

Abstract
Intronic GGGGCC (G4C2) hexanucleotide repeat expansion within the human C9orf72 gene represents the most common cause of familial forms of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (C9ALS/FTD). Repeat‐associated non‐AUG (RAN) translation of repeat‐containing C9orf72 RNA results in the production of neurotoxic dipeptide‐repeat proteins (DPRs). Here, we developed a high‐throughput drug screen for the identification of positive and negative modulators of DPR levels. We found that HSP90 inhibitor geldanamycin and aldosterone antagonist spironolactone reduced DPR levels by promoting protein degradation via the proteasome and autophagy pathways respectively. Surprisingly, cAMP‐elevating compounds boosting protein kinase A (PKA) activity increased DPR levels. Inhibition of PKA activity, by both pharmacological and genetic approaches, reduced DPR levels in cells and rescued pathological phenotypes in a Drosophila model of C9ALS/FTD. Moreover, knockdown of PKA‐catalytic subunits correlated with reduced translation efficiency of DPRs, while the PKA inhibitor H89 reduced endogenous DPR levels in C9ALS/FTD patient‐derived iPSC motor neurons. Together, our results suggest new and druggable pathways modulating DPR levels in C9ALS/FTD.
Keywords: C9ALS/FTD, C9orf72, DPR, PKA, protein clearance
Subject Categories: Molecular Biology of Disease, Neuroscience
Screening small‐molecule agonists for the ability to modulate accumulation of toxic polypeptides suggests PKA signaling, proteasomal and autophagy pathways as potential therapeutic targets for C9orf72‐associated ALS/FTD.

Introduction
Repeat‐associated non‐AUG (RAN) translation is an unconventional translation mechanism associated with several nucleotide‐repeat expansion disorders. The hexanucleotide repeat expansion GGGGCC n , also known as (G4C2) n , is localized in the first intron of the C9orf72 gene (DeJesus‐Hernandez et al, 2011; Renton et al, 2011) and it is the most common genetic cause of familial forms of ALS and FTD (hereafter C9ALS/FTD) (Gijselinck et al, 2012). The pathogenic mechanisms proposed for C9ALS/FTD suggest that sense (G4C2) n ‐ and antisense (C4G2) n ‐containing transcripts cause two different mechanisms of toxicity. The first is mediated by the formation of RNA foci that bind and sequester RNA‐binding proteins, thereby leading to impairment of RNA metabolism (Donnelly et al, 2013; Lee et al, 2013; Mori et al, 2013b; Xu et al, 2013; Cooper‐Knock et al, 2014; Haeusler et al, 2014; Wen et al, 2014; Zhang et al, 2015; Conlon et al, 2016; Swinnen et al, 2018); and the second mediated by their unconventional RAN translation into five different dipeptide‐repeat proteins (DPRs: poly‐GA, poly‐GP, poly‐GR, poly‐PA, poly‐PR) (Ash et al, 2013; Mori et al, 2013a, 2013c). In addition, pathological expansions of (G4C2)n reduce C9orf72 transcription and translation with decreased C9orf72 protein levels (DeJesus‐Hernandez et al, 2011; Renton et al, 2011); this latter event can also be associated with endosomal trafficking, autophagy dysfunction, which synergizes with repeat‐associated toxicity (Shi et al, 2018; Boivin et al, 2020; Zhu et al, 2020). DPR‐induced toxicity has been shown in several cell lines, in iPSC‐derived neurons (May et al, 2014; Su et al, 2014; Yamakawa et al, 2015; Yang et al, 2015a; Westergard et al, 2019), in Drosophila (Mizielinska et al, 2014; Wen et al, 2014; Chew et al, 2015; Freibaum et al, 2015; Yang et al, 2015a; Boeynaems et al, 2016) and in mouse models (Zhang et al, 2016, Zhang et al, 2018, Zhang et al, 2019; Schludi et al, 2017; Choi et al, 2019; Hao et al, 2019). Multiple studies demonstrated proteasome dysfunction due to the sequestration of proteasomal proteins by poly‐GA in both in vitro (May et al, 2014; Yamakawa et al, 2015) and in vivo models (Zhang et al, 2016). RAN translation of (G4C2) n ‐RNAs has been recently shown to require a near‐cognate start codon upstream of the repeat in the +1 frame (Green et al, 2017; Tabet et al, 2018) and to be triggered by stress conditions in a cap‐dependent (Kearse et al, 2016; Green et al, 2017; Tab et et al, 2018) or cap‐independent way (Cheng et al, 2018; Sonobe et al, 2018). However, the mechanisms regulating RAN translation have not yet been completely elucidated.
Antisense oligonucleotides (ASOs) (Jiang et al, 2016; Gendron et al, 2017) and small molecules targeting the (G4C2)n (Su et al, 2014; Simone et al, 2018; Wang et al, 2019) and/or r(CGG)n RNAs (Yang et al, 2015b, 2016; Green et al, 2019) have been proposed as possible therapeutic approaches, but no clinically approved drugs are known to selectively modulate RAN translation. The small molecules available at present alter RNA secondary structures providing a proof of principle as to how their binding to (G4C2)n can inhibit both RNA foci formation and RAN translation. A genetic screen recently identified the ribosomal protein RPS25 as a regulator of RAN translation of different repeat sequence expansions (Yamada et al, 2019). Other modifiers of DPR production were identified by two independent genome‐wide CRISPR‐Cas9 screens (Kramer et al, 2018; Cheng et al, 2019; Wilson et al, 2019). In addition, the RNA helicase DHX36 has been shown to favour (G4C2)n and FMR1‐associated RAN translation (Tseng et al, 2021), whereas the RNA helicase DDX3X inhibits (G4C2)n RAN translation (Cheng et al, 2019) but it promotes FMR1‐associated RAN translation (Linsalata et al, 2019).
Here, we used a chemical genomic approach to identify small molecules and relative molecular targets. These small molecules modulate DPR levels by either increasing protein clearance or inhibiting translation of (G4C2)n‐containing RNAs. Among these small molecules, we found that Geldanamycin (GELD, an inhibitor of Heat Shock Protein 90, HSP90) increases proteasome activity and that Spironolactone (SPL, an aldosterone antagonist) modulates DPR autophagy degradation. Moreover, we found for the first time that cAMP‐elevating compounds increase DPR levels by boosting protein kinase A (PKA) activity, while PKA silencing, or inhibition reverted these effects. This suggests a novel mechanism in which PKA is involved in pathways that aberrantly enhance the translation of C9orf72 (G4C2)n mRNA to neurotoxic DPRs.
Results
Development of a HTS assay for identifying modulators of C9‐DPR levels
We set up a cell‐based high‐throughput screen (HTS) (full HTS protocol in Appendix Materials and Methods), to find small molecules capable of modulating DPR levels. In the HTS we used an artificial reporter containing 58 G4C2 repeats outside of the native C9orf72 sequence, and with the GFP sequence in the GP frame (Freibaum et al, 2015) (hereafter polyGP‐GFP) (Appendix Tables S1 and S2). We obtained a consistent signal of polyGP‐GFP across experimental repeats using a reverse transfection approach in HEK293T cells (Fig EV1A and B). In the HTS we also co‐transfected an AUG‐RFP plasmid (Fig 1A) to report on AUG‐mediated translation. In the screen, Cycloheximide (CHX) was used to model general translation inhibition. We used the variation in the number of GFP‐ or RFP‐positive cells as the read‐out of the assay. Due to the lack of a positive control, small molecule RAN translation inhibitor, the variability and the robustness of the assay were optimized to perform a HTS based on the effect of CHX on RFP‐expressing cells (Z′‐factor = 0.5) (Zhang et al, 1999) (Fig EV1C). We observed that CHX did not decrease the fluorescent intensity or the number of cells expressing polyGP‐GFP (Fig EV1D), consistent with a recent report showing non‐AUG translation to be resistant to elongation inhibitors (Kearse et al, 2019).
Figure EV1. Set up of a High‐Throughput Screen (HTS) for identifying modulators of DPR levels.
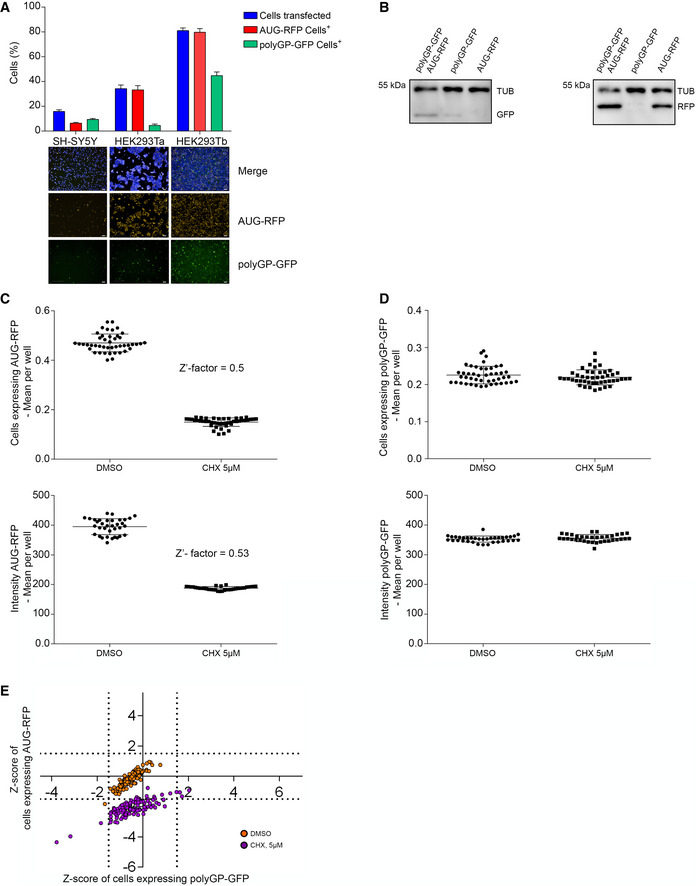
- SH‐SY5Y and HEK293T cells were co‐transfected with AUG‐RFP and polyGP‐GFP plasmids by standard (SH‐SY5Y and HEK293Ta) or reverse (HEK293Tb) transfection method. Images were acquired 24 h after using Operetta High‐Content Imaging System and transfection efficiency were calculated on the ratio of cells expressing RFP/tot and cells expressing GFP/tot. Data are mean ± SD from three biological replicates. Images in HEK293Tb are from DMSO in Fig 2A. Scale bars 200 μm.
- Immunoblot for testing the level of polyGP‐GFP and AUG‐RFP in lysates from HEK293T cells co‐transfected with both plasmids or with polyGP‐GFP or AUG‐RFP and mock.
- Distribution of cells expressing AUG‐RFP (upper inset) or AUG‐RFP fluorescent intensity (lower inset) in negative (DMSO) and positive (CHX, 5 μM) controls. Data used to calculate Z′‐factor. Data are mean ± SD from 45 (upper inset) and 35 (lower inset) technical replicates.
- Distribution of cells expressing polyGP‐GFP (upper inset) and polyGP‐GFP fluorescent intensity (lower inset) in negative (DMSO) and positive (CHX, 5 μM) controls. Data are mean ± SD from 45 (upper inset) and 35 (lower inset) technical replicates.
- HTS. HEK293T cells were co‐transfected with the constructs shown in Fig 1A. Scatter plot shows the distribution of negative (DMSO, orange dots) and positive (CHX 5 μM, purple dots) controls added 3 h after reverse co‐transfection. On the Y‐axis reported the Z‐score values of cells expressing AUG‐RFP and on the X‐axis the Z‐score values of cells expressing polyGP‐GFP. Grid lines represent the thresholds arbitrarily set up around the DMSO distribution (polyGP‐GFP ± 1.5 on the X‐axis and ± 1.5 for AUG‐RFP on the Y‐axis).
Source data are available online for this figure.
Figure 1. Primary and confirmatory screening for identifying modulators of C9orf72‐derived DPR levels.
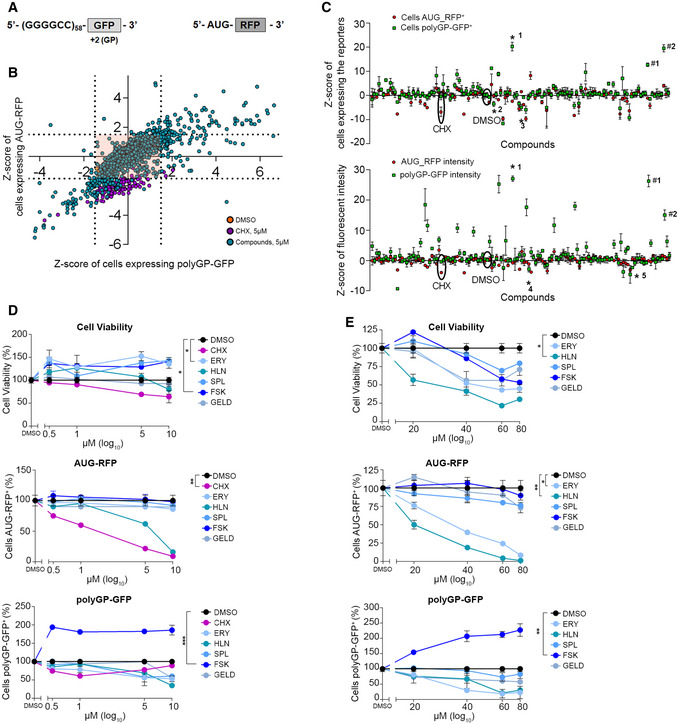
-
ASchematic representation of the constructs utilized in (B) and (C). The first construct contains 58 (G4C2) repeats outside of the native C9orf72 sequence and GFP in the GP frame (polyGP‐GFP). The start codon of GFP was removed. The second construct AUG‐RFP is used as a positive control of canonical translation.
-
BHTS. HEK293T cells were co‐transfected with the constructs in (A) and, negative (DMSO, orange dots) and positive (CHX 5 µM, purple dots) controls added 3 h after reverse co‐transfection with compound‐libraries (5 µM). Images and data acquisition collected after about 30 h of treatment. Scatter plot shows the distribution of compounds. On the Y‐axis reported the Z‐score values of cells expressing AUG‐RFP and on the X‐axis the Z‐score values of cells expressing polyGP‐GFP. Grid lines represent the thresholds arbitrarily set up around DMSO distribution (polyGP‐GFP ± 1.5 on the X‐axis and ± 1.5 for AUG‐RFP on the Y‐axis) to select compounds for the confirmatory screening and eliminate the ones without effect (orange square).
-
CConfirmatory screening performed as described above. Schematic distribution of compounds based on Z‐score values of cells expressing polyGP‐GFP and AUG‐RFP (above) and on Z‐score values of the fluorescent intensity of the two reporters per each compound (below). Data are from four technical replicates, boxes are the Z‐score mean value and whiskers represent ± SD. Baseline of Z‐score = 0 indicates that Z‐score is identical to the mean score. Asterisk (*) represents the selected compounds: 1 Forskolin (FSK), 2 Erysolin (ERY), 3 Geldanamycin (GELD), 4 Helenin (HLN) and 5 Spironolactone (SPL). Number sign (#) represents cellular stress inducers: #1 Thapsigargin and #2 Tunicamycin.
-
D, EDose‐response analysis of ERY, HLN, SPL, FSK and GELD. Cells were co‐transfected with AUG‐RFP and polyGP‐GFP plasmids and treated with two concentration ranges 0.5, 1, 5 and 10 µM (D) and 20, 40 and 60 µM (E) for 24 h. CHX used only low dosages. Data are mean ± SD from three biological replicates. One‐way ANOVA followed by Dunnett’s multiple comparison tests: *P < 0.5; **P < 0.01; ***P < 0.001.
We screened approximately 2,500 compounds with biological activity from different chemical libraries (see Materials and Methods). The compounds were added 3 h after plasmid reverse co‐transfection. GFP and RFP reporter signals were measured approximately 30 h later. Plotting the Z‐score of the number of cells expressing AUG‐products (RFP, Y‐axis) versus the Z‐score of the number of cells expressing DPR‐products (polyGP‐GFP, X‐axis), we obtained a graphical representation of the simultaneous effect of the small molecules on both AUG and RAN translation‐dependent products (Fig 1B). The majority of tested compounds did not modify the levels of the reporters. Their signals overlapped with the distribution of negative controls (Figs 1B and EV1E), indirectly proving the assay quality. We selected effective compounds using an arbitrary threshold of ± 1.5 Z‐score for cells expressing polyGP‐GFP and ± 1.5 for AUG‐derived positive cells. These thresholds gave a significant separation of DMSO treated from CHX‐treated samples (see Appendix for full details on thresholding calculations). We excluded highly toxic compounds using a threshold Z‐score nuclei ≤ −2, indicating that < 50% of cells survived. A confirmatory screen was next performed as described above but increased the number of replicates from one to four. As expected, while only a few compounds were able to reduce levels of the RAN products, many others had the opposite effect (Fig 1C). This comes as no surprise because many RAN‐increasing compounds were cellular stressors (Thapsigargin, (Green et al, 2017; Westergard et al, 2019) and Tunicamycin (Green et al, 2017; Westergard et al, 2019)) present in the chemical library (Fig 1C). We selected three small molecules according to their capability to specifically reduce or increase the number of cells expressing polyGP‐GFP and/or the fluorescent intensity of GFP (Fig 1C, Table 1 and Dataset EV1). GELD and SPL reduced RAN products, whilst cAMP‐elevating compounds, with Forskolin (FSK) being the most potent one, increased them. Interestingly, FSK, which activates adenylyl cyclase (AC) and enhances intracellular cAMP levels, triggers a multitude of PKA‐dependent and/or ‐independent pathways resulting in pleiotropic effects on cells. These events include the activation of many intracellular signalling cascades and of the cAMP Response Elements Binding (CREB) family of transcription factors that, upon phosphorylation, regulate the expression of genes containing cAMP Response Elements (CREs) in their promoters (Seamon et al, 1981; Sapio et al, 2014; Kanne et al, 2015). We also identified two phytochemicals with undefined mechanisms of action, Erysolin (ERY) and Helenin (HLN) that effectively reduced RAN products. These results were then validated in the confirmatory screen (Dataset EV1).
Table 1.
List of the small molecules selected from the confirmatory screening.
| Small molecules | Cells expressing polyGP‐GFP | Cells expressing AUG‐RFP | polyGP‐GFP intensity | AUG‐RFP intensity | Number of cells |
|---|---|---|---|---|---|
| DMSO | −0.06 | −0.2 | 0.3 | −0.12 | −0.2 |
| Erysolin (ERY) | −3.5 | −0.2 | −1.34 | −0.7 | −1.1 |
| Forskolin (FSK) | 20.3 | −2.4 | 27 | −0.8 | 0.05 |
| Geldanamycin (GELD) | −5 | 1.6 | 0.002 | 3 | −2.6 |
| Helenin (HLN) | −1.05 | −0.7 | −2.8 | −1.1 | −1.3 |
| Spironolactone (SPL) | −0.5 | 0.7 | −2.3 | 0.6 | −0.9 |
Mean of compounds from four biological replicates. All the compounds were added after 3h of reverse co‐transfection and used at the final concentration of 5 µM for approximately 30 h. Mean of DMSO from 64 biological replicates (32 replicates per each 384‐well plate). Data are reported as Z‐score values.
The entire list of small molecules used is reported in Dataset EV1.
To obtain a confidence interval of safe utilization of the selected compounds, we then performed dose–response experiments by treating cells with two concentration ranges for each compound (Fig 1D and E), simultaneously checking their toxicity and the effects on DPR levels. All the compounds confirmed their activity in modulating the number of polyGP‐GFP‐positive cells, although to various extents. The most potent compound was FSK that selectively increased polyGP‐GFP‐positive cells compared to AUG‐RFP‐positive cells. HLN decreased both products, whereas ERY, GELD and SPL decreased more efficiently the polyGP‐GFP products than the AUG‐RFP. All these compounds were moderately toxic at concentrations higher than 40 μM, with HLN the most toxic. Therefore, we excluded HLN due to its toxicity and proceeded with the other four molecules to gain information about their molecular mechanism of action.
GELD, SPL and FSK modulate DPR levels independently of the near‐cognate CUG start codon
To understand whether the four selected DPR modulators (Fig 2A) affect general transcription and translation, we used the incorporation of the modified nucleoside 5‑ethynyl uridine (EU) to evaluate general RNA transcription. In parallel, we took advantage of O‐propargyl‐puromycin (OPP) incorporation assay to evaluate de novo protein synthesis. GELD marginally induced general transcription (Fig 2B). None of the compounds modulated translation (Fig 2C). The molecular mechanism of RAN translation initiation (Kearse et al, 2016) is still a matter of debate and a near‐cognate CUG start codon within C9orf72 first intron 1A has been suggested to play a key role in (G4C2)n RAN translation (Green et al, 2017; Tab et et al, 2018). The polyGP‐GFP reporter used in the HTS did not contain the native sequence upstream of the repeat. Therefore, to evaluate whether the effect of these compounds was CUG independent, we used the (G4C2)x66 construct (hereafter 66R) that contains repeats within the native C9orf72 sequence and a specific C‐term tag for each frame (Gendron et al, 2013). To prove the efficacy of GELD and SPL in motor neuron‐like cells, we used NSC34 cells (Appendix Fig S1A). To ensure the proper evaluation of the total amounts of DPRs produced in cells, we quantified both the soluble DPR levels and the PBS‐insoluble DPR aggregate fraction by immunoblot analysis and filter retardation assay, respectively. GELD and SPL significantly reduced the accumulation of DPRs, while FSK significantly increased poly‐GA in HEK293T (Fig 2D), poly‐GP and its PBS‐insoluble fraction in NSC34 cells (Fig 2E and F), despite the presence of the upstream CUG codon. In contrast, ERY did not show any effect in modulating DPR levels in either cell line (Fig 2D–F). We further challenged the selected compounds using C9RAN NLuc reporters (in the GA frame) with the native (CUG) or mutated (CCC) start codon (Green et al, 2017). Consistent with the results obtained with the 66R construct, GELD and SPL led to a marked reduction, while FSK increased the expression of C9RAN NLuc reporters in either condition (Fig 2G). Together, these data suggest that the effect of the compounds is not dependent on the upstream CUG codon. Moreover, the modulation of DPR levels was not dependent on different amounts of G4C2 mRNA (Appendix Fig S1B and C). Finally, we observed that GELD, SPL and FSK did not change the level of AUG‐GFP (Fig EV2A and B).
Figure 2. GELD, SPL and FSK modulate DPR levels independently of the near cognate CUG start codon.
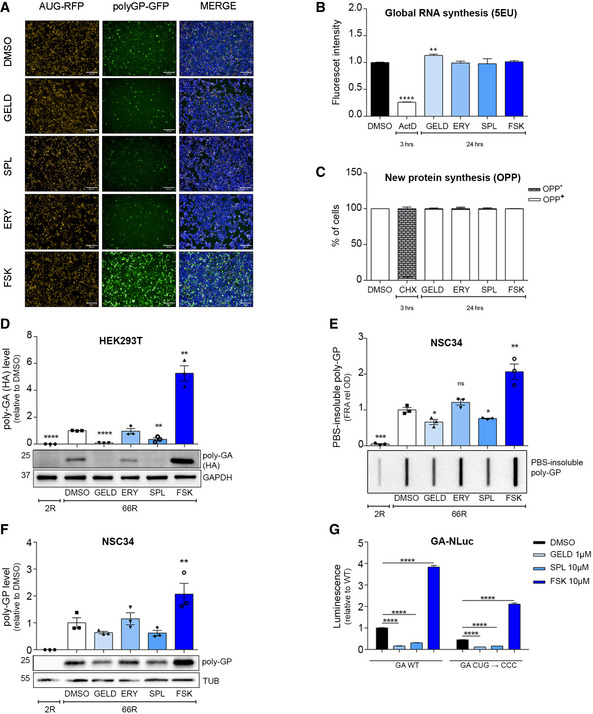
-
AImages of the HTS representing the effect of the selected small molecules (5 µM). Scale bar, 200 μm.
-
B, CHEK293T cells were treated for 24 h and general RNA and protein synthesis were monitored by the incorporation of 5‐ethynyl uridine (EU) and O‐propargyl‐puromycin (OPP), respectively. ActD (5 μM) and CHX (350 μM) were used for 3 h as positive control to block RNA and protein synthesis, respectively. Data are from three biological replicates and represented as mean ± SEM. (B) Two‐tailed, unpaired t‐test; ***P < 0.001, **P = 0.0047.
-
DLysates from HEK293T cells, transfected with (G4C2)x2 hereafter 2R or (G4C2)x66, hereafter 66R vectors and treated for 24 h, immunoblotted using antibodies for poly‐GA expression (HA tagged). GAPDH was used as a loading control. Quantification from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test; ****P < 0.0001, **P = 0.0052 for SPL versus DMSO, **P = 0.0016 for FSK versus DMSO.
-
E, FLysates from NSC34 cells, transfected with 2R or 66R vectors and treated for 24 h, immunoblotted for poly‐GP expression. α‐Tubulin was used as a loading control. (E) Quantification of PBS‐insoluble poly‐GP (FRA) from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test; ***P < 0.001, **P = 0.0087, *P = 0.0256 for GELD versus DMSO, *P = 0.0229 for SPL versus DMSO. (F) Quantification poly‐GP from three biological replicates. Data are mean ± SEM. No significant results using two‐tailed, unpaired t‐test. One‐way ANOVA followed by Uncorrected Fisher's LSD, P** = 0.0034 for FSK versus DMSO.
-
GLuminescence derived from HEK293T transfected with GA‐NLuc reporters WT or mutated and treated for 24 h. Mutational analysis of near‐cognate CUG start codon from three biological replicates and four technical replicates, n = 12. Data are mean ± SEM. Two‐way ANOVA followed by Dunnett’s multiple comparisons test, ****P < 0.0001.
Data information: Concentration used: 1 μM GELD, 10 μM ERY, SPL and FSK.
Source data are available online for this figure.
Figure EV2. Effect of the compounds and PKA‐knockdowns on AUG‐GFP levels.
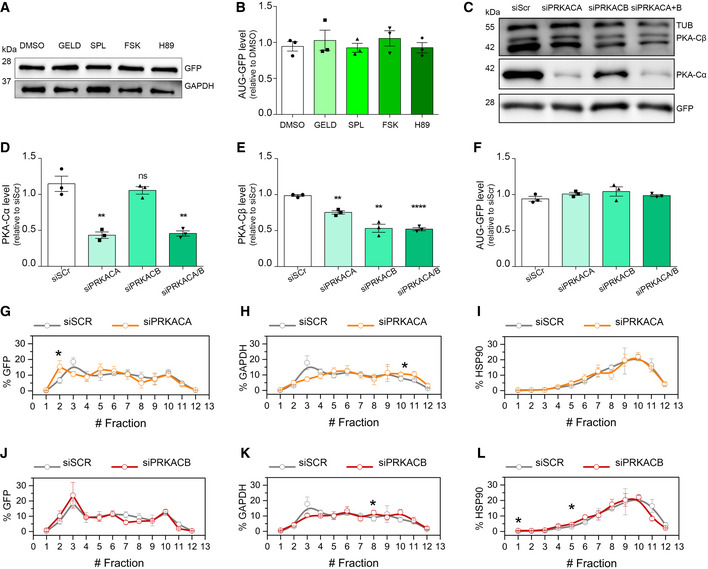
-
A, BLysates from HEK293T cells, transfected with AUG‐GFP and treated for 24 h, immunoblotted using antibodies for GFP expression and GAPDH used as a loading control. (B) Quantification of (A) from three biological replicates. Data are mean ± SEM. No significant results using two‐tailed, unpaired t‐test.
-
C–FLysates from HEK293T cells, transfected with RNAi scrambled, PRKACA, PRKACB and PRKACA/B (72 h) and with AUG‐GFP (24 h), immunoblotted using antibodies for PKA‐Cα, PKA‐Cβ and GFP expression. GAPDH used as a loading control (C). (D) Quantification of PKA‐Cα expression from three biological replicates. Two‐tailed, unpaired t‐test, **P = 0.0334 siPRKACA versus siScr, **P = 0.0336 siPRKACA/B versus siScr. (E) Quantification of PKA‐Cβ expression from three biological replicates. Two‐tailed, unpaired t‐test, **P = 0.0010 siPRKACA versus siScr, **P = 0.0014 siPRKACB versus siScr, ****P < 0.0001 siPRKACA/B versus siScr. (F) Quantification of GFP expression from three biological replicates. No significant results using two‐tailed, unpaired t‐test. Data are mean ± SEM.
-
G–ICo‐sedimentation profile of GFP (G), GAPDH (H) and HSP90 (I) mRNAs along the sucrose gradient fractions of polysomal profiles from control (grey lines) and PRKACA‐depleted (orange lines) cells. Data are mean ± SEM from three independent biological replicates. One‐tailed t‐test, *P < 0.05.
-
J–LCo‐sedimentation profile of GFP (J), GAPDH (K) and HSP90 (L) mRNAs along the sucrose fractions of polysomal profiles from control (grey lines) and PRKACB‐depleted (red lines) cells. Data are mean ± SEM from three independent biological replicates. One‐tailed t‐test, *P < 0.05.
Source data are available online for this figure.
GELD and SPL enhance the degradation of DPRs
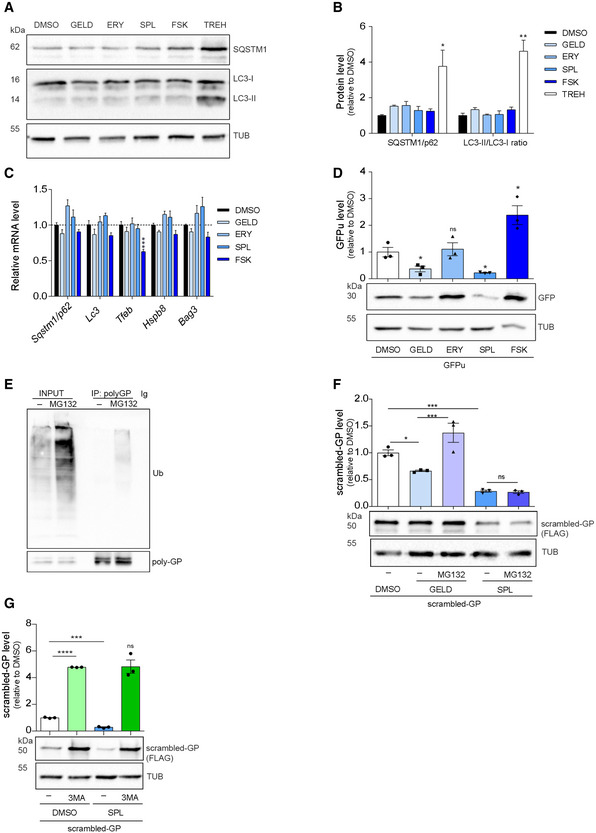
Among the possible post‐transcriptional/post‐translational mechanisms underlying the modulatory effect of selected small molecules, we investigated their activity on DPR clearance. DPR degradation mainly relies on autophagic clearance, while, under basal conditions, only poly‐GP is additionally degraded by the ubiquitin–proteasome system (UPS) (Yamakawa et al, 2015; Cristofani et al, 2017, 2018). Dysfunction in the protein quality control machinery has been demonstrated in ALS, C9ALS/FTD as well as in other neurodegenerative disorders (May et al, 2014; Zhang et al, 2014; Yamakawa et al, 2015). Therefore, we evaluated the expression level of autophagy markers (p62/SQSTM1, LC3, HSPB8, BAG3) using trehalose as a positive control (Rusmini et al, 2019). None of the compounds increased either transcriptional expression or protein levels of autophagy markers in NSC34 (Fig 3A–C) and SH‐SY5Y cells (Appendix Fig S2A). Only FSK induced a modest decrease in TFEB mRNA, an autophagy master regulator (Rusmini et al, 2019) in NSC34 cells (Fig 3C). Immunofluorescence analysis also showed that the compounds did not modify the intracellular distributions of p62/SQSTM1 and LC3 in NSC34 cells (Appendix Fig S2B and C). Moreover, we did not observe nuclear translocation of TFEB or changes in its protein levels (Appendix Fig S3A–D). These data ruled out TFEB‐mediated autophagy activation as the mechanism of action of our compounds. To investigate a possible involvement of the proteasome in DPR degradation, we transfected NSC34 and SH‐SY5Y cells with a GFP tagged with a short degron (CL1) (GFPu) that directs the reporter protein to the proteasome for clearance (Bence et al, 2001; Cristofani et al, 2017); so that proteasome impairment correlates with GFPu accumulation. GELD and SPL increased the functionality of the proteasome, ERY did not, whereas FSK led to a marked GFPu accumulation in both cell lines (Fig 3D, Appendix Fig S2D), contrary to expectations (Huang et al, 2013; Lokireddy et al, 2015). GFPu accumulation by FSK was not due to a block of the proteasome, as suggested by a CHX chase experiment (Appendix Fig S4A and B). In addition, we did not observe an increase in GFPu mRNA and ruled out promoter‐dependent effects (Appendix Fig S4C–E).
Figure 3. GELD and SPL enhance the degradation of DPRs.
-
A, BLysates from NSC34 cells treated for 24 h and immunoblotted for SQSTM1/p62 and LC3 expression. (B) Quantification from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test, **P = 0.0047, *P = 0.0393.
-
CRelative expression level of Sqstm1/p62, Lc3, Tfeb, Hspb8 and Bag3 mRNAs in NSC34 cells treated for 24 h. Data are mean ± SEM from three biological replicates. Two‐way ANOVA followed by Dunnett’s multiple comparisons test, ***P < 0.001. Dotted lines represent RNA expression in the DMSO control.
-
DLysates from NSC34 cells transfected with a proteasome activity reporter (GFPu), treated for 24 h and immunoblotted for GFP expression. Quantification from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test, *P = 0.0398 GELD versus DMSO, *P = 0.0124 SPL versus DMSO, *P = 0.0253 FSK versus DMSO.
-
ECo‐immunoprecipitation (IP) analysis on 66R‐derived poly‐GP and ubiquitin interaction. Lysates from NSC34 cells transfected with 66R and treated with 10 µM MG132 for 16 h, immunoprecipitated for poly‐GP and immunoblotted for ubiquitin and poly‐GP.
-
FLysates from NSC34 cells transfected with scrambled‐GPx100, treated for 24 h with GELD, SPL and/or 16 h with MG132 and immunoblotted for scrambled‐GP expression. Quantification from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by Uncorrected Fisher's LSD, P* = 0.0186 for GELD versus DMSO, ***P = 0.0001 GELD + MG132 versus GELD and SPL versus DMSO.
-
GLysates from NSC34 cells transfected with scrambled‐GPx100, treated for 24 h with SPL and/or 10 mM 3MA and immunoblotted for scrambled‐GP expression. Quantification from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test, ****P < 0.0001 for SPL versus DMSO and 3MA versus DMSO, ***P = 0.0008 SPL + 3MA versus SPL.
Data information: Concentration used: 1 μM GELD, 10 μM ERY, SPL, FSK.
Source data are available online for this figure.
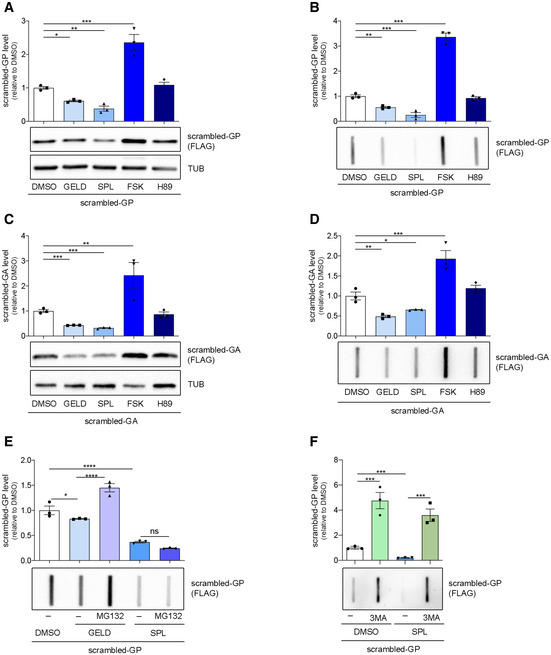
Since 66R‐derived poly‐GP was poly‐ubiquitinated (Fig 3E) we further investigated DPR levels using ATG initiated, codon‐optimized (scrambled (Lee et al, 2017)) repeats for poly‐GA and poly‐GP (Yamakawa et al, 2015) (see Appendix Tables S1 and S2). We observed that GELD and SPL reduced the soluble and PBS‐insoluble level of either scrambled‐DPR while FSK increased scrambled‐DPR levels (Fig EV3A–D). Inhibition of proteasome activity by MG132 counteracted the GELD effect on scrambled‐GP levels (Figs 3F and EV3E). SPL activity was not blocked by MG132 (Figs 3F and EV3E), while it was counteracted by the autophagy inhibitor 3‐MethylAdenine (3MA) (Figs 3G and EV3F), although SPL was not able to increase TFEB dependent autophagic flux (Appendix Fig S3E).
Figure EV3. Effects of selected compounds on scrambled‐GP and ‐GA levels.
-
A, BLysates from NSC34 cells transfected with scrambled‐GPx100 and treated for 24 h with DMSO, GELD, SPL, FSK, H89. (A) Immunoblot for poly‐GP expression. Tubulin was used as a loading control. Quantification from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by Uncorrected Fisher's LSD, *P < 0.05; **P < 0.01; ***P < 0.001. (B) Quantification of PBS‐insoluble poly‐GP (FRA). Quantification from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by Uncorrected Fisher's LSD, **P < 0.01; ***P < 0.001.
-
C, DLysates from NSC34 cells transfected with scrambled‐GAx100 and treated for 24 h with DMSO, GELD, SPL, FSK, H89. (C) Immunoblot for poly‐GA expression. Tubulin was used as a loading control. Quantification from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by Uncorrected Fisher's LSD, **P < 0.01; ***P < 0.001. (D) Quantification of PBS‐insoluble poly‐GA (FRA) from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by Uncorrected Fisher's LSD, *P < 0.05; **P < 0.01; ***P < 0.001.
-
ELysates from NSC34 cells transfected with scrambled‐GPx100, treated for 24 h with GELD, SPL and/or 16 h with 10 µM MG132. Quantification of PBS‐insoluble poly‐GP (FRA) from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by Uncorrected Fisher's LSD, *P < 0.05; ****P < 0.0001.
-
FLysates from NSC34 cells transfected with scrambled‐GPx100, treated for 24 h with SPL and/or 10 mM 3MA. Quantification of PBS‐insoluble poly‐GP (FRA) from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by Uncorrected Fisher's LSD, ***P < 0.001.
Source data are available online for this figure.
Together, these results suggest that GELD and SPL reduce DPR levels by enhancing protein degradation mediated by proteasome and autophagy, respectively, although we cannot exclude further mechanisms of action, at present. ERY did not reduce DPR levels nor trigger degradative pathways and we thus did not proceed with its analysis, considering it a false positive. As FSK did not inhibit the proteasome, we further investigated the mechanism of action elicited by this molecule.
The PKA inhibitor H89 reduces DPR levels in cell lines
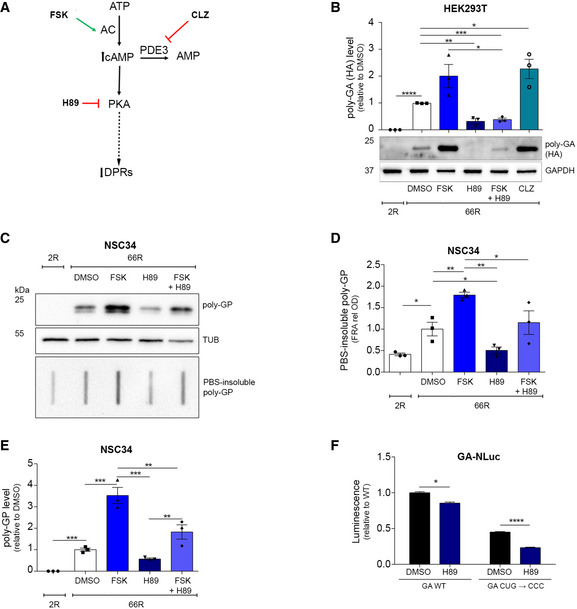
FSK is a derivative of the geranylgeranyl pyrophosphate (GGPP), characterized by the insertion of a tetrahydropyran‐derived heterocyclic ring (Fuse et al, 2004), and differs from the analogous geranylgeranyl acetone (GGA), which is a potent inducer of HSP70, HSPB8 and HSPB1 (Hoshino et al, 2013; Marunouchi et al, 2014) (modulator of the protein quality control system and autophagy). As an internal, additional validation of the screening results, we observed that other cAMP‐increasing compounds (Dibutyryl cAMP·Na, Desacetylcolforsin, Colforsin and Cilostazol) similarly increased polyGP‐GFP levels (Appendix Fig S5A). The increase in intracellular cAMP activates PKA that in turn phosphorylates numerous substrates, positively or negatively modulating their activity. These events include the activation of many intracellular signalling cascades and transcription factors including CREB that regulates the expression of genes containing the CREs (Mayr & Montminy, 2001). To prove the importance of the adenylate cyclase (AC)/PKA pathway in increasing RAN products, we re‐tested Cilostazol (CLZ), a phosphodiesterase III (PDE III) inhibitor that increases cAMP level by reducing its degradation and we tested the PKA inhibitor H89 (Fig 4A).
Figure 4. PKA inhibition decreases DPR levels.
-
ASchematic representation showing where the compounds act along the AC/PKA signalling pathway.
-
BLysates from HEK293T cells, transfected with 2R or 66R and treated for 24 h, immunoblotted using antibody for poly‐GA expression (HA–tagged). Quantification from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test, ****P < 0.0001, **P = 0.0027 H89 versus DMSO, ***P = 0.0004 FSK + H89 versus DMSO, *P = 0.0234 CLZ versus DMSO, *P = 0.0209 FSK + H89 versus FSK.
-
C–ELysates from NSC34 cells, transfected with 2R or 66R and treated for 24 h, immunoblotted for poly‐GP expression. (D) Quantification of PBS‐insoluble poly‐GP from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by uncorrected Fisher’s LSD, *P = 0.0196 2R versus 66R DMSO, **P = 0.0039 FSK versus DMSO, *P = 0.0418 H89 versus DMSO, **P = 0.0017 H89 versus FSK, *P = 0.0364 FSK + H89 versus FSK. (E) Quantification of poly‐GP from three biological replicates. Data are mean ± SEM. One‐way ANOVA followed by uncorrected Fisher’s LSD, *P = 0.0112 2R versus 66R DMSO, ****P < 0.0001 FSK versus DMSO, ns (not significant) H89 versus DMSO, *P = 0.0281 FSK + H89 versus DMSO, ****P < 0.0001 H89 versus FSK, ***P = 0.0004, FSK + H89 versus FSK, **P = 0.0030 H89 versus FSK +H89.
-
FLuminescence derived from HEK293T transfected with GA‐NLuc reporters WT or mutated and treated with H89 for 24 h. Mutational analysis of near‐cognate CUG start codon from three biological replicates and four technical replicates, n = 12. Data are mean ± SEM. Two‐way ANOVA followed by Dunnett’s multiple comparisons test, *P = 0.0130 WT H89 versus WT DMSO, ****P < 0.0001, CUG mutated H89 versus CUG mutated DMSO.
Data information: Concentration used: 2.5 μM H89, 10 μM FSK, CLZ.
Source data are available online for this figure.
While CLZ showed a similar effect to FSK, H89 reduced 66R‐derived poly‐GA in HEK293T cells and, as suggested by the co‐administration of FSK and H89, blocked the cAMP‐stimulated increase in RAN products (Fig 4B). H89 reduced 66R‐derived poly‐GP both in its soluble and PBS‐insoluble forms in NSC34 cells (Fig 4C–E). Notably, the increase in RAN products due to FSK was dampened by H89 (Fig 4C–E), suggesting that the PKA role in accumulating DPRs in cells is more important that of cAMP.
H89 also reduced the expression of C9RAN NLuc reporters irrespective of the presence of the intronic CUG (Fig 4F) and it did not change the protein level of AUG‐driven fluorescent reporters (Fig EV2A and B, Appendix Fig S5B and C). H89 slightly reduced the expression of RAN transcripts (Appendix Fig S6A and B) and did not induce an autophagy response, as it did not modulate SQSTM1/p62 or LC3 levels in NSC34 and SH‐SY5Y cells (Appendix Fig S6C–H). Finally, H89 did not induce the degradation of the GFPu reporter containing the CL1 degron (Appendix Fig S6I and J) and did not change the soluble and insoluble fractions of scrambled DPRs (Fig EV3A and B), supporting the hypothesis that FSK and H89 may exert their effect independently to protein degradation.
Knockdown of PKA represses (G4C2)n translation
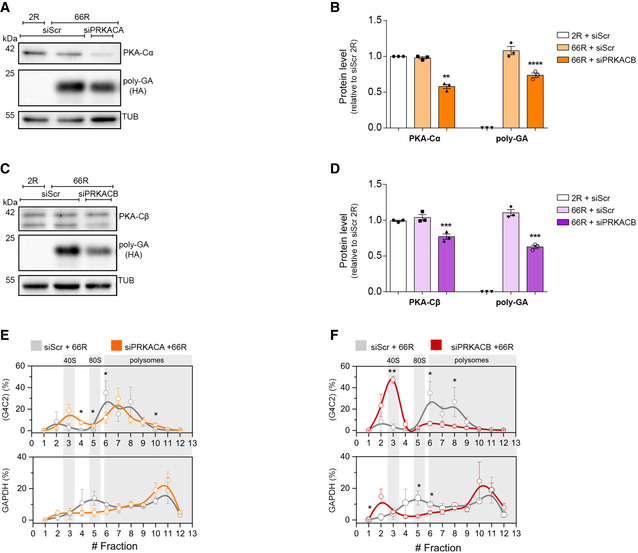
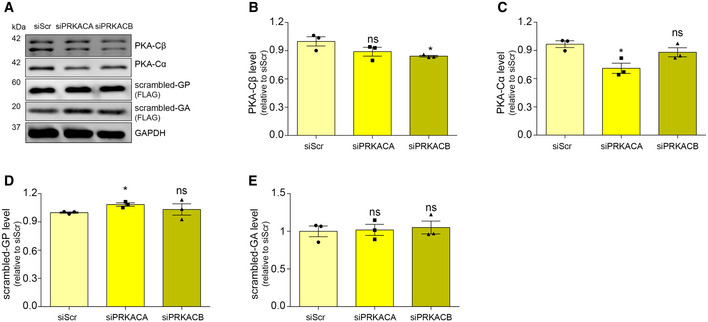
We next wondered whether PKA was crucial in modulating the level of DPRs. H89 is a well‐known and specific PKA inhibitor, but when used at concentrations ≥ 10 μM, it loses specificity showing some PKA‐independent activities, complicating the interpretation of its cellular effects (Davies et al, 2000; Lochner & Moolman, 2006; Murray, 2008; Limbutara et al, 2019). In line with these observations, H89 reduces the phosphorylation of PKA substrates in a dose‐dependent manner (Appendix Fig S6K), and it loses its PKA‐specificity at high doses, even becoming cytotoxic (IC50 20 μM on HEK293T cells) (Appendix Fig S6L). Therefore, to further investigate the role of PKA, we assessed 66R‐derived DPR levels in PKA‐silenced cells. In humans, PRKACA and PRKACB are the genes encoding for the PKA catalytic subunits α (PKA‐Cα) and β (PKA‐Cβ) respectively. Using RNA interference, we genetically downregulated the level of both PKA‐Cα and PKA‐Cβ subunits (Appendix Fig S7A and B). We found that a marked reduction in PKA‐Cα level (42%) slightly decreased the level of poly‐GA (26.4%) but not the RNA expression (Fig 5A and B, Appendix Fig S7C). In contrast, even a low reduction in PKA‐Cβ level (23%) significantly reduced the level of poly‐GA (37.4%) (Fig 5C and D) and the knockdown of both subunits further reduced poly‐GA (Appendix Fig S7D–G). The level of AUG‐driven GFP was not reduced in either condition or in double silencing (Fig EV2C–F) and similar results were obtained using scrambled DPR plasmids (Fig EV4). Moreover, while we observed a marked reduction in PRKACA mRNA upon PRKACB silencing, we did not observe the contrary (Appendix Fig S7A and B), suggesting a stronger inhibition of PKA activity during PRKACB silencing. The total level of the (G4C2)n transcript did not change in either condition (Appendix Fig S7C). Therefore, we reasoned that PKA could act at the translational level and we checked the polysomal loading of (G4C2)n mRNA under either silencing.
Figure 5. RNA interference of PKA catalytic subunit β reduces DPR levels by repressing (G4C2)n translation.
-
A, BLysates from HEK293T cells, transfected with RNAi scrambled or RNAi PRKACA (72 h) and 2R or 66R (24 h), immunoblotted for PKA‐Cα and poly‐GA (HA‐tag) expression. α‐Tubulin was used as a loading control. (B) Quantification of PKA‐Cα and poly‐GA expression from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test. PKA‐Cα: ns siScr 66R versus siScr 2R, ***P = 0.0002 siPRKACA 66R versus siScr 2R, ***P = 0.0003 siPRKACA 66R versus siScr 66R. poly‐GA expression: ****P < 0.0001 siScr 66R versus siScr 2R, ****P < 0.0001 siPRKACA 66R versus siScr 2 R, **P = 0.0062 siPRKACA 66R versus siScr 66R.
-
C, DLysates from HEK293T cells, transfected with RNAi scrambled or RNAi PRKACB (72 h) and 2R or 66R (24 h), immunoblotted PKA‐Cβ and poly‐GA (HA tag) expression. α‐Tubulin was used as a loading control. (D) Quantification of PKA‐Cβ and poly‐GA expression from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test. PKA‐Cβ: ns siScr 66R versus siScr 2R, **P = 0.0049 siPRKACB 66R versus siScr 2R, **P = 0.0091 siPRKACB 66R versus siScr 66R. poly‐GA expression: ****P < 0.0001 siScr 66R versus siScr 2R, ****P < 0.0001 siPRKACB 66R versus siScr 2R, ***P = 0.0007 siPRKACB 66R versus siScr 66R.
-
ECo‐sedimentation profile of (G4C2) (upper panel) and GAPDH (lower panel) mRNAs along the sucrose gradient fractions of polysomal profiles from control (grey lines) and PRKACA‐depleted (orange lines) cells. Data are mean ± SEM from three independent biological replicates. One‐tailed t‐test, *P < 0.05.
-
FCo‐sedimentation profile of (G4C2) (upper panel) and GAPDH (lower panel) mRNAs along the sucrose fractions of polysomal profiling from control (grey lines) and PRKACB‐depleted (red lines) cells. Data are mean ± SEM from three independent biological replicates. One‐tailed t‐test, *P < 0.05; **P < 0.01.
Data information: ns = not significant.
Source data are available online for this figure.
Figure EV4. Knockdowns of PKA do not change scrambled‐DPR levels.
-
A–ELysates from HEK293T cells, transfected with RNAi scrambled, PRKACA, PRKACB (72 h) and with scrambled‐GAx100 or ‐GPx100 (24 h), immunoblotted using antibodies for PKA‐Cα, PKA‐Cβ and FLAG‐tag for GA and GP expression. GAPDH used as a loading control (A). (B) Quantification of PKA‐Cβ expression from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test, *P = 0.0357 siPRKACB versus siScr. (C) Quantification of PKA‐Cα expression from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test, *P = 0.0169 siPRKACA versus siScr. (D) Quantification of scrambled‐GP (FLAG) expression from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test,*P = 0.0129 siPRKACA versus siScr. (E) Quantification of scrambled‐GA (FLAG) expression from three biological replicates. Data are mean ± SEM. Two‐tailed, unpaired t‐test, no significant results (ns) observed in siPRKACA versus siScr and in siPRKACB versus siScr.
Surprisingly, while the knockdown of PRKACA slightly shifted the (G4C2)n mRNA from heavy to light polysomes (Fig 5E, upper inset) compared to the control condition, the knockdown of PRKACB strongly repressed the translation of (G4C2)n transcript (Fig 5F, upper inset). On the other hand, the silencing of PRKACB marginally changed the polysomal loading of GAPDH (Fig 5F, lower inset and Fig EV2K) and HSP90 (Fig EV2L) mRNAs, and the silencing of PRKACA showed a non‐significant change in polysomal loading of the GAPDH (Fig 5E, lower inset and Fig EV2H) and HSP90 mRNAs (Fig EV2I). In addition, we did not observe any significant change in polysomal loading of AUG‐GFP mRNA (Fig EV2G and J) or in de novo protein synthesis under either silencing (Appendix Fig S7H–K). Collectively, these results demonstrate that PKA plays a crucial role in modulating DPR levels, by affecting the translation efficiency of the aberrantly expanded (G4C2)n transcript.
PKA inhibition rescues motility defects and increases lifespan in a Drosophila model of C9ALS/FTD
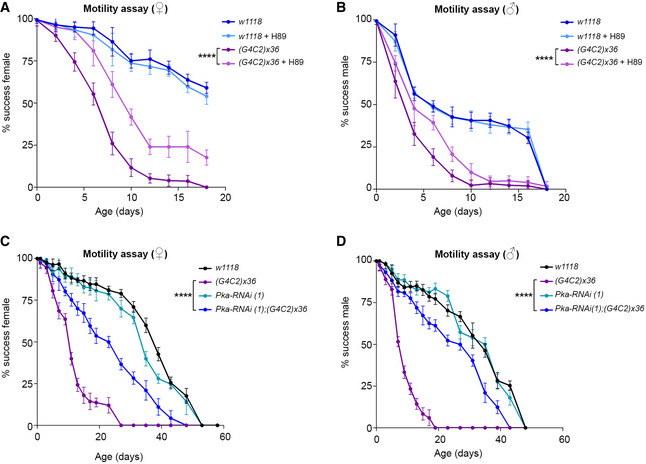
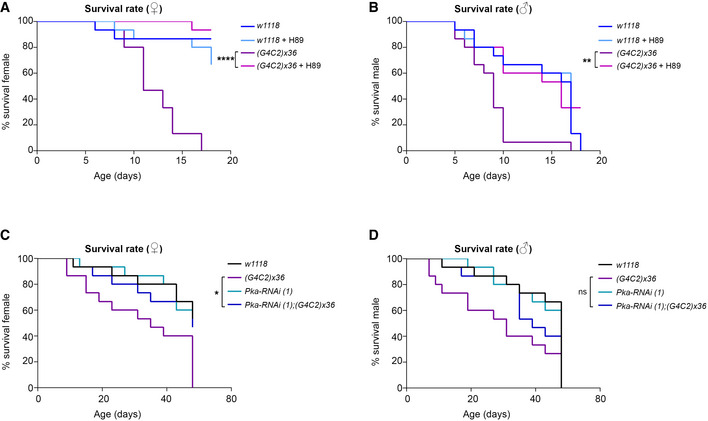
To assess in vivo the effect of PKA inhibition, we used a well‐established Drosophila model of C9ALS/FTD expressing an artificial sequence of (G4C2)x36 repeats in neurons using the Elav‐Gal4 driver (Mizielinska et al, 2014). These transgenic flies (hereafter C9 flies) show a reduction in lifespan (Mizielinska et al, 2014) and a progressive loss of motility (Fig 6A and B). Adult flies were treated daily with 10 µM H89 from 1 day after eclosion (DAE). Their ability to climb was tested every two days (Zhang et al, 2009). H89 significantly improved the motility of C9 flies starting from 3 DAE, with no significant effect in wild‐type flies (Elav‐w1118 ) (Fig 6A and B). C9 female flies showed a significant improvement in their climbing activity, with 50% of flies still successfully completing the assay at 10 DAE in the presence of H89, whereas vehicle‐treated flies declined to a 50% success rate by 7 DAE. In control females, H89 treatment did not show any significant effect (Fig 6A). In males, the ability of H89 to ameliorate the motility of C9 flies was also significant, but more limited in time (Fig 6B). Moreover, a 2‐week treatment with H89 increased the viability in both female and male C9 flies compared to the mutants treated with vehicle (Fig EV5A and B), suggesting a potential beneficial effect also on animal survival. To investigate the effect of FSK, it was administered daily at two different doses (40 and 100 µM). Toxicity and climbing impairment were observed at the highest dose in C9 and control fly lines starting from 9 DAE and 14 DAE respectively (Appendix Fig S8).
Figure 6. H89 and PKA knockdown improve motility defects in a Drosophila model of C9ALS/FTD.
-
A, BRepresentative graphs showing the climbing ability flies carrying the UAS‐(G4C2)x36 construct in neurons using the Elav‐gal4 promoter upon treatment with H89 10 µM diluted with 0.1% of DMSO and 5% sucrose in PBS. The wild‐type (w1118 ) line was used as a control. (A) Female flies (♀). Data are mean ± SD. Two‐way ANOVA followed by Tukey's multiple comparisons, significance is reported for the 6th day of treatment ****P < 0.0001 (G4C2)x36 versus (G4C2)x36 + H89. (B) Male flies (♂). Data are mean ± SD. Two‐way ANOVA followed by Tukey's multiple comparisons, significance is reported for the 6th day of treatment. ****P < 0.0001 (G4C2)x36 versus (G4C2)x36 + H89. Experiments were performed three times, N = 15 adult flies were used for each genotype, sex and treatment (N tot = 45).
-
C, DRepresentative graphs showing climbing activity of flies co‐expressing the UAS‐Pka‐RNAi construct (1, v330111) in combination with UAS‐(G4C2)x36 in neurons using the Elav‐gal4 promoter. The wild‐type (w1118 ) line was used as a control. (C) Female flies (♀). Data are mean ± SD. Two‐way ANOVA followed by Tukey's multiple comparisons, significance is reported for the 9th day ****P < 0.0001 (G4C2)x36 versus Pka‐RNAi (1);(G4C2)x36. (D) Male flies (♂). Data are mean ± SD. Two‐way ANOVA followed by Tukey's multiple comparisons, significance is reported for the 7th day ****P < 0.0001 (G4C2)x36 versus Pka‐RNAi (1);(G4C2)x36. Experiments were performed twice with different RNA interfering sequence, N = 15 adult flies were used for each genotype and sex (N tot = 30).
Figure EV5. H89 and PKA knockdown extend lifespan in a Drosophila model of C9ALS/FTD.
-
A, BRepresentative graphs showing the survival curves of flies carrying the UAS‐(G4C2)x36 construct in neurons using the Elav‐gal4 promoter upon treatment with H89 10 µM diluted with 0.1% of DMSO and 5% sucrose in PBS. The wild‐type (w1118 ) line was used as control. Relative motility experiments are reported in Fig 6A and B. (A) Female flies (♀). N = 15 adult flies were used for each genotype and treatment. Survival analysis: log‐rank (Mantel‐Cox) test; ****P < 0.0001 (G4C2)x36 versus (G4C2)x36 + H89. (B) Male flies (♂). N = 15 adult flies were used for each genotype and treatment. Survival analysis: log‐rank (Mantel‐Cox) test; **P = 0.0021 (G4C2)x36 versus (G4C2)x36 + H89.
-
C, DGraphs showing the survival curves of flies co‐expressing the UAS‐Pka‐RNAi (1, v330111) construct in combination with UAS‐(G4C2)x36 in neurons using the Elav‐gal4 promoter. The wild‐type (w1118 ) line was used as a control. Relative motility experiments are reported in Fig 6C and D. (C) Female flies (♀). N = 15 adult flies were used for each genotype. Survival analysis: log‐rank (Mantel‐Cox) test; *P = 0.0106 (G4C2)x36 versus Pka‐RNAi (1);(G4C2)x36. (D) Male flies (♂). N = 15 adult flies were used for each genotype. Survival analysis: log‐rank (Mantel‐Cox) test shows no significant differences in (G4C2)x36 versus Pka‐RNAi (1);(G4C2)x36.
To further prove the PKA relevance in mediating the G4C2 expansion toxicity, we genetically reduced the level of Pka‐C1 (the PRKACB human homolog), using two UAS‐RNAi lines against the catalytic subunit of PKA in combination with the expression of (G4C2)x36 (Appendix Fig S9A). The data clearly demonstrated that the downregulation of Pka‐C1 significantly ameliorated both motility defects (Fig 6C and D and Appendix Fig S9B and D) and lethality (Fig EV5C and D and Appendix Fig S9C and E) in both genders of C9 flies, strongly indicating the relevance of PKA in mediating the toxicity of the (G4C2)n expansion.
PKA inhibitor H89 reduces endogenous DPR levels in C9orf72 patient‐derived iPSC‐motor neurons
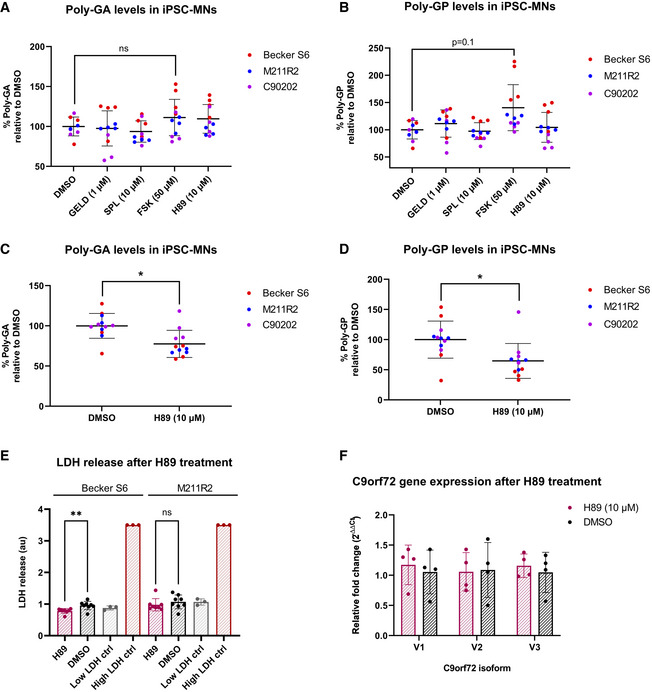
Next, we tested the effect of our DPR modulating compounds on endogenously produced DPRs in patient‐derived iPSC spinal motor neurons (C9 iPSC‐MNs). As DPRs can have long half‐lives in vitro (Westergard et al, 2019), 7‐day treatments were trialled in the first instance. The highest non‐toxic doses of GELD, SPL, FSK and H89 were selected using a lactate dehydrogenase (LDH) toxicity assay (Appendix Fig S10). Three independent C9 iPSC‐MN donor lines were treated, followed by MSD immunoassay for poly‐GA and poly‐GP, the most abundant DPRs found in C9orf72 ALS/FTD patient brains (Gendron et al, 2013; Mori et al, 2013a, 2013c). After 7 days, none of the compounds had a significant effect on poly‐GA levels, when all donor lines were analysed together (Fig 7A). When examining the individual poly‐GA levels of the donor lines (Appendix Fig S11A–C), FSK significantly increased poly‐GA levels in one of the lines, while SPL significantly reduced levels in another line. Interestingly, FSK treatment showed a trend towards increased poly‐GP levels, when the poly‐GP levels of all lines were analysed together (Fig 7B), which was significant in two of the three lines when the data were analysed individually (Appendix Fig S11D–F).
Figure 7. H89 reduces DPR levels in C9 patient‐derived iPSC‐MNs.
-
A, BThree independent C9 patient iPSC‐MN lines (Becker S6, M211R2, C90202) were treated with GELD, SPL, FSK, H89 or DMSO vehicle control and levels of poly‐GA (A) and poly‐GP (B) measured after 7 days. Values are displayed as percentages of mean DMSO signal. No significant changes in either DPR were observed (linear mixed effects model, all not significant, P > 0.05; P = 0.1 DMSO versus FSK). Treatments were performed in four technical replicates.
-
C, DThree independent C9 patient iPSC‐MN lines were treated with 10 µM H89 and levels of poly‐GA (C) and poly‐GP (D) were measured after 14 days. Values are displayed as percentages of mean DMSO signal. H89 significantly reduced poly‐GA *P = 0.039 and poly‐GP levels *P = 0.018 (linear mixed effects model). Treatments were performed in four technical replicates.
-
ENo increased LDH release was observed after 14‐day 10 µM H89 treatment compared to vehicle treatment in two independent C9 patient iPSC‐MN lines (unpaired t‐tests, Becker S6: significant decrease in LDH, **P = 0.0064; M211R2: not significant, P > 0.05). In Becker S6, mean LDH release was 22% lower after H89 treatment compared to vehicle. Treatments were performed in eight technical replicates.
-
FRelative fold changes of C9orf72 variant RNA levels normalized to RPL13A, after 14‐day 10 µM H89 treatment versus vehicle treatment (N = 1 C9 patient line, four technical replicates). No significant changes were observed between H89 and vehicle for any of the isoforms (2‐way ANOVA with Šídák's multiple comparisons test, all not significant, P > 0.05).
Data information: Error bars reported as ± SD. Not significant = P > 0.05, *P < 0.05, **P < 0.01.
We hypothesized that decreasing DPR levels may take longer than increasing them in our C9 iPSC‐MNs, due to the long half‐life of DPRs. Thus, H89 treatment of the three independent lines was extended to 14 days. Significant reductions in both poly‐GA and poly‐GP levels were observed (Fig 7C and D and Appendix Fig S12A–F). Neither LDH assay (Fig 7E) nor confluence analysis (Appendix Fig S13A and B) revealed any cytotoxicity after 14 days of H89 treatment. Furthermore, H89 treatment for 14 days did not impact the RNA levels of any of the C9orf72 variants (V1, V2 nor V3) in the iPSC‐MNs (Fig 7F), nor the levels of G4C2 RNA foci (Appendix Fig S13C and D). These findings suggest that the observed decrease in DPRs was due to neither cytotoxicity nor altered transcription. We confirmed that the dose of H89 that reduced DPR levels also inhibited PKA activity in the iPSC‐MNs (Appendix Fig S13E); therefore, PKA inhibition might contribute to the reduction in endogenously translated DPRs in patient C9 iPSC‐MNs, while being well tolerated by RAN translating neurons.
Discussion
We performed a chemical screen by using libraries of small molecules with biological activity and used the level of polyGP‐GFP as a readout. Contrary to other efforts (Cheng et al, 2019), we were not able to obtain cell clones stably overexpressing the polyGP‐GFP products. We succeeded in the optimization of a robust assay using an artificial reporter containing the G4C2 repeats out of their native context. A small number of small molecules that modulated the level of the RAN products were identified and three of them, GELD, SPL and FSK, were validated in downstream experiments, using the native genetic context. Among the internal controls, we observed that inhibitors of general translation, such as mTOR inhibitors (e.g. Torin, Rapamycin) and ribosome inhibitors (such as Puromycin), were able to decrease the level of both RAN‐ and AUG‐dependent products. GELD, and SPL decreased the overall level of DPRs. In contrast, FSK markedly increased DPR levels. SPL is an aldosterone antagonist that targets the mineralocorticoid receptor (MR). Its antihypertensive effect was thought to primarily depend on its diuretic and saluretic action. However, it appears that part of its mechanism of action is due to the blockade of the MR in tissues other than the kidneys, including cardiac tissue and the central nervous system (CNS) (Gomez‐Sanchez, 2016). Indeed, SPL is able to induce ubiquitin‐activating enzyme and proteasome‐mediated degradation of the helicase Xeroderma Pigmentosum group B complementing protein (XPB), independently of its MR blocking effect, in HeLa (Alekseev et al, 2014) and in human pulmonary artery endothelial cells (PAECs) (Elinoff et al, 2018). Moreover, it is known that SPL’s diuretic effect can act on Na+ and Ca2+ metabolism and it has been shown that SPL can increase the survival rate of flies carrying CUG‐repeat expansions, suppressing their semi‐lethal phenotypes (Garcia‐Lopez et al, 2008). Our results showed that the effect of SPL was counteracted by blocking autophagy, suggesting that SPL most likely acts via a TFEB independent mechanism of autophagy‐mediated DPR degradation. Given the ability of SPL to cross the blood–brain barrier (Gomez‐Sanchez, 2016), its activity should be evaluated in an in vivo model to reduce the levels of RAN products in the CNS. Notably, SPL is a commercially available drug, recently also introduced as an oral suspension to aid patients with difficult swallowing, including ALS patients. Therefore, it might be interesting to evaluate whether disease progression in C9ALS/FTD patients under SPL treatment is altered, compared to non‐treated patients.
GELD is the “first in class” discovered HSP90 inhibitor (Whitesell et al, 1994). GELD inhibits HSP90 binding to its amino terminus impeding its binding to misfolded proteins, and thereby inducing their proteasomal degradation (Ochel et al, 2001). Since many of these clients are oncogenic proteins, the antitumoral effect of GELD is due to the degradation of these oncodrivers (Sidera & Patsavoudi, 2013). Unfortunately, no HSP90 inhibitors have been approved for clinical use, even if HSP90 inhibitors, including some GELD derivatives like 17‐AAG and 17‐DMAG, are presently in advanced phase II clinical trials for a variety of cancers. Their clinical use is based on their capability to induce the Heat Shock Response (HSR), a pro‐survival response that upregulates the expression of the protective‐stress induced small HSPs, like HSPB1 and HSP70 (Parcellier et al, 2003; Manaenko et al, 2010; Putcha et al, 2010). This response is, however, detrimental to anticancer therapy. HSPB1, like its cognate HSPB8 (Cristofani et al, 2017, 2018), shows protective effects by inducing proteasome or autophagy degradation of ubiquitinated and misfolded proteins (Parcellier et al, 2003). A plausible explanation of our data might be that RAN products are clients of the HSP90 protein and GELD inhibits the stabilization effect of the HSP90, by also triggering the HSR and enhancing proteasome‐mediated clearance of DPRs in cells, as already shown in our previous work (Cristofani et al, 2018). To note, the activation of the proteasome by GELD triggered the degradation of poly‐GA that is not normally degraded via this mechanism (Cristofani et al, 2018).
Our screening also identified FSK as a potent inducer of polyGP‐GFP levels. FSK acts as a stimulator of the AC enzyme thereby increasing intracellular cAMP levels and regulating several downstream pathways by PKA activation (Seamon et al, 1981; Kanne et al, 2015). The mechanism by which FSK increases DPR levels could not be related to proteasome inhibition. We cannot exclude an inhibitory effect on autophagy given the induced decrease in TFEB mRNA. However, this seems unlikely, as we did not observe any apparent change on the TFEB protein level and on other autophagy markers. On the other hand, the PKA inhibitor, H89, reduced RAN product level, in different cell models, C9 iPSC‐MN lines included, and improved motor dysfunction in a Drosophila model of C9ALS/FTD, showing a coherent beneficial effect in different experimental models. We also noted potential gender‐related effects in C9 flies (Vegeto et al, 2020), but this may be explained by the dosage compensation effect of the X chromosome (Lucchesi & Kuroda, 2015) where the Elav‐Gal4 transgene is inserted. H89 is thought to inhibit PKA by competitively binding the ATP site on its catalytic subunit (Engh et al, 1996). Several studies demonstrated that H89 also shows PKA independent effects.
To provide evidence of PKA involvement in our models, we knocked down its catalytic subunits in vitro and in vivo with the simultaneous expression of (G4C2)n repeat sequence. Depletion of PKA‐Cβ showed a marked reduction in poly‐GA protein level, compared to PKA‐Cα reduction. No changes in (G4C2)n mRNA expression were observed. In addition, silencing of PRKACB caused a reduction in PRKACA mRNA expression, but not vice versa. Moreover, reduction in PRKACB strongly repressed the translation of (G4C2)n transcripts in vitro, and knock‐down of its homolog Pka‐C1 in Drosophila significantly ameliorated both motility defects and lethality in C9 flies. However, as we could not detect DPRs in Drosophila heads, we cannot exclude that PKA inhibition may alleviate the detrimental phenotype by different mechanisms than by reducing DPR levels. Of note, PKA‐Cβ is more specifically expressed in neuronal tissue (Ørstavik et al, 2001; Kvissel et al, 2004) than PKA‐Cα, and depletion of PRKACB in mice led to protection against age‐related effects (Enns et al, 2010). Interestingly, there is evidence that PKA is involved in translation regulation at different levels. In fungi, PKA: i) activates decapping enzymes Dcp1/2, via Pat1 (deadenylation factor) phosphorylation, thereby allowing the association of the translational machinery to mRNA and protecting it from degradation and ii) regulates the abundance of eukaryotic Initiation Factor 4 G (eIF4G) (Tudisca et al, 2012; Leipheimer et al, 2019). Moreover, PKA has been reported to regulate translation elongation by phosphorylating the eukaryotic elongation factor 2 kinase (eEF2K), which in turn phosphorylates eEF2, thereby slowing down the elongation phase (Liu & Proud, 2016). An intriguing hypothesis would be that PKA‐Cβ regulates translation of specific transcripts and that the mechanism of (G4C2)n mRNA regulation can depend on PKA‐driven translation. Finally, we obtained a strong rescue of motility defects in C9 affected flies using either H89 or a reduction in PKA in the nervous tissue, suggesting that PKA‐Cβ is a previously unidentified target for C9ALS/FTD disease. A plausible hypothesis explaining the selective involvement of PKA in mediating neuronal toxicity by the (G4C2)n expansion could be related to cross‐talk with neuronal excitatory signalling, leading to cAMP formation that activates the PKA pathway(s). For example, calcium‐permeable AMPA (CP‐AMPA) receptors are normally expressed at low levels at synapses and their expression on the membrane requires the phosphorylation of GluA1 serine 845 by PKA (He et al, 2009). Interestingly, the increase in intracellular oligomeric amyloid β promotes the synaptic level of CP‐AMPA in a PKA‐ and CAMKII‐dependent manner (Whitcomb et al, 2015), even if the mechanism remains undefined. Therefore, given the involvement of PKA in mediating different signalling pathways in neuronal tissues, it is important to further elucidate its potential role in C9ALS/FTD and in other neurodegenerative diseases characterized by repeat sequence expansion. To note, FSK is widely sold as a dietary supplement for weight loss and muscle building, but, according to our data, it could lead to long‐term adverse events.
In contrast, H89, a PKA inhibitor, significantly reduced endogenous RAN products, in patient C9 iPSC‐MNs, in both the poly‐GA and poly‐GP frames, although we cannot exclude PKA independent mechanisms contribute to this effect. In this model, significant reduction in DPR levels was observed after 14 days of treatment, confirming the previously reported long half‐lives of DPRs (Gendron et al, 2017; Westergard et al, 2019). On the other hand, treatment with FSK, a cAMP elevating compound, for 7 days, revealed a non‐significant increase in endogenous DPR levels (P = 0.1), detected in the poly‐GP frame. Notably, the independent iPSC‐MN lines responded differently to FSK or H89 treatment. This could be due to several reasons. The iPSC‐MN lines have diverse baseline DPR levels. Interestingly, the line which has the highest baseline levels of DPRs (Becker S6) also showed the greatest increase after FSK treatment, while the line with the lowest baseline DPR levels (M211R2) had the largest decrease in DPR levels after H89 treatment, with no correlation between G4C2 repeat length and DPR expression levels. Endogenous DPR levels depend on many factors, including RAN translation rates and protein degradation capacities. These data highlight the importance of testing multiple donor iPSC lines for response to RAN translation modifying treatments. DPRs have been shown to drive toxicity of C9orf72‐ALS/FTD in multiple models. The potential for PKA inhibition to reduce DPR levels should therefore be further investigated.
In summary, we identified three small molecules, GELD, SPL and H89 that reduced DPR levels in several models of C9ALS/FTD. We found that GELD acts by enhancing the activity of the proteasome, and SPL enhances the function of the autophagy pathway, further confirming degradative machineries as a valid therapeutic approach to tackle this pathology (Cristofani et al, 2017, 2018; Boivin et al, 2020). FSK and H89 identify the AC/PKA‐Cβ pathway as a potential new regulator of RAN translation, grounding the work for further investigation of downstream PKA pathways and exploration of new molecular targets for C9ALS/FTD.
Materials and Methods
Chemicals
Chemicals utilized in this study are as follows: Geldanamycin (GELD; InvivoGen, ant‐gl), Erysolin (ERY; Santa Cruz Biotechnology, sc‐205679), Spironolactone (SPL; Santa Cruz Biotechnology, sc‐204294), Forskolin (FSK; SelleckChem, S2449), Cilostazol (CLZ; Cayman Chemical, 15035), H89 hydrochloride (H89; Cayman Chemical, 10010556), Cycloheximide (CHX; Sigma‐Aldrich, C1988), ActinomycinD (ACTD; Sigma‐Aldrich, A9400), Dimethyl sulfoxide (DMSO; Sigma‐Aldrich, 41639), Trehalose (TREH, Sigma‐Aldrich, T9531), Ammonium chloride (NH4Cl, EuroClone, EMR089500), Z‐Leu‐Leu‐Leu‐al (MG132; Sigma‐Aldrich, C2211), 3‐ Methyladenine (3MA; SelleckChem, S2767).
Compound‐libraries screened: (i) Spectrum Collection library (MicroSource, USA) including 60% of FDA/EMA‐approved drugs, 25% of natural products and 15% of molecules in preclinical stages for a total of 2,000 compounds; (ii) Anti‐cancer compound‐library (Selleck), including 349 bioactive compounds; (iii) NIH Clinical Collection assembled by the National Institutes of Health (NIH) comprised of 450 molecules having a history of use in human clinical trials; (iv) Screen‐Well® Autophagy library (Enzo Life Science) including 94 compounds with defined autophagy‐inducing or ‐inhibitory activity.
Cell cultures and transfections
Human embryo kidney HEK293T (STR performed in 2015) cell line was cultured in standard conditions using in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma‐Aldrich) with 10% FBS (Sigma‐Aldrich), 1% l‐glutamine (Sigma‐Aldrich) and 1% penicillin–streptomycin (Sigma‐Aldrich). HEK293T cells were plated at 1 × 105 or 5 × 104 cells (for siRNA experiments) per well in 12‐well plates for RT‐qPCR and immunoblotting assay; 1.2 × 104 or 2.5 × 103 cells (for siRNA experiments) per well in 96‐well plates for viability cell assay and dose–response assay; 1.2 × 104 cells per well in 384‐well plates for primary and secondary screens.
The immortalized mouse motor neuronal cell line NSC34 (Cashman et al, 1992; Durham et al, 1993; Simeoni et al, 2000) was cultured in standard condition using DMEM (Euroclone) with 5% FBS (Sigma‐Aldrich), 1 mM l‐glutamine (EuroClone) and antibiotics (penicillin, SERVA; streptomycin, SERVA). NSC34 cells were plated at 8 × 104 cells per well in 12‐well plates for RT‐qPCR, immunoblotting and filter retardation (FRA) assays.
Human neuroblastoma SH‐SY5Y (Crippa et al, 2016) cell line was cultured in DMEM/F12 (EuroClone) supplemented with 10% FBS (Sigma‐Aldrich), 1 mM l‐glutamine (EuroClone) and antibiotics (penicillin, SERVA; streptomycin, SERVA). SH‐SY5Y cells were plated at 1.5 × 105 cells per well in 12‐well plates for RT‐qPCR and immunoblotting assay. All cell lines were tested for mycoplasma contamination.
Plasmid and siRNA transfections were performed using Lipofectamine 3000 and Opti‐MEM (Thermo Scientific) following the manufacturer’s instructions. Plasmid transfections in 384‐well plates were performed using the reverse protocol.
Plasmids and siRNAs
The pcDNA3.1‐CMV‐(G4C2)x58‐GFP vector was kindly provided by Professor J Paul Taylor (Freibaum et al, 2015) (St. Jude Children’s Research Hospital, Memphis, TN, USA). The (G4C2)x58‐GFP plasmid does not contain the native intronic sequence above the repeats. GFP sequence is in the GP frame. The pAG3‐CAG‐(G4C2)x2 or (G4C2)x66 (2R or 66R) vectors with three epitope tags one in each frame were kindly provided by Professor Leonard Petrucelli (Gendron et al, 2013) (Mayo Clinic, Jacksonville, FL, USA). Both constructs contain the native intronic sequence above the repeat. The pSIN. Lenti.H2B‐RFP vector was kindly provided by Professor Anna Cereseto (Francis et al, 2014) (University of Trento, Trento, Italy). pcDNA3.1‐CMV‐(G4C2)x58‐GFP and AUG‐RFP were utilized to perform the primary and confirmatory screens and dose–response assay. C9RAN NLuc reporters, with native CUG or mutated (CCC) codon, were kindly provided by Professor Peter K Todd (University of Michigan, Ann Arbor, MI, USA) (Green et al, 2017). These RAN C9NLuc reporters contain (G4C2)x70 repeats and NLuc sequence is placed in the GA frame. Plasmid expressing the proteasome reporter GFPu was kindly provided by Professor Ron Kopito (Bence et al, 2001) (Stanford University, Stanford, CA, USA). The pCMV‐β (Clontech) and pSV‐β‐gal (Promega) plasmids encoding for β‐galactosidase. The FLAG‐tagged plasmids coding for scrambled‐GA and scrambled‐GP (100 repeats) were kindly provided by Professor Daisuke Ito (Keio University School of Medicine, Tokyo, Japan) (Yamakawa et al, 2015). The plasmid pcDNA3.1 (ThermoFisher) has been used to normalize the total DNA amount in each transfection.
To silence endogenous human PKA catalytic isoforms we used siRNAs targeting PKA catalytic subunit α (esiRNA human PRKACA, EHU132541) and PKA catalytic subunit β (esiRNA human PRKACB, EHU075621) purchased from Sigma‐Aldrich.
Cell viability assays
In vitro drug sensitivity was assessed in HEK293T cells by the fluorescent and colorimetric OZBlue CellViability kit (OZbiosciences). HEK293T cells were plated at 1.2 × 104 in 96‐well plates and treated for 24 h with different concentrations of compounds (and DMSO as vehicle), and at 2.5 × 103 for siRNA experiments (72 h). Eight microliters of OZBlue was added to the media and incubated for 3 h. Fluorescence measuring (560 nm Ex/590 nm Em) was determined using a plate reading at the following time‐point 1, 2 and 3 h.
The 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5 diphenyl‐2H‐tetrazolium bromide (MTT; Sigma‐Aldrich)‐based cell proliferation assay (MTT assay) was carried out on NSC34 cells 24 h with selected compounds and performed in 24‐well plates at 4 × 104 cells/well (6 wells for each condition to be tested; n = 6). MTT solution was prepared at 1.5 mg/ml in DMEM without phenol red and was filtered through a 0.2‐mm filter. Then, the culture medium was removed from the plate and 300 µl of MTT solution was added into each well. Cells were incubated for 30 min at 37°C with 5% CO2, 95% air and complete humidity, then 500 μl of 2‐propanol was added into each well and the precipitates were suspended. The optical density (OD) of the wells was determined using a plate reader at a wavelength of 550 nm.
Dose–Response assay
HEK293T cells (1.2 × 104/well in 96‐well plates) were co‐transfected with polyGP‐GFP and AUG‐RFP, treated with different concentrations of compounds for 24 h and Hoechst 33342 was added to the living cells for nuclear staining. Image acquisition was performed using an Operetta High‐Content Imaging System (Perkin Elmer). Three channels were acquired: Hoechst33342 Ex 380/40 nm, Em 445/70 nm; GFP Ex 475/30nm, Em 525/50 nm; RFP Ex 535/30 nm, Em 595/70 nm. Image analysis was performed using Harmony 4.1 software (Perkin Elmer). Cell number was estimated by counting the nuclei Hoechst positive and the transfected cells were defined as GFP‐ or RFP‐positive cells.
RNA and protein click‐iT imaging
Click‐iT RNA Alexa Fluor 488 Imaging Kit (Thermo Scientific) was used to quantify the level of global RNA synthesis by imaging. HEK293T cells were treated for 24 h with compounds (or DMSO) and for 3 h with 5 μM ActD (used as a positive control of inhibitor of RNA synthesis), and then incubated for 1 h with 1 mM 5‐ethynyl uridine (EU) working solution without removing the drug‐containing media. EU detection was performed following the manufacturer’s protocol after cell fixation and permeabilization by imaging acquisition with Operetta. Hoechst 33342 was used for DNA staining.
Click‐iT Plus OPP Alexa Fluor 488 Protein Synthesis Assay Kit (Thermo Scientific) was used to measure the rate of translation. HEK293T cells were treated for 24 h with compounds (or DMSO) and for 3 h with 350 μM CHX (used as positive control to inhibit translation), and then incubated for 30 min with 20 μM O‐propargyl‐puromycin (OPP) in working solution without removing the drug‐containing media. After fixation and permeabilization, OPP incorporation was assessed by imaging acquisition with Operetta. NuclearMask Blue Stain was used for DNA staining.
RT‐qPCR
HEK293T cells were seeded in 12‐well plates. Transfection with (G4C2)x66 was performed 24 h after and treatments immediately after. Cells were harvested 24 h after treatments and centrifuged 5 min at 350 × g at 4°C; pellets were resuspended in 300 µl of TRI Reagent (Thermo Scientific) and total RNA isolated according to manufacturer’s instructions. RNA quantification was carried out by absorbance at 260 nm.
The retro‐transcription reaction was performed starting from 1 μg of RNA using the RevertAid First Strand cDNA synthesis kit (Thermo Scientific) according to the manufacturer’s protocol using 0.5 µl random primers and 0.5 µl Oligo(dT) primers (Invitrogen). qPCR was carried out using the CFX Connect Real‐Time PCR Detection System (BioRad) using Kapa Syber Fast qPCR Mastermix (Kapa Biosystems).
The primers used for the qPCR are listed in Appendix Materials and Methods Table S1. Relative mRNA quantification was obtained with the ΔCt method using GAPDH as a reference gene. Relative mRNA quantification of (G4C2)n was obtained by normalizing for the amount of transfected plasmid. NSC34 and SH‐SY5Y cells were plated in 12‐well plates, allowed to grow for 24 h and then treated with compounds or DMSO (used as negative control). Cells were harvested 24 h after treatments and centrifuged 5 min at 100 × g at 4°C; the pellets were resuspended in 300 µl of TRI Reagent (Sigma‐Aldrich) and total RNA isolated according to manufacturer’s instructions. RNA quantification was carried out by absorbance at 260 nm. Total RNA (1 μg) was treated with DNAse I (Sigma‐Aldrich), and reverse transcribed into cDNA using the High‐Capacity cDNA Archive Kit (Life Technologies) according to the manufacturer’s protocol. qPCR was carried out using the CFX 96 PCR Detection System (BioRad) using iTaq Universal SYBR® Green Supermix (Biorad). The primers used for the qPCR are listed in Appendix Materials and Methods Table S1. Relative mRNA quantification was obtained with the ΔCt method using Rplp0/RPLP0 as reference genes.
Lysates preparation and immunoblotting assay
HEK293T cells were plated in 12‐well plates (3 wells for each condition to be tested; n = 3). 24 h after plating, cells were transfected with (G4C2)x2 or (G4C2)x66 plasmids and treated immediately after for 24 h. For the silencing of PRAKCA and PRKACB, cells were siRNA transfected 24 h after plating and transfected with plasmid 48 h after the siRNA transfection. 24 or 72 h after plating, HEK293T were harvested and centrifuged 5 min at 350 × g at 4°C, then cells were lysed for 15 min in RIPA lysis buffer supplemented with the Protease Inhibitor Cocktail (Sigma‐Aldrich). Supernatants were collected after centrifugation at 1,380 × g for 20 min, and protein concentration was determined using the Bradford method (Sigma‐Aldrich). Equal amounts of total proteins extract were separated on 12% or 15% SDS–PAGE gels, transferred onto PVDF membranes (Amersham Hybond, GE Healthcare) and blocked with 5% (v/v) non‐fat‐dried milk powder (EuroClone) in Tris‐buffered saline with Tween 20 0.1% (Sigma‐Aldrich) (TBS‐T; pH 7.5). The membranes were then incubated overnight at 4°C in TBS‐T with 5% (v/v) BSA with one of the primary antibodies (Appendix Materials and Methods Table S2) washed twice with TBS‐T, incubated with horseradish peroxidase (HRP)‐conjugated secondary antibodies (donkey anti‐rabbit GE Healthcare Life Sciences #NA934, sheep anti‐mouse GE Healthcare Life Sciences #NA931) and washed with TBS‐T. Signal was revealed with chemiluminescence detection kit reagents (Amersham ECL Select, GE Healthcare).
NSC34 or SH‐SY5Y cells were plated in 12‐well plates (3 wells for each condition to be tested; n = 3). Twenty‐four hour after plating, cells were transfected as previously described and treated with compounds for 24 h. In experiments involving autophagy induction 100 mM Trehalose for the last 48 h were added to the cells. Forty‐eight or seventy‐two hour after plating, cells were harvested and centrifuged 5 min at 100 × g at 4°C; the cell pellets were resuspended in PBS (Sigma‐ Aldrich) supplemented with the Protease Inhibitor Cocktail (Sigma‐Aldrich) and homogenized using slight sonication to lyse cells and nuclei as previously described (Rusmini et al, 2007). Total proteins were determined with the bicinchoninic acid method (QPRO BCA assay; Cyanagen). An equal amount of total proteins extract was separated on 10 or 15% SDS–PAGE gels, transferred onto PVDF membranes (polyscreen transfer membrane; Amersham) and blocked with 5% (v/v) non‐fat‐dried milk powder (EuroClone) in Tris‐buffered saline with 0.1% Tween 20 (TBS‐T; pH 7.5). The membranes were then incubated overnight at 4°C with the primary antibodies (Appendix Materials and Methods Table S3), washed twice with TBS‐T, incubated with horseradish peroxidase (HRP)‐conjugated secondary antibodies (goat anti‐rabbit Jackson Immunoresearch Laboratories 111–035‐003, goat anti‐mouse Jackson Immunoresearch Laboratories 115–035‐003). Signal was revealed with chemiluminescence detection kit reagents (WESTAR ANTARES Western ECL Blotting Substrate; Cyanagen, XLS142). Membranes were subsequently processed with different antibodies to detect the levels of different proteins in the same sample, after stripping for 20 min at RT (Renew Stripping Buffer, Cyanagen).
Filter retardation assay (FRA)
FRA was used to analyse PBS‐insoluble fraction accumulating in DPR expressing cells as previously described (Cristofani et al, 2018). FRA was performed using a Bio‐Dot SF Microfiltration Apparatus (Bio‐Rad). 8 μg of the total proteins were filtered through a 0.2‐μm cellulose acetate membrane (Whatman). Slot‐blots were probed as described for immunoblotting assay. ChemiDoc XRS System (Bio‐Rad, Hercules, California, USA) was used for the image acquisition of FRA. Optical density (OD) of samples assayed with FRA was detected and analysed using the Image Lab software (Bio‐Rad). Statistical analyses have been performed using the relative optical densities defined as the ratio between optical densities of each independent biological sample (n = 3) and the mean optical density.
Luminescence assay
HEK293T cells were seeded in 96‐well plates at 1.2 × 104 cells/well. Cells were co‐transfected in 1:1 ratio with C9RAN NLuc reporters and pGL4.13 FLuc reporter (used as internal control) and treated for 24 h. Luminescence measurements were performed in white 384‐well plates (1 well of 96‐well plate was divided into 4 wells of 384‐well plate) using Nano‐Glo® Dual‐Luciferase® Reporter Assay System (Promega) according to manufacturer’s instructions. An experiment was performed in biological triplicate.
Immunoprecipitation
Cells were collected and centrifuged at 100 × g for 5 min then cells were resuspended in RIPA buffer added with protease inhibitors cocktail (Complete, Roche) and then centrifuged at 16,100 × g. Supernatant was used for immunoprecipitation with SureBeads Protein A Magnetic Beads (BIO‐RAD, 161‐4013) following the manufacturers’ instructions. Five microlitres of poly‐GP antibody (Merck, ABN455) was used for immunoprecipitation. Samples were then run on SDS‐PAGE and immunoblot analysis was performed (see immunoblot section).
Immunostaining and confocal microscope analysis
NSC34 cells were seeded on coverslips at a density of 3 × 104 cells/well (in 24‐well plates), and the day after plating were transfected and/or treated. After 24 h of treatments, cells were fixed at 37°C for 25 min using a solution 1:1 of 4% paraformaldehyde (Sigma‐Aldrich) in PB 0.2 M [solution made of KH2PO4 (0.06 M) and Na2HPO4 (0.26 M)] and 4% (v/v) sucrose (Sigma‐Aldrich). Then, the fixing solution was removed and cold methanol was added for 10 min to complete the fixation. Cell permeabilization was performed using a solution of 0.2% TRITON X100 (Sigma‐Aldrich) followed by incubation for 1 h in blocking solution. Incubation with the primary antibody was kept overnight at 4°C. Incubation with the fluorescent‐tagged secondary antibody was proceeded by three PBS washes. Nuclei were stained with DAPI (Sigma‐Aldrich). The primary antibodies used are listed in Appendix Materials and Methods Table S2. The following secondary antibodies were used: goat anti‐rabbit Alexa 594 (Thermo Scientific, A‐11012; dilution 1:1,000). All the primary and secondary antibodies were diluted in blocking solution (5% non‐fat‐dried milk in 1X PBS‐T). Coverslips were mounted on a glass support using MOWIOL and images were acquired using Eclipse Ti2 (Nikon, Netherlands) confocal microscope equipped with A1 plus camera (Nikon) and processed with the NIS‐Elements software (Nikon) processed with Fiji ImageJ distribution (Schindelin et al, 2012) based on ImageJ version: 2.0.0‐rc‐69/1.52p. Intensity Ratio Nuclei Cytoplasm Tool (RRID:SCR_018573) was used to measure TFEB nuclear translocation.
β‐galactosidase assay
NSC34 cells were plated in 24‐well plate at density of 4 × 104 cells/well (6 wells for each condition to be tested; n = 3) and transfected with 0.4 μg of pCMV‐βgal or pSV‐βgal plasmids. After 48 h of transfection, cells were lysed in 250 μl of lysis buffer (Promega) and 100 μl of samples was added to 750 μl of assay buffer (60 mM Na2HPO4 40 mM NaH2PO4 10 mM KCl, 1 mM MgSO4, pH 7.0) in presence of 4 mg/ml of β‐galactosidase substrate o‐nitrophenyl‐b‐D‐galactopyraniside (ONPG; Sigma‐Aldrich,) and incubated at 37°C until yellow colour appearance. Then, 500 μl of 1 M Na2CO3 was added and 200 μl of the final solution was transferred into a 96‐well plate and 420‐nm absorbance was evaluated using Enspire plate‐reader (PerkinElmer).
Polysomal profiling
Polysomal profiling was performed according to previously described protocols (Bernabò et al, 2017). Briefly, cells were seeded in 10‐cm dishes, treated with CHX (10 µg/ml) for 4 min and then lysed in 300 µl of cold hypotonic lysis buffer [10 mM NaCl, 10 mM MgCl2•6H2O, 10 mM Tris–HCl, pH 7.5, 1% Triton X‐100, 0.2 U/μl Ribolock RNase inhibitor (Thermo Scientific), 0.0005 U/μl DNaseI (Thermo Scientific), CHX 10 μg/ml and 1 mM dithio‐threitol, 1% sodium deoxycholate]. The lysate was centrifuged at 4°C for 5 min at 1,620 × g to pellet cell debris. The cytoplasmic lysates loaded on a linear 10–40% [w/v] sucrose gradient and centrifuged in a SW41Ti rotor (Beckman) for 1 h 30 min at 187,813 × g at 4°C in a Beckman Optima XPN‐100 Ultracentrifuge. Fractions of 1 ml of volume were then collected monitoring the absorbance at 254 nm with the UA‐6 UV/VIS detector (Teledyne Isco).
Extraction of polysomal RNA and RT‐qPCR analysis
Polysomal RNAs were isolated from single fractions along sucrose gradient as described (Tebaldi et al, 2012). Collected fractions (polysomal and subpolysomal) were incubated with proteinase K (Thermo Scientific) and 1% SDS for 1 h 45 min at 37°C. After acid phenol–chloroform extraction (Ambion) and isopropanol precipitation, polysomal RNA was re‐suspended in 20 μl of water and RNA quantification was determined by 260/280 absorbance ratios using NanoDrop 2000 spectrophotometer (Thermo Scientific). The retro‐transcription reaction was performed starting from 1 μl of RNA using the RevertAid First Strand cDNA synthesis kit (Thermo Scientific). qPCR was carried out using the CFX Connect Real‐Time PCR Detection System (BioRad) using Kapa Syber Fast qPCR Mastermix (Kapa Biosystems). qPCRs were run in three biological and three technical replicates. The percentage of each transcript distribution along the profile was obtained using the following formula:
where “n” is the number of the fraction, %[mRNA]n is the percentage of mRNA of choice in each fraction.
Fly husbandry and lines
Animals were raised at low density, at 25°C, on a standard food medium containing 9 g/l agar (ZN5 B&V, Italy), 75 g/l corn flour, 60 g/l white sugar, 30 g/l brewer yeast (Acros Organic), 50 g/l fresh yeast, and 50 ml/l molasses (Biosigma), along with nipagin and propionic acid (Acros Organic). The fly lines obtained from the Bloomington Drosophila Stock Center p{UAS‐GGGGCC.36} attP40 (B58688) (Mizielinska et al, 2014) and from Vienna Drosophila Research Center w1118 (VDRC60000) and UAS\Pka‐ RNAi (VDRC330111, 1st line), (VDRC101524, 2nd line). Flies have the same isogenic background.
H89 and FSK treatment in Drosophila
One‐day‐old animals were transferred in a plastic vial (15 animals for each genotype) containing a Whatmann 3MM paper disc imbibed with 200 μl of H89 diluted to 10 μM final concentration, or FSK to 40 or 100 μM final concentration in a 5% sucrose in PBS with 0.1% DMSO or sucrose and DMSO alone.
Motility assays
One‐day‐old adult animals of each genotype were transferred in a plastic vial without food (15 animals for each genotype), and their ability to climb up the empty vial after a knock‐down to the bottom was analysed as previously described (Zhang et al, 2009; Vernizzi et al, 2020). The number of flies that were able to climb half of the tube in 15 s was recorded. The total number of flies alive was counted every day. Values were expressed as a percentage of success with respect to the total number of flies in the vial. For each genotype, the test was repeated 20 times for each time point. After the test, adults were transferred in vials with food (in RNAi experiments) or with 5% sucrose/PBS (in H89 treatment experiments) and vials were changed every 2 days. In the motility assay, the number of surviving flies was scored every day. Data are represented as a curve of progressive motility impairment and as a percentage of survival over time. H89 treatment was repeated three times, males and females separated. In the first experiment out of three, animals were randomized to treatment and the researcher was blind. RNAi experiments were repeated twice with two different interference sequences, males and females separated. The statistical analysis of variance (2‐way ANOVA, Tukey correction) was performed using PRISM GraphPad Software (CA). Bars represent the standard deviation (SD) in climbing assays. Experiments were performed with 15 adult flies for each genotype (n tot = 60 animals for experiment, 45 for genotype, sex and treatment summing the three experiments). Using G*power software, F test, two‐way ANOVA, assuming effect size 0.4, α error = 0.05, power 0.8, numerator df = 1, number of groups = 4, the total sample size is 52. The statistical analysis of survival distribution during motility assays (log‐rank test) was performed using PRISM GraphPad Software (CA).
RNA extraction from Drosophila larvae and RT‐qPCR
Total RNAs were isolated, using 1ml TRI Reagent (Thermo Scientific), from larvae (n = 5 for each genotype) of the following genotypes: w1118 wild‐type control and Actin‐Gal4; UAS‐Pka‐C1‐RNAi. The retro‐transcription reaction was performed starting from 1 μg of RNA using the RevertAid First Strand cDNA synthesis kit (Thermo Scientific) according to the manufacturer’s protocol. PCR primers were designed as follows: Pka 5′‐ TTCAGTTCCCCTTCCTCGTC −3′ (forward), 5′‐ GAGGTCCAAGTAGTGCAGGT −3′ (reverse); Actin 5′‐ CAGATCATGTTCGAGACCTTCAAC −3′ (forward), 5′‐ ACGACCGGAGGCGTACAG −3′ (reverse).
Statistical analysis
Values are expressed as mean ± SD or ± SEM of three independent biological experiments conducted in three technical replicates. Student’s t‐test, one‐way and two‐way ANOVA were employed to determine statistical significance between control and test groups. Values of *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 were considered significant. Data were plotted by GraphPad Prism 6 Software (CA).
Differentiation and treatment of iPSC‐spinal cord motor neurons
C9orf72 patient iPSC lines were kindly provided by Professor Siddharthan Chandran (Selvaraj et al, 2018) and Professor Kevin Talbot (Ababneh et al, 2020). See Appendix Materials and Methods Table S3 for further details. iPSCs were cultured on Geltrex‐coated cell‐culture dishes and fed daily with Essential 8 medium. iPSCs were differentiated into ventral spinal cord motor neurons as previously described (Simone et al, 2018). See Appendix Materials and Methods Tables S4 and S5 for details of the cell culture media and product concentrations. After 18 days of differentiation, mitotic motor neuron progenitors were expanded in medium supplemented with FGF (10 ng/ml). For compound treatment experiments, iPSC‐motor neuron progenitors were dissociated with StemPro Accutase (Gibco) and plated on polyethylenimine (Sigma) and Geltrex‐coated cell‐culture dishes. Progenitors were matured into spinal cord motor neurons using Compound E supplemented medium for 3 days prior to treatment. Every 3–4 days’ post‐treatment, a half‐medium change was carried out, containing Compound E and the same initial compound concentration or vehicle.
MSD immunoassays
The iPSC‐motor neurons were lysed in cold RIPA buffer containing 2% SDS (Fisher Bioreagents) and 2× Complete Mini Protease Inhibitor Cocktail (Roche). Samples were lysed on ice for ten minutes and then scraped into Eppendorf tubes. Samples were then sonicated (3 × 5 s) at 4°C and centrifuged at 17,000 × g for 20 min at 16°C. The supernatant was collected and frozen at −80°C. An aliquot was taken from each sample prior to freezing to perform the detergent compatible (DC) protein assay (Bio‐Rad). Meso Scale Discovery (MSD) immunoassay was performed in singleplex to quantify endogenous poly‐GP and poly‐GA expression levels in the iPSC‐motor neurons. The assays were performed as previously described (Simone et al, 2018). For poly‐GP immunoassay, samples were loaded at 30 µg protein per well, while for poly‐GA, 16 µg protein was loaded. See Appendix Materials and Methods Table S6 for reagents and antibodies used.
iPSC‐MN toxicity assays
Cytotoxicity was measured using the Cytotoxicity Detection Kit PLUS (LDH) from Roche, following manufacturers’ instructions. Additionally, confluence and cytotoxicity were assessed using IncuCyte® Live‐Cell Analysis System (Sartorius). Two fields of view from four replicate wells were analysed at the beginning and end of treatment. Confluence pre and post 14‐day treatment with H89 was quantified using IncuCyte software.
C9orf72 isoform qPCR
RNA was extracted using RNeasy Mini Kit (QIAGEN) following manufacturers’ instructions. Samples were treated with DNase I (Invitrogen) and cDNA synthesis was performed using the SuperScript® IV Reverse Transcriptase Kit (Invitrogen). TaqMan qPCR was carried out in singleplex with previously validated FAM‐labelled probes and primers targeting the three C9orf72 isoforms (Fratta et al, 2013). The gene RPL13A was used as a reference gene. Relative abundance was calculated using the 2−ΔΔCt method. See Appendix Materials and Methods Table S7 for probe and primer sequences.
Protein kinase A activity assay
iPSC‐motor neuron samples were prepared and assayed with the Protein Kinase A (PKA) Colorimetric Activity Kit (Invitrogen) following the manufacturer’s instructions. Briefly, samples were prepared in four replicate wells in 12‐well cell culture plates, lysed in the provided cell lysis buffer prepared according to instructions and subsequently clarified by centrifugation at 960 × g for 10 min at 4°C. Samples were diluted 1:2 before loading onto the PKA substrate‐coated assay plate alongside known PKA standards. The plate was subsequently incubated with adenosine triphosphate (ATP), followed by a two‐antibody system for the detection of phosphorylated PKA. Lastly, a chromogen was added before reading the absorbance at 450 nm on a FLUOstar Omega microplate reader (BMG Labtech). One unit of PKA activity is defined as the amount of PKA needed to catalyse the transfer of 1 pmol phosphate from ATP to substrate at 30°C.
RNA‐fluorescence in situ hybridization of iPSC‐MNs
Sense RNA foci load in iPSC‐MNs was assayed through RNA‐fluorescence in situ hybridization (RNA‐FISH). A previously published protocol was used with modifications (Simone et al, 2018). Briefly, iPSC‐MNs were seeded on a clear bottom 96‐well plate (Perkin Elmer) and treated with H89 or DMSO. After 14 days of treatment, the cells were fixed in 4% PFA in PBS for 7 min, followed by dehydration in a graded series of alcohols. After re‐hydration in 70% ethanol, cells were washed in hybridization block solution (40% formamide, 2xSSC, 10% dextran sulphate, 2 mM ribonucleoside vanadyl complex) for 5 min, then permeabilized in 0.2% Triton X‐100 in PBS for 10 min. Cells were incubated in hybridization block solution for another 30 min, followed by a 3‐hour incubation with 40 nM sense probe (CCCCGGCCCCGGCCCC LNA probe, 5′TYE563‐labelled, Qiagen) in hybridization block solution at 66°C covered from light. After hybridization, cells were washed once with high‐stringency buffer (0.2% Triton X‐100 in 2xSSC) and incubated for 30 min at 66°C, followed by incubation in 0.2xSSC for 20 min at 66°C. Lastly, incubation with Hoechst (1:5,000 in 0.2xSSC) was performed for 10 mins for nuclear staining and the plate was kept in 0.2xSSC at 4°C covered from light until imaging.
Nuclear RNA foci quantification
RNA foci were imaged on an Opera Phenix High Content Screening confocal microscope (Perkin Elmer). 18 z‐stacks were acquired for each field and four to seven independent fields were imaged per well. Columbus 2.8 software (Perkin Elmer) was used to calculate RNA foci load as RNA foci intensity x RNA foci area.
Statistical analyses for iPSC experiments
GraphPad Prism version 9 or R were used for all analyses. For analyses of DPR levels in iPSC‐motor neurons following compound treatment experiments, a linear mixed effects model was fitted to the log DPR values, testing the intercepts of the treatment groups to control for the different baseline levels of the cell lines.
Author contributions
APo and APr designed research; NVL, RC, SS, KMW, LK, DV, VGD, PB, GV, AR, AQ, AMI, APo and APr analysed the data; NVL, RC, SS, KMW, LK, DV, VGD, DP, RL, MP, VA, PB and GV performed research; NVL, RC and SS prepared the figures; NVL, RC, APo and Apr wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Professor Leonard Petrucelli (Mayo Clinic, Jacksonville, FL, USA) for kindly provided (G4C2)x2 or x66 plasmids, Professor Taylor (St. Jude Children’s Research Hospital, Memphis, TN, USA) for kindly provided (G4C2)x58 plasmid, Professor Anna Cereseto (University of Trento, Trento, Italy) for kindly provided AUG‐RFP plasmid and Professor Ron Kopito (Stanford University, Stanford, CA, USA) for kindly provided the GFPu proteasome reporter. A special thanks to Professor Peter K Todd (University of Michigan, Ann Arbor, MI, USA) who kindly provided RAN NLuc reporters. We thank Dr Vincent Plagnol for his advice on the statistical analysis of iPSC‐MN data. We thank the Bloomington Drosophila Stock Center and the VDRC Stock Center for fly lines. Part of this work was carried out at NOLIMITS, an advanced imaging facility established by the Università degli Studi di Milano. This work was supported by grants from: Fondazione Cassa di Risparmio di Trento e Rovereto (Drug repositioning project #40102838) to APr and AQ; Fondazione ARISLA (project TARGET RAN # 40103385 to APr and APo and project MLOpathy to APo); Fondazione Cariplo, Italy (n. 2014‐0686, # 40102636) to APo; Fondazione Telethon, Italy (GGP19128 to APo); Kennedy’s disease association (2018 grant to RC); Italian Ministry of University and Research (MIUR); PRIN ‐ Progetti di ricerca di interesse nazionale (n. 2017F2A2C5 to APo); Agenzia Italiana del Farmaco (AIFA) (Co_ALS to APo); Fondazione Regionale per la Ricerca Biomedica (FRRB) (Regione Lombardia, TRANS_ALS, project nr. 2015‐0023, to APo and AR). AMI received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (648716 ‐ C9ND), the Motor Neuron Disease Association and the UK Dementia Research Institute which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. Open Access Funding provided by Universita degli Studi di Trento within the CRUI‐CARE Agreement.
The EMBO Journal (2022) 41: e105026
Contributor Information
Angelo Poletti, Email: angelo.poletti@unimi.it.
Alessandro Provenzani, Email: alessandro.provenzani@unitn.it.
Data availability
This study includes no data deposited in external repositories.
References
- Ababneh NA, Scaber J, Flynn R, Douglas A, Barbagallo P, Candalija A, Turner MR, Sims D, Dafinca R, Cowley SA et al (2020) Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell‐derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology‐directed repair. Hum Mol Genet 29: 2200–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseev S, Ayadi M, Brino L, Egly J‐M, Larsen AK, Coin F (2014) A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem Biol 21: 398–407 [DOI] [PubMed] [Google Scholar]
- Ash P, Bieniek K, Gendron T, Caulfield T, Lin W‐L, DeJesus‐Hernandez M, van Blitterswijk M, Jansen‐West K, Paul J, Rademakers R et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77: 639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin‐proteasome system by protein aggregation. Science 292: 1552–1555 [DOI] [PubMed] [Google Scholar]
- Bernabò P, Tebaldi T, Groen EJN, Lane FM, Perenthaler E, Mattedi F, Newbery HJ, Zhou H, Zuccotti P, Potrich V et al (2017) In vivo translatome profiling in spinal muscular atrophy reveals a role for SMN protein in ribosome biology. Cell Rep 21: 953–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I et al (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 6: 20877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin M, Pfister V, Gaucherot A, Ruffenach F, Negroni L, Sellier C, Charlet‐Berguerand N (2020) Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J 39: e100574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP (1992) Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn 194: 209–221 [DOI] [PubMed] [Google Scholar]
- Cheng W, Wang S, Mestre AA, Fu C, Makarem A, Xian F, Hayes LR, Lopez‐Gonzalez R, Drenner K, Jiang J et al (2018) C9ORF72 GGGGCC repeat‐associated non‐AUG translation is upregulated by stress through eIF2α phosphorylation. Nat Commun 9: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Wang S, Zhang Z, Morgens DW, Hayes LR, Lee S, Portz B, Xie Y, Nguyen BV, Haney MS et al (2019) CRISPR‐Cas9 screens identify the RNA helicase DDX3X as a repressor of C9ORF72 (GGGGCC)n repeat‐associated non‐AUG translation. Neuron 72: 885–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang Y‐J, Castanedes‐Casey M, Lee CW, Jansen‐West K, Kurti A, Murray ME et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP‐43 pathology, neuronal loss, and behavioral deficits. Science 348: 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Lopez‐Gonzalez R, Krishnan G, Phillips HL, Li AN, Seeley WW, Yao W‐D, Almeida S, Gao F‐B (2019) C9ORF72‐ALS/FTD‐associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo . Nat Neurosci 22: 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL, Miller S, Cunningham K, Vidensky S et al (2016) The C9ORF72 GGGGCC expansion forms RNA G‐quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 5: 1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper‐Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J et al (2014) Sequestration of multiple RNA recognition motif‐containing proteins by C9orf72 repeat expansions. Brain 137: 2040–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa V, D’Agostino VG, Cristofani R, Rusmini P, Cicardi ME, Messi E, Loffredo R, Pancher M, Piccolella M, Galbiati M et al (2016) Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci Rep 6: 22827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofani R, Crippa V, Rusmini P, Cicardi ME, Meroni M, Licata NV, Sala G, Giorgetti E, Grunseich C, Galbiati M et al (2017) Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 13: 1280–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofani R, Crippa V, Vezzoli G, Rusmini P, Galbiati M, Cicardi ME, Meroni M, Ferrari V, Tedesco B, Piccolella M et al (2018) The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72‐related neurodegenerative diseases. Cell Stress Chaperones 23: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M, Mackenzie I, Boeve B, Boxer A, Baker M, Rutherford N, Nicholson A, Finch NiCole A, Flynn H, Adamson J et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly C, Zhang P‐W, Pham J, Haeusler A, Mistry N, Vidensky S, Daley E, Poth E, Hoover B, Fines D et al (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80: 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham HD, Dahrouge S, Cashman NR (1993) Evaluation of the spinal cord neuron X neuroblastoma hybrid cell line NSC‐34 as a model for neurotoxicity testing. Neurotoxicology 14: 387–395 [PubMed] [Google Scholar]
- Elinoff JM, Chen L‐Y, Dougherty EJ, Awad KS, Wang S, Biancotto A, Siddiqui AH, Weir NA, Cai R, Sun J et al (2018) Spironolactone‐induced degradation of the TFIIH core complex XPB subunit suppresses NF‐κB and AP‐1 signalling. Cardiovasc Res 114: 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engh RA, Girod A, Kinzel V, Huber R, Bossemeyer D (1996) Crystal structures of catalytic subunit of cAMP‐dependent protein kinase in complex with isoquinolinesulfonyl protein kinase inhibitors H7, H8, and H89. Structural implications for selectivity. J Biol Chem 271: 26157–26164 [DOI] [PubMed] [Google Scholar]
- Enns LC, Pettan‐Brewer C, Ladiges W (2010) Protein kinase A is a target for aging and the aging heart. Aging 2: 238–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis AC, Di Primio C, Quercioli V, Valentini P, Boll A, Girelli G, Demichelis F, Arosio D, Cereseto A (2014) Second generation imaging of nuclear/cytoplasmic HIV‐1 complexes. AIDS Res Hum Retroviruses 30: 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, Ryan N, Hensman D, Mizielinska S, Waite AJ et al (2013) Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol 126: 401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez‐Gonzalez R, Kim NC, Almeida S, Lee K‐H, Badders N, Valentine M, Miller BL, Wong PC et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525: 129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuse M, Tanaka T, Shibata T, Yoshida T, Noguchi Y, Misawa N, Yasuda T, Saito Y, Kohn LD, Tatsuno I (2004) Regulation of geranylgeranyl pyrophosphate synthase in the proliferation of rat FRTL‐5 cells: involvement of both cAMP‐PKA and PI3‐AKT pathways. Biochem Biophys Res Commun 315: 1147–1153 [DOI] [PubMed] [Google Scholar]
- Garcia‐Lopez A, Monferrer L, Garcia‐Alcover I, Vicente‐Crespo M, Alvarez‐Abril MC, Artero RD (2008) Genetic and chemical modifiers of a CUG toxicity model in Drosophila . PLoS One 3: e1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Bieniek KF, Zhang Y‐J, Jansen‐West K, Ash PEA, Caulfield T, Daughrity L, Dunmore JH, Castanedes‐Casey M, Chew J et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat‐associated non‐ATG translation in c9FTD/ALS. Acta Neuropathol 126: 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Chew J, Stankowski JN, Hayes LR, Zhang Y‐J, Prudencio M, Carlomagno Y, Daughrity LM, Jansen‐West K, Perkerson EA et al (2017) Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72‐associated amyotrophic lateral sclerosis. Sci Transl Med 9: eaai7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S et al (2012) A C9orf72 promoter repeat expansion in a Flanders‐Belgian cohort with disorders of the frontotemporal lobar degeneration‐amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11: 54–65 [DOI] [PubMed] [Google Scholar]
- Gomez‐Sanchez EP (2016) Third‐generation mineralocorticoid receptor antagonists: why do we need a fourth? J Cardiovasc Pharmacol 67: 26–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KM, Glineburg MR, Kearse MG, Flores BN, Linsalata AE, Fedak SJ, Goldstrohm AC, Barmada SJ, Todd PK (2017) RAN translation at C9orf72‐associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun 8: 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KM, Sheth UJ, Flores BN, Wright SE, Sutter AB, Kearse MG, Barmada SJ, Ivanova MI, Todd PK (2019) High‐throughput screening yields several small‐molecule inhibitors of repeat‐associated non‐AUG translation. J Biol Chem 294: 18624–18638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EAJ, Shaw PG, Kim M‐S, Maragakis NJ, Troncoso JC, Pandey A, Sattler R et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507: 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z, Liu L, Tao Z, Wang R, Ren H, Sun H, Lin Z, Zhang Z, Mu C, Zhou J et al (2019) Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly‐PR. Nat Commun 10: 2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee H‐K (2009) Stabilization of Ca2+‐permeable AMPA receptors at perisynaptic sites by GluR1‐S845 phosphorylation. Proc Natl Acad Sci USA 106: 20033–20038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino T, Suzuki K, Matsushima T, Yamakawa N, Suzuki T, Mizushima T (2013) Suppression of Alzheimer’s disease‐related phenotypes by geranylgeranylacetone in mice. PLoS One 8: e76306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Wang H, Figueiredo‐Pereira ME (2013) Regulating the ubiquitin/proteasome pathway via cAMP‐signaling: neuroprotective potential. Cell Biochem Biophys 67: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Zhu Q, Gendron T, Saberi S, McAlonis‐Downes M, Seelman A, Stauffer J, Jafar‐nejad P, Drenner K, Schulte D et al (2016) Gain of toxicity from ALS/FTD‐linked repeat expansions in C9ORF72 Is alleviated by antisense oligonucleotides targeting GGGGCC‐containing RNAs. Neuron 90: 535–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanne H, Prasanna V, Burte N, Gujjula R (2015) Extraction and elemental analysis of Coleus forskohlii extract. Pharmacognosy Res 7: 237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, Goldman DH, Choi J, Nwaezeapu C, Liang D, Green KM, Goldstrohm AC, Todd PK, Green R, Wilusz JE (2019) Ribosome queuing enables non‐AUG translation to be resistant to multiple protein synthesis inhibitors. Genes Dev 33: 871–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MGG, Green KMM, Krans A, Rodriguez CMM, Linsalata AEE, Goldstrohm ACC, Todd PKK (2016) CGG repeat‐associated non‐AUG translation utilizes a cap‐dependent scanning mechanism of initiation to produce toxic proteins. Mol Cell 62: 314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer NJ, Haney MS, Morgens DW, Jovičić A, Couthouis J, Li A, Ousey J, Ma R, Bieri G, Tsui CK et al (2018) CRISPR–Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide‐repeat‐protein toxicity. Nat Genet 50: 603–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvissel A‐K, Ørstavik S, Øistad P, Rootwelt T, Jahnsen T, Skålhegg BS (2004) Induction of Cβ splice variants and formation of novel forms of protein kinase A type II holoenzymes during retinoic acid‐induced differentiation of human NT2 cells. Cell Signal 16: 577–587 [DOI] [PubMed] [Google Scholar]
- Lee Y‐B, Baskaran P, Gomez‐Deza J, Chen H‐J, Nishimura AL, Smith BN, Troakes C, Adachi Y, Stepto A, Petrucelli L et al (2017) C9orf72 poly GA RAN‐translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum Mol Genet 26: 4765–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y‐B, Chen H‐J, Peres João N, Gomez‐Deza J, Attig J, Štalekar M, Troakes C, Nishimura A, Scotter E, Vance C et al (2013) Hexanucleotide repeats in ALS/FTD form length‐dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5: 1178–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipheimer J, Bloom ALM, Panepinto JC (2019) Protein kinases at the intersection of translation and virulence. Front Cell Infect Microbiol 9: 318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbutara K, Kelleher A, Yang C‐R, Raghuram V, Knepper MA (2019) Phosphorylation changes in response to kinase inhibitor H89 in PKA‐null cells. Sci Rep 9: 2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsalata AE, He F, Malik AM, Glineburg MR, Green KM, Natla S, Flores BN, Krans A, Archbold HC, Fedak SJ et al (2019) DDX 3X and specific initiation factors modulate FMR 1 repeat‐associated non‐AUG‐initiated translation. EMBO Rep 20: e47498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Proud CG (2016) Eukaryotic elongation factor 2 kinase as a drug target in cancer, and in cardiovascular and neurodegenerative diseases. Nat Publ Gr 37: 285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochner A, Moolman JA (2006) The many faces of H89: a review. Cardiovasc Drug Rev 24: 261–274 [DOI] [PubMed] [Google Scholar]
- Lokireddy S, Kukushkin NV, Goldberg AL (2015) cAMP‐induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci USA 112: E7176–E7185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi JC, Kuroda MI (2015) Dosage compensation in Drosophila . Cold Spring Harb Perspect Biol 7: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manaenko A, Fathali N, Chen H, Suzuki H, Williams S, Zhang JH, Tang J (2010) Heat shock protein 70 upregulation by geldanamycin reduces brain injury in a mouse model of intracerebral hemorrhage. Neurochem Int 57: 844–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marunouchi T, Inomata S, Sanbe A, Takagi N, Tanonaka K (2014) Protective effect of geranylgeranylacetone via enhanced induction of HSPB1 and HSPB8 in mitochondria of the failing heart following myocardial infarction in rats. Eur J Pharmacol 730: 140–147 [DOI] [PubMed] [Google Scholar]
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf‐Strathmann J et al (2014) C9orf72 FTLD/ALS‐associated Gly‐Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128: 485–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation‐dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609 [DOI] [PubMed] [Google Scholar]
- Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 345: 1192–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, Weng S‐M, Schludi MH, van der Zee J, Cruts M et al (2013a) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126: 881–893 [DOI] [PubMed] [Google Scholar]
- Mori K, Lammich S, Mackenzie IRA, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M et al (2013b) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62‐positive/TDP43‐negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125: 413–423 [DOI] [PubMed] [Google Scholar]
- Mori K, Weng S‐M, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C et al (2013c) The C9orf72 repeat is translated into aggregating dipeptide‐repeat proteins in FTD/ALS. Sci Mag 339: 1335–1338 [DOI] [PubMed] [Google Scholar]
- Murray AJ (2008) Pharmacological PKA inhibition: all may not be what it seems. Sci Signal 1: re4 [DOI] [PubMed] [Google Scholar]
- Ochel HJ, Eichhorn K, Gademann G (2001) Geldanamycin: the prototype of a class of antitumor drugs targeting the heat shock protein 90 family of molecular chaperones. Cell Stress Chaperones 6: 105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørstavik S, Reinton N, Frengen E, Langeland BT, Jahnsen T, Skålhegg BS (2001) Identification of novel splice variants of the human catalytic subunit cβ of cAMP‐dependent protein kinase. Eur J Biochem 268: 5066–5073 [DOI] [PubMed] [Google Scholar]
- Parcellier A, Schmitt E, Gurbuxani S, Seigneurin‐Berny D, Pance A, Chantôme A, Plenchette S, Khochbin S, Solary E, Garrido C (2003) HSP27 is a ubiquitin‐binding protein involved in I‐kappaBalpha proteasomal degradation. Mol Cell Biol 23: 5790–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha P, Danzer KM, Kranich LR, Scott A, Silinski M, Mabbett S, Hicks CD, Veal JM, Steed PM, Hyman BT et al (2010) Brain‐permeable small‐molecule inhibitors of Hsp90 prevent alpha‐synuclein oligomer formation and rescue alpha‐synuclein‐induced toxicity. J Pharmacol Exp Ther 332: 849–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A, Majounie E, Waite A, Simón‐Sánchez J, Rollinson S, Gibbs J, Schymick J, Laaksovirta H, van Swieten J, Myllykangas L et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusmini P, Cortese K, Crippa V, Cristofani R, Cicardi ME, Ferrari V, Vezzoli G, Tedesco B, Meroni M, Messi E et al (2019) Trehalose induces autophagy via lysosomal‐mediated TFEB activation in models of motoneuron degeneration. Autophagy 15: 631–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusmini P, Sau D, Crippa V, Palazzolo I, Simonini F, Onesto E, Martini L, Poletti A (2007) Aggregation and proteasome: the case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol Aging 28: 1099–1111 [DOI] [PubMed] [Google Scholar]
- Sapio L, Di Maiolo F, Illiano M, Esposito A, Chiosi E, Spina A, Naviglio S (2014) Targeting protein kinase A in cancer therapy: an update. EXCLI J 13: 843–855 [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: An open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schludi MH, Becker L, Garrett L, Gendron TF, Zhou Q, Schreiber F, Popper B, Dimou L, Strom TM, Winkelmann J et al (2017) Spinal poly‐GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol 134: 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamon KB, Padgett W, Daly JW (1981) Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci USA 78: 3363–3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj BT, Livesey MR, Zhao C, Gregory JM, James OT, Cleary EM, Chouhan AK, Gane AB, Perkins EM, Dando O et al (2018) C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+‐permeable AMPA receptor‐mediated excitotoxicity. Nat Commun 9: 347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang W‐H, Hung S‐T, Hendricks E, Linares GR, Wang Y, Son EY et al (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 24: 313–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidera K, Patsavoudi E (2013) HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov 9: 1–20 [PubMed] [Google Scholar]
- Simeoni S, Mancini MA, Stenoien DL, Marcelli M, Weigel NL, Zanisi M, Martini L, Poletti A (2000) Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum Mol Genet 9: 133–144 [DOI] [PubMed] [Google Scholar]
- Simone R, Balendra R, Moens TG, Preza E, Wilson KM, Heslegrave A, Woodling NS, Niccoli T, Gilbert‐Jaramillo J, Abdelkarim S et al (2018) G‐quadruplex‐binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo . EMBO Mol Med 10: 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonobe Y, Ghadge G, Masaki K, Sendoel A, Fuchs E, Roos RP (2018) Neurobiology of disease translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol Dis 116: 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Zhang Y, Gendron T, Bauer P, Chew J, Yang W‐Y, Fostvedt E, Jansen‐West K, Belzil V, Desaro P et al (2014) Discovery of a biomarker and lead small molecules to target r(GGGGCC)‐associated defects in c9FTD/ALS. Neuron 83: 1043–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B, Bento‐Abreu A, Gendron TF, Boeynaems S, Bogaert E, Nuyts R, Timmers M, Scheveneels W, Hersmus N, Wang J et al (2018) A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol 135: 427–443 [DOI] [PubMed] [Google Scholar]
- Tabet R, Schaeffer L, Freyermuth F, Jambeau M, Workman M, Lee C‐Z, Lin C‐C, Jiang J, Jansen‐West K, Abou‐Hamdan H et al (2018) CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat Commun 9: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebaldi T, Re A, Viero G, Pegoretti I, Passerini A, Blanzieri E, Quattrone A (2012) Widespread uncoupling between transcriptome and translatome variations after a stimulus in mammalian cells. BMC Genom 13: 220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng Y‐J, Sandwith SN, Green KM, Chambers AE, Krans A, Raimer HM, Sharlow ME, Reisinger MA, Richardson AE, Routh ED et al (2021) The RNA helicase DHX36–G4R1 modulates C9orf72 GGGGCC hexanucleotide repeat–associated translation. J Biol Chem 297: 100914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudisca V, Simpson C, Castelli L, Lui J, Hoyle N, Moreno S, Ashe M, Portela P (2012) PKA isoforms coordinate mRNA fate during nutrient starvation. J Cell Sci 125: 5221–5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegeto E, Villa A, Della Torre S, Crippa V, Rusmini P, Cristofani R, Galbiati M, Maggi A, Poletti A (2020) The role of sex and sex hormones in neurodegenerative diseases. Endocr Rev 41: 273–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernizzi L, Paiardi C, Licata G, Vitali T, Santarelli S, Raneli M, Manelli V, Rizzetto M, Gioria M, Pasini ME et al (2020) Glutamine synthetase 1 increases autophagy lysosomal degradation of mutant huntingtin aggregates in neurons, ameliorating motility in a Drosophila model for huntington’s disease. Cells 9: 196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z‐F, Ursu A, Childs‐Disney JL, Guertler R, Yang W‐Y, Bernat V, Rzuczek SG, Fuerst R, Zhang Y‐J, Gendron TF et al (2019) The hairpin form of r(G4C2)exp in c9ALS/FTD Is repeat‐associated non‐ATG translated and a target for bioactive small molecules. Cell Chem Biol 26: 179–190.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB et al (2014) Antisense proline‐arginine RAN dipeptides linked to C9ORF72‐ALS/FTD form toxic nuclear aggregates that initiate invitro and invivo neuronal death. Neuron 84: 1213–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergard T, McAvoy K, Russell K, Wen X, Pang Y, Morris B, Pasinelli P, Trotti D, Haeusler A (2019) Repeat‐associated non‐AUG translation in C9orf72‐ALS/FTD is driven by neuronal excitation and stress. EMBO Mol Med 11: e9423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb DJ, Hogg EL, Regan P, Piers T, Narayan P, Whitehead G, Winters BL, Kim D‐H, Kim E, St George‐Hyslop P et al (2015) Intracellular oligomeric amyloid‐beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci Rep 5: 10934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM (1994) Inhibition of heat shock protein HSP90‐pp60v‐src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA 91: 8324–8328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KM, Muralidharan B, Isaacs AM (2019) Relax, don’t RAN translate it. Neuron 104: 827–829 [DOI] [PubMed] [Google Scholar]
- Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS et al (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci USA 110: 7778–7783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada SB, Gendron TF, Niccoli T, Genuth NR, Grosely R, Shi Y, Glaria I, Kramer NJ, Nakayama L, Fang S et al (2019) RPS25 is required for efficient RAN translation of C9orf72 and other neurodegenerative disease‐associated nucleotide repeats. Nat Neurosci 22: 1383–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa M, Ito D, Honda T, Kubo K‐I, Noda M, Nakajima K, Suzuki N (2015) Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet 24: 1630–1645 [DOI] [PubMed] [Google Scholar]
- Yang D, Abdallah A, Li Z, Lu Y, Almeida S, Gao F‐B (2015a) FTD/ALS‐associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol 130: 525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, He F, Strack RL, Oh SY, Frazer M, Ja SR, Todd PK, Disney MD (2016) Small molecule recognition and tools to study modulation of r(CGG)(exp) in Fragile X‐associated tremor ataxia syndrome. ACS Chem Biol 11: 2456–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W‐YY, Wilson HD, Velagapudi SP, Disney MD (2015b) Inhibition of non‐ATG translational events in cells via covalent small molecules targeting RNA. J Am Chem Soc 137: 5336–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J‐H, Chung TDY, Oldenburg KR (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4: 67–73 [DOI] [PubMed] [Google Scholar]
- Zhang KE, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525: 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Feany MB, Saraswati S, Littleton JT, Perrimon N (2009) Inactivation of Drosophila Huntingtin affects long‐term adult functioning and the pathogenesis of a Huntington’s disease model. Dis Model Mech 2: 247–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐J, Gendron TF, Grima JC, Sasaguri H, Jansen‐West K, Xu Y‐F, Katzman RB, Gass J, Murray ME, Shinohara M et al (2016) C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 19: 668–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐J, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen‐West K, Zhang XU, Prudencio M, Chew J, Cook CN et al (2018) Poly(GR) impairs protein translation and stress granule dynamics in C9orf72‐associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med 24: 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐J, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen‐West K, O’Raw AD, Pickles SR, Prudencio M, Carlomagno Y et al (2019) Heterochromatin anomalies and double‐stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363: eaav2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐J, Jansen‐West K, Xu Y‐F, Gendron TF, Bieniek KF, Lin W‐L, Sasaguri H, Caulfield T, Hubbard J, Daughrity L et al (2014) Aggregation‐prone c9FTD/ALS poly(GA) RAN‐translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128: 505–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Jiang J, Gendron TF, McAlonis‐Downes M, Jiang L, Taylor A, Diaz Garcia S, Ghosh Dastidar S, Rodriguez MJ, King P et al (2020) Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD‐causing repeat expansion in C9orf72. Nat Neurosci 23: 615–624 [DOI] [PMC free article] [PubMed] [Google Scholar]