

Abstract
Metabolic-associated fatty liver disease (MAFLD) is the most common metabolic disease with a global prevalence of 25%. While MAFLD is serious and incurable at the later stage, it can be controlled or reversed at the early stage of hepatosteatosis originating from unhealthy diets. Recent laboratory evidence implicates a critical role of the mammalian target of rapamycin (mTOR)-autophagy signaling pathway in the pathogenesis of MAFLD induced by a high-fructose diet mimicking the overconsumption of sugar in humans. This review discusses the possible molecular mechanisms of mTOR-autophagy-endoplasmic reticulum (ER) stress in MAFLD. Based on careful analysis of recent studies, we suggest possible new therapeutic concepts or targets that can be explored for the discovery of new anti-MAFLD drugs.
Keywords: mTOR, autophagy, ER stress, MAFLD, metabolic syndrome, high-fructose diet
Introduction
Metabolic-associated fatty liver disease (MAFLD) is a new nomenclature recently endorsed by the international consensus panel to replace the previously well-known term nonalcoholic fatty liver disease (NAFLD) [1]. This updated definition encapsulates the central role of metabolic syndrome (MetS) as its main pathogenesis rather than alcohol consumption, as previously recognized [1]. While the subclassification of this broad range of liver metabolic disorders remains to be defined by the international consensus panel [1, 2], this review will consider literature reports on the pathogenesis and progression of NAFLD under the new nomenclature MAFLD.
MAFLD is a pathological manifestation of MetS in the liver [1–4]. MetS is a set of metabolic disorders involving multiple organs and is characterized as central obesity, hypertension, hyperglycemia, and dyslipidemia [5–7]. MAFLD is expected to increase in parallel with the increase in MetS (particularly central obesity and hypertriglyceridemia). Currently, the global prevalence of MAFLD is ~25% [2, 3], which is similar to that of MetS, with 20%–25% in adults worldwide. The population with MAFLD is estimated to increase to 590 million by 2035, including 300 million in China alone [3, 8].
The earliest stage of MAFLD is an excess accumulation of lipids in the liver (simple fatty liver or hepatosteatosis), which by itself is asymptomatic [5, 6]. However, hepatosteatosis predisposes the liver to other etiologies, which can exacerbate liver inflammation, injury and even fibrosis, called nonalcoholic steatohepatitis (NASH). NASH is a critical turning point in the progression of MAFLD from a relatively benign and reversible stage toward liver damage and even cirrhosis and hepatocellular carcinoma [2, 4, 9]. MAFLD has imposed a very large healthcare and economic burden worldwide. Since fully established NASH (particularly with fibrosis) is difficult to treat [2, 9, 10], early intervention to control or reduce lipid accumulation in the liver is critical for the management of MAFLD [11, 12].
It is now well recognized that MAFLD is heterogeneous, involving multiple etiologies and mechanisms in different populations or ethnicities [1–4]. It has been shown from a recent review [13] and experimental studies [14] that MAFLD is closely associated with the overconsumption of fructose. For example, the prevalence of MAFLD (18.0% in 1988–1991 to 31.0% in 2011–2012) as well as that of obesity (from 22.5% to 34.6%) continuously increased along with the excessive consumption of added sugars in the same period (average 200 kCal/day vs 325 kCal/day) [13]. This review will focus on one major type of dietary factor, sugar (namely, sucrose or fructose), either in the diet or drink, which is believed to be an important risk factor for the pathogenesis of MAFLD in the early stage in the general population [3, 7, 13, 14].
Recent laboratory studies have shown that changes in mammalian target of rapamycin (mTOR)-autophagy-endoplasmic reticulum (ER) stress are causally related to the pathological process of MAFLD [12, 15–17]. However, the role of mTOR-autophagy-ER stress in liver metabolic diseases has not been fully explored. In this review, we will evaluate how the mTOR-autophagy-ER stress pathway mediates the pathogenesis of dietary fructose-induced MALFD and reveal possible new therapeutic concepts or targets that can be explored for the discovery of new anti-MAFLD drugs.
mTOR signaling pathway
mTOR is an important eukaryotic signal with multiple functions, including the regulation of protein synthesis, apoptosis, and autophagy [18, 19]. mTOR is a very sensitive sensor for cell energy and nutrition metabolism and plays an important role in the regulation of liver cell metabolism. It has been shown that mTOR hyperactivity is closely associated with fatty acid synthesis, hepatic insulin resistance, and type 2 diabetes [18, 20, 21]. Consistent with this, mTOR inhibition (such as with rapamycin and its analogs) has been found to alleviate MAFLD in animal studies [16, 17]. mTOR consists of two different signaling complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which bind with multiple companion proteins. mTORC1 activity is inhibited by adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and its inhibitors (such as rapamycin) but activated by increased nutrient consumption (amino acids and fructose) or insulin [12, 18, 19].
mTORC1 mainly affects protein synthesis and cell proliferation by regulating the phosphorylation of ribosomal protein S6 kinase 1 (S6K1) and eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4EBP1) [18, 19, 22]. Activation of mTORC1 can promote de novo lipogenesis by stimulating sterol-regulatory element binding protein (SREBP), a master transcription factor regulating the expression of multiple lipogenic enzymes, including acetyl-CoA carboxylase, fatty acid synthase (FAS), stearoyl-CoA desaturase 1, and glycerol-3-phosphate acyltransferase [18, 21]. Interestingly, our recent study on liver ex vivo clearly demonstrated that mTORC1 can be activated by fructose independent of other known stimuli [12], providing direct evidence to indicate a role of mTORC1 in fructose-induced lipid metabolism in the liver (as illustrated in Fig. 1). Apart from promoting hepatic lipogenesis, mTORC1 plays an important role in regulating the activity of autophagy, which can also influence lipogenesis via the ER stress pathway and lipid droplet hydrolysis (as reviewed below).
Fig. 1. Upstream regulation of mTORC1 activity and resultant downstream changes in autophagy and lipid synthesis.
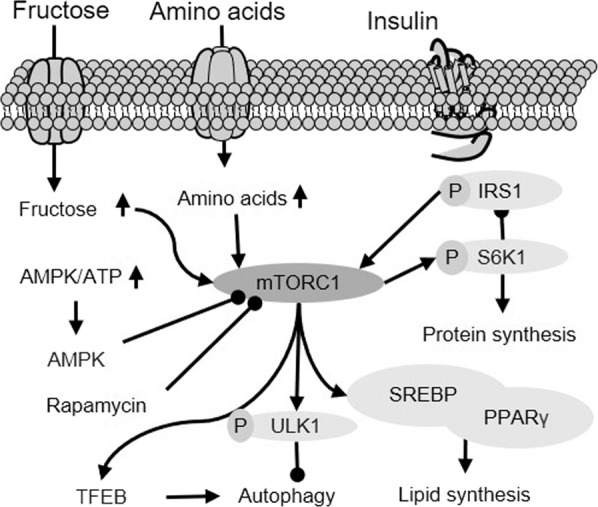
As a nutrient sensor in cells, mTORC1 activity is inhibited by AMPK but activated by increased nutrients (amino acids and fructose) or insulin stimulation. Once activated, mTORC1 mediates lipid synthesis via SREBP1 and inhibits autophagy. Created based on references [12, 18, 21, 23]. IRS1 insulin receptor substrate 1, ATP adenosine triphosphate, TFEB transcription factor EB, ULK1 unc-51 like autophagy activating kinase 1, PPRAγ peroxisome proliferator-activated receptor γ.
Role of autophagy by mTOR in regulating lipid metabolism and ER stress
Autophagy is a hydrolysis process that decomposes and clears intracellular metabolites and molecular debris [23, 24], and can degrade intracellular lipid droplets (lipophagy). Lysosomes are responsible for the catabolism of impaired cytoplasmic proteins and organelles [25–27]. In the process of autophagy, autophagosomes with double membranes wrap and isolate intracellular components and then fuse with lysosomes to form autophagosomes [23, 25]. Autophagosomes degrade their contents to regenerate nutrients.
The activation of mTOR is an important mechanism for inhibiting autophagy (Fig. 2). mTORC1 can phosphorylate Unc-51-like autophagy activating kinase 1 (ULK1) and autophagy-related protein 13 to inhibit autophagy [28]. Recent studies, including studies from our laboratory, showed that during high-fructose feeding, mTOR was activated to inhibit hepatic autophagy, thereby activating ER stress and promoting fatty acid synthesis [12, 27, 29], leading to fatty liver [30, 31]. Enhanced autophagy can alleviate liver metabolic diseases through the following mechanisms: phagocytosis of liposomes, clearance of damaged cell fragments, and elimination of ER stress, thereby inhibiting lipid toxicity caused by fatty acid synthesis [32, 33].
Fig. 2. Regulation of autophagy on ER stress and lipid metabolism.
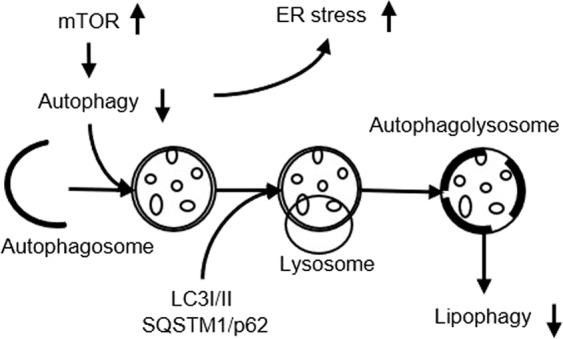
Autophagy and ER stress can interact with each other. The suppressed autophagy can lead to increased ER stress to stimulate lipogenesis and reduce lipid hydrolysis by suppressing lipophagy. Created based on references [12, 23, 30]. LC3 light chain 3.
Studies have found that inhibited autophagy can also activate a series of downstream FASs of SREBP through ER stress inositol-requiring enzyme 1 (IRE1)/X-Box binding protein 1 (XBP1) signal transduction to increase the synthesis of fatty acids, leading to increased triglyceride content in liver cells [34, 35]. At the same time, it can activate the c-Jun N-terminal kinase (JNK)/IκB kinase (IKK) inflammatory pathway [36]. In contrast, autophagy inducers can effectively reverse the liver ER stress induced by high-fructose feeding [12]. Hepatic autophagy can also induce fibrosis [37]. Studies in rat models have shown that inducing the expression of heat shock protein 72 (HSP72) can protect cells from lipopolysaccharide-induced peritonitis injury by increasing autophagy, further confirming the regulatory effect of HSP72 on autophagy activity [38]. It is worth pointing out that the phosphorylation-mediated activation of mTOR is an important mechanism that inhibits autophagy and contributes to the pathogenesis of MAFLD [34].
Role of ER stress in lipid synthesis, inflammation, and apoptosis
The ER is the main organelle for protein synthesis and lipid synthesis in cells. ER stress is usually caused by the accumulation of unfolded or misfolded proteins within the ER, and it triggers an unfolded protein response (UPR), which consists of three major downstream signaling pathways [39–41]: (1) the IRE1/XBP1 signaling pathway, (2) the protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2α (eIF2α) signaling pathway and (3) the activating transcription factor 6 signaling pathway. Activation of the IRE1/XBP1 and PERK/eIF2α signal transduction pathways promotes fatty acid synthesis, inflammatory cytokine production, and apoptosis [36, 42, 43]. Activation of the PERK/eIF2 signaling pathway promotes the expression of the proapoptotic protein C/EBP homologous protein (CHOP) and damages cells. Inhibition of PERK/eIF2 can reduce CHOP expression and liver damage [44].
As illustrated in Fig. 3, prolonged ER stress has been suggested not only to promote hepatosteatosis but also to promote the progression of hepatosteatosis to NASH by IRE1 and PERK downstream signaling pathways. ER stress can activate SREBP1c to upregulate lipogenic genes [36, 45]. It has been shown that reduced eIF2α phosphorylation or ATF4 mutation also reduces hepatosteatosis induced by a high-fat diet [31, 35]. These studies suggest that ER stress may induce hepatosteatosis. Saturated fatty acids can induce ER stress and apoptosis in hepatocytes, and activation of the PERK arm in the UPR pathway can also activate IKK and induce inflammation [35]. In addition, the activation of IRE1 in the UPR pathway may lead to hepatic insulin resistance to accelerate the development of MAFLD [46]. This may be due to phosphorylation of IKK and JNK by activated IRE1, which then impairs insulin signal transduction by interrupting phosphorylation of the serine residue of insulin receptor substrate (IRS) [40]. In summary, these studies suggest that ER stress may play an important role in the development of MAFLD.
Fig. 3. Role of ER stress in MAFLD.
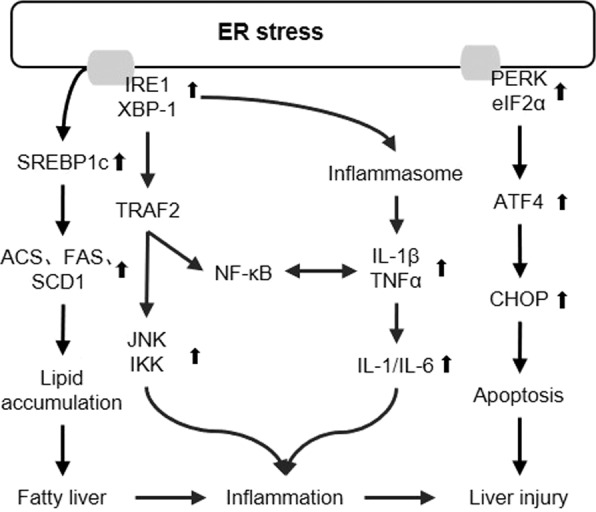
ER stress can induce hepatosteatosis and promote the progression hepatosteatosis to NASH by IRE1 and PERK downstream signaling pathways. Created based on references [12, 33, 43, 44]. TRAF2 TNF receptor-associated factor 2, NF-κB nuclear factor κB, TNFα tumor necrosis factor α, IL-1β interleukin-1β, IL-1 interleukin-1, ATF4 activating transcription factor 4.
Role of mTOR-autophagy-ER stress in the pathogenesis of MAFLD
In recent years, with continuous in-depth research on liver metabolic diseases, the mechanism of the occurrence and development of MAFLD has been gradually clarified. MAFLD may be caused by autophagy/mTOR, ER stress, insulin resistance, inflammation, and mitochondrial dysfunction (as illustrated in Fig. 4). Recent studies have shown that the activation of ER stress and dysfunction of mTOR-autophagy can induce lipid metabolism, inflammation, and fibrosis [24, 27, 41, 47].
Fig. 4. Mechanism of high fructose (HFru)-induced MetS and MAFLD via the mTOR-autophagy-ER stress pathway.
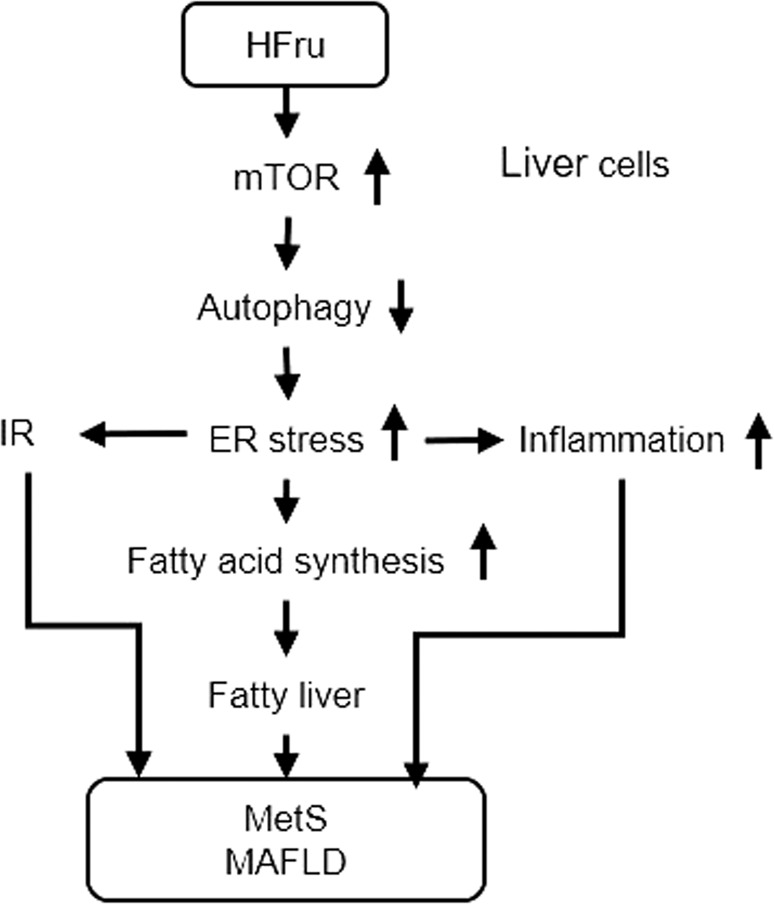
This proposed mechanism is supported by the sequential changes of the events and interrogated with mTOR-dependent and independent inhibitors and rescue of suppressed autophagy. Based on references [12, 29, 31, 48]. IR Insulin resistance.
A large number of studies have shown that autophagy can be activated as a protective mechanism under ER stress, and the decrease in autophagy activity is related to ER stress [12, 16]. First, the inhibition of autophagy can be mediated by the activation of mTOR, which is independent of insulin and amino acids [10]. Second, high fructose-induced autophagy inhibition may be one of the causes of ER stress and may lead to changes in the JNK/IKK and insulin signaling pathways [12, 16]. These changes may occur in the absence of lipid accumulation or during the upregulation of lipid production [12, 48]. Finally, regardless of mTOR activity, restoring autophagy through pharmacological agents can improve ER stress and correct related events [49, 50].
Targeting mTOR-autophagy-ER stress with new anti-MAFLD drugs
Currently, multiple targets have been discovered for the development of anti-MAFLD drugs, including peroxisome proliferator-activated receptor, farnesoid X receptor, C–C chemokine receptor type 2/C–C chemokine receptor type 5, fibroblast growth factor 19, and thyroid hormone receptor β [35, 48], which mainly aim to target a single protein, and drugs for the treatment of NASH. The difficulty is that the development of MAFLD involves several factors, and it is difficult to block them individually to achieve the desired effect, that is, the alleviation of lipid accumulation, liver inflammation, liver injury, and liver fibrosis. Experimental studies have shown that inhibition of mTOR or activation of autophagy can help eliminate the related mechanisms of MAFLD [12, 48]. During the pathogenesis of NASH, mTOR is activated, and autophagy is inhibited [12, 16, 51].
As summarized in Table 1, a number of drugs with anti-MALFD properties have been shown to act on the mTOR-autophagy-ER stress pathway at different sites in various animal models. In the MAFLD model of high-fructose diet-fed mice, rapamycin improves hepatosteatosis by selectively inhibiting mTOR, blocking the activation of p70 S6 kinase, and activating ER stress [12]. Simultaneously, rapamycin also showed a tendency to prevent the development of hepatosteatosis in a fructose-fed zebrafish model [16, 51]. In a high-fat diet-fed mouse model, caffeine reduced liver lipid accumulation and improved liver steatosis induced by high fat by inducing autophagy and downregulating mTOR and lipid uptake in lysosomes [52]. Additionally, caffeine enhances autophagy activity, reduces fatty acid intake and lipid synthesis, improves stress and reduces the inflammatory response to liver injury in rats treated with carbon tetrachloride [53]. Metformin activates AMPK to inhibit mTOR, improve liver steatosis induced by high fat, and reduce inflammation and fibrosis [54]. Metformin reduces hepatic insulin resistance in high fructose-fed mice and can reduce hepatic gluconeogenesis and glucose production in a mouse MAFLD model [29, 55]. Matrine [29], ezetimib and simvastatin [56] can inhibit ER stress and reduce lipid accumulation in the liver. Studies on high fructose-fed mice have shown that the mechanism of resveratrol is relatively complex; it can inhibit IL-6 and reduce lipid accumulation [46, 57].
Table 1.
Anti-MAFLD drugs with effects on the mTOR-autophagy-ER stress pathway.
| Drugs | Effects on mTOR-autophagy-ER stress | Animal models | Effects on MAFLD | References |
|---|---|---|---|---|
| Rapamycin |
Inhibit mTOR Enhance autophagy Inhibit ER stress |
HFru fed mice Fructose fed Zebrafish |
Reduce inflammation and improve insulin signaling transduction Reduce hepatosteatosis |
[12] [16] |
| Caffeine |
Inhibit mTOR Enhance autophagy |
High-fat (HF) fed mice | Ameliorate HF-induced hepatosteatosis |
[52] [53] |
| Metformin |
Enhance autophagy Inhibit mTOR Inhibit ER stress |
HF fed mice HFru fed mice |
Reduce hepatic insulin resistance Ameliorate HF-induced hepatosteatosis Reduce inflammation and fibrosis |
[54] [60] |
|
Ezetimibe Simvastatin |
Inhibit ER stress | High-fat cholesterol (HFC) fed zebrafish | Ameliorate HFC-induced hepatosteatosis | [56] |
| Matrine |
Down-regulate mTOR Inhibit ER stress Up-regulate HSP72 |
HFru fed mice Methionine and choline deficiency (MCD) fed mice |
Abolish HFru-induced hepatosteatosis (reduce TG content) MCD-induced hepatic injury (reduced plasma ALT and AST levels) Suppress MCD-induced hepatic inflammation (reduced TNFα, CD68, MCP-1, and NLRP3 levels) and fibrosis (reduced collagen 1, TGFβ, Smad3, and Sirius red staining) |
[12] [29] [55] |
| Resveratrol |
Enhance autophagy Inhibit ER stress |
Egg yolk powder fed zebrafish HFru fed mice |
Relieve destroyed hepatic structure (fatty infiltration, hepatocyte ballooning, and irregular arrangement and rupture of hepatocyte) in overfed zebrafish Reduce inflammation and improve insulin signaling transduction |
[12] [46] |
TG triglyceride, ALT alanine aminotransferase, AST aspartate aminotransferase, CD68 cluster of differentiation 68, MCP-1 monocyte chemotactic protein-1, NLRP3 NLR family pyrin domain containing 3, TGFβ transforming growth factor-β.
Recent studies from our laboratory suggest that matrine (a hepatoprotective drug identified from the traditional Chinese medicine Kushen) can inhibit SREBP by inhibiting IRE1/XBP1 signal transduction in ER stress to reduce lipid accumulation, inflammation, and fibrosis in the liver [29, 48, 55]. Oral matrine could effectively inhibit the expression of mTOR in the liver as well as restore HSP72 content and its transcription factor heat shock factor 1 [47, 48, 58], which has been shown to eliminate ER stress, promote autophagy clearance [38], and alleviate inflammation [59] and apoptosis. Interestingly, matrine does not seem to act on any other reported targets for the treatment of MAFLD. These findings further suggest that mTOR-autophagy-ER stress can be targeted for the discovery of new anti-MAFLD drugs.
Conclusion
Based on a literature analysis of the pathological effects of mTOR, autophagy, and ER stress on lipid metabolism, inflammatory factors, and cell apoptosis, this review reveals the possible important role of mTOR-autophagy-ER stress in the pathogenesis of MAFLD driven by overconsumption of dietary fructose. Studies have shown that mTOR activation can inhibit autophagy and then activate ER stress. ER stress, on the one hand, can promote the synthesis of fatty acids to aggravate lipid accumulation in the liver; on the other hand, it can increase the expression of inflammatory factors to promote the development of NASH in the long term [31]. This mechanism is particularly obvious in MAFLD caused by high fructose and has been validated by experimental treatment [12]. However, the subtypes of MAFLD have yet to be defined in line with their heterogeneous etiologies [1, 2, 36, 40]. The role of the proposed mTOR-autophagy-ER stress mechanism in MAFLD caused by other etiologies needs further elucidation. It is expected that the study of mTOR-autophagy-ER stress as a guide pathway will provide new insights into the pathological mechanism of fatty liver disease and provide new possibilities for revealing new drug targets for MAFLD driven by overconsumption of dietary fructose (or sugar).
Acknowledgements
The authors would like to acknowledge Prof Li-hong Hu and Prof Hua-liang Jiang (Shanghai Institute of Materia Medica, China) for collaborative research on matrine. We would also like to thank Dr. Hao Wang (RMIT University, Australia), who significantly contributed to our initial study on the relationship among mTOR, autophagy and ER stress in high fructose-induced hepatosteatosis. This review was supported in part by the National Natural Science Foundation of China (81870608) and Jiangmen Innovation Research Team Project Fund (2018630100180019806).
Competing interests
The authors declare no competing interests.
References
- 1.Eslam M, Sanyal AJ, George J, International Consensus P. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158:1999–2014 e1. doi: 10.1053/j.gastro.2019.11.312. [DOI] [PubMed] [Google Scholar]
- 2.Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology. 2020;158:1851–64. doi: 10.1053/j.gastro.2020.01.052. [DOI] [PubMed] [Google Scholar]
- 3.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11–20. doi: 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- 4.Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69:2672–82. doi: 10.1002/hep.30251. [DOI] [PubMed] [Google Scholar]
- 5.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 6.Goedeke L, Perry RJ, Shulman GI. Emerging pharmacological targets for the treatment of nonalcoholic fatty liver disease, insulin resistance, and type 2 diabetes. Annu Rev Pharmacol Toxicol. 2019;59:65–87. doi: 10.1146/annurev-pharmtox-010716-104727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romero-Gomez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67:829–46. doi: 10.1016/j.jhep.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J, Zhou F, Wang W, Zhang XJ, Ji YX, Zhang P, et al. Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology. 2020;71:1851–64. doi: 10.1002/hep.31150. [DOI] [PubMed] [Google Scholar]
- 9.Spengler EK, Loomba R. Recommendations for Diagnosis, Referral for liver biopsy, and treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mayo Clin Proc. 2015;90:1233–46. doi: 10.1016/j.mayocp.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner N, Zeng XY, Osborne B, Rogers S, Ye JM. Repurposing drugs to target the diabetes epidemic. Trends Pharmacol Sci. 2016;37:379–89. doi: 10.1016/j.tips.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the american association for the study of liver diseases. Hepatology. 2018;67:328–57. doi: 10.1002/hep.29367. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Sun RQ, Zeng XY, Zhou X, Li S, Jo E, et al. Restoration of autophagy alleviates hepatic ER stress and impaired insulin signalling transduction in high fructose-fed male mice. Endocrinology. 2015;156:169–81. doi: 10.1210/en.2014-1454. [DOI] [PubMed] [Google Scholar]
- 13.Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;68:1063–75. doi: 10.1016/j.jhep.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. 2017;127:4059–74. doi: 10.1172/JCI94585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki A, Diehl AM. Nonalcoholic steatohepatitis. Annu Rev Med. 2017;68:85–98. doi: 10.1146/annurev-med-051215-031109. [DOI] [PubMed] [Google Scholar]
- 16.Sapp V, Gaffney L, EauClaire SF, Matthews RP. Fructose leads to hepatic steatosis in zebrafish that is reversed by mechanistic target of rapamycin (mTOR) inhibition. Hepatology. 2014;60:1581–92. doi: 10.1002/hep.27284. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Tschumi BO, Lopez-Mejia IC, Oberle SG, Meyer M, Samson G, et al. Mammalian target of rapamycin complex 2 controls CD8 T cell memory differentiation in a Foxo1-dependent manner. Cell Rep. 2016;14:1206–17. doi: 10.1016/j.celrep.2015.12.095. [DOI] [PubMed] [Google Scholar]
- 18.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–71. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 19.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 20.Zhou X, Fouda S, Zeng XY, Li D, Zhang K, Xu J, et al. Characterization of the therapeutic profile of albiflorin for the metabolic syndrome. Front Pharmacol. 2019;10:1151. doi: 10.3389/fphar.2019.01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–20. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–71. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 23.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–6. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–78. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yim WW, Mizushima N. Lysosome biology in autophagy. Cell Discov. 2020;6:6. doi: 10.1038/s41421-020-0141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu L. A special review collection on autophagy. Cell Res. 2020;30:553. doi: 10.1038/s41422-020-0361-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61:585–96. doi: 10.1042/EBC20170021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng XY, Wang H, Bai F, Zhou X, Li SP, Ren LP, et al. Identification of matrine as a promising novel drug for hepatic steatosis and glucose intolerance with HSP72 as an upstream target. Br J Pharmacol. 2015;172:4303–18. doi: 10.1111/bph.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–95. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren LP, Chan SM, Zeng XY, Laybutt DR, Iseli TJ, Sun RQ, et al. Differing endoplasmic reticulum stress response to excess lipogenesis versus lipid oversupply in relation to hepatic steatosis and insulin resistance. PLoS One. 2012;7:e30816. doi: 10.1371/journal.pone.0030816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Musso G, Cassader M, Paschetta E, Gambino R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology. 2018;155:282–302 e8. doi: 10.1053/j.gastro.2018.06.031. [DOI] [PubMed] [Google Scholar]
- 33.Zhou X, Fouda S, Li D, Zhang K, Ye JM. Involvement of the autophagy-ER stress axis in high fat/carbohydrate diet-induced nonalcoholic fatty liver disease. Nutrients. 2020;12:2626. [DOI] [PMC free article] [PubMed]
- 34.Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. 2016;15:249–74. doi: 10.1038/nrd.2015.3. [DOI] [PubMed] [Google Scholar]
- 35.Sun RQ, Wang H, Zeng XY, Chan SM, Li SP, Jo E, et al. IRE1 impairs insulin signaling transduction of fructose-fed mice via JNK independent of excess lipid. Biochim Biophys Acta. 2015;1852:156–65. doi: 10.1016/j.bbadis.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 36.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–46. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S, Zhou Y, Fan J, Cao S, Cao T, Huang F, et al. Heat shock protein 72 enhances autophagy as a protective mechanism in lipopolysaccharide-induced peritonitis in rats. Am J Pathol. 2011;179:2822–34. doi: 10.1016/j.ajpath.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–73. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 40.Arab JP, Arrese M, Trauner M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol. 2018;13:321–50. doi: 10.1146/annurev-pathol-020117-043617. [DOI] [PubMed] [Google Scholar]
- 41.Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol. 2010;8:e1000410. doi: 10.1371/journal.pbio.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang K. Endoplasmic reticulum stress response and transcriptional reprogramming. Front Genet. 2014;5:460. doi: 10.3389/fgene.2014.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, et al. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell. 2018;175:133–45 e15. doi: 10.1016/j.cell.2018.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao Y, Lu YT, Li C, Song QQ, Xu YH, Xu Z, et al. Bouchardatine analogue alleviates non-alcoholic hepatic fatty liver disease/non-alcoholic steatohepatitis in high-fat fed mice by inhibiting ATP synthase activity. Br J Pharmacol. 2019;176:2877–93. doi: 10.1111/bph.14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 46.Ran G, Ying L, Li L, Yan Q, Yi W, Ying C, et al. Resveratrol ameliorates diet-induced dysregulation of lipid metabolism in zebrafish (Danio rerio) PLoS One. 2017;12:e0180865. doi: 10.1371/journal.pone.0180865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 48.Mahzari A, Li S, Zhou X, Li D, Fouda S, Alhomrani M, et al. Matrine protects against MCD-induced development of NASH via upregulating HSP72 and downregulating mTOR in a manner distinctive from metformin. Front Pharmacol. 2019;10:405. doi: 10.3389/fphar.2019.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol. 2010;2010:930509. doi: 10.1155/2010/930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H, Sun RQ, Camera D, Zeng XY, Jo E, Chan SM, et al. Endoplasmic reticulum stress up-regulates Nedd4-2 to induce autophagy. FASEB J. 2016;30:2549–56. doi: 10.1096/fj.201500119. [DOI] [PubMed] [Google Scholar]
- 51.Holtta-Vuori M, Salo VT, Nyberg L, Brackmann C, Enejder A, Panula P, et al. Zebrafish: gaining popularity in lipid research. Biochem J. 2010;429:235–42. doi: 10.1042/BJ20100293. [DOI] [PubMed] [Google Scholar]
- 52.Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59:1366–80. doi: 10.1002/hep.26667. [DOI] [PubMed] [Google Scholar]
- 53.Lee KJ, Terada K, Oyadomari S, Inomata Y, Mori M, Gotoh T. Induction of molecular chaperones in carbon tetrachloride-treated rat liver: implications in protection against liver damage. Cell Stress Chaperones. 2004;9:58–68. doi: 10.1379/459.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rai RC, Bagul PK, Banerjee SK. NLRP3 inflammasome drives inflammation in high fructose fed diabetic rat liver: effect of resveratrol and metformin. Life Sci. 2020;253:117727. doi: 10.1016/j.lfs.2020.117727. [DOI] [PubMed] [Google Scholar]
- 55.Li S, Zeng XY, Zhou X, Wang H, Jo E, Robinson SR, et al. Dietary cholesterol induces hepatic inflammation and blunts mitochondrial function in the liver of high-fat-fed mice. J Nutr Biochem. 2016;27:96–103. doi: 10.1016/j.jnutbio.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 56.Dai W, Wang K, Zheng X, Chen X, Zhang W, Zhang Y, et al. High fat plus high cholesterol diet lead to hepatic steatosis in zebrafish larvae: a novel model for screening anti-hepatic steatosis drugs. Nutr Metab. 2015;12:42. doi: 10.1186/s12986-015-0036-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oka T, Nishimura Y, Zang L, Hirano M, Shimada Y, Wang Z, et al. Diet-induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC Physiol. 2010;10:21. doi: 10.1186/1472-6793-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chou SD, Prince T, Gong J, Calderwood SK. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS One. 2012;7:e39679. doi: 10.1371/journal.pone.0039679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dokladny K, Lobb R, Wharton W, Ma TY, Moseley PL. LPS-induced cytokine levels are repressed by elevated expression of HSP70 in rats: possible role of NF-kappaB. Cell Stress Chaperones. 2010;15:153–63. doi: 10.1007/s12192-009-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ha BG, Park JE, Shin EJ, Shon YH. Modulation of glucose metabolism by balanced deep-sea water ameliorates hyperglycemia and pancreatic function in streptozotocin-induced diabetic mice. PLoS One. 2014;9:e102095. doi: 10.1371/journal.pone.0102095. [DOI] [PMC free article] [PubMed] [Google Scholar]