

Abstract
Patient-derived orthotopic xenograft (PDOX) models have been verified as a useful method for studying human cancers in mice. Previous studies on the extent of metastases in these models have been limited by the necessity of welfare euthanasia (primary tumors reaching threshold size), at which point metastases may only be micrometers in diameter, few in number, and solely identified by step-sectioning of formalin-fixed paraffin-embedded tissue. These small micro-metastases are less suitable for many downstream molecular analyses than macro-metastases. Resection of the primary tumor by survival surgery has been proven to allow further time for metastases to grow. Although PDOX models of triple-negative breast cancer (TNBC) shed circulating tumor cells (CTCs) into the bloodstream and metastasize, similar to human TNBC, little data has been collected in these TNBC PDOX models regarding the association between CTC characteristics and distant metastasis following excision of the primary tumor xenograft. This study assembles a timeline of PDOX tumor shedding and metastatic tumor progression before and after tumor excision surgery. We report the ability to use tumorectomies to increase the lifespan of TNBC PDOX models with the potential to obtain larger metastases. CTC clusters and CTCs expressing a mesenchymal marker (vimentin) were associated with metastatic burden in lung and liver. The data collected through these experiments will guide the further use of PDOX models in studying metastatic TNBC.
Keywords: Patient-derived orthotopic xenograft (PDOX), circulating tumor cells (CTCs), triple-negative breast cancer (TNBC), epithelial-to-mesenchymal transition (EMT), tumorectomy, metastasis
Introduction
Breast cancer accounts for the highest percentage of all new cancer diagnoses in women and is responsible for the second largest number of deaths from cancer in the United States [1]. Triple-negative breast cancers (TNBCs) are distinguished as lacking or having low expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor 2 (HER2) expression [2]. TNBCs make up 10–17% of breast cancer occurrences and cause more aggressive tumor growth, larger tumors, and a greater likelihood of metastasis when compared to other breast cancers [2–5]. Unfortunately, there are currently no targeted therapies for the treatment of TNBC. Higher mortality rates and shorter disease-free survival are both seen in TNBCs, with the majority of these deaths being a result of metastatic disease [4–7]. Distant metastases are usually seen in the bone, brain, lung, liver, or distant lymph nodes [5, 8, 9].
Accurate models of TNBC and its metastatic pathway are needed to better understand this subtype and develop new therapies. Human cancers can be studied in vivo through the successful transplantation and propagation of human cells and tissues into several strains of immunodeficient mice [10–13]. NOD scid gamma (NSG) mice lack the IL-2 γ-chain and therefore do not develop functional natural killer (NK) cells. They are also “non-leaky” and have longer lifespans than NOD/SCID mice due to the absence of thymic lymphomas [14]. The lack of functional B cells, T cells, and NK cells, in addition to the non-leakiness and increased lifespan of NSG mice, make them ideal recipients of xenografts and have been shown to have higher engraftment rates and faster tumor growth than other strains [15–17].
Patient-derived xenograft (PDX) models are advantageous because tumor tissue is taken from the human patient and directly implanted subcutaneously into the mouse, allowing for the convenient study of patient tumors and the ability to anticipate treatment response [18]. However, subcutaneous implantation of human tumor tissue rarely leads to metastasis and fails to recapitulate the original tumor microenvironment [19, 20]. Patient-derived orthotopic xenograft (PDOX) models differ from PDX models in that the patient-derived tissue is implanted into the mouse in the same anatomical location from which the tumor was derived in the patient, rather than subcutaneously [12]. As well described by Hoffman in xenograft models of breast and other cancers, orthotopic implantation better recapitulates the natural course of disease and metastasis when compared to subcutaneous implantation [20–23]. PDOX models retain characteristics, such as histopathology, gene expression, and copy number variation, of the human primary tumor, even through multiple passages [16, 24, 25].
Patient-derived tissue engraftment has been proven to be a reliable model of human cancers including lung cancer, pancreatic cancer, colorectal cancer, prostate cancer, ovarian cancer, and breast cancer [19, 24]. Mouse models of cancer using patient-derived tissue can be used to test new therapies, study tumor progression in depth, find biomarkers of drug response and resistance, and provide an individualized assessment of chemosensitivity of a patient’s tumor [19, 21, 24–26]. However, these models do come with challenges, such as relatively low rates of primary tumor establishment [27]. Additionally, the study of metastases is restricted by the growth of primary tumors and the need to euthanize mice when primary tumor burden exceeds established welfare criteria, i.e. largest tumor diameter reaches 1.75 cm. Therefore, euthanasia may be required prior to the detection of metastases [28]. This restriction has been circumvented by resecting primary tumors, thus removing the primary tumor burden and allowing the metastases to continue growing in a living model [29]. However, primary tumor regrowth at the excision site has limited the ability to study distant metastases in previous studies [30].
Circulating tumor cells (CTCs) are one mechanism through which metastases form. Their presence is clinically significant in patients with metastatic breast cancer [31]. CTCs are cancer cells that are shed from primary tumors or metastases, travel through the bloodstream, and potentially form metastases at distant sites [32, 33]. They are a clinically useful tool, in part because of the correlation between CTCs and disease progression as well as the ability to track CTCs serially in minimally invasive patient blood draws [33, 34]. CTCs can be an accurate predictor of overall survival and progression-free survival in women with metastatic breast cancer, including TNBC [31, 35].
Epithelial-to-mesenchymal transition (EMT) is one method through which epithelial circulating tumor cells can invade and metastasize. During EMT, epithelial cells lose their cell-to-cell adhesions and other epithelial characteristics and gain mesenchymal features that confer increased motility and invasive potential, thus enabling them to invade through endothelial cell tight junctions and enter the circulatory system [36–38]. Eventually, the cancer cells may then undergo a reverse process, called mesenchymal-to-epithelial transition (MET), to form metastases in distant sites [36–38]. The ability to undergo these transitions is thought to be an essential characteristic of successful metastatic cells [38].
Our study aimed to investigate the effect of interval tumor excision in PDOX models of TNBC on CTCs and metastases. We aimed to further characterize the timeline of tumor shedding and its correlation with CTC enumeration and characteristics. We have previously shown that metastases in multiple TNBC PDOX models, if present, are microscopic at the time of welfare euthanasia [39]. The lack of gross metastases limits the interrogation of metastases to histological analyses. Surgical removal of the primary tumor may allow metastases to develop longer and grow larger, potentially enabling far more extensive downstream molecular studies, including analysis of mutations, copy number alterations, and gene and protein expression among potentially heterogeneous metastases in the same or in multiple metastatic sites. These downstream molecular studies could then be correlated with the evolution of single CTCs and CTC clusters to better understand the progression of TNBC and determine accurate methods of detection, prognosis, and treatment.
Results
1. Validation of the TNBC Model
A single-cell suspension of tumor SUTI151, previously obtained from a human TNBC patient, was injected into the mammary fat pad of 30 mice. Two weeks following injection of SUTI151 tumor cells, 23 of 30 mice had palpable primary tumors. Of the seven mice that lacked palpable primary tumors within two weeks, one failed to grow tumor throughout the study duration; two had late-growing tumors that only became palpable 11 weeks post-injection and too late for a tumorectomy at week six; and four developed peritoneal carcinomatosis, likely due to deep injection of the tumor cell suspension with inadvertent intra-abdominal seeding. Thus, in all, 25 of 30 mice (83.3%) had mammary fat pad engraftment of human TNBC, but only 23 had tumors at week two and eligible for randomization into no treatment and treatment groups at week 6. As a control, five age-matched mice were injected with 100μl of pure PBS and 100μl of Matrigel combined. None of these control mice developed tumors.
2. Tumor Excision and Primary Tumor Analysis
Primary tumors were measured twice a week with primary tumor volume plotted as a function of time (Figure 1A).
Figure 1.

Primary tumor, CTC, and metastasis data for mice engrafted with a single-cell suspension from TNBC patient SUTI151(N=19). (A) Tumor volume is graphed as a function of days post-implant. Each graph corresponds to a single group with each line representing a separate mouse. (B) Raw data for primary tumors, metastatic tumors, and CTCs for individual mice. Mice in Group A did not receive tumorectomies, and therefore did not have tumor regrowth. Mouse C5 was the only mouse that received a tumor excision surgery and did not have regrowth at the site of tumor resection - days until regrowth has thus been indicated as N/A. For the one mouse from which we were unable to collect cardiac blood (ID: C4), N/A has been indicated in columns for CTC clusters and characteristics. Control mice did not grow primary or metastatic tumors, relevant values have been marked as N/A. Large tumors were considered those with a volume greater than 2000mm3, medium tumors had volumes between 1000mm3 and 2000mm3, and small tumors had volumes less than 1000mm3. CK+ -cytokeratin positive, VIM+ -vimentin positive, CK+VIM+ -cytokeratin and vimentin positive.
To account for variation in tumor size at the time of tumor excision at week 6, mice with small, medium, and large tumors were randomly assigned to three groups. Of the 23 mice undergoing randomization at week six, four were excluded from further study due to regional extension of primary tumor within the abdominal cavity that precluded tumor resection (n=2) or because of rapid and aggressive regrowth of primary tumor following tumorectomy (n=2), with all four mice requiring early humane euthanasia. The remaining 19 mice were divided into three groups: the no-tumorectomy Group A (n=7) and mice who underwent tumor resections at week 6. Mice receiving tumorectomies were further divided into two groups based on the time at which they were euthanized (Group B: 13–14 weeks post-implantation, n=6; Group C = 15–16 weeks post-implantation, n=6) (Figure 2). Of the 12 mice undergoing tumorectomy, 11 had tumor regrowth at the excision site requiring early euthanasia. Six mice had tumor regrowth that appeared between 16 and 20 days following surgery and were euthanized between 13 and 14 weeks post-implantation; these mice are categorized as Group B (Figure 1B, Figure 2). The remaining mice were euthanized between 15 and 16 weeks and labeled as Group C (Figure 1B, Figure 2). Within Group C, tumor regrowth occurred more slowly and appeared between 30 and 40 days following surgery. One mouse did not show local tumor regrowth following excision.
Figure 2.
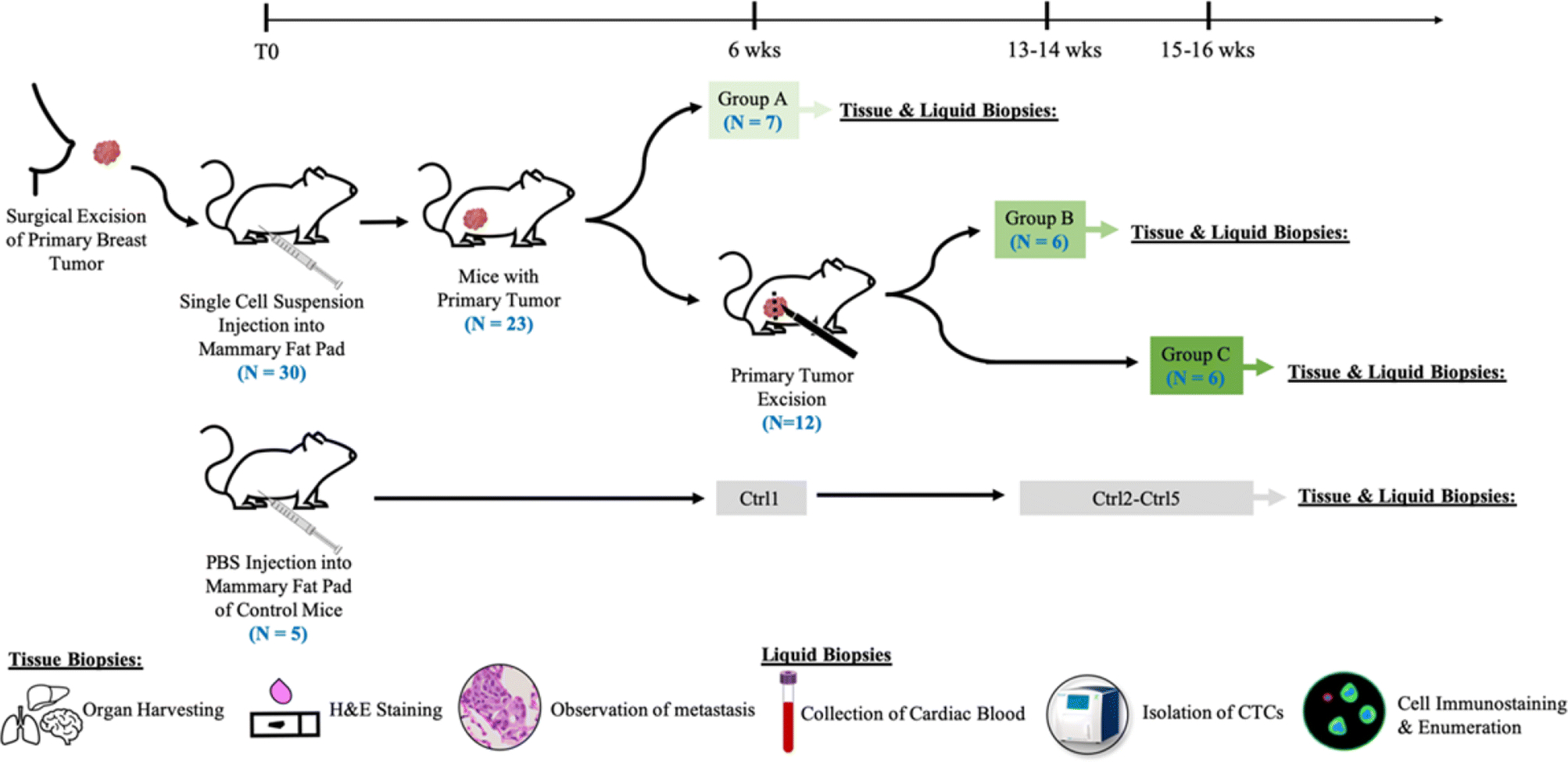
Workflow schematic of the study. Experimental mice (N=30) were injected with single-cell suspension of patient SUTI151 and primary tumors were allowed to grow. Age matched control mice (N=5) were injected with PBS. At six weeks post-implant, seven mice with intact primary tumors were euthanized (Group A). The remaining mice (N=12) received tumorectomies and were euthanized at 13–14 weeks post-implant (Group B) and 15–16 weeks post implant (Group C) respectively. For each cohort, at the time of euthanasia, blood was drawn for CTC immunostaining and enumeration, while organs were collected for H&E analysis of metastases.
3. CTC Analysis
To study the application of tumor excision surgery in PDOX models of TNBC, we analyzed the CTCs and metastases of mice across all groups. In total, 23 experimental (n=18) and control (n=5) mice underwent CTC analysis. CTC-like baseline counts in naïve age-matched control mice ranged from 21–40 CTCs/ml, with 40 CTC-like cells/ml serving as a lower limit threshold. One mouse (C4) did not have an adequate blood sample to perform CTC enumeration. One mouse (A3) did not exceed the criterion CTC threshold of 40 CTCs/ml. The highest number of CTCs was 3349 CTCs/ml in mouse A1 from Group A (Figure 1B, Figure 3A). The median number of CTCs/ml increased as the mice aged, with the median increasing by more than 50 cells in each subsequent group (Figure 3A). However, there was no significant correlation between the number of CTCs/ml and metastatic variables. CTC clusters were found in 12 of the 18 mice, all of whom had metastases in both the lung and the liver (Figure 1B). No CTC clusters were found in control mice.
Figure 3.
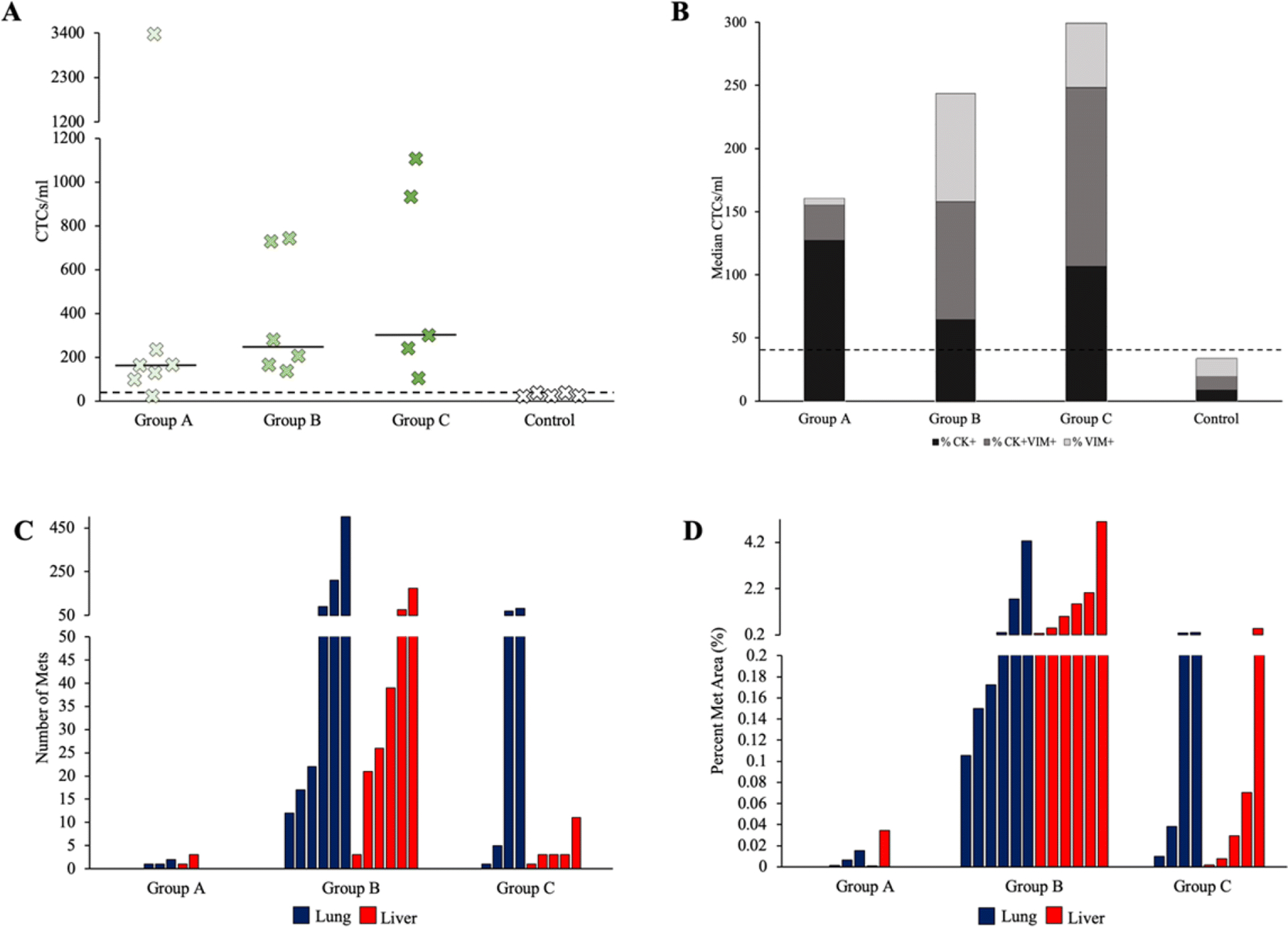
CTC and metastasis data for PDOX models of TNBC. (A) The number of CTCs/ml for each mouse studied. The median for each group is represented by solid horizontal lines. The control threshold of 40 CTCs/ml is represented by a dashed horizontal line. (B) Percent of total CTCs/ml expressing different phenotypes in each group. Control mice had between 21 and 40 cells that stained similarly to CTCs, so a threshold of 40 CTCs/ml is set and represented by a dashed horizontal line. CK+ -cytokeratin positive, VIM+ -vimentin positive, CK+VIM+ -cytokeratin and vimentin positive. (C) The number of metastases found on the organ section with the largest metastatic burden for each mouse. Blue bars indicate lung data and red bars indicate liver data. (D) Percent area of metastases for each mouse studied. Areas were calculated as a percent of the organ tissue on the section with the largest metastatic burden.
We chose to characterize EMT in CTCs using human cytokeratin (CK) and vimentin (VIM) markers. CK staining is a commonly used and accepted marker for epithelial CTC classification and enumeration and VIM has been shown to be among the most frequent and representative marker of EMT in human tumors and CTCs [40–42]. The proportion of CTCs expressing cytokeratin, vimentin, or both, varied between groups (Figure 1B, Figure 3B). The presence of cytokeratin expression was highest in Group A, combined phenotype expression (cytokeratin- and vimentin-positive) was highest in Group C, and vimentin expression was highest in Group B. Neither cytokeratin expression nor combined vimentin and cytokeratin expression had a significant correlation to metastatic variables. Overall, vimentin expression had a moderately positive correlation with both the number of lung slides with metastases (Pearson correlation coefficient: r=0.5633) and the number of liver slides with metastases (Pearson correlation coefficient: r=0.4958).
4. Metastasis analysis
Hematoxylin and eosin (H&E) analysis showed metastases in both the lung and the liver (Figure 4 and Figure 5). Overall, 14 out of 19 (73.7%) mice had metastases, with 12 of those having metastases to both the lung and the liver (Figure 1B). Two of the 12 mice (16.7%) that received tumor excision surgery had gross metastases found during necropsy at 14 weeks post-implant. One mouse had a grossly-visible hepatic metastasis while the other mouse had hepatic and pulmonary metastases. Grossly visible tumors were less than 2mm in diameter. Both mice were in Group B and had regrowth at the primary tumor site following excision.
Figure 4.
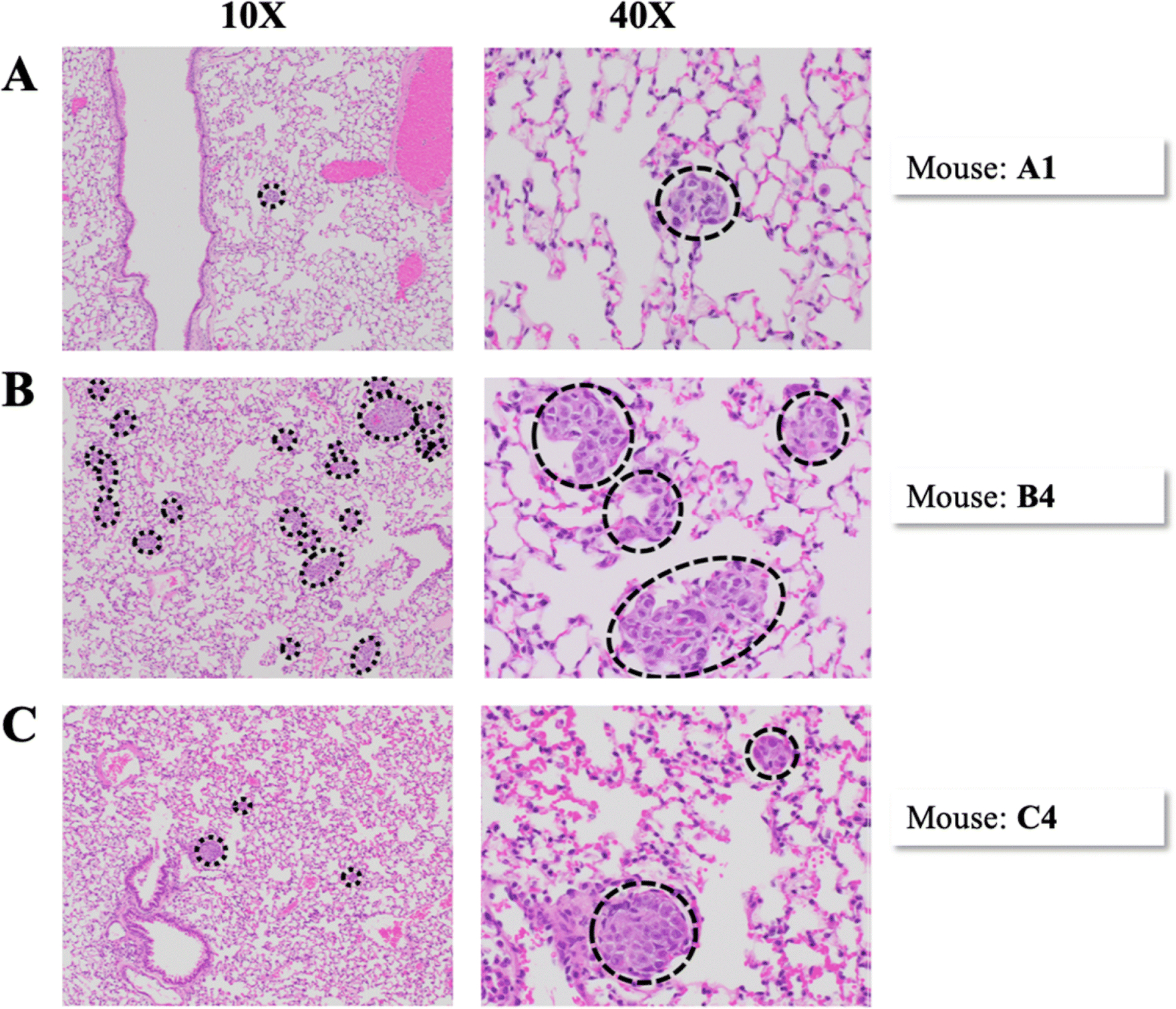
Representative H&E images of metastases in the lung. Each row of images represents a separate mouse. Images in the left column were taken at 10x magnification and images in the right column were taken at 40x magnification. Dashed lines encircle metastases. (A) Mouse A1, Group A. (B) Mouse B4, Group B. (C) Mouse C4, Group C.
Figure 5.
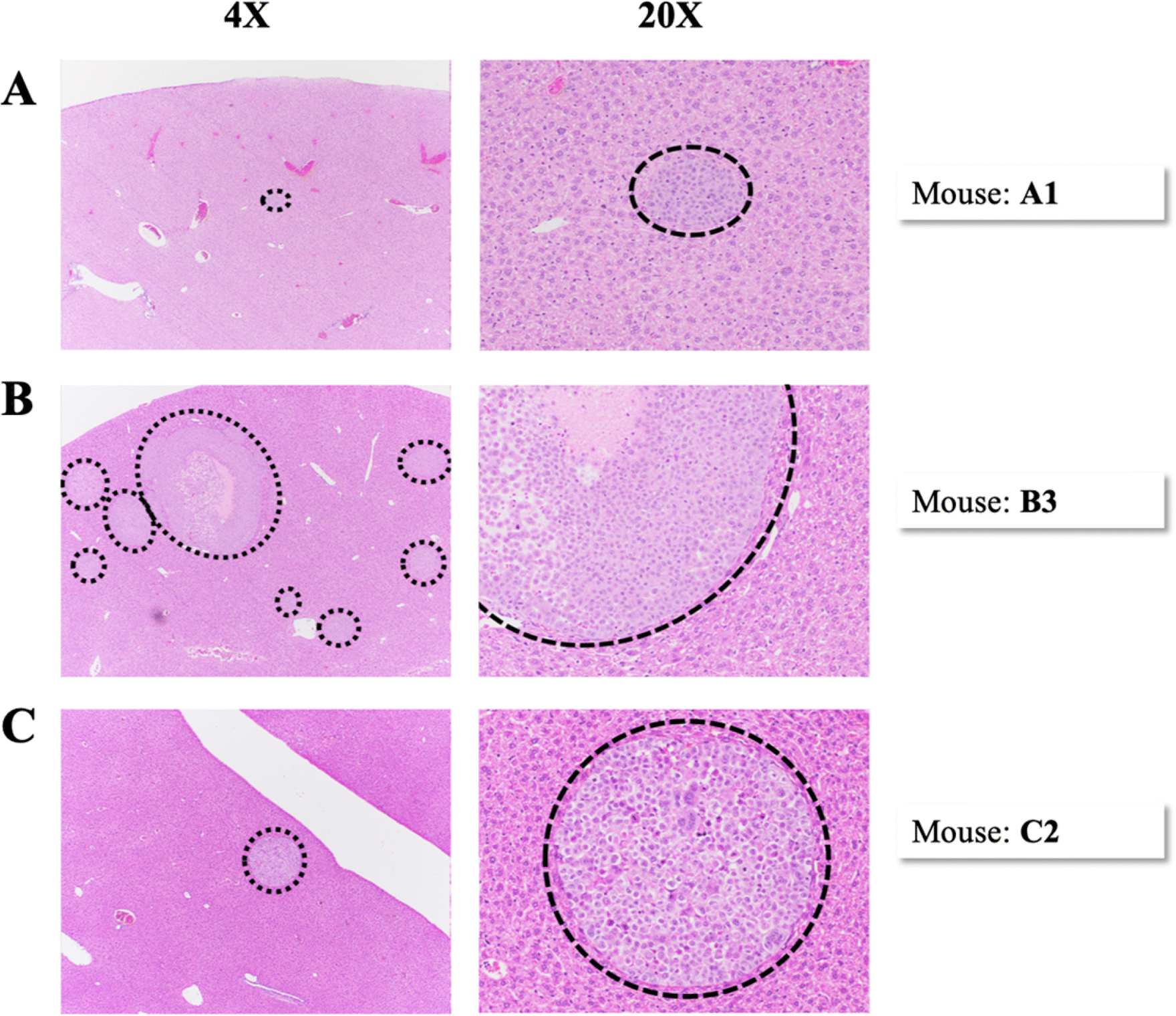
Representative H&E images of metastases in the liver. Each row of images represents a separate mouse. Images in the left column were taken at 4x magnification and images in the right column were taken at 20x magnification. Dashed lines encircle metastases. (A) Mouse A1, Group A. (B) Mouse B3, Group B. (C) Mouse C2, Group C.
Metastases were not seen or detected histologically in the brain sections of any mice for this particular PDOX model SUTI151, similar to our previous findings [39]. Overall, the number of lung slides with metastatic foci was similar to the number of liver slides, 103 out of 210 (49%) and 95 out of 210 (45%) respectively. The number of lung and liver slides with metastases present was greatest in Group B, with pulmonary and hepatic metastases found in all six mice. The median number of lung and liver metastases was also greatest in Group B (Figure 3C), with one mouse having over 500 lung metastases. The median area of metastases in the slide with the most metastatic burden was greatest in Group B and lowest in Group A for both the lungs and livers (Figure 3D). No correlation was found between the size of the original tumor at six weeks (either small, medium, or large), and the number of CTCs, CTC marker expression, number of slides with metastases, number of metastases, or area of metastases. Within individual metastases, there was a moderate amount of cellular and nuclear pleomorphism. Nuclei were large and irregularly shaped with relatively frequent mitotic figures. Several metastases had large areas of ischemic necrosis, which is indicative of rapid growth (Figure 5B).
Discussion
Our study reports the ability to use tumor excision surgery to increase the lifespan of PDOX models of TNBC and follow disease progression through EMT marker expression in CTCs and metastatic characteristics. By removing primary tumor burden, we successfully increased the lifespan of the mice from six weeks post implant to 16 weeks post implant and saw rare incidences of gross metastases in both the lung and the liver. Groups of mice were compared using the number of CTCs/ml, presence of CTC clusters, cytokeratin and vimentin expression, the number of organ sections with metastases, the area of metastases, and the number of metastases.
While the median number of CTCs/ml generally increased with time, CTC expression of vimentin proved to possibly be a better predictor of metastatic burden than CTCs/ml and was considerably greater in mice with higher metastatic burden. Though CTCs have meaningful prognostic potential, 30–70% of patients with tumor cells in their bone marrow or lymph nodes never develop metastases [43]. Metastasis is an inefficient process with many obstacles that may stop a cancer cell from successfully forming a metastasis [9]. Metastasis-initiating cells make up an extremely small percentage of the total CTCs [44], meaning the large majority of cells in circulation never become metastases and may not have the ability to form metastases. This could be the reason why CTCs/ml generally increased as time increased but did not correlate with metastases in this study. This is also consistent with the results of our prior study showing that some mice with CTCs did not have metastases [39]. It should be noted that because mice in both Groups B and C had local tumor regrowth, a portion of the CTCs could have been shed from the tumor regrowth rather than from the original primary tumor or metastases, and may have altered CTC counts.
The advancement of CTC isolation technologies has allowed for the capture of CTC clusters as well as single CTCs and led to studies showing that CTC clusters have a dramatically higher potential to become metastases compared to single CTCs in PDOX models of breast cancer [45, 46]. In human breast cancer patients, CTC clusters add prognostic value and are more strongly correlated with decreased progression-free survival than single CTCs [45, 47]. Similarly, our own study showed that all mice with both pulmonary and hepatic metastases had CTC clusters present in the blood. Mice without metastases did not have clusters, which is consistent with previous studies indicating that over 97% of metastases were the result of multicellular seeds rather than single cells [48]. The absence of CTC clusters in mice with metastases to a single organ defends the ability of single CTCs to develop into metastases, but this route may be a slower and more inefficient process. It is necessary to note that, in the present study, CTCs were isolated from terminal blood rather than serial survival blood draws - making it possible that clusters or variable expression profiles were present prior to the euthanasia timepoint. Additionally, CTC clusters increase significantly as cancer progresses [49], so it follows that CTC clusters would be seen in mice with greater disease progression. Mesenchymal markers have also been associated with CTC clusters, indicating that EMT could be occurring in clusters within the bloodstream [50]. Our own study found that both vimentin-expressing CTCs and CTC clusters were associated with more widespread metastases in the lung and liver, supporting the role of EMT and group dissemination in the metastatic process.
We found that the percent of vimentin expression, a mesenchymal marker suggestive of the occurrence of EMT, was correlated with a greater distribution of metastases in the lung and liver. The expression of combined vimentin and cytokeratin increased with time and was greater in mice that lived the longest. This would indicate that mesenchymal expression is a possible prognostic factor in predicting metastasis and is consistent with available data showing that disease progression is correlated with an increase in mesenchymal CTCs in human patients with TNBC [50]. In this particular study, it is important that tumor regrowth could affect CTC phenotype, as cells that regrow following resection are biologically invasive and may be more likely to shed CTCs that have undergone EMT with increased vimentin expression. Association between mesenchymal CTCs and more aggressive characteristics is generally seen across breast cancers, with TNBC patients having CTCs that are predominantly mesenchymal [50]. More invasive breast cancers are associated with higher levels of vimentin, while epithelial marker expression is associated with more weakly invasive breast cancers [51].
Mice receiving tumorectomies were divided into two groups, Group B and Group C. Though mice in Group C lived slightly longer, mice in Group B had larger percent areas of metastases, higher number of metastases, and greater distributions of metastases in organ sections. Faster local tumor regrowth in Group B compared to Group C may point to the tumors being more aggressive and account for the differences in metastatic burden. Through the process of self-seeding, CTCs can leave the metastatic site and enter circulation with the ability to re-infiltrate the original tumor or new sites [52]. The process self-selects for more aggressive cells that have already been able to disseminate and could therefore lead to increased tumor growth [52]. This may explain why regrowth at the site of original primary tumor resection was faster and grew larger than the original primary tumors, especially in Group B. It is also interesting to note that mouse C5 in Group C is the only mouse that did not develop tumor recurrence and did not have any metastases. This may suggest that regrowth is a symptom of systemic disease in PDOX mouse models rather than due to limitations of the tumor resection.
In total, mice with a larger number of metastases in the lung had larger areas of lung metastases, but the number of metastases and the size of metastases did vary (Figure 4 and Figure 5). For example, mouse B3 had 210 individual metastases that took up over 4% of the lung section, but mouse B4 had less than half that area and over 500 individual metastases. Mouse B2 had just 22 lung metastases which accounted for 0.15% of the tissue but was the only mouse with grossly visible metastases to the lung. The reason that mice with gross metastases might have a lower number of micro-metastases or area of micro-metastases than mice that did not have gross metastases could be due to tumor mass latency. Tumor latency describes when small colonies of cancer cells exist as micro-metastases and, due to competing rates of proliferation and cell death, never become macro-metastases unless they acquire the ability to colonize by possibly gaining genetic or epigenetic changes [9, 53]. Some of the micro-metastases seen in this study may be oscillating in the tumor latency state and will never become gross metastases or may take much longer to grow. Gross metastases are generally rare, as seen in a similar study which resected primary tumors of mice implanted with human TNBC tissue and only found overt metastases in the lungs of 3 of 144 mice examined (2.1%) [30]. This is somewhat less than our own study, which found gross metastases in 2 of 12 mice (16.7%) that received tumor excision surgery and were euthanized 14 weeks post-implant due to local tumor regrowth. More gross metastases may have been found if the mice in this study were able to live longer.
One of our challenges was the high incidence of tumor regrowth after resection, which has also been seen in other studies of breast cancer mouse models [30, 54, 55]. Local or regional recurrence of breast cancer occurs in 5–30% of patients who receive radical or modified radical mastectomy [53]. A higher rate of locoregional recurrence after undergoing mastectomy is seen in women with TNBC compared to other subtypes [56]. In the context of an aggressive cancer such as TNBC, there is likely a higher risk of recurrence in PDOX models as well. A possible causal factor for the recurrence of primary tumors is lympho-vascular invasion, the spread of cancer cells into the lymphatics or blood vessels. TNBC has comparable or lower rates of lymphatic invasion and nodal involvement compared to other subtypes, but lympho-vascular invasion is still a significant risk factor for decreased disease-free survival and is linked to greater possibility of local recurrence and poorer prognosis in human patients with TNBC [57–59]. In this study, skin overlying the tumor was replaced after the excision and may have had dermal lympho-vascular invasion and/or residual tumor cells, so removing overlying skin during a tumorectomy may decrease the incidence of local recurrence. However, this technique would not have been possible in many of these xenograft tumors without imposing undue tension on the remaining skin surrounding the tumor.
Conclusion
This study reviews the use of survival surgery to extend the lifespan of PDOX models of TNBC by removing the primary tumor burden. Single-cell suspension of an aggressive patient-derived TNBC tumor was injected orthotopically and resulted in an overall engraftment rate of 83.3%. CTC expression of the mesenchymal marker vimentin, an indication of the occurrence of EMT, and the presence of CTC clusters were associated with metastases and may be better indicators of metastatic burden than enumeration of single CTCs as a continuous variable. To our knowledge, this study is the first to report a correlation between mesenchymal expression in CTCs and metastases in PDOX models of TNBC after use of tumor excision surgery to extend the lifespan of the models. However, due to the high rate of primary tumor recurrence following resection, further investigation is recommended to substantiate these associations. This data support a significant role of EMT and CTC clusters in the metastatic process and has the potential to guide future use of PDOX models to study TNBC.
Methods
Patient-Derived Orthotopic Xenografts
This study was completed according to the ethical protocol approved by Stanford’s Administrative Panel on Laboratory Animal Care (APLAC Protocol #12809). Female, 4-week-old NSG mice were obtained from The Jackson Laboratory – JAX stock #005557 [14, 60]. Mice were kept in pathogen-free housing and permitted to acclimate for three weeks prior to the implantation procedure. At experimental endpoints, or when the largest diameter of the tumor reached 1.75cm, mice were humanely euthanized with CO2 and cardiac exsanguination.
A single-cell suspension of tumor SUTI151, previously obtained from a woman’s aggressive TNBC and sterilely frozen [25], was thawed and resuspended in sterifiltered PBS. Live cells were then counted using a hemocytometer and volume adjusted so that approximately 1.1 million live cells were present per 100μl. The 100μl of tumor cell suspension was mixed with 100μl of Matrigel (Corning® Matrigel® Matrix High Concentration (HC), Phenol-Red Free *LDEV-Free) and drawn into a syringe for injection into the fourth mammary fat pad of 30 experimental mice at seven weeks of age. Matrigel is a matrix of proteins which increases the take rate, decreases the latency period, and increases tumor growth rate [61, 62]. Five control mice were injected with 100μl of pure PBS and 100μl of Matrigel combined. Mice were briefly placed in 1–3% of isoflurane before injection of the single-cell suspension into the mammary pad. Primary tumors were measured using calipers and their volume was calculated using the formula: (a2b)/2, where a is the shortest diameter and b is the longest diameter. Large tumors were considered those with a volume greater than 2000mm3, medium tumors had volumes between 1000mm3 and 2000mm3, and small tumors had volumes less than 1000mm3.
Of the 30 mice initially injected with the single-cell tumor suspension, only 19 mice were eligible for tumor follow-up or resection at week six and included in our experimental group analyses. Eleven mice were excluded for the following reasons: four had peritoneal carcinomatosis without mammary fat pad tumor growth, indicating possible injection depth error (the mammary fat pad in a non-pregnant mouse is only 1–2mm thick and tumor cells may have been injected intraperitoneally); two had primary tumors growing through the abdominal wall and resection could not be performed; two mice had immediate aggressive regrowth of tumor at the tumorectomy site requiring early euthanasia prior to the length of post-surgical survival time needed to be included in experimental groups; two mice had extremely slow-growing tumors that did not become palpable until well after the week six tumorectomy timepoint; and one mouse never grew a primary tumor.
Tumor Excision
At week six, mice in Groups B and C were anesthetized by inhalation of 1–3% isofluorane and given one subcutaneous dose of Carprofen (5–10 mg/kg) for analgesia. Lubricant was applied to the eye to avoid corneal desiccation. Hair was removed using an electric shaver and the surgical area was disinfected with alternating iodine and alcohol scrubs. Toe-pinch reflex was confirmed negative before incision, and the procedure was done using the tips-only aseptic technique. A skin incision was made over the mass, and the tumor was bluntly dissected away from surrounding tissue. After excision, half of the tumor tissue was fixed in formalin for histological analysis while the other half was placed into sterile Eppendorf tubes and stored at −80C for use in future studies. The incisions were closed using either sutures or surgical staples, which were removed one week after the procedure.
CTC Isolation, Enumeration and Expression Analysis
At the time of euthanasia, approximately 750μl of blood was collected via cardiac puncture for analysis of CTCs. A small skin incision was made to open the skin over the xyphoid process prior to blood draw to avoid contamination of skin epithelial cells. Cardiac blood was inserted into EDTA tubes and immediately taken to Vortex Biosciences laboratory so that blood processing could begin within 2–3 hours. Cardiac blood (500μl) was diluted 40x in PBS, transferred to a Vortex VTX-1 cartridge, and inserted into the VTX-1 Liquid Biopsy System [63] (Vortex Biosciences, Inc.). The VTX-1 system is an automated label-free platform which efficiently isolates CTCs based on physical characteristics such as size and deformability, without the use of biomarkers [63]. The blood underwent 3 cycles, and the resulting cells were collected in a cell culture-treated 96-well plate (Nunc), fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 10 minutes, permeabilized with 0.4% v/v Triton X-100 (Sigma Aldrich) for 7 minutes, blocked with 10% goat serum (Invitrogen) for 30 minutes, and labeled for 1 hour at room temperature (RT) with 4,6-diamidino-2-phenylin- dole (DAPI; Life Technologies), anti-CD45–phycoerythrin antibody (CD45-PE, clone HI30, BD Biosciences), a cocktail of primary antibodies labeled with fluorescein isothiocyanate to identify human cytokeratin (CK) positive cells (clone CK3–6H5, Miltenyi Biotec, and clone CAM5.2, BD Biosciences), an anti-CK-AlexaFluor (AF) 488 antibody (clone AE1/AE3, eBioscience) and an anti-vimentin-AF647 antibody reactive to human vimentin and not to mouse vimentin (clone V9, Abcam) [39, 63]. Cells were then imaged using a Zeiss Z1 microscope and counted manually by two different observers to account for inter-reader variability. The average difference between reader counts was less than 4 CTCs/ml and the maximum difference was 12 CTCs/ml (Supp. Figure 1). CTC counts were averaged between the two readers and calculated per ml (Figure 1B). Cells were identified as CTCs if they stained positive for DAPI, negative for CD45 (to rule out white blood cells), and positive for cytokeratin and/or vimentin [39, 63, 64]. CTC clusters were identified as groups of two or more CTCs; some clusters showed only CTCs and other clusters showed CTCs and WBCs. All CD45− and DAPI+ CTCs, whether single CTCs or CTCs in clusters, were counted and placed into one of three groups: those expressing only cytokeratin, those expressing only vimentin, and those expressing both cytokeratin and vimentin. Note that the cytokeratin and vimentin antibody stains used were previously shown to detect human but not mouse antigens [39- Figure S1]. Marker expression was calculated as a percent of the total number of CTCs/ml (Figure 1B, Figure 3B). Age-matched control mice had 40 or fewer CTC-like cells/ml, and 40 was used as the healthy threshold. Any mouse with CTC counts that fell below the threshold was designated as having zero CTCs/ml during statistical analysis, the counts of mice with greater than 40 CTC-like cells/ml were not changed (Supp. Figure 1).
Histology
After humane euthanasia via CO2, a necropsy was performed to identify any gross metastases. Upon identification of gross metastases, tissue was removed and placed into sterile Eppendorf tubes for future studies. Lungs, livers, and brains were extracted and placed into 10% neutral buffered formalin (NBF). Lungs were inflated with 10% NBF prior to submersion. After at least 48 hours, tissues and organs were processed routinely, embedded in paraffin, and sectioned at 5μm. For each paraffin-embedded block, 10 step sections were taken 100μm apart before routine staining with H&E. Each mouse had 30 slides screened for tumor metastases, 10 slides from each of the three organs. Since the number of organ sections that had metastases varied between mice, the number of slides containing metastases was counted out of the 10 examined (Figure 1B). In order to quantify the size and number of metastases in a single slide, while also considering the variation in the area of tissue present in the section, area of metastases was measured as a percent of the tissue area on the slide. Groups of 8 or more cancer cells were identified as metastases. The slide with the largest metastatic burden was used for enumeration and area analysis (Figure 1B).
Statistics
Number of CTCs/ml, percent of CTCs staining for cytokeratin and/or vimentin expression, number of organ sections with metastases, area of metastases, and number of metastases across all groups were analyzed. The strength of a linear relationship between continuous variables was determined by the Pearson Correlation Coefficient using JMP Pro (JMP®, Version 13. SAS Institute Inc., Cary, NC, 1989–2019). R-values less than 0.3 were considered to have negligible correlation.
Supplementary Material
Acknowledgments
The authors would like to thank the Department of Comparative Medicine, the MLAS Training Program, and the Veterinary Service Center (VSC) at Stanford University for their support of this project. We would especially like to acknowledge and thank Elias Godoy of the VSC for his assistance. This project was funded through the Stanford MLAS Graduate Student Fund (AMR) and the John and Marva Warnock Research Fund (SSJ).
Footnotes
Competing Interests
CAL and ES have financial interests in Vortex Biosciences and intellectual property described herein. SSJ serves as an expert advisor for Ravel Biotechnology, which is developing an analytic platform for early cancer detection using cell-free DNA. All other authors declare no competing interests.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on request.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. [DOI] [PubMed] [Google Scholar]
- 2.Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol. 2011;24(2):157–67. [DOI] [PubMed] [Google Scholar]
- 3.Haffty BG, Yang QF, Reiss M, Kearney T, Higgins SA, Weidhaas J, et al. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006;24(36):5652–7. [DOI] [PubMed] [Google Scholar]
- 4.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15):4429–34. [DOI] [PubMed] [Google Scholar]
- 5.Rakha EA, El-Sayed ME, Green AR, Lee AHS, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109(1):25–32. [DOI] [PubMed] [Google Scholar]
- 6.Sharma P, López-Tarruella S, García-Saenz JA, Khan QJ, Gómez HL, Prat A, et al. Pathological response and survival in triple-negative breast cancer following neoadjuvant carboplatin plus docetaxel. Clin Cancer Res. 2018;24(23):5820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–81. [DOI] [PubMed] [Google Scholar]
- 8.Kennecke H, Yerushalmi R, Woods R, Cheang MCU, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28(20):3271–7. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–U65. [DOI] [PubMed] [Google Scholar]
- 10.Morton CL, Houghton PJ. Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc. 2007;2(2):247–50. [DOI] [PubMed] [Google Scholar]
- 11.Bosma MJ, Carroll AM. The SCID mouse mutant: definition, characterization, and potential uses. Annu Rev of Immunol. 1991;9:323–50. [DOI] [PubMed] [Google Scholar]
- 12.Manzotti C, Audisio RA, Pratesi G. Importance of orthotopic implantation for human tumors as model systems: relevance to metastasis and invasion. Clin Exp Metastasis. 1993;11(1):5–14. [DOI] [PubMed] [Google Scholar]
- 13.Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to “Nude” mice. Apmis. 2007;115(5):604–6. [DOI] [PubMed] [Google Scholar]
- 14.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen XH, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma(null) mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477–89. [DOI] [PubMed] [Google Scholar]
- 15.Simpson-Abelson MR, Sonnenberg GF, Takita H, Yokota SJ, Conway TF, Kelleher RJ, et al. Long-term engraftment and expansion of tumor-derived memory T cells following the implantation of non-disrupted pieces of human lung tumor into NOD-scid IL2R gamma(null) mice. J Immunol. 2008;180(10):7009–18. [DOI] [PubMed] [Google Scholar]
- 16.DeRose YS, Wang GY, Lin YC, Bernard PS, Buys SS, Ebbert MTW, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17(11):1514–U227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iorns E, Drews-Elger K, Ward TM, Dean S, Clarke J, Berry D, et al. A new mouse model for the study of human breast cancer metastasis. Plos One. 2012;7(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin KT, Teng LS, Shen YP, He KF, Xu ZZ, Li GL. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol. 2010;12(7):473–80. [DOI] [PubMed] [Google Scholar]
- 20.Hoffman RM. Patient-derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat Rev Cancer. 2015;15(8):451–2. [DOI] [PubMed] [Google Scholar]
- 21.Hoffman RM. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: A bridge to the clinic. Invest New Drugs. 1999;17(4):343–59. [DOI] [PubMed] [Google Scholar]
- 22.Fu X, Le P, Hoffman RM. A metastatic orthotopic-transplant nude-mouse model of human patient breast cancer. Anticancer Res. 1993;13(4):901–4. [PubMed] [Google Scholar]
- 23.Eccles SA. Models for evaluation of targeted therapies of invasive and metastatic disease. In: Teicher B, editors. Tumor Models in Cancer Research. Cancer Drug Discovery and Development. Humana Press, Totowa, NJ; 2011. pp. 447–95. [Google Scholar]
- 24.Kawaguchi T, Foster BA, Young J, Takabe K. Current update of patient-derived xenograft model for translational breast cancer research. J Mammary Gland Biol Neoplasia. 2017;22(2):131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang HY, Cohen AL, Krishnakumar S, Wapnir IL, Veeriah S, Deng G, et al. Patient-derived xenografts of triple-negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res. 2014;16(2):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang XF, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12(15):4652–61. [DOI] [PubMed] [Google Scholar]
- 27.Kanaya N, Somlo G, Wu J, Frankel P, Kai M, Liu XL, et al. Characterization of patient-derived tumor xenografts (PDXs) as models for estrogen receptor positive (ER+HER2−and ER+HER2+) breast cancers. J Steroid Biochem Mol Biol. 2017;170:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bibby MC. Orthotopic models of cancer for preclinical drug evaluation: advantages and disadvantages. Eur J Cancer. 2004;40(6):852–7. [DOI] [PubMed] [Google Scholar]
- 29.Gast CE, Shaw AK, Wong MH, Coussens LM. Surgical procedures and methodology for a preclinical murine model of de novo mammary cancer metastasis. J. Vis. Exp. 2017;(125). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paez-Ribes M, Man S, Xu P, Kerbel RS. Development of patient derived xenograft models of overt spontaneous breast cancer metastasis: a cautionary note. Plos One. 2016;11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cristofanilli M Circulating tumor cells, disease progression, and survival in metastatic breast cancer. Semin Oncol. 2006;33(3):S9–S14. [DOI] [PubMed] [Google Scholar]
- 32.Sethi N, Kang YB. Unravelling the complexity of metastasis - molecular understanding and targeted therapies. Nat Rev Cancer. 2011;11(10):735–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004;10(20):6897–904. [DOI] [PubMed] [Google Scholar]
- 34.Hayes DF, Smerage J. Is there a role for circulating tumor cells in the management of breast cancer? Clin Cancer Res. 2008;14(12):3646–50. [DOI] [PubMed] [Google Scholar]
- 35.Budd GT, Cristofanilli M, Ellis MJ, Stopeck A, Borden E, Miller MC, et al. Circulating tumor cells versus imaging - predicting overall survival in metastatic breast cancer. Clin Cancer Res. 2006;12(21):6403–9. [DOI] [PubMed] [Google Scholar]
- 36.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 37.Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology. 2007;39(3):305–18. [DOI] [PubMed] [Google Scholar]
- 38.Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, et al. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213(2):374–83. [DOI] [PubMed] [Google Scholar]
- 39.Ramani VC, Lemaire CA, Triboulet M, Casey KM, Heirich K, Renier C, et al. Investigating circulating tumor cells and distant metastases in patient-derived orthotopic xenograft models of triple-negative breast cancer. Breast Cancer Res. 2019;21(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, Agelaki S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011;13(3):R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polioudaki H, Agelaki S, Chiotaki R, Politaki E, Mavroudis D, Matikas A, et al. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer. 2015;15:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowes LE, Allan AL. Circulating tumor cells and implications of the epithelial-to-mesenchymal transition. Adv Clin Chem. 2018;83:121–81. [DOI] [PubMed] [Google Scholar]
- 43.Klein CA. The systemic progression of human cancer: a focus on the individual disseminated cancer cell - the unit of selection. Adv Cancer Res. 2003;89:35–67. [DOI] [PubMed] [Google Scholar]
- 44.Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31(6):539–U143. [DOI] [PubMed] [Google Scholar]
- 45.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giuliano M, Herrera S, Christiny P, Shaw C, Creighton CJ, Mitchell T, et al. Circulating and disseminated tumor cells from breast cancer patient-derived xenograft-bearing mice as a novel model to study metastasis. Breast Cancer Res. 2015;17:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larsson AM, Jansson S, Bendahl PO, Jorgensen CLT, Loman N, Graffman C, et al. Longitudinal enumeration and cluster evaluation of circulating tumor cells improve prognostication for patients with newly diagnosed metastatic breast cancer in a prospective observational trial. Breast Cancer Res. 2018;20(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A. 2016;113(7):E854–E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suo YZ, Xie CY, Zhu X, Fan ZC, Yang ZR, He H, et al. Proportion of circulating tumor cell clusters increases during cancer metastasis. Cytometry A. 2017;91(3):250–253. [DOI] [PubMed] [Google Scholar]
- 50.Yu M, Bardia A, Wittner B, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zajchowski DA, Bartholdi MF, Gong Y, Webster L, Liu HL, Munishkin A, et al. Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res. 2001;61(13):5168–78. [PubMed] [Google Scholar]
- 52.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XHF, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139(7):1315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Demicheli R Tumour dormancy: findings and hypotheses from clinical research on breast cancer. Semin Cancer Biol. 2001;11(4):297–305. [DOI] [PubMed] [Google Scholar]
- 54.Doornebal CW, Klarenbeek S, Braumuller TM, Klijn CN, Ciampricotti M, Hau CS, et al. A preclinical mouse model of invasive lobular breast cancer metastasis. Cancer Res. 2013;73(1):353–63. [DOI] [PubMed] [Google Scholar]
- 55.Keller PJ, Lin AF, Arendt LM, Klebba I, Jones AD, Rudnick JA, et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res. 2010;12(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dominici LS, Mittendorf EA, Wang XM, Liu J, Kuerer HM, Hunt KK, et al. Implications of constructed biologic subtype and its relationship to locoregional recurrence following mastectomy. Breast Cancer Res. 2012;14(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pistelli M, Pagliacci A, Battelli N, Santinelli A, Biscotti T, Ballatore Z, et al. Prognostic factors in early-stage triple-negative breast cancer: lessons and limits from clinical practice. Anticancer Res. 2013;33(6):2737–42. [PubMed] [Google Scholar]
- 58.Mohammed RAA, Ellis IO, Mahmmod AM, Hawkes EC, Green AR, Rakha EA, et al. Lymphatic and blood vessels in basal and triple-negative breast cancers: characteristics and prognostic significance. Mod Pathol. 2011;24(6):774–85. [DOI] [PubMed] [Google Scholar]
- 59.Ugras S, Stempel M, Patil S, Morrow M. Estrogen receptor, progesterone receptor, and HER2 status predict lymphovascular invasion and lymph node involvement. Ann Surg Oncol. 2014;21(12):3780–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coughlan AM, Harmon C, Whelan S, O’Brien EC, O’Reilly VP, Crotty P, et al. Myeloid engraftment in humanized mice: impact of granulocyte-colony stimulating factor treatment and transgenic mouse strain. Stem Cells Dev. 2016;25(7):530–41. [DOI] [PubMed] [Google Scholar]
- 61.Noel A, Depauwgillet MC, Purnell G, Nusgens B, Lapiere CM, Foidart JM. Enhancement of tumorigenicity of human breast adenocarcinoma cells in nude mice by matrigel and fibroblasts. Br J Cancer. 1993;68(5):909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fridman R, Kibbey MC, Royce LS, Zain M, Sweeney TM, Jicha DL, et al. Enhanced tumor-growth of both primary and established human and murine tumor-cells in athymic mice after coinjection with matrigel. J Natl Cancer Inst. 1991;83(11):769–74. [DOI] [PubMed] [Google Scholar]
- 63.Lemaire CA, Liu SZ, Wilkerson CL, Ramani VC, Barzanian NA, Huang KW, et al. Fast and label-free isolation of circulating tumor cells from blood: from a research microfluidic platform to an automated fluidic instrument, VTX-1 liquid biopsy system. Slas Technol. 2018;23(1):16–29. [DOI] [PubMed] [Google Scholar]
- 64.Che J, Yu V, Dhar M, Renier C, Matsumoto M, Heirich K, et al. Classification of large circulating tumor cells isolated with ultra-high throughput microfluidic Vortex technology. Oncotarget. 2016;7(11):12748–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on request.