

Abstract
Metagenome-assembled genomes (MAGs) are microbial genomes reconstructed from metagenome data. In the last few years, many thousands of MAGs have been reported in the literature, for a variety of environments and host-associated microbiota, including humans. MAGs have helped us better understand microbial populations and their interactions with the environment where they live; moreover most MAGs belong to novel species, therefore helping to decrease the so-called microbial dark matter. However, questions about the biological reality of MAGs have not, in general, been properly addressed. In this review, I define the notions of hypothetical MAGs and conserved hypothetical MAGs. These notions should help with the understanding of the biological reality of MAGs, their worldwide occurrence, and the efforts to improve MAG recovery processes.
The first bacterial genome was sequenced and published in 1995 (Fleischmann et al. 1995). This was a landmark achievement, both in terms of the sequence itself and in terms of the computational techniques used to assemble and annotate it. Following the model established in that work, several other prokaryotic genomes were sequenced and published in subsequent years. In 1997, Tatusov, Koonin, and Lipman (Tatusov et al. 1997) foresaw that “the number of sequenced genomes” would “grow exponentially for at least the next few years.” Based on the seven complete genomes available at the time (six prokaryotes and Saccharomyces cerevisiae), they created and made available the COG database, a hugely useful resource. In 2003 the first update of the COG database was published (Tatusov et al. 2003), and it included information from 63 prokaryotic genomes. The jump from six genomes to 10 times that in 6 years confirmed Tatusov, Koonin, and Lipman’s prediction, but I think even they would not have predicted that 24 years later, the number would be more than 350,000 (Sayers et al. 2019). This of course happened because of the astonishing improvements in DNA sequencing technology, starting with the 454 DNA sequencing machine around 2004 (Margulies et al. 2005).
And yet, now in 2021, 350,000 prokaryotic genomes seem puny compared to the numbers we should see in the near future. The reason is that in the past few years, we have been experiencing another step-up of the growth rate of available microbial genomes, but this time the driving factor is not sequencing technology per se, but the advent of the metagenome-assembled genome, or MAG, made possible by metagenomics and associated bioinformatics.
I define metagenomics as the technique of extracting DNA from an environmental (or host-associated) sample and sequencing it. A metagenome is the collection of reads obtained by such sequencing. Metagenomics has allowed the study of microbial populations that were until a few years ago basically unreachable, because of the well-known difficulty in cultivating in the lab the vast majority of prokaryotes, a majority that is estimated to be around 99% (Rinke et al. 2013).
If the read coverage of a metagenome is deep enough, it will contain DNA of all or nearly all microorganisms living in the sampled environment. The microbiome is the collection of genomes and genes that can be derived from metagenome data. “Genome” in this context is the MAG.
How can we obtain genomes from metagenomes? Very briefly, this can be done by assembling reads and binning the results. The assembly phase has as its main aim the generation of contigs, which are contiguous genomic fragments longer than reads. In the binning phase, we try to determine patterns that can tell whether two contigs belong to the same genome or not, primarily based on k-mer profiles. We then use these patterns to separate contigs into bins. Each bin thus obtained corresponds to a MAG. Additional details can be found, for example, in (Sangwan et al. 2016; Perez-Cobas et al. 2020). MAGs are now routinely assembled from metagenome data and reported in the literature, in many cases by the thousands in single papers (Tully et al. 2018; Pasolli et al. 2019; Campanaro et al. 2020).
This deluge of MAGs has created the need for some quality standards. This need was addressed by Bowers et al. (Bowers et al. 2017), who proposed four quality categories; high-quality draft MAGs, for example, should be those that are more than 90% complete and have less than 5% contamination. Completeness and contamination are generally estimated by the program checkM (Parks et al. 2015). The issue of contamination is naturally a concern. When working with MAGs, one should always ask: is a given MAG real? Or is it an amalgam of parts from different genomes? In this context, it is useful to take into account what I call the “genome heterogeneity spectrum” (Fig. 1). My aim in presenting this concept is to state, on the one hand, that a MAG sequence almost always will not be as free of contamination as a genome sequence from an isolate; but on the other hand, depending on its quality, it may still be a valid approximation of the genomes of the microorganisms in the sample. Support for this statement is provided in what follows.
Fig. 1.
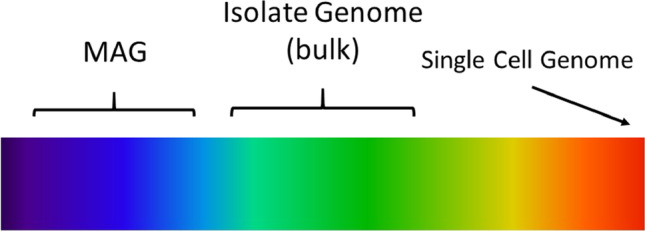
Genome heterogeneity spectrum. On the right are single-cell genomes, those that have no DNA heterogeneity from different cells. In the middle are isolate (bulk) genomes, those that may have some DNA heterogeneity from different cells, assuming all cells from which the genome was sequenced are from the same isolate. On the left are MAGs, which usually have DNA heterogeneity derived from genomes of different strains of the same species, when such strains are present in the sample from which the MAG was reconstructed
The study of prokaryotic MAGs leads us to establish two categories: MAGs for which a species can be assigned (let us call them SMAGs) and MAGs for which this is not possible, because they supposedly are genomes of novel species (let us call them HMAGs, for reasons that will become clear in a moment). How can we determine that a MAG is an SMAG? Establishing that a MAG belongs to species s will in general require the alignment of the MAG with the genome sequence of an isolate of s. And this is the evidence for the reality of an SMAG: if a bin of sequences obtained from a metagenome M, by applying, for example, the MetaWRAP pipeline (Uritskiy et al. 2018), without any reference whatsoever to any database sequence or genome, neatly aligns with the genome sequence of an isolate obtained independently, perhaps from a source on the other side of the planet, then we can be reasonably sure that the MAG in question is real. By “neatly align” I mean that the average nucleotide identity between the two sequences should be at least 97% and should have at least 90% coverage in both query and subject sequences. As an example, Braga et al. (Braga et al. 2021) described several SMAGs obtained from composting samples that “neatly aligned” with isolate genomes obtained by other research groups from a wide variety of sources but all of them in some way related to composting.
Currently there is no consensus on the threshold of similarity to be used in determining whether a MAG is an SMAG; some authors use 95%, and others use larger values. Even more rigorous than simply using alignments and similarity thresholds is the use of statistical techniques, as in GGDC (Meier-Kolthoff et al. 2013) or phylogenetic placement, as in GTDB-tk (Parks et al. 2018). By saying that an SMAG that satisfies these requirements is real, we do not claim that it is not a composite of different strains from the same species; it may well be and that is another way of explaining why I have placed MAGs on the left end of the spectrum presented in Fig. 1.
The question of whether an HMAG is real is more delicate. Here we do not have a reference genome to compare to; all we have is the set of contigs. A radical critique published by Garg et al. (Garg et al. 2021) does not see any biological reality in certain groups of HMAGs presented in the literature (“Asgard and CPR MAGs are unnatural constructs, genome-like patchworks of genes that have been stitched together into computer files by binning.”). Meziti et al. (Meziti et al. 2021) have also shown problems in MAG reconstruction by careful comparison between MAGs and their corresponding isolate genomes. The biological reality of HMAGs is particularly pressing, because most MAGs are HMAGs (Lloyd et al. 2018).
I believe there is one argument that can be put forward in favor of the reality of high-quality HMAGs. The argument is an extrapolation of the validity of SMAGs. If the same methodology applied to a given dataset yields both real high-quality SMAGs and high-quality HMAGs, then this seems to support the reality of these HMAGs. In addition, in some cases additional evidence can be obtained. To explain this I offer an analogy between MAGs and the annotation of protein-coding genes.
The issue of determining whether a given protein-coding gene in a newly sequenced genome has homologs in sequence databases is also done with alignments, similarity and coverage thresholds, and phylogenetic placements (Setubal and Stadler 2018). This is the reason why I chose HMAGs as the acronym for those MAGs for which we cannot assign a species: these would be the hypothetical MAGs, analogously to the practice of saying that a protein-coding gene codes for a hypothetical protein when we cannot assign a function to it (it does not have an ortholog with a functional assignment). Additional evidence for the reality of an HMAG may come from searches against MAG catalogs, as I now explain.
Suppose we have obtained a high-quality HMAG from a given sample, and we find a significant hit for it in a MAG catalog (using the same criteria as that used for establishing that a MAG is an SMAG, or using a tool such as GTDB-tk). This means that our query has also shown up in another independent sample (and possibly another environment). This is additional confirmation for the reality of the HMAG, and hence we could now say that our hypothetical MAG is also a Conserved hypothetical MAG, or CHMAG, in analogy to the practice of differentiating between hypothetical proteins (no hits) and conserved hypothetical proteins (those that have a significant hit in a BLAST search, although the hit itself is annotated as a hypothetical protein).
For environmental MAGs, the best catalog at this point is GEM (Nayfach et al. 2021), with more than 50,000 entries. GTDB-tk may also point to “ortholog MAGs” for a given MAG query, but it is not clear how extensive the MAG catalog of GTDB is (Parks et al. 2018). For MAGs retrieved from human metagenomes, there is another, specific catalog (Almeida et al. 2021). One wishes, however, that there was a unified catalog of all MAGs, regardless of source, to facilitate these comparisons. This is another analogy to protein-coding genes. Until a few years ago, one could search NCBI’s nr database using BLAST and be fairly confident that one would find out all significant similarities to known proteins for a given query sequence. However, we no longer can have such confidence, in part because of the surge in genome sequences caused by the “MAG revolution” that I am discussing here.
I should also like to add that a desirable feature of MAG catalogs would be their classification of MAGs using the three categories here proposed. This in turn might allow the monitoring of HMAG status over time: my expectation is that, as more and more MAGs are made available, many HMAGs will be found to have become CHMAGs.
Assuming the biological reality of SMAGs and CHMAGs opens up interesting investigation avenues. One of them is what I call the cosmopolitism of bacterial and archaeal species: Given a MAG, we know the environment where it came from (its sample). Where else in the world has this species also been found? To answer this question, we need to have access to the metadata associated with isolate genomes of the same species, for the case of SMAGs. Here, the GOLD database (Mukherjee et al. 2021) and the NCBI genome records (Sayers et al. 2019) are valuable resources, although in many instances the desired metadata (isolation source and location) is lacking. For the case of CHMAGs, we need to rely on metadata provided by MAG catalogs.
I believe research on MAGs has two main challenges. The first is in the improvement of reconstruction methods. Efforts are underway to address this (Chen et al. 2020; Lui et al. 2021), and surely more will follow. Methods that can help identify strains in metagenomes (Segata 2018; Quince et al. 2021) are also a step in the direction of bringing MAGs towards the right-hand side of the genome heterogeneity spectrum. The second challenge is experimentally verifying the biological reality of hypothetical MAGs. This will probably remain a problem for many years to come, since it is unlikely that current and new cultivation techniques (Lagier et al. 2018) will be able to keep pace with the exponential rate of MAG discovery.
In sum, MAGs have become a powerful tool to explore all kinds of microbiota. MAGs have helped us better understand microbial populations and their interactions with the environment where they live. Moreover, as most MAGs belong to novel species, their discovery helps decrease the so-called microbial dark matter. We are still in the early stages of tool and resource development to support MAG reconstruction and analysis. One can expect that many new MAG-related tools and resources will become available over the next several years, thus helping turn MAGs into first-class citizens in microbiological research.
Glossary
- Genome completeness
The completeness of MAGs and draft isolate genomes can be estimated by determining the fraction of certain marker genes present in the genome for the particular prokaryotic clade to which the MAG or the isolate belongs. These marker genes are assumed to be required in all members of the clade.
- Genome contamination
For a given isolate genome or MAG sequence, the percentage of the sequence that is estimated to belong to a different species.
- Genome
The set of all DNA molecules in a cell.
- Genome alignment
This is a particular case of DNA sequence alignment. A pairwise alignment algorithm seeks to establish a correspondence between positions in one sequence with positions in the other sequence, in order to maximize the matches between positions. When two sequences have 95% identity, this means that matches were found between 95% of the positions participating in the alignment. Because prokaryotic genomes have usually more than a million base pairs, and in some cases surpass ten million base pairs, their alignments require special programs, different from those employed to align shorter sequences. One popular program to align genomes is MUMmer (Kurtz et al. 2004).
- Homology and orthology
Two DNA sequences (in particular, two gene sequences) are homologous if they share a common ancestor. Homology is therefore a biological concept. In practice, one has to resort to sequence similarity in order to infer homology. This has led to widespread misleading statements in the literature, where it is easy to find expressions such as “sequence X and Y have 55% homology”; what the authors of such statements mean is that sequence X and Y, when aligned, display 55% of sequence identity. When a homology relationship can be inferred between two DNA sequences in the absence of the complicating factor of duplications, the term orthology can be used. The expression “ortholog MAGs” is not standard and has been used in the spirit of the analogy between annotation of protein-coding genes and MAG similarity relationships proposed in the text.
- Reads
The output of a DNA sequencing machine. The length of a read can vary from 50 bp to thousands of kbp, depending on the sequencing technology.
Funding
The author was funded in part by a CNPq Senior Researcher Fellowship.
Declarations
Conflict of interest
The author declares no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, Segata N, Kyrpides NC, Finn RD. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol. 2021;39(1):105–114. doi: 10.1038/s41587-020-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glockner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu WT, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, Genome Standards C, Lapidus A, Meyer F, Yilmaz P, Parks DH, Eren AM, Schriml L, Banfield JF, Hugenholtz P, Woyke T. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35(8):725–731. doi: 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga LPP, Pereira RV, Martins LF, Moura LMS, Sanchez FB, Patane JSL, da Silva AM, Setubal JC. Genome-resolved metagenome and metatranscriptome analyses of thermophilic composting reveal key bacterial players and their metabolic interactions. BMC Genomics. 2021;22(1):652. doi: 10.1186/s12864-021-07957-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanaro S, Treu L, Rodriguez RL, Kovalovszki A, Ziels RM, Maus I, Zhu X, Kougias PG, Basile A, Luo G, Schluter A, Konstantinidis KT, Angelidaki I. New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol Biofuels. 2020;13:25. doi: 10.1186/s13068-020-01679-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LX, Anantharaman K, Shaiber A, Eren AM, Banfield JF. Accurate and complete genomes from metagenomes. Genome Res. 2020;30(3):315–333. doi: 10.1101/gr.258640.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269(5223):496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Garg SG, Kapust N, Lin W, Knopp M, Tria FDK, Nelson-Sathi S, Gould SB, Fan L, Zhu R, Zhang C, Martin WF (2021) Anomalous phylogenetic behavior of ribosomal proteins in metagenome-assembled Asgard Archaea. Genome Biol Evol 13(1). 10.1093/gbe/evaa238 [DOI] [PMC free article] [PubMed]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier JC, Dubourg G, Million M, Cadoret F, Bilen M, Fenollar F, Levasseur A, Rolain JM, Fournier PE, Raoult D. Culturing the human microbiota and culturomics. Nat Rev Microbiol. 2018;16:540–550. doi: 10.1038/s41579-018-0041-0. [DOI] [PubMed] [Google Scholar]
- Lloyd KG, Steen AD, Ladau J, Yin J, Crosby L (2018) Phylogenetically novel uncultured microbial cells dominate earth microbiomes. mSystems 3(5). 10.1128/mSystems.00055-18 [DOI] [PMC free article] [PubMed]
- Lui LM, Nielsen TN, Arkin AP. A method for achieving complete microbial genomes and improving bins from metagenomics data. PLoS Comput Biol. 2021;17(5):e1008972. doi: 10.1371/journal.pcbi.1008972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Kolthoff JP, Auch AF, Klenk HP, Goker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meziti A, Rodriguez RL, Hatt JK, Pena-Gonzalez A, Levy K, Konstantinidis KT (2021) The reliability of metagenome-assembled genomes (MAGs) in representing natural populations: insights from comparing MAGs against isolate genomes derived from the same fecal sample. Appl Environ Microbiol 87(6). 10.1128/AEM.02593-20 [DOI] [PMC free article] [PubMed]
- Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Sundaramurthi JC, Lee J, Kandimalla M, Chen IA, Kyrpides NC, Reddy TBK. Genomes OnLine Database (GOLD) vol 8: overview and updates. Nucleic Acids Res. 2021;49(D1):D723–D733. doi: 10.1093/nar/gkaa983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Roux S, Seshadri R, Udwary D, Varghese N, Schulz F, Wu D, Paez-Espino D, Chen IM, Huntemann M, Palaniappan K, Ladau J, Mukherjee S, Reddy TBK, Nielsen T, Kirton E, Faria JP, Edirisinghe JN, Henry CS, Jungbluth SP, Chivian D, Dehal P, Wood-Charlson EM, Arkin AP, Tringe SG, Visel A, Consortium IMD. Woyke T, Mouncey NJ, Ivanova NN, Kyrpides NC, Eloe-Fadrosh EA. A genomic catalog of Earth's microbiomes. Nat Biotechnol. 2021;39(4):499–509. doi: 10.1038/s41587-020-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36(10):996–1004. doi: 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, Collado MC, Rice BL, DuLong C, Morgan XC, Golden CD, Quince C, Huttenhower C, Segata N. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell. 2019;176(3):649–662. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cobas AE, Gomez-Valero L, Buchrieser C (2020) Metagenomic approaches in microbial ecology: an update on whole-genome and marker gene sequencing analyses. Microb Genom 6(8). 10.1099/mgen.0.000409 [DOI] [PMC free article] [PubMed]
- Quince C, Nurk S, Raguideau S, James R, Soyer OS, Summers JK, Limasset A, Eren AM, Chikhi R, Darling AE. STRONG: metagenomics strain resolution on assembly graphs. Genome Biol. 2021;22(1):214. doi: 10.1186/s13059-021-02419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu WT, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499(7459):431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- Sangwan N, Xia F, Gilbert JA. Recovering complete and draft population genomes from metagenome datasets. Microbiome. 2016;4:8. doi: 10.1186/s40168-016-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I. GenBank. Nucleic Acids Res. 2019;47(D1):D94–D99. doi: 10.1093/nar/gky989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N (2018) On the road to strain-resolved comparative metagenomics. mSystems 3(2). 10.1128/mSystems.00190-17 [DOI] [PMC free article] [PubMed]
- Setubal JC, Stadler PF. Gene phylogenies and orthologous groups. Methods Mol Biol. 2018;1704:1–28. doi: 10.1007/978-1-4939-7463-4_1. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278(5338):631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tully BJ, Graham ED, Heidelberg JF. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci Data. 2018;5:170203. doi: 10.1038/sdata.2017.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6(1):158. doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]