

Abstract
Cancer genome-sequencing studies have revealed a remarkably high prevalence of mutations in genes encoding subunits of the SWI/SNF chromatin-remodelling complexes, with nearly 25% of all cancers harbouring aberrations in one or more of these genes. A role for such aberrations in tumorigenesis is evidenced by cancer predisposition in both carriers of germline loss-of-function mutations and genetically engineered mouse models with inactivation of any of several SWI/SNF subunits. Whereas many of the most frequently mutated oncogenes and tumour-suppressor genes have been studied for several decades, the cancer-promoting roles of mutations in SWI/SNF genes has been recognized only more recently, and thus comparatively less is known about these alterations. Consequently, increasing research interest is being focused on understanding the prognostic and, in particular, the potential therapeutic implications of mutations in genes encoding SWI/SNF subunits. Herein, we review the burgeoning data on the mechanisms by which mutations affecting SWI/SNF complexes promote cancer and describe promising emerging opportunities for targeted therapy, including immunotherapy with immune-checkpoint inhibitors, presented by these mutations. We also highlight ongoing clinical trials open specifically to patients with cancers harbouring mutations in certain SWI/SNF genes.
Introduction
The high prevalence of mutations in genes encoding chromatin regulatory proteins is one of the most notable insights into cancer biology that has been revealed by systematic sequencing of cancer genomes. Of these aberrations, mutations in genes encoding subunits of SWI/SNF chromatin-remodelling complexes are among the most frequent, collectively occurring in nearly 25% of all cancers1,2. Whereas most genes that are mutated at such high frequencies in cancer have been studied for many decades, recognition of a prominent role for SWI/SNF mutations is much more recent. Bi-allelic inactivating mutations in SMARCB1 were identified in 1998 in a rare but highly aggressive type of paediatric soft-tissue sarcoma termed rhabdoid tumour 3. Subsequently, genetically engineered mice with inactivation of SMARCB1 were shown to rapidly develop cancer, with 100% penetrance4. However, it was not until the advent of results from cancer genome-sequencing studies that it became clear that SMARCB1 was not alone5–7. It is now clear that at least nine genes encoding subunits of the SWI/SNF complexes are recurrently mutated in cancer, across a wide variety of tumour types1,2. Therefore, key questions emerge: what are the mechanisms by which these mutations contribute to cancer development? Do mutations affecting SWI/SNF subunits confer vulnerabilities upon these cancers? If so, are the vulnerabilities specific to the subunit mutated, or do any apply more broadly to cancers with diverse SWI/SNF aberrations? Most importantly, are any such vulnerabilities currently therapeutically actionable? In this Review, we provide insights into the mechanisms by which SWI/SNF mutations drive cancer and discuss some of the latest discoveries relating to therapeutic vulnerabilities, to both small-molecule drugs and immunotherapies, that are garnering substantial research and, in some cases, have led to the initiation of clinical trials.
SWI/SNF — a chromatin remodeller
Within cells, DNA does not exist as a naked chain of nucleotides; rather, the ~3 billion base pairs of the human genome are tightly associated with histones and other proteins in a structure termed chromatin. Within chromatin, organization and compaction of the human genome are achieved by wrapping 146 base pairs of DNA around histone protein octamers, forming structures termed nucleosomes, thus enabling ~3 meters of DNA to be encased within a nucleus with an average diameter of only 5 μm. Additionally, nucleosomes have a central role in controlling gene expression, as their presence generally prevents the binding of transcription factors, the proteins responsible for activating or inactivating the expression of specific genes. Consequently, an elaborate cellular machinery works in concert with transcription factors to mobilize nucleosomes in order to control gene expression, a process termed chromatin remodelling8,9. The SWI/SNF family of chromatin-remodelling complexes, also known as BRG1/BRM-associated factor (BAF) complexes (BOX 1), are key regulators of nucleosome positioning10. SWI/SNF complexes use the energy generated through hydrolysis of ATP to slide or eject nucleosomes11. Belying this seemingly straightforward role, SWI/SNF complexes are complicated macromolecular assemblies consisting of many diverse and variable subunits. Mammalian SWI/SNF complexes belong to three broad subfamilies: canonical BAF (cBAF)8,12; polybromo-associated BAF (PBAF)13,14; and the GLTSCR1 or GLTSCR1L-containing and BRD9-containing (GBAF) complex, which was only discovered in 2018 and is also known as non-canonical BAF (ncBAF)15–18. All three complexes contain the core subunits including SMARCC1, SMARCC2, and either of the ATPases SMARCA4 or SMARCA2, but also contain numerous variable subunits that provide each of the complexes with a distinct identity19 (BOX 1). Additional heterogeneity occurs within each subfamily owing to the differential use of related subunits, which are often encoded by multi-gene families, such that hundreds or even thousands of subtly different SWI/SNF complexes might exist within each broad subfamily.
Box 1 |. Nomenclature of SWI/SNF complexes.
The numerous nomenclature systems currently in use for the switch/sucrose non-fermentable (SWI/SNF) complexes and their subunits can create substantial challenges for neophytes. The SWI/SNF name itself is derived from original discoveries that orthologous complexes in yeast are required for mating-type switching and sucrose fermentation (with mutations affecting the complex leading to a sucrose non-fermenting phenotype). As the composition of SWI/SNF complexes becomes more diverse with evolution to higher-order organisms, the BRG1/BRM-associated factor (BAF) nomenclature was developed (BRG1 and BRM are core ATPases of SWI/SNF complexes). Subsequently, the SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin (SMARC) naming system was developed and became part of official Human Genome Organisation (HUGO) nomenclature, although evidence supporting matrix association of the complexes is limited. Consequently, owing to the varied evolutionary biology, compositional diversity and myriad different proposed functions of SWI/SNF complexes, the nomenclature remains problematic and is often variably used throughout literature. Commonly used HUGO gene names and interchangeable synonyms of SWI/SNF components are indicated below, with the subunits listed according to the SWI/SNF complexes subfamilies- canonical BAF (cBAF), polybromo-associated BAF (PBAF) and non-canonical BAF (ncBAF) -with which they are most frequently associated, although additional variability exists.
Shared subunits
SMARCC1 (BAF155)
SMARCC2 (BAF170)
SMARCA4 (BRG1 or BAF190A)
SMARCA2 (BRM, BAF190B or SNF2L2)
SMARCB1 (BAF47, INI1 or SNF5)
SMARCE1 (BAF57)
SMARCD1, SMARCD2 and SMARCD3 (BAF60A, BAF60B and BAF60C, respectively)
ACTB
ACTL6A and ACTL6B (BAF53A and BAF53B, respectively)
BCL7A, BCL7B and BCL7C
SS18 (SYT or SSXT)
cBAF subunits
ARID1A (BAF250A or SMARCF1)
ARID1B (BAF250B)
DPF2 and DPF3 (BAF45D and BAF45C, respectively)
BCL11A and BCL11B
PBAF subunits
ARID2 (BAF200)
PBRM1 (BAF180)
BRD7
PHF10 (BAF45A)
ncBAF subunits
BRD9
BICRA and BICRAL (GLTSCR1 and GLTSCR1L)
cBAF, canonical BAF; ncBAF, non-canonical BAF; PBAF, polybromo-associated BAF.
The SWI/SNF complexes are evolutionarily conserved and were first discovered through pioneering studies in yeast, in which mutations affecting various component subunits were found to result in defective transcriptional control of mating-type switching (the SWI phenotype) or, in independent studies, of sucrose fermentation (the sucrose non-fermenting, or SNF, phenotype)20–23. Mammalian SWI/SNF complexes serve broad roles in transcriptional regulation and have been implicated in the facilitation of specific transcriptional programs such as in differentiation and lineage specification. Regions of the genome where the complexes have a clear role include enhancers, which are short non-protein-coding DNA elements that form binding sites for transcription factors and thereby regulate the transcriptional activity of adjacent genes. Enhancers constitute only a tiny percentage of the genome, but SWI/SNF complexes are highly enriched at these sites and have essential roles in modulating enhancer accessibility that is required for transcription factors to activate gene expression24–28. The functional implications of the hyper-diversity of SWI/SNF subunit composition is not entirely clear, but the different subfamilies have distinct location profiles across enhancers, promoters and gene bodies (FIG. 1), and their distinctive compositions are thought to provide specificity in interactions with transcription factors and other chromatin regulators17,19,25,29.
Fig. 1|. Function of SWI/SNF chromatin-remodelling complexes.
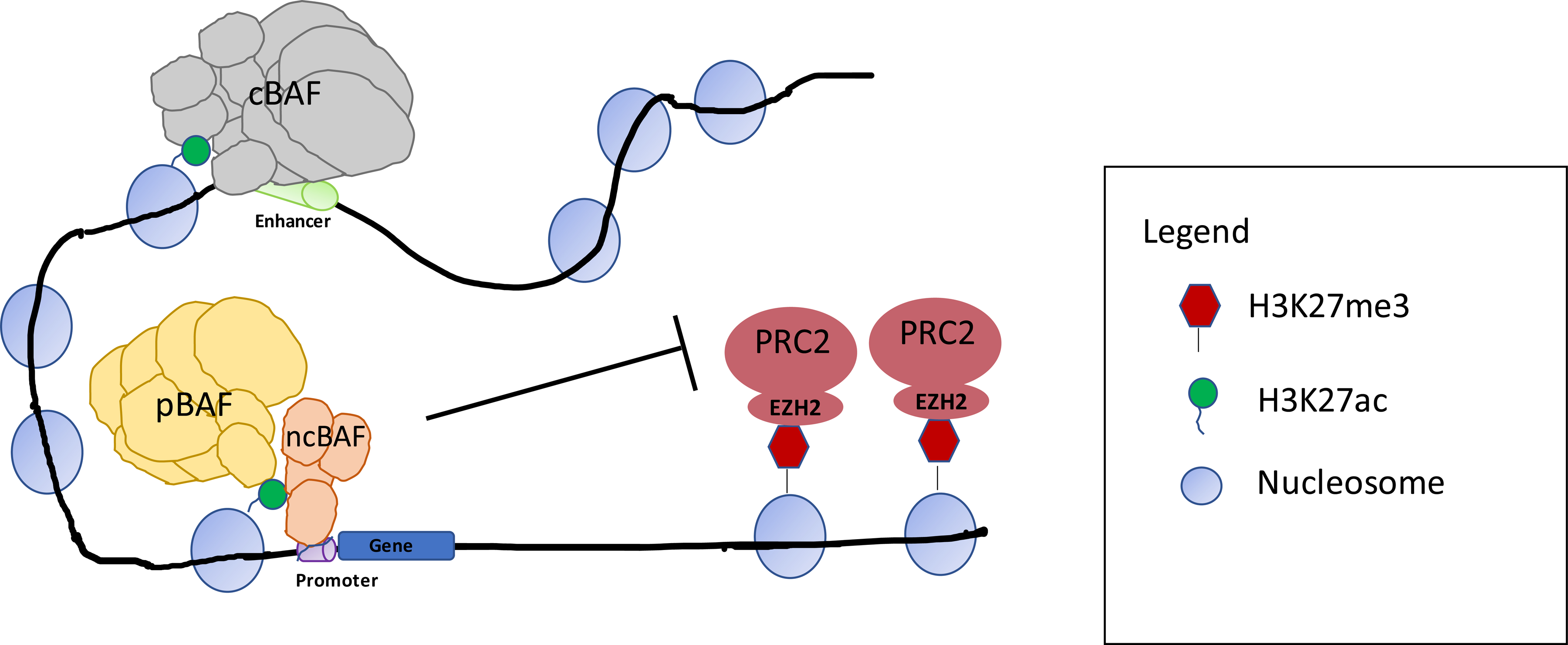
Illustration of SWI/SNF complex subfamilies and their genomic localization with respect to gene promoters and enhancers in nonmalignant cells. SWI/SNF complexes frequently localize at sites marked by histone H3 lysine 27 acetylation (H3K27ac), which is associated with active transcription, and cooperate with transcription factors to establish an open chromatin state25,26. This activity can be opposed by that of the Polycomb repressor complexes (PRCs), particularly PRC2 that places the repressive H3K27 trimethylation (H3K27me3) mark via its enzymatic subunit, enhancer of Zeste homologue 2 (EZH2)115,117. Canonical BAF (cBAF), polybromo-associated BAF (PBAF) and the most recently discovered non-canonical BAF (ncBAF, also known as GLTSCR1-containing BAF (GBAF)) are the three major SWI/SNF complex subfamilies17,18,29. cBAF activity might occur most strongly at enhancers, whereas PBAF and ncBAF are reported to be enriched at promoters, although also bind to some enhancers18,26,29,174. Understanding of the distinct functions of these three SWI/SNF subfamilies is limited and requires further study.
Transcription is not the only cellular process that requires access to specific stretches of DNA, and SWI/SNF complexes have been implicated in several mechanisms of DNA-damage repair (DDR)30. Different SWI/SNF family members have been shown to have distinct roles in DDR, ranging from modifying chromatin structure around sites of DNA damage to directly recruiting proteins required for DDR31–33. cBAF and PBAF complexes have been implicated in both non-homologous end joining (NHEJ) and homologous recombination (HR) repair processes31,34,35. Indeed, both SMARCA4 and the cBAF-specific subunit ARID1A have been shown to be recruited to sites of DNA damage and assist in HR-mediated DNA repair and NHEJ at double-strand breaks32,36. SMARCA4 has been reported to interact with poly(ADP-ribose) polymerase 1 (PARP1) at sites of DNA damage and to remodel chromatin in order to reduce nucleosome density at such sites, as well as to induce phosphorylation of histone H2AX (to produce the γH2AX mark) that promotes DNA double-strand break (DSB) repair33,37. Similarly, the authors of another report identified a role for ARID1A in promoting DSB repair38. Furthermore, the loss of either SMARCA4 or ARID1A expression has been associated with delayed mitosis and abnormal chromosomal segregation, linked to roles in DNA decatenation or telomere cohesion39,40. The PBRM1 subunit of PBAF has also been implicated in DDR, with evidence suggesting a function for this protein in transcriptional silencing at DSBs, which facilitates the repair of these DNA lesions, as well as in the maintenance of centromeric cohesion that is important for the maintenance of genomic stability41,42. As discussed further below, the degree to which the tumour-suppressor activity of SWI/SNF complexes is derived from their roles in transcriptional regulation versus DDR is the subject of ongoing debate and continued investigation.
SWI/SNF subunit mutations in cancer
SWI/SNF complexes were first implicated in oncogenesis upon the discovery that SMARCB1 (also known as INI1, SNF5 and BAF47; BOX 1) is inactivated by biallelic mutations in nearly all cases of rhabdoid tumour (FIG. 2)3,43,44. Rhabdoid tumours typically develop in children <3 years of age, often before the age of 1 year, and have a notably poor prognosis, with patients often surviving for <1 year after diagnosis 45. Studies in genetically engineered mouse models (GEMM) demonstrated that Smarcb1 is a bona-fide tumour-suppressor gene, with 10–30% of heterozygous mice developing cancers, mostly nervous-system and soft-tissue sarcomas (consistent with rhabdoid tumours), at a median age of 11 months46–48. Although germline homozygous inactivation of Smarcb1 results in early embryonic lethality46–48, induced somatic homozygous inactivation results in the rapid onset of lymphomas and sarcomas in 100% of mice at a median of only 11 weeks, which is less than half the time required for cancer to form following Tp53 inactivation4. Similarly, inactivation of Smarcb1 in the brain of developing mice results in nearly all mice rapidly developing intracranial rhabdoid tumours49. In children, two-thirds of rhabdoid tumour arise in CNS sites (termed atypical teratoid rhabdoid tumor (ATRT), with the remainder being in kidney (termed malignant rhabdoid tumor (MRT)), or in soft tissues at other anatomical locations (sometimes termed extra-renal extra-cranial rhabdoid tumours (EERT)). 45 These findings clearly linked SMARCB1 to cancer; however, as mentioned previously, not until the dawn of systematic cancer genome-sequencing studies was the breadth of SWI/SNF subunit mutations recognized.
Fig. 2. Frequency and pattern of SWI/SNF subunit mutations across human cancers.
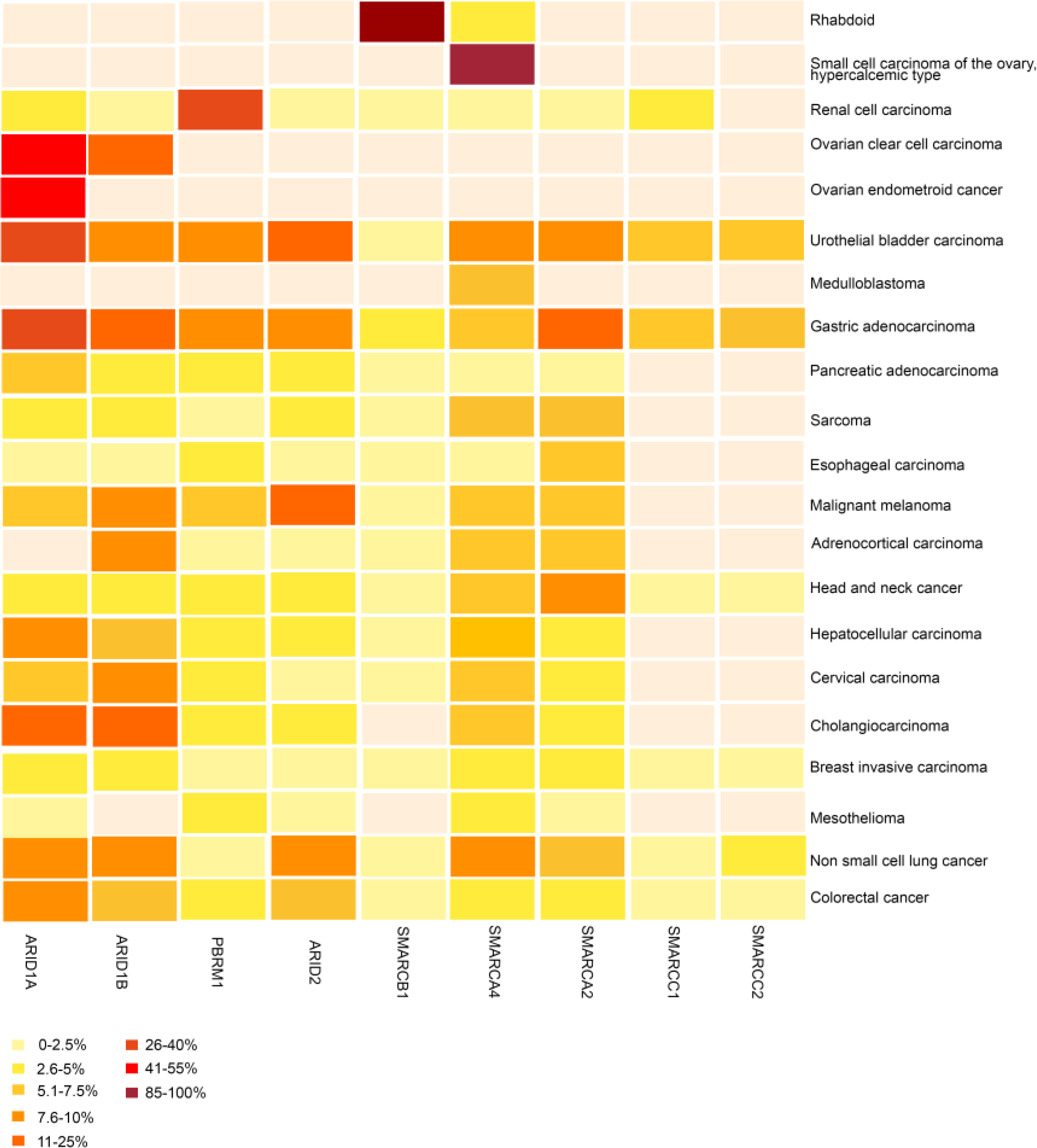
The heatmap depicts the frequency of non-synonymous mutations and deletions in select genes encoding components of SWI/SNF complexes across cancer types. Overall, the figure depicts the high prevalence of mutations affecting nine SWI/SNF subunits and the context-specificity of these mutations, with most being highly enriched in certain paediatric and adult malignancies. ARID1A is the most frequently mutated SWI/SNF complex gene, followed by SMARCA4 and PBRM1. The heatmap was compiled using data generated by the The Cancer Genome Atlas (TCGA) Research Network, accessible through cBioPortal55, as well as datasets sourced from various publications1–3,5,6,35,56. The white background color indicates tumors in which less than 2.5% of tumors had mutations in the subunit.
In 2010, ARID1A was discovered to be mutated in nearly 50% of all ovarian clear cell carcinomas (OCCCs) and ovarian endometrioid carcinomas (OECs)5,6. Similarly, PBRM1 mutations were identified in 41% of patients with clear-cell renal cell carcinoma (ccRCC)7. Subsequently, myriad cancers were found to harbour mutations in genes that encode SWI/SNF subunits. In total, at least nine different SWI/SNF subunits have been identified as being recurrently mutated in various cancers (FIG. 2), and such mutations are collectively found in nearly 25% of all cancers1,2. Interestingly, SWI/SNF subunit mutations are not randomly distributed across cancer types but rather exhibit patterns of association. For example, >95% of rhabdoid tumours have SMARCB1 mutations, with the other 5% harbouring SMARCA4 mutations50,51. Conversely, >90% of cases of small-cell carcinoma of the ovary, hypercalcemic type (SCCOHT), a rare form of ovarian cancer that predominantly occurs in young women with a median onset around 25 years of age52,53. SCCOHTs have been referred to as malignant rhabdoid tumour of the ovary and are the most common type of ovarian cancer in women aged <40 years. SCCOHTs are often characterized by biallelic SMARCA4 inactivating mutations but rarely SMARCB1 mutations53. Of note, SMARCA2 is either epigenetically silenced and/or transcriptionally inactive in both rhabdoid tumour and SCCOHT, and transcriptional reactivation of this gene inhibits the proliferation of cell lines derived from these two tumour types52,54. Whether the absence of SMARCA2 expression is pathogenic or simply a reflection of a non-SMARCA2-expressing cell of origin is unclear. Overall, ARID1A is the most frequently mutated SWI/SNF subunit across cancer types; however, PBRM1 mutations are much more common than ARID1A mutations in ccRCC (FIG. 2). Mutations affecting components of the ncBAF complex have been less frequently identified in cancer, although focal amplifcations of BRD9 have been identified in several cancer types55–57. Owing to the fact that mutations in SMARCB1 were identified in cancer more than a decade before mutations in genes encoding other SWI/SNF subunits, and because Smarcb1 inactivation leads to the rapid onset of genomically simple cancers in mice, preclinical models with SMARCB1 mutation have been central to the study of SWI/SNF function in cancer.
Mutations in genes encoding SWI/SNF subunits, which include nonsense, frameshift and deletion mutations, are often suggestive of loss-of-function phenotypes. Missense mutations might be the most common type of mutation and are preferentially located in conserved domains of SWI/SNF subunits, for example, within the enzymatic ATPase domain of SMARCA4 58. A substantial body of data from in vitro and in vivo studies supports the notion that SWI/SNF subunit mutations are indeed tumour-promoting. For example, studies in GEMMs have shown that inactivation of Smarcb1, Arid1a, Smarca4 or Pbrm1 results in cancer phenotypes24,59,60. Thus, these genes clearly qualify as tumour suppressors, but the cancer-driving mechanism involved might be somewhat complicated. For example, although genetic mutations result in the absence of SMARCB1 protein in rhabdoid tumours, residual SWI/SNF complexes are present and are essential for the survival of rhabdoid tumour cell lines15,17,18. This finding raises the possibility that SMARCB1-mutant cancers, rather than being driven by the absence of SMARCB1 protein per se, are driven by aberrant functioning of residual SWI/SNF complexes, thereby blurring the line between loss-of-function and gain-of-function phenotypes. Further adding to this complexity, SWI/SNF aberrations that result in gain-of-function chromatin phenotypes have also been identified; one such aberration is the SS18–SSX fusion found in synovial sarcoma, which confers SWI/SNF complexes with increased nucleosome mobilization activity61,62.
Within the spectrum of human disease, the pathogenic role of SWI/SNF subunit mutations is not confined to cancer. Germline heterozygous mutations of genes encoding specific SWI/SNF subunits form the aetiological basis of the rare neurodevelopmental disorders known as Coffin-Siris-syndrome (CSS; OMIM #135900) and Nicolaides-Baraitser syndrome (OMIM #601358). Patients with CSS harbour mutations in an array of genes belonging to the cBAF subfamily of SWI/SNF complexes: the most commonly mutated subunit is ARID1B, followed by ARID1A, SMARCB1, SMARCA4, SMARCA2 and SMARCE163. By contrast, Nicolaides-Baraitser syndrome is exclusively associated with missense mutations in SMARCA264. Notably, specific missense mutations in SMARCA4 (R885H and L921F) and SMARCB1 (K364del and R377H) are found in both patients with CSS and those with cancer, suggesting that the CSS phenotype can provide insights into the functional consequences of the cancer-driving mutations55,65.
Tumorigenic mechanism of aberrant SWI/SNF
Whether the tumour-suppressive effect of SWI/SNF complexes is a consequence of aberrant transcriptional regulation or impaired DDR, or both, has been the subject of longstanding debate. Understanding the central mechanism of tumour suppression is important because this knowledge can inform therapeutic approaches. The contributions of defective DDR to oncogenesis have been widely recognized, from Boveri’s original reports of rearranged chromosomes in cancer, to discoveries that defective telomere maintenance leads to both genome instability and cancer, followed by demonstration of the integral roles of genes encoding components of various DDR machineries in tumour suppression66–68. Given the identified roles of SWI/SNF complexes in DDR, a simple model can be postulated: SWI/SNF complexes are essential for the maintenance of genome integrity and, thus, dysfunction of these complexes leads to high rates of genetic mutations, which in turn drive cancer development. Numerous findings, however, are not easy to reconcile with such a model. Notably, rhabdoid tumors that result from germline SMARCB1 mutation, despite being highly aggressive and lethal, have remarkably simple diploid genomes69,70. The same is true for SCCOHTs, which are often associated with germline SMARCA4 mutations, and for ccRCCs with PBRM1 mutations71,72. Some cancers with SWI/SNF subunit mutations do indeed have high mutational burdens, but in these cases, the SWI/SNF gene mutations are rarely, if ever, germline. Consequently, it is difficult to determine in these cases whether the SWI/SNF gene mutation predisposed cells to genomic instability or whether a pre-existing genomically unstable neoplastic cell subsequently acquired a SWI/SNF gene mutation that facilitated clonal expansion and cancer progression. Clear evidence of the latter scenario has been reported, with mutation of ARID1A being identified as one of the few genes that is more commonly mutated in metastatic than in primary endometrial or breast cancers73,74.
An additional finding that is challenging to reconcile with DDR as the central tumour-suppressive mechanism of SWI/SNF complexes relates to reports that restoration of SWI/SNF subunits in mutant cell lines typically results in cell-cycle arrest or death of the cells52,75. Expanding on this paradox, if aberrations of SWI/SNF subunits simply enable the generation of cancer-promoting mutations in DNA owing to genetic instability, such mutations would not be reversed upon restoration of SWI/SNF expression, and the cancer cells should continue to grow. Indeed, ongoing genetic instability can confer toxicity and negative selection on cancer cells, as illustrated in cancers driven by telomerase mutations wherein, after cancer development, the cancer cells often evolve mechanisms that restore telomerase function and maintain genome integrity67. Additionally, whereas agents that cause non-specific genomic toxicity, such as radiation, can result in many types of cancer, mutations in SWI/SNF subunits are often associated with remarkably specific cancer phenotypes. For example, inactivation of SMARCB1 is largely confined to the aforementioned rare paediatric rhabdoid tumours and a few other rare types of cancer (FIG. 2). Consistent with such specificity, Smarcb1 inactivation in mice results in the rapid development of lymphomas that are derived from a highly specific subset of memory CD8+ T cells76. Notably, loss of SMARCB1 expression does not give rise to cancers derived from CD4+ T cells, immature T cells, B cells or myeloid cells — only from CD8+ memory cells76. Thus, although cancers that occur after SMARCB1 inactivation arise rapidly, they are derived from exquisitely specific cell types, which argues against a non-specific promotion of cancer through genomic instability. Also inconsistent with DDR being the central tumour-suppressive function of SWI/SNF complexes, rhabdoid tumours caused by SMARCB1 mutation are largely restricted to children aged < 3 years50. In older children, the risk of rhabdoid tumours decreases precipitously, 50.
Although DDR defects are often assumed to be a central cause of cancer, the links between defective transcriptional control and malignant transformation are remarkably strong. Studies reported in the 1980s and 1990s revealed that recurrent chromosomal translocations in acute leukaemias do not perturb DDR or alter genome integrity, but rather cause aberrant expression of lineage-specifying master transcription factors77. Such cancers typically have simple genomes, suggesting that, in the setting of perturbation of the transcriptional control of lineage specification, chromosomal instability is not required — and might even be selected against — for cancer formation70,78,79. Moreover, mouse models of several cancers have been used to demonstrate that the disruption of transcriptional regulation can lead to remarkably rapid cancer onset, faster than that of cancers driven by mutations in genes encoding mediators of DDR80–83. Collectively, these findings allude to the power of dysfunctional transcriptional regulation in promoting cancer. Although SWI/SNF complexes have been linked to DDR pathways, their links to transcriptional regulation are more extensive25,84. Ultimately, the activities of SWI/SNF in DDR and transcriptional regulation both have the potential to suppress cancer, and disruption of each function might contribute to SWI/SNF subunit-mutant cancers; however, supported by the rationale described above, in our view, the evidence seems most consistent with disruption of the function of lineage-specifying transcription factors as the central mechanism.
Prognosis of SWI/SNF-defective cancers
With mutations in SWI/SNF gene being found in nearly 25% of cancers, the question naturally arises whether such mutations have prognostic significance. These mutations have indeed been linked to a worse prognosis across several cancer types85. Of note, not only mutations but also overall changes in the expression of specific SWI/SNF subunits are implicated as prognostic markers for survival. In patients with hepatocellular carcinoma or cervical cancer, for example, loss of SMARCA2 and ARID1A expression, respectively, is associated with unfavourable overall survival86,87. However, the association between reduced expression and a poor prognosis is not universal: low expression of SMARCA4 and ARID1A proteins is associated with favourable outcomes in both patient with breast cancer and those with bladder cancer88–90. Conversely, high expression of SMARCA4 is associated with an unfavourable prognosis in patients with hepatocellular carcinoma86,90,91. These findings suggest that the effects of SWI/SNF abberations on prognosis are subunit-specific and/or context-specific.
One of the starkest examples of the context-specific effects of SWI/SNF aberrations is provided by the distinct phenotypes of tumours associated with SMARCB1 mutations92. SMARCB1-deficient rhabdoid tumours are among the most aggressive and lethal paediatric cancers; however, mutations in SMARCB1 — although probably hypomorphic — also form the aetiological basis of familial schwannomatosis, which is characterized by a predisposition to benign tumours3,93,94–96. Thus, distinct mutations in the same gene can lead to markedly different tumour types and prognoses65,85,97,98.
Collectively, these findings indicate that the consequences of mutations and alterations of SWI/SNF subunit expression are highly context-specific and do not universally confer a poor prognosis. Such complexities are not unique to cancers with SWI/SNF aberrations. For example, mutation of the gene encoding enhancer of Zeste homologue 2 (EZH2), which is the catalytic histone-lysine N-methyltransferase subunit of the Polycomb repressive complex 2 (PRC2) and thus another chromatin-remodelling factor, is also associated with cancer in discrepant contexts. Loss-of-function mutations of EZH2 are found in T cell acute lymphoblastic leukaemia (T-ALL), whereas gain-of-function EZH2 mutations are found in diffuse large B cell lymphoma (DLBCL)99,100. Mutations in EZH2 also constitute the aetiological basis of Weaver syndrome (OMIM# 277590), a rare overgrowth syndrome that is reported to be associated with cancer predisposition101. Together, these data suggest that mutations affecting chromatin-remodelling factors, such as SWI/SNF complexes, do not directly dictate the level of aggressiveness or prognosis of cancers. Rather, we propose a model in which SWI/SNF gene mutations simply impair transcriptional control, and the variable consequences of that impairment are largely dictated by the underlying biology of cell in which the mutation arises and, in particular, the transcription factors expressed therein.
Emerging therapeutic vulnerabilities
The fact that genes encoding SWI/SNF components are widely mutated in cancer raises several key questions, including whether such mutations, despite promoting cancer growth, result in synthetic lethal dependencies and, if so, are these dependencies therapeutically tractable. If tractable, defining whether any such dependencies are specific to the particular subunit that is mutated and/or the tissue of origin, or whether the mutations confer shared synthetic lethal dependencies regardless of which subunit is mutated, will be important. Emerging data indicate that mutations in SWI/SNF gene do indeed result in vulnerabilities in cancers, some of which are subunit and/or cell-type specific, although others are potentially more broadly applicable. The pursuit of therapeutic translation is underway for several of these vulnerabilities (FIG. 3), with a number of treatment approachs being tested in ongoing clinical trials involving patients whose cancers harbour SWI/SNF aberrations (TABLE 1).
Fig. 3. Translational science of cancers with SWI/SNF complex aberrations.
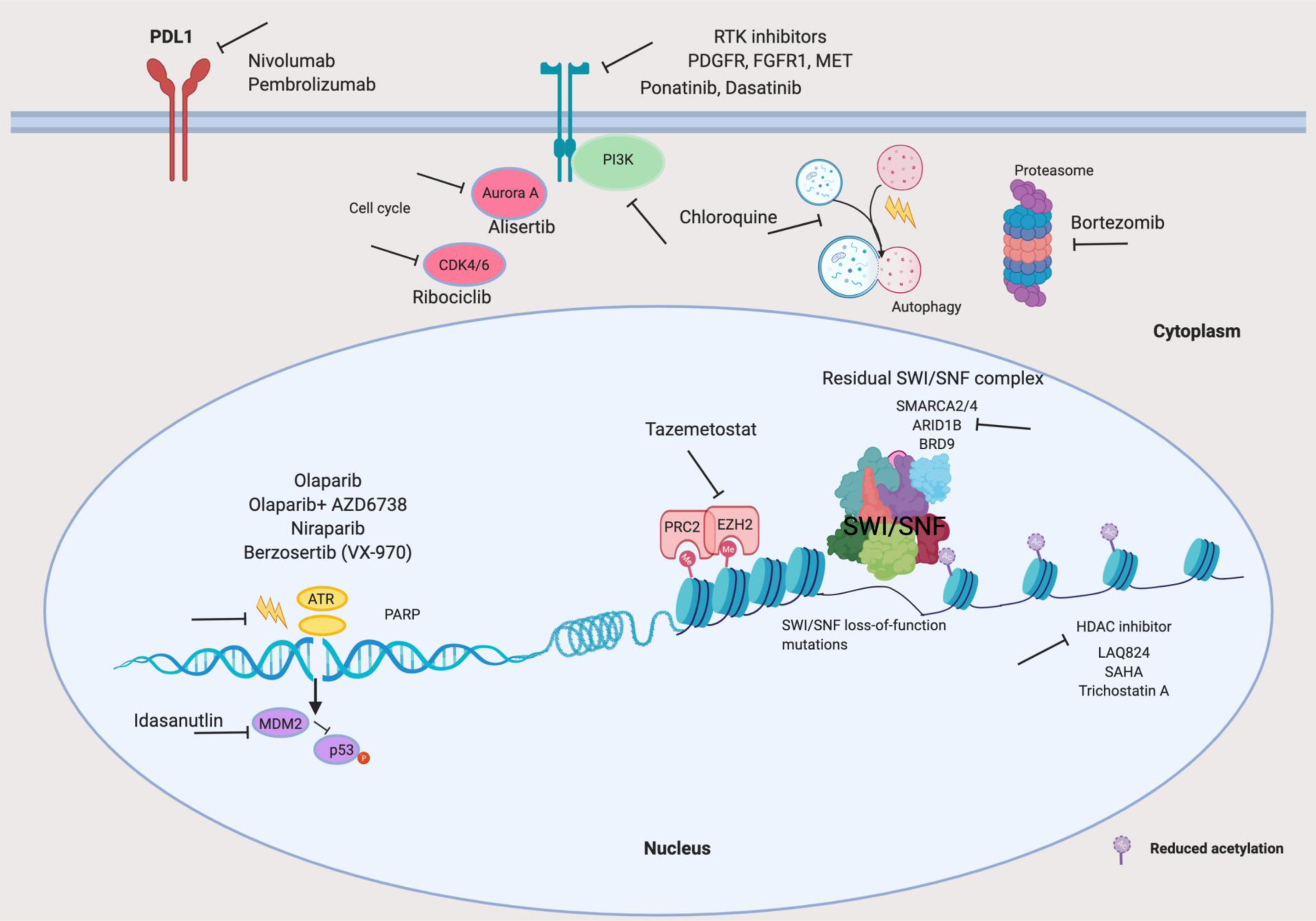
Illustration of reported vulnerabilities of cancers with loss-of-function mutations in SWI/SNF-complex genes, depicting both therapeutic opportunities supported only by preclinical evidence and treatments currently being evaluated in ongoing clinical trials (TABLE 1). The therapeutic targets include: residual SWI/SNF complexes17,18,102,104; Polycomb repressive complex 2 (PRC2), mainly its enzymatic subunit, enhancer of Zeste homologue 2 (EZH2), and predominantly in SMARCB1-mutant or SMARCA4-mutant120,122; components of the DNA damage repair pathway, in particular, poly(ADP-ribose) polymerase (PARP) and ATR in ARID1A-mutant cancers159,160; and receptor tyrosine kinases (RTKs) in several cancers enriched for mutations in SWI/SNF-complex genes, in a context-specific manner144,145,147,148. Targeting of Aurora A or CDK4/6 (cell-cyle kinases), MDM2 (a negative regulator of the tumour suppressor p53), autophagy or the proteasome could also be of potential therapeutic benefit in patients with cancers harbouring particular SWI/SNF-complex abberations138,142,157,158. Additionally, mutations in several genes encoding SWI/SNF-complex subunits have been associated with sensitivity to immune-checkpoint inhibitors targeting programmed cell death 1 (PD-1) or programmed cell death 1 ligand 1 (PD-L1)161,163–166,168,172. ARID1A, AT-rich interactive domain-containing protein 1A; bromodomain-containing protein 9, BRD9; FGFR1, fibroblast growth factor receptor 1; HDACs, histone deacetylases; PDGFR, platelet-derived growth factor receptor; SMARCA2/4, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2 or 4.
Table 1 ׀.
Ongoing interventional clinical trials involving cancers harbouring mutated SWI/SNF components
| Interventional agents (targets) | ClinicalTrial.gov ID | Disease setting | Study phase | Enrolment target | End points or objectives |
|---|---|---|---|---|---|
| SMARCB1-mutant, SMARCA4-mutant or SS18–SSX rearranged cancers | |||||
| Tazemetostat (EZH2) | NCT01897571 | Adults with advanced-stage solid tumours or B cell lymphomas, including DLBCL | I/II | 420 | MTD, bioavailability, DDI, efficacy and safety of monotherapy or combination with prednisolone. |
| NCT02601937 | Children with R/R MRT, ATRT, RTK and other selected tumours with rhabdoid features and loss of SMARCB1 or SMARCA4, other SMARCB1-negative tumours, or synovial sarcoma with SS18–SSX rearrangement, | I | 82 | Dose escalation to MTD and dose expansion at MTD | |
| NCT03213665 | Children with R/R advanced-stage solid tumours, non-Hodgkin lymphoma or histiocytic disorders with EZH2, SMARCB1 or SMARCA4 mutations | II | 49 | ORR, PFS, tolerability of tazemetostat in children with R/R cancer | |
| NCT02601950 | Adults with MRT, ATRT, RTK and other selected tumours with rhabdoid features and loss of SMARCB1 or SMARCA4 (including SCCOHT), SMARCB1-negative tumours or tumours with with an EZH2 gain-of-function mutation, refractory synovial sarcoma with SS18–SSX rearrangement, renal medullary carcinoma, epithelioid sarcoma, or poorly differentiated chordoma | II | 250 | ORR, DoR, PFS, and effect of tazemetostat on immune priming | |
| NCT02875548 | Adults with various cancers (including MRT, ATRT, RTK, synovial or epitheliod sarcoma, mesothelioma and DLBCL) who have completed an antecedent tazemetostat study |
II | 300 | Long-term safety profile, TTF and OS | |
| Alisertib (Aurora A) | NCT02114229 | Children and young adults (aged <22 years) with newly diagnosed or R/R ATRT and/or extra-CNS MRT (with loss of SMARCB1 or SMARCA4) | II | 180 | Sustained ORR, PFS, and PK and PD of alisertib in paediatric patients142,143 |
| ARID1A-mutant cancers | |||||
| Niraparib (PARP) | NCT03207347 | Adults with BAP1-mutant and other DDR-deficient neoplasms, including tumours with ARID1A mutation | II | 57 | ORR, PFS and OS |
| Olaparib (PARP) | NCT04042831 | Adults with billiary tract cancer with DDR gene aberrations, including ARID1A mutations | II | 36 | ORR, DoR and OS |
| Olaparib (PARP) + capivasertib (also known as AZD5363; AKT) |
NCT02576444 | Adults with cancer containing mutations in homologous DNA repair or other DDR genes, including ARID1A | II | 64 | ORR |
| Olaparib (PARP) + Ceralasertib (also known as AZD6738; ATR) |
NCT04065269 | Adults with relapsed gynaecological cancers with or without loss of ARID1A | II | 40 | ORR, PFS, TTP and OS |
| Berzosertib (also known as VX-970 and M6620; ATR) | NCT03718091 | Adults with advanced-stage solid tumours, including an ARID1A-mutant cohort | II | 223 | Validation of anticancer effect of VX-970 observed in preclinical studies: changes in Phospho-CHK1, γH2AX levels and DCR |
| Dasatinib (various tyrosine kinase) | NCT02059265 | Adults with recurrent or persistent ovarian, fallopian tube, endometrial or peritoneal cancer with and without loss of ARID1A | II | 35 | ORR, PFS, OS and toxicity profile |
| Dasatinib + PD-1 | NCT04284202 | Adults with advanced NSCLC with ARID1A mutation as the third line of treatment. | I | 30 | PFS, ORR, OS and toxicity profile |
Information on the clinical trials was obtained from https://clinicaltrials.gov on 15/03/2020. γH2AX, phosphorylated histone H2AX; ATRT, atypical teratoid rhabdoid tumour; CNS, central nervous system; DCR, disease control rate; DDI, drug–drug interactions; DDR, DNA damage response and/or repair; DLBCL, diffuse large B cell lymphoma; DoR, duration of response; EZH2, enhancer of Zeste homologue 2; MRT, malignant rhabdoid tumour; MTD, maximum tolerated dose; ORR, objective response rate; OS, overall survival; PD, pharmacodynamics; PFS, progression-free survival; PK, pharmacokinetics; R/R relapsed and/or refractory; RTK, rhabdoid tumour of kidney; SCCOHT, small cell carcinoma of the ovary, hypercalcemic type; TTP, time to progression; NSCLC, non-small cell lung cancer
Directly targeting SWI/SNF complexes
One type of vulnerability has been clearly recognized based on findings that mutations in certain genes encoding SWI/SNF subunits often create specific dependencies on genes encoding other SWI/SNF subunits. Such data suggests a model whereby subunit mutations do not fully inactivate SWI/SNF function but rather result result in aberrant cell function owing to a reliance on the activity of alternative residual SWI/SNF complexes102,103. For example, screens revealing that ARID1A-mutant cell lines are specifically dependent on its paralogue, ARID1B102 were among the first studies to illustrate a broader concept of paralogue dependencies. Similarly, SMARCA4-mutant cell lines have been shown to be enriched for dependence on its paralogue, SMARCA2102,104,105. These findings suggest a mechanism whereby loss of a SWI/SNF subunit is partially compensated for by a paralogue, making the paralogue a particular vulnerability. Such intra-complex dependencies are not restricted to paralogous subunits, as SMARCB1-mutant cell lines have been shown to have an increased dependence on the non-paralogous SWI/SNF subunit BRD9 17,18. Indeed, the identification of this dependency contributed to the recent discovery of the third subfamily of SWI/SNF complexes, that is, the BRD9-containing ncBAF complexes that lack SMARCB1 15. Thus, disruption of the two SMARCB1-containing subfamilies of SWI/SNF complexes (cBAF and PBAF) might increase dependence on this third subfamily. Collectively, the discovery of these dependencies has led of testing of whether they can be exploited therapeutically.
Several molecules capable of inhibiting SWI/SNF ATPase activity have been identified. For example, an orally available allosteric inhibitor of both SMARCA2 and SMARCA4 has been discovered and has demonstrated antiproliferative activity in a mouse xenograft model of SMARCA4-mutant lung cancer106. Protein degraders are being pursued as another means of targeting SMARCA2 and SMARCA4107 (FIG. 3). In particular, the development of proteolysis targeting chimeras (PROTACs) has enabled targeting of previously intractable targets108–110. PROTACs are bifunctional molecules that use either the Von Hippel-Lindau (VHL) or celebron E3 ubiquitin ligases, covalently linked to a target-binding ligand, to directly target the protein of interest for proteosomal degradation108–110. The structure-based design of a PROTAC targeting bromodomains present in SWI/SNF components resulted in the development of ACBI1, which binds to and mediates the degradation of SMARCA4, SMARCA2, and PBRM1 and has demonstrated anti-proliferative effects in SMARCA4-mutant cancer cell lines in vitro at concentrations ranging from 30 nM to 400nM107. PROTACs as a class of drugs have entered clinical trials in 2019, and, therefore, much remains unknown about their potential for therapeutic translation.
Importantly, the development of therapeutic strategies to target aberrant residual SWI/SNF complex warrants consideration since some SMARCA4-mutant cancers, such as SCCOHTs and a subset of non-small-cell lung carcinomas, lack expression of SMARCA2 and can, therefore, grow in the absence of both ATPase subunits52,111. Given the high degree of context-specificity in the association of SWI/SNF aberrations with specific types of cancer, further study will be required to determine whether cancers deficient in one subunit paralogue are addicted to that state and thus vulnerable to agents targeting the other paralogue, or whether they can evolve paralogue independence and adapt to a dual-deficient state. Notably, however, a dual-deficient state might in turn result in specific dependencies, as a demonstrated preclinically by the high sensitivity of lung and ovarian cancers lacking both SWI/SNF ATPases to bromodomain inhibitors112.
Similar to targeting of the residual SWI/SNF in other cancers, the potential to target BRD9 in SMARCB1-mutant cancers is also of interest, particularly as small-molecule inhibitors of BRD9 (such as BI-7273 and I-BRD9), with half maximal inhibitory concentrations of <50 nM, have been developed and have anticancer activity in models of AML113,114. Given the finding of preferential dependence upon BRD9 in SMARCB1-mutant cancers, these molecules have been tested in rhabdoid tumour and synovial sarcoma cell lines. Whereas knockdown or deletion of BRD9 impaired proliferation, inhibitors that bind to the bromodomain had no effect17,18,113,114. By contrast, a ‘degron’ compound that causes the degradation of BRD9 (dBRD9) was effective in such models16,17. Together, these findings indicate that simply blocking the bromodomain of BRD9 might be sufficient for anticancer activity against AML, but degradation of BRD9 is likely to be required in SMARCB1-mutant cancers perhaps suggesting a need for structural disruption of ncBAF in the latter case (FIG. 3).
Targeting PRC2 via EZH2
The relationship between SWI/SNF complexes and Polycomb repressive complexes and, in particular, whether cancers with mutations affecting select SWI/SNF subunits are sensitive to inhibition of EZH2, the enzymatic subunit of PRC2, have been the subjects of a substantial body of research. Upon the discovery of the tumour-suppressor activity of SWI/SNF, the Polycomb repressive complexes became of immediate interest because earlier genetic studies in fruit flies had revealed that SWI/SNF complexes and Polycomb repressor complexes have opposing gene-regulatory functions (FIG. 1)115,116. Subsequent investigations of this phenomenon in mammalian systems and models of cancer revealed that the functional antagonism between SWI/SNF and Polycomb repressive complexes was evolutionarily conserved117. For genes bound by SWI/SNF complexes, inactivation of SMARCB1 was shown to result in increased levels of histone H3 lysine-27 trimethylation (H3K27me3), which is a repressive chromatin mark written by PRC2118. Elegant work in model systems subsequently elucidated the mechanism involved, with recruitment of SWI/SNF complexes resulting in rapid displacement of both PRC2 and PRC1 that was abolished by mutation of SMARCB1 (REF.119). In some models, mutations in SWI/SNF genes also resulted in increased sensitivity to EZH2 inhibition120,121 (FIG. 3). Additional complexity of this relationship was unveiled when a study of human SMARCB1-mutant primary rhabdoid tumour specimens revealed that levels of H3K27me3 were increased at SWI/SNF-target genes, but were extremely low elsewhere in the tumour genome118. This finding suggests that SMARCB1 mutation results in the hyper-deposition of Polycomb repressive complexes locally but, potentially as a consequence, leads to the downregulation of the overall activity of these complexes as the cells attempt to restore a balanced gene-expression profile. Thus, tumour cells might have hyper-repression at some genes but hypo-repression at others, a finding that complicates prediction of the effects of EZH2 inhibition in these cancers.
The EZH2 inhibitor tazemetostat is currently being tested in several phase I–II trials involving adults with DLBCL or SMARCB1-negative or SMARCA4-negative solid tumours and in phase I−II trials involving paediatric patients with rhabdoid tumours, synovial sarcoma, epithelioid sarcomas or other cancers harbouring SMARCB1, SMARCA4 or EZH2 mutations (TABLE 1)120,122–126. One trial is in patients with epithelioid sarcomas, a cancer type in which nearly all tumors have homozygous deletion of SMARCB1127. Interim data from this trial demonstrated an overall response rate of 15% with 1.6% (2 of 43) of patients having a complete response and 13% (8 of 62) a partial response. Of the patients who responded, 67% had a response lasting six months or longer. In January of 2020, based in part on these data, FDA accelerated approval was granted for the use of tazemetostat in patients aged ≥16 years with metastatic or unresectable epithelioid sarcomas. Additional clinical studies evaluating alternative strategies to inhibit PRC2 with either a novel allosteric inhibitor of EED or with a compound capable of dually inhibiting both EZH2 and its paralogue, EZH1 are currently ongoing128,129.
Separately from the investigation of specific hypothesized dependencies of SWI/SNF-aberrant cancers, several broad screening endeavours have been pursued with the goal of identifying either genetic or pharmacological vulnerabilities of these cancer. Although still in their early stages, these projects have yielded mechanistic insights into SWI/SNF function and have identified several novel avenues for therapeutic investigation.
Targeting downstream vulnerabilities
Given the findings that SWI/SNF complexes contribute to the regulation of enhancer function and, as part of this role, facilitate the acetylation of H3K27 (FIG. 1), investigation of compounds that alter histone acetylation levels are obviously of interest in the context of SWI/SNF-aberrant cancers25–27. Several studies have demonstrated the activity of histone deacetylase (HDAC) inhibitors LAQ824, SAHA and trichostatin A in ATRT cell lines130,131 (FIG. 3).
In 2016, Torchia et al.130 and Johann et al.132 independently demonstrated that ATRT can be classified into three major groups according to their distinct epigenetic and molecular profiles: ATRT-MYC, ATRT-Sonic hedgehog (SHH) and ATRT-tyrosinase (TYR)133. In the same year, Chun et al.134 reported that extra-CNS RTs could similarly be stratified into two main subgroups. Mechanistically, several independent lines of investigation have linked the loss of SMARCB1 expression with aberrant activation of the MYC or non-canonical Hedgehog (GLI1) signalling pathways, consistent with the molecular characterization of ATRT subgroups135,136. These findings have generated interest in applying targeted therapies according to the dominant molecular networks of each particular subgroup, including the bromodomain and extra-terminal motif (BET) protein inhibitor JQ1 for the MYC subgroup and Smoothened (SMO) inhibitors for the ATRT-SHH subgroup137.
Drug screens coupled with functional genomic depletion screens in rhabdoid tumour, SCCOHT and non-small-cell lung carcinoma cell lines have identified specific dependencies on kinases involved in the cell cycle, such as cyclin dependent kinase 4 and/or 6 (CDK4/6) and Aurora A, owing to disrupted transcriptional control of these pathways upon mutation of SWI/SNF genes (FIG. 3)138–141. Data from a phase I trial of the CDK4/6 inhibitor ribociclib has shown some benefit, in the form of stable disease, in paediatric patients with rhabdoid tumours142,143, and the Aurora A kinase inhibitor alisertib is being testing in an ongoing phase II trial involving children and young adults with such tumours (TABLE 1)142,143.
Similar to the cell-cycle kinases, studies have revealed the dependency of rhabdoid tumours and ARID1A-mutant ovarian clear cell carcinomas on several receptor tyrosine kinases (RTKs), including platelet-derived growth factor receptors (PDGFRs), fibroblast growth factor receptor 1 (FGFR1) and MET130,144–147. Thus, co-targeting of multiple RTKs using either a single multi-kinase inhibitor, such as dasatinib or ponatinib, or a combination of inhibitors might also hold promise in patients with rhabdoid tumours, SCCOHT or OCCC (FIG. 3)144,146–148. A phase II trial of dasatinib in patients with ovarian cancer, including ARID1A-mutant disease, is ongoing (TABLE 1).
Loss-of-function mutations in ARID1A often co-occur with activating mutations in PI3K, AKT or mTOR or with loss of PTEN, which all result in upregulation of PI3K–AKT signalling149,150. Several independent lines of investigation, including initial genome-wide RNA interference dropout (negative-selection) screens conducted as part of Project Achilles and mutational analyses of OCCC and OEC specimens, have revealed PIK3CA, which encodes the PI3K catalytic subunit α isoform, to be one of the most predominant vulnerabilities of ARID1A-mutant cancers151–153. Consistent with these findings, data from drug screens demonstrate increased sensitivity to PI3K and AKT inhibitors in subsets of breast and endometrial cancers harbouring loss-of-function ARID1A mutations152,154. These findings raise the possibility of clinical benefit from PI3K and/or AKT inhibition in patients with ARID1A-mutant cancers. Importantly, this approach is likely to be clinically feasible considering that >30 different inhibitors of the PI3K pathway are currently being tested in clinical trials155. However, the finding that the small-molecule SMARCA4/2 inhibitor PFI-3 has promising therapeutic activity in preclinical models of PTEN-deficient prostate cancer suggests additional complexity in the relationship between the PI3K–AKT axis and SWI/SNF function156.
MDM2 has been identified as another, probably rhabdoid tumour-specific, vulnerability: although rhabdoid tumours are typically p53 wild type, a heightened dependence on MDM2 has been demonstrated in SMARCB1-mutant cancers157. The mechanism of dependence remains unclear, but might relate to the role of p53 in proteostasis in SMARCB1-deficient cancers, as evidenced by the sensitivity of rhabdoid tumours to the proteasome and autophagy inhibitors bortezomib and chloroquine, respectively158 (FIG. 3).
Inhibitors of DNA damage repair
Therapeutic vulnerabilities relating to the role of SWI/SNF complexes in facilitating DDR have also been identified. Preclinical data demonstrate the synergy between ARID1A loss and VX-970)159, an inhibitor of the DNA damage-checkpoint kinase ATR, as well as the efficacy of PARP inhibitors in concert with radiation in ARID1A-mutant tumours38 (FIG. 3). Indeed, long-term remissions of ARID1A-mutant patient-derived xenograft tumours in mice after combined treatment with radiation and PARP inhibition support the translation of this approach into clinical studies160. PARP or ATR inhibitors are currently being evaluated in several trials involving patients with ARID1A-mutant cancers (TABLE 1). Data from these trials will be key to understanding the feasibility and efficacy of this therapeutic strategy across different ARID1A-mutant cancers. Further study is required to determine whether vulnerabilities derived from impairment of DDR extend to other forms of SWI/SNF aberration.
Immune-checkpoint inhibition
One of the most exciting vulnerabilities associated with mutations in SWI/SNF genes that has emerged to date relates to sensitivity to immune-checkpoint inhibition (ICI). In a clinical trial involving 35 patients with metastatic ccRCC who underwent ICI with anti-programmed cell death 1 (PD-1) or anti-programmed cell death 1 ligand 1 (PD-L1) antibodies, loss-of-function mutations in the PBAF-specific gene PBRM1 were correlated with clinical benefit, defined as an complete or partial response, or a smaller reduction in tumour burden lasting >6 months (odds ratio (OR) 12.93, 95% confidence interval (CI) 1.54–190.8; P = 0.012)161. This finding was validated in an independent expansion cohort of 63 patients treated with anti-PD-1 or anti-PD-L1 antibodies alone or in combination with anti-cytotoxic T lymphocyte protein 4 (CTLA-4) antibodies (OR 6.10, 95% CI 1.42–32.64; P = 0.0071)161. Accompanying in vitro studies demonstrated that loss of PBRM1 expression facilitates broad transcriptional changes of genes involved in the JAK–STAT and other immune-related signalling pathways, as well as hypoxia-responsive genes, that might underlie the improved responsiveness to ICI161. Separately but contemporarily, a genome-scale CRISPR screen revealed that inactivation of Pbrm1 or either of two other genes of the PBAF SWI/SNF subfamily, Arid2 and Brd7, sensitized mouse melanoma cells to T cell-mediated cytotoxicity in vitro162. These mutations were also associated with enhanced chemokine secretion by the tumour cells in response to IFNγ, as well as higher levels of cytotoxic T cell infiltration into tumours and sensitivity to ICI with anti-PD-1 plus anti-CTLA-4 antibodies in mouse models162. PBRM1 is mutated in 40% of patients with ccRCC7, at lower frequencies in a range of other tumour types, and ARID2 mutations occur in cancers including urothelial carcinoma and melanoma (FIG. 2); thus, a substantial number of patients could potentially benefit from ICI.
In a preclinical study, mice bearing Arid1a-deficient ovarian cancers treated with an anti-PD-L1 antibody had substantially reduced tumour burdens and prolonged survival compared with mice treated with a control antibody163. Additionally, such effects were not observed in treatment of Arid1a-wild-type tumors with anti-PD-L1 antibody, although those tumors were inherently less aggressive. Similar to the Pbrm1-deficient tumours, the Arid1a-mutant tumours contained increased numbers of cytotoxic T cell, when compared with Arid1a-wild-type tumours, as well as higher levels of PD-L1 expression, with similar association observed in patient-derived ovarian cancer specimens163. However, the reported mechanisms underlying the sensitivity to ICI differ between Pbrm1-mutant and Arid1a-mutant tumours. Whereas Pbrm1 mutation enhances the immunogenicity of tumour cells via increased expression of immune-related genes. ARID1A was found to interact with the mismatch repair protein MSH2 and inactivation of Arid1a compromising DNA mismatch repair, resulting in increases in tumour mutational load, cytotoxic T cell infitration and PD-L1 expression163. Other studies have revealed that levels of PD-L1 are increased in patient-derived ARID1A-mutant gastric cancer specimen and in ARID1A-mutant cell lines, relative to their wild-type counterparts, and have linked this finding to observed benefits of ICI164–166. Of note, the link between ARID1A mutation and immune activation may be context dependent rather than universal as a 2020 analysis found that ARID1A mutations correlated with markedly higher immune infiltrates in endometrial, stomach and colon cancer but dramatically lower CD8+ T cell infiltrations in renal clear cell carcinoma167.
The association between SWI/SNF aberrations and immunogenicity has been extended with the finding that human SMARCB1-mutant rhabdoid tumours are infiltrated by clonally expanded subpopulations of T cells, suggesting a tumour-specific immune response168–170. Moreover, ICI resulted in tumour regression in up to 70% mice harbouring Smarcb1-mutant rhabdoid tumours168. In this model, the implicated mechanism was somewhat distinct from those proposed for other SWI/SNF aberrations. Loss of SMARCB1 caused expression of endogenous retroviruses triggering double-stranded RNA-sensing pathway and subsequent induction of interferon signalling168. Another study in four patients with SMARCA4-mutant SCCOHT showed that these tumours also have an immune-active microenvironment and are responsive to ICI, despite having a low mutational burden171.
Thus, emerging and increasing evidence links mutations in several genes encoding SWI/SNF subunits with sensitivity to ICI. However, in the context of treatment of current patients, it should be kept in mind that almost all of the studies involved small cohorts of patients, that not all of the preclinical findings have been corroborated clinically and that the proposed mechanisms do not clearly align172,173. Further evidence from experimental models and clinical studies will be required to determine the extent to which mutations in SWI/SNF genes correlate with, and confer, susceptibility to ICI.
Conclusions
The discovery that genes encoding subunits of SWI/SNF complexes are mutated at high frequency across a wide variety of cancers was largely unanticipated at the time when these complexes were identified. In contrast to mutations in classical oncogenes and tumour-suppressor genes, such as MYC, RAS and TP53, the roles of which have been studied in cancers for >40 years, the discovery of widespread SWI/SNF gene mutations in cancer is only a decade old. Consequently, our understanding of the mechanisms and any corresponding therapeutic implications remains in its infancy. Although it is now clear that inactivating mutations affecting individual SWI/SNF subunits can confer specific dependencies on other genes or pathways, whether any broad dependencies extend across all SWI/SNF cancers remains a key question and an active area of research.
With repeated discoveries that mutations in SWI/SNF subunits often increase dependencies on other components of residual SWI/SNF complexes, growning interest surrounds the possibility to therapeutically target the SWI/SNF complex itself. The potential for such an approach is enhanced by the existence of domains within these proteins, such as bromodomains and enzymatic ATPase domains, that can be targeted using small molecules. A point of caution relates to the fact that these complexes have bona fide tumour-suppressor activity and, specifically, whether inhibiting certain subunits could actually promote cancer development and/or growth, rather than suppressing these processes. Although tumour-promoting effects are likely to be reversible with cessation of therapy, this risk needs to be monitored in trials of such agents, particularly with long-term use.
The discovery of vulnerabilities, both genetic and pharmacological, has been accelerated by the advances in high-throughput screening assays. Such assays have proved powerful for the identification of synthetic lethality and other similar relationships; however, whether dependencies identified using cell lines will be sufficiently robust to translate into true therapeutic vulnerabilities in clinical trials remains to be determined.
Key points.
At least nine different genes encoding subunits of the SWI/SNF family of chromatin-remodelling complexes are recurrently mutated in cancer and these mutations are collectively present in nearly 25% of cancers.
Mutations in specific SWI/SNF genes are enriched in particular cancer types, suggesting differential roles for individual SWI/SNF components; consistent with this hypothesis, different SWI/SNF gene mutations confer distinct cancer vulnerabilities in mouse models.
The tumour-suppressor activity of the SWI/SNF chromatin-regulatory complexes is most likely attributable to their roles in facilitating transcription factor function, which is central to cell-fate specification; however, roles of the complexes in facilitating DNA repair might also contribute.
The identification of potential therapeutic vulnerabilities that arise from SWI/SNF gene mutations is leading to new areas of clinical investigation, including studies of immunotherapy in addition to kinase inhibitors and agents targeting mediators of DNA damage repair.
Acknowledgements
The work of C.W.M.R. is supported by grants from the US National Institutes of Health (R01CA172152 and R01CA113794), Cure AT/RT Now, the Avalanna Fund, the Garrett B. Smith Foundation and American Lebanese Syrian Associated Charities (ALSAC) of the St. Jude Children’s Research Hospital. The authors thank Stacy Throm of the St. Jude Children’s Research Hospital for her insights and Keith A. Laycock, also of the St. Jude Children’s Research Hospital, for scientific editing of the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Clinical Oncology thanks Michael C. Frühwald, and three other anonymous reviewers, for their contribution to the peer review of this work.
RELATED LINKS
The Cancer Genome Atlas (TCGA) Research Network: www.cancer.gov/tcga
cBioPortal: https://www.cbioportal.org/
References
- 1.Shain AH & Pollack JR The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One 8, e55119, doi: 10.1371/journal.pone.0055119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadoch C et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45, 592–601, doi: 10.1038/ng.2628 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Versteege I et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394, 203–206, doi: 10.1038/28212 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Roberts CW, Leroux MM, Fleming MD & Orkin SH Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2, 415–425, doi: 10.1016/s1535-6108(02)00185-x (2002). [DOI] [PubMed] [Google Scholar]
- 5.Wiegand KC et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 363, 1532–1543, doi: 10.1056/NEJMoa1008433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones S et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231, doi: 10.1126/science.1196333 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varela I et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469, 539–542, doi: 10.1038/nature09639 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clapier CR & Cairns BR The biology of chromatin remodeling complexes. Annu Rev Biochem 78, 273–304, doi: 10.1146/annurev.biochem.77.062706.153223 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Wilson BG & Roberts CW SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11, 481–492, doi: 10.1038/nrc3068 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Euskirchen G, Auerbach RK & Snyder M SWI/SNF chromatin-remodeling factors: multiscale analyses and diverse functions. J Biol Chem 287, 30897–30905, doi: 10.1074/jbc.R111.309302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon H, Imbalzano AN, Khavari PA, Kingston RE & Green MR Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 370, 477–481, doi: 10.1038/370477a0 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Wang W et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J 15, 5370–5382 (1996). [PMC free article] [PubMed] [Google Scholar]
- 13.Lemon B, Inouye C, King DS & Tjian R Selectivity of chromatin-remodelling cofactors for ligand-activated transcription. Nature 414, 924–928, doi: 10.1038/414924a (2001). [DOI] [PubMed] [Google Scholar]
- 14.Raab JR, Resnick S & Magnuson T Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes. PLoS Genet 11, e1005748, doi: 10.1371/journal.pgen.1005748 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alpsoy A & Dykhuizen EC Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J Biol Chem 293, 3892–3903, doi: 10.1074/jbc.RA117.001065 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brien GL et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife 7, doi:ARTN e41305 10.7554/eLife.41305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michel BC et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol 20, 1410–1420, doi: 10.1038/s41556-018-0221-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat Commun 10, 1881, doi: 10.1038/s41467-019-09891-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mashtalir N et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288 e1220, doi: 10.1016/j.cell.2018.09.032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cairns BR, Kim YJ, Sayre MH, Laurent BC & Kornberg RD A multisubunit complex containing the SWI1/ADR6, SWI2/SNF2, SWI3, SNF5, and SNF6 gene products isolated from yeast. Proc Natl Acad Sci U S A 91, 1950–1954, doi: 10.1073/pnas.91.5.1950 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neigeborn L & Carlson M Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics 108, 845–858 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stern M, Jensen R & Herskowitz I Five SWI genes are required for expression of the HO gene in yeast. J Mol Biol 178, 853–868, doi: 10.1016/0022-2836(84)90315-2 (1984). [DOI] [PubMed] [Google Scholar]
- 23.Martens JA, Wu PY & Winston F Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae. Genes Dev 19, 2695–2704, doi: 10.1101/gad.1367605 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathur R et al. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat Genet 49, 296–302, doi: 10.1038/ng.3744 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet 49, 289–295, doi: 10.1038/ng.3746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakayama RT et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet 49, 1613–1623, doi: 10.1038/ng.3958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alver BH et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun 8, 14648, doi: 10.1038/ncomms14648 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu C & Allis CD SWI/SNF complex in cancer. Nat Genet 49, 178–179, doi: 10.1038/ng.3779 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gatchalian J et al. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat Commun 9, 5139, doi: 10.1038/s41467-018-07528-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodges C, Kirkland JG & Crabtree GR The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Csh Perspect Med 6, doi:ARTN a026930 10.1101/cshperspect.a026930 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogiwara H et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene 30, 2135–2146, doi: 10.1038/onc.2010.592; 10.1038/onc.2010.592 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Qi W et al. BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. J Cell Sci 128, 317–330, doi: 10.1242/jcs.159103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y et al. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res 47, 8563–8580, doi: 10.1093/nar/gkz592 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brownlee PM, Meisenberg C & Downs JA The SWI/SNF chromatin remodelling complex: Its role in maintaining genome stability and preventing tumourigenesis. DNA Repair (Amst) 32, 127–133, doi: 10.1016/j.dnarep.2015.04.023 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Chabanon RM, Morel D & Postel-Vinay S Exploiting epigenetic vulnerabilities in solid tumors: Novel therapeutic opportunities in the treatment of SWI/SNF-defective cancers. Semin Cancer Biol, doi: 10.1016/j.semcancer.2019.09.018 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Watanabe R et al. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res 74, 2465–2475, doi: 10.1158/0008-5472.CAN-13-3608 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Park JH et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J 25, 3986–3997, doi: 10.1038/sj.emboj.7601291 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen J et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov 5, 752–767, doi: 10.1158/2159-8290.CD-14-0849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dykhuizen EC et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIalpha. Nature 497, 624–627, doi: 10.1038/nature12146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao B et al. ARID1A promotes genomic stability through protecting telomere cohesion. Nat Commun 10, 4067, doi: 10.1038/s41467-019-12037-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kakarougkas A et al. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol Cell 55, 723–732, doi: 10.1016/j.molcel.2014.06.028 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brownlee PM, Chambers AL, Cloney R, Bianchi A & Downs JA BAF180 promotes cohesion and prevents genome instability and aneuploidy. Cell Rep 6, 973–981, doi: 10.1016/j.celrep.2014.02.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biegel JA et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59, 74–79 (1999). [PubMed] [Google Scholar]
- 44.Sevenet N et al. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65, 1342–1348, doi: 10.1086/302639 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finetti MA, Grabovska Y, Bailey S & Williamson D Translational genomics of malignant rhabdoid tumours: Current impact and future possibilities. Semin Cancer Biol, doi: 10.1016/j.semcancer.2019.12.017 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Roberts CW, Galusha SA, McMenamin ME, Fletcher CD & Orkin SH Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci U S A 97, 13796–13800, doi: 10.1073/pnas.250492697 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guidi CJ et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol 21, 3598–3603, doi: 10.1128/MCB.21.10.3598-3603.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klochendler-Yeivin A et al. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep 1, 500–506, doi: 10.1093/embo-reports/kvd129 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han ZY et al. The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat Commun 7, 10421, doi: 10.1038/ncomms10421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brennan B, Stiller C & Bourdeaut F Extracranial rhabdoid tumours: what we have learned so far and future directions. Lancet Oncol 14, e329–336, doi: 10.1016/S1470-2045(13)70088-3 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Hasselblatt M et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128, 453–456, doi: 10.1007/s00401-014-1323-x (2014). [DOI] [PubMed] [Google Scholar]
- 52.Karnezis AN et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J Pathol 238, 389–400, doi: 10.1002/path.4633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu B & Shi H An In-Depth Look at Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT): Clinical Implications from Recent Molecular Findings. J Cancer 10, 223–237, doi: 10.7150/jca.26978 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kahali B et al. The silencing of the SWI/SNF subunit and anticancer gene BRM in Rhabdoid tumors. Oncotarget 5, 3316–3332, doi: 10.18632/oncotarget.1945 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404, doi: 10.1158/2159-8290.CD-12-0095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sima X et al. The genetic alteration spectrum of the SWI/SNF complex: The oncogenic roles of BRD9 and ACTL6A. PLoS One 14, e0222305, doi: 10.1371/journal.pone.0222305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu Z et al. Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat Commun 10, 733, doi: 10.1038/s41467-019-08554-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hodges HC et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat Struct Mol Biol 25, 61–72, doi: 10.1038/s41594-017-0007-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gu YF et al. Modeling Renal Cell Carcinoma in Mice: Bap1 and Pbrm1 Inactivation Drive Tumor Grade. Cancer Discov 7, 900–917, doi: 10.1158/2159-8290.CD-17-0292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bultman SJ et al. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene 27, 460–468, doi: 10.1038/sj.onc.1210664 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Kadoch C & Crabtree GR Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85, doi: 10.1016/j.cell.2013.02.036 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McBride MJ et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 33, 1128–1141 e1127, doi: 10.1016/j.ccell.2018.05.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsurusaki Y et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 44, 376–378, doi: 10.1038/ng.2219 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Van Houdt JK et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet 44, 445–449, S441, doi: 10.1038/ng.1105 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Orlando KA, Nguyen V, Raab JR, Walhart T & Weissman BE Remodeling the cancer epigenome: mutations in the SWI/SNF complex offer new therapeutic opportunities. Expert Rev Anticancer Ther 19, 375–391, doi: 10.1080/14737140.2019.1605905 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stern C Boveri and the early days of genetics. Nature 166, 446, doi: 10.1038/166446a0 (1950). [DOI] [PubMed] [Google Scholar]
- 67.Lazzerini-Denchi E & Sfeir A Stop pulling my strings - what telomeres taught us about the DNA damage response. Nat Rev Mol Cell Biol 17, 364–378, doi: 10.1038/nrm.2016.43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li SKH & Martin A Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol Med 22, 274–289, doi: 10.1016/j.molmed.2016.02.003 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Lee RS et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122, 2983–2988, doi: 10.1172/JCI64400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lawrence MS et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218, doi: 10.1038/nature12213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Creighton CJ et al. Comprehensivemolecular characterization of clear cell renal cell carcinoma. Nature 499, 43-+, doi: 10.1038/nature12222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ramos P et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet 46, 427–429, doi: 10.1038/ng.2928 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yates LR et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 32, 169–184 e167, doi: 10.1016/j.ccell.2017.07.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gibson WJ et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat Genet 48, 848–855, doi: 10.1038/ng.3602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Betz BL, Strobeck MW, Reisman DN, Knudsen ES & Weissman BE Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 21, 5193–5203, doi: 10.1038/sj.onc.1205706 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Wang X et al. TCR-dependent transformation of mature memory phenotype T cells in mice. J Clin Invest 121, 3834–3845, doi: 10.1172/JCI37210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Look AT Oncogenic transcription factors in the human acute leukemias. Science 278, 1059–1064, doi: 10.1126/science.278.5340.1059 (1997). [DOI] [PubMed] [Google Scholar]
- 78.Andersson AK et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47, 330–337, doi: 10.1038/ng.3230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huether R et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun 5, 3630, doi: 10.1038/ncomms4630 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van der Weyden L et al. Somatic drivers of B-ALL in a model of ETV6-RUNX1; Pax5(+/−) leukemia. BMC Cancer 15, 585, doi: 10.1186/s12885-015-1586-1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aster JC et al. Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1. Mol Cell Biol 20, 7505–7515, doi: 10.1128/mcb.20.20.7505-7515.2000 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adams JM et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318, 533–538, doi: 10.1038/318533a0 (1985). [DOI] [PubMed] [Google Scholar]
- 83.Condorelli GL et al. T-cell-directed TAL-1 expression induces T-cell malignancies in transgenic mice. Cancer Res 56, 5113–5119 (1996). [PubMed] [Google Scholar]
- 84.Peterson CL & Herskowitz I Characterization of the yeast SWI1, SWI2, and SWI3 genes, which encode a global activator of transcription. Cell 68, 573–583, doi: 10.1016/0092-8674(92)90192-f (1992). [DOI] [PubMed] [Google Scholar]
- 85.Savas S & Skardasi G The SWI/SNF complex subunit genes: Their functions, variations, and links to risk and survival outcomes in human cancers. Crit Rev Oncol Hematol 123, 114–131, doi: 10.1016/j.critrevonc.2018.01.009 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Endo M et al. Alterations of the SWI/SNF chromatin remodelling subunit-BRG1 and BRM in hepatocellular carcinoma. Liver Int 33, 105–117, doi: 10.1111/liv.12005 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Cho H et al. Loss of ARID1A/BAF250a expression is linked to tumor progression and adverse prognosis in cervical cancer. Hum Pathol 44, 1365–1374, doi: 10.1016/j.humpath.2012.11.007 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Faraj SF et al. ARID1A immunohistochemistry improves outcome prediction in invasive urothelial carcinoma of urinary bladder. Hum Pathol 45, 2233–2239, doi: 10.1016/j.humpath.2014.07.003 (2014). [DOI] [PubMed] [Google Scholar]
- 89.Bai J et al. BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PLoS One 8, e59772, doi: 10.1371/journal.pone.0059772 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaufmann B et al. BRG1 promotes hepatocarcinogenesis by regulating proliferation and invasiveness. PLoS One 12, e0180225, doi: 10.1371/journal.pone.0180225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu P et al. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat Commun 7, 13608, doi: 10.1038/ncomms13608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Agaimy A SWI/SNF Complex-Deficient Soft Tissue Neoplasms: A Pattern-Based Approach to Diagnosis and Differential Diagnosis. Surg Pathol Clin 12, 149–163, doi: 10.1016/j.path.2018.10.006 (2019). [DOI] [PubMed] [Google Scholar]
- 93.Hadfield KD et al. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J Med Genet 45, 332–339, doi: 10.1136/jmg.2007.056499 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Smith MJ et al. Expression of SMARCB1 (INI1) mutations in familial schwannomatosis. Hum Mol Genet 21, 5239–5245, doi: 10.1093/hmg/dds370 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith MJ, Wallace AJ, Bowers NL, Eaton H & Evans DG SMARCB1 mutations in schwannomatosis and genotype correlations with rhabdoid tumors. Cancer Genet 207, 373–378, doi: 10.1016/j.cancergen.2014.04.001 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Bourdeaut F et al. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17, 31–38, doi: 10.1158/1078-0432.CCR-10-1795 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Anaya J, Reon B, Chen WM, Bekiranov S & Dutta A A pan-cancer analysis of prognostic genes. Peerj 3, e1499, doi: 10.7717/peerj.1499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agaimy A & Foulkes WD Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol 35, 193–198, doi: 10.1053/j.semdp.2018.01.002 (2018). [DOI] [PubMed] [Google Scholar]
- 99.Morin RD et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42, 181–185, doi: 10.1038/ng.518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ntziachristos P et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18, 298–301, doi: 10.1038/nm.2651 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gibson WT et al. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet 90, 110–118, doi: 10.1016/j.ajhg.2011.11.018 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Helming KC et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med 20, 251–254, doi: 10.1038/nm.3480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang X et al. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res 69, 8094–8101, doi: 10.1158/0008-5472.CAN-09-0733 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hoffman GR et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A 111, 3128–3133, doi: 10.1073/pnas.1316793111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oike T et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res 73, 5508–5518, doi: 10.1158/0008-5472.CAN-12-4593 (2013). [DOI] [PubMed] [Google Scholar]
- 106.Papillon JPN et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J Med Chem 61, 10155–10172, doi: 10.1021/acs.jmedchem.8b01318 (2018). [DOI] [PubMed] [Google Scholar]
- 107.Farnaby W et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol 15, 672–680, doi: 10.1038/s41589-019-0294-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Winter GE et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381, doi: 10.1126/science.aab1433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zengerle M, Chan KH & Ciulli A Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 10, 1770–1777, doi: 10.1021/acschembio.5b00216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lu J et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 22, 755–763, doi: 10.1016/j.chembiol.2015.05.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reisman DN, Sciarrotta J, Wang W, Funkhouser WK & Weissman BE Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res 63, 560–566 (2003). [PubMed] [Google Scholar]
- 112.Shorstova T et al. SWI/SNF-Compromised Cancers Are Susceptible to Bromodomain Inhibitors. Cancer Res 79, 2761–2774, doi: 10.1158/0008-5472.CAN-18-1545 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Hohmann AF et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nature Chemical Biology 12, 672-+, doi: 10.1038/nchembio.2115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Theodoulou NH et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J Med Chem 59, 1425–1439, doi: 10.1021/acs.jmedchem.5b00256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kennison JA & Tamkun JW Dosage-dependent modifiers of polycomb and antennapedia mutations in Drosophila. Proc Natl Acad Sci U S A 85, 8136–8140, doi: 10.1073/pnas.85.21.8136 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tamkun JW et al. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 68, 561–572, doi: 10.1016/0092-8674(92)90191-e (1992). [DOI] [PubMed] [Google Scholar]
- 117.Wilson BG et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18, 316–328, doi: 10.1016/j.ccr.2010.09.006 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Erkek S et al. Comprehensive Analysis of Chromatin States in Atypical Teratoid/Rhabdoid Tumor Identifies Diverging Roles for SWI/SNF and Polycomb in Gene Regulation. Cancer Cell 35, 95-+, doi: 10.1016/j.ccell.2018.11.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kadoch C et al. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet 49, 213–222, doi: 10.1038/ng.3734 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim KH et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med 21, 1491–1496, doi: 10.1038/nm.3968 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim KH & Roberts CW Targeting EZH2 in cancer. Nat Med 22, 128–134, doi: 10.1038/nm.4036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knutson SK et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8, 890–896, doi: 10.1038/nchembio.1084 (2012). [DOI] [PubMed] [Google Scholar]
- 123.Italiano A et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19, 649–659, doi: 10.1016/S1470-2045(18)30145-1 (2018). [DOI] [PubMed] [Google Scholar]
- 124.Kawano S et al. Preclinical Evidence of Anti-Tumor Activity Induced by EZH2 Inhibition in Human Models of Synovial Sarcoma. PLoS One 11, e0158888, doi: 10.1371/journal.pone.0158888 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kurmasheva RT et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 64, doi: 10.1002/pbc.26218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Knutson SK et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110, 7922–7927, doi: 10.1073/pnas.1303800110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Le Loarer F et al. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosomes Cancer 53, 475–486, doi: 10.1002/gcc.22159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maruyama D et al. First-in-Human Study of the EZH1/2 Dual Inhibitor DS-3201b in Patients with Relapsed or Refractory Non-Hodgkin Lymphomas — Preliminary Results. Blood 130, 4070–4070, doi: 10.1182/blood.V130.Suppl_1.4070.4070 (2017). [DOI] [Google Scholar]
- 129.Mohammad HP, Barbash O & Creasy CL Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med 25, 403–418, doi: 10.1038/s41591-019-0376-8 (2019). [DOI] [PubMed] [Google Scholar]
- 130.Torchia J et al. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 30, 891–908, doi: 10.1016/j.ccell.2016.11.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Knipstein JA et al. Histone deacetylase inhibition decreases proliferation and potentiates the effect of ionizing radiation in atypical teratoid/rhabdoid tumor cells. Neuro Oncol 14, 175–183, doi: 10.1093/neuonc/nor208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Johann PD et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29, 379–393, doi: 10.1016/j.ccell.2016.02.001 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Ho B et al. Molecular subgrouping of Atypical Teratoid / Rhabdoid Tumors (ATRT) - a reinvestigation and current consensus. Neuro Oncol, doi: 10.1093/neuonc/noz235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chun HE et al. Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell 29, 394–406, doi: 10.1016/j.ccell.2016.02.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weissmiller AM et al. Inhibition of MYC by the SMARCB1 tumor suppressor. Nat Commun 10, 2014, doi: 10.1038/s41467-019-10022-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jagani Z et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med 16, 1429–1433, doi: 10.1038/nm.2251 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Alimova I et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int J Cancer 144, 1983–1995, doi: 10.1002/ijc.31873 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Venkataraman S et al. Targeting Aurora Kinase A enhances radiation sensitivity of atypical teratoid rhabdoid tumor cells. J Neurooncol 107, 517–526, doi: 10.1007/s11060-011-0795-y (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xue Y et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat Commun 10, 557, doi: 10.1038/s41467-019-08380-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xue Y et al. CDK4/6 inhibitors target SMARCA4-determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat Commun 10, 558, doi: 10.1038/s41467-018-06958-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tagal V et al. SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat Commun 8, 14098, doi: 10.1038/ncomms14098 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Geoerger B et al. A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin Cancer Res 23, 2433–2441, doi: 10.1158/1078-0432.CCR-16-2898 (2017). [DOI] [PubMed] [Google Scholar]
- 143.Mosse YP et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin Cancer Res 25, 3229–3238, doi: 10.1158/1078-0432.CCR-18-2675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Oberlick EM et al. Small-Molecule and CRISPR Screening Converge to Reveal Receptor Tyrosine Kinase Dependencies in Pediatric Rhabdoid Tumors. Cell Rep 28, 2331–2344 e2338, doi: 10.1016/j.celrep.2019.07.021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chauvin C et al. High-Throughput Drug Screening Identifies Pazopanib and Clofilium Tosylate as Promising Treatments for Malignant Rhabdoid Tumors. Cell Rep 21, 1737–1745, doi: 10.1016/j.celrep.2017.10.076 (2017). [DOI] [PubMed] [Google Scholar]
- 146.Wong JP et al. Dual Targeting of PDGFRalpha and FGFR1 Displays Synergistic Efficacy in Malignant Rhabdoid Tumors. Cell Rep 17, 1265–1275, doi: 10.1016/j.celrep.2016.10.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Miller RE et al. Synthetic Lethal Targeting of ARID1A-Mutant Ovarian Clear Cell Tumors with Dasatinib. Mol Cancer Ther 15, 1472–1484, doi: 10.1158/1535-7163.MCT-15-0554 (2016). [DOI] [PubMed] [Google Scholar]
- 148.Lang JD et al. Ponatinib Shows Potent Antitumor Activity in Small Cell Carcinoma of the Ovary Hypercalcemic Type (SCCOHT) through Multikinase Inhibition. Clin Cancer Res 24, 1932–1943, doi: 10.1158/1078-0432.CCR-17-1928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yamamoto S, Tsuda H, Takano M, Tamai S & Matsubara O PIK3CA mutations and loss of ARID1A protein expression are early events in the development of cystic ovarian clear cell adenocarcinoma. Virchows Arch 460, 77–87, doi: 10.1007/s00428-011-1169-8 (2012). [DOI] [PubMed] [Google Scholar]
- 150.Bosse T et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol 26, 1525–1535, doi: 10.1038/modpathol.2013.96 (2013). [DOI] [PubMed] [Google Scholar]
- 151.St Pierre R & Kadoch C Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev 42, 56–67, doi: 10.1016/j.gde.2017.02.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Samartzis EP et al. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget 5, 5295–5303, doi: 10.18632/oncotarget.2092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dal Molin M et al. Loss of expression of the SWI/SNF chromatin remodeling subunit BRG1/SMARCA4 is frequently observed in intraductal papillary mucinous neoplasms of the pancreas. Hum Pathol 43, 585–591, doi: 10.1016/j.humpath.2011.06.009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang Q et al. Chromatin remodeling gene AT-rich interactive domain-containing protein 1A suppresses gastric cancer cell proliferation by targeting PIK3CA and PDK1. Oncotarget 7, 46127–46141, doi: 10.18632/oncotarget.10060 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yap TA, Bjerke L, Clarke PA & Workman P Drugging PI3K in cancer: refining targets and therapeutic strategies. Curr Opin Pharmacol 23, 98–107, doi: 10.1016/j.coph.2015.05.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ding Y et al. Chromatin remodeling ATPase BRG1 and PTEN are synthetic lethal in prostate cancer. J Clin Invest 129, 759–773, doi: 10.1172/JCI123557 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Howard TP et al. MDM2 and MDM4 Are Therapeutic Vulnerabilities in Malignant Rhabdoid Tumors. Cancer Res 79, 2404–2414, doi: 10.1158/0008-5472.CAN-18-3066 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Carugo A et al. p53 Is a Master Regulator of Proteostasis in SMARCB1-Deficient Malignant Rhabdoid Tumors. Cancer Cell 35, 204–220 e209, doi: 10.1016/j.ccell.2019.01.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Williamson CT et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun 7, 13837, doi: 10.1038/ncomms13837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Park Y et al. Loss of ARID1A in Tumor Cells Renders Selective Vulnerability to Combined Ionizing Radiation and PARP Inhibitor Therapy. Clin Cancer Res 25, 5584–5594, doi: 10.1158/1078-0432.CCR-18-4222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Miao D et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806, doi: 10.1126/science.aan5951 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pan D et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775, doi: 10.1126/science.aao1710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shen J et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med 24, 556–562, doi: 10.1038/s41591-018-0012-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ashizawa M et al. Prognostic role of ARID1A negative expression in gastric cancer. Sci Rep 9, 6769, doi: 10.1038/s41598-019-43293-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Buglioni S et al. The clinical significance of PD-L1 in advanced gastric cancer is dependent on ARID1A mutations and ATM expression. Oncoimmunology 7, e1457602, doi: 10.1080/2162402X.2018.1457602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim YB, Ahn JM, Bae WJ, Sung CO & Lee D Functional loss of ARID1A is tightly associated with high PD-L1 expression in gastric cancer. Int J Cancer 145, 916–926, doi: 10.1002/ijc.32140 (2019). [DOI] [PubMed] [Google Scholar]
- 167.Jiang T, Chen X, Su C, Ren S & Zhou C Pan-cancer analysis of ARID1A Alterations as Biomarkers for Immunotherapy Outcomes. J Cancer 11, 776–780, doi: 10.7150/jca.41296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Leruste A et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell, doi: 10.1016/j.ccell.2019.10.008 (2019). [DOI] [PubMed] [Google Scholar]
- 169.Chun HE et al. Identification and Analyses of Extra-Cranial and Cranial Rhabdoid Tumor Molecular Subgroups Reveal Tumors with Cytotoxic T Cell Infiltration. Cell Rep 29, 2338–2354 e2337, doi: 10.1016/j.celrep.2019.10.013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Forrest SJ et al. Genomic and immunologic characterization of INI1-deficient pediatric cancers. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-19-3089 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jelinic P et al. Immune-Active Microenvironment in Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Rationale for Immune Checkpoint Blockade. J Natl Cancer Inst 110, 787–790, doi: 10.1093/jnci/djx277 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.McDermott DF et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 24, 749–757, doi: 10.1038/s41591-018-0053-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Keenan TE, Burke KP & Van Allen EM Genomic correlates of response to immune checkpoint blockade. Nat Med 25, 389–402, doi: 10.1038/s41591-019-0382-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Schick S et al. Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat Genet 51, 1399–1410, doi: 10.1038/s41588-019-0477-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]