

Abstract
RNA-based gene therapy requires therapeutic RNA to function inside target cells without eliciting unwanted immune responses. RNA can be ferried into cells using non-viral drug delivery systems, which circumvent the limitations of viral delivery vectors. Here, we review the growing number of RNA therapeutic classes, their molecular mechanisms of action, and the design considerations for their respective delivery platforms. We describe polymer-based, lipid-based, and conjugate-based drug delivery systems, differentiating between those that passively and those that actively target specific cell types. Finally, we describe the path from preclinical drug delivery research to clinical approval, highlighting opportunities to improve the efficiency with which new drug delivery systems are discovered.
Subject terms: Nucleic-acid therapeutics, Nanobiotechnology
RNA therapies can be used to manipulate gene expression or produce therapeutic proteins. Here, the authors describe the growing number of RNA therapies and their molecular mechanisms of action. They also discuss the path from preclinical drug delivery research to clinical approval of these drugs.
Introduction
RNA therapies can manipulate gene expression or produce therapeutic proteins, making these drugs suitable for pathologies with established genetic targets, including infectious diseases, cancers, immune diseases and Mendelian disorders (including neurological disorders). Moreover, the ability to sequence hundreds of thousands of genomes, analyse gene expression at the single-cell level, and manipulate genes with programmable nucleases is driving the discovery of new targets for gene therapies. Yet the ability to manipulate these targets, especially non-coding DNA and the 85% of the genome that might be undruggable using small molecules1, is lessened without the capacity to deliver therapeutic RNA to diseased cells. In this Review, therapeutic RNA refers to antisense oligonucleotides (ASOs), such as gapmers, which contain DNA nucleotides flanked by RNA2, small interfering RNAs (siRNAs), or large RNAs, such as messenger RNA (mRNA) (Fig. 1). These RNA therapies act by targeting RNA or proteins, by encoding missing or defective proteins, or by mediating DNA or RNA editing. Irrespective of their therapeutic mechanism of action, the large size of some therapeutic RNAs, such as mRNAs, their anionic charge, and their susceptibility to RNases present in both the bloodstream and tissues make it difficult for therapeutic RNA to enter cells efficiently and function on its own.
Fig. 1. The expanding universe of therapeutic RNA payloads.

a | One class of RNA therapeutics requires delivery of small RNA molecules. Small interfering RNAs (siRNAs) can reduce gene expression via RNA-induced silencing complex (RISC)-mediated mRNA degradation, antisense oligonucleotides (ASOs) can alter isoforms by binding to splice sites, and adenosine deaminase acting on RNA ASOs (ADAR-oligonucleotides) can edit RNA. In all three cases, these small RNAs can be designed with site-specific chemical modifications using solid-phase synthesis and can be delivered using nanoparticles or conjugate delivery systems. In this figure, the blue molecule represents the small therapeutic RNA being ferried into the cell. b | A second class of RNA therapeutics requires delivery of large RNA molecules. In vitro transcribed mRNA consists of a 5′ cap, 5′ and 3′ untranslated regions (UTRs), an open reading frame encoding antigen(s), and a 3′ poly(A) tail. c | mRNA payloads can encode nucleases that mediate DNA or RNA editing. mRNA can also be used to replace dysfunctional protein or encode antigens that confer longer-term immunity to a pathogen, such as SARS-CoV-2. mRNAs are transcribed in vitro and thus cannot currently be made with site-specific chemical modifications. In this figure, the blue molecule represents the protein encoded by the mRNA. Cas, CRISPR-associated protein; crRNA, CRISPR RNA; dCas9, dead Cas9; GFP, green fluorescent protein; pegRNA, prime editing guide RNA; sgRNA; single-guide RNA.
To overcome the barriers to safe and effective RNA delivery, scientists have developed both viral-vector-based and non-viral delivery systems that protect the RNA from degradation, maximize delivery to on-target cells and minimize exposure to off-target cells. Viral gene therapies3 have generated successful clinical readouts4–9, but the effectiveness of these approaches can be limited by pre-existing immunity10, viral-induced immunogenicity11, unwanted genomic integration12, payload size constraints13, the inability to re-dose, complications involved in upscaling14, and expensive vector production. Although scientists are overcoming some of these limitations15, they have fuelled the search for alternative drug delivery vehicles. Concurrent advances in the development of synthetic materials that encapsulate RNA, such as polymers, lipids and lipid nanoparticles (LNPs), have invigorated research into non-viral-based delivery systems, leading to US Food and Drug Administration (FDA) approval of subcutaneously administered N-acetylgalactosamine (GalNAc)–siRNA conjugates that target hepatocytes16–18, intravenously administered LNP-based siRNA drugs that target hepatocytes19, and emergency use authorization (EUA) and FDA approval20 for intramuscularly administered LNP-based mRNA COVID vaccines21,22. These approvals suggest that improved delivery to non-liver tissues (also known as extrahepatic tissues) as well as local delivery to the central nervous system, eye and ear could result in new drugs. Nanoparticle-based drug delivery systems may also be useful for non-viral DNA delivery, which has been reviewed elsewhere23.
Here, we detail the expanding number of therapeutic RNA payloads and highlight how the downstream biochemical mechanism of action influences delivery. After reviewing the chemistries commonly used in drug delivery systems as well as the approaches to targeting specific cells, we describe the series of experiments used to characterize nanoparticles in preclinical studies (hereafter referred to as the nanoparticle discovery pipeline) and identify opportunities to improve the efficiency of this pipeline. Finally, we describe the advantages, disadvantages and hallmarks of existing FDA-approved and EUA-approved RNA delivery systems.
Therapeutic RNA payloads
RNA drugs are often classified by the biochemical mechanism of action used to manipulate genes or gene expression (Fig. 1). When designing drug delivery vehicles, it is important to consider how these mechanisms of action influence the requirements for clinically relevant drug delivery. Oligonucleotide drugs, such as ASOs and siRNAs, that utilize enzymes endogenous to eukaryotic cells, such as RNAse H1 or the RNA-induced silencing complex (RISC), respectively, facilitate delivery by not requiring the delivery of large enzymes. Important improvements have been made in recent years in terms of the delivery of small molecules and macromolecules24, but most therapeutic oligonucleotides must still be maintained at high concentrations over time in order to manipulate the target gene25. For example, givosiran16 is administered subcutaneously at 2.5 mg/kg monthly, and lumasiran18 is administered subcutaneously at 3.0 mg/kg monthly for 3 months, then 3 mg/kg once every 3 months. More promising, however, is inclisiran26, which is administered as a 284 mg dose given subcutaneously as a single injection on day 1, day 90 and every 6 months thereafter. With additional improvements to delivery or siRNA design, it may become feasible to inject patients annually, which could coincide with a yearly checkup. By contrast, DNA nucleases, including many clustered regularly interspaced short palindromic repeat (CRISPR)-based systems, can cause long-term effects in cells even if the construct is only expressed transiently27. MicroRNAs (miRNAs) recruit RISC to complementary mRNA sequences, thereby facilitating targeted RNA interference. As a result, miRNA mimics, which are designed to increase native miRNA activity, and anti-miRNAs or antago-miRNAs, which inhibit miRNA activity, have been studied in animal models and used in clinical trials28. One phase I clinical trial investigated the use of MRX34, which uses liposomes to deliver a double-stranded miRNA-34a mimic, for the treatment of advanced solid tumours29. In a phase II clinical trial, the anti-miRNA-122 miravirsen, which binds miRNA-122 and leads to its subsequent inactivity, was tested for the treatment of hepatitis C30. Using a similar approach, RG-101, which antagonizes miRNA-122, was reported to reduce viral load by several logs in patients with chronic hepatitis C in a phase Ib clinical trial31. However, RG-101 was discontinued after the drug was found to cause hyperbilirubinaemia32. Finally, mRNA drugs promise a simple and flexible way to treat a litany of diseases; these therapeutics encompass antigen production, including the COVID-19 vaccine, protein replacement therapies and genome engineering.
siRNA
One biochemical mechanism of action that has been safely used in humans is siRNA-mediated gene silencing. These double-stranded RNAs with a molecular weight of approximately 13 kDa suppress protein translation by recruiting RISC to mRNA via Watson–Crick base pairing (Fig. 1a). Through the action of the catalytic RISC protein Ago2, a member of the Argonaute family, the target mRNA is cleaved. Alternatively, other Ago proteins (Ago1, Ago3 and Ago4) catalyse endonuclease-mediated nonspecific mRNA degradation by localizing the bound mRNA in processing (P)-bodies33,34. siRNA can reduce the expression of any protein-coding gene and has been approved by the FDA or the European Medicines Agency (EMA) in the form of the following drugs: patisiran, which is used to treat hereditary transthyretin-mediated amyloidosis (hATTR)19; givosiran, which is used to treat acute hepatic porphyria16; lumasiran, which is used to treat primary hyperoxaluria type 118; and inclisiran, which is used to treat hypercholesterolaemia17. In addition, the FDA has accepted a new drug application for vutrisiran35, an investigational RNA interference (RNAi) therapeutic for the treatment of hATTR amyloidosis with polyneuropathy in adults, following a successful phase III clinical trial36. The FDA approved the first siRNA drug 20 years19 after the first report of RNAi in eukaryotic cells37. This speedy clinical implementation of siRNA has been made easier by three traits. First, its small number of nucleotides enables scientists to use solid-phase synthesis to manufacture siRNA with site-specific chemical modifications, usually in the phosphate backbone and sugar rings. A whole suite of chemistries has been developed for a variety of ribose modifications, such as 2′-O-methyl (2′-OMe), 2′-methoxyethyl (2′-MOE), 2′-fluor (2′-F), locked nucleic acid oligonucleotides, constrained ethyl oligonucleotides (cEt), and tricyclo-DNA oligonucleotides (tc-DNA)38. Complementing these modifications are phosphate backbone modifications including phosphorothioates, phosphorodiamidate morpholino oligonucleotides (PMO), peptide nucleic acid oligonucleotides (PNA), and nucleobase modifications, such as 5-methylcytosine (m5C). Changing the combination of different chemical modifications of the siRNA has enabled scientists to improve the pharmacokinetic properties, innate immune response and stability39. Second, siRNA utilizes RISC, which is endogenous to eukaryotic cells and thus does not require the delivery of large enzymes with nuclease domains. Finally, given that siRNA interferes with mature mRNA, it requires only cytoplasmic delivery, which is easier to achieve than nuclear delivery.
Antisense oligonucleotides
ASOs are a second class of RNA therapeutics, and are oligonucleotides with a molecular weight of 6–9 kDa (ref.40). ASOs have the same manufacturing advantages as siRNA and have been approved by the FDA to treat familial hypercholesterolaemia41, hATTR amyloidosis with polyneuropathy42, specific subtypes of Duchenne muscular dystrophy43,44, and infantile-onset spinal muscular atrophy45. ASOs can act through three mechanisms of action (Fig. 1a). First, similar to siRNAs, ASOs bind mRNA via Watson–Crick base pairing, but unlike siRNAs, the ASO DNA–RNA heteroduplex recruits RNase H1 rather than RISC. RNase H1-dependent ASOs are also known as gapmers and lead to cleavage of the target RNA46. Second, ASOs can also interfere with splicing machinery by interacting with pre-mRNA, thereby promoting alternative splicing46 and increasing target protein expression47. Thus, unlike siRNA, which silences target genes, ASOs can be used to increase protein activity in diseases including Duchenne muscular dystrophy and spinal muscular atrophy. However, these two mechanisms require nuclear delivery so that the ASO–RNase H1 complex can interact with pre-mRNA. The third mode, which causes downregulation of mRNA expression, is the translational arrest of the targeted protein through binding with the translation initiation codon of the target mRNA48.
ASO chemical modification can influence the mechanism of action, as well as the target sequence binding affinity. Gapmers have a general chimeric structure of regions of five nucleotides of RNA-like residues flanking a central 10-nucleotide DNA region49. The gapmer can improve hybridization and nuclease resistance, while still retaining RNase H1 activation. As described above, other modifications, such as locked nucleic acid oligonucleotides, can be used instead of the 2′-modified designs, but although these modifications can improve potency, they can also increase toxicity50,51. The ASOs that involve steric blocking are RNAse H1-independent and are entirely made of RNA bases, and not DNA. Notably, ASOs often have a full phosphorothioate backbone, which can facilitate their transport into the nucleus52,53.
Chemically modified oligonucleotides with an antisense target binding motif have been designed with an engineered hairpin domain that recruits the endogenous RNA-editing enzyme adenosine deaminase acting on RNA (ADAR)54,55 (Fig. 1a). These ADAR-oligonucleotides, which have a molecular weight of approximately 10–35 kDa and can be manufactured with site-specific chemical modifications, use their single-stranded RNA domain to bind a target mRNA via Watson–Crick base pairing. The other domain recruits ADAR to the RNA, where ADAR converts adenosines to inosines; the inosines are subsequently read as guanosine by the endogenous translational machinery, leading to A to I to G RNA editing. A to I to G editing can occur on mature mRNA, suggesting that cytoplasmic delivery of ADAR-oligonucleotides can be sufficient to achieve an effect. Thus, ADAR-oligonucleotides represent an emerging class of oligonucleotides for treating genetic disease56.
mRNA
Another type of RNA therapeutic is mRNA, which can encode proteins that have therapeutic activity. Because of their size, mRNAs are in vitro transcribed and cannot currently be made with site-specific chemical modifications using solid-state synthesis (Fig. 1b). mRNA can be used to replace protein, using replacement therapies57; to reduce protein levels, using Cas9 cutting approaches58; or to repair protein mutations at the DNA level, using base editing59,60. In 2021, researchers reported the successful use of LNPs encapsulating Streptococcus pyogenes Cas9 (CRISPR-associated endonuclease Cas9) mRNA and a CRISPR guide RNA in six patients with hATTR amyloidosis with polyneuropathy; a single 0.3 mg/kg dose resulted in a mean reduction from baseline of blood transthyretin (TTR) levels of 87% at 28 days post-dose58. The gene product TTR is responsible for transporting vitamin A and thyroxine throughout the body, and mutations in this gene cause hATTR58. This milestone for a new therapeutic modality comes alongside the remarkable success of the now FDA-approved20, mRNA-based vaccine against SARS-CoV2 (Box 1)61,62. Other clinical examples of mRNA-mediated protein replacement include efforts to treat cystic fibrosis as well as ornithine transcarbamylase deficiency. Translate Bio is continuing its trial of mRNA-mediated protein replacement for cystic fibrosis, despite not showing much improved lung function in patients with cystic fibrosis63, and discontinuing its trial for ornithine transcarbamylase deficiency, owing to an undesirable pharmacokinetic and safety profile64. Similarly, Arcturus Therapeutics have received regulatory approval to initiate a phase II clinical trial of an mRNA therapeutic to treat ornithine transcarbamylase deficiency65. In addition to gene replacement, transient expression of myelin oligodendrocyte glycoprotein has led to immunological tolerance and subsequent treatment of experimental autoimmune encephalomyelitis in mice66. The antithesis of this tolerance mechanism is mRNA-mediated expression of an antigen to generate long-lasting immunity against the antigen, the so-called mRNA vaccines. Fundamental mRNA vaccine research67 focused on viruses such as Zika68, HIV69 and influenza70, or diseases such as melanoma71.
Intramuscular injections of human carcinoembryonic antigen mRNA have been used to induce antigen-specific immune responses in mice as far back as 1995 (ref.72). Since then, strides have been made and multiple therapeutic cancer vaccines are currently undergoing clinical trials. BNT111 from BioNTech’s FixVac platform targets a fixed combination of four mRNA-encoded melanoma-associated antigens (NY-ESO-1, MAGEA3, tyrosinase and TPTE). Phase I trials of BNT111 in patients with advanced melanoma showed partial responses and shrinkage of metastases after eight vaccinations given via intravenous injections73. Aside from vaccination, mRNA can be used to deliver co-stimulatory immune checkpoint molecules, such as OX40L for the treatment of solid tumours74. mRNA-2416 from Moderna was injected intratumorally every 2 weeks for up to 12 doses and was shown to be tolerable with increased OX40L protein expression and elevated pro-inflammatory activity and PD-L1 levels in patients with locally advanced, recurrent or metastatic solid malignancy or lymphoma74.
mRNA can also transiently express zinc finger nucleases, transcription activator-like nucleases, or nucleases derived from CRISPR–Cas systems75. Nucleases that are designed to manipulate DNA are particularly well suited for mRNA-based therapeutics, which produce protein for hours to days76 instead of weeks, as would be the case with DNA-based therapeutics; a short-lived DNA nuclease can create a long-lived gene editing event77. A clinical trial of an adeno-associated viral vector-based SaCas9 has been initiated78, but two arguments suggest that mRNA-based or protein-based transient Cas expression might be preferable in cases when the enzyme is an active DNA nuclease. First, long-term expression of an active DNA nuclease might lead to increased editing events in off-target loci27. Second, adeno-associated viral vector-based Cas9 delivery has been reported to lead to vector integration following DNA double-stranded breaks79.
Cas enzymes are also amenable to three types of biochemistry-driven improvements that expand their possible therapeutic potential80. In the first type of improvement, Cas enzymes can be rationally designed81 or evolved82 to target DNA next to different protospacer-adjacent motif sites. A second type of improvement is that Cas enzymes evolved to make double-stranded cuts in target DNA can be mutated in one or two inactive nuclease domains, leading to ‘nickases’ or ‘dead’ Cas (dCas9) enzymes, respectively. A third type of improvement is that Cas enzymes can be appended with domains that lead to transcriptional activation83, epigenome editing84,85, base editing86, changes to mitochondrial DNA87, reverse transcriptase-mediated gene insertion88, and transposition89. Notably, these changes can also be made to Cas12a enzymes90, which can require a shorter guide RNA and lead to staggered double-stranded cuts.
Complementing these DNA nucleases are RNA nucleases that bind and cleave RNA91,92 or, alternatively, can be engineered with an ADAR domain to enable RNA base editing93,94 (Fig. 1c). Compared with DNA-editing nucleases, non-viral-mediated delivery of an RNA nuclease facilitates transient, shorter-lived changes in gene expression93. Thus, whereas DNA nucleases may be well suited for chronic diseases, RNA nucleases may be more useful for short-term diseases, such as transient inflammation, or diseases driven by RNA pathogens such as coronaviruses95,96. Delivering CRISPR therapeutics is challenging because both the Cas protein and the single-guide RNA (sgRNA) must be present at sufficient concentrations to form intracellular ribonucleoproteins. In preclinical studies, this two-payload problem has been solved by concurrently delivering mRNA and sgRNA in nanoparticles77,96–104, by constitutively expressing sgRNA via adeno-associated virus (AAV) before injecting LNPs loaded with Cas9 mRNA105, or by injecting pre-complexed RNPs106–108. An alternative solution is to reduce the size of the Cas enzyme, which might make it easier to package alongside the sgRNA in the same delivery system. Several compact nucleases have been reported, including a Cas12j (termed Casϕ)109, Cas12f110,111, Cas13bt112 and Cas13ct112.
Box 1 mRNA vaccines against SARS-CoV-2.
Intramuscular injections of lipid nanoparticles (LNPs) carrying mRNA encoding full-length, stabilized SARS-CoV-2 spike proteins reduce infection, morbidity and spread caused by COVID-19 (refs61,62). Both of the COVID-19 mRNA vaccines, created by Moderna and Acuitas/BioNTech/Pfizer, are groundbreaking for three reasons. First, they were developed in months rather than the years that is typical for drug development255. Second, the vaccines were tremendously effective, reducing viral load in patients infected after vaccination256, reducing infection by as much as 95% in clinical trials61,62, and substantially reducing disease under real-world conditions257. The mechanisms responsible for this efficacy require further study, but intramuscular administration can result in LNPs transfecting high numbers of immune cells, including antigen-presenting cells258. Once antigen-presenting cells transfected by LNPs produce the SARS-CoV-2 spike protein, the antigen is degraded and presented in complex with major histocompatibility complex (MHC) class I or class II, thereby activating T cell subsets259 and promoting B cell-mediated humoral immunity260. Third, evidence suggests that mRNA vaccines protect against emerging SARS-CoV-2 variants, including those with viral spike protein mutations261. Given that the LNP remains the same, it has been estimated that mRNA-based vaccines encoding a novel variant could be produced in as little as 6 weeks262. This ability to respond to an emerging SARS-CoV-2 variant without the need to massively re-engineer the LNP–mRNA formulation, manufacturing or distribution procedures is critical, especially as evidence of COVID vaccine breakthrough infections appears263 and, more generally, as the frequency with which zoological viruses infect humans increases264.
Synthetic vehicles for RNA delivery
Although different RNA payloads can have different biochemical mechanisms of action, all of them must avoid clearance by off-target organs, must access the correct tissue, must interact with the desired cell type in a complex tissue microenvironment, must be taken up by endocytosis, and must exit the endosome, without eliciting a deleterious immune response113. Although small oligonucleotide RNA therapeutics, including ASOs, siRNAs and ADAR-oligonucleotides, can be modified using stable chemistries and delivered using conjugates, mRNA-based and DNA-based therapeutics require a vehicle for entry into a cell. To facilitate this process, scientists have developed several RNA delivery systems using a range of materials, including polymers and LNPs.
Lipids and lipid-based nanoparticles
LNPs are a key class of drug delivery system that includes nanoparticles approved by the FDA for liver siRNA delivery19 and for mRNA vaccine delivery61,62. On the basis of the size of the hydrophilic head group relative to the size of the hydrophobic tail or tails114, lipids form distinct structures including micelles, liposomes and LNPs (Fig. 2a). FDA-approved LNPs contain variations of four basic components: a cationic or ionizable lipid, cholesterol, a helper lipid, and a poly(ethylene glycol) (PEG)-lipid (Fig. 2b,c). Scientists have investigated the structure of lipid-based delivery systems complexed with nucleic acid115,116 and demonstrated that lipid structure alters how LNPs interact with cells117. Given that lipid structure influences delivery and that lipids can be easily synthesized using chemistries including Michael addition-based, epoxide-based and alcohol-based reactions, scientists have created libraries of dozens to thousands of chemically distinct lipid delivery systems118,119. Many of these efforts focused on improving siRNA delivery to hepatocytes in mice120. This work, along with a more rational approach to lipid design117, reduced the dose required for robust in vivo hepatocyte gene silencing in mice from approximately 1.0 mg/kg (ref.121) to 0.002 mg/kg (ref.122). Key lipids that delivered siRNA in non-human primates included C12-200, which was synthesized using epoxide–amine chemistry120; cKK-E12, a peptide-like lipid compound122; DLin-KC2-DMA, an ionizable lipid identified using rational design117; and DLin-MC3-DMA123, which was used in patisiran to treat hATTR19 (Fig. 2c,d).
Fig. 2. FDA-approved lipid-based structures contain some variation of the four basic components: cholesterol, a helper lipid, a PEG-lipid, and a cationic or ionizable lipid.

a | Lipid-based structures can include micelles, which consist of a lipid monolayer, or liposomes, which consist of a bilayer. Lipid nanoparticles are composed of multiple lipid layers as well as microdomains of lipid and nucleic acid. b,c | In addition to the RNA payload, LNPs often consist of cholesterol, a helper lipid, a PEG-lipid (all shown in part b), and a cationic or ionizable lipid (part c). Despite the variety of chemical structures and lipid lengths, all the lipids in part c contain amine groups, which become positively charged at lower pH; this charge binds the anionic backbone of the RNA, providing a driving force for the formation of a stable LNP. d | The molar ratios of the four components making up the FDA-approved Acuitas/BioNTech/Pfizer COVID vaccine and patisiran, which delivers siRNA to the liver.
LNPs have also delivered mRNA to the liver in mice, in non-human primates and in humans. In some cases, LNPs utilized lipids previously developed for siRNA delivery. For example, in mice, LNPs formulated with cKK-E12124,125, C12-200126, and DLin-MC3-DMA127 delivered mRNA to the liver. Newer lipids reported, such as LP0177(Intellia Therapeutics), Lipid H128(Moderna), and FTT5103(Ohio State and Beam Therapeutics), have also delivered mRNA to the mouse liver. Recently, two LNPs formulated with an unreported cationic or ionizable lipid, PEG-lipid, cholesterol and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) delivered mRNA encoding a base-editing Cas9 and sgRNA targeting PCSK9 to the liver in non-human primates59,60. A single LNP administration led to months of sustained PCSK9 silencing. Moreover, long-term PCSK9 silencing mediated by antibodies129 or siRNA17 has shown beneficial effects in cardiovascular disease in humans. Separately, Beam Therapeutics reported that LNP-mediated delivery of base editors led to sustained effects in the liver of non-human primates130. In addition to these preclinical studies, Intellia has released data in patients dosed with its NTLA-2001, used to inactivate the TTR gene. Inactivation of TTR has also previously been validated in humans; siRNA or ASOs targeting TTR slowed the progression of hATTR amyloidosis with polyneuropathy36,42.
In addition to the RNA payload, the Alnylam, Moderna and Pfizer/BioNTech/Acuitas LNPs comprise four components: the cationic or ionizable lipids DLin-MC3-DMA (Alnylam), SM-102 (Moderna), or ALC-0315 (Pfizer/BioNTech/Acuitas), cholesterol, the PEG-lipids PEG-2000-C-DMG (Alnylam), PEG-2000-DMG (Moderna), or ALC-0159 (Pfizer/BioNTech/Acuitas), and DSPC (Fig. 2b–d). Although most preclinical studies have evaluated how the structure of the cationic or ionizable lipid influences delivery, the other three components can also affect delivery131,132. For example, by changing the cholesterol, PEG-lipid or ‘helper’ lipid, an LNP that delivered siRNA to pulmonary and cardiovascular endothelial cells in mice133 and non-human primates134 was retargeted to deliver siRNA135, sgRNA136 or mRNA99 to bone marrow, hepatic and splenic endothelial cells after intravenous administration as well as lung epithelial cells after nebulization137. In additional examples, changing the PEG-lipid structure or its molar percentage altered LNP pharmacokinetics and liver siRNA delivery138 in mice and affected delivery within the eye139. Both the PEG and lipid components of the PEG-lipid affect how it interacts with the LNP and cells: the lipid ‘anchors’ the PEG-lipid into the LNP, while the hydrophilic PEG interacts with water in the blood, thereby creating an aqueous barrier similar to other PEGylated nanomedicines140. Likewise, although most LNPs have been formulated with unmodified cholesterols, LNP delivery in cell cultures and in mice has been shown to be improved by use of oxidized cholesterols124, esterified cholesterols136, or cholesterol analogues such as phytosterols116. Although the mechanism behind cholesterol-mediated improvements in delivery remains unknown, incorporating modified cholesterol into LNPs can change their structure141. Researchers have demonstrated that replacing DSPC with other lipids can promote LNP delivery to the spleen or lungs125,142. Similarly, by adding another lipid to the LNP, thereby changing the LNP from a four-component system to a five-component system, LNPs were targeted to the lung and spleen in a process termed selective organ targeting101. Finally, both Intellia143 and Beam Therapeutics130 have shared information that LNPs can be made to deliver mRNA to haematopoietic stem and progenitor cells in mice, with the hope of developing in vivo haematopoietic stem cell-targeting therapies.
Polymers and polymer-based nanoparticles
Many non-viral RNA delivery systems also utilize polymers and polymeric nanoparticles144 (Fig. 3a). Chemists can vary polymer traits including charge, degradability and molecular weight, all of which influence how polymers deliver RNA into cells145,146. One frequently used polymer is poly(lactic-co-glycolic acid) (PLGA). PLGA drug delivery systems have been approved by the FDA for the delivery of small-molecule drugs but not for the delivery of nucleic acids147. At neutral pH, PLGA does not have the positive charge required to complex the anionic RNA phosphodiester backbone. Thus, to utilize PLGA as an RNA delivery system, scientists have added cationic chemical groups such as chitosan to deliver siRNA in mice148.
Fig. 3. RNA can be delivered using nanoparticles formulated with polymers or dendrimers.

a | Polymeric nanoparticles and polymers based on poly(ethylenimine) (PEI), poly(l-lysine) (PLL), and poly(beta-amino-ester) (PBAE) use cationic amine groups to complex the anionic phosphodiester backbone of RNA. Polymers based on poly(lactic-co-glycolic acid) (PLGA) are typically engineered to contain separate cationic groups. b | Dendrimers are polymeric structures with a defined number of molecules emanating from a core. PAMAM, poly(amidoamine).
Polymers that contain amine groups that can become cationic, such as polyethylenimine (PEI) and poly(l-lysine) (PLL), can complex with RNA via electrostatic interactions and deliver it into cells149,150. However, unmodified PEI and PLL are not always well tolerated151, and PEI transfection capability and toxicity increase with molecular weight152. Thus, PEI and PLL have been chemically modified to improve in vivo efficacy and tolerability. For example, nanoparticles made with PEG-grafted PEI have been used to deliver mRNA to immune cells in the lungs153, and cyclodextrin-PEI conjugates have been used to deliver an mRNA vaccine in vivo154. Similarly, iron oxide nanoparticles were surface-modified with PLL to deliver genes to the central nervous system in mice155.
Another cationic polymer class is the poly(beta-amino ester)s (PBAEs), which are synthesized by conjugating amine monomers to diacrylates. These polymers, which were designed to have improved biodegradation and cytotoxicity relative to PEI and PLL156, contain cationic amines as well as biodegradable ester bonds157. Early research used Michael addition chemistry to synthesize hundreds of chemically distinct PBAEs and then evaluated how these nanoparticles delivered DNA158,159 and RNA in cell culture160. These PBAE ‘libraries’ enabled researchers to study how PBAE chemical structure influences drug delivery, thereby generating design rules for subsequent PBAEs161,162. Initial design rules suggested that effective polymers were almost always hydrophobic, had monoalcohol or dialcohol side groups, and had linear bis (secondary) amines161. A follow-up study using these design criteria showed that top polymers were all formed from amino alcohols and were similar chemically, only differing by one carbon162. More generally, these studies established the feasibility of high-throughput chemical synthesis followed by high-throughput drug delivery studies, which has been applied to other nanoparticle chemical classes, including LNPs118. PBAEs have been used for the delivery of DNA vectors to pulmonary cells after nebulization163, the delivery of mRNA intranasally164, and the delivery of siRNA to a human orthotopic glioblastoma tumour model in mice165. More recently, researchers have also used PBAE-based polymers to deliver Cas13a mRNA and to guide RNA to the respiratory tract of mice and hamsters via nebulization for the treatment of SARS-Cov-2 (ref.96).
Researchers have also synthesized lipid–polymer hybrids and found that adding lipids to PBAEs improved serum stability and delivery166,167. An additional polymer class used for RNA delivery is dendrimers, which consist of a defined number of branched monomers emanating from a central core molecule (Fig. 3b). Dendrimers synthesized with cationic groups, such as poly(amidoamine) (PAMAM) or PLL, can form complexes and deliver RNA into cells. Dendrimers have delivered RNA to the central nervous system168, acted as intramuscular vaccines against the Ebola and H1N1 viruses169, and delivered siRNA to hepatic endothelial cells170. Dendrimer structure has also been modified to protect nucleic acids from enzymatic degradation171 and to enhance endosomal escape172.
Active versus passive tissue targeting
Passive tissue targeting
As outlined in the section ‘Lipids and lipid-based nanoparticles’, research has demonstrated that LNPs originally developed for liver siRNA or mRNA delivery can be redirected to other organs without the need for antibody fragments, peptides, aptamers or other active targeting ligands that bind specific receptors on the surface of target cells173. For the purposes of this Review, we define such retargeting, probably driven by interactions between the nanoparticle and serum proteins in endogenous trafficking pathways, as a process termed ‘passive targeting’ or ‘endogenous targeting’ (Fig. 4a). As a sphere becomes smaller, its surface area to volume ratio increases; as a result, nanoparticles have large surface areas174 and when a nanoparticle comes into contact with a biological milieu, many biomolecules can coat its surface175. By covering the nanoparticle surface, these coronas (proteins that bind to the nanoparticle surface) change the chemical and biological molecules at the surface of the nanoparticle, thereby altering how the nanoparticle interacts with immune cells176 and on-target tissues177. In one clinical example178, apolipoprotein E (ApoE) adsorption was required for ionizable, but not cationic, lipids to deliver siRNA to hepatocytes179. Researchers have found LNPs that can be trafficked via an albumin-dependent mechanism180 as well as LNPs with variable dependence on low-density lipoprotein (LDL), very low-density lipoprotein (VLDL) and caveolin-1 receptors136,181. In addition to the effect that the protein corona can have on LNP tropism, LNP size can also contribute to passive tissue targeting. In one example, nanoparticle size influenced the potency of siRNA-based LNPs in vivo182. In another example, nanoparticle size governed the frequency with which an LNP was cleared by immune cells in the lymph node183. Similarly, several groups have demonstrated that LNP charge can affect delivery. In one example, scientists developed a lipoplex that delivered mRNA to the spleen by altering its charge142. Similar systems have delivered therapeutic mRNA in different animal models66,184. Likewise, by adding a cationic lipid to the LNP, scientists retargeted an LNP with liver tropism to the lung125. These splenic and pulmonary delivery datasets were subsequently observed using LNPs that facilitated mRNA delivery and Cas9-mediated gene editing101.
Fig. 4. Drug delivery vehicles can use two mechanisms of action to reach their target cell type.

a–d | Delivery vehicles can reach desired cells using passive or endogenous targeting (part a), which leads to adsorption of serum biomolecules onto the outside of the lipid nanoparticle (LNP) in the bloodstream. For example, the serum lipoprotein ApoE binds to LNPs, leading to delivery via low-density lipoprotein receptor (LDLR) expressed on hepatocytes. Active targeting employs a ligand directly conjugated to a nucleic acid (part b), an antibody directly conjugated to a nucleic acid (part c) or a ligand or antibody conjugated to a nanoparticle to target a receptor expressed on the cell (part d). ASGPR, asialoglycoprotein receptor; GalNAc, N-acetylgalactosamine. ApoE, apolipoprotein E.
Active targeting
RNA can also be ferried into on-target cells using ‘active targeting’. In this strategy, a ligand that binds a specific biomolecule is added to the delivery system (Fig. 4b–d). The most clinically validated examples are GalNAc–siRNA and GalNAc–ASO conjugates, which have led to the FDA-approved drugs givosiran16 and lumasiran18, as well as the EMA-approved drug inclisiran17. GalNAc is a carbohydrate-derived trivalent ligand that binds the asialoglycoprotein receptor (ASGPR) (Fig. 4b). ASGPR is an ideal receptor for active targeting: it is highly expressed on target cells (in this case hepatocytes), not expressed on other cell types, leads to rapid endocytosis upon GalNAc binding, and is rapidly recycled to the cell surface following endocytosis. GalNAc also exhibits several traits that make it an ideal targeting ligand. First, it has a molecular weight of <2 kDa, which is many times less than the molecular weight of the ASO or siRNA it delivers185. This low molecular weight ensures that most of the drug being administered is siRNA and not GalNAc. Second, improvements can be made to its chemical structure to enhance silencing in vivo185,186, and its interactions with serum proteins are well understood187; in addition, site-specific modifications made to the ribose and phosphate moieties have reduced nuclease-mediated siRNA degradation as well as off-target mRNA binding187–189. The combination of this ideal receptor and optimized ligand has resulted in subcutaneous delivery of siRNA and ASO at doses far below those that cause toxicity in large-animal models190,191.
Small ligands have also been developed for extrahepatic delivery. Specifically, by measuring the biodistribution and delivery mediated by a library of different siRNA–lipid conjugates, researchers found that hydrophobic conjugates accumulated in the liver, whereas less hydrophobic conjugates accumulated in the kidneys192. In the same study, the authors found that when compared with cholesterol (a well studied hepatic conjugate), both dichloroacetic acid alone and dichloroacetic acid with a phosphocholine polar head group improved siRNA delivery to extrahepatic tissues such as the lung and heart and, to a lesser degree, improved siRNA delivery to skeletal muscle and fat. In a follow-up study, researchers identified two lipid transport pathways that could be guiding delivery: hydrophobic siRNA conjugates tended to spontaneously associate with LDL, an association known to improve trafficking to the liver, whereas less-hydrophobic conjugates bound to high-density lipoprotein (HDL), part of the reverse cholesterol transport pathway193. A 2021 study reported that cholesterol-functionalized DNA–RNA heteroduplexes were capable of crossing the blood–brain barrier in mice and rats after systemic administration. Once again, the chemical structure of the lipid and conjugate was critical for cell targeting, gene silencing and pharmacokinetics194. RNA aptamers, which are RNAs that fold into defined three-dimensional structures195, have also emerged as ligands for specific cell-targeted delivery of RNAi-based therapeutics. In one example, researchers conjugated an siRNA-targeting STAT3, a key regulator of glioblastoma, to the receptor tyrosine kinase anti-PDGFRα RNA aptamer. This delivery system inhibited the expression of target genes and showed strong reduction of cell viability in vitro196.
A distinct targeting mechanism requires the use of antibody fragments or antibodies to target cell types. For example, siRNA and ASO have also been delivered to extrahepatic tissues using antibody–siRNA conjugates (Fig. 4c). In one example, an anti-CD71 antibody fragment delivered siRNA to the heart and skeletal muscle, leading to extended on-target gene silencing197. Data using either anti-CD71 fragments197 or monoclonal antibodies198 have demonstrated long-term muscle silencing in preclinical models, including 12 weeks of silencing in non-human primates after a single administration198. As a result, Avidity Biosciences initiated a phase I/II clinical trial delivering siRNA against myotonic dystrophy protein kinase to treat myotonic dystrophy type I in 2021.
Although ASOs and siRNAs are small enough to be delivered using a small conjugate or antibody, mRNAs are too large. For this reason, mRNA has been formulated inside nanoparticles that are decorated with antibodies (Fig. 4d). Researchers have developed a cell-targeting platform known as Anchored Secondary scFv Enabling Targeting (ASSET) that used monoclonal antibody-coated LNPs to deliver siRNA or mRNA. This system relies on a membrane-anchored lipoprotein within the LNP that binds to an antibody’s fragment (Fc) domain; switching out the variable region enables specific targeting to different cell subsets199. ASSET was used to facilitate LNP binding to extrahepatic cell types for the purpose of treating inflammatory bowel disease200. These approaches can also be targeted to receptors in specific conformations, as demonstrated by recent data using ASSET to deliver LNPs to a specific, high-affinity conformation of α4β7 (ref.201). Finally, LNPs covalently conjugated to an antibody that binds to plasmalemma vesicle-associated protein have been used to target lung cells by facilitating specific caveolin-mediated endocytosis202, and LNPs conjugated to mannose have been used to immunize mice against H1N1203.
The pathway to clinical RNA delivery
Independently of their chemical structure or targeting mechanism, nanoparticle-based RNA delivery systems are selected for clinical trials using a series of preclinical experiments here termed ‘the nanoparticle discovery pipeline’. This pipeline often starts with initial high-throughput studies in cell culture. Based on these in vitro delivery data, which are used to optimize nanoparticle traits, a small number of nanoparticles is selected for mouse studies before a smaller number is tested in rats and, finally, an even smaller number in non-human primates (Fig. 5a). If the nanoparticle has a sufficiently safe toxicity profile in non-human primates, the delivery system can then be considered for subsequent clinical trials.
Fig. 5. The preclinical nanoparticle discovery pipeline.

a | Thousands of nanoparticles are often synthesized and tested in vitro before a few nanoparticles are evaluated in mice. However, in vitro delivery can be a poor predictor of in vivo delivery. A smaller number of nanoparticles are then often tested in rats or non-human primates (NHPs). Given that NHPs are the best preclinical models for human delivery, one key opportunity to improve the efficiency of this pipeline is to understand which small-animal model is most predictive of NHP efficacy and tolerability; this species-to-species drug delivery relationship is understudied. b | Tissue weight as a percentage of total animal body weight (bw) in female animals. Relative organ size varies across species, which may alter nanoparticle on-target and off-target delivery.
High-throughput nanoparticle discovery pipeline
Although the nanoparticle discovery pipeline has identified nanoparticles that deliver RNA to immune cells after intramuscular administration21,22 and hepatocytes after systemic administration19, opportunities do exist to improve the efficiency of the pipeline. For example, chemists can synthesize thousands of nanoparticles, but running a multi-thousand-mouse experiment is infeasible. As a result, nanoparticles are first evaluated in cell culture, even though delivery in cell culture can poorly predict delivery in vivo204. To address this problem, DNA barcoding assays to test thousands of nanoparticles in vivo have been developed205–208. In these assays, LNP-1 is formulated to carry DNA barcode 1 and a nucleic acid (such as siRNA or mRNA), whereas LNP-N is formulated to carry DNA barcode N and the same nucleic acid. After pooling the LNPs together and administering them to mice, cells in which functional RNA delivery occurs — such as mRNA-mediated protein production99 or siRNA-mediated gene silencing135 — are isolated and all N barcodes are quantified. This approach has been used to deliver RNA to non-hepatocytes without targeting ligands99,124,136,209–211. A second way to improve the nanoparticle discovery pipeline is to understand which small animals best predict delivery in a non-human primate. Species variability can derive from physiological differences in basal metabolic rates212, which can affect nanoparticle delivery213–215, or differences in serum lipids216, which can affect the colloidal stability, biodistribution, cellular interactions, toxicity and clearance of the resulting nanoparticle coronas217. Additional species variability probably derives from the mass of a given tissue, relative to the total animal weight, which is not conserved (Fig. 5b). For example, mouse218–222 and rat223 livers are larger, normalized to overall body mass, than those of non-human primates224 and humans225–228. These differences may increase the effective off-target liver delivery in mice and rats, relative to non-human primates and humans. To address some of these concerns, researchers have developed species-agnostic nanoparticle delivery screening (SANDS)229. Using SANDS, the authors tested how dozens of LNPs delivered mRNA to normal mice, mice with primatized livers, and mice with humanized livers. After quantifying the correlation between delivery in murine, non-human primate and human cells in vivo, and observing species-dependent delivery, the authors used RNA sequencing to identify genes that might differentiate delivery across species. This approach might enable scientists to compare how many chemically distinct nanoparticles deliver drugs across different species.
Hallmarks of clinically relevant delivery systems
As the nanoparticle pipeline continues to improve, next-generation delivery vehicles might benefit from recapitulating traits shared among current clinical RNA delivery vehicles. For example, as part of its clinical trials, detailed pharmacokinetic and clearance studies of patisiran in humans have been published230,231. In addition to describing sustained gene silencing over several months, these studies demonstrated that patisiran had consistent mean serum concentration and dynamics over 18 months with repeated administration. Similarly, the FDA Center for Drug Evaluation and Research report on patisiran232 describes the accepted animal models, toxicology readouts and time points used in the approval process. These data provide a roadmap for the clinical characterization of nanoparticle RNA drugs. More generally, the three RNA drug delivery systems approved by the FDA19,20 or granted an EUA21 to date tend to share six characteristics that taken together could constitute hallmarks of approved delivery vehicles (Fig. 6).
Fig. 6. The hallmarks of a clinically relevant delivery system.
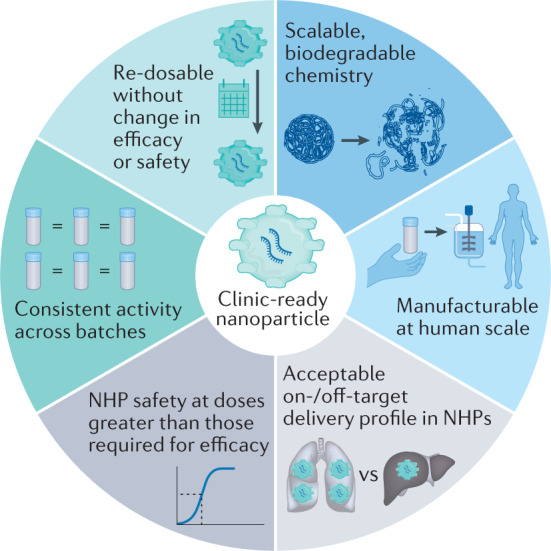
These six traits, which can be studied early in the development of the drug delivery vehicle, can help to increase the odds that a drug delivery system is approved by the FDA. NHP, non-human primate.
First, clinical delivery systems tend to be synthesized using scalable chemistry that is often biodegradable. For example, adding ester bonds, which can degrade in water, to ionizable lipids improved LNP safety233; the Moderna, Acuitas and Alnylam lipids used in humans contain esters (Fig. 2c). Second, the drug delivery system should be chemically simple enough to be manufactured at human scale; for example, in a hypothetical clinical trial injecting 100 kg patients with 6 mg/kg lipid and 0.3 mg/kg RNA (that is, a lipid:RNA mass ratio of 20:1), assuming lipid loss during the synthesis and formulation process, nearly 1 g of lipid would need to be synthesized per injection. GalNAc conjugates can be synthesized and, separately, conjugated to siRNAs or ASOs at human scale (that is, with large-batch manufacturing capability that is compliant with Current Good Manufacturing Practice). We note that clinically approved LNPs have so far included four lipid components and have not included targeting ligands. Determining the advantages and disadvantages of adding targeting ligands in clinical nanoparticles will be important234. Third, the drug delivery system must have an acceptable ratio of on-target to off-target delivery. On-target and off-target delivery should be measured both as biodistribution (that is, where does the delivery system go?) and as function (that is, where does the payload affect cell function?). As over 95% of RNA can be retained in endosomes235, biodistribution is necessary, but not sufficient, for functional cytoplasmic RNA delivery. Research has also shown that small copy numbers of siRNAs are sufficient for gene knockdown in vitro236, implying that siRNA acts in a catalytic manner. Fourth, the dose required for RNA efficacy must be substantially lower than the dose at which toxicity occurs. Ideally, this finding is observed in non-human primates, as mice have historically not been rigorous models of RNA toxicity, for yet-to-be-determined reasons. Fifth, the activity of the drug should be consistent across many batches, even after shipping. To this end, scientists are developing methods of stabilizing mRNA drugs; a CureVac mRNA platform was active after lyophilization and storage at 40 °C for up to 6 months and room temperature for 3 years237. However, for LNP-encapsulated mRNA, lyophilization might decrease stability by promoting lipid crystallization, unless cryoprotectants such as sucrose are used238. For example, by adding 10% sucrose, lipid nanoparticles complexed with RNA can be lyophilized and stored at room temperature for ≥8 months239. Sixth, in most clinical settings, re-dosing the RNA drug without losing efficacy or safety will be necessary to maintain the biological effect or ‘dose to effect’. siRNA drugs have been safely re-dosed in patients when doses have been given 3 weeks apart19. mRNA vaccines have been safely dosed twice, with doses either 3 weeks or 4 weeks apart21,22. Re-dosing mRNA weekly has been reported to reduce its efficacy in mice240, with this effect being driven by a subset of B lymphocytes.
FDA-approved and EMA-approved RNA therapeutics
Although the criteria constituting hallmarks of approved delivery vehicles make clinically relevant delivery challenging, the number of successful RNA drugs is increasing. A leading example is the GalNAc–siRNA conjugates, which have demonstrated efficacy and safety in patients. Like the mRNA-LNP vaccines, GalNAc–siRNA conjugates can easily be redesigned to treat different diseases, since only the siRNA sequence needs to change. As a result, the GalNAc–siRNA conjugates givosiran, which treats acute intermittent hepatic porphyria, and lumasiran, which treats primary hyperoxaluria type 1, have been approved by the FDA. Inclisiran, a subcutaneous therapeutic used to treat hypercholesterolaemia or mixed dyslipidaemia by inhibiting hepatic synthesis of proprotein convertase subtilisin–kexin type 9 (PCSK9)17, has been approved by the EMA, but not by the FDA (which delayed approval owing to issues at a manufacturing plant). The GalNAc–siRNA conjugates fitusiran and vutrisiran have also been used to generate promising clinical data. Fitusiran is used to treat haemophilia A or B241 by targeting antithrombin mRNA in the liver241. Vutrisiran (see above) is used to treat hATTR amyloidosis with polyneuropathy in adults, and generated positive top-line results in a phase III study242. Arrowhead, Silence Therapeutics and Dicerna are also utilizing GalNAc–siRNA conjugates in ongoing phase I/II trials for the treatment of hepatitis B, hereditary haemochromatosis, and primary hyperoxaluria, among other diseases62, and Ionis Pharmaceuticals is using GalNAc to deliver ASOs243. For all of these drugs, long-term gene silencing mediated by GalNAc conjugates is important for their eventual clinical use and to improve patient compliance. For example, inclisiran reduces PCSK9 levels for up to 6 months after administration in humans26.
Conclusions
In the past decade, preclinical and clinical data have hinted at the potential for RNA therapies to treat disease. However, to fully reach their potential, several advances are needed. One advance will be to understand how RNA payloads interact with delivery vehicles and how these interactions affect targeting and tolerability. For example, emerging evidence suggests that nanoparticle tropism can change with RNA payload99. This effect may be caused by changes in the nanoparticle and therefore the biomolecules with which it interacts in the body. Alternatively, payload-dependent tropism may be affected by cell state244, since a cell that is optimized for the production of exogenous mRNA might not be optimized for siRNA-based mRNA silencing. Similarly, although LNP interactions with ApoE are known to drive liver delivery178, a need remains to identify trafficking mechanisms that promote delivery to extrahepatic tissues; it is feasible that disease-specific trafficking could be exploited to enhance extrahepatic delivery. One potential approach is to use naturally occurring systems such as PEG10, which package mRNA in human cells245 or extracellular vesicles, which are outside the scope of this manuscript and have been reviewed elsewhere246. Another related advance is the need to understand how chemical modifications made to the RNA payload influence RNA stability, avoidance of intracellular off-target effects, such as binding based on partial complementarity247, or activation of the innate immune system248. For example, CureVac recently reported that its mRNA vaccine, which utilizes unmodified mRNA, did not provide robust protection against COVID-19249. Unmodified mRNA programmes led by Translate Bio have also been ineffective when nebulized to treat cystic fibrosis63 or systemically administered to treat ornithine transcarbamylase deficiency64. These findings, alongside evidence that chemical modification patterns boost siRNA and ASO efficacy250 and that the untranslated regions of mRNA can be engineered for improved251 or cell-type-specific activity252, suggest that mRNA payloads can be improved in future therapies. Finally, we need an improved understanding of how efficacy and tolerability in smaller animal models (such as mice and rats) predict efficacy and tolerability in non-human primates and humans. Given the major ethical issues with testing many potential drug delivery systems in non-human primates, one key advance would be the identification of smaller animal models that are maximally predictive of delivery in non-human primates253. In cancer biology, researchers identified genetically engineered mice that more accurately recreated clinical outcomes from human trials carried out in parallel254. If we are thereby able to expand our understanding of how the RNA drug, drug delivery system and body all interact with one another, patients will benefit from effective next-generation gene therapies.
Acknowledgements
The authors thank K. Tiegren at the Georgia Institute of Technology for copyediting the manuscript.
Author contributions
The authors contributed equally to all aspects of the article.
Competing interests
J.E.D. is a consultant for GV. The other authors declare no competing interests.
Footnotes
Peer review information
Nature Reviews Genetics thanks Yizhou Dong and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hopkins AL, Groom CR. The druggable genome. Nat. Rev. Drug. Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 2.Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020;19:673–694. doi: 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.High KA, Roncarolo MG. Gene therapy. N. Engl. J. Med. 2019;381:455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 4.Pasi KJ, et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 5.Mendell JR, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 6.Frangoul H, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 7.Esrick EB, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N. Engl. J. Med. 2021;384:205–215. doi: 10.1056/NEJMoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohn DB, et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N. Engl. J. Med. 2021;384:2002–2013. doi: 10.1056/NEJMoa2027675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russell S, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aronson SJ, et al. Prevalence and relevance of pre-existing anti-adeno-associated virus immunity in the context of gene therapy for Crigler–Najjar syndrome. Hum. Gene Ther. 2019;30:1297–1305. doi: 10.1089/hum.2019.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bryson TE, Anglin CM, Bridges PH, Cottle RN. Nuclease-mediated gene therapies for inherited metabolic diseases of the liver. Yale J. Biol. Med. 2017;90:553–566. [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen GN, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandler M, Panigaj M, Rolband LA, Afonin KA. Challenges to optimizing RNA nanostructures for large scale production and controlled therapeutic properties. Nanomedicine. 2020;15:1331–1340. doi: 10.2217/nnm-2020-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leborgne C, et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020;26:1096–1101. doi: 10.1038/s41591-020-0911-7. [DOI] [PubMed] [Google Scholar]
- 16.Balwani M, et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Engl. J. Med. 2020;382:2289–2301. doi: 10.1056/NEJMoa1913147. [DOI] [PubMed] [Google Scholar]
- 17.Ray KK, et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N. Engl. J. Med. 2020;382:1507–1519. doi: 10.1056/NEJMoa1912387. [DOI] [PubMed] [Google Scholar]
- 18.Garrelfs SF, et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N. Engl. J. Med. 2021;384:1216–1226. doi: 10.1056/NEJMoa2021712. [DOI] [PubMed] [Google Scholar]
- 19.Adams D, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 20.Parums DV. Editorial: first full regulatory approval of a COVID-19 vaccine, the BNT162b2 Pfizer-BioNTech vaccine, and the real-world implications for Public Health Policy. Med. Sci. Monit. 2021;27:e934625. doi: 10.12659/MSM.934625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baden LR, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021;384:403–416. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polack FP, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buck J, Grossen P, Cullis PR, Huwyler J, Witzigmann D. Lipid-based DNA therapeutics: hallmarks of non-viral gene delivery. ACS Nano. 2019;13:3754–3782. doi: 10.1021/acsnano.8b07858. [DOI] [PubMed] [Google Scholar]
- 24.Vargason AM, Anselmo AC, Mitragotri S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021;5:951–967. doi: 10.1038/s41551-021-00698-w. [DOI] [PubMed] [Google Scholar]
- 25.Watts JK, Corey DR. Silencing disease genes in the laboratory and the clinic. J. Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosmas CE, et al. Inclisiran for the treatment of cardiovascular disease: a short review on the emerging data and therapeutic potential. Ther. Clin. Risk Manag. 2020;16:1031–1037. doi: 10.2147/TCRM.S230592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen F, Alphonse M, Liu Q. Strategies for nonviral nanoparticle-based delivery of CRISPR/Cas9 therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020;12:e1609. doi: 10.1002/wnan.1609. [DOI] [PubMed] [Google Scholar]
- 28.Hanna J, Hossain GS, Kocerha J. The potential for microRNA therapeutics and clinical research. Front. Genet. 2019;10:478. doi: 10.3389/fgene.2019.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong DS, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer. 2020;122:1630–1637. doi: 10.1038/s41416-020-0802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Ree MH, et al. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment. Pharmacol. Ther. 2016;43:102–113. doi: 10.1111/apt.13432. [DOI] [PubMed] [Google Scholar]
- 31.van der Ree MH, et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet. 2017;389:709–717. doi: 10.1016/S0140-6736(16)31715-9. [DOI] [PubMed] [Google Scholar]
- 32.Regulus announces pipeline updates and advancements. Regulushttp://ir.regulusrx.com/news-releases/news-release-details/regulus-announces-pipeline-updates-and-advancements (2017).
- 33.Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013;42:217–239. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 2005;7:719–723. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alnylam announces U.S. Food and Drug Administration acceptance of new drug application for investigational vutrisiran for the treatment of the polyneuropathy of hereditary ATTR amyloidosis. Alnylamhttps://investors.alnylam.com/press-release?id=25811 (2021).
- 36.HELIOS-A: 9-month results from the phase 3 study of vutrisiran in patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy. Alnylamhttps://www.alnylam.com/wp-content/uploads/2021/04/Adams_HELIOS-A-9-Month-Results.pdf (2021).
- 37.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 38.Adachi H, Hengesbach M, Yu YT, Morais P. From antisense RNA to RNA modification: therapeutic potential of RNA-based technologies. Biomedicines. 2021;9:550. doi: 10.3390/biomedicines9050550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphreys SC, et al. Emerging siRNA design principles and consequences for biotransformation and disposition in drug development. J. Med. Chem. 2020;63:6407–6422. doi: 10.1021/acs.jmedchem.9b01839. [DOI] [PubMed] [Google Scholar]
- 40.Evers MM, Toonen LJ, van Roon-Mom WM. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015;87:90–103. doi: 10.1016/j.addr.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Santos RD, et al. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: results of 4 phase III trials. Arterioscler. Thromb. Vasc. Biol. 2015;35:689–699. doi: 10.1161/ATVBAHA.114.304549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benson MD, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:22–31. doi: 10.1056/NEJMoa1716793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel Ther. 2017;11:533–545. doi: 10.2147/DDDT.S97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frank DE, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94:e2270–e2282. doi: 10.1212/WNL.0000000000009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finkel RS, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017;377:1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 46.Crooke ST. Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Ther. 2017;27:70–77. doi: 10.1089/nat.2016.0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim KH, et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020;11:3501. doi: 10.1038/s41467-020-17093-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kilanowska A, Studzińska S. In vivo and in vitro studies of antisense oligonucleotides — a review. RSC Adv. 2020;10:34501–34516. doi: 10.1039/D0RA04978F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 2017;57:81–105. doi: 10.1146/annurev-pharmtox-010716-104846. [DOI] [PubMed] [Google Scholar]
- 50.Burdick AD, et al. Sequence motifs associated with hepatotoxicity of locked nucleic acid — modified antisense oligonucleotides. Nucleic Acids Res. 2014;42:4882–4891. doi: 10.1093/nar/gku142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamamoto T, et al. Highly potent GalNAc-conjugated tiny LNA anti-miRNA-122 antisense oligonucleotides. Pharmaceutics. 2021;13:817. doi: 10.3390/pharmaceutics13060817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen W, et al. Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat. Biotechnol. 2019;37:640–650. doi: 10.1038/s41587-019-0106-2. [DOI] [PubMed] [Google Scholar]
- 53.Miller CM, et al. Stabilin-1 and stabilin-2 are specific receptors for the cellular internalization of phosphorothioate-modified antisense oligonucleotides (ASOs) in the liver. Nucleic Acids Res. 2016;44:2782–2794. doi: 10.1093/nar/gkw112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merkle T, et al. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019;37:133–138. doi: 10.1038/s41587-019-0013-6. [DOI] [PubMed] [Google Scholar]
- 55.Qu L, et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019;37:1059–1069. doi: 10.1038/s41587-019-0178-z. [DOI] [PubMed] [Google Scholar]
- 56.Aquino-Jarquin G. Novel engineered programmable systems for ADAR-mediated RNA editing. Mol. Ther. Nucleic Acids. 2020;19:1065–1072. doi: 10.1016/j.omtn.2019.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Da Silva Sanchez A, Paunovska K, Cristian A, Dahlman JE. Treating cystic fibrosis with mRNA and CRISPR. Hum. Gene Ther. 2020;31:940–955. doi: 10.1089/hum.2020.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gillmore JD, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 59.Musunuru K, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434. doi: 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- 60.Rothgangl T, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021;39:949–957. doi: 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thompson MG, et al. Interim estimates of vaccine effectiveness of BNT162b2 and mRNA-1273 COVID-19 vaccines in preventing SARS-CoV-2 infection among health care personnel, first responders, and other essential and frontline workers — eight U.S. locations, December 2020-March 2021. MMWR. 2021;70:495–500. doi: 10.15585/mmwr.mm7013e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobrowolsk C, Paunovska K, Hatit MZC, Lokugamage MP, Dahlman JE. Therapeutic RNA delivery for COVID and other diseases. Adv. Health. Mater. 2021;10:e2002022. doi: 10.1002/adhm.202002022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Translate Bio announces results from second interim data analysis from ongoing phase 1/2 clinical trial of MRT5005 in patients with cystic fibrosis (CF). Translate Biohttps://investors.translate.bio/news-releases/news-release-details/translate-bio-announces-results-second-interim-data-analysis (2021).
- 64.Translate Bio announces pipeline program update. Translate Biohttps://investors.translate.bio/news-releases/news-release-details/translate-bio-announces-pipeline-program-update (2021).
- 65.Arcturus Therapeutics announces first quarter 2021 company overview and financial results and provides new clinical data. Arcturus Therapeuticshttps://ir.arcturusrx.com/news-releases/news-release-details/arcturus-therapeutics-announces-first-quarter-2021-company (2021).
- 66.Krienke C, et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science. 2021;371:145–153. doi: 10.1126/science.aay3638. [DOI] [PubMed] [Google Scholar]
- 67.Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines — a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luisi K, et al. Development of a potent Zika virus vaccine using self-amplifying messenger RNA. Sci. Adv. 2020;6:eaba5068. doi: 10.1126/sciadv.aba5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leal L, et al. Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS. 2018;32:2533–2545. doi: 10.1097/QAD.0000000000002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feldman RA, et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine. 2019;37:3326–3334. doi: 10.1016/j.vaccine.2019.04.074. [DOI] [PubMed] [Google Scholar]
- 71.Sahin U, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 72.Conry RM, et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995;55:1397–1400. [PubMed] [Google Scholar]
- 73.Sahin U, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585:107–112. doi: 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- 74.Jimeno A, et al. Abstract CT032: A phase 1/2, open-label, multicenter, dose escalation and efficacy study of mRNA-2416, a lipid nanoparticle encapsulated mRNA encoding human OX40L, for intratumoral injection alone or in combination with durvalumab for patients with advanced malignancies. Cancer Res. 2020;80:CT032. [Google Scholar]
- 75.Zhang HX, Zhang Y, Yin H. Genome editing with mRNA encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019;27:735–746. doi: 10.1016/j.ymthe.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pardi N, et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Controlled Rel. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Finn JD, et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 78.Allergan and Editas Medicine announce dosing of first patient in landmark phase 1/2 clinical trial of CRISPR medicine AGN-151587 (EDIT-101) for the treatment of LCA10. Editas Medicinehttps://ir.editasmedicine.com/news-releases/news-release-details/allergan-and-editas-medicine-announce-dosing-first-patient (2020).
- 79.Hanlon KS, et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiang F, Doudna JA. CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017;46:505–529. doi: 10.1146/annurev-biophys-062215-010822. [DOI] [PubMed] [Google Scholar]
- 81.Slaymaker IM, et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kleinstiver BP, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thakore PI, Black JB, Hilton IB, Gersbach CA. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat. Methods. 2016;13:127–137. doi: 10.1038/nmeth.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nuñez JK, et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184:2503–2519.e2517. doi: 10.1016/j.cell.2021.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Porto EM, Komor AC, Slaymaker IM, Yeo GW. Base editing: advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020;19:839–859. doi: 10.1038/s41573-020-0084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mok BY, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583:631–637. doi: 10.1038/s41586-020-2477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anzalone AV, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saito M, et al. Dual modes of CRISPR-associated transposon homing. Cell. 2021;9:2441–2453.e18. doi: 10.1016/j.cell.2021.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zetsche B, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abudayyeh OO, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Özcan A, et al. Programmable RNA targeting with the single-protein CRISPR effector Cas7-11. Nature. 2021;597:720–725. doi: 10.1038/s41586-021-03886-5. [DOI] [PubMed] [Google Scholar]
- 93.Cox DBT, et al. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–1027. doi: 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abudayyeh OO, et al. A cytosine deaminase for programmable single-base RNA editing. Science. 2019;365:382–386. doi: 10.1126/science.aax7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abbott TR, et al. Development of CRISPR as an antiviral strategy to combat SARS-CoV-2 and influenza. Cell. 2020;181:865–876.e812. doi: 10.1016/j.cell.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blanchard EL, et al. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat. Biotechnol. 2021;39:717–726. doi: 10.1038/s41587-021-00822-w. [DOI] [PubMed] [Google Scholar]
- 97.Miller JB, et al. Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Edn Engl. 2017;56:1059–1063. doi: 10.1002/anie.201610209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang C, et al. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017;27:440–443. doi: 10.1038/cr.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sago CD, et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl Acad. Sci. USA. 2018;115:E9944–E9952. doi: 10.1073/pnas.1811276115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yin H, et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017;35:1179–1187. doi: 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cheng Q, et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020;15:313–320. doi: 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rosenblum D, et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020;6:eabc9450. doi: 10.1126/sciadv.abc9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang X, et al. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci. Adv. 2020;6:eabc2315. doi: 10.1126/sciadv.abc2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qiu M, et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl Acad. Sci. USA. 2021;118:e2020401118. doi: 10.1073/pnas.2020401118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yin H, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee B, et al. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat. Biomed. Eng. 2018;2:497–507. doi: 10.1038/s41551-018-0252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee K, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017;1:889–901. doi: 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wei T, Cheng Q, Min Y-L, Olson EN, Siegwart DJ. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020;11:3232. doi: 10.1038/s41467-020-17029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pausch P, et al. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science. 2020;369:333–337. doi: 10.1126/science.abb1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim DY, et al. Efficient CRISPR editing with a hypercompact Cas12f1 and engineered guide RNAs delivered by adeno-associated virus. Nat. Biotechnol. 2021 doi: 10.1038/s41587-021-01009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu X, et al. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol. Cell. 2021;81:4333–4345.e4. doi: 10.1016/j.molcel.2021.08.008. [DOI] [PubMed] [Google Scholar]
- 112.Kannan S, et al. Compact RNA editors with small Cas13 proteins. Nat. Biotechnol. 2021 doi: 10.1038/s41587-021-01030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cheng CJ, Tietjen GT, Saucier-Sawyer JK, Saltzman WM. A holistic approach to targeting disease with polymeric nanoparticles. Nat. Rev. Drug Discov. 2015;14:239–247. doi: 10.1038/nrd4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Israelachvili JN, Mitchell DJ, Ninham BW. Theory of self-assembly of lipid bilayers and vesicles. Biochim. Biophys. Acta. 1977;470:185–201. doi: 10.1016/0005-2736(77)90099-2. [DOI] [PubMed] [Google Scholar]
- 115.Kulkarni JA, et al. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano. 2018;12:4787–4795. doi: 10.1021/acsnano.8b01516. [DOI] [PubMed] [Google Scholar]
- 116.Herrera M, Kim J, Eygeris Y, Jozic A, Sahay G. Illuminating endosomal escape of polymorphic lipid nanoparticles that boost mRNA delivery. Biomater. Sci. 2021;9:4289–4300. doi: 10.1039/D0BM01947J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Semple SC, et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 118.Altınoglu S, Wang M, Xu Q. Combinatorial library strategies for synthesis of cationic lipid-like nanoparticles and their potential medical applications. Nanomedicine. 2015;10:643–657. doi: 10.2217/nnm.14.192. [DOI] [PubMed] [Google Scholar]
- 119.Zhang Y, Sun C, Wang C, Jankovic KE, Dong Y. Lipids and lipid derivatives for RNA delivery. Chem. Rev. 2021;121:12181–12277. doi: 10.1021/acs.chemrev.1c00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Love KT, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl Acad. Sci. USA. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zimmermann TS, et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 122.Dong Y, et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl Acad. Sci. USA. 2014;111:3955–3960. doi: 10.1073/pnas.1322937111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jayaraman M, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Edn Engl. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Paunovska K, et al. Nanoparticles containing oxidized cholesterol deliver mRNA to the liver microenvironment at clinically relevant doses. Adv. Mater. 2019;31:1807748. doi: 10.1002/adma.201807748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kauffman KJ, et al. Rapid, single-cell analysis and discovery of vectored mRNA transfection in vivo with a loxP-flanked tdtomato reporter mouse. molecular therapy. Nucleic Acids. 2018;10:55–63. doi: 10.1016/j.omtn.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kauffman KJ, et al. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 2015;15:7300–7306. doi: 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- 127.Sedic M, et al. Safety evaluation of lipid nanoparticle-formulated modified mRNA in the Sprague–Dawley rat and cynomolgus monkey. Vet. Pathol. 2018;55:341–354. doi: 10.1177/0300985817738095. [DOI] [PubMed] [Google Scholar]
- 128.ModernaTx. Compounds and compositions for intracellular delivery of therapeutic agents. US patent US20170210697A1 (2021).
- 129.Sabatine MS, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 130.Beam Therapeutics announces updated preclinical data highlighting optimized LNP delivery approaches for in vivo base editing to the liver and other tissues. Beam Therapeuticshttps://investors.beamtx.com/news-releases/news-release-details/beam-therapeutics-announces-updated-preclinical-data (2021).
- 131.Kulkarni JA, Cullis PR, van der Meel R. Lipid nanoparticles enabling gene therapies: from concepts to clinical utility. Nucleic Acid. Ther. 2018;28:146–157. doi: 10.1089/nat.2018.0721. [DOI] [PubMed] [Google Scholar]
- 132.Cheng X, Lee RJ. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016;99:129–137. doi: 10.1016/j.addr.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 133.Dahlman JE, et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat. Nano. 2014;9:648–655. doi: 10.1038/nnano.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Khan OF, et al. Endothelial siRNA delivery in nonhuman primates using ionizable low-molecular weight polymeric nanoparticles. Sci. Adv. 2018;4:eaar8409. doi: 10.1126/sciadv.aar8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sago CD, et al. Nanoparticles that deliver RNA to bone marrow identified by in vivo directed evolution. J. Am. Chem. Soc. 2018;140:17095–17105. doi: 10.1021/jacs.8b08976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Paunovska K, et al. Analyzing 2000 in vivo drug delivery data points reveals cholesterol structure impacts nanoparticle delivery. ACS Nano. 2018;12:8341–8349. doi: 10.1021/acsnano.8b03640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lokugamage MP, et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat. Biomed. Eng. 2021;5:1059–1068. doi: 10.1038/s41551-021-00786-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mui BL, et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol. Ther. Nucleic acids. 2013;2:e139. doi: 10.1038/mtna.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ryals RC, et al. The effects of PEGylation on LNP based mRNA delivery to the eye. PLoS ONE. 2020;15:e0241006. doi: 10.1371/journal.pone.0241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016;99:28–51. doi: 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Eygeris Y, Patel S, Jozic A, Sahay G. Deconvoluting lipid nanoparticle structure for messenger RNA delivery. Nano Lett. 2020;20:4543–4549. doi: 10.1021/acs.nanolett.0c01386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kranz LM, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 143.Intellia Therapeutics presents preclinical proof of concept for CRISPR-based in vivo editing of bone marrow at Keystone eSymposium. Intellia Therapeuticshttps://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-presents-preclinical-proof-concept-crispr (2021).
- 144.Rai R, Alwani S, Badea I. Polymeric nanoparticles in gene therapy: new avenues of design and optimization for delivery applications. Polymers. 2019;11:745. doi: 10.3390/polym11040745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem. Rev. 2016;116:2602–2663. doi: 10.1021/acs.chemrev.5b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Crucho CIC, Barros MT. Polymeric nanoparticles: A study on the preparation variables and characterization methods. Mater. Sci. Eng. C. 2017;80:771–784. doi: 10.1016/j.msec.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 147.Zhong H, Chan G, Hu Y, Hu H, Ouyang D. A comprehensive map of FDA-approved pharmaceutical products. Pharmaceutics. 2018;10:263. doi: 10.3390/pharmaceutics10040263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xiao B, et al. Combination therapy for ulcerative colitis: orally targeted nanoparticles prevent mucosal damage and relieve inflammation. Theranostics. 2016;6:2250–2266. doi: 10.7150/thno.15710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Harada-Shiba M, et al. Polyion complex micelles as vectors in gene therapy — pharmacokinetics and in vivo gene transfer. Gene Ther. 2002;9:407–414. doi: 10.1038/sj.gt.3301665. [DOI] [PubMed] [Google Scholar]
- 150.Ewe A, et al. Optimized polyethylenimine (PEI)-based nanoparticles for siRNA delivery, analyzed in vitro and in an ex vivo tumor tissue slice culture model. Drug Deliv. Transl. Res. 2017;7:206–216. doi: 10.1007/s13346-016-0306-y. [DOI] [PubMed] [Google Scholar]
- 151.Gao X, et al. The association of autophagy with polyethylenimine-induced cytotoxicity in nephritic and hepatic cell lines. Biomaterials. 2011;32:8613–8625. doi: 10.1016/j.biomaterials.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 152.Breunig M, Lungwitz U, Liebl R, Goepferich A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc. Natl Acad. Sci. USA. 2007;104:14454–14459. doi: 10.1073/pnas.0703882104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ke X, et al. Surface-functionalized PEGylated nanoparticles deliver messenger RNA to pulmonary immune cells. ACS Appl. Mater. Interf. 2020;12:35835–35844. doi: 10.1021/acsami.0c08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tan L, et al. Optimization of an mRNA vaccine assisted with cyclodextrin–polyethyleneimine conjugates. Drug. Deliv. Transl. Res. 2020;10:678–689. doi: 10.1007/s13346-020-00725-4. [DOI] [PubMed] [Google Scholar]
- 155.Xiang JJ, et al. IONP-PLL: a novel non-viral vector for efficient gene delivery. J. Gene Med. 2003;5:803–817. doi: 10.1002/jgm.419. [DOI] [PubMed] [Google Scholar]
- 156.Yin H, et al. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 157.Choi J, et al. Nonviral polymeric nanoparticles for gene therapy in pediatric CNS malignancies. Nanomedicine. 2020;23:102115. doi: 10.1016/j.nano.2019.102115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Akinc A, Lynn DM, Anderson DG, Langer R. Parallel synthesis and biophysical characterization of a degradable polymer library for gene delivery. J. Am. Chem. Soc. 2003;125:5316–5323. doi: 10.1021/ja034429c. [DOI] [PubMed] [Google Scholar]
- 159.Green JJ, Langer R, Anderson DG. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc. Chem. Res. 2008;41:749–759. doi: 10.1021/ar7002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vandenbroucke RE, et al. Prolonged gene silencing in hepatoma cells and primary hepatocytes after small interfering RNA delivery with biodegradable poly(beta-amino esters) J. Gene Med. 2008;10:783–794. doi: 10.1002/jgm.1202. [DOI] [PubMed] [Google Scholar]
- 161.Anderson DG, Lynn DM, Langer R. Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angew. Chem. Int. Edn Engl. 2003;42:3153–3158. doi: 10.1002/anie.200351244. [DOI] [PubMed] [Google Scholar]
- 162.Anderson DG, Akinc A, Hossain N, Langer R. Structure/property studies of polymeric gene delivery using a library of poly(beta-amino esters) Mol. Ther. 2005;11:426–434. doi: 10.1016/j.ymthe.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 163.Mastorakos P, et al. Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene therapy. Proc. Natl Acad. Sci. USA. 2015;112:8720–8725. doi: 10.1073/pnas.1502281112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Su X, Fricke J, Kavanagh DG, Irvine DJ. In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol. Pharm. 2011;8:774–787. doi: 10.1021/mp100390w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kozielski KL, et al. Cancer-selective nanoparticles for combinatorial siRNA delivery to primary human GBM in vitro and in vivo. Biomaterials. 2019;209:79–87. doi: 10.1016/j.biomaterials.2019.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Eltoukhy AA, Chen D, Alabi CA, Langer R, Anderson DG. Degradable terpolymers with alkyl side chains demonstrate enhanced gene delivery potency and nanoparticle stability. Adv. Mater. 2013;25:1487–1493. doi: 10.1002/adma.201204346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kaczmarek JC, et al. Polymer–lipid nanoparticles for systemic delivery of mRNA to the lungs. Angew. Chem. Int. Edn Engl. 2016;55:13808–13812. doi: 10.1002/anie.201608450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Xu L, Zhang H, Wu Y. Dendrimer advances for the central nervous system delivery of therapeutics. ACS Chem. Neurosci. 2014;5:2–13. doi: 10.1021/cn400182z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chahal JS, et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl Acad. Sci. USA. 2016;113:E4133–E4142. doi: 10.1073/pnas.1600299113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Khan OF, et al. Ionizable amphiphilic dendrimer-based nanomaterials with alkyl-chain-substituted amines for tunable siRNA delivery to the liver endothelium in vivo. Angew. Chem. Int. Edn Engl. 2014;53:14397–14401. doi: 10.1002/anie.201408221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bielinska AU, Kukowska-Latallo JF, Baker JR., Jr The interaction of plasmid DNA with polyamidoamine dendrimers: mechanism of complex formation and analysis of alterations induced in nuclease sensitivity and transcriptional activity of the complexed DNA. Biochim. Biophys. Acta. 1997;1353:180–190. doi: 10.1016/S0167-4781(97)00069-9. [DOI] [PubMed] [Google Scholar]
- 172.Sonawane ND, Szoka FC, Jr, Verkman AS. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J. Biol. Chem. 2003;278:44826–44831. doi: 10.1074/jbc.M308643200. [DOI] [PubMed] [Google Scholar]
- 173.Yoo J, Park C, Yi G, Lee D, Koo H. Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers. 2019;11:640. doi: 10.3390/cancers11050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nel AE, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 175.Dawson KA, Yan Y. Current understanding of biological identity at the nanoscale and future prospects. Nat. Nanotechnol. 2021;16:229–242. doi: 10.1038/s41565-021-00860-0. [DOI] [PubMed] [Google Scholar]
- 176.Schöttler S, et al. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016;11:372–377. doi: 10.1038/nnano.2015.330. [DOI] [PubMed] [Google Scholar]
- 177.Salvati A, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013;8:137–143. doi: 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- 178.Akinc A, et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019;14:1084–1087. doi: 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- 179.Akinc A, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010;18:1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Miao L, et al. Synergistic lipid compositions for albumin receptor mediated delivery of mRNA to the liver. Nat. Commun. 2020;11:2424. doi: 10.1038/s41467-020-16248-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sago CD, et al. Modifying a commonly expressed endocytic receptor retargets nanoparticles in vivo. Nano Lett. 2018;18:7590–7600. doi: 10.1021/acs.nanolett.8b03149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chen S, et al. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J. Control. Rel. 2016;235:236–244. doi: 10.1016/j.jconrel.2016.05.059. [DOI] [PubMed] [Google Scholar]
- 183.Nakamura T, et al. The effect of size and charge of lipid nanoparticles prepared by microfluidic mixing on their lymph node transitivity and distribution. Mol. Pharm. 2020;17:944–953. doi: 10.1021/acs.molpharmaceut.9b01182. [DOI] [PubMed] [Google Scholar]
- 184.Reinhard K, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367:446–453. doi: 10.1126/science.aay5967. [DOI] [PubMed] [Google Scholar]
- 185.Nair JK, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 186.Prakash TP, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42:8796–8807. doi: 10.1093/nar/gku531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Agarwal S, et al. Impact of serum proteins on the uptake and RNA interference activity of N-acetylgalactosamine-conjugated small interfering RNAs. Nucleic Acid Ther. 2021;31:309–315. doi: 10.1089/nat.2020.0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Foster DJ, et al. Advanced siRNA designs further improve in vivo performance of GalNAc-siRNA conjugates. Mol. Ther. 2018;26:708–717. doi: 10.1016/j.ymthe.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nair JK, et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017;45:10969–10977. doi: 10.1093/nar/gkx818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zanardi TA, et al. Safety, pharmacokinetic, and pharmacodynamic evaluation of a 2′-(2-methoxyethyl)-d-ribose antisense oligonucleotide-triantenarry N-acetyl-galactosamine conjugate that targets the human transmembrane protease serine 6. J. Pharmacol. Exp. Ther. 2021;377:51–63. doi: 10.1124/jpet.120.000222. [DOI] [PubMed] [Google Scholar]
- 191.Janas MM, et al. The nonclinical safety profile of GalNAc-conjugated RNAi therapeutics in subacute studies. Toxicol. Pathol. 2018;46:735–745. doi: 10.1177/0192623318792537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Biscans A, et al. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 2019;47:1082–1096. doi: 10.1093/nar/gky1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Osborn MF, et al. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 2019;47:1070–1081. doi: 10.1093/nar/gky1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nagata T, et al. Cholesterol-functionalized DNA/RNA heteroduplexes cross the blood–brain barrier and knock down genes in the rodent CNS. Nat. Biotechnol. 2021 doi: 10.1038/s41587-021-00972-x. [DOI] [PubMed] [Google Scholar]
- 195.Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2017;16:181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yoon S, Wu X, Armstrong B, Habib N, Rossi JJ. An RNA aptamer targeting the receptor tyrosine kinase PDGFRα induces anti-tumor effects through STAT3 and p53 in glioblastoma. Mol. Ther. Nucleic Acids. 2019;14:131–141. doi: 10.1016/j.omtn.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sugo T, et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Controlled Rel. 2016;237:1–13. doi: 10.1016/j.jconrel.2016.06.036. [DOI] [PubMed] [Google Scholar]
- 198.Avidity corporate presentation. Avidity Bioscienceshttps://aviditybiosciences.investorroom.com/events-and-presentations (2021).
- 199.Kedmi R, et al. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol. 2018;13:214–219. doi: 10.1038/s41565-017-0043-5. [DOI] [PubMed] [Google Scholar]
- 200.Veiga N, et al. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018;9:4493. doi: 10.1038/s41467-018-06936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dammes N, et al. Conformation-sensitive targeting of lipid nanoparticles for RNA therapeutics. Nat. Nanotechnol. 2021 doi: 10.1038/s41565-021-00928-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Li Q, et al. Engineering caveolae-targeted lipid nanoparticles to deliver mRNA to the lungs. ACS Chem. Biol. 2020;15:830–836. doi: 10.1021/acschembio.0c00003. [DOI] [PubMed] [Google Scholar]
- 203.Zhuang X, et al. mRNA vaccines encoding the HA protein of influenza A H1N1 virus delivered by cationic lipid nanoparticles induce protective immune responses in mice. Vaccines. 2020;8:123. doi: 10.3390/vaccines8010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Paunovska K, et al. A direct comparison of in vitro and in vivo nucleic acid delivery mediated by hundreds of nanoparticles reveals a weak correlation. Nano Lett. 2018;18:2148–2157. doi: 10.1021/acs.nanolett.8b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Paunovska K, Loughrey D, Sago CD, Langer R, Dahlman JE. Using large datasets to understand nanotechnology. Adv. Mater. 2019;31:e1902798. doi: 10.1002/adma.201902798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lokugamage MP, Sago CD, Dahlman JE. Testing thousands of nanoparticles in vivo using DNA barcodes. Curr. Opin. Biomed. Eng. 2018;7:1–8. doi: 10.1016/j.cobme.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yaari Z, et al. Theranostic barcoded nanoparticles for personalized cancer medicine. Nat. Commun. 2016;7:13325. doi: 10.1038/ncomms13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Dahlman JE, et al. Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proc. Natl Acad. Sci. USA. 2017;114:2060–2065. doi: 10.1073/pnas.1620874114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lokugamage MP, Sago CD, Gan Z, Krupczak BR, Dahlman JE. Constrained nanoparticles deliver siRNA and sgRNA to T cells in vivo without targeting ligands. Adv. Mater. 2019;31:e1902251. doi: 10.1002/adma.201902251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lokugamage MP, et al. Mild innate immune activation overrides efficient nanoparticle-mediated RNA delivery. Adv. Mater. 2019;32:1904905. doi: 10.1002/adma.201904905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Riley RS, et al. Ionizable lipid nanoparticles for in utero mRNA delivery. Sci. Adv. 2021;7:eaba1028. doi: 10.1126/sciadv.aba1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Havel PJ, Kievit P, Comuzzie AG, Bremer AA. Use and importance of nonhuman primates in metabolic disease research: current state of the field. ILAR J. 2017;58:251–268. doi: 10.1093/ilar/ilx031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Paunovska K, et al. Increased PIP3 activity blocks nanoparticle mRNA delivery. Sci. Adv. 2020;6:eaba5672. doi: 10.1126/sciadv.aba5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Li R, et al. Therapeutically reprogrammed nutrient signalling enhances nanoparticulate albumin bound drug uptake and efficacy in KRAS-mutant cancer. Nat. Nanotechnol. 2021;16:830–839. doi: 10.1038/s41565-021-00897-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Patel S, et al. Boosting intracellular delivery of lipid nanoparticle-encapsulated mRNA. Nano Lett. 2017;17:5711–5718. doi: 10.1021/acs.nanolett.7b02664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yin W, et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 2012;53:51–65. doi: 10.1194/jlr.M019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rampado R, Crotti S, Caliceti P, Pucciarelli S, Agostini M. Recent advances in understanding the protein corona of nanoparticles and in the formulation of “stealthy” nanomaterials. Front. Bioeng. Biotechnol. 2020;8:166. doi: 10.3389/fbioe.2020.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Delprato A, et al. Systems genetic analysis of hippocampal neuroanatomy and spatial learning in mice. Genes Brain Behav. 2015;14:591–606. doi: 10.1111/gbb.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Harrill AH, et al. A mouse diversity panel approach reveals the potential for clinical kidney injury due to DB289 not predicted by classical rodent models. Toxicol. Sci. 2012;130:416–426. doi: 10.1093/toxsci/kfs238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Church RJ, et al. A systems biology approach utilizing a mouse diversity panel identifies genetic differences influencing isoniazid-induced microvesicular steatosis. Toxicol. Sci. 2014;140:481–492. doi: 10.1093/toxsci/kfu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Leist SR, et al. Influenza H3N2 infection of the collaborative cross founder strains reveals highly divergent host responses and identifies a unique phenotype in CAST/EiJ mice. BMC Genomics. 2016;17:143. doi: 10.1186/s12864-016-2483-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Jaxpheno2 project protocol: morphometric (organ weight) survey of 11 strains of mice (2006). Mouse Phenome Database at the Jackson Laboratoryhttps://phenome.jax.org/projects/Jaxpheno2/protocol?method=organ+weights (2006).
- 223.Sugimoto, K. et al. Background data on organ weights and histopathological lesions in Cej:CD(SD)IGS rats for 4-, 13- and 26-weeks repeated-dose toxicity studies. Biological reference data on CD(SD)IGS rats. In IGS Databook 2000 79–87 (Charles River Laboratory, 2000).
- 224.Durbin, P. W., Jeung, N., Williams, M. H., Kullgren, B. & Parrott, M. W. Weights of bones and tissues at maturity and growth of the skeleton of rhesus (Macaca mullata and cynomolgus (Macaca fascicularis) monkeys. escholarshiphttps://escholarship.org/content/qt6kw7682s/qt6kw7682s.pdf (1996).
- 225.Molina DK, DiMaio VJ. Normal organ weights in men. Part II — the brain, lungs, liver, spleen, and kidneys. Am. J. Forensic Med. Pathol. 2012;33:368–372. doi: 10.1097/PAF.0b013e31823d29ad. [DOI] [PubMed] [Google Scholar]
- 226.Molina DK, DiMaio VJ. Normal organ weights in women. Part II — the brain, lungs, liver, spleen, and kidneys. Am. J. Forensic Med. Pathol. 2015;36:182–187. doi: 10.1097/PAF.0000000000000175. [DOI] [PubMed] [Google Scholar]
- 227.Molina DK, DiMaio VJ. Normal organ weights in women. Part I — the heart. Am. J. Forensic Med. Pathol. 2015;36:176–181. doi: 10.1097/PAF.0000000000000174. [DOI] [PubMed] [Google Scholar]
- 228.Molina DK, DiMaio VJ. Normal organ weights in men. Part I — the heart. Am. J. Forensic Med. Pathol. 2012;33:362–367. doi: 10.1097/PAF.0b013e31823d298b. [DOI] [PubMed] [Google Scholar]
- 229.Hatit MZC, et al. Species-dependent in vivo mRNA delivery and cellular responses to nanoparticles. Nat. Nanotechnol. 2021 doi: 10.1038/s41565-021-01030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhang X, Goel V, Robbie GJ. Pharmacokinetics of patisiran, the first approved RNA interference therapy in patients with hereditary transthyretin-mediated amyloidosis. J. Clin. Pharmacol. 2019;60:573–585. doi: 10.1002/jcph.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zhang X, et al. Patisiran pharmacokinetics, pharmacodynamics, and exposure-response analyses in the phase 3 APOLLO trial in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. J. Clin. Pharmacol. 2020;60:37–49. doi: 10.1002/jcph.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Center for Drug Evaluation and Research application number: 210922Orig1s000. FDAhttps://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922Orig1s000MultiR.pdf (2018).
- 233.Maier MA, et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 2013;21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gilleron J, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013;31:638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 236.Wittrup A, et al. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 2015;33:870–876. doi: 10.1038/nbt.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Alberer M, et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet. 2017;390:1511–1520. doi: 10.1016/S0140-6736(17)31665-3. [DOI] [PubMed] [Google Scholar]
- 238.Zhao P, et al. Long-term storage of lipid-like nanoparticles for mRNA delivery. Bioact. Mater. 2020;5:358–363. doi: 10.1016/j.bioactmat.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Gerhardt A, et al. A thermostable, flexible RNA vaccine delivery platform for pandemic response. bioRxiv. 2021 doi: 10.1101/2021.02.01.429283y. [DOI] [Google Scholar]
- 240.Besin G, et al. Accelerated blood clearance of lipid nanoparticles entails a biphasic humoral response of B-1 followed by B-2 lymphocytes to distinct antigenic moieties. Immunohorizons. 2019;3:282–293. doi: 10.4049/immunohorizons.1900029. [DOI] [PubMed] [Google Scholar]
- 241.Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J. Blood Med. 2018;9:135–140. doi: 10.2147/JBM.S159297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Habtemariam BA, et al. Single-dose pharmacokinetics and pharmacodynamics of transthyretin targeting N-acetylgalactosamine–small interfering ribonucleic acid conjugate, vutrisiran, in healthy subjects. Clin. Pharmacol. Ther. 2021;109:372–382. doi: 10.1002/cpt.1974. [DOI] [PubMed] [Google Scholar]
- 243.Wang Y, Yu RZ, Henry S, Geary RS. Pharmacokinetics and clinical pharmacology considerations of GalNAc(3)-conjugated antisense oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2019;15:475–485. doi: 10.1080/17425255.2019.1621838. [DOI] [PubMed] [Google Scholar]
- 244.Ferguson CM, Echeverria D, Hassler M, Ly S, Khvorova A. Cell type impacts accessibility of mRNA to silencing by RNA interference. Mol. Ther. Nucleic Acids. 2020;21:384–393. doi: 10.1016/j.omtn.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Segel M, et al. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science. 2021;373:882. doi: 10.1126/science.abg6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021;16:748–759. doi: 10.1038/s41565-021-00931-2. [DOI] [PubMed] [Google Scholar]
- 247.Dhuri K, et al. Antisense oligonucleotides: an emerging area in drug discovery and development. J. Clin. Med. 2020;9:2004. doi: 10.3390/jcm9062004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Hung G, et al. Characterization of target mRNA reduction through in situ RNA hybridization in multiple organ systems following systemic antisense treatment in animals. Nucleic Acid. Ther. 2013;23:369–378. doi: 10.1089/nat.2013.0443. [DOI] [PubMed] [Google Scholar]
- 249.CureVac provides update on phase 2b/3 trial of first-generation COVID-19 vaccine candidate, CVnCoV. CureVachttps://www.curevac.com/en/2021/06/16/curevac-provides-update-on-phase-2b-3-trial-of-first-generation-covid-19-vaccine-candidate-cvncov/ (2021).
- 250.McKenzie LK, El-Khoury R, Thorpe JD, Damha MJ, Hollenstein M. Recent progress in non-native nucleic acid modifications. Chem. Soc. Rev. 2021;50:5126–5164. doi: 10.1039/D0CS01430C. [DOI] [PubMed] [Google Scholar]
- 251.Orlandini von Niessen AG, et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3′ UTRs identified by cellular library screening. Mol. Ther. 2019;27:824–836. doi: 10.1016/j.ymthe.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Jain R, et al. MicroRNAs enable mRNA therapeutics to selectively program cancer cells to self-destruct. Nucleic Acid Ther. 2018;28:285–296. doi: 10.1089/nat.2018.0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hrkach J, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci. Transl. Med. 2012;4:128ra139. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 254.Chen Z, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–617. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kojima N, Turner I, Klausner JD. The Covid-19 vaccine-development multiverse. N. Engl. J. Med. 2021;384:681–682. doi: 10.1056/NEJMc2034838. [DOI] [PubMed] [Google Scholar]
- 256.Levine-Tiefenbrun M, et al. Initial report of decreased SARS-CoV-2 viral load after inoculation with the BNT162b2 vaccine. Nat. Med. 2021;27:790–792. doi: 10.1038/s41591-021-01316-7. [DOI] [PubMed] [Google Scholar]
- 257.Dagan N, et al. BNT162b2 mRNA Covid-19 vaccine in a nationwide mass vaccination setting. N. Engl. J. Med. 2021;384:1412–1423. doi: 10.1056/NEJMoa2101765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lindsay KE, et al. Visualization of early events in mRNA vaccine delivery in non-human primates via PET-CT and near-infrared imaging. Nat. Biomed. Eng. 2019;3:371–380. doi: 10.1038/s41551-019-0378-3. [DOI] [PubMed] [Google Scholar]
- 259.Pardi N, et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018;215:1571–1588. doi: 10.1084/jem.20171450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Laczkó D, et al. A single immunization with nucleoside-modified mRNA vaccines elicits strong cellular and humoral immune responses against SARS-CoV-2 in mice. Immunity. 2020;53:724–732.e727. doi: 10.1016/j.immuni.2020.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Wu K, et al. Serum neutralizing activity elicited by mRNA-1273 vaccine. N. Engl. J. Med. 2021 doi: 10.1056/NEJMc2102179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Callaway E, Ledford H. How to redesign COVID vaccines so they protect against variants. Nature. 2021;590:15–16. doi: 10.1038/d41586-021-00241-6. [DOI] [PubMed] [Google Scholar]
- 263.Hacisuleyman E, et al. Vaccine breakthrough infections with SARS-CoV-2 variants. N. Engl. J. Med. 2021 doi: 10.1056/NEJMoa2105000. [DOI] [PubMed] [Google Scholar]
- 264.Han BA, Kramer AM, Drake JM. Global patterns of zoonotic disease in mammals. Trends Parasitol. 2016;32:565–577. doi: 10.1016/j.pt.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]