

Significance Statement
Accumulating evidence implicates microRNAs in IgA nephropathy (IgAN), which is a common primary GN in which aberrant IgA aggregated with complement 3, with or without IgG/IgM is deposited in glomerular mesangium. We showed, in a mouse model, that miR-23b deficiency induced an IgA nephropathy-like disease, marked by mesangial IgA and C3 deposition, increased proteinuria, elevated serum IgA levels, and development of high blood pressure. Elevations in serum IgA levels and mesangial IgA deposition were associated with dysregulated mucosal IgA synthesis through targeting the activation-induced cytidine deaminase gene in the gut. miR-23b deficiency-induced dysfunctional kidney effects were likely mediated through regulation of expression of gremlin 2 (Grem2) and the transferrin receptor (Tfrc), which weexamined in human mesangial cells.
Keywords: chronic kidney disease, IgA, immunology
Visual Abstract

Abstract
Background
IgA nephropathy (IgAN) is the most common primary GN worldwide. Circulating immune complexes form that are prone to deposition in the mesangium, where they trigger glomerular inflammation. A growing body of evidence suggests that dysregulated expression of microRNAs in IgAN may play a significant role in establishing the disease phenotype.
Methods
We generated single miR-23b-3p(miR-23b) knockout mice using CRISPR-Cas9.
Results
In humans, miR-23b levels are downregulated in kidney biopsies and sera of patients with IgAN, and serum miR-23b levels are negatively correlated with serum IgA1 levels. We show that miR-23b−/− mice develop an IgAN-like phenotype of mesangial IgA and C3 deposition associated with development of albuminuria, hypertension, an elevated serum creatinine, and dysregulated mucosal IgA synthesis. Dysregulation of IgA production is likely mediated by the loss of miR-23b–mediated suppression of activation-induced cytidine deaminase in mucosal B cells. In addition, we show that loss of miR-23b increases the susceptibility of the kidney to progressive fibrosis through loss of regulation of expression of gremlin 2 and IgA accumulation through downregulation of the transferrin receptor.
Conclusions
Our findings suggest an indispensable role for miR-23b in kidney disease, and in particular, IgAN. miR-23b may in the future offer a novel therapeutic target for the treatment of IgAN.
MicroRNAs (miRs) are short nucleotides that suppress gene expression by hybridizing to the 3′ untranslated region of mRNA, promoting mRNA degradation or disrupting translation. miRs have been implicated in a number of chronic conditions, including cancer, heart disease, and diabetes.1–3 Accumulating evidence also supports a role for miRs in the pathogenesis of a number of different kidney diseases, including AKI, diabetic nephropathy, and multiple glomerulonephritides.4–6
We have previously shown that the peripheral blood from patients with diabetes and the kidneys of animals with type 1 or 2 diabetes have low levels of miR-23b compared with those of their nondiabetic counterparts.7 Furthermore, exposure to high glucose downregulated miR-23b in cultured kidney cells. In vivo, overexpression of miR-23b in db/db mice reversed development of albuminuria and kidney fibrosis, whereas miR-23b antagomir treatment promoted kidney fibrosis and increased albuminuria in wild-type (WT) mice. Importantly, manipulation of miR-23b did not affect serum glucose levels in these experiments, suggesting that miR-23b may have a much broader role outside of diabetic nephropathy in regulating the kidney response to injury. As in vivo delivery of LNA-modified anti-miR oligonucleotides has been associated with off-target effects compared with miR-null mice,8 we elected to generate a miR-23b knockout mouse to more precisely delineate the intrakidney pathways regulated by miR-23b.
miR-23b null mice developed an IgA nephropathy (IgAN)–like disease, characterized by mesangial IgA and C3 deposition associated with elevated serum IgA levels and increased intestinal IgA production. Although this was unexpected, it is consistent with earlier reports of miR-23a cluster-deficient mice having increased numbers of B lymphocytes and miR-23b being linked to regulation of the innate immune response.9 Reflecting these observations in humans, we found reduced levels of miR-23b in the kidneys and sera of patients with IgAN and that levels of miR-23b in the serum negatively correlated with serum IgA1 levels in IgAN.
Methods
Human Samples
For Chinese samples (kidney and serum), the study was approved bythe Ethics Review Board of Mudanjiang Medical University, Heilongjiang (kidney) and Peking University, Beijing (serum) (Supplemental Table 1). For United Kingdom (UK) samples (kidney only), ethical approval was obtained from the Northamptonshire, Leicestershire, and Rutland Ethics Committee (University Hospitals of Leicester 09873).10 Formalin-fixed paraffin-embedded and frozen kidney biopsy cores were obtained with written informed consent from the University Hospitals of Leicester National Health Services Trust and the Affiliated Hongqi Hospital, Mudanjaing Medical University kidney biopsy archives. IgAN cases with rapidly progressive/crescentic GN were excluded from the study. MiR profiles from patients with membranous nephropathy (MN) and thin membrane nephropathy (TMN) were also included to serve as positive CKD and “normal” controls, respectively. In all patients, none were receiving immunosuppression at the time of kidney biopsy. The healthy subject (HS) total RNA was from Biochain Newark, CA (Cat R1234142-P).
Animal Treatment
miR-23b−/− mice were generated by the Model Animal Research Center of Nanjing University, China, using CRISPR-Cas9 technology; a 92 bp fragment of genomic DNA containing the miR-23b was deleted (Supplemental Figure 1, A–C). The miR-23b genotypes were confirmed by PCR using the following primers: forward TGGTCCCTAAGGTATTGGTCTCAT, reverse TTGTTTCCAAAGAGCCACAAGG. All animal experiments were conducted in accordance with protocols approved by Beihua University Animal Care and Veterinary Services. All procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. All mice were housed in sterile individual ventilated cages in specific pathogen-free conditions, and fed water and sterile growth and reproduction food (KeAoXieLi Feed Co., Ltd., Beijing, China). Same-sex and aged 3- to 7-month-old male and female miR-23b−/− mice (littermates) were used in the studies unless otherwise stated. Echocardiography (VEVO 2100, STTARR, Toronto, Canada) and magnetic resonance imaging (MRI) (BioSpec, Bruker, Ettlingen, Germany) were performed on 3- to 5-month-old male mice, before sacrificing the animals. BP measurements (MRBP System, IITC Life Science, USA) were performed by pressing mice tails. After collection, kidney tissue and serum were stored at −80°C.
Cell Culture
Human mesangial cells (HMC) were obtained from ScienCell Research Laboratories and grown in mesangial cell growth supplement (ScienCell Research Laboratories, Cat 4252) with 10 ml of FBS, 5% CO2/95% air. IgA1-positive human B lymphoma cell line, DAKIKI, cells were from American Type Culture Collection. DAKIKI cells were cultured in 75 cm2 flasks in RPMI 1640 (Gibco, Beijing, USA) medium supplemented with 10% FCS (HyClone, USA), 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified environment of 95% atmospheric air and 5% CO2. Transfection of DAKIKI cells and HMC was performed over 48 hours using Lipofectamine2000 (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions with miRNA-23b/24/27b agomir (50 nmol/L; Rabio Co. Guangzhou, China); miR-23b antagomir (100 nmol/L; Rabio Co. Guangzhou, China) and Grem2 siRNA (100 nmol/L; Rabio Co. Guangzhou, China). Scrambled controls were used in parallel. rhPRDC/Grem2 protein (250 ng/ml, R&D Systems, USA) was directly added to HMC. For SMAD4 gain of function experiments, SMAD4-Cas9 activation and control plasmids (3 μg/ml, Santacruz, Texas, USA) were transfected into HMC.
Luciferase Reporter Assay
Aid, Grem2, and Tfrc or Jarid2 gene 3′-UTR luciferase vector containing the miR-23b response elements were amplified by PCR from mouse cDNA. Plasmid DNA and miR-23b agomir (50 nmol/L) and antagomir (100 nmol/L) were cotransfected into HEK293A cells for 48 hours. Luciferase activity was measured using a SpectraMax M5 (Molecular Devices, Sunnyvale, CA, USA) and normalized by measuring β-galactosidase activity. The primers used to generate specific fragments for the mouse AID, GREM2, TFRC and JARID2-related gene 3′-UTR are listed in Supplemental Table 2.
Histologic Analysis of the Kidney and Intestine Tissue
For immunofluorescence and immunochemistry, mouse monoclonal antibodies against IgA (GeneTex, USA), CD19 (Santacruz, Shanghai, China), AID (Invitrogen, Shanghai, China), early B cell factor 1 (EBF1) (Bioss, Beijing, China), IgG, C3, and IgM (Abcam, Shanghai, China) were used. For quantitative histomorphometry, cells stained in ten randomly selected micrographs were counted using Image ProPlus software (Image-Pro Plus, Media Cybernetics, Rockville, MD, USA). Kidney paraffin sections (3 μm) were stained using periodic acid–Schiff (Abcam Hong Kong Ltd., Hong Kong, China) kits, according to the manufacturer’s protocols.
Western Blotting and ELISA
The kidney tissues were homogenized in lysis buffer (ThermoFisher Scientific, Shanghai, China) containing 0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, 5% glycerol, pH 7.4, protease inhibitor (Roche Hong Kong Ltd., Hong Kong, China). Total protein in each sample was quantified using the BCA assay kit (Thermo Fisher Scientific, IL, USA). Samples (20 µg/lane) were resolved by SDS-PAGE, blotted, and probed with the following primary antibodies: anti-Grem2 (GeneTex, CA, USA), anti-BMP2/4/7 (Cell Signaling Technology, Shanghai, China), anti-SMAD1/4/5 (Cell Signaling Technology, Shanghai, China), anti–p-SMAD1/5 (Cell Signaling Technology, Shanghai, China). anti-TFRC (Abcam Hong Kong Ltd., Hong Kong, China). β-actin (Abcam Hong Kong Ltd., Hong Kong, China) staining was used to correct for lane loading, and was performed on each stripped membrane. For serum polymeric and monomeric IgA measurement, we performed a nonreduced SDS-PAGE and Western blot using anti-human IgA antibody (Southern Biotech, AL, USA). ELISA for human IgA1 (Abcam Hong Kong Ltd., Hong Kong, China) and murine IgA (GeneTex, USA), AID (Thermo Fisher Scientific, IL, USA), IgG, and IgM (Abcam Hong Kong Ltd., Hong Kong, China) were performed according to the manufacturer’s instructions. An inhouse ELISA was performed for murine IgA-IgG immune complexes using the same murine IgA and IgG reagents.
Ultrastructural Analysis
Kidney tissues were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at 4°C for 24 h. The samples were then washed with phosphate buffer (0.1 M, pH 7.4) for 12 hours and postfixed for 20 minutes in 1% OsO4 in 0.1 M phosphate buffer (pH 7.4). The samples were then washed with phosphate buffer (0.1 M, pH 7.4) for 30 minutes, dehydrated, and embedded in Epon. Thin sections (50 nm) were placed on copper grids and stained for 30 minutes with a 2% uranyl acetate solution and a 1% solution of lead citrate. A JEM-1010 transmission electron microscope was used to visualize the ultrastructure. Ten randomly selected areas from each specimen were photographed and analyzed using Image ProPlus software (Image-Pro Plus, Media Cybernetics, USA).
MRI
The animals were anesthetized by inhalation of 2% isoflurane and a mixture of O2 and N2O. Bed temperature was maintained at 37.5°C by applying warm water circulation. All MRI data were collected at 9.4T (Bruker Biospec 94/20 USR; Bruker Biospin, Ettlingen, Germany). Mice were placed in the prone position. Then, a 1H volume coil (Bruker Biospin) was used for both radio frequency transmission and signal reception and tuned to 1H resonance frequencies (400.31 MHz). Scout images were acquired using a gradient echo sequence with the following imaging parameters: field of view (FOV), 40 × 40 mm2; image size, 256 × 256; repetition time/echo time, 4/1.5 ms; flip angle, 8°; number of slices, three (axial), eight (coronal), and three (sagittal); slice thickness, 1 mm; and six signal averages. For T1 imaging, IG_FLASH was used and the parameters were echo time, 3.0 ms; repetition time, 202.235 ms; flip angle, 40°; oversampling, 15; image size, 320 × 320; FOV, 40 × 40 mm2; 15 slices were acquired (coronal). For T2 imaging, TurboRARE was used and the parameters were echo time, 30 ms; repetition time, 1800 ms; averages, nine; echo spacing, 10 ms; rare factor, eight; image size, 256 × 256; FOV, 40 × 40 mm2, 15 (coronal) and 20 (axial) slices were acquired, respectively.
RNA Extraction and Analysis
For the tissue extraction, RNA was extracted from the kidneys using a miRNA isolation kit (Ambion Inc., Austin, TX, USA) to separate large and small RNAs, according to the manufacturer’s instructions. miR-23b quantification was carried out by quantitative RT-PCR (RT-qPCR). RT-qPCR (China) was performed using a final reaction volume of 20 μl containing 9 μl Fast Start Universal SYBR Green Master Mix (Roche, Beijing, China), 7.4 μl nuclease-free water, 0.8 μl miRNA primers (Rabio Co. Guangzhou, China), and a 2 μl RT product. The data were normalized to RNU6B small nuclear RNA by a standard curve method to account for differences in reverse transcription efficiencies and the amount of template in the reaction mixtures. RT-qPCR (UK) was performed using TaqMan miR PCR assays on RNA extracted from three pools of at least three randomly selected frozen biopsy cores from the Leicester IgAN, MN, and TMN cohorts. RNA samples (2 ng/μl) were reverse-transcribed using TaqMan microRNA reverse-transcription kit. RT-qPCR was performed using TaqMan universal mastermix with UNG using Taqman PCR assays. miR-191 was used as the housekeeping miR to normalize for expression.
For serum miRNA extraction, three volumes of TRIzol solution (Invitrogen Life Technologies) were added to one volume of serum, vortexed, and then incubated at room temperature for 5 minutes. All IgAN serum samples were from Peking University First Hospital (Supplemental Table 4). Next, 5 μl of 20 pM ath-miR-156a mimic were added to each sample to provide a normalized control. Aqueous and organic phases were separated by adding of molecular grade chloroform (0.2 vol/vol). After vortexing at the maximum setting for 30 seconds and centrifugation at 12,000 rpm for 15 minutes at 4°C, the aqueous phase was rapidly transferred to a new tube, and the TRIzol protocol continued. Finally, total RNA was resuspended in 15 μl of RNase-free water.
Whole Genome Sequencing
The genomic DNA was fragmented and a library constructed according to the BGI protocol. The constructed library was sequenced in MGI200 by PE100, with 32× coverage to the genome. The data were filtered on the basis of quality using the BGI inhouse pipeline. The sites of mutation, insertion, and deletion were called using GATK software with default settings. We did not find any significant off-target genomic changes in this experiment. The whole genome sequencing data from miR-23b−/− mice kidneys are available from Sequence Read Archive (SRA) (accession number SRR8468084).
RNA-sequencing
For murine kidneys, all large and small RNAs were isolated using the mirPremie microRNA Isolation Kit (Ambion Inc., USA). The mRNA libraries were constructed using the TruSeq Stranded Total RNA Library Prep Kit (NEB Inc., Ipswich, MA, USA). Sequencing was conducted using Hi-Seq 2000 sequencers with PE50 at Beijing Genomics Institution (BGI) (Shenzhen, China). The whole kidney RNA-sequencing (RNA-seq) data for 3- and 5-month-old male WT (3 month, n=4; 5 month, n=4) and miR-23b−/− mice (3 month, n=6; 5 month, n=5) are available from SRA (accession numbers listed in Supplemental Table 3).
For human kidney tissue (China), small RNAs were prepared using the microRNA Isolation Kit (Ambion, USA); miRNA-Seq data are available at China National GeneBank (accession number CNP0000326). For kidney tissue (UK) 100 ng of total RNA was converted into microRNA libraries using NEBNEXT library generation kit (New England Biolabs Inc.) and Next Generation Sequencing performed by Exiqon (Vedbaek, Denmark) with samples sequenced on the Illumina NextSeq 500 system. Sequencing data are available at Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) accession number GSE141344. miR sequence data for UK samples were analyzed as previously reported.10
RNA-seq Data Analysis
The raw RNA-Seq data were filtered by trimmomatic (v0.33).11 The clean reads were mapped using Tophat2 (v2.1.1) then counted into different gene regions as corresponding GTF files (mm9, GRCh38 or hg19, GRCh37 genome assembly) by featureCount (v1.5.3).12 The counts of mapped genes were normalized by DEGseq (v1.32.0) with default parameters.13 The significant genes were selected as suggested value P<0.001 and abs(log2foldchange) >0.5 or the program default significance criteria. miRNA target genes were determined using the Bioconductor Package: targetscan.Mm.eg.db and targetscan.Hs.eg.db.14
Results
miR-23b Is Dysregulated in Human IgAN
Next-generation sequencing studies of kidney biopsy tissue from patients with IgAN, MN, and TMN from China and the UK were performed. We found that miR-23b expression was decreased in IgAN cases from China (Figure 1A) and the UK (Figure 1B), compared with HS and patients with MN and TMN. Subsequent RT-qPCR validation studies confirmed downregulation of miR-23b in the kidneys of UK patients with IgAN (Figure 1B, lower panel). In an independent Chinese IgAN cohort, we also found that serum miR-23b levels were significantly lower in patients with IgAN compared with HS (Figure 1C), and negatively correlated with serum IgA1 levels (Figure 1D).
Figure 1.

miR-23b is dysregulated in human IgAN. Next-generation sequencing of kidney biopsy tissue showed that miR-23b was downregulated in kidneys from (A) Chinese and (B, upper panel) UK patients with IgAN, compared with healthy subjects and patients with MN and TMN. RT-qPCR studies (B, lower panel) confirmed downregulation of miR-23b in the kidneys of UK patients with IgAN (n=13) compared with MN (n=6) and TMN (n=7). Furthermore, serum miR-23b levels were significantly lower in patients with IgAN compared with (C) healthy subjects and (D) negatively correlated with serum IgA1 levels in an independent Chinese IgAN cohort. *P<0.05. Data are shown as the mean±SEM.
These data suggested that miR-23b may have a role both in modulating the kidney response to mesangial IgA accumulation in IgAN, consistent with our previous observation of a role for miR-23b in determining the kidney response to hyperglycemia, and may also have a more IgAN-specific effect in influencing the production of pathogenic IgA.
miR-23b−/− Mice Develop an IgAN-like Disease
To characterize the role of miR-23b in kidney disease, we generated miR-23b−/− mice (Supplemental Figure 1, A–C). MiR-23b−/− mice were born at a Mendelian ratio with normal body weight (Figure 2A) and exhibited a natural survival rate. miR-23b expression was absent in 11 organs, confirming complete inactivation of miR-23b (Figure 2B, Supplemental Figure 1, A–C), which was also confirmed by whole genome sequencing (Figure 2C). We noted a concomitant downregulation in expression of miR-23a (Figure 2D) and miR-24/27b (Figure 2E), both of which are located on chromosome 13 near to the miR-23b locus, in miR-23b−/− mice. There was, therefore, no evidence of a compensatory response by miR-23b family members in the miR-23b−/− mice.
Figure 2.
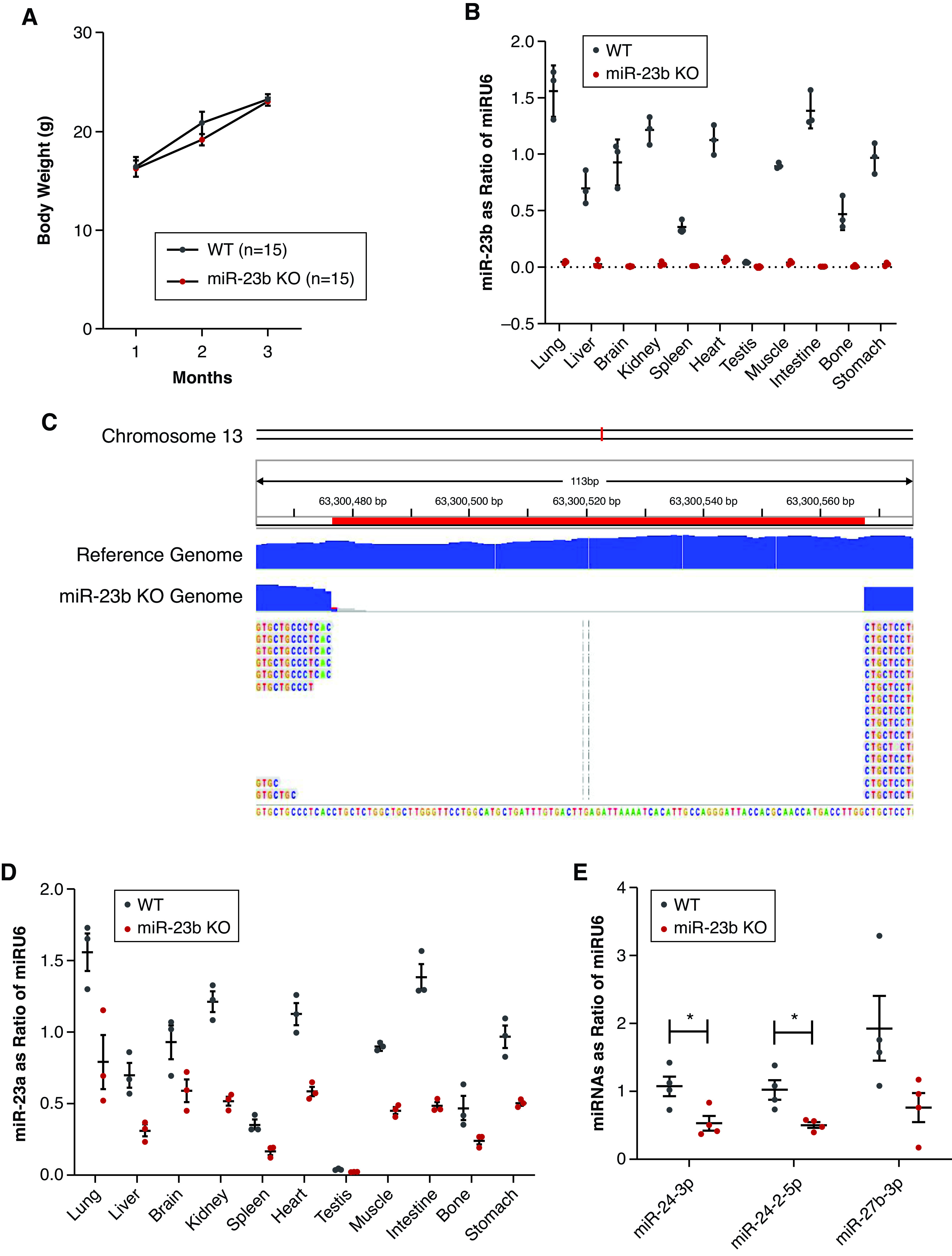
Characterization of the miR-23b−/− mouse. (A) Changes in the body weight of miR-23b−/− and WT mice over the first 3 months. (B) Quantitative analysis of miR-23b in miR-23b−/− mice (n=3) in different tissues by RT-qPCR. (C) Whole-genome sequencing confirmed deletion of the miR-23b sequence. (D) Quantitative analysis of miR-23a in miR-23b−/− mice (n=3) in different tissues by RT-qPCR indicated deletion of miR-23b did not result in cluster compensation. (E) Quantitative analysis of miR-24 and -27 in kidney, two miRs closely located (<1000 bp) to miR-23b on chromosome 13, again indicated an absence of cluster compensation in miR-23b−/− mice (n=3). *P<0.05. Data are shown as the mean±SEM.
Over time, miR-23b−/− mice developed increasing albuminuria and renal impairment (Figure 3, A and B). The 5-month-old miR-23b−/− mice displayed mild segmental multifocal expansion of the mesangial matrix and tubular atrophy on light microscopy (Figure 3, C and D). Electron microscopy (EM) revealed significant mesangial cell proliferation (Figure 3, E and F), an increase in glomerular basement membrane thickening and partial podocyte foot process effacement in miR-23b−/− compared with WT mice (Figure 3, E, G, and H). Immunofluorescence demonstrated IgA and C3 deposition in the glomerular mesangium of miR-23b−/− mice, which was absent in age-matched WT mice (Figure 4A). Immune complex deposits were confirmed by EM (Figure 4A). Older mice (5–7 months) displayed more severe and diffuse mesangial IgA and C3 deposition, again confirmed by EM. miR-23b−/− kidneys also had weak glomerular deposition of IgG and IgM; however, this was not significantly different from that observed in WT mice (Figure 4A).
Figure 3.

miR-23b−/− mice develop a mesangioproliferative GN. (A) Quantification of 24-hour albumin excretion in 5-month-old miR-23b−/− (n=8) and WT (n=4) mice. (B) Quantification of serum creatinine in 5-month-old miR-23b−/− (n=8) and WT (n=5) mice. (C) Representative images of periodic acid–Schiff stained kidney biopsy sections from 5-month-old miR-23b−/− and WT mice. (D) Quantification of the ratio of the mesangial matrix to total glomerular area in miR-23b−/− and WT mice. (E) Representative images of transmission and scanning electron microscope (TEM and SEM) images from 5-month-old miR-23b−/− and WT mice. Red arrows indicate mesangial cells, white arrows indicate foot process fusion, asterisk indicates basement membrane, the bars indicate 2 μm. (F) Quantification of mesangial cell number using the TEM images in miR-23b−/− and WT mice. (G) and (H) Quantification of “tight” pores in podocyte foot processes and glomerular basement membrane thickness in 3-month-old miR-23b−/− and WT mice. **P<0.01 and ***P<0.001. Data are shown as mean±SEM.
Figure 4.

miR-23b−/− mice develop an IgAN-like disease. (A) Representative images of IgA, C3, IgG, and IgM immunofluorescence staining of kidney sections and TEM. TEM images demonstrate the presence of electron-dense mesangial deposits (red arrows) consistent with mesangial IgA immune complex deposition in miR-23b−/− mice. Quantification of (B) serum IgA, (C) IgG, and (D) IgM levels in 5-month-old WT (n=5) and miR-23b−/− (n=8) mice. SDS-PAGE and IgA western blotting of serum IgA from (E) 5-month-old WT (n=11) and miR-23b−/− (n=11) mice with (F) densitometric analysis of polymeric IgA: total serum IgA and monomeric IgA: (G) total serum IgA. (H) Levels of IgA-IgG immune complexes in the sera of 5-month-old WT (n=9) and miR-23b−/− (n=10) mice.
miR-23b−/− Is Associated with Elevated Levels of Serum IgA
Having identified miR-23b−/− was associated with an IgAN-like kidney disease, we went on to measure serum IgA levels, which were elevated 1.5- to two-fold in miR-23b−/− mice by the age of 5 months, compared with WT mice (Figure 4B). We also detected minor changes in serum IgG and IgM levels (Figure 4, C and D). Moreover, we assessed the size distribution of serum IgA by SDS-PAGE and Western blotting and found that, compared with WT mice, there was an increase in the polymeric IgA fraction in the serum of miR-23b−/− mice (Figure 4, E–G). Consistent with this increase we also observed an increase in the amount of IgA-IgG immune complexes in the serum of miR-23b−/− mice, despite there being no increase in total serum IgG (Figure 4H).
miR-23b−/− Is Associated with Dysregulated Mucosal Immunity In The Gut
An increasing understanding of the importance of the gut-kidney axis in IgAN and the relative abundance of miR-23b expression in the intestine (Figure 2B) prompted us to examine intestinal mucosal IgA synthesis in miR-23b−/− mice. We performed immunofluorescence staining of the small intestine, which revealed that the number and percentage of CD19+ IgA+ cells were significantly increased in the small intestine of miR-23b−/− compared with WT mice (Figure 5A). In addition, CD19+ IgA+ cells were also increased in the Peyer’s patches, the ileocecal junction, and the colon in miR-23b−/− mice (Supplemental Figure 2, A and B; Supplemental Figure 3, A and B).
Figure 5.

miR-23b−/− mice develop dysregulated mucosal immunity in the gut. (A) Representative images of CD19 and IgA immunofluorescence and AID immunohistochemistry staining of the intestinal mucosa from 5-month-old WT and miR-23b−/− mice. (B) Representative images of AID, CD19, and IgA immunofluorescence staining of the intestinal mucosal of 5-month-old WT and miR-23b−/− mice. (C) Luciferase activity in HEK 293A cells transfected with an AID 3′-UTR reporter construct demonstrates binding of miR-23b with the 3′-UTR of each reporter. *P<0.05, **P<0.01. Data are shown as mean±SEM.
As part of our bioinformatics analysis to identify putative miR-23b target genes, we identified activation-induced cytidine deaminase (AICDA or AID). AID is a member of the cytidine deaminase family, is mainly expressed in B cells, and is intimately involved in somatic hypermutation and IgA class switch recombination.15,16 We found increased expression of AID in the small intestine tissue of miR-23b−/− mice (Figure 5A). Importantly, AID was mainly expressed in CD19+ IgA+ cells and the number of these cells was increased in the small intestine, Peyer’s patches, and ileocecum in miR-23b−/− mice compared with WT mice (Figure 5B, Supplemental Figure 2, A and B). Separately, we confirmed that miR-23b could suppress AID activity by binding to the 3′UTR using a luciferase assay (Figure 5C).
To further understand the potential drivers of mucosal B cell proliferation in the miR-23b−/− mouse, we examined the expression of EBF1. EBF1 is a key transcriptional regulator of B cell differentiation, and has previously been shown to be negatively regulated by miR-23a.9 miR-23b−/− mice displayed increased numbers of CD19+ EBF1+ cells in the small intestine compared with WT controls (Supplemental Figure 2C).
Finally, to confirm that the observations in the miR-23b−/− mouse were relevant to the human, we demonstrated a direct effect of miR-23b on B cell IgA synthesis by transfecting the surface IgA1-positive human B lymphoma DAKIKI cell line with miR-23b. Transfection resulted in reduced expression of both AID and IgA1, confirming that miR-23b is capable of regulating human B cell synthesis of IgA1 (Supplemental Figure 3, C–F). Suppression of IgA1 synthesis was not observed after transfection with miR-24 and miR-27b, both of which are located on chromosome 13 near to the miR-23b locus (Supplemental Figure 3G).
Deletion of miR-23b Resulted in Structural Kidney Changes Consistent with Development of Progressive Glomerular Disease
To assess kidney development, 3- to 5-month-old miR-23b−/− mice underwent noninvasive MRI and kidney ultrasonography. Consistent with the observed development of albuminuria and renal impairment miR-23b−/− mice had significantly thinner kidney cortices by ultrasonography that was confirmed with MRI (Figure 6, A–C). In addition, the cortico-medullary junction was indistinct in miR-23b−/− mice compared with WT mice (Figure 6A). Although the cortical structure changed noticeably, the thickness of the medulla was not significantly different in miR-23b−/− mice (Figure 6, A, D, and E). This is consistent with the observed kidney weight and the ratio of kidney weight to body weight in miR-23b−/− mice (Supplemental Figure 1, E and F).
Figure 6.
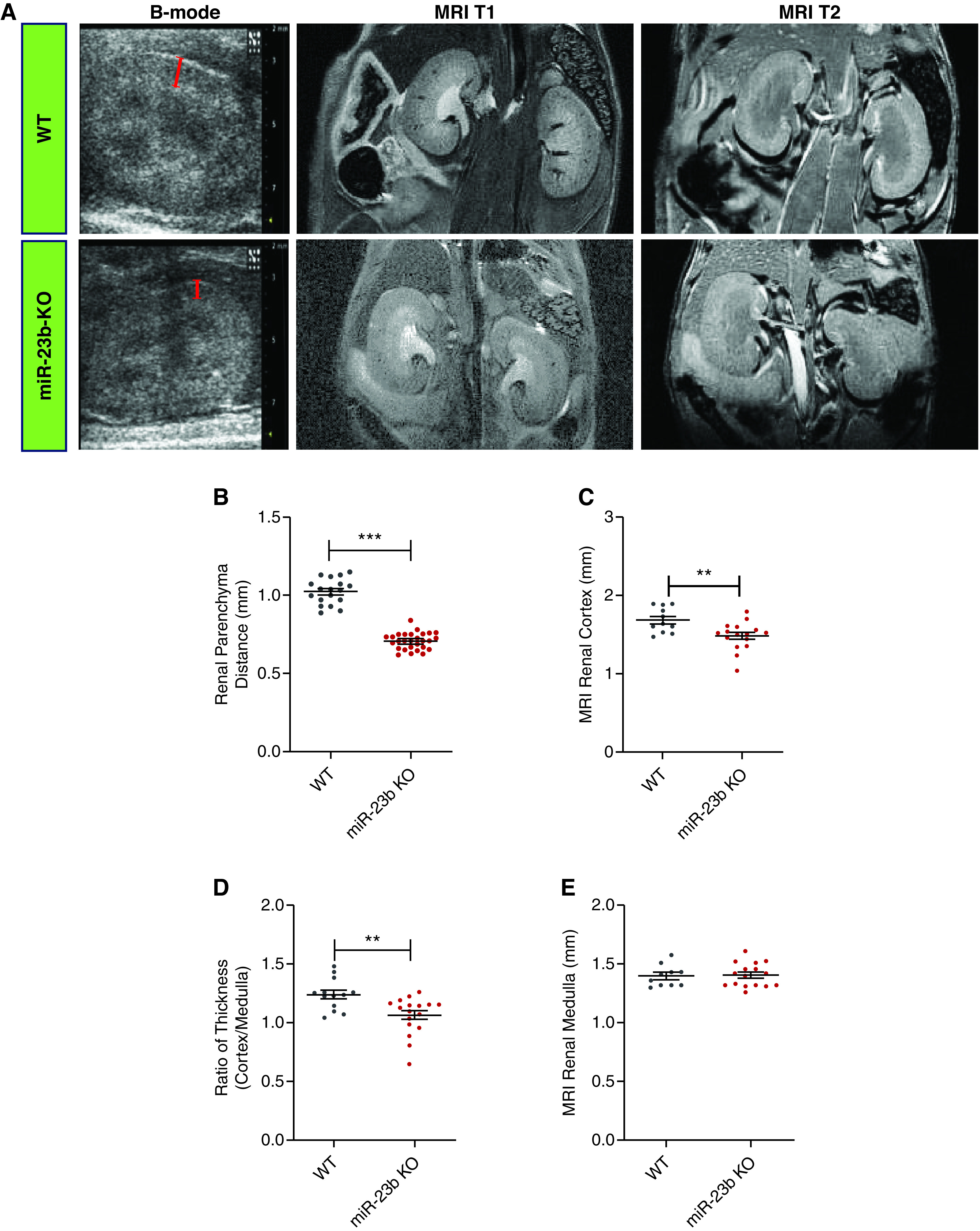
Deletion of miR-23b induces structural kidney changes. (A) Representative images of B-mode ultrasonography of the kidney (first panel, bars estimating the renal parenchyma distance) in WT (n=5) and miR-23b−/− (n=7); MRI T1/2 images (second and third panels) in WT (n=10), and miR-23b−/− (n=12) 5-month-old male mice. (B) Quantification of the renal parenchymal thickness measured from kidney ultrasound images. (C) Quantification of the renal cortical thickness measured from MRI T2 images. (D) Quantification of the ratio of MRI T2 cortex to medulla thickness. (E) Quantification of MRI T2 medulla thickness; *P<0.05, **P<0.01, and ***P<0.001. Data are shown as the mean±SEM.
miR-23b−/− Is also Associated with The Development of Hypertension
The kidney artery resistance index (the peak systolic and end-diastolic blood velocities divided by the peak systolic velocity) was measured by color-Doppler in the intrarenal artery. We observed a progressive increase in kidney artery resistance in miR-23b−/− mice detected by ultrasound from 2 to 6 months (Figure 7, A and B, Supplemental Figure 4A). In parallel, progressive blood pressure elevation was seen in 1- to 5-month-old miR-23b−/− mice relative to WT mice (Figure 7C). In an attempt to understand the molecular effect of this elevation in blood pressure on the kidneys, we performed RNA sequencing of 3- and 5-month-old miR-23b−/− kidneys (Supplemental Tables 1 and 2). KEGG analysis of the 3- and 5-month miR-23b−/− RNA-seq data identified the renin angiotensin system as a significantly enriched term (Figure 7D).17 We went on to show that the renin angiotensin system–related protein renin was significantly upregulated in 3- and 5-month-old miR-23b−/− kidneys (Figure 7E, Supplemental Figure 4, B and C).
Figure 7.
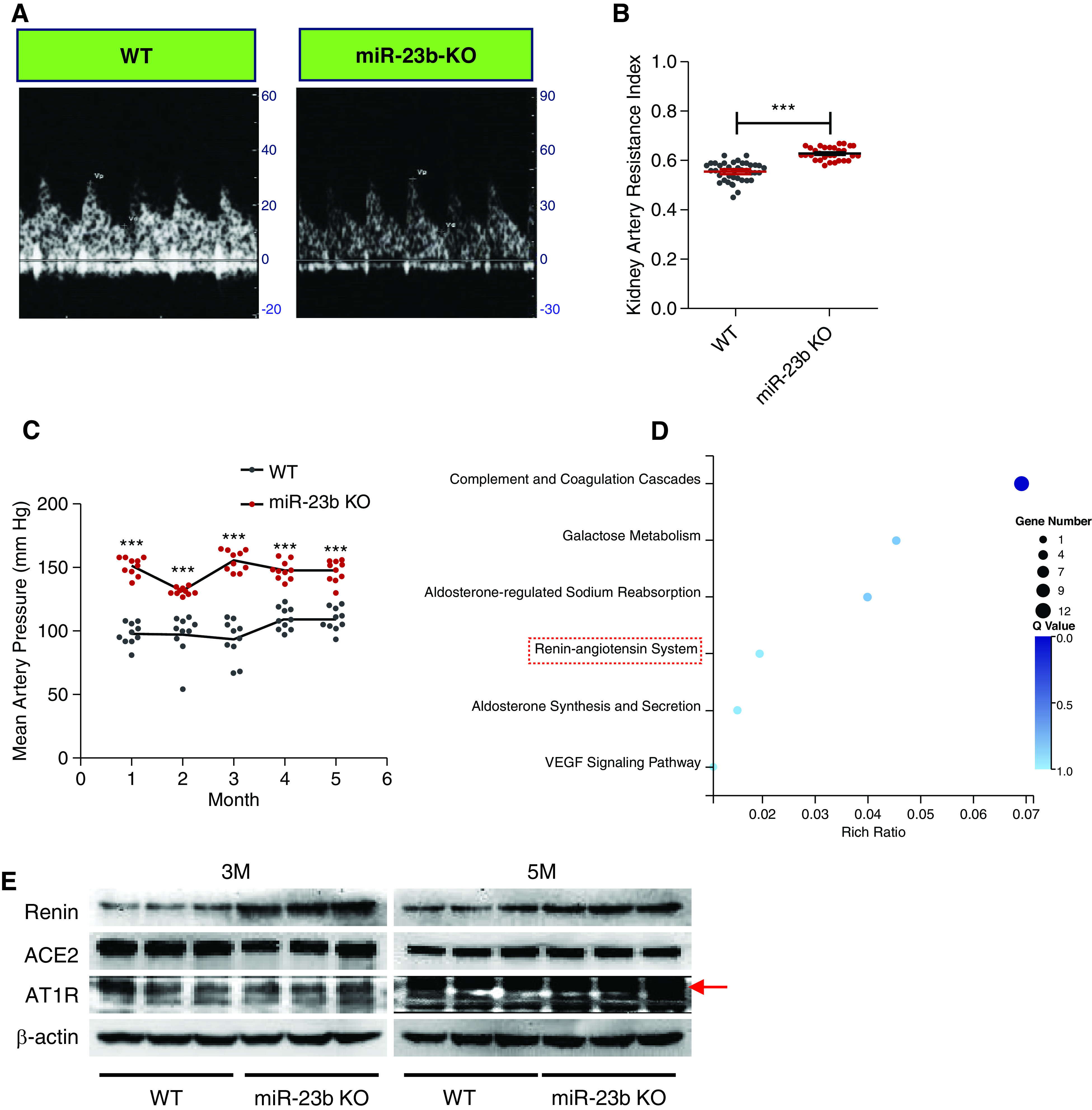
miR-23b−/− mice develop hypertension. (A) Representative Doppler flow images of the intrarenal artery in 5-month-old WT (left image) and miR-23b−/− (right image) mice. (B) Quantification of the kidney artery resistance index (KRI, the peak systolic and end-diastolic blood velocities divided by the peak systolic velocity) in 5-month-old male WT (n=18) and miR-23b−/− (n=21) mice. (C) Quantification of mean arterial blood pressure in WT (n=10) and miR-23b−/− (n=10) mice. (D) KEGG analysis of the 3-month miR-23b−/− RNA-seq data highlighted the complement and coagulation cascades and renin angiotensin system as significantly enriched terms. (E) Western blot analyses for renin, angiotensin converting enzyme 2, and angiotensin II receptor type 1, in 3- and 5-month-old miR-23b−/− and WT mice. **P<0.01, ***P<0.001. Data are shown as the mean±SEM.
miR-23b Deficiency Leads to Kidney Dysfunction through Bone Morphogenetic Protein Signaling
Having shown that miR-23b is likely to play a role in human IgAN, we analyzed potential downstream transcriptional targets for miR-23b in the kidneys of miR-23b−/− mice. Analysis of the 3- and 5-month miR-23b−/− kidney RNA-seq data revealed distinct patterns of transcript expression (Figure 8A). The majority of transcripts that were increased in 3-month miR-23b−/− kidneys were subsequently decreased in 5-month miR-23b−/− kidneys (Figure 8, B and C). Such transcriptional diversification was more apparent in potential miR-23b targets (14 genes, predicted using the Targetscan database) (Figure 8, B and D). We performed GO term analysis for these significantly differentially expressed 14 genes and found bone morphogenetic protein (BMP) signaling was the most enriched term (Figure 8E).
Figure 8.
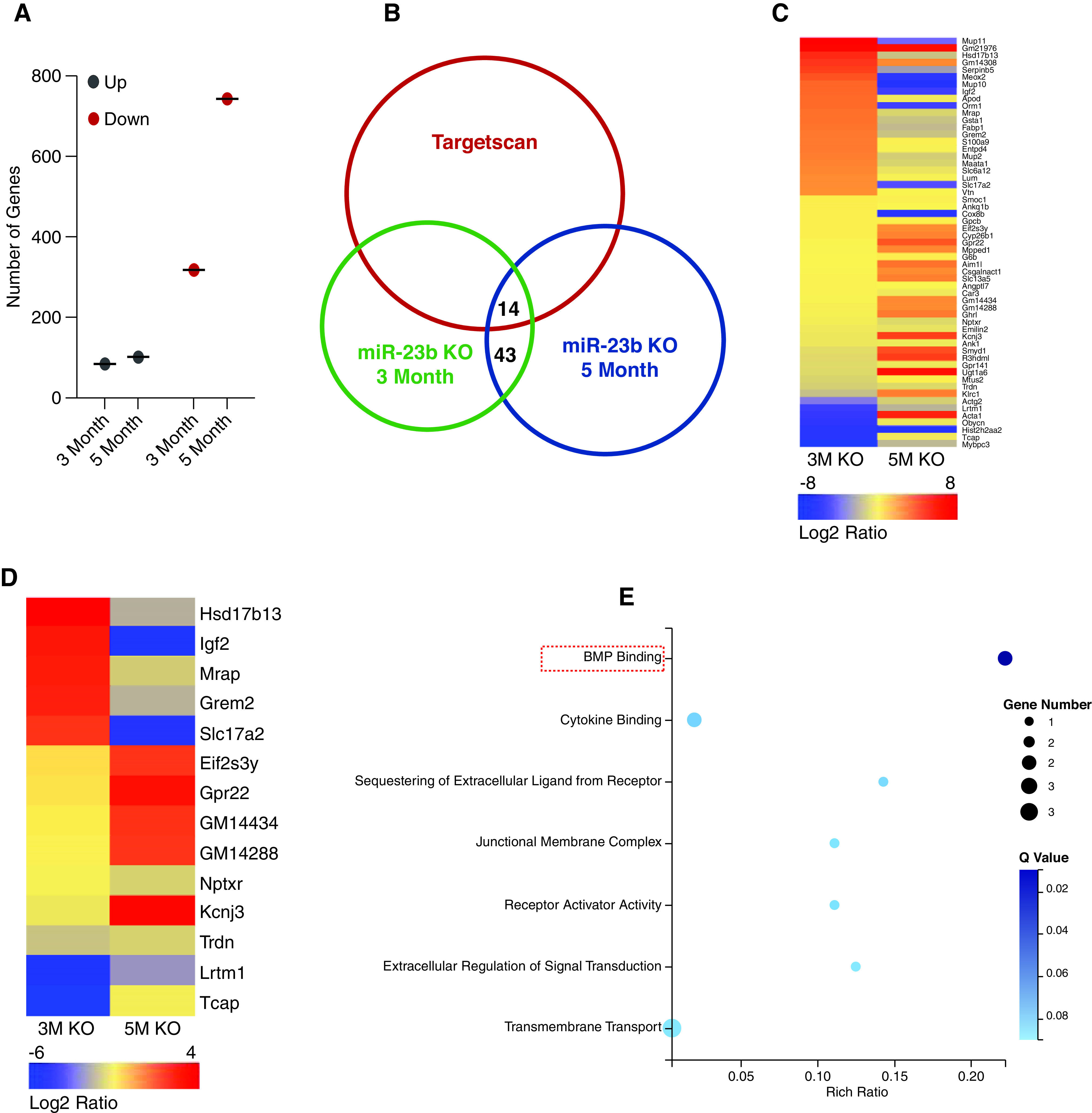
Analysis of the 3- and 5-month miR-23b −/− kidney RNA-seq data revealed distinct patterns of transcript expression. (A) Quantification of dysregulated genes in the kidneys of 3- and 5-month-old miR-23b−/− mice. (B) Venn diagram showing the number of genes in the kidneys with significant changes in both 3- and 5-month-old miR-23b−/− mice and those identified as potential miR-23b targets using the Targetscan database. (C) and (D) Heatmaps of differentially expressed genes and potential miR-23b targets in the kidneys of 3- and 5-month-old miR-23b−/− mice for all genes (C) and only those potential miR-23b targets (D), (the color gradation indicates a log2 fold change in gene expression). (E) GO term analysis for those genes dysregulated in the kidneys of both 3- and 5-month-old miR-23b−/− mice and potential miR-23b targets.
The miR-23b target gene GREM2 encodes a member of the BMP antagonist family, with the antagonistic effect of the secreted glycosylated protein encoded by this gene likely mediated by direct binding to BMP proteins. Using a luciferase assay we first confirmed miR-23b was capable of specifically suppressing GREM2 activity (Figure 9A). Consistent with GREM2 suppression by miR-23b, we observed elevated Grem2 expression in 3-month-old miR-23b−/− kidneys, which was associated with downregulation of BMP pathway–related proteins, reflecting in vivo Grem2-mediated antagonism of BMP signaling in the kidney (Figure 9B, Supplemental Figure 5). By contrast, at 5 months, the level of Grem2 in the miR-23b−/− kidneys had reduced with consequent upregulation of BMP pathway–related proteins. This later suppression of Grem2 may reflect in vivo upregulation of miR-23b–independent transcriptional regulatory pathways.
Figure 9.
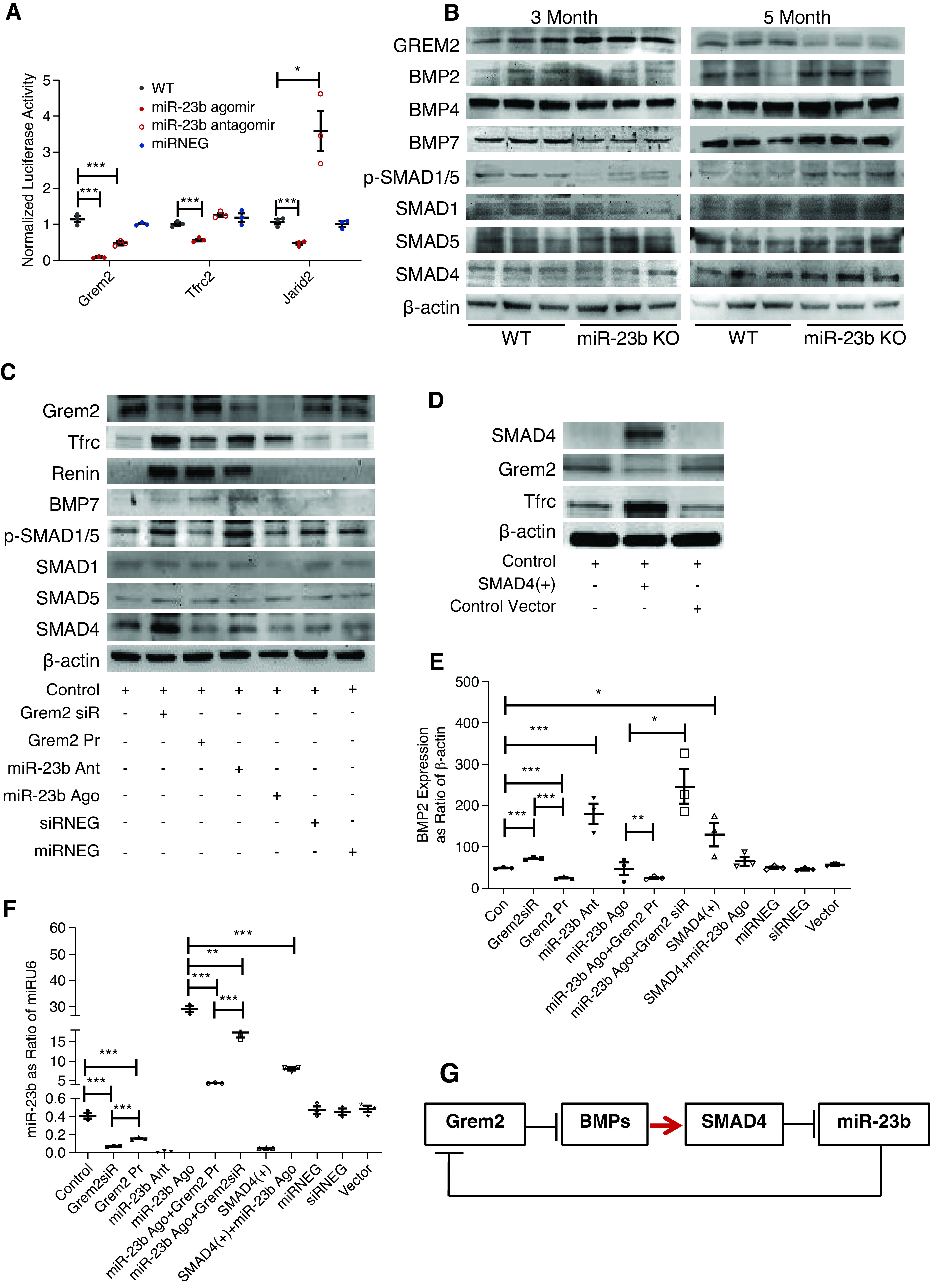
miR-23b regulates the BMP pathway through Grem2. (A) Luciferase activity in HEK 293A cells transfected with GREM2, TFRC, and JARID2 3′-UTR reporter constructs demonstrated binding of miR-23b to the 3′-UTR of each reporter. (B) Western blot analysis for Grem2 and BMP pathway related proteins in kidneys of 3- and 5-month-old WT and miR-23b−/− mice. (C) Western blot analysis of BMP pathway–related proteins and TFRC in HMC driven to overexpress or downregulate Grem2 and miR-23b. (D) Western blot analysis of SMAD4, Grem2, and TFRC in HMC driven to overexpress SMAD4; RT-qPCR quantification of BMP2 (E) and miR-23b (F) expression in HMC treated with Grem2 siRNA and protein, miR-23b agomir, and antagomir, miR-23b agomir combined with Grem2 siRNA or protein, SMAD4-activated plasmid, SMAD4-activated plasmid combined with miR-23b agomir, and miRNA or siRNA-negative control. (G) A schematic model of potential regulation and function of miR-23b. *P<0.05, **P<0.01, ***P<0.001. Data are shown as the mean±SEM.
To study the effects of miR-23b–mediated regulation of Grem2 protein expression in more detail, we established an in vitro system to study the downstream signaling pathways in HMC. In this system there was constitutive expression of Grem2 by HMC (Figure 9C). Addition of a miR-23b mimic or a Grem2 siRNA resulted in reduced Grem2 protein expression and an increase in expression of BMP pathway related proteins (Figure 9C, Supplemental Figure 6). This likely reflects both BMP-dependent (antagonism of HMC-derived BMPs) and BMP-independent cellular actions of Grem2. Addition of a miR-23b antagomir did not appreciably affect levels of Grem2 protein, suggesting in this system constitutive expression of miR-23b was likely to be low.
miR-23b−/− was also associated with the development of hypertension and KEGG analysis of miR-23b−/− RNA-seq data had highlighted the renin angiotensin system as a significantly enriched term (Figure 7D). We found low basal expression of renin in HMC; however, levels increased after exposure to a miR-23b antagomir consistent with miR-23b–mediated suppression of renin synthesis in HMC (Figure 9C, Supplemental Figure 6). This offers a plausible explanation for why miR-23b−/− mice developed high kidney artery resistance index and hypertension. Paradoxically, both Grem2 siRNA and exogenous Grem2 protein also induced renin expression in HMC.
The transferrin receptor (TFRC) is one of the IgA receptors expressed by HMC and is responsible for mesangial IgA clearance and HMC activation. In light of the IgAN-like phenotype of miR-23b−/− mice, we wanted to determine if there was a relationship between Grem2, BMP signaling, and HMC TFRC expression. SMAD4 is a key molecule in the BMP signaling cascade and, because we had already shown that suppression of Grem2 was associated with increased SMAD4 expression, we elected to overexpress SMAD4 in vitro to mimic BMP pathway activation. We observed both an upregulation in TFRC protein and a reduction in Grem2 (Figure 9D, Supplemental Figure 6G), suggesting that mesangial IgA accumulation in miR-23b−/− mice may have been triggered by an early suppression of HMC TFRC expression due to Grem2-mediated suppression of BMP signaling, resulting in impaired mesangial IgA clearance by TFRC-mediated endocytosis. However, at 5 months the development of compensatory Grem2 suppression and increased BMP signaling and TFRC expression is likely to have contributed to the glomerular injury secondary to mesangial IgA accumulation.
Finally, when we measured the levels of BMP2 in HMC, we found that miR-23b constitutively suppressed BMP2 and this suppression was lost when a Grem2 siRNA was simultaneously applied to HMC (Figure 9E). By contrast, SMAD4 overexpression resulted in a significant increase in BMP2 expression. SMAD-4–induced activation of the BMP pathway significantly decreased expression of miR-23b expression (Figure 9F). Paradoxically, both exogenous Grem2 and Grem2 siRNA suppressed miR-23b levels in HMC.
Overall these results indicate that miR-23b is regulated by the Grem2, BMP, and SMAD4 cascade, probably through a miR-23b/Grem2 feedback loop (Figure 9G).
Discussion
In this study, we found that deletion of miR-23b resulted in a novel murine phenotype characterized by development of albuminuria, impairment of kidney function, hypertension and an IgA-dominant mesangioproliferative GN which could not be explained by miRNA cluster member compensation. We went on to demonstrate that these mice also developed increased circulating levels of IgA and that this is likely to be driven by excessive mucosal IgA synthesis, specifically intestinal IgA synthesis. These observations are consistent with dysregulation of putative miR-23b target genes and in particular dysregulation of intestinal IgA synthesis.18 The unbiased mRNA profiling of the kidney and GO and KEGG term enrichment analysis combined with identification of potential miR-23b targets using the Targetscan database showed that a separate miR-23b/Grem2/BMP axis could in part explain the kidney-specific action of miR-23b observed in this and other studies.
There is increased recognition of the importance of the gut-kidney axis in IgAN, with the Peyer’s patch–rich small intestine being hypothesized to be a major source of pathogenic IgA.19 This pathogenic IgA is polymeric, poorly O-galactosylated, and prone to form high molecular weight immune complexes, both through self-aggregation and interaction with IgA and IgG antibodies with specificity for the IgA1 hinge region. Mesangial deposition of these IgA-containing immune complexes triggers a variable mesangioproliferative GN, resulting in development of progressive CKD in many patients.20 In this study, deletion of miR-23b resulted in elevated levels of serum IgA, specifically polymeric IgA, and a clear intestinal phenotype with proliferation of mucosal CD19+ IgA+ EBF1+ AID+ B cells, and in particular, an increase in CD19+ IgA+ B cells in the Peyer’s patches of the small intestine. We hypothesize that miR-23b may, such as miR-23a, be acting through EBF1, to regulate B cell proliferation and through AID to regulate IgA class-switch recombination and affinity maturation. Intriguingly, a recent report describes a hα1+/+ murine IgAN model in which deletion of AID resulted in a more severe IgAN phenotype, characterized by significantly greater IgA deposition and complement activation.21 Importantly, when we overexpressed miR-23b in human DAKIKI cells, we found downregulation of IgA and AID, consistent with our hypothesis that the observed miR-23b−/− phenotype is in part mediated through AID. Further elucidation of the pathways involved is required.
The miR-23b −/− mouse resembles human IgAN in a number of ways, and may provide a novel murine model to facilitate better understanding of the drivers for both mucosal dysregulation and kidney fibrosis in IgAN. Previously, miR-23b has been shown to play a role in development of autoimmune disease through regulation of TGF-β–activated kinase 1/MAP3K7 binding protein 2, TGF-β–activated kinase 1/MAP3K7 binding protein 3, and inhibitor of nuclear factor κ-B kinase subunit a.22 In this study, miR-specific bioinformatic analysis of RNA-seq data from the kidneys of 3- and 5-month-old miR-23b−/− mice identified the BMP pathway, regulated by Grem2, which is a putative miR-23b target, as one of the most enriched terms and likely to be responsible in part for the observed kidney phenotype. BMP-signaling related proteins have been both associated with protection of mesangial cells from polymeric IgA–induced injury and shown to play broader roles in the development of kidney fibrosis and hypertension.23,24 The TFRC is a key HMC receptor for IgA and plays a role in both the clearance of mesangial IgA deposits and HMC activation in IgAN. The TFRC gene contains a putative SMAD4 binding site and we observed that overexpression of SMAD4, as a surrogate for enhanced BMP signaling, led to TFRC upregulation in HMC. This raises the possibility that mesangial accumulation in the miR-23b −/− mice was due to both excess mucosal IgA synthesis, combined with Grem2-mediated suppression of BMP/SMAD4 signaling in the kidney and reduced TFRC-mediated IgA mesangial clearance.
An additional broader role for miR-23b in development of progressive kidney failure was suggested in our previous study, where we showed that overexpression of miR-23b in db/db mice could reverse the albuminuria and kidney fibrosis associated with development of diabetes, independent of any effect on glycemic control. In addition, miR-23b antagomir treatment promoted renal fibrosis and increased albuminuria in WT mice, again in the absence of any effect on glycemic control. The actions of miR-23b in this model of diabetic nephropathy were largely mediated through a miR-23b/G3BP2 feedback circuit involving p38MAPK and p53. In this study, we have again shown that deletion of miR-23b results in kidney fibrosis that is independent of changes in serum glucose. Thus, it appears that one role of miR-23b in the kidney is to regulate the development of kidney fibrosis.
Intriguingly, when we analyzed the RNA-seq data from 3- and 5-month miR-23b−/− kidneys, we saw definite changes in the pattern of transcript expression with age, and this was particularly marked for putative miR-23b target genes. We speculate that this transcriptional diversification likely represents the differential intra- and extrarenal effect of miR-23b loss during the natural history of disease development. One of the early manifestations of loss of miR-23b was the development of hypertension and, therefore, early transcript changes likely reflect hemodynamic-induced changes in the kidneys. By contrast, the later transcript changes were likely related to the direct effects of mesangial immune complex accumulation, which increased with age as a result of elevated serum polymeric IgA levels due to excess intestinal IgA synthesis secondary to extrarenal loss of miR-23b. Furthermore, intrarenal loss of miR-23b compounded the effects of hypertension and immune complex deposition by amplifying generic downstream signaling pathways, including the BMP pathway, that are intimately involved in kidney fibrosis.
In summary, our findings suggest an indispensable role for miR-23b in kidney disease, and in particular, IgAN. Although further dissection of the molecular mechanisms mediating the intra- and extrarenal actions of miR-23b are required, miR-23b may in the future offer a novel therapeutic target for the treatment of IgAN.
Disclosures
J. Barratt reports having consultancy agreements with Astellas, Alnylam, BioCryst, Calliditas, Chinook, Dimerix, Novartis, Omeros, Travere Therapeutics, Vera Therapeutics, and Visterra; reports receiving research funding from argenx, Calliditas, Chinook, Galapagos, GlaxoSmithKline, Novartis, Omeros, Travere Therapeutics, and Visterra; and reports being a scientific advisor or member via the Editorial Board of CJASN, Glomerular Diseases & Clinical Science, and Kidney International. All remaining authors have nothing to disclose.
Funding
This work was supported, in part, by the Natural Science Foundation of China (NSFC/81770856 and NSFC/81970644 to B. Zhao, NSFC/81601541 to K. Wang, NSFC/81925006 to J. Lv) and Heilongjiang Province Natural Science Foundation (H201496) and (LH2020H067) to B. Zhao and K. Wang, respectively. This work was supported by Jilin Province Innovation Centre for Science and Technology Program (JPIC/20190902013TC to B. Zhao). Work in the UK was supported by Kidney Research UK and the Mayer Family Foundation.
Acknowledgments
B. Zhao designed the experiments, analyzed the data, and contributed to the discussion; J. Barratt, J. Lv, and B. Zhao reviewed and edited the manuscript; W. Chen, Z. Chen, and M. Shi performed the bioinformatics analysis; K. Wang performed the MRI research; S. Zhang performed the transmission electron microscope, H. Li, and I.Z.A. Pawluczyk experiments; and the other authors performed research and collected the data. J. Li performed the immunofluorescence experiments and contributed to the manuscript discussion. All authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2021010133/-/DCSupplemental
Supplemental Figure 1. The generation and phenotypic characterization of miR-23b KO mice.
Supplemental Figure 2. miR-23b−/− mice develop B cell proliferation in the gut.
Supplemental Figure 3. Overexpression of miR-23b inhibits IgA1 production in DAKIKI cells.
Supplemental Figure 4. Deletion of miR-23b induces activation of the renin angiotensin aldosterone system.
Supplemental Figure 5. 3- and 5-month-old miR-23b−/− kidneys display distinct patterns of BMP transcript expression.
Supplemental Figure 6. miR-23b and its target Grem2 regulate the BMP pathway.
Supplemental Table 1. Clinical characteristics of IgAN patients in the China Cohort.
Supplemental Table 2. miR-23b target gene 3′-UTR primers.
Supplemental Table 3. RNA sequencing upload data accession numbers in SRA.
Supplemental Table 4. Number of up- and down-regulated mRNAs and predicted miRNA targets in 5-months old miR-23b-/- mice.
Supplemental Table 5. Number of up- and down-regulated mRNAs and predicted miRNA targets in 3-months old miR-23b-/- mice.
References
- 1.Gebert LFR, MacRae IJ: Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 20: 21–37, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Creemers EE, Tijsen AJ, Pinto YM: Circulating microRNAs: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res 110: 483–495, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Guay C, Regazzi R: Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat Rev Endocrinol 9: 513–521, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Collino F, Bruno S, Incarnato D, Dettori D, Neri F, Provero P, et al. : AKI recovery induced by mesenchymal stromal cell-derived extracellular vesicles carrying microRNAs. J Am Soc Nephrol 26: 2349–2360, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, et al. : Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol 23: 252–265, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trionfini P, Benigni A, Remuzzi G: MicroRNAs in kidney physiology and disease. Nat Rev Nephrol 11: 23–33, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Zhao B, Li H, Liu J, Han P, Zhang C, Bai H, et al. : MicroRNA-23b targets Ras GTPase-activating protein SH3 domain-binding protein 2 to alleviate fibrosis and albuminuria in diabetic nephropathy. J Am Soc Nephrol 27: 2597–2608, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, et al. : Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Invest 120: 3912–3916, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurkewich JL, Bikorimana E, Nguyen T, Klopfenstein N, Zhang H, Hallas WM, et al. : The mirn23a microRNA cluster antagonizes B cell development. J Leukoc Biol 100: 665–677, 2016 [DOI] [PubMed] [Google Scholar]
- 10.Pawluczyk IZA, Didangelos A, Barbour SJ, Er L, Becker JU, Martin R, et al. : Differential expression of microRNA miR-150-5p in IgA nephropathy as a potential mediator and marker of disease progression. Kidney Int 99: 1127–1139, 2021 [DOI] [PubMed] [Google Scholar]
- 11.Bolger AM, Lohse M, Usadel B: Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao Y, Smyth GK, Shi W: featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930, 2014 [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Feng Z, Wang X, Wang X, Zhang X: DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26: 136–138, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. : Combinatorial microRNA target predictions. Nat Genet 37: 495–500, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T: Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563, 2000 [DOI] [PubMed] [Google Scholar]
- 16.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. : Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell 102: 565–575, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Passos-Silva DG, Brandan E, Santos RA: Angiotensins as therapeutic targets beyond heart disease. Trends Pharmacol Sci 36: 310–320, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Petersen C, Bell R, Klag KA, Lee SH, Soto R, Ghazaryan A, et al. : T cell-mediated regulation of the microbiota protects against obesity. Science 365: eaat9351, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Floege J, Feehally J: The mucosa-kidney axis in IgA nephropathy. Nat Rev Nephrol 12: 147–156, 2016 [DOI] [PubMed] [Google Scholar]
- 21.Wehbi B, Oblet C, Boyer F, Huard A, Druilhe A, Paraf F, et al. : Mesangial deposition can strongly involve innate-like IgA molecules lacking affinity maturation. J Am Soc Nephrol 30: 1238–1249, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novak J, Rizk D, Takahashi K, Zhang X, Bian Q, Ueda H, et al. : New insights into the pathogenesis of IgA nephropathy. Kidney Dis 1: 8–18, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y, et al. : The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat Med 18: 1077–1086, 2012 [DOI] [PubMed] [Google Scholar]
- 23.Hwangbo C, Lee HW, Kang H, Ju H, Wiley DS, Papangeli I, et al. : Modulation of endothelial bone morphogenetic protein receptor type 2 activity by vascular endothelial growth factor receptor 3 in pulmonary arterial hypertension. Circulation 135: 2288–2298, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan WL, Leung JC, Chan LY, Tam KY, Tang SC, Lai KN: BMP-7 protects mesangial cells from injury by polymeric IgA. Kidney Int 74: 1026–1039, 2008 [DOI] [PubMed] [Google Scholar]