce the fusion proteins. WCEs were prepared in lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM KCl, 2.5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, protease inhibitors, complete protease inhibitor tablets without EDTA) by sonication six times for 2 min each with 30-s intervals on ice. Glutathione-Sepharose beads (Amersham) were prewashed in binding buffer (BB; same as lysis buffer but including 0.1% Triton X-100, 5 mM NaF, and 0.1 mM ATP) and 30 μl of a 50% slurry was mixed with WCE in BB for 40 min at 4°C on a nutator. The beads were washed thrice with 500 μl of ice-cold BB and resuspended in 50 μl of BB. The immobilized fusion proteins were incubated with or without PKR for 10 min at room temperature. In control assays, 5 to 10 μCi of [γ32P]ATP was added prior to addition of PKR. The kinase reaction was stopped by addition of 150 μl of BB on ice. Radioactive beads were washed with BB, mixed with 12 μl of 2× Laemmli's sample buffer (NOVEX), boiled, resolved by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), stained with Coomassie blue, dried, and exposed to X-ray film, and the film was developed.
The binding studies were carried out by addition of partially purified eIF2B or yeast WCEs to the phosphorylated or unphosphorylated immobilized fusion proteins and incubation for 2 h at 4°C in BB. The beads were washed thrice with 500 μl of BB, resuspended in 12 μl of 2× Laemmli's sample buffer, and separated by SDS-PAGE using 10 to 20% gradient gels. Proteins were transferred to a nitrocellulose membrane (NOVEX) at 25 V for 2 h and probed with the appropriate antisera. Immune complexes were visualized with a horseradish peroxidase-conjugated anti-rabbit secondary antibody and an enhanced chemiluminescence kit from Amersham.
Nickel pull-down assays with His-tagged eIF2.
His6-tagged eIF2 (2.5 μg) was purified as described above and incubated in 50 μl of modified binding buffer BB-1 (in which 5 mM 2-mercaptoethanol replaced 1 mM DTT) with or without PKR (1 μg) for 10 min at RT. The reaction was stopped by addition of 50 μl of BB-1 on ice and added to 100 μg of WCEs prepared from yeast transformants overexpressing the appropriate eIF2B subunits and incubated for 10 min at 10°C. The His6-tagged eIF2 and bound proteins were recovered using 10 μl of Ni-silica beads (prewashed in BB-1) as described previously (21) except that BB-1 was used instead of PD (21). The samples were resolved by SDS-PAGE on a 10 to 20% gradient gel and subjected to immunoblot analysis as described above.
RESULTS
GST-SUI2 binds purified eIF2B in vitro dependent on Ser-51 phosphorylation
To determine whether the α subunit of yeast eIF2 interacts directly with eIF2B, we purified a full-length GST-SUI2 fusion protein from E. coli by using glutathione-Sepharose beads and tested the immobilized protein for interaction with partially purified yeast eIF2B. In parallel, we examined a mutant fusion protein containing Ala in place of Ser at position 51 (GST-SUI2-S51A). Prior to the binding reactions, both immobilized fusion proteins were treated with purified human PKR and ATP to phosphorylate Ser-51 in the wild-type protein. In control experiments where [γ-32P]ATP was included in the kinase reactions, we confirmed that GST-SUI2, but not GST-SUI2-S51A, was phosphorylated by PKR in vitro, confirming PKR's specificity for Ser-51 (Fig. 1A). The eIF2B used in the binding assays was purified by nickel chelation chromatography from a yeast strain expressing a polyhistidine-tagged form of eIF2Bγ (GCD1). The purification was carried out at a high salt concentration to dissociate eIF2 from eIF2B, as the presence of eIF2 would complicate the interpretation of binding data. Western analysis confirmed the absence of eIF2 subunits in the His6-eIF2B preparation (Fig. 1B, lane I; Fig. 1C). After incubation of purified His6-eIF2B with the immobilized fusion proteins, with or without pretreatment with PKR, the beads were washed extensively and the bound proteins were analyzed by Western blotting using antibodies against eIF2B subunits. As shown in Fig. 1B, His6-eIF2B bound to wild-type GST-SUI2 in a manner stimulated by incubation with PKR. In contrast, little or no His6-eIF2B bound to GST-SUI2-S51A regardless of PKR treatment. These results provide strong evidence that eIF2B interacts directly with the α subunit of eIF2 and that this interaction is greatly enhanced by phosphorylation of Ser-51.
FIG. 1.

GST-SUI2 binds to purified eIF2B in a manner stimulated by phosphorylation of Ser-51. GST-SUI2, GST-SUI2 S51A, or GST alone was expressed in E. coli, immobilized on glutathione-Sepharose beads, and incubated with (+) or without (−) 1 μg of purified PKR in buffer BB. In panel A, 5μCi of [γ-32P]ATP was added with the PKR, and the reactions were resolved by SDS-PAGE, stained with Coomassie blue (lower panel), and subjected to autoradiography (upper panel, 32P). In panel B, 2.0 μg of partially purified His6-eIF2B was incubated with the immobilized GST-SUI2, GST-SUI2-S51A, or GST proteins treated with or without PKR. After extensive washing, the bound proteins were resolved by SDS-PAGE and analyzed by Western blotting using antibodies against GCD6, GCD7, and GCD11 (GST pull-down assay). Two different amounts of bound proteins differing by a factor of 3 were loaded in successive lanes for each fusion protein. The input (I) lane contains 25% of the input amount of purified eIF2B used in the pull-down assays shown in lanes 1 to 10. (C) Western blot comparing the levels of SUI2 and GCD6 in 1 μg of yeast WCE (lane 1) and 3 μg of the purified His6-eIF2B (lane 2) used in panel B. wt, wild type.
In an effort to map the minimal binding domain for eIF2B in SUI2, we produced GST-SUI2 fusion proteins truncated to different extents from the C terminus and tested them for binding to His6-eIF2B. The results showed that removing 59 residues from the C terminus of GST-SUI2 had little effect on its phosphorylation by PKR or binding to His6-eIF2B (Fig. 2, 1-245aa). Interestingly, the GST-SUI2 protein truncated to position 197 was phosphorylated by PKR and bound His6-eIF2B at high levels; however, the binding was not dependent on Ser-51 phosphorylation. These last findings may indicate that SUI2 residues 197 to 245 contain a domain that interferes with binding of eIF2B to full-length SUI2 in a way that can be overcome by Ser-51 phosphorylation. The GST-SUI2 fusions that were truncated to position 140 or 100 were not phosphorylated by PKR and no longer bound to His6-eIF2B.
FIG. 2.

(A) SUI2 residues 1 to 245 are sufficient for binding of GST-SUI2(P) to eIF2B stimulated by phosphoserine 51. Full-length GST-SUI2 (wild type [wt]) and the indicated derivatives truncated at the C terminus (designated by the amino acids [aa] remaining) were immobilized on glutathione-Sepharose beads, treated with (+) or without (−) 3 μg of PKR in buffer BB, and incubated with 4 μg of purified eIF2B. Binding of eIF2B to the GST-SUI2 fusions in pull-down assays was analyzed by SDS-PAGE and Western blotting (upper panel) as described in Fig. 1B. The lower panel shows Ponceau S staining of the bound proteins, with asterisks indicating the full length GST-SUI2 fusions. The input (I) lane contained 50% of the eIF2B used in each reaction. (B) Deletion of the C terminus (amino acids 140 to 304) of SUI2 abolished phosphorylation of GST-SUI2 by PKR. The experiment was carried out exactly as described for the upper panel of Fig. 1A for the indicated GST-SUI2 proteins, using the same amounts designated 3X in panel A.
GST-SUI2(P) binds to the regulatory subcomplex of eIF2B.
The eIF2B regulatory subcomplex can be overexpressed in yeast from a high-copy-number plasmid bearing GCD2, GCD7, and GCN3 (under the control of their native promoters) and coimmunoprecipitated with eIF2 from WCEs (29). Previously, we showed that the overexpressed regulatory subcomplex in WCEs could bind to exogenously added eIF2 holoprotein in a manner stimulated ∼3-fold by prior phosphorylation of the eIF2 on Ser-51 (21). The catalytic subcomplex, comprised of GCD1 and GCD6, also bound to eIF2 but did not discriminate between phosphorylated and unphosphorylated eIF2 (21). To determine whether SUI2 alone can interact with these eIF2B subcomplexes, and in a manner stimulated by phosphorylation on Ser-51, we incubated the immobilized GST-SUI2 and GST-SUI2-S51A fusions (pretreated with PKR) with WCEs containing the appropriate overexpressed eIF2B subunits and analyzed the bound proteins by Western blotting.
Using a control extract from a wild-type strain containing an empty vector (designated vector extract), we observed binding of the eIF2B subunits to GST-SUI2(P) but not to GST-SUI2-S51A (Fig. 3A, lanes 2 and 3 versus 16 and 17). We attribute these interactions to binding of the native eIF2B holocomplex in the vector extract to GST-SUI2(P), as shown above for purified His6-eIF2B (Fig. 1). For the extract containing the overexpressed eIF2B regulatory subunits (designated h.c.GCD2-GCD7-GCN3), we observed increased binding of these three subunits compared to that seen with an equivalent amount of vector extract, whereas the amounts of bound GCD6 and GCD1 were indistinguishable between the two extracts (Fig. 3A, lanes 4 to 6 versus 1 to 3). Much lower amounts of GCD2, GCD7, and GCN3 in the h.c.GCD2-GCD7-GCN3 extract bound to GST-SUI2-S51A than to wild-type GST-SUI2(P) (lanes 18 to 20 versus 4 to 6). These findings indicate that the overexpressed regulatory subcomplex bound to GST-SUI2 stimulated by phosphorylation of Ser-51.
FIG. 3.

The eIF2B regulatory subcomplex in cell extracts binds to GST-SUI2(P). Wild-type (wt) GST-SUI2 and GST-SUI2-S51A fusions were immobilized on glutathione-Sepharose beads and treated with 1 μg of PKR in buffer BB, followed by incubation with the appropriate yeast WCE and 800 μg of bovine serum albumin in buffer BB. (A) The pull-down assays contained 100 or 200 μg of GST-SUI2 (lanes 1 to 14), or 150 or 300 μg of GST-SUI2-S51A (lanes 15 to 20), and 600 μg of WCE from transformants of yeast strain BJ1995 overexpressing the regulatory subcomplex GCD2-GCD7-GCN3 (from plasmid p1871; lanes 5, 6, 19, and 20), GCD2 (from plasmid p2297; lanes 8 and 22), GCD7 (from plasmid p2305; lanes 10, 11, 24, and 25), or GCN3 (from plasmid p2304; lanes 13, 14, 27, and 28) or carrying the empty vector (from plasmid pRS426; lanes 2, 3, 16, and 17). The bound proteins were analyzed by SDS-PAGE (on an 8 to 16% gradient gel) and Western blotting as described for Fig. 1B. Input (I) lanes contained 10% of the WCE used in each reaction. For the binding reactions in lanes 8 and 22, only the larger amounts of the GST-SUI2 fusion proteins described above were used. In panel B, the pull-down assays contained 25 or 100 μg of wild-type GST-SUI2 or of GST-SUI2-S51A and 200 μg of WCE from transformants of yeast strain BJ1995 overexpressing GCD1 and GCD6 (from plasmid p2302; lanes 7 to 10) or carrying the empty vector (from plasmid pRS426; lanes 2 to 5). The bound proteins were analyzed as described for Fig. 1B. Input (I) lanes contained 5% of the WCE used in each reaction.
We did not observe increased binding of GCD2, GCD7, or GCN3 to GST-SUI2 when these subunits were overexpressed individually (Fig. 3A, lanes 7 to 14 versus 1 to 3), although the degree of GCD2 and GCD7 overexpression was less than when all three regulatory subunits were cooverexpressed (Fig. 3A, lanes 7, 9, and 12 versus 4). Overexpression of GCD2 alone did result in greater binding of this subunit to GST-SUI2-S51A versus that seen with the vector extract (Fig. 3A, lanes 22 versus 16 and 17), at a level comparable to that seen when all three subunits were cooverexpressed (Fig. 3A, lanes 22 versus 19 and 20). However, because this GCD2 binding was independent of phosphoserine 51, it may not be physiologically relevant. In any case, it was much lower than the amount of GCD2 that bound to GST-SUI2(P) when all three regulatory subunits were cooverexpressed (Fig. 3A, lanes 22 versus 5 and 6).
Interestingly, overexpression of GCD2, GCD7, or GCN3 individually reduced the binding of native eIF2B holocomplex to GST-SUI2(P) (Fig. 3A, lanes 7 to 14 versus 1 to 3). One interpretation of this result could be that overexpression of the individual subunits titrates other subunits away from the native eIF2B holoprotein, leaving partial eIF2B subcomplexes that do not bind efficiently to GST-SUI2(P). Consistent with this interpretation, we showed previously that overexpression of GCD2 alone reduced eIF2B activity in vivo. Similarly, overexpression of GCD7 titrated GCN3 away from eIF2B and made the latter less susceptible to inhibition by eIF2(αP) (29).
We found previously that the GCD1-GCD6 catalytic subcomplex bound eIF2 holoprotein independently of Ser-51 phosphorylation (21). To determine whether SUI2 can interact with the catalytic subcomplex, we compared the binding of GCD6 to GST-SUI2(P) in the vector extract (containing GCD1 and GCD6 in native eIF2B holoprotein) to that given by an extract containing overexpressed GCD1 and GCD6. The results in Fig. 3B show that there was considerably more GCD6 in the extract overexpressing GCD1 and GCD6 (compare lanes 1 and 6); however, the excess GCD6 did not bind to GST-SUI2(P) (lanes 3 versus 8). The fact that the S51A substitution greatly reduced the binding of GCD6 to GST-SUI2 in both extracts (Fig. 3B, lanes 4, 5, 9, and 10) indicates that GCD6 bound to GST-SUI2 only as a subunit of native eIF2B holoprotein. We conclude that the GCD1-GCD6 catalytic subcomplex does not form a stable complex with SUI2 alone, regardless of Ser-51 phosphorylation.
Gcn− regulatory mutations near Ser-51 eliminate binding of GST-SUI2(P) to both eIF2B and the GCD2-GCD7-GCN3 regulatory subcomplex.
Previously, we isolated point mutations in the amino terminus of SUI2 that suppressed the growth-inhibitory effects of eIF2 phosphorylation and prevented derepression of GCN4 translation (Gcn− phenotype) (27). Recently, additional Gcn− mutations were isolated in SUI2, and it was shown that they did not diminish Ser-51 phosphorylation by GCN2 in vivo or in vitro (T. E. Dever, unpublished results). Thus, the latter mutations most likely abrogate the inhibitory effect of eIF2(αP) on eIF2B activity in vivo. One possibility is that these mutations weaken the direct interaction between phosphorylated SUI2 and the eIF2B regulatory subcomplex. To test this idea, we introduced the newly identified Gcn− mutations into the GST-SUI2 fusion and investigated whether they reduce binding of GST-SUI2(P) to eIF2B. The GST-SUI2 fusion proteins containing the mutations indicated in Fig. 4A were purified, phosphorylated with PKR, and incubated with purified His6-eIF2B. Except for GST-SUI2-D83A, the mutant proteins were phosphorylated efficiently by PKR (Fig. 4A, 32P); however, none bound to eIF2B at the high levels observed for wild-type GST-SUI2(P). In fact, all of them displayed the low-level nonspecific binding characteristic of GST alone (Fig. 4A, Western).
FIG. 4.

Gcn− mutations proximal and distal to phosphoserine 51 in SUI2 disrupt binding of eIF2B holoprotein and the eIF2B regulatory subcomplex to GST-SUI2(P). (A) Wild-type GST-SUI2 and the indicated mutant derivatives were immobilized on glutathione-Sepharose beads and treated with 1 or 2 μg of PKR in buffer BB. The immobilized proteins were incubated with His6-eIF2B (4 μg) and bovine serum albumin (1 mg) in buffer BB, and the pull-down assays were analyzed as described for Fig. 1B, with the results shown in the upper two panels (Western). The input (I) lane contained 25% of the His6-eIF2B used in each reaction. The results in the lower two panels were obtained exactly as described for the panels labeled 32P and Coomassie in Fig. 1A, respectively. Amounts of the fusion proteins used for the pull-down assays were the same as shown in the bottom panel (Coomassie) used for the kinase assays. (B) Pull-down assays were carried out using 84 or 168 μg of GST, 100 or 200 μg of GST-SUI2, 150 or 300 μg of either GST-SUI2-E49N or GST-SUI2-R88T fusion protein, and 800 μg of WCE from transformants of strain BJ1995 overexpressing the regulatory subcomplex GCD2-GCD7-GCN3 (from plasmid p1871; lanes 11 to 17) or carrying the empty vector (from plasmid pRS426; lanes 2 to 9). Bound proteins were analyzed as described for Fig. 1B. Input (I) lanes contained 10% of the WCE used in each reaction. wt, wild type.
We wished to confirm that the SUI2 Gcn− mutations also weaken the interaction of phosphorylated SUI2 with the GCD2-GCD7-GCN3 regulatory subcomplex. To do so, we conducted binding assays with the mutant and wild-type GST-SUI2(P) fusion proteins using the WCE containing overexpressed GCD2, GCD7, and GCN3 and the vector WCE containing only native eIF2B holoprotein. In agreement with results described above, greater amounts of the three regulatory subunits in the h.c.GCD2-GCD7-GCN3 compared to the vector extract bound to wild-type GST-SUI2(P) (Fig. 4B, compare lanes 10 to 12 versus 1 to 3). In contrast, no binding of the native eIF2B or overexpressed regulatory subunits above background levels was observed for GST-SUI2-E49N(P) or GST-SUI2-R88T(P) (Fig. 4B, lanes 4 through 7 and 13 to 17). We conclude that the SUI2 Gcn− mutations abolish interaction of SUI2(P) with both eIF2B holoprotein and the GCD2-GCD7-GCN3 regulatory subcomplex.
Gcn− mutations in GCD7 reduce binding of the eIF2B regulatory subcomplex to GST-SUI2(P)
Previously, we described mutant alleles of GCD7 with a Gcn− phenotype (22, 28) and showed that the substitutions in two such alleles (GCD7-S119P and GCD7-I118T, D178Y [Fig. 5A]) allowed eIF2B to accept eIF2(αP)-GDP as a substrate using in vitro assays for guanine nucleotide exchange (21). Accordingly, we investigated here whether these GCD7 mutations would weaken association between GST-SUI2(P) and the eIF2B holoprotein. We prepared three different WCEs containing overexpressed amounts of all five eIF2B subunits, containing either wild-type GCD7, GCD7-S119P (mutant *M1), or GCD7-I118T,D178Y (*M2), and incubated them with GST-SUI2(P) and GST-SUI2-S51A. As shown in Fig. 5B, the eIF2B*M1 and eIF2B*M2 complexes showed moderate (*M1) or severe (*M2) reductions in binding to GST-SUI2(P) compared to the wild-type eIF2B complex (lanes 4 to 6 versus 7 to 12). As expected, the two mutant eIF2B complexes did not bind to GST-SUI2-S51A (data not shown).
FIG. 5.

Gcn− mutations in GCD7 decrease binding of the eIF2B holoprotein and the eIF2B regulatory subcomplex to GST-SUI2(P). (A) Schematic showing the sequence similarities among the eIF2B regulatory subunits and the point mutations in GCD7 that were analyzed in this study. (B) Wild-type (wt) GST-SUI2 fusion was immobilized on glutathione-Sepharose beads and treated with 1 μg of PKR in buffer BB, followed by incubation with the appropriate yeast WCE and 800 μg of bovine serum albumin in buffer BB. The pull-down assays were carried out with 100 or 200 μg of GST-SUI2 and 600 μg of yeast WCE from transformants of strain BJ1995 overexpressing all five wild-type eIF2B subunits (from plasmids p1873 and p1871; lanes 5 and 6), wild-type GCD1-GCD6-GCD2-GCN3 and GCD7-S119P (from plasmids p1873 and pAV1139, designated eIF2B*M1; lanes 8 and 9), or wild-type GCD1-GCD6-GCD2-GCN3 and GCD7-I118T, D178Y (from plasmids p1873 and pAV1140; designated eIF2B*M2; lanes 11 and 12) or carrying the empty vectors (from plasmids pRS425 and pRS426; lanes 2 and 3). Bound proteins were analyzed as described for Fig. 1B. Input (I) lanes contained 10% of the WCE used in each reaction. (C) Pull-down assays were done exactly as described for panel B except that the WCEs were from transformants of strain BJ1995 overexpressing wild-type GCD2-GCD7-GCN3 (from plasmid p1871; lanes 1 to 3), wild-type GCD2-GCN3 and GCD7-S119P (from plasmid pAV1139; lanes 4 to 6), or wild-type GCD2-GCN3 and GCD7-I118T, D178Y (from plasmid pAV1140; lanes 7 to 9). Input (I) lanes contained 10% of the WCE used in each reaction. (D) Histograms showing the results of densitometric quantification of the binding data in panel C relative to the input signal in percentage.
We also carried out binding reactions using WCEs containing overexpressed amounts of the three regulatory subunits, again including either wild-type GCD7, GCD7-S119P (mutant *M1), or GCD7-I118T,D178Y (*M2) (Fig. 5C). The GCD7 Gcn− mutations led to reductions in binding of all three subunits to GST-SUI2(P) (Fig. 5C and D). The residual binding of GCD2 and GCD7 observed for the *M1 and *M2 extracts may be nonspecific because we observed similar amounts of low-level binding of these proteins to the GST-SUI2-S51A fusion for the wild-type, *M1, and *M2 extracts (data not shown). We conclude that the *M1 and ∗M2 Gcn− mutations in GCD7 impair binding of the eIF2B regulatory subcomplex to SUI2(P).
Gcn− mutations in GCD7 decrease interaction between the eIF2B and eIF2 holoproteins.
GCD6, the principal catalytic subunit of yeast eIF2B, has a strong binding domain for eIF2β (1); hence, it was unclear how much the enhanced interaction between SUI2(P) and the eIF2B regulatory subunits would contribute to the stability of the complex formed between the eIF2 and eIF2B holoproteins. To address this issue, we purified eIF2 holoprotein containing a polyhistidine-tagged form of the γ subunit (His6-eIF2), phosphorylated the eIF2 in vitro with PKR, and incubated it with WCEs containing overexpressed amounts of all five eIF2B subunits, containing either wild-type GCD7, GCD7-*M1, or GCD7-*M2. Following incubation, the His6-eIF2 was recovered on nickel-silica resin and probed for bound eIF2B subunits by Western blotting. The overexpressed wild-type eIF2B holoprotein bound to His6-eIF2 in a manner enhanced by phosphorylation of Ser-51 (Fig. 6A, lanes 7 and 8), to a degree similar to that observed previously (21). Interestingly, binding of eIF2B to both phosphorylated and unphosphorylated His6-eIF2 was reduced by the *M1 and *M2 mutations (Fig. 6A, lanes 11, 12, 15, and 16 versus 7 and 8; Fig. 6B). These data imply that the GCD7 Gcn− mutations impair the interaction between SUI2 and the eIF2B regulatory subunits in the context of the eIF2-eIF2B holocomplex. Apparently, loss of these contacts destabilizes the eIF2-eIF2B holocomplex even when SUI2 is unphosphorylated.
FIG. 6.

Binding of wild-type and mutant eIF2B holoproteins to His-tagged eIF2 holoprotein. (A) WCEs from transformants of strain BJ1995 overexpressing all five wild-type eIF2B subunits (from plasmids p1873 and p1871, designated h.c.eIF2B wt; lanes 5 to 8), wild-type subunits GCD1-GCD6-GCD2-GCN3 and mutant subunit GCD7-S119P (from plasmids p1873 and pAV1139. designated h.c. eIF2*M1; lanes 9 to 12), or wild-type subunits GCD1-GCD6-GCD2-GCN3 and mutant subunit GCD7-I118T,D178Y (from plasmids p1873 and pAV1140, designated h.c. eIF2*M2; lanes 13 to 16) or carrying the empty vectors (from plasmids pRS425 and pRS426; lanes 1 to 4) were incubated with purified eIF2 phosphorylated in vitro with PKR [eIF2(αP)] (lanes 4, 8, 12, and 16), unphosphorylated eIF2 (lanes 3, 7, 11, and 15), or no eIF2 (lanes 2, 6, 10, and 14). The proteins that bound to eIF2 were purified by Ni-silica affinity chromatography and analyzed by SDS-PAGE and Western blotting using antibodies against the proteins listed to the right of each panel. For SUI2(P), antibodies specific for eIF2α phosphorylated on Ser-51 were employed. Input lanes contained 20% of the WCE used in each reaction. (B) Histograms showing densitometry of signals for each eIF2B antibody shown in panel A as a percentage of the input.
Genetic evidence that SUI2 binds to the eIF2B regulatory subcomplex in vivo.
To obtain evidence that SUI2 on its own can interact with the eIF2B regulatory subcomplex in vivo, we exploited our previous finding (29) that cooverexpression of GCD2, GCD7, and GCN3 alleviates the slow-growth phenotype associated with the constitutively activated kinase encoded by GCN2c-M719V-E1537G (Fig. 7A, compare strains 1 and 2). We previously provided evidence that the GCD2-GCD7-GCN3 subcomplex sequestered eIF2(αP) and prevented it from inhibiting native eIF2B holoprotein, freeing the latter to catalyze GDP-GTP exchange on the unphosphorylated pool of eIF2-GDP (21, 29) (Fig. 7C, strains 1 and 2). It is known that excess SUI2 in cells overexpressing only this eIF2 subunit is phosphorylated to high levels by GCN2 in vivo (8, 9). Hence, we predicted that if SUI2 and the eIF2B regulatory subcomplex were cooverexpressed in cells containing GCN2c-M719V-E1537G, the SUI2(P) would compete with eIF2(αP) for binding to the regulatory subcomplex. In this event, the eIF2(αP) would be released from the overexpressed eIF2B regulatory subcomplex and become available to inhibit the GEF activity of native eIF2B holoprotein (Fig. 7C, strain 3). In accordance with this prediction, overexpressing SUI2 from a high-copy-number plasmid restored the slow-growth phenotype conferred by GCN2c-M719V-E1537G in the strain cooverexpressing the eIF2B regulatory subunits (Fig. 7A, compare strains 2 and 3). Importantly, overexpressing SUI2 alone did not exacerbate the slow-growth phenotype of the GCN2c-M719V-E1537G mutant (Fig. 7A). We also verified that overexpressing SUI2 did not reduce the degree of GCD2, GCD7, and GCN3 overexpression (Fig. 7B). These findings provide strong evidence that SUI2(P) can compete effectively with eIF2(αP) holoprotein for interaction with the eIF2B regulatory subcomplex in vivo, implying that SUI2(P) on its own makes strong contacts with the regulatory subunits of eIF2B.
FIG. 7.

Genetic evidence that SUI2 binds individually to the regulatory subcomplex of eIF2B in vivo. (A) Strain H1608 bearing the chromosomal GCN2c-M719V,E1537G allele was transformed with high-copy-number (H.C.) plasmids encoding GCD2, GCD7, and GCN3 (p1871) or SUI2 (pTK29) or with empty vectors (V, pRS425 and pRS426). Isogenic GCN2 strain H1402 was transformed with the empty vectors to provide a wild-type control (WT). The transformants were streaked on SD medium supplemented with inositol and incubated at 30°C for 4 days. (B) WCEs were prepared from the transformants of strain H1608 overexpressing GCD2-GCD7-GCN3 or GCD2-GCD7-GCN3-SUI2 as described for panel A. Forty micrograms of each WCE was resolved by SDS-PAGE and subjected to immunoblot analysis using antibodies against the indicated proteins. (C) A model explaining the possible protein-protein interactions occurring in the transformants described in panel A. (Strain 1) eIF2B holoprotein (labeled 2, 7, 3, 6, 1) interacts with unphosphorylated eIF2 holoprotein (α, β, γ) to exchange the GDP (▴) present on eIF2 for GTP. As these cells contain an activated GCN2c kinase, much of the eIF2 is phosphorylated (●, labeled ∼P) and forms inactive complexes with eIF2B, impeding GDP-GTP exchange on the unphosphorylated eIF2-GDP. This leads to a slow-growth phenotype. (Strain 2) In GCN2c cells overexpressing the GCD2-GCD7-GCN3 regulatory subcomplex of eIF2B (labeled 2, 3, 7), the latter competes with eIF2B holoprotein for the inhibitor, eIF2(αP)-GDP, allowing the eIF2B to exchange GDP for GTP on unphosphorylated eIF2. This suppresses the slow-growth phenotype associated with the GCN2c allele. (Strain 3) Overexpressed SUI2 is phosphorylated in GCN2c cells and competes with eIF2(αP) holoprotein for binding to the eIF2B regulatory subcomplex. This releases eIF2(αP) and reinstates inhibition of eIF2B and the attendant slow-growth phenotype of GCN2c cells. (See Fig. 9 for additional details on the relative orientations of eIF2 and eIF2B subunits in the different complexes.)
DISCUSSION
In previous experiments, we provided genetic and biochemical evidence that eIF2B contains two independent binding sites for eIF2. The GCD1-GCD6 subcomplex was found to be sufficient to catalyze nucleotide exchange on eIF2-GDP. Unlike eIF2B holoprotein, however, its affinity for eIF2 was not increased, and its GEF activity was not inhibited, by phosphorylation of eIF2 on Ser-51. The GCD2-GCD7-GCN3 regulatory subcomplex had no GEF activity, but it bound to eIF2 holoprotein in a manner stimulated by phosphorylation of Ser-51 (21). Hence, we proposed that interaction of eIF2(αP)-GDP with its binding site in the eIF2B regulatory subcomplex would interfere with the ability of the GCD1-GCD6 subcomplex to catalyze nucleotide exchange (22). Consistent with this model, Gcn− mutations were obtained in all three regulatory subunits of eIF2B that abolished the inhibitory effect of eIF2 phosphorylation on eIF2B function in vivo (22, 28). Moreover, several such mutations allowed eIF2B to catalyze nucleotide exchange on eIF2(αP)-GDP in vitro (22). These findings, combined with the identification of additional Gcn− mutations in the N terminus of SUI2 (27), led us to propose that SUI2 interacts directly with the eIF2B regulatory subcomplex dependent on Ser-51 phosphorylation, and that this interaction impedes GDP-GTP exchange by the eIF2B catalytic subcomplex (21). In the context of this model, the Gcn− mutations in SUI2 or the eIF2B regulatory subunits might weaken interaction between SUI2(P) and the regulatory subcomplex, neutralizing the effect of Ser-51 phosphorylation on the GEF activity of eIF2B.
A different view of the effect of eIF2α phosphorylation on interaction between eIF2 and eIF2B was proposed by Kimball and colleagues (17). These workers found that the α subunit of rat eIF2 did not interact with any individual subunits of rat eIF2B in vitro, even when the eIF2α was phosphorylated on Ser-51. In contrast, the C-terminal portion of eIF2β bound to the δ and ɛ subunits of rat eIF2B. Accordingly, they proposed that eIF2α does not directly interact with eIF2B subunits and that its phosphorylation leads to a conformational change in the eIF2 holoprotein that enhances interactions of eIF2β with eIF2Bδ and eIF2ɛ (17). Presumably, this enhanced interaction would be responsible for inhibiting the GEF activity of eIF2B.
In accordance with our model, we found that the α subunit of eIF2 can interact directly with the regulatory subcomplex of eIF2B. As reported by Kimball et al. (17), we observed no stable association between a recombinant form of eIF2α, GST-SUI2, and any individual subunits of eIF2B, with the possible exception of GCD2 (eIF2Bδ). On the other hand, we found that GST-SUI2 formed a tight complex with the eIF2B holoprotein, and also with the GCD2-GCD7-GCN3 regulatory subcomplex, and that both interactions were strongly stimulated by phosphorylation of Ser-51. Hence, we propose that the binding domain for SUI2 in eIF2B requires contributions from all three regulatory subunits. These eIF2B subunits are related in sequence, and most of the Gcn− mutations in these proteins are clustered in two regions of strong similarity (22). Single amino acid substitutions in any one of these proteins is sufficient to overcome the effects of Ser-51 phosphorylation on eIF2B function in vivo. Thus, the homologous segments in GCN3, GCD2, and GCD7 that are altered by these Gcn− mutations may form a multivalent binding surface for SUI2 rather than providing alternative, redundant binding sites. Presumably, SUI2(P) makes more extensive molecular contacts with this binding surface than does unphosphorylated SUI2.
In contrast to its stable interaction with the regulatory subcomplex, we detected no interaction between GST-SUI2 and the catalytic subunits of eIF2B, irrespective of Ser-51 phosphorylation. Although these are negative results, they are in keeping with our proposal that SUI2 interacts primarily with the regulatory subunits of eIF2B. Previously, we detected stable interaction between the N-terminal half of eIF2β and the C-terminal domain of eIF2Bɛ (GCD6) that was important for association between the eIF2 and eIF2B holoproteins in vivo and also for eIF2B function (1). Thus, it appears that eIF2β interacts directly with the catalytic subcomplex in eIF2B. It remains to be seen whether eIF2γ, which contains conserved motifs for guanine nucleotide binding, also interacts directly with the eIF2B catalytic subunits, or whether interaction of the latter with eIF2β leads to a conformational change in eIF2γ that stimulates dissociation of GDP.
We showed previously that overexpression of the GCD2-GCD7-GCN3 regulatory subcomplex overcomes the growth-inhibitory effects of high-level eIF2α phosphorylation in GCN2c cells (29). Based on the tighter binding of eIF2(αP) versus unphosphorylated eIF2 to the regulatory subcomplex (21), we proposed that GCD2-GCD7-GCN3 sequestered eIF2(αP)-GDP and prevented it from competing with unphosphorylated eIF2-GDP for binding to native eIF2B. Here we showed that cooverexpressing SUI2 with GCD2, GCD7, and GCN3 neutralized the ability of the eIF2B subcomplex to rescue growth in cells containing high-level eIF2(αP). These data are consistent with our in vitro binding data showing that SUI2(αP) interacted strongly with GCD2-GCD7-GCN3 independently of the β and γ subunits of eIF2. Hence, we propose that the overexpressed SUI2(αP) sequestered GCD2-GCD7-GCN3 and reduced its association with eIF2(αP) holoprotein, reinstating the inhibition of native eIF2B by eIF2(αP)-GDP. These data provide in vivo evidence that the interaction between GST-SUI2(P) and the eIF2B regulatory subcomplex is an important aspect of the regulatory mechanism. Additional support for this conclusion came from the fact that binding of GST-SUI2(P) to eIF2B in vitro was impaired by all of the Gcn− mutations that we tested. These included mutations mapping in the N-terminal third of SUI2 and GCD7 mutations shown previously to permit eIF2B to catalyze nucleotide exchange on phosphoryated eIF2(αP)-GDP (21, 28). These last results provide strong evidence that tight binding of SUI2(P) to the eIF2B regulatory subunits (in the context of the two haloproteins) is required for inhibition of eIF2B activity by phosphorylated eIF2.
Evidence for multiple contacts between SUI2(P) and the eIF2B regulatory subcomplex.
The Gcn− mutations in SUI2 that weakened binding of GST-SUI2(P) to eIF2B mapped in Glu-49, two residues away from the phosphorylation site, and in Lys-79, Gly-80, and Arg-88, located 30 or more residues away from Ser-51. These findings suggest that two noncontiguous segments in the N terminus of SUI2 are involved in binding to the regulatory subunits of eIF2B. Interestingly, recent findings indicate that eIF2α kinases also have a bipartite binding domain in the N terminus of SUI2. The K3L protein is a pseudosubstrate inhibitor of PKR encoded by vaccinia virus that is 28% identical to the N-terminal one-third of SUI2 and contains a perfect match to residues 79KGYID83 in eIF2α. Truncations or mutations of the 79KGYID83 sequence in K3L abolished its PKR inhibitory activity (16), suggesting that 79KGYID83 is an important binding determinant in SUI2 for its interaction with PKR. In accordance with this hypothesis, mutations that block phosphorylation by GCN2 both in vivo and in vitro have been identified at Glu-49 and 79KGYID83 of SUI2 (Dever, unpublished results). Our finding that SUI2 mutations in residues 49, 80, 83, and 88 impaired interaction between GST-SUI2(P) and eIF2B suggests that there is considerable overlap between the binding domains for eIF2B and eIF2α kinases in SUI2. At the same time, the requirements for binding to eIF2B and eIF2α kinases cannot be identical because most of the SUI2 mutations analyzed here impaired its interaction with eIF2B but did not reduce phosphorylation by eIF2α kinases.
The N terminus of SUI2 (residues 2 to 87) and the K3L protein share sequence similarity with the so-called S1 domain of E. coli ribosomal protein S1 (2, 10) and E. coli polynucleotide phosphorylase (PNPase), whose solution structure is comprised of a five-stranded antiparallel β barrel (2) (Fig. 8). An alignment of the SUI2 and S1 domain sequences suggests that Ser-51 of SUI2 is located in the loop region connecting β strands 3 and 4, whereas 79KGYID83 would reside in the loop between β strands 4 and 5 and extend into the fifth β strand (2). Hence, the two parts of the overlapping binding domains for eIF2B and eIF2α kinases described above may reside within flexible loops located at the N terminus of SUI2 (Fig. 8).
FIG. 8.
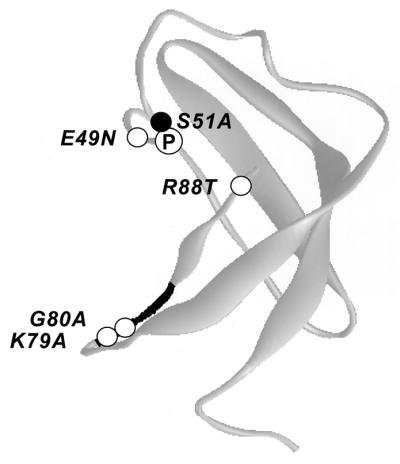
Locations of regulatory mutations in a hypothetical structure of the N-terminal region of SUI2 predicted from the structure of ribosomal protein S1 domain of E. coli PNPase. The three-dimensional structure of the S1 domain of E. coli PNPase (2) is depicted in grey, using the accession code 1SR0 and the program WebLab ViewerLite from Molecular Simulation Inc. Based on a sequence alignment of eIF2α residues 2 to 87 and the S1 domain of PNPase, the Ser-51 phosphorylation site (●, labeled with a circled P) falls in the loop connecting β strands 3 and 4, while the eIF2α kinase recognition motif 79KGYID83 (shown in black) resides in the loop connecting β strands 4 and 5 and extending into strand 5. Indicated in the structure are the predicted locations of Gcn− mutations in SUI2 (O, labeled with amino acid substitutions) that reduce the inhibition of eIF2B by eIF2(αP) in vivo and decrease binding of GST-SUI2(αP) to the eIF2B regulatory subcomplex in vitro.
The model shown in Fig. 9 is an attempt to explain how binding of SUI2(P) to the eIF2B regulatory subunits would impede guanine nucleotide exchange on eIF2-GDP by eIF2B. As suggested previously (21), eIF2B can bind to eIF2-GDP in two ways. In the productive mode of binding, interaction between the catalytic subunit of eIF2B (GCD6) and eIF2β brings GCD6 into proximity with eIF2γ and the bound GDP, and nucleotide exchange occurs (Fig. 9A). (Recall that the C terminus of GCD6 binds to multiple lysine-rich stretches in the N terminus of eIF2β [1]). The productive mode of binding is favored when eIF2-GDP is unphosphorylated and the interaction between SUI2 and the eIF2B regulatory subunits is relatively weak (Fig. 9A). Phosphorylation of SUI2 would lead to new contacts between phosphoserine 51 and the regulatory subunits of eIF2B. It may also produce a conformational change in SUI2 that creates a more extensive interface with the regulatory subcomplex. The resultant tight binding between SUI2(P) and the regulatory subunits disrupts the proper juxtaposition of GCD6 with eIF2βγ and the bound GDP, preventing nucleotide exchange (Fig. 9B). The Gcn− mutations in SUI2 and GCD7 weaken contacts between SUI2 and the eIF2B regulatory subunits and restore the productive interaction of GCD6 with eIF2βγ and the bound GDP when SUI2 is phosphorylated. This allows GDP-GTP exchange on eIF2(αP)-GDP (Fig. 9C).
FIG. 9.

A mechanistic model for negative regulation of the guanine nucleotide exchange activity of eIF2B by eIF2(αP). (A) Unphosphorylated SUI2 promotes the GDP-GTP exchange activity of eIF2B. The heterotrimeric eIF2 (shown as α, β, γ) complexed with GDP (▴) has two binding sites in eIF2B. The GCD2-GCD7-GCN3 regulatory subcomplex in wild-type (WT) eIF2B (labeled 2, 3, 7) binds to the α subunit of eIF2 (SUI2), while the GCD1-GCD6 catalytic subcomplex in eIF2B (labeled 1, 6) interacts with the β and γ subunits of eIF2. Based on results with rat proteins, the GCD2 (δ) subunit of eIF2B may also interact with eIF2β. The binding interactions shown here position the catalytic subunit of eIF2B (GCD6) in proximity to the bound GDP in the manner required to catalyze exchange of GDP for GTP (■) on eIF2. (B) SUI2(P) inhibits the GDP-GTP exchange activity of eIF2B. Phosphorylation of SUI2 [●, labeled ∼P in eIF2(αP)-GDP] leads to more extensive interactions between SUI2 and the eIF2B regulatory subcomplex, preventing productive interactions between GCD6 and the β and γ subunits of eIF2, inhibiting nucleotide exchange. The arrow depicts the proposed shift in eIF2-eIF2B interactions elicited by phosphorylation. (C) A Gcn− mutation in the GCD7 regulatory subunit of eIF2B weakens interaction between SUI2(P) and the regulatory subcomplex of the mutant eIF2B complex (eIF2B*), permitting the interaction between GCD6 and eIF2(αP)-GDP necessary for GDP-GTP exchange.
In the model shown in Fig. 9, the eIF2B regulatory subunits are in contact with eIF2α even in the unphosphorylated state. This can account for our finding that the *M1 and *M2 mutations in GCD7 decreased binding of eIF2B to both eIF2(αP) and eIF2 holoprotein (Fig. 5 and 6), implying that contacts between unphosphorylated SUI2 and the regulatory subunits contribute to the binding energy of the eIF2-GDP-eIF2B complex. This interpretation is consistent with recent findings by Nika et al. (19) that eIF2B will catalyze nucleotide exchange on an eIF2βγ dimer but that the absence of SUI2 increased the Km for eIF2βγ-GDP by an order of magnitude. They concluded that SUI2 is required for structural interactions between the eIF2 and eIF2B holoproteins needed for wild-type rates of nucleotide exchange. Our data suggest that these interactions occur between SUI2 and the eIF2B regulatory subunits. The model in Fig. 9 also provides a reasonable explanation for the fact that phosphorylation of Ser-51 produces a relatively small increase in the stability of the eIF2-eIF2B complex. Although phosphorylation will strengthen association of SUI2 with the regulatory subcomplex, it will simultaneously weaken interaction between eIF2βγ and the catalytic subunits of eIF2B, impeding nucleotide exchange (Fig. 9).
In summary, our results provide strong evidence that SUI2 interacts directly with the eIF2B regulatory subcomplex, independently of the other two subunits of eIF2, and that this interaction is stimulated by Ser-51 phosphorylation. This interaction was disrupted by Gcn− mutations in GCD7 that permit eIF2B to utilize eIF2(αP)-GDP as a substrate and by mutations in SUI2 that abrogate the inhibitory effect of eIF2 phosphorylation on eIF2B function in vivo. Hence, we conclude that tight binding between SUI2 and the eIF2B regulatory subunits is essential for the inhibition of eIF2B activity by phosphorylation of Ser-51. Future experiments will be aimed at defining the binding pocket for phosphoserine 51 in the eIF2B regulatory subcomplex.
ACKNOWLEDGMENTS
We thank Weimin Yang for yeast strains and plasmids. We also thank present and past members of the Hinnebusch and Dever groups for advice and assistance in carrying out this study. We are grateful to Evelyn Sattlegger for comments on the manuscript.
REFERENCES
- 1.Asano K, Krishnamoorthy T, Phan L, Pavitt G D, Hinnebusch A G. Conserved bipartite motifs in yeast eIF5 and eIF2Bɛ, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J. 1999;18:1673–1688. doi: 10.1093/emboj/18.6.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bycroft M, Hubbard T J P, Proctor M, Freund S M V, Murzin A G. The solution structure of the S1 RNA binding domain: a member of an ancient nucleic acid-binding fold. Cell. 1997;88:235–242. doi: 10.1016/s0092-8674(00)81844-9. [DOI] [PubMed] [Google Scholar]
- 3.Chen J-J. Heme-regulated eIF2α kinase. In: Sonenberg N, Hershey J W B, Mathews M B, editors. Translational control of gene expression. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2000. pp. 529–546. [Google Scholar]
- 4.Choi S Y, Scherer B J, Schnier J, Davies M V, Kaufman R J. Stimulation of protein synthesis in COS cells transfected with variants of the α-subunit of initiation factor eIF-2. J Biol Chem. 1992;267:286–293. [PubMed] [Google Scholar]
- 5.Christianson T W, Sikorski R S, Dante M, Shero J H, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 6.Cigan A M, Foiani M, Hannig E M, Hinnebusch A G. Complex formation by positive and negative translational regulators of GCN4. Mol Cell Biol. 1991;11:3217–3228. doi: 10.1128/mcb.11.6.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies M V, Furtado M, Hershey J W B, Thimmappaya B, Kaufman R J. Complementation of adenovirus-associated RNA I gene deletion by expression of a mutant eukaryotic translation initiation factor. Proc Natl Acad Sci USA. 1989;86:9163–9167. doi: 10.1073/pnas.86.23.9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dever T E, Feng L, Wek R C, Cigan A M, Donahue T D, Hinnebusch A G. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992;68:585–596. doi: 10.1016/0092-8674(92)90193-g. [DOI] [PubMed] [Google Scholar]
-
9.Dever T E, Yang W, Åström S, Byström A S, Hinnebusch A G. Modulation of tRNA
 , eIF-2, and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2 · GTP · Met-tRNAiMet ternary complexes. Mol Cell Biol. 1995;15:6351–6363. doi: 10.1128/mcb.15.11.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
, eIF-2, and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2 · GTP · Met-tRNAiMet ternary complexes. Mol Cell Biol. 1995;15:6351–6363. doi: 10.1128/mcb.15.11.6351. [DOI] [PMC free article] [PubMed] [Google Scholar] - 10.Gribskov M. Translational initiation factors IF-1 and eIF-2 alpha share an RNA-binding motif with prokaryotic ribosomal protein S1 and polynucleotide phosphorylase. Gene. 1992;119:107–111. doi: 10.1016/0378-1119(92)90073-x. [DOI] [PubMed] [Google Scholar]
- 11.Hannig E M, Hinnebusch A G. Molecular analysis of GCN3, a translational activator of GCN4: evidence for posttranslational control of GCN3 regulatory function. Mol Cell Biol. 1988;8:4808–4820. doi: 10.1128/mcb.8.11.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinnebusch A G. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. In: Sonenberg N, Hershey J W B, Mathews M B, editors. Translational control of gene expression. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2000. pp. 185–243. [Google Scholar]
- 13.Jones E W. Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol. 1991;194:428–453. doi: 10.1016/0076-6879(91)94034-a. [DOI] [PubMed] [Google Scholar]
- 14.Kaufman R. Double-stranded RNA-activated protein kinase PKR. In: Sonenberg N, Hershey J W B, Mathews M B, editors. Translational control of gene expression. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2000. pp. 503–527. [Google Scholar]
- 15.Kaufman R J, Davies M V, Pathak V K, Hershey J W B. The phosphorylation state of eucaryotic initiation factor 2 alters translational efficiency of specific mRNAs. Mol Cell Biol. 1989;9:946–958. doi: 10.1128/mcb.9.3.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawagishi-Kobayashi M, Silverman J B, Ung T L, Dever T E. Regulation of the protein kinase PKR by the vaccinia virus pseudosubstrate inhibitor K3L is dependent on residues conserved between the K3L protein and the PKR substrate eIF2α. Mol Cell Biol. 1997;17:4146–4158. doi: 10.1128/mcb.17.7.4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimball S R, Heinzinger N K, Horetsky R L, Jefferson L S. Identification of interprotein interactions between the subunits of eukaryotic initiation factors eIF2 and eIF2B. J Biol Chem. 1998;273:3039–3044. doi: 10.1074/jbc.273.5.3039. [DOI] [PubMed] [Google Scholar]
- 18.Murtha-Riel P, Davies M V, Scherer B J, Choi S Y, Hershey J W B, Kaufman R J. Expression of a phosphorylation-resistant eukaryotic initiation factor 2α-subunit mitigates heat shock inhibition of protein synthesis. J Biol Chem. 1993;268:12946–12951. [PubMed] [Google Scholar]
- 19.Nika J, Rippel S, Hannig E M. Biochemical analysis of the eIF2βγ complex reveals a structural function for eIF2α in catalyzed nucleotide exchange. J Biol Chem. 2001;276:1051–1056. doi: 10.1074/jbc.M007398200. [DOI] [PubMed] [Google Scholar]
- 20.Nika J, Yang W, Pavitt G D, Hinnebusch A G, Hannig E M. Purification and kinetic analysis of eIF2B from Saccharomyces cerevisiae. J Biol Chem. 2000;275:26011–26017. doi: 10.1074/jbc.M003718200. [DOI] [PubMed] [Google Scholar]
- 21.Pavitt G D, Ramaiah K V A, Kimball S R, Hinnebusch A G. eIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange. Genes Dev. 1998;12:514–526. doi: 10.1101/gad.12.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavitt G D, Yang W, Hinnebusch A G. Homologous segments in three subunits of the guanine nucleotide exchange factor eIF2B mediate translational regulation by phosphorylation of eIF2. Mol Cell Biol. 1997;17:1298–1313. doi: 10.1128/mcb.17.3.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Proud C G. Protein phosphorylation in translational control. Curr Top Cell Regul. 1992;32:243–369. doi: 10.1016/b978-0-12-152832-4.50008-2. [DOI] [PubMed] [Google Scholar]
- 24.Ramirez M, Wek R C, Vazquez de Aldana C R, Jackson B M, Freeman B, Hinnebusch A G. Mutations activating the yeast eIF-2α kinase GCN2: isolation of alleles altering the domain related to histidyl-tRNA synthetases. Mol Cell Biol. 1992;12:5801–5815. doi: 10.1128/mcb.12.12.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ron D, Harding H P. PERK and translational control by stress in the endoplasmic reticulum. In: Sonenberg N, Hershey J W B, Mathews M B, editors. Translational control of gene expression. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2000. pp. 547–560. [Google Scholar]
- 26.Sudhakar A, Krishnamoorthy T, Jain A, Chatterjee U, Hasnain S E, Kaufman R J, Ramaiah K V. Serine 48 in initiation factor 2α (eIF2α) is required for high-affinity interaction between eIF2 α(P) and eIF2B. Biochemistry. 1999;38:15398–15405. doi: 10.1021/bi991211n. [DOI] [PubMed] [Google Scholar]
- 27.Vazquez de Aldana C R, Dever T E, Hinnebusch A G. Mutations in the α subunit of eukaryotic translation initiation factor 2 (eIF-2α) that overcome the inhibitory effects of eIF-2α phosphorylation on translation initiation. Proc Natl Acad Sci USA. 1993;90:7215–7219. doi: 10.1073/pnas.90.15.7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vazquez de Aldana C R, Hinnebusch A G. Mutations in the GCD7 subunit of yeast guanine nucleotide exchange factor eIF-2B overcome the inhibitory effects of phosphorylated eIF-2 on translation initiation. Mol Cell Biol. 1994;14:3208–3222. doi: 10.1128/mcb.14.5.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang W, Hinnebusch A G. Identification of a regulatory subcomplex in the guanine nucleotide exchange factor eIF2B that mediates inhibition by phosphorylated eIF2. Mol Cell Biol. 1996;16:6603–6616. doi: 10.1128/mcb.16.11.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]