

Abstract
Summary
We present HoPhage (Host of Phage) to identify the host of a given phage fragment from metavirome data at the genus level. HoPhage integrates two modules using a deep learning algorithm and a Markov chain model, respectively. HoPhage achieves 47.90% and 82.47% mean accuracy at the genus and phylum levels for ∼1-kb long artificial phage fragments when predicting host among 50 genera, representing 7.54–20.22% and 13.55–24.31% improvement, respectively. By testing on three real virome samples, HoPhage yields 81.11% mean accuracy at the genus level within a much broader candidate host range.
Availability and implementation
HoPhage is available at http://cqb.pku.edu.cn/ZhuLab/HoPhage/data/
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
With the help of metagenomics technology, a wealth of novel phages that cannot be cultured are identified. Compared to the traditional culturing-based approach which naturally carries direct host information, the metagenomic method, especially metavirome, lacks the links between phages and their bacteria hosts (Edwards et al., 2016), thus brings the increasing demand to develop computational tools for host identification of short phage fragments.
Recently, several strategies, mainly based on abundance profiles, genetic homology, CRISPRs, exact matches and oligonucleotide profiles, have been proposed to predict phage–host relationships (Edwards et al., 2016). Subsequently, many tools for phage host prediction have been developed. HostPhinder (Villarroel et al., 2016) assigns the host species of a query phage as that of the reference phage which is most genomically similar to the query one. Since the microbial community contains a large number of novel phages with low similarity to the known phages, this approach cannot handle the task of host prediction of novel phages. Four tools, VirHostMatcher (Ahlgren et al., 2017), WIsH (Galiez et al., 2017), VirHostMatcher-Net (Wang et al., 2020) and PHP (Lu et al., 2021), were developed mainly based on sequence signatures. To select the most probable host, they calculated the similarity between the phage sequence and each candidate host genome by oligonucleotide frequency, Markov chain model or Gaussian model. However, the performance of these tools in short DNA fragments generated by large-scale sequencing technology is rather unsatisfactory. For example, WIsH only reaches an accuracy at the genus level of about 60% for 3000-bp fragments among 20 candidate host genera, while a considerable proportion of assembled contigs in metagenomic data obtained by next-generation sequencing are shorter than 3000 bp.
Considering that the phage fragments in the metagenomic data of real community are short in length, as well as the taxonomic composition of microbial community is complex, we developed HoPhage (Host of Phage) and demonstrated its good performance in identifying the hosts of short phage fragments within a much wider candidate host range.
2 Materials and methods
The data set used in HoPhage includes 20 003 complete prokaryotic (bacterial and archaea) genomes from the NCBI RefSeq database, 4498 phage genomes recorded in the Virus-Host DB (Mihara et al., 2016) and 404 prophages from two manually verified datasets. The details of constructing the benchmark of short phage fragments as well as the training and test sets are provided in Supplementary Section S2.1.
Since most phages evolve to adapt host codon usage to evade host immunity and to ensure translational efficiency (Carbone, 2008), HoPhage is designed mainly based on the signatures of coding sequence (CDS). HoPhage consists of two modules, HoPhage-G (genus) and HoPhage-S (strain), at both genus and strain levels. The HoPhage-G module performs host identification based on deep learning that has been widely employed in inferring interactions between biological components (Yang et al., 2018). By constructing pairs of phage fragments and prokaryotes at the genus level, host identification is transformed from a complex multiclass prediction issue to a binary classification task of judging whether there is an infection relationship between a pair. For the HoPhage-S module, the CDS of each candidate host genome is used to construct a Markov chain model and then calculate the likelihood of query phage fragments. Subsequently, two modules are integrated by calculating their weighted average score. Detailed methods and workflow are described in Supplementary Section S2. As HoPhage is designed for host prediction of the phage fragments (including assembled contigs) usually from metavirome, when the contigs are derived from metagenomes, the phage fragments should be preidentified by tools such as PPR-Meta (Fang et al., 2019), DeepVirFinder (Ren et al., 2020) and VirSorter2 (Guo et al., 2021).
3 Results
To evaluate the prediction performance of HoPhage on short phage fragments, the benchmark datasets of artificial short contigs with three different lengths, 100–400, 401–800 and 801–1200 bp, were generated.
We first assessed the performance of host prediction using HoPhage-G and HoPhage-S individual alone. Results showed that HoPhage-G outperforms other existing tools and achieves AUCs (area under ROC curve) of 0.988–0.993, which were evidently higher than that of other tools (Supplementary Fig. S3B). As for the HoPhage-S, the prediction accuracy is higher than that of PHP and WIsH, except VHM-Net (Supplementary Figs S6 and S7). Besides, it is necessary to point out that the performance of VHM-Net is unavoidably overestimated. Because it integrates phage-phage similarity and we cannot remove the phages in our test set from its phage library when using it.
We set the weight of HoPhage-G/HoPhage-S to 0.50/0.50 and compared the performance of incorporated prediction of HoPhage with other tools. The details on the weight selection are described in Supplementary Section S3.3. Although the advantages of HoPhage-S compared with other tools were not significant, after narrowing the host range by HoPhage-G in advance, HoPhage which integrates these two modules achieved significantly better performance compared with all other tools. The prediction accuracy was calculated as the percentage of phage fragments whose predicted hosts had the same taxonomy as their respective annotated hosts. As a result, the average accuracies of HoPhage for groups ‘100–400’, ‘401–800’ and ‘801–1200’ were 11.94%, 8.48% and 7.54% higher than that of VHM-Net at the genus level (Fig. 1A), respectively. We further tested the generalization ability of HoPhage by eliminating the phage fragments with high genomic similarity between the training set and the test set. The results showed that HoPhage still has significant advantages over other related tools in those test phage fragments that have low genomic similarity with the phage fragments in the training set (Supplementary Table S5). Moreover, we evaluated HoPhage’s performance on phage fragments with longer lengths, including 1201–3000, 3001–5000, 5001–10 000 and 10 001–20 000 bp. Although the deep learning models in HoPhage-G were trained on the above three groups of fragments shorter than 1200 bp, our results demonstrated that HoPhage-G can achieve the best performance on longer phage fragments (Supplementary Figs S8 and S9), suggesting that HoPhage can well handle long input fragments with high performance.
Fig. 1.
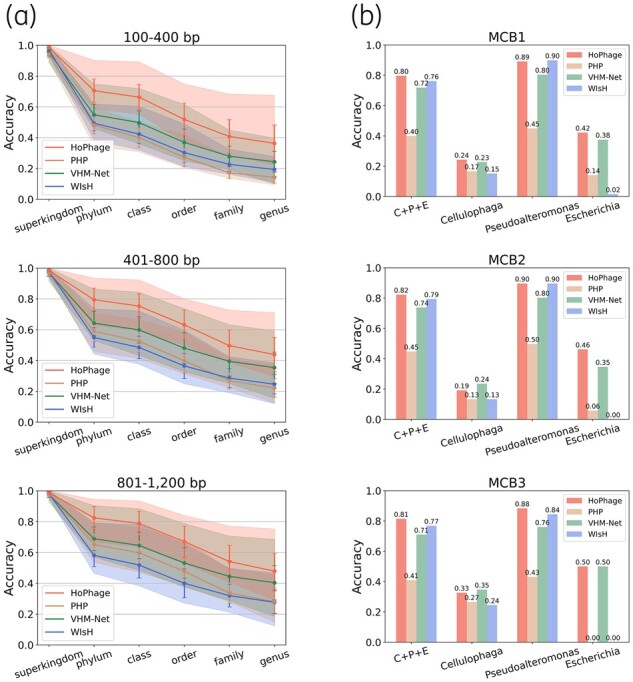
Performance of HoPhage. (a) Prediction accuracies of HoPhage at different taxonomic levels and comparisons with related tools. VHM-Net: VirHostMatcher-Net. The solid lines with error bars are the average accuracy of 20 randomly selected data. The light-colored area indicates the range of prediction accuracies. (b) Genus accuracies of HoPhage and related tools on contigs from three real virome samples. ‘C + P + E’ indicates the overall accuracy of all three genera, while ‘Cellulophaga’, ‘Pseudoalteromonas’ and ‘Escherichia’ are calculated separately
We further used the real virome data to evaluate the performance of HoPhage. These data come from the mock virus communities including 12 specific phages that grow on Pseudoalteromonas, Cellulophaga baltica and Escherichia coli (Roux et al., 2016). Details on data preprocessing refer to Supplementary Section S3.8. For each sample, the host prediction accuracies of HoPhage, PHP, VHM-Net and WIsH at the genus level are shown in Figure 1B. Among the host range of all 1353 candidate genera in our data, the average overall accuracy of HoPhage at the genus level was 81.11% while WIsH was 77.42%, VHM-Net was 72.19% and PHP was 41.90%. Although VHM-Net may overestimate the performance, the prediction accuracy of HoPhage is still significantly higher than VHM-Net, and the advantage of HoPhage on Pseudoalteromonas is greater. What’s more, the overall accuracy of WIsH is only about 4% lower than HoPhage. This is because the contigs that can infect Pseudoalteromonas account for the majority of these real samples while the performance of HoPhage and WIsH on Pseudoalteromonas is basically the same. Actually, HoPhage has significant advantages on these two genera Cellulophaga and Escherichia, and WIsH hardly correctly predicts any phage contig whose host belonging to Escherichia. Furthermore, we also found that HoPhage has a higher probability of obtaining accurate host prediction than WIsH among the incomplete candidate hosts (Supplementary Fig. S10), which is conducive to the research of the relationship between phages and the prokaryotic genera that are not sufficiently studied.
We further explored the marker genes of phages based on the potential of the single phage gene in identifying its host. We found that the potential of genes that encode infection-related proteins is 3–17 times that of other genes (Supplementary Table S6 and Fig. S11). Since phages lack conservative genes like 16S rRNA in bacteria and the taxonomic classification of phages often depends on their morphology, genes with high potential in host identification may be potential markers for taxonomic classification (Supplementary Fig. S12).
In conclusion, by integrating a deep learning-based module and a Markov chain model-based module, HoPhage simultaneously utilizes the sequence signatures of both phages and their host prokaryotes to predict hosts of phage fragments and demonstrates a satisfactory performance on short fragments. Therefore, we expect HoPhage to play a role in identifying hosts of novel phages and help researchers to explore the underlying ecological impact of phages in a community.
Funding
This work was supported by the National Key Research and Development Program of China [2017YFC1200205] and the National Natural Science Foundation of China [32070667, 31671366].
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Jie Tan, State Key Laboratory for Turbulence and Complex Systems, Department of Biomedical Engineering, College of Engineering and Center for Quantitative Biology, Peking University, Beijing 100871, China.
Zhencheng Fang, State Key Laboratory for Turbulence and Complex Systems, Department of Biomedical Engineering, College of Engineering and Center for Quantitative Biology, Peking University, Beijing 100871, China.
Shufang Wu, State Key Laboratory for Turbulence and Complex Systems, Department of Biomedical Engineering, College of Engineering and Center for Quantitative Biology, Peking University, Beijing 100871, China.
Qian Guo, State Key Laboratory for Turbulence and Complex Systems, Department of Biomedical Engineering, College of Engineering and Center for Quantitative Biology, Peking University, Beijing 100871, China.
Xiaoqing Jiang, State Key Laboratory for Turbulence and Complex Systems, Department of Biomedical Engineering, College of Engineering and Center for Quantitative Biology, Peking University, Beijing 100871, China.
Huaiqiu Zhu, State Key Laboratory for Turbulence and Complex Systems, Department of Biomedical Engineering, College of Engineering and Center for Quantitative Biology, Peking University, Beijing 100871, China.
References
- Ahlgren N.A. et al. (2017) Alignment-free oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucleic Acids Res., 45, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone A. (2008) Codon bias is a major factor explaining phage evolution in translationally biased hosts. J. Mol. Evol., 66, 210–223. [DOI] [PubMed] [Google Scholar]
- Edwards R.A. et al. (2016) Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev., 40, 258–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z. et al. (2019) PPR-Meta: a tool for identifying phages and plasmids from metagenomic fragments using deep learning. GigaScience, 8, giz066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiez C. et al. (2017) WIsH: who is the host? Predicting prokaryotic hosts from metagenomic phage contigs. Bioinformatics, 33, 3113–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. et al. (2021) VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome, 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C. et al. (2021) Prokaryotic virus host predictor: a Gaussian model for host prediction of prokaryotic viruses in metagenomics. BMC Biol., 19, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara T. et al. (2016) Linking virus genomes with host taxonomy. Viruses, 8, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J. et al. (2020) Identifying viruses from metagenomic data using deep learning. Quant. Biol., 8, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S. et al. (2016) Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ, 4, e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel J. et al. (2016) HostPhinder: a phage host prediction tool. Viruses, 8, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (2020) A network-based integrated framework for predicting virus-prokaryote interactions. NAR: Genomics Bioinf., 2, lqaa044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. et al. (2018) LncADeep: an ab initio lncRNA identification and functional annotation tool based on deep learning. Bioinformatics, 34, 3825–3834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.