

Significance statement
AKI has been recognized as a common complication of coronavirus disease 2019 (COVID-19) and is associated with disease severity and mortality. The mechanisms behind these associations remain obscure, due, in part, to unsuccessful attempts to consistently detect the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in urine, despite evidence of kidney tropism. This study consistently quantifies the SARS-CoV-2 genome via quantitative RT-PCR in cells of urine sediments from patients with COVID-19. It was found that viral load in urine sediment was higher within 2 weeks of the AKI event among patients with COVID-19, and it correlated with increased risk of death. Quantification of viral load in urine sediment offers a noninvasive approach that could help identify and care for those patients with COVID-19 who are at higher risk of kidney injury and poor outcome.
Keywords: acute renal failure, virology, renal injury, mortality risk, COVID-19
Visual Abstract

Abstract
Background
AKI is a complication of coronavirus disease 2019 (COVID-19) that is associated with high mortality. Despite documented kidney tropism of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), there are no consistent reports of viral detection in urine or correlation with AKI or COVID-19 severity. Here, we hypothesize that quantification of the viral load of SARS-CoV-2 in urine sediment from patients with COVID-19 correlates with occurrence of AKI and mortality.
Methods
The viral load of SARS-CoV-2 in urine sediments (U-viral load) was quantified by qRT-PCR in 52 patients with PCR-confirmed COVID-19 diagnosis, who were hospitalized between March 15 and June 8, 2020. Immunolabeling of SARS-CoV-2 proteins Spike and Nucleocapsid was performed in two COVID-19 kidney biopsy specimens and urine sediments. Viral infectivity assays were performed from 32 urine sediments.
Results
A total of 20 patients with COVID-19 (39%) had detectable SARS-CoV-2 U-viral load, of which 17 (85%) developed AKI with an average U-viral load four-times higher than patients with COVID-19 who did not have AKI. U-viral load was highest (7.7-fold) within 2 weeks after AKI diagnosis. A higher U-viral load correlated with mortality but not with albuminuria or AKI stage. SARS-CoV-2 proteins partially colocalized with the viral receptor ACE2 in kidney biopsy specimens in tubules and parietal cells, and in urine sediment cells. Infective SARS-CoV-2 was not detected in urine sediments.
Conclusion
Our results further support SARS-CoV-2 kidney tropism. A higher SARS-CoV-2 viral load in urine sediments from patients with COVID-19 correlated with increased incidence of AKI and mortality. Urinary viral detection could inform the medical care of patients with COVID-19 and kidney injury to improve prognosis.
The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in late 2019, and the associated coronavirus disease 2019 (COVID-19) rapidly became a pandemic.1 Patients with severe COVID-19 present with dyspnea and hypoxemia, leading to respiratory failure. However, SARS-CoV-2 does not exclusively infect respiratory tract cells, as documented in recent studies in enterocytes, endothelial cells, and kidney cells.2–5 It is not clear to what extent this multiorgan tropism contributes to poor COVID-19 outcome, mainly due to lack of conclusive studies addressing viral presence in target organs in relation with COVID-19 complications. One of the most common complications of COVID-19 is AKI, which occurs in up to 49% of patients, with higher rates in minority groups,6–11 and is linked to poor COVID-19 prognosis and mortality.12–14 Despite the documented presence of SARS-CoV-2 in kidney tissue,2–5,15,16 efforts to detect viral genetic material or infective particles in urine have been inconclusive. Most studies reported an absent or very low detection rate of viral genetic material in whole urine, and only few laboratories have examined the presence of infective SARS-CoV-2.17–21 This lack of in-depth urine analysis has prevented the establishment of an association between urine SARS-CoV-2 load and kidney injury, precluding us from taking advantage of an easily accessible, noninvasive sample as a diagnostic tool for patients with COVID-19. It is unclear why virus detection in urine is so challenging. Possible reasons could be related to variability in urine volume, incomplete replication or release by infected kidney cells, or virus destruction in urine by excreted proteases and RNAses. All of these factors could be minimized using detection methodologies with increased sensitivity that measure the presence of SARS-CoV-2 directly in cells of renal origin, such as the cells shed in urine sediments. Here, we tested the hypothesis that the viral load of SARS-CoV-2 can be quantified in urine sediment and that it correlates with occurrence of AKI and risk of death among patients with COVID-19. A better understanding of the effect of SARS-CoV-2 presence in the kidney, its detection in urine, and its relationship to kidney damage could better inform medical decisions to treat patients with COVID-19.
Methods
Patients
During the time frame of our study, 1320 patients were admitted to the Henry Ford Hospital in Detroit, Michigan, with a clinical diagnosis of COVID-19. Between March 15 and June 8, 2020, a cohort of 652 patients enrolled in the Translational and Clinical Research Center biobank at the Henry Ford Hospital. From our biobank, 52 patients were enrolled with a COVID-19 diagnosis confirmed by laboratory RT-PCR of nasopharyngeal swabs or tracheal aspirates. Clinical history, course of COVID-19, and clinical chart diagnosis of AKI were abstracted from the electronic medical record. Subsequently, the presence of AKI, its Kidney Disease Improving Global Outcomes stage, and the presence of RRT was adjudicated by two independent and blinded nephrologists. Any discrepancy was reconciled by a third nephrologist. The results of the adjudication were used for all analyses reported (Figure 1). The institutional review board at Henry Ford Health System approved specimen collection and biobanking. Each patient, or their legal representative, provided informed consent before participation, and only deidentified data were analyzed. Urine specimens were collected from 14 consented, healthy, control volunteers; 18 patients with COVID-19 who had no AKI; and 33 patients with COVID-19 in the following weeks after an AKI episode (up to 8 weeks). Only one patient in the cohort was not available for specimen collection after the AKI episode and was excluded from the quantitative RT-PCR (qRT-PCR) analysis.
Figure 1.
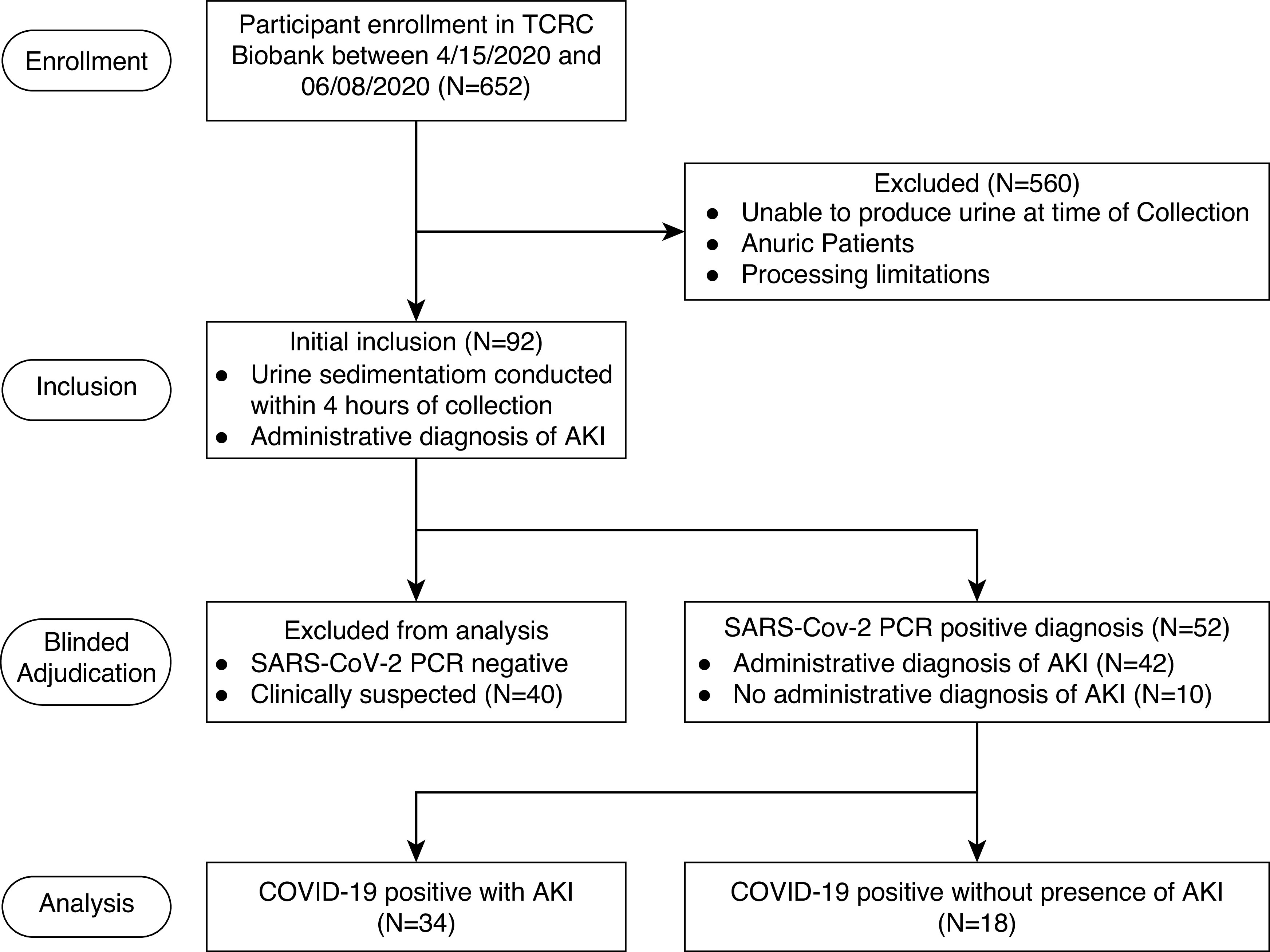
Study design and inclusion criteria. Flowchart showing the exclusion and inclusion criteria used in this study to select the study population of patients with COVID-19 who did or did not have AKI during their COVID-19–related hospital stay. TCRC, Translational and Clinical Research Center.
Urine Sediment Specimen Collection
Urine samples were obtained as spontaneously voided or bladder-catheterized specimens, and only included in the study if fresh urine sample could be obtained and processed immediately by study staff. Urine sediments were obtained within 2–4 hours of sampling by centrifugation at 1000 × g for 10 minutes, followed by two washes with cold PBS buffer. All samples were handled in a biosafety level 2 laboratory after approval from the internal Institutional Recombinant DNA and Biosafety Committee. Viral RNA was extracted by resuspending urine sediments in lysis buffer from the viral RNA extraction kit (Takara Bio USA, Ann Arbor, MI), following the manufacturer’s instructions. RNA was quantified in a Qubit 4 Fluorometer using the Qubit RNA High Sensitivity Assay Kit (Thermo Fisher Scientific, Waltham, MA). To estimate RNA sample purity, the ratios of the absorbance at 260 nm and 280 nm were measured by NanoDrop (Gen5 Synergy H1 plate reader; BioTek, Winooski, VT) in separate aliquots of ten samples of the highest RNA concentration, and were determined to be, on average, 2.3±0.3.
Real-Time RT-PCR for SARS-CoV-2 in Urine Sediments
Viral RNA was extracted from urine sediments using a commercial kit (Takara Bio USA). Between 10 and 500 ng of RNA were reverse transcribed using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) and random hexamers. Of the resulting cDNA from this reaction, 5 μl were used for real-time PCR in a ViiA 7 Real-Time PCR System (Applied Biosystems, Foster City, CA). The cDNA was diluted at 1:10, 1:20, or 1:50 to ensure the measurements were within the linear range of the standard curve (Supplemental Figure 1). Oligonucleotide primers specific for the SARS-CoV-2 Spike (S) and Nucleocapsid (N) genes were previously developed. The S primer set22 had the following sequences: forward, 5′-CAATGGTTTAACAGGCACAGG-3′; reverse, 5′-CTCAAGTGTCTGTGGATCACG-3′. The N primers corresponded to the N2 set developed by the Centers for Disease Control (CDC; https://www.cdc.gov/coronavirus/2019-ncov/downloads/rt-pcr-panel-primer-probes.pdf). All primers were synthetized by Eurofins Genomics (Louisville, KY). Real-Time SYBR Green PCR mix was from Bio-Rad (Hercules, CA). Urine sediments from 14 healthy, nonsymptomatic, controls, with no history of COVID-19 or AKI, were analyzed to establish background RT-PCR signal. Viral RNA copies were quantified with a standard curve of heat-inactivated SARS-CoV-2 complete genome (catalog number NR-52347, lot number 70033926; National Institutes of Health [NIH], National Institute of Allergy and Infectious Diseases, BEI Resources, Manassas, VA). The number of viral copies per nanograms of RNA was calculated as: viral copies×PCR dilution factor/nanograms RNA.
The intra- and interassay coefficients of variation were 0.9%±0.1% and 1.8%±0.3%, respectively, as determined by triplicates that were run in parallel and sequentially. Single-band amplification was verified by gel electrophoresis.
SARS-CoV-2 Infectivity Assay
Virus infectivity was assessed at a biosafety level 3 facility at Michigan State University Veterinary Diagnostic Laboratory. African green monkey Vero E6 cells (ATCC, Manassas, VA) were grown at 80% confluency in Medium 199 with Earle Balanced Salts (MilliporeSigma, St. Louis, MO) supplemented to 6% US origin FBS (Millipore Sigma). Urine sediment lysate was brought to 1 ml with Bovarnick buffer (pH 7.0), and then centrifuged at 8,000 × g for 5 minutes to pellet debris. The clarified liquid was passed through a 0.22-μm syringe filter directly onto cell cultures. Cells were cultured and monitored daily for cytopathic effects for up to 10 days. At the end of this period, RNA was extracted and real-time qRT-PCR was performed for SARS-CoV-2 genes N and S to confirm presence or absence of viral replication.
Immunofluorescence and Confocal Microscopy
Kidney biopsy sample slides were obtained from two patients with RT-PCR–confirmed COVID-19 and two patients negative for COVID-19 (negative controls) by the Translational and Clinical Research Center. Biopsies were indicated for unexplained nephrotic-range proteinuria with clinical concern for rapidly progressing GN in patients with COVID-19. Formalin-fixed, paraffin-embedded slices were first deparaffinized with xylene and then rehydrated gradually through 100% ethanol to distilled water. Slides were incubated for 10 minutes with 0.05% Triton X-100 at room temperature (RT) for permeabilization followed by antigen retrieval using citric acid buffer (10 mM, pH 6.0). Slides were then blocked with 5% BSA to block nonspecific binding. Slides were incubated with primary antibodies, either a 1:200 dilution of SARS-CoV-2 spike mAB (clone 1A9; Gentex, Irvine, CA) or SARS-CoV-2 nucleocapsid antibody (clone 6H3; Gentex), at 4°C overnight. This was followed by 1:100 of Alexa Fluor 647 donkey anti-mouse IgG (Molecular Probes, Eugene, OR) for 1 hour at RT. To label angiotensin-converting enzyme 2 (ACE2), slides were then incubated with a 1:50 dilution of ACE2 rabbit mAB (clone EPR4435; Abcam, Cambridge, MA) after blocking with 1% BSA, followed by incubation with 1:100 Cyanine3 donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA) for 1 hour at RT. Finally, slides were counterstained with 4′,6-diamidino-2-phenylindole at a dilution of 1:2000 for 5 minutes at RT. Controls for nonspecific secondary antibody binding were performed without primary antibodies, and were completely negative. COVID-19–negative and COVID-19–positive slides were imaged the same day, with identical settings, using a Leica TCS SP8 MP confocal/multiphoton microscopy system (Leica Microsystems, Wetzlar, Germany), with a 63×/1.40 oil CS2 Harmonic Compound Plan Apochromat lens. We also tested antibodies against SARS-CoV-2 nucleocapsid protein (A50, 1:250; Rockland Antibodies, Limerick, PA) or SARS-CoV-2 membrane protein (A55, 1:200; Rockland Antibodies).23 All of these antibodies have recently been used by other investigators to detect SARS-CoV-2 in respiratory tissue and placenta,5,24 and in human liver duct organoids.25
Albumin-Creatinine Ratio
Spontaneously voided or bladder-catheterized urine samples were obtained from hospitalized patients and briefly centrifuged (1000 × g, 10 minutes) to eliminate particulate material. Supernatants were used for determination of urine albumin (catalog number ab108788; Abcam) and creatinine (BioAssay, Hayward CA) concentration with ELISA kits. The measured urine albumin and creatinine concentrations were used to calculate the albumin-creatinine ratios (ACRs) in milligrams per gram.
Statistical Analyses
For descriptive purposes, continuous variables were summarized using means and SDs, and categoric variables are summarized using frequency counts and percentages. Between-group differences in continuous variables were assessed using the nonparametric Kruskal–Wallis test. For categoric variables, between-group frequency differences were assessed using the Fisher exact test. Correlations between continuous variables were tested using Spearman rank correlations. All statistical analyses were performed using GraphPad Prism 8.2.1 (GraphPad Software, San Diego, CA), with the exception of the analysis for inpatient mortality. The association between the viral load of SARS-CoV-2 in urine sediments (U-viral load) and inpatient COVID-19 mortality was performed using the Fine and Gray competing risk model with the R statistical programming language package “cmprsk.” In this analysis, inpatient mortality was modeled as the primary hazard, and alive at hospital discharge was modeled as the secondary hazard. Patients who were still in the hospital and alive as of June 8, 2020 were censored. To account for multiple admissions, the total number of inpatient days across all admissions for a patient was considered the inpatient time to event. Hazard ratios (HRs) and 95% CIs for the association of U-viral load with mortality were calculated both unadjusted and adjusted for age, sex, and Black race/ethnicity. P values <0.05 were considered statistically significant.
Results
SARS-CoV-2 Detection in Urine Sediments from Patients with COVID-19 and AKI
The baseline characteristics of our study cohort of 52 patients with a confirmed COVID-19 diagnosis are summarized in Table 1. The mean±SD follow-up time from COVID-19–related admission was 53.7±17.9 days. Two SARS-CoV-2 genes (S and N) were successfully detected by RT-PCR and quantified by extrapolation to a standard curve of the SARS-CoV-2 genome (Supplemental Figure 1). A mean±SD U-viral load of 1730±344 copies/ng RNA was measured in non-AKI COVID-19 specimens (NS versus background in COVID-19–negative controls, P=0.38; Figure 2A). In specimens from patients with COVID-19 who developed AKI, the U-viral load was 3.9-times higher (P=0.001; mean±SD, 6779±1479 copies/ng RNA) compared with background in COVID-19–negative controls (Figure 2A). In patients where urine was collected within the first 2 weeks after an AKI episode, the U-viral load was significantly higher (13,266±3033 copies/ng RNA, P=0.004 versus non-AKI; Figure 2B), returning to levels comparable with non-AKI after 2 weeks (3212±897 copies/ng RNA, P>0.99 versus non-AKI). To assess the prevalence of high U-viral titers among patients with AKI, we set a threshold of three SDs of the background signal from noninfected controls. Approximately half of the patients with AKI had U-viral loads above this threshold, compared with only three out of 18 (17%) of the non-AKI group (P=0.02; Figure 2C), indicating a larger proportion of patients with AKI who had a higher SARS-CoV-2 viral load in urine sediments. The amount of RNA purified from patients with COVID-19 with or without AKI was not significantly different (P>0.99; Supplemental Figure 2) and there was no correlation between U-viral load and starting amount of RNA in the RT-PCR reaction (Spearman ρ correlation coefficient=0.08, R2= 0.006), indicating that viral load in patients with AKI is not artificially high due to increased cell shedding. To rule out additional possible confounding factors, we further determined that the method of urine collection (spontaneous voiding versus catheterization), the presence of blood in urine, or requirement of mechanical ventilation were not associated with higher U-viral loads (Supplemental Figure 3).
Table 1.
Baseline characteristics of the study cohort
| Characteristics | COVID-19 | COVID-19 and AKI | COVID-19 and No AKI | P Value |
|---|---|---|---|---|
| Total | 52 (100) | 34 (65.4) | 18 (34.6) | |
| Age (years) | 62±13 | 64±12 | 58±15 | 0.24 |
| Sex, n (%) | 0.02 | |||
| Male | 30 (57.7) | 24 (80.0) | 6 (20.0) | |
| Female | 22 (42.3) | 10 (45.5) | 12 (54.5) | |
| Race, n (%) | 0.42 | |||
| Black | 37 (71.2) | 23 (62.2) | 14 (37.8) | |
| White | 11 (21.2) | 8 (72.7) | 3 (27.3) | |
| Hispanic | 2 (3.8) | 2 (100) | 0 | |
| Asian | 1 (1.9) | 1 (100) | 0 | |
| Unknown | 1 (1.9) | 0 | 1 (100) | |
| Height (m), mean±SD | 1.72±0.11 | 1.75±0.09 | 1.67±0.09 | 0.008 |
| Weight (kg), mean±SD | 94.9±26.4 | 97.1±25.3 | 90.9±28.7 | 0.34 |
| BMI (kg/m2), mean±SD | 32.04±8.65 | 31.47±6.98 | 33.11±11.30 | 0.90 |
| Comorbidities, n (%) | ||||
| Hypertension | 41 (33.1) | 28 (68.3) | 13 (31.7) | 0.48 |
| Heart Failure | 12 (9.7) | 6 (50.0) | 6 (50.0) | 0.30 |
| Diabetes | 27 (21.8) | 21 (77.8) | 6 (22.2) | 0.08 |
| COPD | 9 (7.2) | 5 (55.6) | 4 (44.4) | 0.70 |
| CKDa | 35 (28.2) | 28 (80.0) | 7 (20.0) | 0.004 |
| Urine volume (ml), mean±SD | 55.2±24.3 | 55.0±17.6 | 55.7±34.2 | 0.66 |
| Serum creatinine (mg/dl), mean±SD | 1.9±1.8 | 2.2±1.8 | 1.7±2.0 | 0.02 |
| Albuminuria (ACR>30 mg/g), n (%) | 33 (63.5) | 20 (60.6) | 13 (39.4) | 0.38 |
| CRP >3 mg/L (34 on record; 65.4%), n (%) | 23 (67.6) | 15 (65.2) | 8 (34.8) | 0.46 |
| Urinary tract infection, n (%) | 12 (23.1) | 8 (66.7) | 4 (33.3) | >0.99 |
| Catheterized, n (%) | 27 (51.9) | 22 (81.5) | 5 (18.5) | 0.02 |
| Blood in urine, n (%) | 12 (23.1) | 8 (66.7) | 4 (33.3) | >0.99 |
| On ventilator, n (%) | 30 (57.7) | 25 (83.3) | 5 (16.7) | 0.003 |
| Mortality, n (%) | 12 (23.1) | 10 (83.3) | 2 (16.7) | 0.18 |
Continuous variables are summarized as means±SDs. P values were calculated using the Kruskal–Wallis test (for continuous variables) or the Fisher exact test (for categoric variables). BMI, body mass index; COPD, chronic obstructive pulmonary disorder; CRP, C-reactive protein.
CKD presence determined by chart diagnosis during medical record abstraction. A breakdown of CKD stages follows: stage 1, n=1; stage 2, n=9; stage 3a/b, n=11; stage 4, n=9; stage 5/ESKD, n=5.
Figure 2.

Higher presence of SARS-CoV-2 genetic material was measured in urine sediments of patients with COVID-19 who developed AKI. (A) U-viral load was higher in patients with COVID-19 and AKI, whereas it was not significantly different from background in patients with COVID-19 without AKI. (B) U-viral load was significantly higher in patients with COVID-19 and AKI during the first 2 weeks after the AKI diagnosis. (C) A larger percentage of patients with COVID-19 and AKI had detectable U-viral loads compared with patients with COVID-19 without AKI (P<0.05, Fisher exact test). For patients with COVID-19, n=51; for healthy controls, n=14. Each dot represents an individual data point, average viral copies per nanogram RNA is indicated (dotted line) and SEM. Dark-colored dots represent the same individuals in (A) and (B) with the highest viral loads. #P<0.05 versus background; *P<0.05 versus non-AKI, Kruskal–Wallis test.
Altogether, these results indicate higher levels of SARS-CoV-2 genetic material in urine sediments from patients with COVID-19 who develop AKI.
U-Viral Load and COVID-19 Severity
U-viral load was associated with COVID-19 inpatient mortality (P<0.001), with each increase of 10,000 viral copies/ng RNA being associated with a 2.87-fold (95% CI, 1.76 to 4.68) increased risk of death, and this increase in risk was largely unchanged after adjustment for age, sex, and Black race/ethnicity (HR, 3.28; 95% CI, 1.77 to 6.06; P<0.001). Among subjects who died, the U-viral load reached a high of 11,073±2816 copies/ng RNA at an average of 9 days before death. This viral load was three times higher than the average 3377±860 copies/ng RNA (P=0.03) recorded in subjects that were still alive at time of cutoff (Figure 3A). Figure 3B displays the cumulative incidence of inpatient mortality in those patients below and above the average of the highest recorded U-viral load (5141 copies/ng RNA) across all patients. At 60 days, those above and below this threshold had cumulative mortality rates of 62% and 9%, respectively, with a corresponding HR of 11.5 (95% CI, 3.27 to 40.30; P<0.001). Table 2 puts some clinical perspective on the subgroup of patients who died. A large majority had developed AKI (83.3%), and only two were in the non-AKI group. As expected, the predominant cause of death was respiratory and systemic complications from COVID-19. The only non-COVID-19–related death on record was one case of metastatic cancer. This patient was in the non-AKI group and had low measurable U-viral load (539 copies/ng RNA), suggesting that the poor outcome was not related to COVID-19. Besides the higher U-viral load associated with subsequent risk of death, we did not observe any correlation between mortality and duration of COVID-19, need of ventilation, sepsis, or changes in BP (Table 2). However, because the number of patients who died (n=12) was relatively small, the statistical power was low. Similarly, we did not observe any strong evidence of risk factors associated with patients presenting with the highest U-viral loads >5141 copies/ng RNA in our cohort (Supplemental Table 1).
Figure 3.
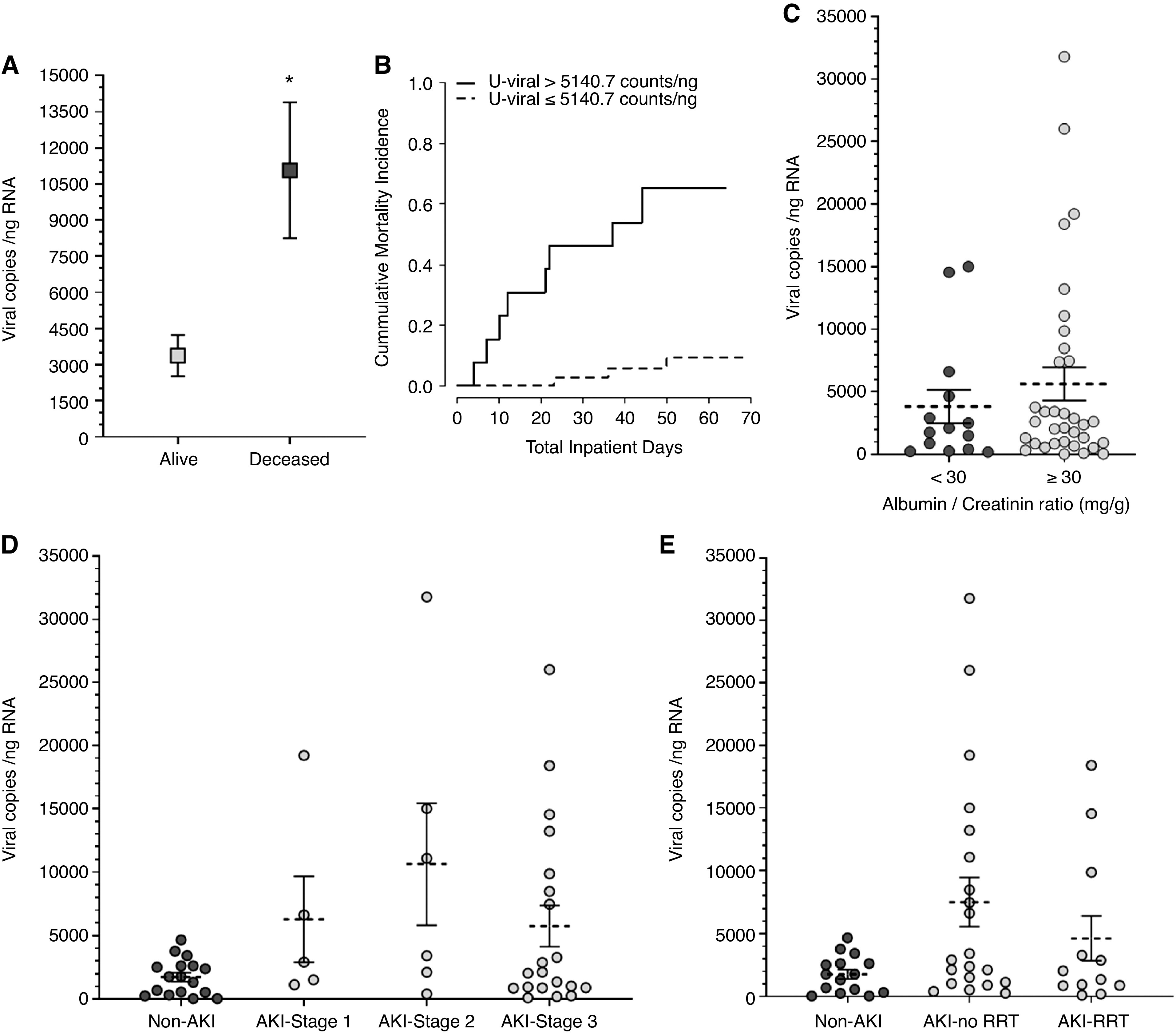
Higher SARS-CoV-2 U-viral load was associated with higher risk of mortality. (A) Increased U-viral load among patients who died. (B) Cumulative incidence of inpatient mortality at above and below a viral load of 5141 copies/ng RNA. Sixty day HR, 11.50 (95% CI, 3.27 to 40.30), P<0.001. The probabilities of nonsurvival (at risk) for the duration of the observation period were as follows: <5141 copies/ng RNA, P=0.09; >5141 copies/ng RNA, P=0.62. (C) The average U-viral load was 1.5 times higher in patients with COVID-19 who had albuminuria at the time of sampling, but this was NS (P=0.50, Kruskal–Wallis test). (D) Average U-viral load in patients with COVID-19 at different stages of AKI (P=0.12, Kruskal–Wallis test). (E) Average U-viral load in patients with COVID-19 and AKI who needed RRT or not (P=0.10, Kruskal–Wallis test). Each dot represents an individual data point, average viral copies per nanograms RNA is indicated (dotted line) along with the SEM. n=48. *P=0.03, Kruskal–Wallis test.
Table 2.
Clinical characteristics of deceased COVID-19 patients
| Characteristics | Alive | Deceased | P Value |
|---|---|---|---|
| Total, n (%) | 40 (76.9) | 12 (23.1) | |
| AKI, n (%) | 0.18 | ||
| Yes | 24 (60.0) | 10 (83.3) | |
| No | 16 (40.0) | 2 (16.7) | |
| Cause of death, n (%) | NA | ||
| Acute respiratory failure | — | 7 (58.3) | |
| Multisystem organ failure | — | 1 (8.3) | |
| Arrythmia | — | 1 (8.3) | |
| Metastatic cancer | — | 1 (non-AKI) (8.3) | |
| Not on record | — | 1 (AKI) + 1 (non-AKI) (16.7) | |
| Length of stay until death (d) | — | 23±16 | NA |
| Duration of COVID-19 at sampling (d) | 28±19 | 20±11 | 0.20 |
| Required ventilation, n (%) | 23 (57.5) | 7 (58.3) | >0.99 |
| Developed sepsis, n (%) | 12 (30.0) | 2 (16.7) | 0.48 |
| Systolic BP (mm Hg), mean±SD | 132±21 | 124±25 | 0.34 |
| On BP medication, n (%) | 31 (77.5) | 8 (66.7) | 0.47 |
Continuous variables are summarized as means±SDs. P values were calculated using the Kruskal–Wallis test (for continuous variables) or the Fisher exact test (for categoric variables). NA, not applicable.
In subjects with a urine ACR >30 mg/g, the average U-viral load tended to be higher, but not significantly different (3839±1330 viral copies/ng RNA for ACR<30 mg/g versus 5649±1329 viral copies/ng RNA for ACR≥30 mg/g; P=0.50; Figure 3C). We did not find significant associations of U-viral load with serum creatinine, blood in urine, C-reactive protein (Supplemental Figure 3), BUN, stage of AKI, or need for RRT (Figure 3D, 3E). This weak correlation with renal parameters and markers of injury may be reflective of transient effects related to the time of collection.
Presence of SARS-CoV-2 Proteins in Kidney Tissue
To provide additional evidence of SARS-CoV-2 infection in the kidney, we performed immunolabeling for the SARS-CoV-2 spike and nucleocapsid proteins in kidney biopsy specimens from patients positive and negative for COVID-19 (Figure 4). Positive labeling for viral proteins was detected in different kidney cell types, including proximal tubules (Figure 4, A, C, and D), parietal cells (Figure 4B), and distal tubules (with the appearance and size of collecting ducts; Supplemental Figure 4). Relatively low immunoreactivity was observed in glomerular cells (Figure 4B). Only background signal was observed in biopsy specimens from patients negative for COVID-19 (Figure 4, E–G). ACE2, the receptor for viral entry, was abundant in proximal tubules and, to a lesser extent, parietal cells, and colocalized with intracellular SARS-CoV-2 in some proximal tubule cells (Figure 4, A and B, and Supplemental Figure 4B). We also conducted immunolabeling in fresh urine sediments embedded in matrigel. The SARS-CoV-2 spike protein was detected in single cells from a patient with positive urine RT-PCR (Figure 5A), but was absent in patients negative for COVID-19 (Figure 5B). ACE2 expression was detected in the same cells (Figure 5), but not all cells in urine sediment expressed ACE2. Coexpression of ACE2 and the viral nucleocapsid protein was also observed in urine sediments from three additional patients with urine-positive RT-PCR (Supplemental Figure 5). The origin or identity of these cells was not determined.
Figure 4.

SARS-CoV-2 spike and nucleocapsid proteins (green) are expressed in kidney biopsies, often coexpressed with ACE2 (red) in the same cell. (A) Proximal tubule (PT) in kidney cortex of patient 1 (positive for COVID-19). ACE2 signal was apical, outlining the lumen of proximal tubules, and SARS-CoV-2 spike protein was observed in an intracellular location (arrows). (B) Glomerulus (G) in the same patient as in (A). Inset shows higher magnification of the area in outlined square, where SARS-CoV-2 spike expression was detected in parietal cells. (C) Spike expression in area of tubular necrosis in the kidney cortex of patient 2 (positive for COVID-19). Inset shows higher magnification of area in outlined square where spike expression was detected in cortical tubules (nephron segment outlined by dotted line). (D) SARS-CoV-2 nucleocapsid expression in cortical tubule of patient 2 (positive for COVID-19). Arrowheads indicate cells of the tubular segment expressing nucleocapsid at different intensities. Inset shows higher magnification of area in outlined square. (E) ACE2 expression and absence of spike expression in proximal tubules of patient negative for COVID-19. (F) Absent spike expression in glomerulus of patient negative for COVID-19. (G) Absent nucleocapsid expression in kidney cortex of a patient negative for COVID-19. Blue label is 4′,6-diamidino-2-phenylindole nuclear staining in all panels.
Figure 5.

SARS-CoV-2 spike protein and ACE2 are coexpressed in cells in urine sediment from patients with COVID-19. (A) ACE2 (red) and SARS-CoV-2 spike (green) coexpression in the same cell isolated from urine sediment of patient positive for COVID-19. (B) ACE2 expression and absence of spike in single cell from urine sediment of patient negative for COVID-19. Blue label is 4′,6-diamidino-2-phenylindole nuclear staining.
Infectivity of SARS-CoV-2 Material from Urine
It is unclear whether infective virions are shed in the urine, and a previous study failed to detect this.18 We studied 32 fresh urine sediments, 28 from patients positive for COVID-19 and four from negative controls, for cytopathic viral growth in inoculated Vero E6 cells. Two out of 28 (7.1%) urine sediment specimens generated cytopathic growth (both from patients diagnosed with AKI). However, subsequent RT-PCR failed to amplify any SARS-CoV-2 genetic material isolated from Vero E6 cells, suggesting the cytopathic effect observed was secondary to a viral infection other than SARS-CoV-2.
Discussion
During the time frame of our study, 1320 patients were admitted to our institution in Detroit, Michigan, with a clinical diagnosis of COVID-19. Retrospective review of our medical records showed that 614 patients (46.5%) were also diagnosed with AKI. AKI was present in 172 of 225 (76.4%) patients who died. A high incidence of AKI (up to 49% in US minority groups)11 has been documented in COVID-19 epicenters around the world.6–10 Furthermore, the mortality rate observed among the AKI subset of patients with COVID-19 is disproportionately high.12–14 To gain insight into the effect of COVID-19 in kidney dysfunction, this study was targeted to a cohort of 52 patients with laboratory RT-PCR diagnosis of COVID-19. Here, we report higher viral copy number detected in urine sediment from patients with COVID-19 who developed AKI, and a strong correlation between U-viral load and subsequent mortality.
Urine sediments represent a noninvasive diagnostic tool with potential for assessing patients with COVID-19 presenting with AKI.26 Previous, smaller-sized studies using whole urine detected no SARS-CoV-2–positive urine specimen,17,18 or only one such specimen.19,20 A recent, larger study in 81 patients showed a SARS-CoV-2 detection rate of 7% in whole urine.21 In an effort to maximize detection in urine, in this study we collected urine sediments from patients with COVID-19 and measured SARS-CoV-2 directly from cells contained in the sediment using a sensitive qRT-PCR assay previously developed for respiratory samples.22 We report a detection rate of 39% of tested urine sediment specimens, significantly higher than a 7.5% detection rate previously reported by a similar study in a cohort of 53 patients with COVID-19.27 This increased sensitivity is probably due to different RT-PCR methodologies, because this previous study using a probe-based RT-PCR assay for detection of the SARS-CoV-2 N and Orf-1ab genes.27 In our hands, equivalent probe-based assays for the N and envelope (E) genes28 did not yield positive results. In comparison, the SYBR Green–based assays we adopted for S and N genes22 showed higher sensitivity. Importantly, it has not been possible, so far, to use U-viral load to establish a link between AKI and COVID-19,27 although this association has been widely discussed in the medical literature, mostly on the basis of case reports or autopsy findings.6–11,15 The quantitative approach presented here, using a viral genome standard curve, revealed a positive correlation between SARS-CoV-2 copy number in urine sediment, a noninvasive sample, and higher frequency of AKI and mortality. Because different methodologies result in varying sensitivity, we emphasize the advantages of standard curve–based quantitative methods for better comparisons across different studies to help identifying patients with COVID-19 who are at higher risk associated with AKI.
Due to limited specimen availability, we did not thoroughly investigate the cause of AKI in our cohort. A kidney biopsy specimen from one of our patients positive for COVID-19 shows evidence of tubular and glomerular damage, which is consistent with previous reports (Supplemental Figure 6). To our knowledge, there is no consensus in the literature on how kidney complications develop in patients hospitalized with COVID-19. AKI could arise from a systemic immune response, hemodynamic alterations, hypercoagulability, viral kidney tropism, or a combination of these factors. Although these mechanisms are not mutually exclusive, the evidence for a localized immune response in kidneys of patients with COVID-19 is inconsistent. Recent reports demonstrated autoimmune-like glomerular basement membrane disease,29 lymphocyte infiltration,4 arteriolar thrombosis, and collapsing glomerulopathy in the kidneys of patients with COVID-19. Other studies did not find strong evidence of immune infiltration in kidney tissue during SARS-CoV-2 infection.26,30 These differences may be related to underlying conditions in populations from different ethnic backgrounds. Concerted actions and establishment of a national biopsy tissue repository should be generated to address these questions.
An alternative cause of kidney damage as a consequence of COVID-19 is active SARS-CoV-2 tropism in the kidney. Recent studies reported detectable SARS-CoV-2 proteins in postmortem kidneys,2–5 in addition to electron microscopy detection of virus-like particles.3,4,31,32 However, concerns have been raised about the misinterpretation of electron microscopy evidence on the basis of morphologic features and their similarity to normal intracellular structures.33,34 This study provides additional immunodetection-based evidence of expression of two SARS-CoV-2 proteins, nucleocapsid and spike, in kidney cells from COVID-19 biopsy specimens (Figure 4 and Supplemental Figure 4). Sites with positive SARS-CoV-2 immunolabeling were proximal tubules, distal nephron, parietal cells, and—potentially—inflammatory cells, along with the cells shed in the urine. Expression of ACE2 in kidney proximal tubule cells has been associated with the ability of SARS-CoV-2 to infect cultured proximal tubule cells.35,36 However, whether this results in cytotoxicity remains to be determined. A more recent study showed clinical and histologic manifestations consistent with proximal tubule injury in a group of patients with COVID-19.37 The strong ACE2 labeling we observed in the brush border of proximal tubules in COVID-19–positive (n=2) and –negative (n=2) kidney biopsy specimens is consistent with previous literature.38,39 We observed very low ACE2 immunolabeling in cells other than proximal tubules. However, this does not preclude the expression of very low levels of ACE2, which has been detected in other kidney cells by RNA sequencing.40,41
Despite finding SARS-CoV-2 genetic material and protein in urine sediments, viability assays for cytopathic viral growth were negative, consistent with a previous report.18 The reasons behind these observations are not clear and invite further investigation designed for this purpose. Some possibilities may include the inability of kidney cells to assemble or release infective virions, or subsequent destruction by extracellular proteases in the urine. However, lack of infective material in urine does not preclude from the fact that SARS-CoV-2 may be able to reach and infect kidney cells, as demonstrated by the detection of viral proteins in kidney biopsy specimens in this report and previous studies in postmortem kidney.2,3,5 Absence of infectious SARS-CoV-2 particles in urine is independent of whether viral presence in urine material is associated with AKI and risk of poor COVID-19 outcome. This fact allowed us to present evidence in support of our hypothesis of direct kidney tropism, and detectable and quantifiable SARS-CoV-2 genetic material in urine sediment, providing an accessible specimen with diagnostic and predictive potential in terms of incidence of AKI and COVID-19–related mortality. Our study was not designed to determine whether SARS-CoV-2 kidney tropism directly causes AKI. This is a standing question requiring mechanistic studies in animal models or organoids directly addressing this possibility.
Limitations of the Study
Our cohort was selected to study patients with kidney involvement. Thus, the elevated rate of AKI is not representative of the overall population of patients with COVID-19. Our patient population is largely Black (70%), with high prevalence of comorbidities like CKD, hypertension, diabetes, and high mortality (Table 1). AKI and prior CKD diagnosis are reportedly higher in Black individuals with a COVID-19 diagnosis.42,43 It is possible that our patient population is highly susceptible to developing AKI and this may not be representative of other populations. That said, we relied on adjudicated AKI diagnoses/outcomes for analyses rather than medical record diagnoses, increasing the accuracy of our kidney outcomes. A larger cohort should be studied to determine the influence of ethnic background or prior CKD diagnosis. In addition, a larger cohort can also better address the predictive value of SARS-CoV-2 RT-PCR detection in urine sediments to determine risk of AKI and severity of COVID-19. This predictive potential is also, most likely, tied to parallel monitoring of viral loads in urine and blood. In this regard, a previous study in the related SARS coronavirus found that viral detection in blood seems to peak transiently during the first week of infection, followed by a peak in urine and stool afterward.44 Determining whether SARS-CoV-2 follows a similar dynamic and its relationship to development of AKI could provide a powerful diagnostic tool. These considerations, together with additional variables like sex and age, should be addressed to inform revised guidelines for care of patients with COVID-19 who have kidney involvement, in efforts to identify patients at higher risk and to better allocate scarce resources.
Disclosures
P.A. Ortiz reports receiving research funding from the American Heart Association (AHA), National Institute of Diabetes and Digestive and Kidney Diseases (DK105818A1), and the NIH; and serving as the past chair on the AHA Kidney in Cardiovascular Diseases Council, on the editorial board of the American Journal of Physiology-Renal Physiology, as a scientific advisor for—or member of—Hypertension, and as a permanent member (2013–2019) of the NIH Study Section (HM). K. Umanath reports receiving research funding from AstraZeneca, GlaxoSmithKline, ReCor Medical, and Sentien; having consultancy agreements with AstraZeneca and ICON/Novo Nordisk; Honoraria from Ardelyx; serving on the editorial board of Kidney Medicine, Frontiers in Physiology – Renal and Epithelial Physiology, Journal of the American Association of Physicians of Indian Origin; and serving as vice president and on the board of directors of The Collaborative Study Group. J. Yee reports Consultancy Agreements with AstraZeneca, GLG; Ardelyx, EBSCO/Dynamed, Bayer; Ownership Interest in Vasc-Alert(TM); Honoraria from AstraZeneca, Fresenius Medical Corporation, North America, Cara Therapeutics, Bayer, Gerson Lehman Group, AlphaSights, Ardelyx; Patents and Inventions with Vasc-Alert(TM); Scientific Advisor or Membership AstraZeneca, Ardelyx, Bayer; and Other Interests/Relationships with National Kidney Foundation, Editorial Board for Am J Kidney Dis, EBSCO/DynaMed Editorial Board, Elsevier Clinical Key Author, Elsevier Section Editor, Ferri's Clinical Advisor 2022, BMC Nephrology Editorial Board, Springer Heart Failure Reviews Editorial Board, Am J Nephrol Nephrology Editorial Board, and Journal of Onco-Nephrology Editorial Board. All remaining authors have nothing to disclose.
Funding
P.A. Ortiz is supported, in part, by NIH grant RO1 DK105818A1. This study was supported, in part, by Henry Ford Health System grant A30960.
Acknowledgments
The following reagent was deposited by the CDC and obtained through BEI Resources, NIAID, NIH: Quantitative PCR (qPCR) ControlA from Heat-Inactivated SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR 52347.
P.A. Ortiz formulated the hypothesis; P.S. Caceres and P.A. Ortiz designed the experiments and wrote the manuscript; P.S. Caceres analyzed the data and prepared figures; G. Savickas, S.L. Murray, M.N. Khan and S. Sarkar recruited patients, reviewed medical records, and compiled Table 1; P.S. Caceres, D. Maskey, and J. Fitzgerald performed RT-PCRs; S. Sarkar analyzed ACRs; K. Umanath, J. Uduman, J. Yee, and Y. Sharma performed the AKI adjudications, established the patient selection criteria, and contributed to the outline of the manuscript; T.-D. Liao and P.A. Ortiz performed immunofluorescences; A.M. Levin performed statistical analysis and prepared the inpatient mortality figure; S. Bolin performed virus infectivity assays; A.H. Ormsby processed the kidney biopsy specimens; and all authors contributed to the final edited version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2021010059/-/DCSupplemental.
Supplemental Table 1. Clinical risk factors for patients with U-viral load higher than 5140 copies/ng RNA.
Supplemental Figure 1. RT-PCR amplification in urine sediments and SARS-CoV-2 standard curve.
Supplemental Figure 2. Starting amount of RNA purified from urine sediments for qRT-PCR.
Supplemental Figure 3. U-viral load in relation with method of sampling, ventilation and parameters of kidney injury.
Supplemental Figure 4. SARS-CoV-2 immunofluorescence in kidney biopsy.
Supplemental Figure 5. Expression of nucleocapsid and ACE2 in cells shed in urine sediment from SARS-CoV-2 urine-positive patients.
Supplemental Figure 6. Kidney histopathology of COVID-19 patient with AKI.
References
- 1.World Health Organization: W.H.O. Coronavirus disease 2019 (COVID-19): Situation report, 51, Geneva, Switzerland, World Health Organization, 2020 [Google Scholar]
- 2.Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. : Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med 383: 590–592, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY, et al. : Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int 98: 219–227, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diao B, Wang C, Wang R, Feng Z, Tan Y, Wang H, et al. : Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Nat Commun 12: 2506, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradley BT, Maioli H, Johnston R, Chaudhry I, Fink SL, Xu H, et al. : Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet 396: 320–332, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shao M, Li X, Liu F, Tian T, Luo J, Yang Y: Acute kidney injury is associated with severe infection and fatality in patients with COVID-19: A systematic review and meta-analysis of 40 studies and 24,527 patients. Pharmacol Res 161: 105107, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmieri L, Vanacore N, Donfrancesco C, Lo Noce C, Canevelli M, Punzo O, et al. ; Italian National Institute of Health COVID-19 Mortality Group: Clinical characteristics of hospitalized individuals dying with COVID-19 by age group in Italy. J Gerontol A Biol Sci Med Sci 75: 1796–1800, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohamed MM, Lukitsch I, Torres-Ortiz AE, Walker JB, Varghese V, Hernandez-Arroyo CF, et al. : Acute kidney injury associated with coronavirus disease 2019 in urban New Orleans. Kidney360 1: 614–622, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arentz M, Yim E, Klaff L, Lokhandwala S, Riedo FX, Chong M, et al. : Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington State. JAMA 323: 1612–1614, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cummings MJ, Baldwin MR, Abrams D, Jacobson SD, Meyer BJ, Balough EM, et al. : Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: A prospective cohort study. Lancet 395: 1763–1770, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelayo J, Lo KB, Bhargav R, Gul F, Peterson E, DeJoy Iii R, et al. : Clinical characteristics and outcomes of community- and hospital-acquired acute kidney injury with COVID-19 in a US inner city hospital system. Cardiorenal Med 10: 223–231, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ali H, Daoud A, Mohamed MM, Salim SA, Yessayan L, Baharani J, et al. : Survival rate in acute kidney injury superimposed COVID-19 patients: A systematic review and meta-analysis. Ren Fail 42: 393–397, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Fang X, Cai Z, Wu X, Gao X, Min J, et al. : Comorbid chronic diseases and acute organ injuries are strongly correlated with disease severity and mortality among COVID-19 patients: A systemic review and meta-analysis. Research (Wash D C) 2020: 2402961, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim JH, Park SH, Jeon Y, Cho JH, Jung HY, Choi JY, et al. : Fatal outcomes of COVID-19 in patients with severe acute kidney injury. J Clin Med 9: 1718, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braun F, Lütgehetmann M, Pfefferle S, Wong MN, Carsten A, Lindenmeyer MT, et al. : SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet 396: 597–598, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanley B, Naresh KN, Roufosse C, Nicholson AG, Weir J, Cooke GS, et al. : Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: A post-mortem study. Lancet Microbe 1: e245–e253, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Xu Y, Gao R, Lu R, Han K, Wu G, et al. : Detection of SARS-CoV-2 in different types of clinical specimens. JAMA 323: 1843–1844, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA, et al. : Virological assessment of hospitalized patients with COVID-2019. Nature 581: 465–469, 2020 [DOI] [PubMed] [Google Scholar]
- 19.Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. ; China Medical Treatment Expert Group for Covid-19: Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382: 1708–1720, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng L, Liu J, Xu W, Luo Q, Chen D, Lei Z, et al. : SARS-CoV-2 can be detected in urine, blood, anal swabs, and oropharyngeal swabs specimens. J Med Virol 92: 1676–1680, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frithiof R, Bergqvist A, Järhult JD, Lipcsey M, Hultström M: Presence of SARS-CoV-2 in urine is rare and not associated with acute kidney injury in critically ill COVID-19 patients. Crit Care 24: 587, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. : A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579: 270–273, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, et al. : The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182: 685–712.e19, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosier H, Farhadian SF, Morotti RA, Deshmukh U, Lu-Culligan A, Campbell KH, et al. : SARS-CoV-2 infection of the placenta. J Clin Invest 130: 4947–4953, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao B, Ni C, Gao R, Wang Y, Yang L, Wei J, et al. : Recapitulation of SARS-CoV-2 infection and cholangiocyte damage with human liver ductal organoids. Protein Cell 11: 771–775, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez-Arroyo CF, Varghese V, Mohamed MMB, Velez JCQ: Urinary sediment microscopy in acute kidney injury associated with COVID-19. Kidney360 1: 819–823, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Li X, Chen H, Yan S, Li D, Li Y, et al. : Coronavirus disease 19 infection does not result in acute kidney injury: An analysis of 116 hospitalized patients from Wuhan, China. Am J Nephrol 51: 343–348, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, et al. : Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 25: 2000045, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prendecki M, Clarke C, Cairns T, Cook T, Roufosse C, Thomas D, et al. : Anti-glomerular basement membrane disease during the COVID-19 pandemic. Kidney Int 98: 780–781, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dorward DA, Russell CD, Um IH, Elshani M, Armstrong SD, Penrice-Randal R, et al. : Tissue-specific tolerance in fatal Covid-19. Am J Respir Crit Care Med 203: 192–201, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen CP, Bourne TD, Wilson JD, Saqqa O, Sharshir MA: Collapsing glomerulopathy in a patient with coronavirus disease 2019 (COVID-19). Kidney Int Rep 5: 935–939, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farkash EA, Wilson AM, Jentzen JM: Ultrastructural evidence for direct renal infection with SARS-CoV-2. J Am Soc Neprhol 31: 1683–1687, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller SE, Brealey JK: Visualization of putative coronavirus in kidney. Kidney Int 98: 231–232, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roufosse C, Curtis E, Moran L, Hollinshead M, Cook T, Hanley B, et al. : Electron microscopic investigations in COVID-19: Not all crowns are coronas. Kidney Int 98: 505–506, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia S, Wu M, Chen S, Zhang T, Ye L, Liu J, et al. : Long term culture of human kidney proximal tubule epithelial cells maintains lineage functions and serves as an ex vivo model for coronavirus associated kidney injury. Virol Sin 35: 311–320, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. : Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181: 905–913.e7, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Werion A, Belkhir L, Perrot M, Schmit G, Aydin S, Chen Z, et al. ; Cliniques universitaires Saint-Luc (CUSL) COVID-19 Research Group: SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int 98: 1296–1307, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H: Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203: 631–637, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. : A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 87: E1–E9, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, et al. : The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A 116: 19619–19625, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD: Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell 23: 869–881.e8, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price-Haywood EG, Burton J, Fort D, Seoane L: Hospitalization and mortality among black patients and white patients with Covid-19. N Engl J Med 382: 2534–2543, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirsch JS, Ng JH, Ross DW, Sharma P, Shah HH, Barnett RL, et al. ; Northwell COVID-19 Research Consortium; Northwell Nephrology COVID-19 Research Consortium: Acute kidney injury in patients hospitalized with COVID-19. Kidney Int 98: 209–218, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu D, Zhang Z, Jin L, Chu F, Mao Y, Wang H, et al. : Persistent shedding of viable SARS-CoV in urine and stool of SARS patients during the convalescent phase. Eur J Clin Microbiol Infect Dis 24: 165–171, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]