

Abstract
Neonatal hypoxic-ischemic encephalopathy (HIE) causes significant morbidity despite treatment with therapeutic hypothermia. Mitochondrial dysfunction may drive the mechanisms underlying neuronal cell death, thereby making mitochondria prime targets for neuroprotection. The mitochondrial permeability transition pore (mPTP) is one such target within mitochondria. In adult animal models, mPTP inhibition is neuroprotective. However, evidence for mPTP inhibition in neonatal models of neurologic disease is less certain. We tested the therapeutic efficacy of the mPTP small molecule inhibitor GNX-4728 and examined the developmental presence of brain mPTP proteins for drug targeting in a neonatal piglet model of hypoxic-ischemic brain injury. Male neonatal piglets were randomized to hypoxia-ischemia (HI) or sham procedure with GNX-4728 (15 mg/kg, IV) or vehicle (saline/cyclodextrin/DMSO, IV). GNX-4728 was administered as a single dose within 5 min after resuscitation from bradycardic arrest. Normal, ischemic, and injured neurons were counted in putamen and somatosensory cortex using hematoxylin and eosin staining. In separate neonatal and juvenile pigs, western blots of putamen mitochondrial-enriched fractions were used to evaluate mitochondrial integrity and the presence of mPTP proteins. We found that a single dose of GNX-4728 did not protect putamen and cortical neurons from cell death after HI. However, loss of mitochondrial matrix integrity occurred within 6h after HI, and while mPTP components are present in the neonatal brain their levels were significantly different compared to that of a mature juvenile brain. Thus, the neonatal brain mPTP may not be a good target for current neurotherapeutic drugs that are developed based on adult mitochondria.
Keywords: ANT, CypD, ischemic-necrosis, mitochondria, neonatal brain damage, neuronal cell death, neuroprotection, RRID:AB_478282, RRID:AB_2191808, RRID:AB_2630358, RRID:AB_2756816, RRID:AB_10862212, RRID:AB_10864110, RRID:SCR_002798, RRID:SCR_003210, VDAC
1 |. INTRODUCTION
Neonatal hypoxic-ischemic encephalopathy (HIE) from birth asphyxia is one of the top three leading causes of neonatal death worldwide (Liu et al., 2015). Therapeutic hypothermia is the standard clinical treatment for HIE because it reduces the risk of death. However, many survivors treated with hypothermia still suffer lifelong disabilities from brain injury (Jacobs et al., 2013; Shankara et al., 2012). Adjunct neuroprotective therapies for HIE are being sought to reduce the burden that HIE causes to patients, their families, and society.
Mitochondrial dysfunction is ubiquitously involved in neurodegeneration and neuronal cell death (Hagberg et al., 2014; Lu et al., 2015; Northington et al., 2007; Thornton et al., 2017). Mitochondrial permeability transition is a functional state shift from normal mitochondria to pathological mitochondria (Hunter & Haworth, 1979). The formation of the mitochondrial permeability transition pore (mPTP) is thought to drive this transition and to be a potential mechanism for neuronal cell death (Bernardi et al., 1998; Martin, 2010). The mPTP is a nonspecific pore that can span the mitochondrial membranes. It forms and opens during the intracellular oxidative stress and calcium overload that occurs with hypoxic-ischemic brain injury (Halestrap, 2009). The mPTP opening uncouples oxidative phosphorylation and leads to ATP depletion with necrotic and apoptotic cell death (Hagberg et al., 2014; Halestrap, 2009). In fact, the mPTP may drive the apoptosis-necrosis cell death continuum in neonatal hypoxic-ischemic brain injury (Martin, 2011; Northington et al., 2011). The mPTP is thus a molecular target for neuroprotection, though its complete composition and biophysical properties are still being explored.
Small molecule inhibitors of the mPTP are being developed for human therapeutics. A class of newly synthesized cinnamic anilides has garnered attention (Fancelli et al., 2014). One such molecule, GNX-4728 (Martin et al., 2014; šileikytė & Forte, 2016), blocks mPTP opening by increasing the calcium retention capacity of the mitochondria, thus altering the threshold for membrane permeability transition and preventing mitochondrial damage and cell death signaling (Fancelli et al., 2014). GNX-4728 and a related compound GNX-4975 appear to target the mitochondrial inner membrane’s adenine nucleotide translocator (ANT) and the inorganic phosphate carrier (PiC) (Richardson & Halestrap, 2016), which physically interact during mPTP formation (Leung et al., 2008). GNX-4728 crosses the blood–brain barrier, increases calcium retention capacity of brain mitochondria within 5 min after systemic administration (Martin et al., 2014), and increases the threshold for permeability transition. Thus, GNX-4728 is a potent brain mPTP inhibitor.
GNX-4728 had neuroprotective and cardioprotective benefits when administered before symptoms began in adult mice with amyotrophic lateral sclerosis (ALS) (Martin et al., 2014) and prior to reperfusion injury in rabbits with myocardial infarction (Fancelli et al., 2014). Fang et al published that GNX-4728 may provide sex-dependent mitochondrial protection acutely after hypoxia-ischemia (HI) in neonatal mice; however, it did not protect against regional brain injury in those neonatal mice (Fang et al., 2019). This therapeutic inefficacy suggested neuropathological and developmental differences in neuronal injury after neonatal HI compared to that of adult neurodegeneration. For example, selective motor neuron degeneration occurs in the adult mouse model of ALS (Martin et al., 2014). By contrast, HI in neonatal mice causes a focal lesion without selective vulnerability because territories encompassing the cerebral cortex, hippocampus, striatum, and thalamus are all infarcted (Fang et al., 2019).
We examined GNX-4728 as a therapy in a neonatal piglet model of hypoxic-ischemic brain injury. This gyrencephalic animal develops well-characterized robust neuropathology after global HI with regional and selective vulnerability that mimics HIE in term human newborns (Koehler et al., 2018; Martin et al., 1997; Northington et al., 2011). The putamen and somatosensory cortex are commonly injured in clinical HIE (Fatemi et al., 2009; Lee et al., 2017). We tested the hypothesis that GNX-4728 would protect neurons in the putamen and somatosensory cortex of neonatal piglets after global HI. We additionally evaluated the acute vulnerability of brain mitochondria in this model and the developmental presence of brain mPTP proteins as targets for neuroprotection.
2 |. METHODS
2.1 |. Animal preparation
All procedures were approved by the Animal Care and Use Committee at Johns Hopkins University, and the care and handling of the animals were in accord with the National Institutes of Health guidelines. All experiments were designed and reported in accordance with ARRIVE guidelines. Neonatal male piglets (2–3 days old, 1–2.5 kg) were randomized to HI or sham procedure. Piglets and humans have analogous perinatal growth with similar brain development at term gestation (Dobbing & Sands, 1979) and the brain injury seen in piglets after HI is similar to those of human neonates (Conrad & Johnson, 2015; Koehler et al., 2018). Because of the exploratory proof-of-principle intent of this study, we did not include female animals. Piglets were anesthetized with 5% isoflurane and 50%:50% nitrous oxide:oxygen for intubation. Piglets were then mechanically ventilated to maintain normocapnea. Isoflurane was decreased to 2% for cannulation of the femoral artery and external jugular vein for continuous arterial blood pressure monitoring and intravenous (IV) administration of medications and fluids. After placement of the catheters, which took approximately 10–15 min, the isoflurane was discontinued. The anesthetic was continued using 70%:30% nitrous oxide:oxygen and fentanyl boluses (10 μg/kg IV) in 0.45% saline with 5% dextrose at 4 ml kg−1 h−1. This anesthetic regimen has been examined for brain neurotoxicity in piglets and does not induce neuronal apoptosis or proteostasis cell stress above normal developmental levels (Lee et al., 2016; Santos et al., 2018; Wang et al., 2015, 2016). Normothermia was maintained (goal 38.5-39.5°C rectal; normothermic for swine) using blankets and heat lamps. Arterial blood pressure, heart rate, rectal temperature, and end-tidal CO2 were monitored continuously. During the 5-min administration period of drug or vehicle, operators blinded to treatment group also continuously monitored the arterial blood pressure and recorded the presence or absence of a change in mean arterial blood pressure (MAP) >10 mmHg.
2.2 |. Whole body hypoxic-ischemic injury
All piglets received rocuronium (0.2 mg/kg, IV) to prevent ventilatory effort during hypoxia and asphyxia and to ensure all groups received the same medications. The inspired oxygen concentration was decreased to 10% for 45 min with a goal oxyhemoglobin saturation of 30%. Five minutes of room air were then provided to allow for successful cardiac resuscitation. Next, the endotracheal tube was clamped for 8 min to produce asphyxia. All piglets develop severe bradycardia or hypotension with this protocol with heart rate less than 60 beats per minute or MAP less than 45. We resuscitated all piglets with manual chest compressions, 50% oxygen, and epinephrine (100 μg/kg, IV). Biphasic defibrillation with 10 J/kg was performed infrequently when necessary for ventricular fibrillation. Piglets that did not resuscitate after 3 min of compressions were excluded. After resuscitation, the inspired oxygen was lowered to 30% for the remainder of the experiment. This respiratory support strategy is well below the contraindicated 100% oxygen for term newborns and is still below the cutoff considered for high oxygen concentration for resuscitation (60%). Sham piglets received the same duration of anesthesia, surgical preparation, and 30% inhaled oxygen but without HI. Three hours after HI or sham procedure, the piglets emerged from anesthesia were extubated, and recovered with ad libitum access to milk.
2.3 |. GNX-4728
Piglets were additionally randomized to receive GNX-4728 (15 mg/kg IV; Congenia Srl, Milan, Italy) or vehicle (10% DMSO/40% cyclodextrin/saline IV) after HI or sham procedure. GNX-4728 and vehicle were freshly made for each experiment and administered as a single dose (15 mg/kg or vehicle volume equivalent, IV) over 5 min and within 5 min after the return of spontaneous circulation (ROSC), which was defined by a consistent heart rate above 60 beats per minute. Operators conducting the piglet experiments were blinded to GNX-4728 or vehicle administration.
The GNX-4728 dosing regimen was extrapolated from the neonatal mouse experiments (Fang et al., 2019) which used an intraperitoneal (IP) dose of 30 mg/kg as well as preliminary unpublished piglet experiments with 30 mg/kg versus 15 mg/kg dosing. Higher doses of GNX-4728 were associated with greater systemic blood pressure effects, thus the drug may have blood pressure dose-limiting effects, and did not confer additional neuroprotection in our initial H&E neuropathology screening. Furthermore, since IV administration has a more rapid and higher bioavailability than IP administration, we chose to use the lower 15 mg/kg dose for our experiment.
2.4 |. Neuropathology
After 4 days of recovery when striatal injury is advanced but still incomplete (Martin et al., 1997), piglets were euthanized by intraperitoneal injection of pentobarbital 390 mg/kg and phenytoin 50 mg/kg (SomnaSol, Dublin, OH) and transcardially perfused with chilled phosphate-buffered saline (PBS) for exsanguination followed by 4% paraformaldehyde in PBS for brain fixation. The piglets were decapitated and the brains were allowed to fix in situ in the head overnight at 4°C before removal from the skull. The brains were immersed in 4% paraformaldehyde for 24 hr followed by placement into 20% glycerol in PBS until tissue processing. After midsagittal hemispheric transection, the right hemisphere was cut into evenly spaced slabs that were sampled to generate appropriate anatomic levels of the striatum and cerebral cortex for neuropathological analysis. Brain samples were embedded in paraffin and cut on a rotary microtome into 10 -μm coronal sections. Brain sections at the anterior to middle striatal level were stained in batches with hematoxylin and eosin (H&E) (Figure 1a).
FIGURE 1.
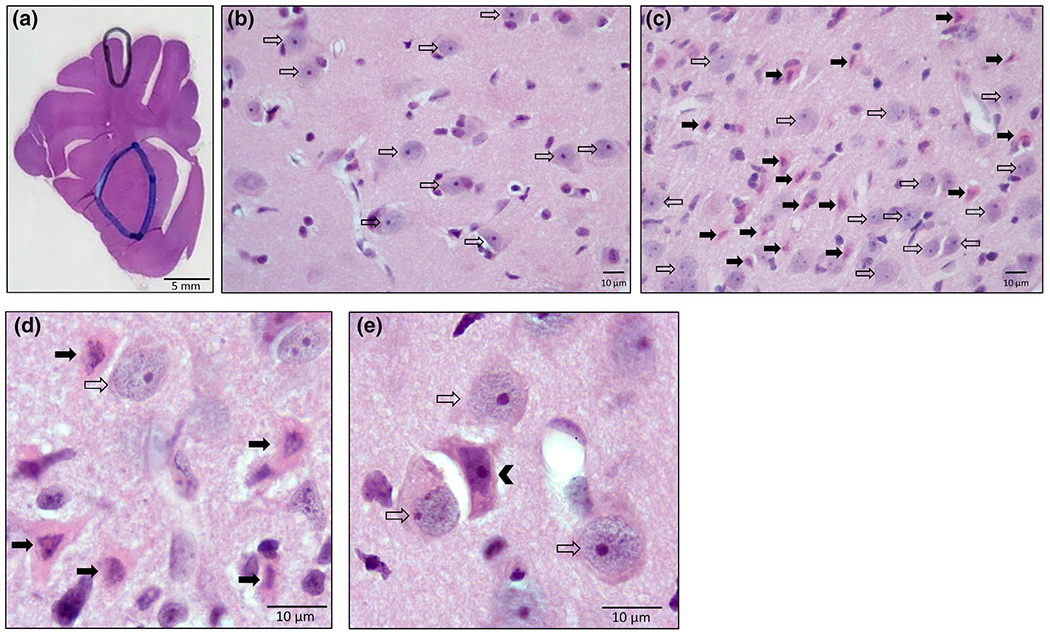
Representative piglet brain neuropathology with H&E staining, (a) Brain section with demarcations of the anatomic regions of putamen (blue outline) and somatosensory cortex (black outline) where neurons were classified and counted. (b) Medium magnification (400×) image of a putamen field in a sham vehicle animal. Normal neurons are present (open arrows). The cell bodies are about 10 μm in diameter and the nucleus is open and round with a prominent nucleolus. (c) Medium magnification (400μ) image of a putamen field in an HI animal. Eosinophilic ischemic-necrotic neurons (solid arrows) are present in addition to normal neurons (open arrows). The degenerating neurons have a bright pink cytoplasm and a dark pyknotic nucleus. (d) Representative ischemic-necrotic (solid arrow) and normal neurons (open arrow) at 1,000× magnification with oil immersion. (e) Representative injured neuron in the center (arrowhead) surrounded by several normal neurons (open arrow) at 1,000× magnification with oil immersion [Color figure can be viewed at wileyonlinelibrary.com]
One investigator (MWC) who was blinded to treatment group identified and counted the cell bodies (soma) of normal, ischemic-necrotic, and injured neurons in putamen and somatosensory cortex (O’Brien et al., 2019; Wang et al., 2015) at 1,000× magnification with oil immersion. These brain regions are readily and consistently identifiable as individual structures in the piglet brain (Figure 1a) and have been shown to exhibit neuronal selective vulnerability to HI (Martin et al., 1997). Normal, ischemic-necrotic, and injured neurons are shown from representative microscopic fields (Figure 1b–e). The cell bodies of normal principal striatal neurons were round, approximately 10 μm in diameter, and flush against the extracellular matrix of the neuropil (Figure 1b–e). In normal striatal neurons, the nucleus, that comprised most of the volume of the soma, had diffuse granular or gritty chromatin in the nucleoplasm (representing the euchromatin) and usually a prominent darkly stained nucleolus (Figure 1b–e). The ischemic-necrotic neurons (Figure 1c,d) were distinguished by their eosinophilic (acidophilic), glassy cytoplasm, angular somal attrition, and nuclear condensation to an irregularly shaped dark (hyperchromic) structure that can be seen as fragments or karyorrhexis. The injured neurons (Figure 1e) had an eosinophilic cytoplasm (less intense that the ischemic-necrotic neurons) and somal shape distortion into an angular cell compared to the normal round cells. The nucleus in injured neurons was hyperchromic and condensed, losing the granular euchromatin, but not displaying the pyknosis or karyorrhexis of ischemic-necrotic neurons. Injured neurons were usually separated from the extracellular matrix. Other forms of cell death such as classic apoptosis and autophagy (Martin, 2010) do not appear to have major contributions to the striatal neuropathology in this model (Martin et al., 1997, 2000). A second investigator (LJM) screened the slides for counter-reliability, but independent assessments were not averaged.
Putamen neurons were counted in 12 distinct non-overlapping fields that spanned dorsal to ventral (corpus callosum/external capsule to basal forebrain) and medial to lateral (internal capsule-external capsule) territories of the putamen. This coverage is necessary because the presence of putamen neuron degeneration can be compartmental and zonal (Martin et al., 1997). Primary somatosensory cortex neurons were counted along the medial bank of the gyrus in 10–12 distinct non-overlapping fields from cortical layers 2 to 6. Counts of degenerating cortical neurons were not layer specific because the lamination is not always distinct, particularly in severely injured piglets (Martin et al., 1997). The average number of neurons per microscopic field and percent total of average counts was used for the analysis.
2.5 |. Western blotting for mitochondrial proteins
To assess mitochondrial integrity and putative mitochondrial targets of GNX-4728, separate neonatal male piglets were randomized to HI or sham procedure. They were euthanized 6 hr after return of spontaneous circulation or time equivalent in sham piglets to harvest fresh brain tissue. For a developmental comparison of brain mPTP proteins, additional neonatal naïve and mature juvenile pigs (30 kg) were used. Three of the juvenile pigs were naïve (did not receive any anesthesia or surgery) and one was used as a sham for a separate adult brain injury experiment. They were perfused transcardially with ice-cold PBS for rapid brain harvest. The putamen was dissected from fresh slabs of neonatal and juvenile brains on a metal plate chilled by wet ice. Samples were flash-frozen in isopentane-chilled dry ice. Samples of unfixed frozen human brain were obtained from the Johns Hopkins University Human Brain Resource Center (Division of Neuropathology). The human brain samples were important for detecting mPTP proteins as a positive control and comparing the mobilities of these proteins on SDS-PAGE to those in piglet brain. We homogenized the tissue and fractionated the samples into nuclear, cytosolic, and mitochondrial-enriched fractions using the NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific, Rockford, IL). This procedure and its variants have been validated (Chestnut et al., 2011; Wong et al., 2013) with confirmatory western blot data shown in Figure S1. Western blotting was conducted as previously published (Santos et al., 2018). We loaded 15–20 μg of protein sample into each lane. Protein loading was verified by membrane Ponceau S stain (Sigma Life Science, St. Louis, MO).
Superoxide dismutase-2 (SOD2) is a highly specific marker for mitochondria because it is a mitochondrial matrix protein. Therefore, the level of SOD2 protein immunoreactivity is a putative biomarker of mitochondrial presence and integrity on western blot and immunohistochemistry (Martin et al., 2007, 2014), and possibly registers mitochondrial swelling previously detected as signal change by MRI (Wu et al., 2019). Immunoblots for SOD2 (mouse monoclonal 1:500; clone E-10; Santa Cruz Biotechnology Cat# sc-137254, RRID:AB_2191808) were performed on all mitochondrial-enriched fractions 6h after HI or sham procedure as well as in neonatal naïve and juvenile pig brain fractions.
For mPTP proteins, we compared neonatal sham or neonatal naïve pig brains to mature juvenile pig brain mitochondrial-enriched fractions by western blot using the primary antibodies listed in Table 1, which include the anti-voltage-dependent anion channel (VDAC also known as porin; mouse monoclonal 1:2,000; clone 20B12F2; MitoScience LLC Cat# MSA03, RRID:AB_478282), antiadenine nucleotide translocases (ANT, mouse monoclonal 1:1,000; clone 5F51BB5AG7; Abcam Cat# ab110322, RRID:AB_10862212), and anti-cyclophilin D (CypD; alias cyclophilin F [gene nomenclature] antibody, mouse monoclonal 1:2,000; clone E11AE12BD4; Abcam Cat# ab110324, RRID:AB_10864110). Several genetically distinct isoforms of human VDAC exist, including VDAC1 (chromosome 5), VDAC2 (chromosome 10), VDAC3 (chromosome 8), and VDAC4 (chromosome 1), and genetically distinct isoforms of human ANT exist, including ANT1 (chromosome 4), ANT2 (chromosome X), ANT3 (chromosome X), and ANT4 (chromosome 4). We were unable to confirm isoform specificity of the antibodies so the target proteins were generically identified as VDAC and ANT. Membranes were then incubated with the appropriate secondary antibody (goat anti-mouse or goat-anti-rabbit IgG; Jackson Immunoresearch, West Grove, PA). Immunoreactive bands were developed with enhanced chemiluminescence (Bio-Rad, Hercules, CA) and imaged with the iBrightCL 1000 Imaging System (Invitrogen) or Amersham Imager 680 (GE Healthcare). We used Mylmage Analysis (Thermo Scientific, Rockford, IL) to analyze the immunoreactive band intensities.
TABLE 1.
Information on primary antibodies used in this study
| Target protein name | Immunogen | Antibody source, catalog number | Antibody registry RRID | Clone ID | Species origin of antibody | Clonality | Molecular weight of target protein | Concentration of primary and secondary used for western blotting |
|---|---|---|---|---|---|---|---|---|
| Adenine Nucleotide Translocase | Recombinant full-length protein corresponding to human Adenine Nucleotide Translocase 1/ANT 1 | Abcam Cat# ab110322 | AB_10862212 | 5F51BB5AG7 | Mouse | Monoclonal | 33 kDa | Primary 1:1,000 Secondary 1:5,000 |
| Cyclophilin F (also known as ppif and CypD) | Recombinant full-length protein corresponding to rat Cyclophilin F aa 1-206 (also known as CypD) | Abcam Cat# ab110324 | AB_10864110 | E11AE12BD4 | Mouse | Monoclonal | 22 kDa | Primary 1:2,000 Secondary 1:5,000 |
| GAPDH | Recombinant fragment within mouse GAPDH aa 100 to the C terminus. The exact sequence is proprietary | Abcam Cat# ab181602 | AB_2630358 | EPR16891 | Rabbit | Monoclonal | 36 kDa | Primary 1:5,000 Secondary 1:5,000 |
| Histone H3 | Peptide specific to the carboxy terminus of human histone H3 protein | Cell Signaling Cat #14269 | AB_2756816 | 1B1B2 | Mouse | Monoclonal | 17 kDa | Primary 1:2,000 Secondary 1:5,000 |
| SOD2 | Specific for an epitope mapping between amino acids 22 and 58 near the N terminus of SOD2 of human origin | Santa Cruz Biotechnology Cat# sc-137254 | AB_2191808 | E-10 | Mouse | Monoclonal | 25 kDa | Primary 1:500 Secondary 1:5,000 |
| Porin (VDAC, Voltage-Dependent Anion Channel) | Recombinant full-length protein. This information is proprietary | MitoScience LLC Cat# MSA03 | AB_478282 | 20B12AF2 | Mouse | Monoclonal | 39 kDa | Primary 1:2,000 Secondary 1:5,000 |
2.6 |. Statistics
Analyses were conducted with SigmaPlot (v. 14.0, Systat Software Inc., San Jose, CA, RRID:SCR_003210), and graphs were generated with GraphPad Prism (v. 8.1.1, GraphPad Software Inc., La Jolla, CA, RRID:SCR_002798). Data are presented as medians and interquartile ranges (IQR). Physiologic variables and histologic neuron counts were analyzed by the Mann-Whitney rank sum test or the Kruskal–Wallis ANOVA on ranks with post hoc Dunn’s pairwise comparisons. Data for western blot immunodensities were normalized to (divided by) the Ponceau S protein load. In the HI versus sham cohort, immunodensities were additionally divided by the median value of the sham procedure piglets to control for gel differences. Western blot data were analyzed using the Mann-Whitney rank sum test or the Kruskal-Wallis ANOVA on ranks with post hoc Dunn’s pairwise comparisons. Statistical significance was set at p < 0.05.
2.7 |. Sample size estimate
A priori data about the effects of GNX-4728 on neonatal piglet brain after HI are not available. However, a similar mPTP inhibitor reduced cardiac ischemia in rabbits by 26% (Fancelli et al., 2014). Using this estimate, n = 4 would permit rejection of the null hypothesis of no protection at an alpha level of 0.05 and power of 0.9. We increased the sample size for the current study to six or seven piglets/group to permit some variability in our initial effect size estimates.
3 |. RESULTS
Twenty-nine piglets underwent HI or sham procedure for planned neuropathologic evaluation with a target survival time of 4 days. Three piglets did not complete the protocol and were excluded from analysis—one from the HI-GNX-4728 group and two from the HI vehicle group. The HI GNX-4728 animal died on recovery day 1 before reaching the 4th recovery day (12% failure). One HI vehicle animal could not be resuscitated from cardiac arrest; the other HI vehicle animal was sacrificed on recovery day 2 for clinical seizures (22% failure). Thus, seven HI GNX-4728, seven HI vehicle, six sham GNX-4728, and six sham vehicle piglets were analyzed by neuropathology. An additional 12 neonatal piglets (HI n = 6, sham n = 6), five neonatal naive piglets, and four juvenile pigs were used for western blotting.
3.1 |. Physiology
At baseline, there were no significant differences in weight, temperature, pH, partial pressure of arterial carbon dioxide, partial pressure of arterial oxygen, oxygen saturation, hemoglobin, MAP, or HR between groups (Table 2). During HI, the HI GNX-4728 and HI vehicle groups had similar levels of oxyhemoglobin saturation, acidosis, and hypercarbia during hypoxia and asphyxia. Two HI GNX-4728 pigs and one HI vehicle pig required defibrillation. Rectal temperature in the HI GNX-4728 group, before drug treatment, was 0.5°C lower than the HI vehicle group at hypoxia 42 min (p < 0.05) but this difference disappeared during asphyxia. During recovery from asphyxia, there were differences between groups in temperature at ROSC 5 min (p < 0.05) and in pH at ROSC 1 hr (p < 0.05) with the HI animals demonstrating lower values, but without significant post hoc pairwise comparisons. There were no differences in oxygen saturation or paO2 values between groups. MAP and HR decreased during asphyxia in both HI groups, then rebounded at ROSC 5 min without differences between the HI groups. At 1 hr, MAP recovered to levels seen in the sham controls.
TABLE 2.
Physiological parameters
| Parameter/Group | n | Baseline (median, IQR) | Hypoxia 42 min (median, IQR) | Asphyxia (median, IQR) | ROSC 5 min (median, IQR) | ROSC 1H (median, IQR) | ROSC 2H (median, IQR) |
|---|---|---|---|---|---|---|---|
| Weight, kg | |||||||
| HI GNX | 7 | 1.72 (1.56, 1.80) | |||||
| HI vehicle | 7 | 1.60 (1.45, 1.76) | |||||
| Sham GNX | 6 | 1.62 (1.55, 1.72) | |||||
| Sham vehicle | 6 | 1.53 (1.49, 1.71) | |||||
|
| |||||||
| Temperature (°C) | |||||||
| HI GNX | 7 | 37.6 (37.5, 37.8) | 38.4 (38.2, 38.7)a | 38.4 (37.8, 38.7) | 37.9 (37.6, 38.2)b | 38.3 (37.6, 39.0) | 38.7 (38.6, 39.0) |
| HI vehicle | 7 | 38.3 (36.0, 38.8) | 38.9 (38.7, 39.2) | 38.1 (37.3, 38.9) | 37.9 (37.7, 38.9) | 38.4 (38.0, 39.0) | 38.9 (38.7, 39.1) |
| Sham GNX | 6 | 38.1 (37.8, 38.5) | 38.7 (38.2, 39.1) | 38.8 (38.5, 39.1) | 39.1 (39.0, 39.3) | ||
| Sham vehicle | 6 | 38.2 (37.5, 38.9) | 38.9 (38.5, 39.1) | 39.2 (38.6, 39.4) | 38.6 (38.3, 39.1) | ||
|
| |||||||
| pH | |||||||
| HI GNX | 7 | 7.39 (7.36, 7.42) | 7.29 (7.26, 7.32) | 6.75 (6.70, 6.85) | 7.40 (7.26, 7.41) | 7.30 (7.20, 7.35)c | 7.38 (7.32, 7.44) |
| HI vehicle | 7 | 7.40 (7.34, 7.41) | 7.25 (7.22, 7.31) | 6.78 (6.74, 6.82) | 7.38 (7.35, 7.49) | 7.33 (7.24, 7.36) | 7.33 (7.33, 7.40) |
| Sham GNX | 6 | 7.39 (7.33, 7.46) | 7.39 (7.31, 7.44) | 7.37 (7.36, 7.44) | 7.35 (7.33, 7.45) | ||
| Sham vehicle | 6 | 7.35 (7.31, 7.37) | 7.33 (7.31, 7.36) | 7.42 (7.32, 7.43) | 7.38 (7.33, 7.48) | ||
|
| |||||||
| paCO2, mmHg | |||||||
| HI GNX | 7 | 37.6 (33.5, 41.7) | 39.6 (38.3, 41.1) | 110.5 (104.1, 114.5) | 38.4 (32.4, 49.4) | 40.1 (35.3, 42.8) | 40.7 (38.8, 41.5) |
| HI vehicle | 7 | 37.6 (36.9, 42.0) | 43.1 (39.2, 47.5) | 114.0 (110.0, 128.0) | 39.3 (38.8, 39.6) | 37.8 (34.7, 46.7) | 40.4 (38.9, 56.8) |
| Sham GNX | 6 | 38.6 (36.8, 43.3) | 39.4 (35.4, 41.4) | 37.1 (32.8, 39.8) | 35.6 (33.1, 45.9) | ||
| Sham vehicle | 6 | 41.0 (39.5, 44.6) | 39.6 (34.4, 42.0) | 40.4 (34.6, 46.9) | 39.5 (31.7, 41.5) | ||
|
| |||||||
| paO2, mmHg | |||||||
| HI GNX | 7 | 144 (138, 191) | 30.4 (27.8, 32.7) | 15.2 (12.7, 18.8) | 147 (140, 165) | 137 (118, 138) | 136 (112, 142) |
| hi vehicle | 7 | 165 (150, 189) | 31.4 (28.2, 32.6) | 18 (15.7, 23.6) | 162 (146, 190) | 146 (126, 165) | 140 (106, 150) |
| Sham GNX | 6 | 162 (131.6, 180.8) | 148 (143.2, 169.8) | 131.5 (130.5, 152.5) | 148 (114, 154.5) | ||
| Sham vehicle | 6 | 144 (131.2, 156.2) | 140 (107.6, 162.5) | 135 (124, 147.5) | 137 (102.9, 172.5) | ||
|
| |||||||
| SaO2, % | |||||||
| HI GNX | 7 | 100 (99.8, 100) | 34.3 (24.2, 37.1) | 4.5 (3.9, 6.5) | 100 (99.2, 100) | 99.7 (98.9, 100) | 99.7 (98.6, 100) |
| HI vehicle | 7 | 100 (99.8, 100) | 30.0 (25.8, 38.5) | 3.6 (3.3, 6.7) | 100 (99.4, 100) | 99.9 (99.5, 100) | 99.4 (98.5, 100) |
| Sham GNX | 6 | 100 (96.7, 100) | 100 (99.8, 100) | 100 (99.7, 100) | 99.6 (99.4, 100) | ||
| Sham vehicle | 6 | 100 (99.5, 100) | 99.5 (98.8, 100) | 99.8 (99.0, 100) | 99.5 (98.9, 100) | ||
|
| |||||||
| Hemoglobin, g/dl | |||||||
| HI GNX | 7 | 6.9 (6.1, 8.4) | 7.1 (6.1, 8.5) | 7.3 (6.7, 8.7) | 6.3 (5.8, 6.3) | 7.6 (5.8, 7.7) | 7.9 (6.8, 8.7) |
| HI vehicle | 7 | 7.7 (6.8, 8.9) | 8.0 (7.4, 9.1) | 8.7 (7.5, 9.1) | 7.0 (6.4, 7.8) | 7.8 (6.8, 8.8) | 8.2 (7.8, 9.5) |
| Sham GNX | 6 | 7.1 (6.7, 8.1) | 7.6 (6.6, 8.7) | 8.4 (7.2, 9.1) | 8.9 (6.9, 14.3) | ||
| Sham vehicle | 6 | 7.6 (6.7, 8.6) | 8.8 (5.5, 9.1) | 8.5 (6.7, 9.4) | 7.9 (6.0, 9.3) | ||
|
| |||||||
| MAP, mmHg | |||||||
| HI GNX | 7 | 75 (64, 78) | 77 (68 84) | 34 (25, 38) | 119 (112, 129)d,†,‡ | 65 (63, 68) | 67 (62, 75) |
| HI vehicle | 7 | 73 (62, 75) | 69 (66, 73) | 25 (16, 48) | 112 (104, 117)‡ | 56 (56, 70) | 63 (59, 73) |
| Sham GNX | 6 | 58 (50, 69) | 60 (51, 88) | 70 (53, 86) | 64 (50, 84) | ||
| Sham vehicle | 6 | 73 (59, 84) | 78 (62, 81) | 84 (67, 94) | 66 (55, 84) | ||
|
| |||||||
| HR, bpm | |||||||
| HI GNX | 7 | 171 (152, 188) | 246 (225, 259) | 55 (43, 73) | 274 (226, 297) | 203 (191, 210) | 204 (188, 242) |
| HI vehicle | 7 | 195 (165, 210) | 225 (222, 253) | 42 (13, 62) | 280 (271, 285) | 210 (207, 230) | 210 (150, 227) |
| Sham GNX | 6 | 192 (144, 224) | 185 (147, 223) | 178 (140, 257) | 199 (171, 238) | ||
| Sham vehicle | 6 | 194 (162, 208) | 192 (170, 270) | 205 (170, 258) | 192 (145, 213) | ||
Abbreviations: bpm, beats per minute; HI, hypoxic-ischemic injury; HR, heart rate;MAP, mean arterial blood pressure; paCO2, partial pressure of carbon dioxide; paO2, partial pressure of oxygen; ROSC, return of spontaneous circulation (ROSC 5 min refers to 5 min after ROSC); SaO2, arterial oxygen saturation.
At 42 min of hypoxia, HI GNX piglets were more hypothermic than the HI vehicle piglets (U = 3.5, p = 0.008) by the Mann-Whiney rank sum test.
At ROSC 5 min, there was a significant difference in temperature between groups (H(3) = 9.536, p = 0.023) by the Kruskal-Wallis ANOVA on ranks test, this did not reach significance on post hoc Dunn’s test (p > 0.05).
At ROSC 1 hr, there was a significant difference in pH between groups (H(3) = 8.017, p = 0.046) by Kruskal-Wallis, this did not reach significance on post hoc Dunn’s test (p > 0.05).
At ROSC 5 min, there was a significant difference in MAPs between groups (H(3) = 17.633, p = 0.001). On post hoc pairwise comparisons, the HI GNX group had significantly higher MAPs than sham GNX (p = 0.003) and sham vehicle (p = 0.01). The HI vehicle group also had significantly higher MAPs than sham GNX (p = 0.05) in post hoc pairwise comparisons
p < 0.05 versus sham vehicle in post hoc Dunn’s test;
p < 0.05 versus sham GNX in post hoc Dunn’s test.
GNX-4728 decreased the MAP more than vehicle. Of the piglets where MAP changes were noted after GNX-4728 or vehicle administration, 60%–75% of those animals given GNX-4728 had a MAP drop > 10 mmHg, while none of the animals given vehicle had such a change (Table 3).
TABLE 3.
Mean arterial blood pressure (MAP) changes after GNX-4728 or vehicle administration
| Number of animals where MAP changes were available | MAP drop > 10 mmHg after drug/vehicle dose (%) | |
|---|---|---|
| HI GNX | 5/7 | 3 (60%) |
| HI vehicle | 3/7 | 0 |
| Sham GNX | 4/6 | 3 (75%) |
| Sham vehicle | 3/6 | 0 |
3.2 |. GNX-4728 does not protect against HI-induced ischemic-necrosis in putamen
The average and percent total number of ischemic-necrotic neurons per field in putamen differed significantly among groups (H(3) = 17.97, p < 0.001; H(3) = 17.825, p < 0.001). In post hoc pairwise tests, the HI vehicle (n = 7) and HI GNX-4728 (n = 7) groups had more ischemic-necrotic neurons in putamen than did sham vehicle piglets (n = 6; p = 0.005; Figure 2a,b). The average and percent total number of injured neurons did not differ among groups (p > 0.05). In contrast, the percentage of normal putamen neurons differed significantly among groups (H(3) = 14.8, p = 0.002) with HI vehicle (n = 7) and HI GNX-4728(n = 7) having less normal neurons than sham vehicle (n = 6; p = 0.008; Figure 2b) in post hoc comparisons. In somatosensory cortex, there were no significant differences in the number of ischemic-necrotic, injured, or normal neurons among the four groups (Figure 2c,d, p > 0.05 for all comparisons).
FIGURE 2.
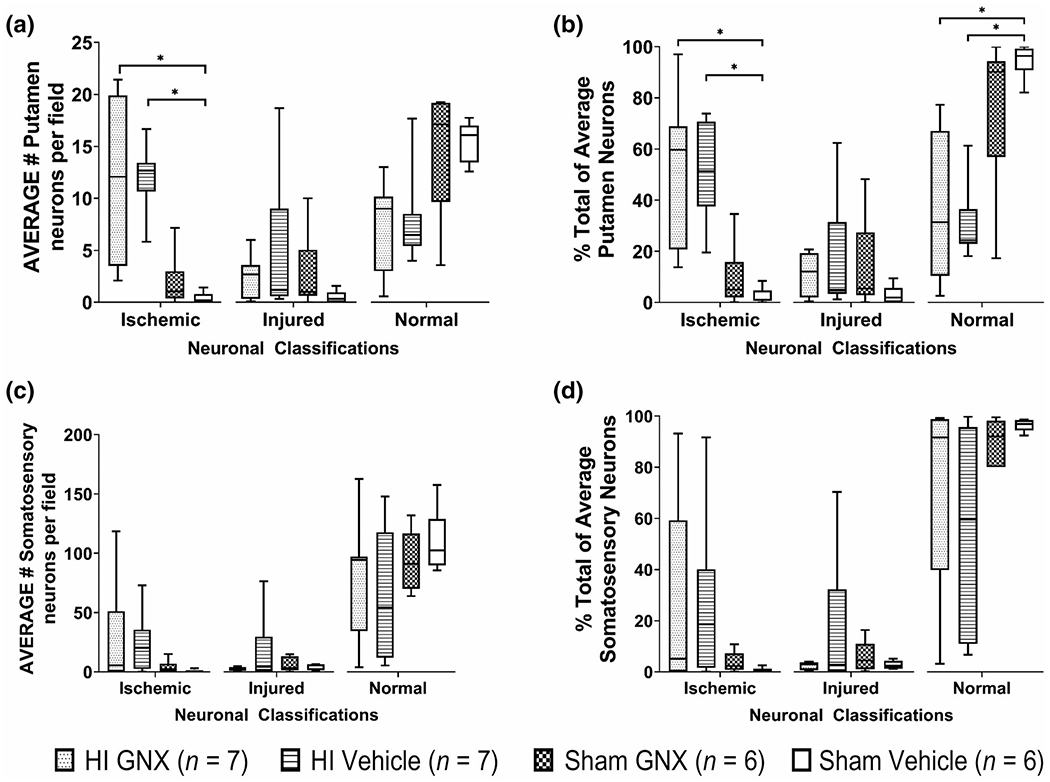
Putamen and somatosensory cortex neuron counts at 1,000× magnification with oil immersion represented as average number of neurons per field and % total of average neuron counts. (a, b) In putamen, the number of ischemic neurons in the HI GNX-4728 (n = 7) and HI vehicle (n = 7) groups were significantly higher compared with sham vehicles (n = 6, *p < 0.05). In contrast, normal neuron percentage was lower in the HI GNX-4728 and HI vehicle groups compared with sham vehicle (*p < 0.05). (c, d) In somatosensory cortex there were no significant differences among groups. Survival was 4 days
3.3 |. HI acutely depletes mitochondria of SOD2 in putamen
In mitochondrial-enriched fractions from putamen (Figure S1), we detected SOD2 immunoreactivity at ~ 23 kD, which is consistent with expectation from rodent western blots (Martin et al., 2007). SOD2 levels at 6h were significantly lower after HI (n = 6) than after sham procedure (n = 6, U = 4, p = 0.026; Figure 3a,b). Putative mPTP proteins (VDAC, ANT, CypD) were all identified in pig brain at molecular weights consistent with observations seen in rodent (Martin et al., 2009) and human brain mitochondria (Wong et al., 2013) (Figure S2). VDAC, ANT, and CypD levels did not differ between HI and sham piglets at 6h (n = 6, p > 0.05; Figure 3c–g).
FIGURE 3.
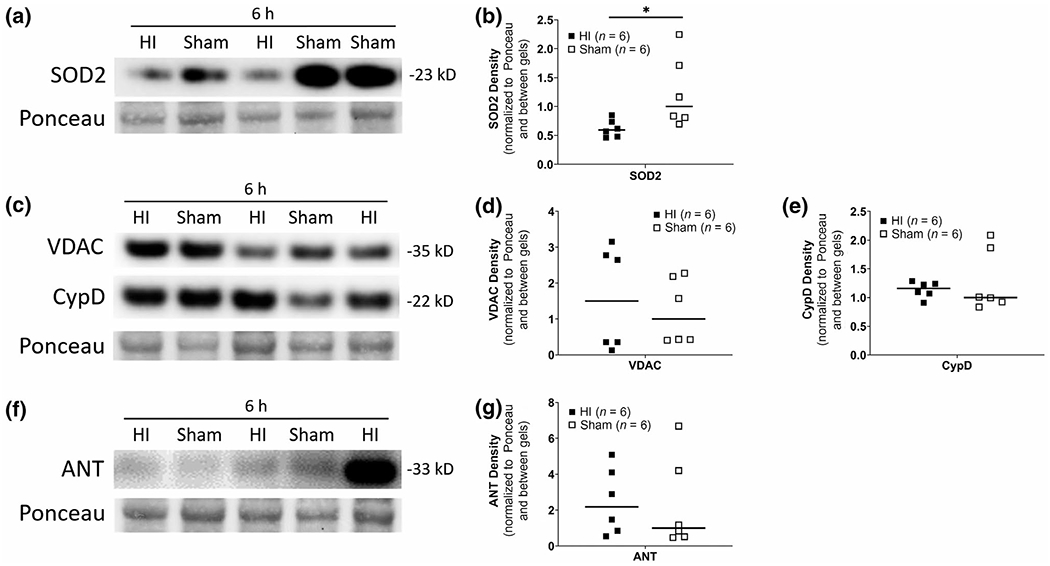
Representative HI versus sham western blots of neonatal putamen mitochondrial-enriched fractions detected by the mitochondrial matrix protein superoxide dismutase-2 (SOD2). See Figure S1 for validation of tissue fractionation method, (a, b) Mitochondrial SOD2 depletion occurs early after HI. SOD2 immunodensity is significantly lower 6h after HI (n = 6) when compared to sham (n = 6) (*p < 0.05). (c-g) There were no significant differences in voltage-dependent anion channel (VDAC), cyclophilin D (CypD), or adenine nucleotide translocase (ANT) immunodensities between HI and sham. Ponceau S-stained membranes of exactly the same blots are shown for protein loading
3.4 |. mPTP proteins are differentially present in the brains of anesthetized neonatal shams relative to mature juvenile brain
SOD2 levels were comparable in mitochondrial-enriched fractions from putamen of neonatal sham (n = 5) and mature juvenile pigs (n = 4, U = 5, p = 0.286, Figure 4a,b). Within this cohort, VDAC levels were lower in the neonatal sham putamen compared to that of juvenile putamen (U = 1, p = 0.032, Figure 4c,d). CypD had lower levels in the neonatal shams compared to juvenile that was not statistically significant (U = 2, p = 0.063, Figure 4c,e). ANT levels were significantly lower in neonatal shams compared to that in the juveniles (U = 1, p = 0.032, Figure 4f,g).
FIGURE 4.
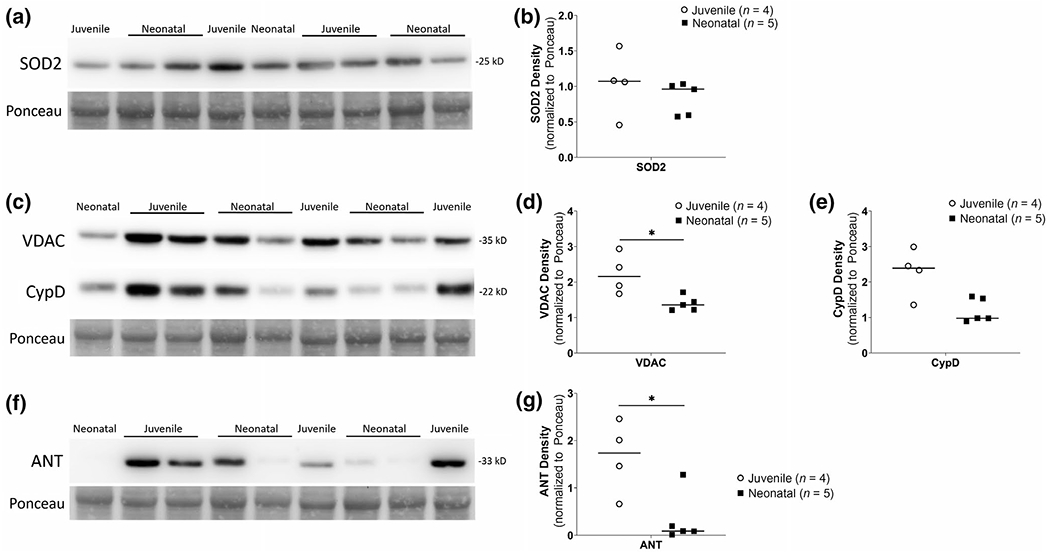
Neonatal sham anesthesia versus juvenile piglet western blots for mPTP proteins in mitochondrial-enriched fractions from putamen. (a, b) No differences in SOD2 immunodensities between groups. (c-e) VDAC showed lower immunodensities in neonatal sham (n = 5) compared to juvenile putamen fractions (n = 4, *p < 0.05). CypD also showed a similar pattern but was not statistically significant (p = 0.063). (f-g) ANT immunodensities were significantly lower in neonatal shams (n = 5) compared to that of juveniles (n = 4, *p < 0.05). Ponceau S-stained membranes of exactly the same blots are shown for protein loading
3.5 |. CypD levels are different in unanesthetized neonatal naïve brain relative to the mature juvenile brain
Separate western blots comparing unanesthetized neonatal naïve piglets (n = 5) to mature juvenile pigs (n = 4) are shown in Figure 5. In mitochondrial-enriched fractions from putamen, SOD2 levels were not significantly different between neonatal naïve (n = 5) and mature juvenile animals (n = 4, U = 5, p = 0.286, Figure 5a,b). VDAC and ANT levels were also not different between the groups (U = 4, p = 0.19 for VDAC, Figure 5c,d; U = 10, p = 1 for ANT, Figure 5f,g); however, CypD levels were lower in the neonatal naïve brain compared to that of the mature juvenile (U = 0, p = 0.016, Figure 5c,e).
FIGURE 5.
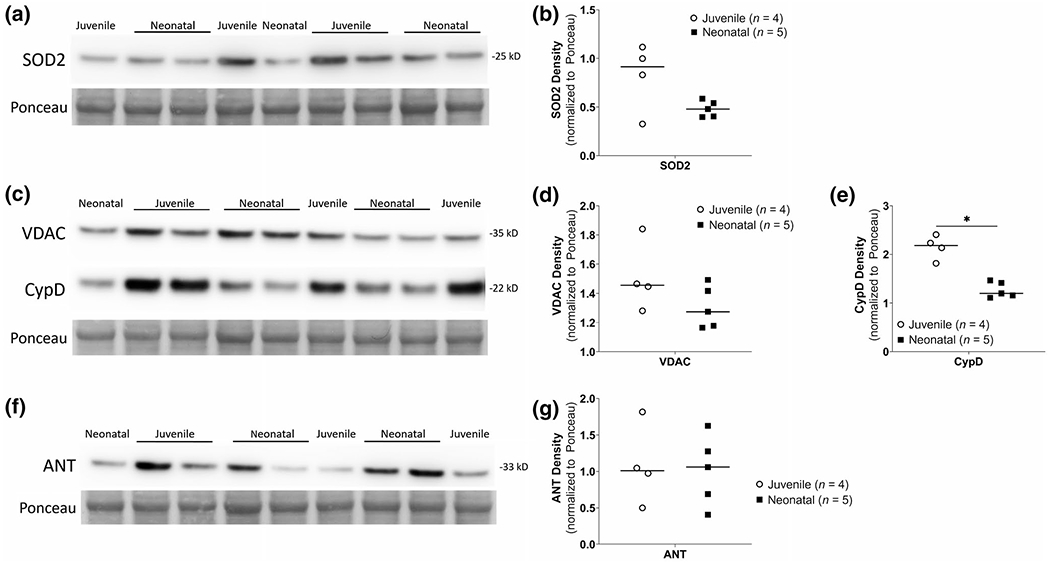
Neonatal naïve unanesthetized versus juvenile piglet western blots for mPTP proteins in mitochondrial-enriched fractions from putamen. (a, b) No differences in SOD2 immunodensities between groups. (c, d) VDAC immunodensities were not significantly different between groups; however, CypD immunodensities were significantly lower in neonatal naive fractions (n = 5) compared to juvenile putamen fractions (n = 4, *p < 0.05). (f, g) ANT immunodensities were similar in neonatal naïve and juvenile fractions. Ponceau S-stained membranes of exactly the same blots are shown for protein loading
4 |. DISCUSSION
Our study focused on three aspects of mitochondria in neonatal piglet brain. We first examined the mPTP as a therapeutic target for neuroprotection in HIE. Secondly, we tested the emergence and acuteness of brain mitochondrial molecular pathology in a piglet model of HI. Finally, we studied the postnatal development of mPTP protein levels in pig brain with and without anesthesia.
We found that a single acutely administered dose of the mPTP inhibitor GNX-4728 was ineffective at preventing neuronal ischemic-necrosis in putamen after 4-day survival from neonatal HI. The HI injury did not severely damage the somatosensory cortex when neurons from cortical layers 2–6 were examined together, and accordingly GNX-4728 did not affect the cortical neuronal counts. We have previously shown that the putamen and primary somatosensory cortex are vulnerable to HI in our piglet model (Agnew et al., 2003; Martin et al., 1997; Wang et al., 2015). The presence of putamen damage at 4-day survival in the current study concurs with our past studies (Martin et al., 1997). In this piglet HI model, cortical layer 5 is typically injured with significant loss of normal neurons (Lee et al., 2012; Martin et al., 1997), but more variable injury in neocortex is also seen (Martin et al., 1997). Lesser injury in the other cortical layers may have decreased the effect of HI on the neuron counts in the current study. Additionally, we classified neurodegeneration as ischemic-necrotic and by a distinct cellular morphology of injured neurons with apparent cytopathology. While the former neuronal cell pathology is the conventional damage that is typical of human neonatal HIE (Northington et al., 2011), the latter category of injured neurons did not meet the criteria of ischemic-necrosis but likely falls along the cell death continuum (Martin, 2001). GNX-4728 did not shift this continuum.
We did not study pharmacokinetics or structure-activity relationships of GNX-4728 in piglets. Nonetheless, we previously showed that GNX-4728 crosses the blood-brain barrier after a single bolus and it enhances brain mitochondrial calcium retention capacity (Martin et al., 2014). Though GNX-4728 has some brain mitoprotective properties in a neonatal mouse model of HI, there was no histopathologic protection (Fang et al., 2019). We conducted the current piglet study to examine the neonatal brain mPTP given the prominent differences in small and large animal models of neonatal HI (Koehler et al., 2018), differences in neuropathology among animal models (Northington et al., 2011), and differences in neuronal cell death mechanisms in mouse, pig, and human neurons (Martin & Chang, 2018).
The benefits and strengths of using a large animal HI model were exemplified by the ability to do detailed physiologic and specifically continuous hemodynamic monitoring and also IV delivery of drugs. The HI piglets had lower rectal temperatures soon after resuscitation likely as a result of poor circulation after asphyxia. Some piglets were acidotic after HI but had normal pH restored by 2h. The higher MAPs in the HI groups immediately after ROSC (and before GNX-4728 or vehicle administration) were likely due to the epinephrine given for resuscitation. Importantly, we identified that GNX-4728 can acutely decrease the MAP by >10 mmHg during drug infusion over 5min. This may account for the toxic effects of GNX-4728 seen in the mouse model of HI at high doses (Fang et al., 2019) where continuous blood pressure monitoring was not possible.
While we did not observe the same protective benefits of GNX-4728 seen in adult animal models (Fancelli et al., 2014; Martin et al., 2014), our results concur with the absence of neuroprotection from GNX-4728 in the neonatal mouse model of HI (Fang et al., 2019). The lack of neuroprotection from GNX-4728 could be due to several reasons. The extra central nervous system (CNS) action of GNX-4728 as shown on systemic blood pressures in Table 3 may have deleteriously effected cerebral blood flow and autoregulation therefore limiting its neuroprotective capacity. Independent of drug treatment, the SOD2 depletion suggests that mitochondria are damaged or have an altered function by 6h after HI and moreover, mPTP molecular components may be lower in neonatal piglet brain relative to the more mature juvenile pig brain. A rapidly evolving mitochondrial pathology after HI in the brain at term gestation alongside the immature properties of neonatal mPTP may make mitochondria a difficult therapeutic target for neonatal HI.
We have also previously found structural evidence of mitochondrial pathology that is present 3h after HI in piglets (Martin et al., 2000), thus supporting our current observation of depletion of SOD2 at 6h on western blot. Early subcellular drug target loss due to rapid mitochondrial destruction may have precluded mitochondria-mediated protection. Similarly in neonatal rodent brain, mitochondrial cell death protein mobilization occurs within 2h of excitotoxic injury (Lok & Martin, 2002) and mitochondrial bioenergetic and structural failure occurs within 3h after HI insult (Chavez-Valdez et al., 2012; Northington et al., 2007). We administered GNX-4728 within 5min after return of spontaneous circulation in an effort to provide mitochondrial protection through inhibition of the mPTP. However, our findings suggest that calcium dysregulation and mitochondrial permeabilization may have already been occurring at this very early time point with engagement of neuronal cell death mechanisms. Using a similar compound, GNX-4975, Richardson and Halestrap established that the mPTP inhibitors work more efficiently in an initially calcium-depleted environment (Richardson & Halestrap, 2016). As a result, GNX-4728 may be less effective in neonatal HI because injury and calcium loading have likely occurred prior to drug administration. We do not yet know precisely when calcium loading occurs in HI piglet striatal neurons.
The SOD2 depletion seen at 6h after HI while other mitochondrial proteins (VDAC, ANT) and even a different matrix protein (CypD) were not depleted at 6h is intriguing. The persistence of the mPTP proteins suggests that mitochondria have not yet undergone complete destruction at 6h. Meanwhile, the SOD2 finding points to a new highly selective mechanism of striatal neuron injury where mitochondrial function is altered in the presence of SOD2 depletion driving unmitigated superoxide generation (normally quenched by SOD2). Evidence of this superoxide pathology has been shown in the putamen of HI piglets with increased levels of peroxynitrite, formed by superoxide and nitric oxide (Martin et al., 2000). Furthermore, an antioxidant with superoxide dismutase mimetic properties, EUK134, can protect striatal neurons in piglets with HI (Ni et al., 2011). It is therefore possible that highly specific mitochondrial-targeted potent SOD2 mimetic drugs acutely administered could be therapeutic in neonatal HIE and warrants further study.
Our finding on the developmental regulation of mPTP protein levels is also new. They anticipate a potential developmentally immature form of the mPTP that may have therapeutic relevance for neonatal and mature brains and perhaps different tissue types. Our mitochondrial isolation and fractionation was completed using flash-frozen tissue, which could fracture the mitochondria and lead to spurious western blot results for mitochondrial proteins. However, we addressed this possibility and found no difference between fresh and flash-frozen putamen mitochondrial fractions and protein expression of VDAC or CypD (Figure S2). The western blot data in neonatal naive unanesthetized piglets showed that CypD, the most convincing regulator of the mPTP, while sustained in neonatal brain, was at low levels. Furthermore, mPTP components may be more sensitive to anesthesia in the neonatal brain relative to the mature juvenile brain. Western blot data in neonatal sham anesthetized piglets showed distinct protein level differences with variability in CypD and significantly lower VDAC and ANT when compared with juvenile animals. A biophysically immature mPTP as well as anesthesia effects might have further reduced the efficacy of GNX-4728. This could explain the potent therapeutic efficacy of GNX-4728 in adult mouse neurodegenerative disease (Martin et al., 2014) and adult rabbit myocardial infarction (Fancelli et al., 2014) and the lack of protection in neonatal mouse (Fang et al., 2019) and piglet HI. Our study highlights the importance of examining developmental differences in mitochondrial physiology and the mPTP (Robertson et al., 2004; Wang et al., 2009) when considering mitochondria-targeted therapies for neonatal brain.
Our study had several limitations. The sample size was small with only male animals but the design was a proof-of-concept study for targeting mitochondrial protection. The timing and administration of a single dose of GNX-4728 after injury could be inadequate for neuroprotection since brain bioavailability of the drug was unknown in the piglet model. We had no published data on drug tolerability in piglets and so the dose was conservative and delivered at a time when mitochondrial pathology was acutely emerging. Mitochondrial failure after HI occurs putatively in two phases (Hagberg et al., 2014). We targeted the primary phase because of its certainty in the involvement in acute neuronal destruction. The identified acute hypotension after GNX-4728 administration and our prestudy limited drug-dosing trial showing greater systemic blood pressure effects with a higher dose suggest that further dose increases and more frequent doses could be detrimental. It is possible that a multi-dose regimen targeting both phases of injury with very slow administration of the drug (slower than 5 min) to avoid hypotension could increase the possibility of neuroprotection. However, given early mitochondrial injury and lower mPTP availability in the neonatal brain that we identified in our study, multidosing could equally be unsuccessful. Lastly, while we were able to demonstrate SOD2 depletion after HI in a similar cohort of piglets, this pilot study, which had a histopathologic endpoint, was not designed for biochemical assays after GNX-4728 treatment. However, future studies should optimize the use of fresh tissue for mitochondrial assays and include additional evaluations such as GNX-4728 levels in the brain and mitochondrial function assays or calcium retention capacity studies.
5 |. CONCLUSION
In our pilot study with a large animal model of neonatal HI, the mPTP inhibitor GNX-4728 was ineffective at neuroprotection in putamen. We advise caution in the translation of adult neuroprotective agents for use in a neonatal population. Furthermore, developmental differences in mitochondrial biology, specifically mPTP composition, may limit the usefulness of mitochondria and the mPTP as organelle and molecular targets for therapeutics in neonatal HIE.
Supplementary Material
Figure S1. Subcellular fractionation in piglet putamen. (a) Representative Ponceau S-stained membrane showing total protein loading of the different fractions using the ThermoFisher NE-PER extraction kit. Fraction selective protein banding can be seen in the different lanes. (b) Western blotting of the fractions for SOD2 verified that the mitochondrial (mito)/organelle fractions used in the paper have a strong mitochondrial (SOD2) signal compared to the other fractions. The mito fraction has minimal cytosolic (GAPDH) and absent nuclear (histone) contamination
Figure S2. Rapid fresh (not frozen) tissue homogenization preparation versus frozen putamen homogenization preparation for isolation of mitochondrial-enriched fractions. (a) Western blots for VDAC and CypD along with (b) band quantification do not show a difference between fresh and flash-frozen samples. A human putamen sample is also shown with a band seen co-migrating at the expected molecular weight for VDAC and CypD with the piglet samples. Human lane was cropped to show a longer exposure time
Transparent Science Questionnaire for Authors
Transparent Peer Review Report
Significance.
Mitochondrial dysfunction in neonatal hypoxic-ischemic encephalopathy (HIE) drives neuronal injury. Targeting of mitochondria and the mitochondrial permeability transition pore (mPTP) are neuroprotective strategies in adult animal models; however, in neonatal brain, data are lacking. We showed that the mPTP inhibitor GNX-4728 does not protect the neonatal piglet brain after hypoxia-ischemia, found mitochondrial matrix injury, and discovered that the neonatal and juvenile brain mPTP could be biophysically different in composition. We conclude that mitochondrial-targeted drugs with efficacy in adults may not translate to the neonatal brain because of organelle vulnerability and differences in mitochondrial properties.
ACKNOWLEDGMENTS
The authors thank Congenia Srl-Genextra Group (Milan, Italy) for providing GNX-4728. They also thank Dr. Rashmi Singh for her assistance in fractionating piglet putamen samples into mitochondrial-enriched fractions.
Funding information
This work was supported by grants from the National Institutes of Health (R01NS107417 [JKL, LJM, RCK], R01NS109029 [JKL, RCK], and R01NS060703 [RCK]), the American Heart Association Transformational Project Award (co-funded by the Lawrence J. and Florence A. DeGeorge Charitable Trust; 18TPA34170077 [JKL, RCK, LJM]), and the Johns Hopkins University Alzheimer’s Disease Research Center (AG005146 [LJM]).
CONFLICT OF INTEREST
Dr. Martin received prior funding for a different project from Congenia Srl-Genextra Group, Milan, Italy. Dr. Lee is currently a paid consultant for Edwards Life Sciences and was a paid advisory board member for Medtronic. Dr. Lee also received a research grant from Medtronic for a separate study. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. The other authors declare no conflict of interest.
Footnotes
DECLARATION OF TRANSPARENCY
The authors, reviewers and editors affirm that in accordance to the policies set by the Journal of Neuroscience Research, this manuscript presents an accurate and transparent account of the study being reported and that all critical details describing the methods and results are present.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Agnew DM, Koehler RC, Guerguerian AM, Shaffner DH, Traystman RJ, Martin LJ, & Ichord RN (2003). Hypothermia for 24 hours after asphyxic cardiac arrest in piglets provides striatal neuroprotection that is sustained 10 days after rewarming. Pediatric Research, 54, 253–262. 10.1203/01.PDR.0000072783.22373.FF [DOI] [PubMed] [Google Scholar]
- Bernardi P, Colonna R, Costantini P, Eriksson O, Fontaine E, Ichas F, Massari S, Nicolli A, Petronilli V, & Scorrano L (1998). The mitochondrial permeability transition pore. BioFactors, 8, 273–281. 10.1002/biof.5520080315 [DOI] [PubMed] [Google Scholar]
- Chavez-Valdez R, Martin LJ, Flock DL, & Northington FJ (2012). Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience, 219, 192–203. 10.1016/j.neuroscience.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, & Martin LJ (2011). Epigenetic regulation of motor neuron cell death through DNA methylation. Journal of Neuroscience, 31, 16619–16636. 10.1523/JNEUROSCI.1639-ll.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad MS, & Johnson RW (2015). The domestic piglet: An important model for investigating the neurodevelopmental consequences of early life insults. Annual Review of Animal Biosciences, 3, 245–264. 10.1146/annurev-animal-022114-111049 [DOI] [PubMed] [Google Scholar]
- Dobbing J, & Sands J (1979). Comparative aspects of the brain growth spurt. Early Human Development, 3, 79–83. 10.1016/0378-3782(79)90022-7 [DOI] [PubMed] [Google Scholar]
- Fancelli D, Abate A, Amici R, Bernardi P, Ballarini M, Cappa A, Carenzi G, Colombo A, Contursi C, Di Lisa F, Dondio G, Gagliardi S, Milanesi E, Minucci S, Pain G, Pelicci PG, Saccani A, Storto M, Thaler F, … Plyte S (2014). Cinnamic anilides as new mitochondrial permeability transition pore inhibitors endowed with ischemia-reperfusion injury protective effect in vivo. Journal of Medicinal Chemistry, 57, 5333–5347. 10.1021/jm500547c [DOI] [PubMed] [Google Scholar]
- Fang J, Chavez-Valdez R, Flock DL, Avaritt O, Saraswati M, Robertson C, Martin LJ, & Northington FJ (2019). An inhibitor of the mitochondrial permeability transition pore lacks therapeutic efficacy following neonatal hypoxia ischemia in mice. Neuroscience, 406, 202–211. 10.1016/j.neuroscience.2019.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi A, Wilson MA, & Johnston MV (2009). Hypoxic-ischemic encephalopathy in the term infant. Clinics in Perinatology, 36, 835–858. 10.1016/j.clp.2009.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Rousset CI, & Thornton C (2014). Mitochondria: Hub of injury responses in the developing brain. The Lancet Neurology, 13, 217–232. 10.1016/S1474-4422(13)70261-8 [DOI] [PubMed] [Google Scholar]
- Halestrap AP (2009). What is the mitochondrial permeability transition pore? Journal of Molecular and Cellular Cardiology, 46, 821–831. 10.1016/j.yjmcc.2009.02.021 [DOI] [PubMed] [Google Scholar]
- Hunter DR, & Haworth RA (1979). The Ca2+ induced membrane transition in mitochondria. Archives of Biochemistry and Biophysics, 195, 453–459. 10.1016/0003-9861(79)90371-0 [DOI] [PubMed] [Google Scholar]
- Jacobs S, Berg M, Hunt R, Tarnow-Mordi W, Inder T, & Davis P (2013). Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database of Systematic Reviews. Art. No. :CD003311. [Google Scholar]
- Koehler RC, Yang ZJ, Lee JK, & Martin LJ (2018). Perinatal hypoxic-ischemic brain injury in large animal models: Relevance to human neonatal encephalopathy. Journal of Cerebral Blood Flow and Metabolism, 38, 2092–2111. 10.1177/0271678X18797328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Poretti A, Perin J, Huisman TAGM, Parkinson C, Chavez-Valdez R, O’Connor M, Reyes M, Armstrong J, Jennings JM, Gilmore MM, Koehler RC, Northington FJ, & Tekes A (2017). Optimizing cerebral autoregulation may decrease neonatal regional hypoxic-ischemic brain injury. Developmental Neuroscience, 39, 248–256. 10.1159/000452833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Wang B, Reyes M, Armstrong JS, Kulikowicz E, Santos PT, Lee JH, Koehler RC, & Martin LJ (2016). Hypothermia and rewarming activate a macroglial unfolded protein response independent of hypoxic-ischemic brain injury in neonatal piglets. Developmental Neuroscience, 38, 277–294. 10.1159/000448585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Yang ZJ, Wang B, Larson AC, Jamrogowicz JL, Kulikowicz E, Kibler KK, Mytar JO, Carter EL, Burman HT, Brady KM, Smielewski P, Czosnyka M, Koehler RC, & Shaffner DH (2012). Noninvasive autoregulation monitoring in a swine model of pediatric cardiac arrest. Anesthesia and Analgesia, 114, 825–836. 10.1213/ANE.0b013e31824762d5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AWC, Varanyuwatana P, & Halestrap AP (2008). The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. Journal of Biological Chemistry, 283, 26312–26323. 10.1074/jbc.M805235200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, Cousens S, Mathers C, & Black RE (2015). Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: An updated systematic analysis. Lancet, 385, 430–440. 10.1016/S0140-6736(14)61698-6 [DOI] [PubMed] [Google Scholar]
- Lok J, & Martin LJ (2002). Rapid subcellular redistribution of bax precedes caspase-3 and endonuclease activation during excitotoxic neuronal apoptosis in rat brain. Journal of Neurotrauma, 19, 815–828. 10.1089/08977150260190410 [DOI] [PubMed] [Google Scholar]
- Lu Y, Tucker D, Dong Y, Zhao N, Zhuo X, & Zhang Q (2015). Role of mitochondria in neonatal hypoxic-ischemic brain injury. Journal of Neuroscience and Rehabilitation, 2, 1–14. [PMC free article] [PubMed] [Google Scholar]
- Martin LJ (2001). Neuronal cell death in nervous system development, disease, and injury (Review). International Journal of Molecular Medicine, 7, 455–478. 10.3892/ijmm.7.5.455 [DOI] [PubMed] [Google Scholar]
- Martin LJ (2010). Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals, 3, 839–915. 10.3390/ph3040839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ (2011). An approach to experimental synaptic pathology using green fluorescent protein-transgenic mice and gene knockout mice to show mitochondrial permeability transition pore-driven excitotoxicity in interneurons and motoneurons. Toxicologic Pathology, 39, 220–233. 10.1177/0192623310389475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Brambrink A, Koehler RC, & Traystman RJ (1997). Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. Journal of Comparative Neurology, 377, 262–285. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Price AC, Kaiser A, Agnew DM, Ichord RN, & Traystman RJ (2000). Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiology of Diseases, 7, 169–191. 10.1006/nbdi.2000.0282 [DOI] [PubMed] [Google Scholar]
- Martin LJ, & Chang Q (2018). DNA damage response and repair, DNA methylation, and cell death in human neurons and experimental animal neurons are different. Journal of Neuropathology and Experimental Neurology, 77(7), 636–655. 10.1093/jnen/nly040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Fancelli D, Wong M, Niedzwiecki M, Ballarini M, Plyte S, & Chang Q (2014). GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Frontiers in Cellular Neuroscience, 8, 433. 10.3389/fncel.2014.00433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, & Chang Q (2009). The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Experimental Neurology, 218, 333–346. 10.1016/j.expneurol.2009.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, & Golden WC (2007). Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. Journal of Comparative Neurology, 500, 20–46. 10.1002/cne.21160 [DOI] [PubMed] [Google Scholar]
- Ni X, Yang ZJ, Carter EL, Martin LJ, & Koehler RC (2011). Striatal neuroprotection from neonatal hypoxia-ischemia in piglets by antioxidant treatment with EUK-134 or edaravone. Developmental Neuroscience, 33, 299–311. 10.1159/000327243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, & Martin LJ (2011). Neuronal cell death in neonatal hypoxia-ischemia. Annals of Neurology, 69, 743–758. 10.1002/ana.22419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Zelaya ME, O’Riordan DP, Blomgren K, Flock DL, Hagberg H, Ferriero DM, & Martin LJ (2007). Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience, 149, 822–833. 10.1016/j.neuroscience.2007.06.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien CE, Santos PT, Kulikowicz E, Reyes M, Koehler RC, Martin LJ, & Lee JK (2019). Hypoxia-ischemia and hypothermia independently and interactively affect neuronal pathology in neonatal piglets with short-term recovery. Developmental Neuroscience, 41, 17–33. 10.1159/000496602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AP, & Halestrap AP (2016). Quantification of active mitochondrial permeability transition pores using GNX-4975 inhibitor titrations provides insights into molecular identity. The Biochemical Journal, 473, 1129–1140. 10.1042/BCJ20160070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Bucci CJ, & Fiskum G(2004). Mitochondrial response to calcium in the developing brain. Developmental Brain Research, 151, 141–148. 10.1016/j.devbrainres.2004.04.007 [DOI] [PubMed] [Google Scholar]
- Santos PT, O’Brien CE, Chen MW, Hopkins CD, Adams S, Kulikowicz E, Singh R, Koehler RC, Martin LJ, & Lee JK (2018). Proteasome biology is compromised in white matter after asphyxic cardiac arrest in neonatal piglets. Journal of the American Heart Association, 7, e009415. 10.1161/JAHA.118.009415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran S, Pappas A, McDonald SA, Vohr BR, Hintz SR, Yolton K, Gustafson KE, Leach TM, Green C, Bara R, Huitema CMP, Ehrenkranz RA, Tyson JE, Das A, Hammond J, Peralta-Carcelen M, Evans PW, Heyne RJ, Wilson-Costello DE, … Higgins RD (2012). Childhood outcomes after hypothermia for neonatal encephalopathy. New England Journal of Medicine, 366, 2085–2092. 10.1056/NEJMoalll2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- šileikytė J, & Forte M (2016). Shutting down the pore: The search for small molecule inhibitors of the mitochondrial permeability transition. Biochimica et Biophysica Acta (BBA)–Bioenergetics, 1857, 1197–1202. 10.1016/j.bbabio.2016.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton C, Leaw B, Mallard C, Nair S, Jinnai M, & Hagberg H (2017). Cell death in the developing brain after hypoxia-ischemia. Frontiers in Cellular Neuroscience, 11, 1–19. 10.3389/fncel.2017.00248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Armstrong JS, Lee JH, Bhalala U, Kulikowicz E, Zhang H, Reyes M, Moy N, Spicer D, Zhu J, Yang ZJ, Koehler RC, Martin LJ, & Lee JK (2015). Rewarming from therapeutic hypothermia induces cortical neuron apoptosis in a swine model of neonatal hypoxic-ischemic encephalopathy. Journal of Cerebral Blood Flow and Metabolism, 35, 781–793. 10.1038/jcbfm.2014.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Armstrong JS, Reyes M, Kulikowicz E, Lee JH, Spicer D, Bhalala U, Yang ZJ, Koehler RC, Martin LJ, & Lee JK (2016). White matter apoptosis is increased by delayed hypothermia and rewarming in a neonatal piglet model of hypoxic ischemic encephalopathy. Neuroscience, 316, 296–310. 10.1016/j.neuroscience.2015.12.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Carlsson Y, Basso E, Zhu C, Rousset CI, Rasola A, Johansson BR, Blomgren K, Mallard C, Bernardi P, Forte MA, & Hagberg H (2009). Developmental shift of Cyclophilin D contribution to hypoxic-ischemic brain injury. Journal of Neuroscience, 29, 2588–2596. 10.1523/JNEUROSCI.5832-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Gertz B, Chestnut BA, & Martin LJ (2013). Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Frontiers in Cellular Neuroscience, 7, 279. 10.3389/fncel.2013.00279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Martin LJ, Northington FJ, & Zhang J (2019). Oscillating-gradient diffusion magnetic resonance imaging detects acute subcellular structural changes in the mouse forebrain after neonatal hypoxia-ischemia. Journal of Cerebral Blood Flow and Metabolism, 39, 1336–1348. 10.1177/0271678X18759859 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Subcellular fractionation in piglet putamen. (a) Representative Ponceau S-stained membrane showing total protein loading of the different fractions using the ThermoFisher NE-PER extraction kit. Fraction selective protein banding can be seen in the different lanes. (b) Western blotting of the fractions for SOD2 verified that the mitochondrial (mito)/organelle fractions used in the paper have a strong mitochondrial (SOD2) signal compared to the other fractions. The mito fraction has minimal cytosolic (GAPDH) and absent nuclear (histone) contamination
Figure S2. Rapid fresh (not frozen) tissue homogenization preparation versus frozen putamen homogenization preparation for isolation of mitochondrial-enriched fractions. (a) Western blots for VDAC and CypD along with (b) band quantification do not show a difference between fresh and flash-frozen samples. A human putamen sample is also shown with a band seen co-migrating at the expected molecular weight for VDAC and CypD with the piglet samples. Human lane was cropped to show a longer exposure time
Transparent Science Questionnaire for Authors
Transparent Peer Review Report
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.