

Abstract

Theoretical studies on molecule–metal surface reactions have so far been limited to small surface unit cells due to computational costs. Here, for the first time molecular dynamics simulations on very large surface unit cells at the level of density functional theory are performed, allowing a direct comparison to experiments performed on a curved crystal. Specifically, the reaction of D2 on a curved Pt crystal is investigated with a neural network potential (NNP). The developed NNP is also accurate for surface unit cells considerably larger than those that have been included in the training data, allowing dynamical simulations on very large surface unit cells that otherwise would have been intractable. Important and complex aspects of the reaction mechanism are discovered such as diffusion and a shadow effect of the step. Furthermore, conclusions from simulations on smaller surface unit cells cannot always be transfered to larger surface unit cells, limiting the applicability of theoretical studies of smaller surface unit cells to heterogeneous catalysts with small defect densities.
Heterogeneous catalysis is vitally important to many industrial processes. To improve these processes, fundamental insights can be gained by performing molecule–metal surface reaction (MMSR) experiments and simulations. For instance, the shape of the metal surface is important for the overall reactivity of an MMSR as different surface facets can yield different elementary reaction rates. Fortunately, the overall reactivity tends to be dominated by one or only a few rate-controlling states such as the dissociative chemisorption transition state (TS) at specific metal surface facets.1−4 Especially defects such as steps and kinks are often dominant locations for reactions in industrial processes.5 However, research involving MMSRs tends to focus on surfaces with small unit cells,6 which are not always the same as the surfaces that are relevant to heterogeneous catalysis. Computational research is particularly limited by the unit cell size of the investigated surface because the computational cost scales rapidly with the cell size and concomitant number of atoms. Experiments, on the contrary, can more easily investigate MMSRs on large unit cells, e.g., by employing well-defined stepped surfaces or curved crystals.7−12
Fortunately, advances in machine learning have enabled previously intractable computational studies. For example, high-dimensional neural network potentials (HDNNPs)13 can be employed to accurately describe reactive scattering of diatomic molecules from flat metal surfaces, while explicitly modeling surface atom motion.14−17 Furthermore, HDNNPs allow the use of more accurate but also computationally more demanding density functionals (DFs) such as meta-generalized gradient approximation (MGGA) DFs,17 which are currently intractable for density functional molecular dynamics (DFMD) studies of MMSRs. Furthermore, polyatomic molecules reacting on metal surfaces can also be accurately described, e.g., CO2 + Ni(100)18 and CHD3 + Cu(111).19 With respect to the metal surface, HDNNPs are accurate for describing not only flat surfaces with small unit cells but also nanoparticles20 and clusters.21
However, so far, reactive scattering of molecules from metal surfaces with large unit cells has not been computationally investigated at the level of density functional theory (DFT).22 The large amount of atoms in such surfaces prohibits the use of DFMD. Furthermore, developing an HDNNP for a large surface unit cell is not straightforward because a large DFT training set is required, which, although cheaper than DFMD, is still a computationally expensive endeavor. Also, such an HDNNP would be able to describe only a single surface facet, precluding a direct comparison with experiments on curved crystals. Recently, it has been shown that the embedded atom neutral network (EANN) approach23 can be transferred between different surface facets by describing reactive scattering of H2 + Cu(100), Cu(111), Cu(110), and Cu(211)24 and of CH4 + Ir(111) and Ir(332).25 Unfortunately, its applicability and accuracy with respect to surface unit cells considerably larger than those included in the training data remain unclear, which is also true for other approaches such as kernel-based regression,26−28 reactive force fields,29−33 and deep neural networks.34,35 Another complicating factor is that neural networks are generally terrible at extrapolating, where the extrapolation is a consequence of evaluating structures in a phase space that is not included in the training data of the HDNNP, although it should also be noted that an HDNNP has been found to be able to extrapolate in the case of protonated water clusters, but still with an accuracy one magnitude lower than in the interpolation regime, i.e., for structures in the phase space that has been included in the development of the HDNNP.36 For a further overview of machine learning approaches for MMSRs, see the reviews in refs (37−47). In this work, I will show that it is possible to develop an accurate HDNNP that is trained on smaller surface unit cells and that can be transferred to considerably larger surface unit cells.
For the reaction of low-energy H2 on stepped Pt surfaces, there has been a long-standing lack of clarity with regard to the reaction mechanism: Does H2 react directly on the step without major trapping, or is the reaction mediated by trapping?48−52 Recently, Lent et al. concluded from reactive scattering of H2 from a curved Pt crystal that low-energy H2 always reacts on the step, either directly on impact or via dynamical trapping in the cusp of the step toward the top step edge, but without long-range diffusion.53 In subsequent experimental work by Jansen and Juurlink, the step sticking cross section has been determined.54 Interestingly, Jansen and Juurlink found that at a low incidence energy the step sticking cross section was dependent on the step density. The reason for this step density dependence was unknown, and it was suggested that theoretical dynamical studies are required to understand the dependence. Therefore, in this work, the HDNNP is developed specifically for the reaction of H2 on a large variety of Pt surface facets, including large surface unit cells. Because high-fidelity experimental sticking probability data are available for the dissociation of H2 on a curved Pt crystal and molecular beam experiments remain the gold standard for benchmarking theory involving MMSRs to experiment,6 this reaction will also serve as an excellent benchmark for the approach presented here.
The HDNNP is trained on Pt surfaces containing (111) terraces and (100) steps, e.g., Pt(211). Specifically, the training data include the (111), (211), (533), (322), (755), (433), and (977) surfaces, adding up to ∼88000 structures (see also section S1 of the Supporting Information). Note that the stepped surfaces are similar, where the step is always the same [i.e., a (100)-like step] but where the (111) terrace length varies. Furthermore, one important detail of the construction of the HDNNP is that surface atom motion is omitted from the training data and concomitantly in the molecular dynamics (MD) simulations, but it is also expected that this omission does not influence the results presented in this work (see section S4). As I will show, the HDNNP is accurate not only for the surface unit cells included in the training but also, more importantly, for quasi-classical trajectory (QCT)55 MD simulations performed for larger surface unit cells not included in the training. This makes (a large number of) MD simulations on very large surface unit cells at the accuracy of DFMD tractable. In this work, QCT-MD simulations are performed up to Pt(171515), which is twice as large as the largest surface unit cell included in the training data [i.e., Pt(977)] and contains 240 “unique” surface atoms in the MD simulations.
The HDNNP reproduces DFT calculations excellently (Figure 1a), where the HDNNP reproduces 99.3% of the DFT energies within chemical accuracy (i.e., 4.2 kJ/mol) and 99.98% within 2-fold chemical accuracy, yielding an RMSE of 0.024 kJ mol–1 atom–1 for the energies. Furthermore, in panels b and c of Figure 1, QCT-MD simulations for H2 + Pt(111) and Pt(211) with the HDNNP yield sticking probabilities in good agreement with MD simulations employing corrugation reducing procedure (CRP)58,59 potential energy surfaces (PESs).56,57 Also, the outstanding fit quality of the HDNNP is underlined by the excellent agreement with DFMD calculations for H2 + Pt(211) using the same computational setup (Figure 1c). Note that the DFT calculations on which the CRP PESs are based employed computational setups slightly different than those of the DFT calculations in this work on which the HDNNP is based. Nevertheless, the same DF is used (i.e., PBEα57-vdW-DF256,60,61) and, therefore, PESs based on DFT calculations from either this work or that of Ghassemi and co-workers56,57 should yield similar sticking probabilities. Moreover, the PBEα57-vdW-DF2 DF has been shown to be chemically accurate for H2 + Pt(111) and Pt(211).56,57 Because these two surfaces can be considered the two extremes of the probed surfaces in this work in terms of the step density, it is likely that the employed DF is chemically accurate for all surfaces considered in this work, which, as I will show below, is found to be the case. For technical details regarding the MD simulations, see section S2.
Figure 1.

(a) Absolute error of the energies in the training and test data predicted by the HD-NNP compared to DFT calculations. The dashed line indicates chemical accuracy, i.e., 4.2 kJ/mol. (b and c) Sticking probability of H2 on Pt(111) and Pt(211), respectively, as a function of incidence energy. The orange circles and green diamonds indicate results obtained from the HDNNP and DFMD, respectively. The blue squares indicate results obtained from the CRP PESs of refs (56) and (57). Note that the DFT calculations used for the CRP PESs employ computational setups slightly different than those in this work, but the same DF, i.e., PBEα57-vdW-DF2.
Figure 2a compares the computed and measured54 sticking probability of D2 on stepped Pt surfaces as a function of step density. Throughout this work, theoretical results correspond to a specific Miller index and concomitant step density, whereas experimental results correspond to an ensemble of facets, yielding an average step density. Fortunately, the experimental variance is sufficiently low to compare experimental results obtained from an ensemble of facets with theoretical results obtained from specific facets.8 At intermediate (2.9 kJ/mol) and high (13.9 kJ/mol) incidence energies, the HDNNP yields sticking probabilities in good agreement with experiment. At a low incidence energy (0.7 kJ/mol), the HDNNP seems to overestimate the sticking probability considerably, but the agreement is actually chemically accurate for a wide variety of step densities and incidence energies (Figure S3). Because the sticking probability decreases rapidly at a low incidence energy, even a small shift along the incidence energy axis can yield large differences in the sticking probability, as is the case here. It should also be emphasized that this shift is considerably smaller than 4.2 kJ/mol; i.e., the experimental sticking probability is reproduced well within chemical accuracy (Figure S3). Furthermore, previous computational work on H2/D2 + Pt(211)57 also yielded sticking probabilities considerably higher than those from experiment at a low incidence energy. Possibly the employed DF or the QCT approach is the cause of the overestimation, but additional future work is required to understand this discrepancy. Nevertheless, the discrepancy shown here between experiment and theory is not an intrinsic part of the HDNNP, nor is it large. Therefore, I can consider the HDNNP to be not only accurate in reproducing DFT calculations but also accurate in reproducing experimental results. This fact is important because even though step densities of <0.53 nm–1 have not been included in the training of the HDNNP, the HDNNP still yields accurate results for low step densities that would have been intractable to simulate with DFMD and difficult to include in the training data of the HDNNP.
Figure 2.

(a) Sticking probability of D2 on stepped Pt surfaces as a function of step density for several incidence energies. The filled (empty) circles with solid (dashed) lines indicate theory (experiment). The incidence energies shown are 0.7 kJ/mol (blue), 2.9 kJ/mol (orange), and 13.9 kJ/mol (green). Note that surfaces with a step density of <0.53 nm–1 are not included in the training data of the HD-NNP. (b) Same as panel a but showing the step sticking cross section (computed with eq 2) instead of the sticking probability.
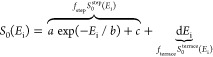
Jansen and Juurlink have also determined the step sticking cross section of the (100)-like step in a curved Pt crystal.54 At a low incidence energy, the sticking probability of D2 on Pt presumably originates solely from the steps and only from molecules impacting the surface on or close to the step53 and rapidly decays with the incidence energy. Therefore, site specific sticking probabilities (i.e., S0step and S0) can be determined from the overall sticking probability S0 as a function of incidence energy Ei as follows:54
 |
1 |
where a–d are parameters fitted to the sticking probability and fstep and fterrace are the fractions of the surface unit cell covered in steps and terraces, respectively. Then, the step sticking cross section (AstepS0step) can be determined as follows:54
| 2 |
where wstep is the width of the unit cell along the step edge and dstep is the step density. Note that the site specific sticking probabilities obtained from eq 1 are employed only when calculating the step sticking cross section with eq 2 (e.g., in Figure 2b). In all other cases, the location at the moment of reaction (i.e., r = 1.5 Å) is taken from the MD simulations and assigned to the step (terrace) as indicated by the (non)shaded areas in Figure 3.
Figure 3.

(a–f) Initial location (i.e., t = 0, left panels) and the location at the moment of reaction (defined as r = 1.5 Å, right panels) of D2 reacting on Pt(433) for several incidence energies. The colors indicate the probability density, where integration over the entire unit cell yields unity. The circles indicate the top layer atoms, where the black circles indicate step atoms and the red and pink circles indicate terrace atoms. Furthermore, the pink (red) circles indicate that the top site (does not) lies in the shadow of the step top sites. The shaded area associated with the step is indicated by the black lines, whereas the nonshaded area is associated with the terrace. (g) Side and (h) top views of Pt(433). The black, red, and pink atoms correspond to the circles in panels a–f. Moreover, the gray atoms correspond to sublayer terrace atoms.
A comparison between the theoretical and experimental step sticking cross section (Figure 2b) yields results similar to those for the sticking probability. At low incidence energies, the agreement is qualitative, and at higher incidence energies, the agreement is improved. In general, the agreement for the step sticking cross section is lower than for the sticking probability, which is caused by the slight overestimation of the sticking probability at low incidence energies. Because the model assumes that at a low incidence energy D2 sticks only near the step, an overestimation of the sticking probability at a low incidence energy causes an overall overestimation of the step sticking cross section, even at higher incidence energies (see also section S5). Fortunately, again this is not an intrinsic problem of the HDNNP (vide supra) and the HDNNP can reproduce experimental trends.
Interestingly, Jansen and Juurlink observed a dependence of the step sticking cross section on the step density at low incidence energies, for which the reason was unclear.54 They mentioned that, among others, short-range diffusion of D2 on the surface might play a role in the step density dependence but that they had no way of confirming this hypothesis. With the simulations presented in this work, the reaction dynamics can be investigated to test this hypothesis, which I will do now. Panels a–f of Figure 3 show the initial location (beginning of the simulation, i.e., t = 0; panels a, c, and e) and the location at the moment of reaction (defined as r = 1.5 Å; panels b, d, and f) of reacting D2 on Pt(433), and panels g and h of Figure 3 show side and top views, respectively, of the Pt(433) surface. Furthermore, Figure 4 shows the step/terrace ratio of the initial and reaction locations as a function of the step density. At the lowest incidence energy (0.7 kJ/mol), the initial location is slightly more concentrated at the step, but most of the distribution is delocalized (Figures 3a and 4a). When the incidence energy increases, the initial location is increasingly more concentrated at either the step or near a top layer surface atom (Figures 3c,e and 4b,c). In contrast, the location at the moment of reaction is always strongly concentrated at the top step edge and top layer surface atoms (Figures 3b,d,f and 4); i.e., the reactive sites are the top step edge and the terrace top sites. Moreover, it is clear that dynamical trapping occurs not only through the cusp near the step, as was previously thought,50,53 but also on the entire surface [which is also confirmed visually for a few trajectories on Pt(877)]. In short, at a low incidence energy a considerable amount of diffusion of D2 can occur prior to the dissociation, whereas at a high incidence energy only a small amount of diffusion is observed, independent of the step density.
Figure 4.

Ratio with which D2 reacts on the step instead of the terrace as a function of the step density for several incidence energies. The blue circles indicate the initial location (i.e., t = 0), whereas the orange circles indicate the location at the moment of reaction (i.e., r = 1.5 Å). The black dashed line indicates the statistical ratio of the unit cell. The error bars represent 68% confidence intervals.
The observations regarding the diffusion of D2 and the reactive sites also offer an explanation for why at a low incidence energy the step sticking cross section depends on the step density. Because the molecule can travel across the terrace and find a favorable location with respect to dissociation (i.e., the top step edge or a terrace top site), the terrace contributes to the overall reactivity, even at a low incidence energy, and effectively increases the step sticking cross section in Figure 2b computed with eq 2 when the step density decreases. Furthermore, even at the lowest incidence energy, the reaction partly takes place on the terrace instead of the step (Figure 4), increasing the step sticking cross section even further (see also section S5). Also, the reactivity of the terrace top sites scales inversely with the step density, again increasing the step sticking cross section for lower step densities. In contrast, the minimum barrier height and geometry on the step, which is an indicator of the reactivity of the step in the absence of diffusion, are hardly affected by the terrace length. For instance, the minimum barrier height on Pt(877) is only 2.2 kJ/mol lower than on Pt(533) (see also section S3). As such, the approximate model overestimates the step sticking cross section (especially for larger surface unit cells) because it relies on the assumption that at a low incidence energy all reaction occurs near the step and that diffusion does not play a significant role. In fact, the step sticking cross section at the moment of reaction even seems visually to be independent of the step density (Figure S5).
As stated earlier, Lent et al. concluded that at a low incidence energy D2 always reacts directly on the step or via dynamical trapping in the cusp of the step toward the top step edge, but without long-range diffusion.53 The significance of the diffusion of D2 across the entire surface in the reaction mechanism found here might lead one to think that both are possible: D2 reacts directly or via long-range diffusion (i.e., trapping-mediated) on the step. However, because the diffusion occurs within a rather short time (<20 ps) and length (<100 Å) scale before the molecule either reacts or desorbs (see also Figure S2), the reaction is not trapping-mediated because this would involve considerably longer time and length scales. Furthermore, Lent et al. also concluded that the reaction at a low incidence energy occurs only on the step. However, reaction takes place at the step and on the terrace. Therefore, I conclude that the reaction of low-energy D2 occurs both on the step and the terrace (although still predominantly on the step), either directly on impact or via short-range diffusion across the terrace.
Another intriguing aspect of the reaction mechanism is that the terrace reactivity at a low incidence energy is not uniform due to a shadow effect of the step. Panels a–c of Figure 5 show that the terrace top sites that lie on the line perpendicular to the step edge “in the shadow” of the step top sites are less reactive than the terrace top sites that do not lie in the shadow of the step top sites [see Figure 3 for a visualization of which terrace top sites are in the shadow of the step top sites; here, the pink (red) terrace circles/atoms (do not) lie in the shadow of the step top sites, i.e., the black circles/atoms]. Although still present for larger surface unit cells, this effect is considerably less pronounced when the terrace length is increased. Also, at higher incidence energies this shadow effect is still noticeable for large step densities (Figure 5b,c). The shadow effect is caused by a considerable difference between the top site barriers that lie in or out of the shadow of the step top sites on smaller stepped surface unit cells, whereas the difference in barrier heights and concomitantly the shadow effect largely disappear on larger stepped surface unit cells (Figure 5d). Furthermore, as mentioned above, in general the terrace top site barrier heights decrease with an increase in terrace length (Figure 5d). As such, one can argue that extrapolation of conclusions from reaction dynamics on small stepped surface unit cells might not always be valid in describing catalysts with low defect densities (i.e., low step density), even though doing so is the present norm, requiring simulations on large unit cells instead.
Figure 5.

(a–c) Ratio of D2 reacting near terrace top sites that lie in the shadow of the top layer step atoms compared to the total sticking probability at the terrace. Blue, orange, and green indicate incidence energies of (a) 0.7, (b) 2.9, and (c) 13.9 kJ/mol, respectively. The black dashed line indicates the statistical ratio of the unit cell. The error bars represent 68% confidence intervals. (d) Terrace top site barriers of the dissociation of H2 on several stepped Pt surfaces. Several step densities are shown (see the legend; the corresponding Miller index is indicated in parentheses). The empty circles are top sites that lie in the shadow of the step top sites, whereas the solid circles do not. The first and last terrace top sites correspond to the sites next to the bottom and top step edge, respectively (see also Figure 3g,h).
In summary, in this work I perform for the first time a large number of MD simulations at the level of DFT that can be directly compared to a molecular beam experiment performed on a curved crystal, i.e., the reaction of D2 on a curved Pt crystal. The MD simulations are made tractable by developing an HDNNP with DFT data consisting of H2 interacting with a flat (111) surface and with several smaller stepped surface unit cells with (100) steps and (111) terraces [from (211) to (977)]. The developed HDNNP can accurately describe not only the reaction of H2 on surface unit cells that have been included in the training data but also on considerably larger surface unit cells that have not been included in the training data, allowing dynamical simulations on very large surface unit cells that otherwise would have been intractable. Moreover, our understanding of the reaction mechanisms of H2 on stepped Pt surfaces is improved. One of the reaction pathways was thought to be dynamical trapping in the cusp of the step before the molecule is attracted by the step and can dissociate, but here it is observed that the dynamical trapping occurs on the entire terrace before the molecule dissociates on the step or, more rarely, on a terrace top site. Furthermore, previous experimental work determined the step sticking cross section, where a dependence of the step sticking cross section on the step density of the surface was observed. With the help of the MD simulations performed in this work, the underlying cause for the step density dependence is identified. At a low incidence energy, H2 can easily travel large distances across the surface prior to the dissociation of the molecule, even at stepped surfaces with very large terrace lengths. Due to assumptions made in the approximate model employed to obtain the step sticking cross section, the computed step sticking cross section increases substantially with a decrease in step density, even though the actual cross section observed in the MD simulations seems to be independent of the step density. Furthermore, the step changes the reactivity across the surface in a complex fashion, of which the effect is dependent on both the incidence energy of the molecule and the step density of the surface. Therefore, reaction dynamics obtained on small stepped surface unit cells are not always the same as those on large stepped surface unit cells. Similarly, fundamental understanding of catalysts with a low defect density might not always be possible through simulations or experiments performed on small stepped surface unit cells, especially when diffusion plays an important role, but requires studies on large stepped surface unit cells instead.
Computational Methods
Here, I provide a short summary of the computational details for the DFT calculations used to construct the HDNNP with RuNNer.38,40,62 All DFT calculations have been performed with a user-modified version of the Vienna Ab-initio Simulation Package (VASP version 5.3.5)63−68 to allow the use of the PBEα57-vdW-DF2 DF.56,60,61 A plane wave kinetic energy cutoff of 400 eV has been used. Furthermore, a Γ-centered k-point grid is employed, ranging from 6 × 6 × 1 [Pt(111)] to 4× 6× 1 [Pt(977)] to ensure that a reasonably consistent basis set is employed between the different facets. For the (111) facet, a 3 × 3 supercell is employed, whereas for the stepped surfaces, a 1 × 3 supercell is employed. A vacuum distance of 15 Å and five layers have been used, where the top three layers have been relaxed in all directions.
Acknowledgments
This work has been financially supported through an NWO Rubicon grant (019.202EN.012). The author thanks Prof. Kroes, Prof. Behler, Dr. Juurlink, and Jansen, MSc. for the useful discussions. Furthermore, the author is grateful to Prof. Kroes for providing computational resources.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.1c03395.
Description of the HDNNP (section S1) and the MD simulations (section S2), minimum barrier heights and geometries (section S3), discussion of the effect of surface atom motion (section S4), comparison of experiment and theory (section S5), trapping probability (Figure S1), average diffusion of D2 (Figure S2), comparison of experimental and theoretical sticking probabilities and concomitant step sticking cross section (Figure S3), site specific sticking probability fits (Figure S4), reactive step cross section on several surfaces (Figure S5), structures in the training and testing data set (Table S1), radial symmetry function parameters (Table S2), D2 and H2 molecular beam parameters (Tables S3 and S4, respectively), and minimum barrier heights and geometries (Table S5) (PDF)
The author declares no competing financial interest.
Supplementary Material
References
- Ertl G. Elementary Steps in Heterogeneous Catalysis. Angew. Chem., Int. Ed. Engl. 1990, 29, 1219–1227. 10.1002/anie.199012191. [DOI] [Google Scholar]
- Somorjai G. A.; York R. L.; Butcher D.; Park J. Y. The Evolution of Model Catalytic Systems; Studies of Structure, Bonding and Dynamics from Single Crystal Metal Surfaces to Nanoparticles, and from Low Pressure (<10–3 Torr) to High Pressure (>10–3 Torr) to Liquid Interfaces. Phys. Chem. Chem. Phys. 2007, 9, 3500–3513. 10.1039/B618805B. [DOI] [PubMed] [Google Scholar]
- Sabbe M. K.; Reyniers M.-F.; Reuter K. First-Principles Kinetic Modeling in Heterogeneous Catalysis: An Industrial Perspective on Best-Practice. Catal. Sci. Technol. 2012, 2, 2010–2024. 10.1039/c2cy20261a. [DOI] [Google Scholar]
- Wolcott C. A.; Medford A. J.; Studt F.; Campbell C. T. Degree of Rate Control Approach to Computational Catalyst Screening. J. Catal. 2015, 330, 197–207. 10.1016/j.jcat.2015.07.015. [DOI] [Google Scholar]
- Xie C.; Yan D.; Li H.; Du S.; Chen W.; Wang Y.; Zou Y.; Chen R.; Wang S. Defect Chemistry in Heterogeneous Catalysis: Recognition, Understanding, and Utilization. ACS Catal. 2020, 10, 11082–11098. 10.1021/acscatal.0c03034. [DOI] [Google Scholar]
- Kroes G.-J. Computational Approaches to Dissociative Chemisorption on Metals: Towards Chemical Accuracy. Phys. Chem. Chem. Phys. 2021, 23, 8962–9048. 10.1039/D1CP00044F. [DOI] [PubMed] [Google Scholar]
- Janlamool J.; Bashlakov D.; Berg O.; Praserthdam P.; Jongsomjit B.; Juurlink L. B. F. Desorption of Water from Distinct Step Types on a Curved Silver Crystal. Molecules 2014, 19, 10845–10862. 10.3390/molecules190810845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter A. L.; Schiller F.; Corso M.; Merte L. R.; Bertram F.; Lobo-Checa J.; Shipilin M.; Gustafson J.; Lundgren E.; Brión-Ríos A. X.; et al. X-Ray Photoemission Analysis of Clean and Carbon Monoxide-Chemisorbed Platinum(111) Stepped Surfaces Using a Curved Crystal. Nat. Commun. 2015, 6, 8903. 10.1038/ncomms9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auras S. V.; van Lent R.; Bashlakov D.; Piñeiros Bastidas J. M.; Roorda T.; Spierenburg R.; Juurlink L. B. F. Scaling Platinum-Catalyzed Hydrogen Dissociation on Corrugated Surfaces. Angew. Chem., Int. Ed. 2020, 59, 20973–20979. 10.1002/anie.202005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roorda T.; Auras S. V.; Juurlink L. B. F. Chiral Surface Characterisation and Reactivity Toward H-D Exchange of a Curved Platinum Crystal. Top. Catal. 2020, 63, 1558–1568. 10.1007/s11244-020-01371-3. [DOI] [Google Scholar]
- Garcia-Martinez F.; Schiller F.; Blomberg S.; Shipilin M.; Merte L. R.; Gustafson J.; Lundgren E.; Ortega J. E. CO Chemisorption on Vicinal Rh(111) Surfaces Studied with a Curved Crystal. J. Phys. Chem. C 2020, 124, 9305–9313. 10.1021/acs.jpcc.0c00039. [DOI] [Google Scholar]
- Auras S. V.; Juurlink L. B. F. Recent Advances in the Use of Curved Single Crystal Surfaces. Prog. Surf. Sci. 2021, 96, 100627. 10.1016/j.progsurf.2021.100627. [DOI] [Google Scholar]
- Behler J.; Parrinello M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98, 146401. 10.1103/PhysRevLett.98.146401. [DOI] [PubMed] [Google Scholar]
- Shakouri K.; Behler J.; Meyer J.; Kroes G.-J. Accurate Neural Network Description of Surface Phonons in Reactive Gas-Surface Dynamics: N2 + Ru(0001). J. Phys. Chem. Lett. 2017, 8, 2131–2136. 10.1021/acs.jpclett.7b00784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Zhou X.; Zhou L.; Zhang Y.; Luo X.; Guo H.; Jiang B. Constructing High-Dimensional Neural Network Potential Energy Surfaces for Gas-Surface Scattering and Reactions. J. Phys. Chem. C 2018, 122, 1761–1769. 10.1021/acs.jpcc.7b12064. [DOI] [Google Scholar]
- Spiering P.; Shakouri K.; Behler J.; Kroes G.-J.; Meyer J. Orbital-Dependent Electronic Friction Significantly Affects the Description of Reactive Scattering of N2 from Ru(0001). J. Phys. Chem. Lett. 2019, 10, 2957–2962. 10.1021/acs.jpclett.9b00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits N.; Geweke J.; Smeets E. W. F.; Voss J.; Wodtke A. M.; Kroes G.-J. Closing the Gap Between Experiment and Theory: Reactive Scattering of HCl from Au(111). J. Phys. Chem. C 2020, 124, 15944–15960. 10.1021/acs.jpcc.0c03756. [DOI] [Google Scholar]
- Zhang Y.; Zhou X.; Jiang B. Bridging the Gap between Direct Dynamics and Globally Accurate Reactive Potential Energy Surfaces Using Neural Networks. J. Phys. Chem. Lett. 2019, 10, 1185–1191. 10.1021/acs.jpclett.9b00085. [DOI] [PubMed] [Google Scholar]
- Gerrits N.; Shakouri K.; Behler J.; Kroes G.-J. Accurate Probabilities for Highly Activated Reaction of Polyatomic Molecules on Surfaces Using a High-Dimensional Neural Network Potential: CHD3 + Cu(111). J. Phys. Chem. Lett. 2019, 10, 1763–1768. 10.1021/acs.jpclett.9b00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich J.; Römer A.; Paleico M. L.; Behler J. Properties of α-Brass Nanoparticles. 1. Neural Network Potential Energy Surface. J. Phys. Chem. C 2020, 124, 12682–12695. 10.1021/acs.jpcc.0c00559. [DOI] [Google Scholar]
- Paleico M. L.; Behler J. Global Optimization of Copper Clusters at the ZnO(10 1̅0) Surface Using a DFT-Based Neural Network Potential and Genetic Algorithms. J. Chem. Phys. 2020, 153, 054704. 10.1063/5.0014876. [DOI] [PubMed] [Google Scholar]
- Kohn W.; Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- Zhang Y.; Hu C.; Jiang B. Embedded Atom Neural Network Potentials: Efficient and Accurate Machine Learning with a Physically Inspired Representation. J. Phys. Chem. Lett. 2019, 10, 4962–4967. 10.1021/acs.jpclett.9b02037. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Zhang Y.; Zhang L.; Zhou X.; Jiang B. Unified and Transferable Description of Dynamics for H2 Dissociative Adsorption on Multiple Copper Surfaces via Machine Learning. Phys. Chem. Chem. Phys. 2020, 22, 13958–13964. 10.1039/D0CP02291H. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Zhang Y.; Guo H.; Jiang B. Towards Bridging the Structure Gap in Heterogeneous Catalysis: The Impact of Defects in Dissociative Chemisorption of Methane on Ir Surfaces. Phys. Chem. Chem. Phys. 2021, 23, 4376–4385. 10.1039/D0CP06535H. [DOI] [PubMed] [Google Scholar]
- Quiñonero-Candela J.; Rasmussen C. E.; Williams C. K. I.. Approximation Methods for Gaussian Process Regression; MIT Press: Cambridge, MA, 2007; pp 203–223. [Google Scholar]
- Bartók A. P.; Payne M. C.; Kondor R.; Csányi G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons. Phys. Rev. Lett. 2010, 104, 136403. 10.1103/PhysRevLett.104.136403. [DOI] [PubMed] [Google Scholar]
- Vandermause J.; Xie Y.; Lim J. S.; Owen C. J.; Kozinsky B. Active Learning of Reactive Bayesian Force Fields: Application to Heterogeneous Hydrogen-Platinum Catalysis Dynamics. arXiv 2021, 2106.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Dong W.; Busnengo H. F. Reactive Force Fields for Surface Chemical Reactions: A Case Study with Hydrogen Dissociation on Pd Surfaces. J. Chem. Phys. 2010, 132, 014704. 10.1063/1.3265854. [DOI] [PubMed] [Google Scholar]
- Lozano A.; Shen X. J.; Moiraghi R.; Dong W.; Busnengo H. F. Cutting a Chemical Bond with Demonas Scissors: Mode- and Bond-Selective Reactivity of Methane on Metal Surfaces. Surf. Sci. 2015, 640, 25–35. 10.1016/j.susc.2015.04.002. [DOI] [Google Scholar]
- Senftle T. P.; Hong S.; Islam M. M.; Kylasa S. B.; Zheng Y.; Shin Y. K.; Junkermeier C.; Engel-Herbert R.; Janik M. J.; Aktulga H. M.; et al. The ReaxFF Reactive Force-Field: Development, Applications and Future Directions. npj Comput. Mater. 2016, 2, 15011. 10.1038/npjcompumats.2015.11. [DOI] [Google Scholar]
- Moiraghi R.; Lozano A.; Peterson E.; Utz A.; Dong W.; Busnengo H. F. Nonthermalized Precursor-Mediated Dissociative Chemisorption at High Catalysis Temperatures. J. Phys. Chem. Lett. 2020, 11, 2211–2218. 10.1021/acs.jpclett.0c00260. [DOI] [PubMed] [Google Scholar]
- Peludhero I. F.; Gutiérrez-González A.; Dong W.; Beck R. D.; Busnengo H. F. Dissociative Sticking Probability of Methane on Pt(110)-(2 × 1). J. Phys. Chem. C 2021, 125, 11904–11915. 10.1021/acs.jpcc.1c00807. [DOI] [Google Scholar]
- Schütt K. T.; Sauceda H. E.; Kindermans P.-J.; Tkatchenko A.; Müller K.-R. SchNet - A Deep Learning Architecture for Molecules and Materials. J. Chem. Phys. 2018, 148, 241722. 10.1063/1.5019779. [DOI] [PubMed] [Google Scholar]
- Schütt K. T.; Gastegger M.; Tkatchenko A.; Müller K.-R.; Maurer R. J. Unifying Machine Learning and Quantum Chemistry with a Deep Neural Network for Molecular Wavefunctions. Nat. Commun. 2019, 10, 5024. 10.1038/s41467-019-12875-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schran C.; Brieuc F.; Marx D. Transferability of Machine Learning Potentials: Protonated Water Neural Network Potential Applied to the Protonated Water Hexamer. J. Chem. Phys. 2021, 154, 051101. 10.1063/5.0035438. [DOI] [PubMed] [Google Scholar]
- Behler J. Representing Potential Energy Surfaces by High-Dimensional Neural Network Potentials. J. Phys.: Condens. Matter 2014, 26, 183001. 10.1088/0953-8984/26/18/183001. [DOI] [PubMed] [Google Scholar]
- Behler J. Constructing High-Dimensional Neural Network Potentials: A Tutorial Review. Int. J. Quantum Chem. 2015, 115, 1032–1050. 10.1002/qua.24890. [DOI] [Google Scholar]
- Jiang B.; Li J.; Guo H. Potential Energy Surfaces from High Fidelity Fitting of Ab Initio Points: The Permutation Invariant Polynomial - Neural Network Approach. Int. Rev. Phys. Chem. 2016, 35, 479–506. 10.1080/0144235X.2016.1200347. [DOI] [Google Scholar]
- Behler J. First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems. Angew. Chem., Int. Ed. 2017, 56, 12828–12840. 10.1002/anie.201703114. [DOI] [PubMed] [Google Scholar]
- Li J.; Song K.; Behler J. A Critical Comparison of Neural Network Potentials for Molecular Reaction Dynamics with Exact Permutation Symmetry. Phys. Chem. Chem. Phys. 2019, 21, 9672–9682. 10.1039/C8CP06919K. [DOI] [PubMed] [Google Scholar]
- Jiang B.; Li J.; Guo H. High-Fidelity Potential Energy Surfaces for Gas Phase and Gas-Surface Scattering Processes from Machine Learning. J. Phys. Chem. Lett. 2020, 11, 5120–5131. 10.1021/acs.jpclett.0c00989. [DOI] [PubMed] [Google Scholar]
- Mueller T.; Hernandez A.; Wang C. Machine Learning for Interatomic Potential Models. J. Chem. Phys. 2020, 152, 050902. 10.1063/1.5126336. [DOI] [PubMed] [Google Scholar]
- Jinnouchi R.; Miwa K.; Karsai F.; Kresse G.; Asahi R. On-the-Fly Active Learning of Interatomic Potentials for Large-Scale Atomistic Simulations. J. Phys. Chem. Lett. 2020, 11, 6946–6955. 10.1021/acs.jpclett.0c01061. [DOI] [PubMed] [Google Scholar]
- Unke O. T.; Koner D.; Patra S.; Käser S.; Meuwly M. High-Dimensional Potential Energy Surfaces for Molecular Simulations: From Empiricism to Machine Learning. Mach. Learn.: Sci. Technol. 2020, 1, 013001. 10.1088/2632-2153/ab5922. [DOI] [Google Scholar]
- Xu J.; Cao X.-M.; Hu P. Perspective on Computational Reaction Prediction Using Machine Learning Methods in Heterogeneous Catalysis. Phys. Chem. Chem. Phys. 2021, 23, 11155–11179. 10.1039/D1CP01349A. [DOI] [PubMed] [Google Scholar]
- Poltavsky I.; Tkatchenko A. Machine Learning Force Fields: Recent Advances and Remaining Challenges. J. Phys. Chem. Lett. 2021, 12, 6551–6564. 10.1021/acs.jpclett.1c01204. [DOI] [PubMed] [Google Scholar]
- Salmeron M.; Gale R. J.; Somorjai G. A. Molecular Beam Study of the H2–D2 Exchange Reaction on Stepped Platinum Crystal Surfaces: Dependence on Reactant Angle of Incidence. J. Chem. Phys. 1977, 67, 5324–5334. 10.1063/1.434711. [DOI] [Google Scholar]
- Gee A. T.; Hayden B. E.; Mormiche C.; Nunney T. S. The Role of Steps in the Dynamics of Hydrogen Dissociation on Pt(533). J. Chem. Phys. 2000, 112, 7660–7668. 10.1063/1.481360. [DOI] [Google Scholar]
- McCormack D. A.; Olsen R. A.; Baerends E. J. Mechanisms of H2 Dissociative Adsorption on the Pt(211) Stepped Surface. J. Chem. Phys. 2005, 122, 194708. 10.1063/1.1900087. [DOI] [PubMed] [Google Scholar]
- Poelsema B.; Lenz K.; Comsa G. The Dissociative Adsorption of Hydrogen on Defect-‘free’ Pt(111.). J. Phys.: Condens. Matter 2010, 22, 304006. 10.1088/0953-8984/22/30/304006. [DOI] [PubMed] [Google Scholar]
- Poelsema B.; Lenz K.; Comsa G. The Dissociative Adsorption of Hydrogen on Pt(111): Actuation and Acceleration by Atomic Defects. J. Chem. Phys. 2011, 134, 074703. 10.1063/1.3530286. [DOI] [PubMed] [Google Scholar]
- van Lent R.; Auras S. V.; Cao K.; Walsh A. J.; Gleeson M. A.; Juurlink L. B. F. Site-Specific Reactivity of Molecules with Surface Defects–the Case of H2 Dissociation on Pt. Science 2019, 363, 155–157. 10.1126/science.aau6716. [DOI] [PubMed] [Google Scholar]
- Jansen C.; Juurlink L. Absolute Dissociation Cross Sections for D2 Dissociation on Pt Steps. Chem. Phys. Lett. 2021, 776, 138679. 10.1016/j.cplett.2021.138679. [DOI] [Google Scholar]
- Karplus M.; Porter R. N.; Sharma R. D. Exchange Reactions with Activation Energy. I. Simple Barrier Potential for (H, H2). J. Chem. Phys. 1965, 43, 3259–3287. 10.1063/1.1697301. [DOI] [Google Scholar]
- Nour Ghassemi E.; Wijzenbroek M.; Somers M. F.; Kroes G.-J. Chemically Accurate Simulation of Dissociative Chemisorption of D2 on Pt(111). Chem. Phys. Lett. 2017, 683, 329–335. 10.1016/j.cplett.2016.12.059. [DOI] [Google Scholar]
- Ghassemi E. N.; Smeets E. W. F.; Somers M. F.; Kroes G.-J.; Groot I. M. N.; Juurlink L. B. F.; Füchsel G. Transferability of the Specific Reaction Parameter Density Functional for H2 + Pt(111) to H2 + Pt(211). J. Phys. Chem. C 2019, 123, 2973–2986. 10.1021/acs.jpcc.8b11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busnengo H. F.; Salin A.; Dong W. Representation of the 6D Potential Energy Surface for a Diatomic Molecule near a Solid Surface. J. Chem. Phys. 2000, 112, 7641–7651. 10.1063/1.481377. [DOI] [Google Scholar]
- Olsen R. A.; Busnengo H. F.; Salin A.; Somers M. F.; Kroes G. J.; Baerends E. J. Constructing Accurate Potential Energy Surfaces for a Diatomic Molecule Interacting with a Solid Surface: H2+Pt(111) and H2+Cu(100). J. Chem. Phys. 2002, 116, 3841–3855. 10.1063/1.1446852. [DOI] [Google Scholar]
- Madsen G. K. H. Functional Form of the Generalized Gradient Approximation for Exchange: The PBEα Functional. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 75, 195108. 10.1103/PhysRevB.75.195108. [DOI] [Google Scholar]
- Lee K.; Murray É. D.; Kong L.; Lundqvist B. I.; Langreth D. C. Higher-Accuracy van Der Waals Density Functional. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 081101. 10.1103/PhysRevB.82.081101. [DOI] [Google Scholar]
- Behler J.RuNNer - A Neural Network Code for High-Dimensional Neural Network Potential-Energy Surfaces; Universität Göttingen: Göttingen, Germany, 2018. (http://www.uni-goettingen.de/de/560580.html).
- Kresse G.; Hafner J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 49, 14251–14269. 10.1103/PhysRevB.49.14251. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47, 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Román-Pérez G.; Soler J. M. Efficient Implementation of a van Der Waals Density Functional: Application to Double-Wall Carbon Nanotubes. Phys. Rev. Lett. 2009, 103, 096102. 10.1103/PhysRevLett.103.096102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.