

Abstract
mRNA display is a powerful biological display platform for the directed evolution of proteins and peptides. mRNA display libraries covalently link the displayed peptide or protein (phenotype) with the encoding genetic information (genotype) through the biochemical activity of the small molecule puromycin. Selection for peptide/protein function is followed by amplification of the linked genetic material and generation of a library enriched in functional sequences. Iterative selection cycles are then performed until the desired level of function is achieved, at which time the identity of candidate peptides can be obtained by sequencing the genetic material.
The purpose of this review is to discuss the development of mRNA display technology since its inception in 1997 and to comprehensively review its use in the selection of novel peptides and proteins. We begin with an overview of the biochemical mechanism of mRNA display and its variants with a particular focus on its advantages and disadvantages relative to other biological display technologies. We then discuss the importance of scaffold choice in mRNA display selections and review the results of selection experiments with biological (e.g., fibronectin) and linear peptide library architectures. We then explore recent progress in the development of “drug-like” peptides by mRNA display through the post-translational covalent macrocyclization and incorporation of non-proteogenic functionalities. We conclude with an examination of enabling technologies that increase the speed of selection experiments, enhance the information obtained in post-selection sequence analysis, and facilitate high-throughput characterization of lead compounds. We hope to provide the reader with a comprehensive view of current state and future trajectory of mRNA display and its broad utility as a peptide and protein design tool.
Keywords: mRNA display, directed evolution, in vitro selection, non-natural amino acids, macrocyclization, next generation sequencing
Graphical Table of Contents

1. Overview
New functional proteins and peptides can aid in the development of new therapeutics to treat human disease or function as molecular tools to help solve biological and chemical problems. Despite recent advances in the rational design of proteins,1 directed evolution remains an important and robust functional solution to engineer new protein and peptide function. In directed evolution, a large library of diverse proteins is first partitioned for a desired function and the functional molecules are subsequently amplified. This process is repeated until most to all of the pool is highly enriched in molecules that perform the desired task (Figure 1).
Figure 1. Directed evolution process.

In directed evolution, a library of molecules is subjected to a selective step that separates functional and non-functional molecules. Functional molecules are then amplified to generate a library for the next round of selection. This process is repeated until the library is dominated by functional molecules. Directed evolution requires that the genotype (e.g., DNA or RNA) is linked to its corresponding phenotype (function).
A key requirement for directed evolution is that the genotype (i.e., DNA or RNA) and phenotype (function) of the molecules must be linked, otherwise amplification of the “fittest” sequences is impossible. The directed evolution of proteins presents a challenge: once a protein is synthesized during translation, its sequence information is lost; no current methods are capable of reverse translation nor can the protein be directly amplified through a process analogous to polymerase chain reaction (PCR). Several “display” methods have been developed to link a protein to its encoding genetic material including phage display (PD)2, 3, mRNA display,4 ribosome display,5 peptides-on-plasmids,6 and CIS display.7 For a comprehensive discussion of biological display technologies, the reader is directed to these reviews.8–12
Of these display methods, mRNA display has several unique advantages over other display techniques including the ability to generate the most diverse peptide libraries and the ability to engineer drug-like peptides containing unnatural amino acids. Over the last 20 years, mRNA display has been used to develop high affinity ligands against a wide range of targets including intracellular proteins, extracellular proteins, membrane proteins, and RNA. In this review, we first introduce basic mRNA display principles, mechanisms, and variants. We define mRNA display as any technique using puromycin to link genetic information to proteins and/or peptides. We discuss the application of mRNA display to engineer new proteins, peptides, and drug-like peptides and efforts to use these new molecules as tools for chemical biology, molecular imaging, and therapy. Finally, we highlight recent technological advances to improve mRNA display, including increasing speed and depth of post-selection analysis.
mRNA display is an elegant solution to the display problem as it involves simply linking a protein to its encoding mRNA.4 This is accomplished by appending puromycin to the 3’ end of an mRNA via a molecular spacer (Figure 2). Puromycin is an antibiotic that mimics the 3’ end of an aminoacylated tRNA (Figure 3) and acts as a bridge between mRNA and protein. The resulting fusion of mRNA and protein (i.e., an mRNA-protein fusion) generates a molecular “Rosetta Stone,” allowing the sequence of the peptide to be recovered by reverse transcription/PCR of the linked nucleic acid followed by DNA sequencing.
Figure 2. mRNA-peptide fusion formation.

(A) puromycin-ligated mRNA is translated by the ribosome (pink) in vitro. (B) When the ribosome reaches the mRNA/DNA junction, puromycin enters the A site of the ribosome, forming a covalent bond with the C-terminus of the nascent peptide, generating an mRNA-peptide fusion. (C) The mRNA strand is reverse transcribed into cDNA, enabling sequence amplification and identification, and eliminating RNA secondary structure.
Figure 3. Structures of Tyrosyl tRNA and puromycin.

Puromycin is a structural mimic of the 3’ end of aminoacylated tRNA. The differences between puromycin and tyrosyl tRNA are highlighted in red
A typical mRNA display in vitro selection cycle is shown in Figure 4. A DNA library, obtained from synthetic or natural sources, is first transcribed into mRNA. A linker containing puromycin is attached to the 3’ end of the mRNA and the resulting mRNA-puromycin template is translated in vitro. The resulting mRNA-protein fusion is typically reverse transcribed into a cDNA/mRNA duplex, which prevents the inadvertent selection of RNA aptamers. The pool of mRNA-peptide fusions is then subjected to a selective step, typically binding to an immobilized macromolecular target, which separates functional and non-functional sequences. Non-functional sequences are washed away and cDNA from functional sequences is amplified by PCR, generating a dsDNA library enriched in functional sequences. This library can either be used for more rounds of selection or sequenced to identify the functional protein sequences.
Figure 4. mRNA display cycle.

A library of dsDNA consisting of a 5’ promoter and 5’ untranslated region (UTR) is PCR amplified and transcribed into an mRNA library which is subsequently ligated to a puromycin-linker. The ligated mRNA library is then translated in vitro to form a library of mRNA-peptide fusion molecules. The mRNA-peptide fusion library is then reverse transcribed and panned against the target of interest to isolate functional sequences. PCR amplification regenerates the DNA library, which can be used for another round of selection or sequenced to identify functional peptides.
2. mRNA Display
2.1. mRNA Display Mechanism
All mRNA display formats require a DNA library with a 5’ promoter for transcription (e.g., for T7 RNA polymerase), a 5’ untranslated region (UTR) for the preferred cell-free translation system, start codon, open reading frame (ORF; usually containing a mix of fixed and random codons), and a 3’ UTR used for RT-PCR amplification. Libraries can be constructed to include an affinity purification tag (e.g., His tag, FLAG tag, hemagglutinin (HA)-tag, etc.) or chemical moiety for purification.
The in vitro translation systems used in mRNA display can be bacterial, eukaryotic, or a synthetic reconstitution of selected proteins from either source. The current model for fusion formation involves translation of an mRNA template until the ribosome reaches the RNA/DNA junction and stalls. There, puromycin enters the A-site and the peptidyl transferase activity of the ribosome catalyzes the formation of an amide bond between the primary amino group of puromycin and the C-terminus of the nascent protein chain in the P-site (Figure 2). This fusion formation reaction must occur “in cis,” i.e., an mRNA template must be fused to the protein that it encodes. A “trans” reaction – an mRNA fused to a protein translated by another mRNA – would result in a mixed message, preventing directed evolution. Early experiments showed that translation of two templates of different lengths resulted in only the correctly fused products. No mixed products were observed, supporting a cis fusion formation model.4, 13 Experiments optimizing the linker length between mRNA and puromycin also support a model for cis fusion formation. Increasing the linker length beyond ~30 nucleotides (which roughly spans a distance corresponding to that between the mRNA decoding site and the peptidyl transferase center) results in decreased fusion formation efficiency, suggesting that trans products would be unlikely to form.13 Lastly, many new ligands have been engineered using mRNA display, which would not be possible unless the fidelity of fusion formation between mRNA and protein within the same ribosome is preserved.
Ribosomal stalling at the mRNA/DNA junction also seems to play an important role in fusion formation. In a study to determine where puromycin can enter the ribosome, Starck et al. used a biotinylated derivative of puromycin to pull-down puromycin-terminated products.14 Rather than observing random puromycin entry at many sites in the sequence, this group observed that puromycin only terminated translation at distinct sites in the protein sequence. This observation suggested that these sites corresponded to ribosomal pause points during translation of the mRNA, supporting the idea that a pause during translation in mRNA display is important for puromycin entry. When the mRNA template is linked to puromycin through a DNA linker (typically poly-dA), the DNA-RNA junction provides an efficient pause site.13 In TRAP display (Transcription–Translation coupled with Association of Puromycin linker), a UAG stop codon is placed at the 3’ end of the ORF to induce ribosomal pausing.15
2.2. mRNA Display Variants
Several variants of mRNA display have been developed, all of which use puromycin as the means to link mRNA and protein (Figure 5). These technologies include Profusion™,16 in vitro virus (IVV) display,17 cDNA display,18 and TRAP display.15 The main difference in these variants lies in the structure of the puromycin linker that bridges mRNA and protein. The original mRNA display linker consisted of a simple deoxyadenosine spacer (5’ – dA27dCdCP – 3’, where P = puromycin),4 which was trademarked as Profusion™ technology.13, 16 Subsequent optimization experiments showed that increased flexibility in the linker, introduced via incorporation of polyethylene glycol spacers (Spacer 9), resulted in higher rates of fusion formation between mRNA and translated protein.13 In both cases, these puromycin linkers were ligated to mRNA using T4 DNA Ligase in the presence of a DNA splint (Figure 5A).19 Alternatively, the puromycin linker can be added to the mRNA using photo-crosslinking by incorporating a psoralen20, 21 (Figure 5B) or a 3-cyanovinylcarbazole nucleoside (CNVK).22 The advantage of psoralen ligation is that the ligated product can be directly translated without a gel purification step, whereas splint-mediated ligation requires gel purification to eliminate the DNA splint that can lead to RNase H-mediated degradation of the mRNA.23 Similarly, a branched puromycin linker containing both psoralen and a reverse transcription primer (Figure 5C) can be used to create a cDNA-protein fusion or a dsDNA-protein fusion, which both show higher stability versus mRNA-protein fusions in environments where RNase is present.20
Figure 5. mRNA Display Variants.

All mRNA display systems use puromycin to conjugate a protein to its encoding mRNA. How the puromycin linker is attached to the mRNA differs between mRNA display variants. DNA is represented by grey lines while RNA is represented by black lines. (A) Classical mRNA display uses a DNA splint in the ligation between mRNA and the linker. The puromycin linker contains a poly dA stretch to stall the ribosome for puromycin entry and serve as an affinity handle for poly-deoxythymidine (dT) purification after fusion formation. Psoralen mediated photo-crosslinking can also be used to form (B) mRNA-protein fusions or (C) cDNA-protein fusions. (D) In TRAP display, the puromycin linker is not covalently attached to the mRNA and the mRNA 3’ UTR contains a UAG codon to stall the ribosome and enhance puromycin entry. (E) In cDNA display, the puromycin linker consists of four main parts including a ligation site, a restriction site, a reverse transcription primer site, and a biotin site.
The IVV approach to mRNA display was named for an mRNA-protein fusion’s similarity to a simple retrovirus.17 In this system, puromycin is attached to mRNA via a DNA/RNA chimera nucleotide termed a “P-acceptor” and a 105 nucleotide DNA spacer linking protein and mRNA.17 A second generation IVV was generated through simple hybridization of a puromycin linker containing 2’-OMe nucleotides, leading to a non-covalent peptide fusion.24 A more recent version, TRAP display, uses a non-covalent hybridized DNA-containing puromycin linker and incorporates a UAG codon into the mRNA to stall the ribosome and enhance puromycin entry (Figure 5D).15 This format was found to reduce the time required for a single selection round to <3 hours.
Several other technologies using splint-less ligation methods have also been developed. A Y-ligation method uses a puromycin linker that hybridizes to the mRNA, after which short single-stranded sections of the mRNA and linker are ligated in the presence of T4 RNA ligase to form a small single-stranded loop.25, 26 This strategy has been further developed to synthesize cDNA-fusions using a linker similar to the branched psoralen linker described above, except that instead of using psoralen crosslinking, the Y-ligation strategy is used.18, 27, 28 This branched Y-ligation linker contains a reverse transcription primer for generation of a covalently linked cDNA-peptide fusion. In some versions of this technique, this linker also incorporates a restriction site and biotin group, allowing for immobilization of the ligated mRNA followed by solid-phase translation and reverse transcription (Figure 5E). This approach effectively eliminates the need for intermediate purification steps27 and the subsequent cDNA-peptide fusions can be released from the solid phase by restriction enzyme cleavage.18 A recent well-controlled study comparing mRNA display and cDNA display showed that both mRNA and cDNA display are comparable in enriching functional sequences, though mRNA display showed higher enrichment versus cDNA display, possibly due to higher non-specific binding of the cDNA display fusions.29 However, this effect was only seen when gel purification steps were included during library synthesis.
3. Advantages and Disadvantages of mRNA Display
As with any technology, mRNA display has a number of inherent advantages and disadvantages that should be considered. As an entirely in vitro technology, virtually any library can be constructed ranging from a totally random peptide library to those based on a complex protein scaffolds (e.g., antibodies. The covalent link between mRNA and protein allows a wider range of temperatures, buffers, binding conditions, and incubation times during selection compared to ribosome display, where a non-covalent ternary complex must be maintained throughout the selection. Flexibility in buffer conditions is also an advantage of in vitro selection systems, as display systems involving cells, such as yeast display (YD) or E. Coli display (ED), require conditions that keep cells viable and intact. Finally, the covalent link between peptide and protein, coupled with the minimized peptide-nucleic acid structure of the mRNA-protein fusion allows for the possibility of post-translational chemical modification which dramatically enhances the potential chemical complexity of mRNA display libraries.
mRNA display also has a number of disadvantages. One general limitation of many mRNA display selections (and all selections using biochemically purified targets) is that targets are often presented on a solid-phase support, which may not recapitulate the conformation of the target in a native biological context. For example, if a target can adopt multiple conformations, not all of these conformations may be sampled on the solid support. Also, by selecting against an immobilized target, in vitro selections partition molecules based purely on their binding affinity or binding kinetics (i.e., KD, kon, and/or koff) as it is difficult to devise a method to partition molecules based on their activity. As a result, some selections result in molecules that can bind to a target in vitro, but show no activity in biological systems or cannot bind the target in a biological context (see, for example, Sections 5.2.2, 5.2.4, or 6.2.1). An alternative strategy is to overexpress a target on the cell surface and select for binding against these cells. However, ensuring that the selection results in ligands to the target of interest is often difficult, even if cells without overexpressed target are used as a negative selection step. Lastly, the mRNA portion of an mRNA-protein fusion can help to solubilize the linked protein, allowing marginally soluble proteins to be selected. As a result, downstream expression or synthesis of the selected proteins or peptides can be challenging.
3.1. Diversity
mRNA display4 and ribosome display libraries30 are effectively the largest peptide libraries that are experimentally accessible. While it is physically possible to construct even larger libraries, such efforts are impractical requiring kilograms of genetic material. Up to 1015 unique sequences can be synthesized using mRNA display (Figure 6) and even a single copy of a functional sequence can be PCR amplified following in vitro selection. In contrast, in vivo display technologies, such as PD, are typically limited to a practical diversity of approximately 109 sequences owing to the obligate in vivo transformation step and resulting loss of diversity. mRNA display libraries are orders of magnitude larger than the immune system (~109 unique molecules), one-bead-one-compound (OBOC) libraries31 (~107 unique molecules), and non-ribosomal peptide synthetase (NRPS) or polyketide synthetase (PKS) libraries32, 33 (~103 unique molecules).
Figure 6. Biological display platforms including one-bead-one-compound (OBOC), yeast display (YD), phage display (PD), E. coli display (ED) and mRNA display libraries.

The platforms are compared based on their complexity and the maximum dissociation constant (KD,max) that can be achieved for the highest affinity ligand for a given library complexity. Ribosome display libraries have the same complexity as mRNA display. KD,max values were roughly estimated according to Reference 34 (not to scale).
In general, library size is directly correlated with the probability of obtaining a functional sequence of desired affinity.34, 35 A theoretical study by Rosenwald36 found that the degree of binding site complementarity is correlated to the size and complexity of the ligand, e.g., the more topologically complex the ligand, the more interactions it can make with a topologically complex binding site, such as a protein surface. There is also a correlation between the initial diversity of the library and the probability of obtaining a ligand of a given dissociation constant (KD), and for every log of increased complexity in the starting library, a log of decreased KD is obtained. An example of the effect of library size on affinity is shown by comparing selections against streptavidin using mRNA display and PD. Using mRNA display, Wilson et al. discovered 20-amino acid peptide sequences that bound to streptavidin with dissociation constants ranging from 110 nM to as low as 2.4 nM.37 In comparison, a PD selection against streptavidin resulted in a single peptide that bound with 13 μM affinity.38, 39 While different libraries were used in these selections, these results suggest that large mRNA display libraries may yield higher affinity compounds than PD.
Despite the advantages of highly diverse libraries, there are some notable drawbacks. For example, if a high diversity library is constructed so that there is only one copy of each sequence, it is possible that functional sequences could be lost stochastically in the first round of selection. Therefore, the copy number of each random sequence in the library should be carefully considered.40 In addition, because there are more total sequences in a high diversity library, more non-functional sequences must be removed in order to identify the functional sequences. This implies more rounds required for convergence in mRNA display (4 to >10) as compared with PD (generally, <3). In addition to requiring more time, additional selection cycles can result in contamination or selection failure. In the end, high diversity display libraries generated by selection platforms like mRNA display offer the potential for identifying high affinity compounds which must be considered alongside the increased selection time and technical difficulty.
3.2. Biases in Display Technology
All display technologies suffer from some bias and no display technology will generate a truly unbiased library. In a translation-based in vitro selection (e.g., mRNA display, ribosome display, PD, YD, etc.) the codon bias of the translation machinery, mRNA secondary structure, and post-translational processing are well-known sources of library bias.41, 42 Finally, differential efficiencies in the amplification and transcription of the fused genetic material may also introduce significant bias into the final protein library.
In PD, proteins are displayed on the surface of a micrometer-long bacteriophage with a negative surface charge [MW > 1 X 108 Da] raising the possibility of non-selective target binding through interactions between the phage and target. In ribosome display, the ribosome, mRNA, and translated peptide form a stable ternary complex. Although smaller than phage, the ribosome is still a ~2,000,000 Da particle, raising the possibility of steric occlusion and/or non-selective binding driven by the nucleic acid and protein components of the ribosome. In other display technologies, peptide or protein libraries are linked to even larger molecules such as cells (ED or YD) or plasmids. mRNA display, on the other hand, only contains mRNA linked to peptide or protein,43 and is the minimal set of elements needed for protein or peptide display. The effect of the display scaffold on the quality of selection results or scaffold bias has been discussed elsewhere44, 45 and is an important source of background binding and false-positive hits.
Display technologies that require an in vivo transformation step (e.g., PD, YD, etc.) introduce additional library biases. In the case of PD, peptides are most commonly displayed as fusions with Gene III (5 copies) or Gene VIII (thousands of copies). Multiple copies of a peptide displayed on phage can result in selection for weaker binding peptides due to avidity effects that prevent discrimination between moderate and high affinity clones.46 In mRNA display, proteins are presented in a strictly monomeric context, avoiding avidity effects and allowing the selection of high affinity sequences. The insertion of a peptide or protein into Gene III can also interfere with the infectivity of the phage particle. If phage amplification is biased by a peptide sequence, the abundance of a clone might reflect a clone’s infectivity and amplification rather than its binding to target. Lastly, during the process of phage assembly and export, known biases against certain sequences prevent display of some peptides (e.g., basic peptides).47–49
3.3. Simultaneous Optimization of Multiple Positions
The stepwise optimization of a peptide sequence is a relatively straightforward strategy – starting with a single functional sequence, the best amino acid at a single position is determined and repeated for each position in the sequence. Once the optimal amino acids are known at each position, they are then combined into one sequence. While a stepwise optimization strategy can be successful, it is often difficult to achieve in practice since changing an amino acid can result in unpredictable changes in binding affinity or specificity. One example is a study where Fiacco and co-workers attempted to introduce N-methyl amino acids into a peptide binding to the G-protein, Gαi1.50 Each position in this peptide was systematically substituted for its corresponding N-methyl amino acid, with many substitutions resulting in dramatic increases in protease resistance. However, almost all substitutions resulted in a decrease in affinity and one substitution resulted in a change in binding specificity from Gαi1 to Gα12.
The best amino acid at one position in a sequence can also change depending on the context of the surrounding positions. mRNA display offers a powerful solution for optimization of a sequence since multiple positions of a sequence can be optimized simultaneously. One example is where mRNA display was used to engineer a peptide that could not be achieved through stepwise modifications.51 In this study, the authors developed a macrocyclic peptide that bound Gαi1. Unlike the parental sequence, this peptide contained two N-methyl Alanine residues and was highly protease resistant. However, substitution of either residue with alanine lead to a 100-1,000-fold decrease in protease stability. In a hypothetical stepwise optimization experiment, adding N-methyl amino acids in a stepwise fashion to the original peptide would have resulted in peptides with no increase in stability and these peptides would have been discarded. As a result, it seems highly unlikely that a stepwise optimization strategy could have resulted in the discovery of the selected Gαi1-binding peptide. Furthermore, only 20% of the original natural amino acids of the parental peptide were retained in the selected peptide. This indicates that a leap of about 8 independent steps would have been required in a stepwise modification strategy beginning with the parental peptide and ending at the protease-resistant selected peptide.51
3.4. Simultaneous Optimization of Multiple Properties
An additional advantage of mRNA display is that it can also be used to optimize multiple biophysical properties of a sequence simultaneously. For example, Howell et al., developed a multi-step selection for binding and for protease resistance.52 This was achieved by first incubating an mRNA display library with chymotrypsin, thereby selecting for sequences that were protease resistant, followed by incubation of the library with target. A 100- to 400-fold increase in protease resistance was observed, while still retaining binding for target. Selections for thermostability have also been performed similarly.53 These examples illustrate the powerful ability of mRNA display to optimize properties along multiple fitness axes.
4. Results from mRNA Display Selections
In the ~20 years since its inception, mRNA display has successfully been used to engineer novel ligands against a wide variety of targets. In this section, we review the results of these selections and observe some common themes and results. These selections fall into two broad categories: 1) Protein selections, where the resulting ligands are folded and contain a buried hydrophobic core, and 2) Peptide selections, where no explicit structural constraints or contexts are imposed on the initial library. We will begin with a discussion of selections in the first category where known biological scaffolds (e.g., fibronectin) are partially randomized to generate mRNA display libraries from which folded, functional ligands can be identified. We will then review the selection of linear, natural peptides by mRNA display and their application as chemical tools. We will conclude by surveying the recent literature describing the use of cyclization and non-proteogenic amino acids to generate non-ribosomally synthesized peptide (NRSP)-like mRNA display libraries and their application to the selection of drug-like peptides. We note that while mRNA display libraries can be constructed from cDNA, we refer the reader to other publications for more information.54, 55
5. Biological Scaffolds in mRNA Display
There is great interest in engineering novel proteins for a number of applications, including therapeutics,56 imaging agents,57 diagnostics,58 and tools for biological/biochemical research. One difficulty encountered when engineering proteins is that it is impossible to exhaustively search the entire sequence space of even the smallest proteins. Sequence space is immeasurably large: the number of possible sequence combinations increases by a factor of 20 with each additional amino acid that is added to a protein chain. Thus, even using libraries of 1015 unique molecules, only a maximum of ~11-12 amino acids can be exhaustively searched by in vitro display technologies. As a result, it is often necessary to design and constrain a library to specific regions within the primary sequence. Many protein libraries are based on a protein “scaffold,” which remains constant and acts to support randomized regions that will be selected to interact with a target of interest. Often, the scaffold’s hydrophobic core is kept constant so that randomization of loops or surface residues will not result in unfolding, aggregation, or loss of solubility.59, 60
The choice of scaffold and if a scaffold is even needed should be carefully considered before embarking on an mRNA display selection. Matching the biophysical properties of a potential scaffold to the final intended end application is important, as each scaffold has both advantages and disadvantages. In general, scaffolds are thought to result in higher affinity ligands relative to peptides. Peptides are generally unstructured in solution and must pay a large entropic penalty upon binding resulting in weaker binding affinities.61 In contrast, random regions within a scaffold are comparatively fixed in place by the scaffold topology and lose less conformational entropy upon binding, resulting in improved affinity.62 Structured proteins are also generally more resistant to proteolytic degradation relative to linear peptides making them an attractive starting point for in vivo applications.63
These advantages must be weighed against the disadvantages of protein scaffolds. In mRNA display, longer proteins have a reduced efficiency of fusion formation,64 which can increase the technical difficulty of successfully completing an mRNA display selection and result in decreased effective library sizes. Likewise, scaffold-based proteins will be selected for both foldedness and function, which can potentially reduce the number of winning sequences. For example, Keefe and Szostak estimated that ATP-binding proteins selected from a random 80-mer library occurred at frequency of roughly 1 in 1011 molecules.65 They hypothesized that this surprising low frequency of ATP binders could have been due to the difficulty of forming a folded protein with a buried hydrophobic core from a totally random library. Additionally, longer random protein libraries often have increased chances of stop codons or frameshifts due to library construction, increasing the probability of aborted translation products.66 Lastly, larger protein scaffolds, such as the fibronectin and single chain fragment variable (scFv) domains, may suffer from reduced tissue/tumor penetration due to poor extravasation rates and rapid systemic clearance.67
In mRNA display, several scaffolds have been used in successful in vitro selections including nanobodies,68 SH3 domains,69 fragment antigen binding (Fab) domains,70 scFv domains, and fibronectin71 scaffolds. Of these, the two most common scaffolds are scFvs (~27 kDa) and fibronectin-based scaffolds (~10 kDa) (Figure 7). While both scaffolds are substantially larger than small peptides (~1-2 kDa), they are still approximately 1/5 to 1/10 the size of monoclonal antibodies (mAbs), facilitating binding to molecular crevices that antibodies are too large to access. Despite their smaller size, however, they are still capable of modulating protein function and can be more effective at blocking protein-protein interactions than small peptides.72, 73
Figure 7. Schematic of various mRNA-protein fusions.

(A) A 13-amino acid peptide attached to its encoding mRNA. Randomized regions (for mRNA and peptide) are red. (B) A fibronectin fusion with three randomized loops (BC (cyan), DE (green), and FG (red) attached to its encoding mRNA. Random regions in the mRNA are colored while the constant scaffold is dark blue. (C) An scFv attached to its encoding mRNA. Constant scaffold regions are dark blue while CDRs are colored accordingly.
5.1. Antibody Scaffolds
Antibodies are the gold standard for protein ligands and can bind to protein targets with high affinities and specificities. mAbs are among the most promising therapeutics and have revolutionized the pharmaceutical industry, with up to $200 billion in annual revenue predicted by 2022.74 Although hybridoma technology has been effectively used to generate thousands of new mAbs,75 this technology is over 40 years old and has a number of disadvantages. mAb production is laborious and little control can be exerted over the binding conditions and the stringency under which a mAb is generated, potentially resulting in mAbs with poor affinity and/or selectivity.76 mRNA display using antibody and antibody-like scaffolds is an attractive alternative for generating novel antibodies and antibody-like compounds, as the larger library size (Figure 6) and precise control over selection conditions can yield antigen-binding ligands with antibody-like affinity and specificity. In the following section, we review the use of mAb scaffolds (antibody) as well as Fab and scFv scaffolds (antibody-like) in mRNA display experiments.
Structurally, immunoglobulin variable domains (Fv) consist of two sandwiched β-sheets. Antigen specificity is conferred by loops that connect β-strands called complementarity determining regions (CDRs). CDRs vary with each antibody and comprise the antigen-binding surface.77 In mRNA display, as with the immune system, CDR loops are commonly randomized in order to create antibody and antibody-like libraries.71 Antibodies are composed of variable heavy (VH) and variable light (VL) chains, which contain both intermolecular (between VH and VL chains) and intramolecular (within a VH or VL chain) disulfide bonds.78 Because these disulfide bonds potentially make antibody folding and assembly difficult, mRNA display methods have been developed to increase the folded fraction of the library.79 Alternative translation conditions can also mitigate folding issues. For example, the PURE (Protein synthesis Using Recombinant Elements) system was used to select a disulfide-containing scFv targeting insulin.80 Reticulocyte-based systems have used protein disulfide isomerase and other disulfide-forming enzymes to translate and properly fold antibodies.81 Lastly, assembling an antibody that retains VH/VL pairing throughout a selection is difficult because the chains are encoded separately. Despite a method using emulsion PCR to retain Fab fragment VH/VL pairs,70 Fab fragments are uncommon in mRNA display.
For the past 20 years, the scFvs have been most frequently used antibody scaffold in mRNA display. The scFv scaffold balances the challenges associated with in vitro library development and expression (e.g., incorporating disulfide bonds) with the benefits of the antibody scaffold. scFvs are derived from VH and VL chains of antibodies where the chains are linked by glycine-serine repeats (Figure 8). Encoding the VH/VL pairs in a single protein chain simplifies mRNA display library construction and selection, making scFvs the preferable scaffold for antibody selection.
Figure 8. Antibody based scaffolds (scFv and Fab) compared to full IgG antibody.

(Fv: immunoglobulin variable domain, Fc: fragment crystallizable domain, Fab: antigen-binding fragment, VH: variable heavy, VL: variable light, CL: constant light, CH: constant heavy, scFv: single chain fragment variable)
mRNA display using scFv scaffolds has resulted in several successful selections. A selection against Mouse Double Minute 2 homolog (MDM2) and p53 combining Surface Plasmon Resonance (SPR) and an off-rate selection converged after two rounds to yield scFvs with 4.3 nM and 12 nM KD values, respectively.82 scFv selections using mRNA display were also performed against Tumor Necrosis Factor α Receptor (TNFR),83 fluorescein (0.95 nM KD),84 and Receptor Activator of Nuclear Factor κB (RANK).85
While scFvs may be better suited for mRNA display, full-length mAbs that include both the Fv and the Fc (Fragment crystallizable) constant regions are important for therapeutic purposes. These regions mediate antibody-dependent cell-mediated toxicity (ADCC), activation of complement, and are responsible for long serum half-lives.86
Unfortunately, as discussed above, full-length mAbs are extremely challenging to adapt to mRNA display library design and selection workflows. One strategy to create full-length mAbs is to first perform an mRNA display selection with an scFv library then graft the selected CDR loop sequences onto a full-length mAb scaffold. In one such study, scFvs obtained by mRNA display were used to design over 100 mAbs against 15 antigens.81 However, this strategy is only successful in some circumstances as many of the grafted antibodies showed loss of specificity/affinity. This phenomenon was also observed when scFvs were selected using PD.87, 88 These observations suggest that the scFv scaffold may not present the CDR loops in the same context/structure as an intact antibody.81
5.2. Fibronectin-Based Scaffolds
While antibody and antibody-like scaffolds are leading sources of biological therapeutics, alternative scaffolds are being developed for the next generation of therapeutic proteins.89 Disulfides in antibodies can cause structural heterogeneity and instability due to isomerization/alternative disulfide pairing, interconversion, and/or reduction.90, 91 Antibodies are also unstable in the reducing intracellular environment, making the use of antibodies inside the cell (i.e., intrabodies) more challenging. During mRNA display, disulfide bond-mediated protein folding also poses an additional challenge that can decrease the library complexity and impair selection robustness. As a result, alternative scaffolds that structurally mimic antibody variable chains without disulfide bonds have been developed for use in mRNA display selections. The most common of these is the fibronectin scaffold.
Fibronectin is a large extracellular matrix protein responsible for interfacing cells with the stromal environment.92 Three types of modules or domains (Type I, II, and III) are found in fibronectin and each type contains two anti-parallel β-sheets that form a β-sandwich. Type I and II domains contain disulfide bonds while the Type III domain lacks disulfide bonds, making the Type III domain an attractive scaffold for mRNA display. The 10th subunit of human Fibronectin is a Type III domain (10Fn3) that is widely used as a scaffold in mRNA display (trademarked as Adnectins™ by Bristol-Myers Squibb, or also called “monobodies”).62 Wild type 10Fn3 has several biophysical properties that are attractive for a biological scaffold: it has high solubility (>15 mg/mL), is highly thermostable (Tm >90 °C) even without disulfide bonds,62, 71, 93 and expresses well in bacterial systems (50 mg/L).94 The 94-amino acid 10Fn3 scaffold contains three loops (BC, FG, DE), analogous to nanobodies, which are based on the single variable domain camelid antibody VHH CDRs (Figure 9A and Figure 9B).95 The FG loop is the longest and most flexible of the three loops and is involved in endogenous RGD-mediated binding of wild type 10Fn3 with integrins.92 Randomization of the BC and FG loops does not affect 10Fn3 stability, allowing the BC and FG loops to form a contiguous molecular surface for target binding.62 mRNA display-generated 10Fn3 ligands have been used to as ligands for extracellular proteins in molecular imaging, immune regulation, crystallization, and therapeutic applications.96–98 10Fn3 probes for intracellular targets (intrabodies) have also been developed for dynamic in vivo imaging.99 A comprehensive list of fibronectin-based ligands selected by mRNA display is presented in Table 1.
Figure 9. The tertiary structure of (A) Llama VHH domain (PDB accession number 1TTG), (B) wild-type human 10Fn3 domain (PDB accession number 1HCV) and (C) truncated human 10Fn3 domain carrying structural mutations (PDB accession number 1HCV).
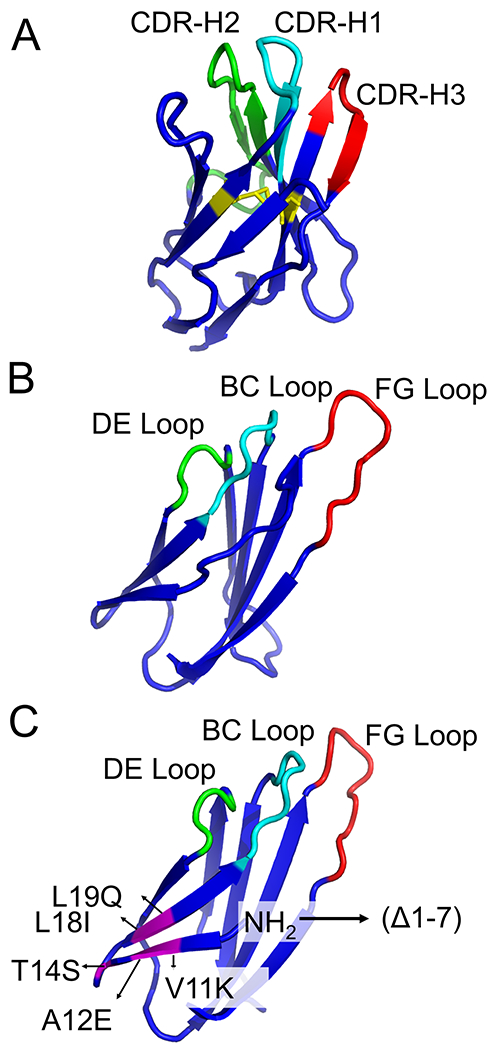
The Llama VHH domain and human 10Fn3 domain show comparable folding into β-sandwiches. (10Fn3: 10th subunit of Fibronectin type III), with the BC, DE, and FG of the 10Fn3 domain resembling the CDR loops of the Llama VHH domain. The complementarity determining regions (CDR) in the VHH domain and the randomized loops in the 10Fn3 domain are showed in green, cyan and red. The disulfide bond formed between cysteines 22 and 92 in the VHH domain is highlighted in yellow. In the truncated human 10Fn3 domain, the mutated residues are highlighted in magenta.
Table 1. Fibronectin Scaffolds Selected with mRNA Display.
Affinities are obtained directly from the corresponding reference and are not normalized for temperature.
| Target | Loop(s) Randomized | Library Diversity* | Rounds Selected | Affinity/ Solubility Maturation | Selected Scaffold Name | Final Loop Sequence(s) | Affinity/IC50/Kinetic Properties | Notes | Ref |
|---|---|---|---|---|---|---|---|---|---|
| TNF-α | BC DE FG |
1012 | 14 | Error-Prone PCR | M12.04 (10Fn3) | BC: SMTPNWP DE: PWAS FG: HRDT (FG truncation) |
KD= 0.02 nM | - | 71 |
| VEGFR2 | BC DE FG |
N/S | - | Thermo-stability optimization | 10Fn3- 159-(A56E) | BC: RHPHFPT DE: LQPP(E) FG: MAQSGHELFT |
KD= 0.59 nM | - | 112 |
| VEGFR2 (KDR); Flk-1 | BC DE FG |
1013 | 6 | Error-prone PCR | VR28-E19 | BC: RHPHFPT DE: LQPP FG: VERNGRHLMTP |
VEGFR2: KD = 0.06 nM Flk-1: KD = 0.34 |
- | 73 |
| Phospho-IκBα | BC FG |
3×1013 | 10 | Error-prone PCR | 10C17C25 (10FnIII) | BC: PASSRWR FG: QQLHQPKWRW |
KD = 18 nM | 1000x more specific for phosphorylated version; Used as intrabody FRET sensor | 113 |
| SARS nucleocapsid protein | BC FG |
1012 | 6 | - | Fn-N22 | BC: LNMWYNV FG: KRSFWSKSAG |
KD = 1.7 nM | Used as Intrabody | 114 |
| EGFR; IGF-1R (bispecific EI-tandem Adnectin) | BC DE FG |
1013 | 4 | - | EI-tandem Adnectin |
EGFR: BC: DSGRGSYQ; DE: GPVH; FG: DHKPHADGPHTYHES IGF-IR: BC: SARLKVAR DE: KNVY FG: RFRDYQ |
EGFR: KD = 10.1 nM IGF-IR: KD = 1.17 nm |
- | 72 |
| IL-6 | BC FG |
1012 | 4 | CFMS | eFn-4B02 | BC: GNGGPLV FG: HAFSGRVAVI |
KD = 21 nM | - | 115 |
| EGFR | BC DE FG |
N/S | - | - | Adnectin1 (EGFR) | BC: DSGRGSY DE: GPVH FG: DHKPHADGPHTYHES |
IC50= 50 nM KD = 2 nM |
IC50 was determined based on inhibition of EGFR phosphorylation | 96 |
| IL-23 | Adnectin 2 (IL-23) | BC: EHDYPYR DE: KDVD FG: SSYKYDMQYS |
IC50= 1 nM KD = 2 nM |
IC50 was determined based on IL-23 receptor competition | |||||
| IgG (Fc) | BC FG |
109 | 1 | CFMS/HTS | eFn-anti-IgG-1 | BC: PATTYTL FG: PCNQCQLRPN |
KD = 28 nM | - | 116 |
| MBP | eFn-anti-MBP-2 | BC: QAPHLFV FG: QFMYLLLPGR |
KD = 130 nM | ||||||
| Human/ mouse DC-SIGN | BC FG |
1012 | Mouse: 3 Human: 4 |
- | eFn-DC6 | BC: PQPWLQV FG: YLPLTYGHYR |
Human: KD = 19 nM; Mouse: KD = 133 nM |
- | 102 |
| PSD-95 and Gephyrin | BC FG |
1012 | PSD-95: 6 Gephyrin: 7 |
- | FingRs | - | - | Used as Intrabody | 117 |
| CAMKIIα | BC FG |
1012 | 7 | - | CAMKIIα FingR clone 1 | BC: IHHAKEI FG: TVSFAWKRVL |
- | Used as intrabody | 118 |
| PXR Ligand Binding Domain (To compete with SRC-1) | BC DE FG |
N/S | - | - | - | BC: PPPYYEGVTV DE: PYWTET FG: EMYPGSPWAGQVMDIQP |
KD = 11 nM | - | 97 |
| IL-23 | BC DE FG |
1012 | - | - | ATI-1221 anti-mouse IL-23p19 Adnectin | - | Parent: KD = 0.245 nM PEGylated form: KD=0.234 nM |
- | 103 |
| H-RAS (G12V) | BC FG |
1012 | 6 | 5 additional rounds for maturation | RasIn2 | BC: SIVFGKHD FG: FRWPKRRLVR |
KD = 120 nM | - | 119 |
| CD4 | NP1 library: BC, DE, FG with N-terminal and flanked randomized residues WS1 library: CD strand FG loop with randomized flanked residues |
N/S | 6 | Stringency and off-rate improvement | Adnectin 6940_B01 | CD strand region: HSYHIQYWPLGSYQRYQVFS FG loop and flank: EYQIRVYAETGGADSDQSFG WIQIGYRTEPES |
KD = 3.9 nM | - | 107 |
| PD-L1 | - | N/S | - | - | Adnectin BMS-986192 | - | KD = 0.035 nM | - | 99 |
| gp41 (N17) | BC DE FG |
N/S | 7 | 3 additional rounds for optimization | Adnectin 6200_A08 | BC: EYKVHPY DE: FVLE FG: VDS |
Parent Adnectin: KD = 0.8 nM | Tethered CD4 and gp41 Adnectin to inhibit HIV (EC50= 37 pM) | 108 |
| Glypican-3 | BC DE FG |
5.3 × 1013 | 7 | - | A2-tub | BC: SDDYHAH DE: GEHV FG: YDAEKAATDWSIS C-terminus: NYRTPC |
KD = 8 nM | - | 110 |
| α211 subunit of α1-Nicotinic Acetylcholine Receptor | BC FG |
≤1014 | 5 | Off-rate optimization/ heat and protease stability selection | α1-FANG3 | BC: PSMFGHV FG: WIHLKWLTNF |
KD = 0.67 nM | - | 53 |
| Subunit S1 of SARS-CoV-2 | BC FG |
3x1013 | 6 | Clone 6 monobody | BC: GGDYVGYY FG: TYNGPWIYGYEEI |
KD = 0.76 nM | 111 |
Refers to the stated complexity of the library in the original reference. N/S is used if this value is not provided.
(Abbreviations: CFMS (Continuous flow magnetic separation); HTS (High throughput sequencing))
5.2.1. Initial mRNA Display Selections with the 10Fn3 Scaffold
In 1998, Koide et al. designed the first 10Fn3-based scaffold library.62 Using PD, they isolated a single clone with moderate specificity and affinity towards ubiquitin (reported IC50: 5 μM). The first mRNA display selection using an 10Fn3 scaffold was against Tumor Necrosis Factor α (TNFα).71 In this selection, three libraries were constructed: one library randomizing the FG loop only, one randomizing the BC and FG loops, and one randomizing the BC, DE, and FG loops. Using these three libraries, the selection yielded ligands with KD values ranging from 1 to 24 nM, with over 90% of the clones originating from the third, three-loop library. Subsequent affinity maturation that included random mutagenesis, high temperature incubations to select for protein stability, and extended washing for increased binding affinity, resulted in improved variants with KD values as low as 20 pM. Biophysical characterization of the two winning sequences showed they were monomeric, could be expressed in E. coli, and were resistant to proteolytic digestion at room temperature. However, the selected molecules showed lower thermostability than wild type 10Fn3 and suffered from low to moderate bacterial expression efficiencies.71 The authors hypothesized that the higher affinity and larger number of functional sequences resulting from the selection (~100 total) was due to the larger mRNA display library (1012 independent sequences) versus the less diverse PD library (108) in the original ubiquitin selection.
A second study described extensive biophysical and biochemical characterization of the fibronectin scaffold. In this study, Olson and co-workers synthesized a library similar to the two-loop library above, where the DE loop was kept constant while the BC and FG loops were randomized. The first seven amino acids of the 10Fn3 scaffold were removed in this library because structural analysis showed these N-terminal residues threaded between the randomized BC and FG loops, potentially affecting binding. Supporting this hypothesis was the observation that removal of the first eight residues of 10Fn3 fibronectins selected against Kinase insert domain receptor (KDR)/vascular endothelial growth factor receptor-2 (VEGFR2) improved binding by ~3-fold on average.73 The wild type and Δ1-7 mutant were equally stable in unfolding studies, suggesting that these seven N-terminal amino acids were not important for folding and stability. The authors also determined the folded fraction of the library by screening clones from the 10Fn3 library expressed with Green Fluorescent Protein (GFP) fused to its C-terminus. GFP fluorescence is highly dependent on the solubility and tertiary stability of the fused N-terminal protein.100 GFP reporter fluorescence showed that ~45% of the naïve fused library was correctly translated in-frame and lacked stop codons. These data supported the hypothesis that the fibronectin library is able to tolerate loop randomization and can display these random loops without significant effects on stability or folded structure.94
5.2.2. Targeting Extracellular Proteins with 10Fn3 Selections
Extracellular proteins are a common target in 10Fn3-based mRNA display selections. Each selection described below employed slightly different selection pressures and antigen presentation strategies. Engineered 10Fn3 scaffolds termed Adnectins were selected against interleukin-23 (IL-23) and epidermal growth factor receptor (EGFR).101 Both Adnectins bound their targets with a KD of 2 nM and inhibited either EGFR receptor phosphorylation (IC50 = 50 nM) or IL-23/IL-23R binding (IC50 = 1 nM).96 Co-crystallization of the Adnectins with their targets revealed they bound epitopes distinct from previously identified mAbs, suggesting their small size allowed them to bind to concave, antibody-inaccessible sites while still being able to sterically hinder receptor/agonist interactions.96 The ability to recognize distinct epitopes from mAbs could make fibronectin scaffolds useful in situations where critical protein surfaces cannot be effectively targeted with larger scaffolds.
The dendritic cell-specific ICAM3-grabbing non-integrin receptor (DC-SIGN) was targeted with a fibronectin mRNA display library containing two random loops. The authors hypothesized a DC-SIGN specific fibronectin would enable targeted delivery of antigens to dendritic cells, promoting a more effective immune response. After seven rounds of selection, several fibronectin ligands were obtained, with the most promising 10Fn3 ligand showing a reported KD of 19 nM.102 While all fibronectins were able to bind to recombinant DC-SIGN in biochemical assays, only one sequence bound to DC-SIGN expressed on the surface of cells. It is unclear if many of these sequences did not bind because the binding epitope for the non-functional sequences is occluded on the cell surface. These results highlight the utility of having multiple potential ligands to screen for function in downstream experiments. Lastly, when this DC-SIGN specific fibronectin was coupled with vaccine antigens, the fibronectin-antigen conjugate resulted in greater DC antigen presentation and enhanced CD8+ cytotoxic T-cell activation.102
Bispecific biologics were also generated using 10Fn3 mRNA display selections. Adnectins were first engineered separately against EGFR and insulin-like growth factor-I receptor (IGF-IR). Optimized 10Fn3 clones were integrated into a bispecific construct through a glycine-serine cross-linker to create the EI-Tandem Adnectin that inhibited EGFR and IGF-IR phosphorylation with IC50 values from 0.1 to 113 nM and triggered receptor degradation. A polyethylene glycol (PEG)ylated version of EI-Tandem increased serum half-life while only minimally impacting affinity for either target. In a preclinical nude mouse model of pancreatic cancer where EGFR and IGF-IR are overexpressed, administration of the EI-Tandem Adnectin resulted in significant tumor growth inhibition.72 Notably, the EI-Tandem Adnectin was more effective than co-administered monomeric EGFR and IGF-IR Adnectins, indicating that bispecific presentation is crucial for enhanced effectiveness. The EGFR binding Adnectin was also adapted for chimeric antigen receptor (CAR) adoptive T-cell transfer.98 This Adnectin demonstrated comparable effectiveness when compared to established scFv molecules derived from EGFR binding mAbs, indicating the 10Fn3 scaffold could provide a simpler and faster route to produce a larger repertoire of CARs.98 Adnectins have also been combined with traditional mAbs or other biologics. For example, a mouse IL-23 specific Adnectin (PEGylated Adnectin KD for IL-23 = 234 pM) was combined with an IL-17 mAb and/or IL-17 decoy receptor.103 Administering both Adnectin and mAb resulted in decreased inflammation with an enduring response in an autoimmune mouse model that was more effective than administration of either inhibitor alone.103
Adnectins targeting immune checkpoint ligands have been developed as well. Notably, the Adnectin BMS-986192 binds programmed death ligand 1 (PD-L1) with a KD = 35 pM even after radiolabeling with 18F.99 18F-BMS-986192 has also been shown to discriminate dynamic levels of PD-L1 in murine xenograft models,104 and was used to assess PD-L1 expression in patients with advanced stage non-small cell lung cancer.105 Alternative radiolabeling (68Ga chelation) was also successful in preclinical mouse models.106 Adnectins were also developed to inhibit human immunodeficiency virus (HIV) infection by binding to CD4 (KD = 3.9 nM).107 In one study, a gp41-binding Adnectin was tethered to a CD4-targeted Adnectin, resulting in enhanced inhibition of HIV replication (EC50 = 37 pM).108 Mutagenesis and addition of a membrane fusion blocking peptide further improved the bispecific Adnectin.109
A two-loop fibronectin library was used to develop ligands against the extracellular domain of the α1-Nicotinic Acetylcholine Receptor. 53 These ligands, called FANGs (Fibronectin Antibody-mimetic Nicotinic acetylcholine receptor Generated ligands) were selected under conditions to improve thermostability (by incubating at 65 °C), protease resistance (by incubating with Proteinase K), and improved affinity (through selection for sequences with slower off-rates). As a result, these FANGs were highly thermostable and bound to the α211 subunit of α1 nicotinic acetylcholine receptor (KD = 670 pM) with the receptor in monomeric and heteropentameric forms in cell culture.53 One of these fibronectins, α1-FANG3, was able to compete directly with the bungarotoxin for nicotinic acetylcholine receptor binding.53
Engineered fibronectins have also been used as targeting agents to deliver cytotoxic drugs in the form of fibronectin-drug conjugates.110 Pharmacokinetically, the smaller size of Adnectins (~10 kDa) results in much faster clearance than antibodies, which could expand the therapeutic window by reducing cytotoxic exposure to normal tissue. In order to test this hypothesis, Lipovsek et al. first engineered an Adnectin targeting Glypican-3, which is overexpressed on hepatocellular carcinoma cells. This Adnectin was then site-specifically conjugated to the cytotoxic drug tubulysin via a C-terminal cysteine on the Adnectin. Conjugation of tubulysin had little impact on Adnectin binding (KD = 8 nM). The group observed that the drug conjugate was effective at the xenograft tumor site, but was cleared rapidly through the kidneys, limiting systemic toxicity. Although this result supports the hypothesis that Adnectin-drug conjugates might display more favorable therapeutic safety profiles relative to antibodies, further studies will be necessary to show this in the clinic.110
Biologics targeting the severe acute respiratory syndrome (SARS) Coronavirus-2 (SARS-CoV-2; the virus that causes COVID-19) were rapidly developed in wake of the pandemic using TRAP display. In this study, a fibronectin library with randomized BC and FG loops was used to target the S1 subunit of SARS-CoV-2 spike protein that contains the receptor binding domain (RBD).111 In the first round of selection, the authors added mRNA coding for the fibronectin library with a puromycin linker instead of the typical TRAP display protocol (where a DNA library is added and transcription/translation occur simultaneously) in order to increase the diversity of the library. In subsequent rounds, TRAP display was used to increase the speed of the selection. Within four days, 10FN3 sequences that bound to the S1 subunit with single-digit nanomolar to sub-nanomolar KD’s were obtained. Several clones were able to block the interaction between the RBD domain of the spike protein and ACE2, some at concentrations as low as 10 nM. The set of clones that were tested also bound to three epitopes and 16 different combinations were suitable reagents for a sandwich enzyme-linked immunosorbent assay (ELISA). Clones also inhibited S1 subunit proteolytic conversion and were able to capture virions from patient nasal swabs. Finally, one clone was able to inhibit SARS-CoV-2 infection in a cell-based assay as a monomer (0.5 nM IC50) or as a tandem dimer (0.4 nM IC50). This work demonstrates that mRNA display-based selections can be used to discover new affinity reagents in exigent public health emergencies.
5.2.3. Clinical Translation of 10Fn3/Adnectin Biologics Targeting Tumor-Promoting Receptors
Fibronectin molecules selected via mRNA display have been translated to the clinic. An Adnectin targeting VEGFR-2 has advanced the furthest along the translational pathway, ending in a Phase 2 clinical trial. Several strategies were used to develop anti-VEGFR-2 10Fn3 scaffolds. Using a combination of mRNA display, affinity maturation and site-directed mutagenesis, an exceptionally stable 10Fn3 derivative capable of binding VEGFR-2 with a 13 nM KD was engineered.112 Stability of the Adnectin was sacrificed in favor of enhanced binding affinity, which was improved by >40-fold through additional screening. Based on these two molecules, a third 10Fn3 scaffold was designed that exhibited a combination of high affinity (KD = 0.59 nM) and improved stability (Tm = 53 °C).112 The dissociation constant was later improved by modifying the FG loop, achieving KD values as low as 0.06 nM. These anti-VEGFR-2 Adnectins were screened against both human and murine VEGFR-2 to ensure that selected Adnectins would bind to VEGFR-2 from both species. This strategy allows the same molecule to be used in mouse preclinical studies and clinical human trials, potentially simplifying translation. These Adnectins significantly inhibited VEGF-mediated cell proliferation in a dose dependent manner.73
CT-322/BMS-844203, a PEGylated anti-VEGFR-2 10Fn3 derivative with a KD of 11 nM, was tested in pre-clinical animal models.120, 121 CT-322 entered the clinic with the goal of blocking angiogenesis in a variety of human cancers.122–124 In a Phase 1 trial, CT-322 was well tolerated up to doses of 2 mg/kg/week, however 82% of patients developed anti-drug antibodies that were directed against the engineered fibronectin loops.123 Surprisingly, these anti-drug antibodies did not have an effect on pharmacokinetics or biological function. CT-322 was then tested in Phase 2 trials assessing efficacy in recurrent glioblastoma as a monotherapy and in combination with camptotechin-11 chemotherapy.125 Unfortunately, CT-322 failed to show efficacy as a monotherapy as measured by six month progression free survival, resulting in termination of the study before completion.125 Despite its patient tolerability and potent VEGFR-2 inhibition, the CT-322 Adnectin has yet to find a clinical role.
5.2.4. Targeting Intracellular Proteins with Fibronectins
The term “intrabodies” refers to antibodies that target intracellular antigens. Intrabodies are useful as reagents for live cell imaging or for the recognition of proteins in real-time, allowing researchers to address temporal and dynamic biological questions. Historically, proteins have been monitored in live cells through fluorescent protein tagging (e.g., GFP). However, overexpressing proteins as fusions with fluorescent proteins can perturb biological systems, resulting in protein mis-localization and unintended functional effects rendering the system incapable of addressing nuanced biology.126 Some intrabodies are based on antibody scaffolds which have been engineered to eliminate redox-labile disulfide bonds.127 However, since the 10Fn3 scaffold is disulfide free, it is well-suited for intrabody applications and these intrabodies can be generated by mRNA display.
The first mRNA display derived intrabody was engineered against the phosphorylated form of IκBα, a negative regulator of nuclear factor-κB signaling. After ten rounds of selection using a 10Fn3 library to target a phosphorylated peptide derived from IκBα, a clone was found to bind phospho-IκBα (KD = 18 nM). This clone showed >1,000-fold greater affinity for the phosphorylated peptide versus the non-phosphorylated version.113 To increase solubility and expression, the authors performed iterative rounds of mutagenic PCR followed by a GFP solubility screen.100 This screen resulted in the replacement of solvent-exposed hydrophobic residues with polar ones, greatly enhancing β-sheet formation and bacterial expression. The intrabody was then adapted into a fluorescence resonance energy transfer (FRET) sensor with the goal of monitoring IκBα phosphorylation within cells.113
A second selection by the same group was directed against the SARS nucleocapsid protein (SARS-N). The selection resulted in at least eight fibronectin intrabodies that recognized different epitopes of SARS-N; six of these recognized the C-terminal domain of SARS-N while two recognized the N-terminal domain. Two were characterized by SPR and found to have low nanomolar dissociation constants (1.7 nM and 72 nM). At least four transfected SARS-N binding intrabodies were capable of selectively disrupting SARS virus replication inside cells.114 These anti-SARS intrabodies have been used as affinity reagents for detection of SARS.128
In a follow up study, anti-SARS fibronectins were engineered to recognize the nucleocapsid protein from SARS-CoV-2.129 The nucleocapsid protein from SARS-CoV-2 and SARS-CoV are highly similar, with 91% identity and 94% similarity. The authors performed an initial doped selection based on one N-terminal domain binding fibronectin (NN1) and one C-terminal domain binding fibronectin (NC1) from the original selection, in order to improve the affinity and stability of these first-generation fibronectins. However, since the nucleocapsid sequence from SARS-CoV-2 (SARS-CoV-2 N) was not known at the beginning of the project, nucleocapsid from SARS-CoV (SARS-N) was instead used as the target. Two second-generation fibronectins, NN2 and NC2 were isolated with binding to both SARS-N (NN2 KD = 3.3 nM; NC2 KD = 390 pM) and to SARS-CoV-2 N (NN2 KD = 28 nM; NC2 KD = 6.7 nM). Both fibronectins retained binding to the respective domain of their parental clone and were able to bind the nucleocapsid protein inside SARS-CoV-2 infected cells. These second-generation fibronectins were once again doped and selected against SARS-CoV-2 N to make third-generation fibronectins. With only a single round of selection coupled with high throughput sequencing and a machine learning approach, several fibronectins were identified. Following expression and purification from E. Coli, one clone, NN3-1, was found to have a KD = 18 nM. Finally, this group engineered a sandwich ELISA-based assay for SARS-CoV-2 N detection using either a fibronectin and an antibody, or using two fibronectins, with sensitivity limits approaching 4 pg/mL in human sera.
A three-loop library was used to produce Adnectins that bound to the ligand-binding domain of a promiscuous nuclear receptor family member, human pregnane X receptor (PXR). Eight Adnectins were identified, with binding affinities (KD) ranging from 100 nM to sub-nanomolar values. These Adnectins bound to different epitopes with one Adnectin (KD = 11 nM) able to compete with the endogenous PXR ligand, steroid receptor co-activator-1 (SRC-1).97 Previously, the authors discovered a small molecule antagonist of CC Chemokine Receptor-1 (CCR1) with undesirable off-target binding to PXR. The anti-PXR Adnectin was used to obtain a co-crystal structure of the small molecule compound in complex with PXR. This co-crystal structure was important for the structure-based design of new small molecule derivatives with reduced PXR binding and off-target effects.97 This work highlights the ability of selected fibronectin ligands to act as crystallization chaperones to enhance target crystallization efforts.130
The 10Fn3 scaffold was also exploited to identify state-selective K- and H-Ras intrabodies. mRNA display was used to select a 10Fn3 that bound GTPγS-K-Ras(G12V), a constitutively active mutant Ras commonly found in cancer. This study included selective pressure for fibronectins that recognized the active, GTP-bound state. The selection resulted in a first-generation antibody called RasIn1 (Ras Intrabody), which had modest binding affinity (KD = 2.1 μM) and was state selective for active GTP-Ras versus inactive GDP-Ras. Affinity maturation of RasIn1 by doping the BC loop of RasIn1 and selecting with a fully randomized FG loop resulted in RasIn2. The second generation fibronectin showed improved affinity (KD = 120 nM) while retaining selectivity towards the GTP bound state of mutant K and H-Ras.119 By designing and expressing RasIn-GFP variants, the authors were able to monitor the activation state of Ras in living cells.119
Lastly, intrabodies to explore neuronal dynamics were engineered with mRNA display. These intrabodies were termed FingRs (Fibronectin Intrabodies Generated with mRNA display) and were first selected against excitatory and inhibitory post-synaptic marker proteins, Post Synaptic Density-95 (PSD-95) and Gephyrin, respectively.117 The authors of this study first selected for fibronectin ligands that recognized each protein. Several clones from each selection were expressed in cells and screened for their ability to colocalize with exogenously expressed target. Surprisingly, only a few fibronectins were able to colocalize in this assay, suggesting that this dual selection/screening strategy was required to identify functional FingRs. Fusing FingRs to GFP enabled live-cell imaging in dynamic protein trafficking and localization experiments. The authors note that use of these FingRs does not lead to observable changes in neurons, such as spine density, size of PSD-95 or Gephryin puncta, morphological, or electrophysiological changes.126 In a complementary study, a similar strategy was used to develop a FingR targeting Ca2+/calmodulin-dependent protein kinase II α (CaMKIIα). Dynamic changes in CaMKIIα clustering and localization in response to pH, Ca2+, or glutamate could be monitored in cortical neuron somata using FingR agents.118 These FingRs have been used in several other follow-up experiments131–133
6. Peptide Evolution by mRNA Display
6.1. Peptides and Peptide-Based Therapeutics
Natural peptides (e.g., hormones, neurotransmitters and growth factors) have critical roles in human physiology, specifically bind to their respective cellular receptors, and trigger a variety of downstream signals. Peptide therapeutics have been developed based on these natural peptides and as a class, have desirable safety and efficacy profiles and can have beneficial outcomes in treating human diseases.134 Peptides have the ability to modulate protein-protein interactions whereas few small molecules that bind tightly or disrupt protein-protein interfaces have been identified, with some exceptions.135 One hypothesis to explain this observation is that protein interaction surfaces are typically large and flat,135, 136 with the interface area per subunit for dimeric protein-protein interactions ranging from 350 to 4,900 angstroms.137 Lastly, peptides also have lower production costs and reduced quality control issues compared to protein-based biopharmaceuticals therapeutics and small molecules, making them nominally attractive molecular architectures for rapid clinical translation.134
Unfortunately, peptides are often seen as poor therapeutic candidates because of the poor stability of unmodified natural peptides and their short circulation times. Rational peptide design approaches to improve the physiochemical properties of peptides include: 1) improving chemical and physical stability by replacing amino acid residues susceptible to oxidation, isomerization, or chemical modifications,134, 138 2) improving solubility by modifying the charge distribution or pI (isoelectric point),134 3) increasing resistance to enzymatic modification by removing potential cleavage sites from the peptide sequence or by addition of non-natural modifications (e.g., cyclization, stapling, N-methylation, etc.), and 4) extending circulation time through PEGylation, acylation or by incorporating albumin binding elements.134, 139–141 However, as discussed above, these rational design approaches can be difficult because of context-dependent effects.
In the context of oncology, peptides are attractive alternatives to existing biologics because their tumor penetration is potentially enhanced relative to larger biologics. Tumor penetration is a complex function of molecular size, vascular permeability, affinity, diffusibility, stability, and plasma clearance.142 A comparison of Her2 targeting therapeutics of vastly differing molecular weights demonstrated that compounds with low effective molecular weight (e.g., affibodies and peptides) and high molecular weight (e.g., antibodies) penetrated tumors much more effectively than compounds with intermediate molecular weights (e.g., Fab fragments and scFvs).67 The higher tumor permeability of small peptides was attributed to their rapid diffusivity and interstitial transport as well as enhanced extravasation through capillary vessels of the tumor tissue.67 High tumor penetration of extremely large molecules, such as antibodies, was attributed to their long serum half-lives (days to weeks) which provides sufficient time for slow, but continuous tumor uptake. In addition, the rate of clearance from the body is roughly inversely proportional to compound size, and as a result, peptides offer different pharmacokinetics than mAbs. Unlike antibodies the small effective molecular weight of peptides generally results in rapid renal clearance within minutes following their administration.143 In some applications – such as imaging or treatment with compounds with small therapeutic windows – fast clearance of a compound could be desirable. Finally, peptides are generally considered less immunogenic than proteins (though peptides may contain some immunogenic sequences143) and therefore are predicted to have more favorable safety profiles.144, 145
Peptides thus offer a different modality for treatment of disease and many efforts have focused on isolating novel peptide sequences that recognize macromolecules or making peptides more drug-like through macrocyclization and introduction of non-natural amino acids. In this section, we review mRNA display selections supporting these efforts.
6.2. Linear Peptides Selected with mRNA Display
6.2.1. RNA Binding Peptides
RNA- and DNA-binding proteins are key regulators of transcription and translation.64 mRNA display has been used to discover novel ligands that recognize nucleic acids with high affinity and specificity.146, 147 Engineering novel nucleic acid-binding ligands is a challenge for mRNA display since the nucleic acid target must be recognized in the context of the fused mRNA/cDNA that could cause off-target binding. Liu et al. showed that the λ N peptide (1-22 amino acids) synthesized as an mRNA-peptide fusion retained its ability to recognize its cognate BoxBR target.13 Selection conditions were also optimized in order to develop a robust protocol for selection of RNA-binding peptides.23 An important result from this work was that increasing the amount of non-specific competitor (i.e., non-target nucleic acid, such as tRNA) increased the enrichment of RNA-binding peptides, probably by increasing the selection stringency for peptides that bound to the target RNA.
Building on this work, Barrick et al. designed several libraries based on the λ N peptide.146 One library randomized two positions and was selected against the BoxBR hairpin target. Surprisingly, few of the resulting peptides contained wild type residues at the randomized positions, with arginine favored at position 15 instead of the wild type glutamine. Although these peptides still bound the BoxBR target, they did so using a distinct recognition mode relative to wild type λ N peptide. A second library of 9 trillion sequences where the first ~11 amino acids of λ N peptide were fixed were while the last 10 C-terminal amino acids were randomized. This library was panned against three targets: 1) the cognate BoxBR RNA hairpin, 2) a non-cognate GNRA tetraloop, or 3) a non-cognate P22 hairpin. These targets all share the same stem as BoxBR, but differ in the hairpin loop by a single nucleotide.23, 146 More than 80 different peptide sequences were isolated which had few similarities with the wild type sequence or to each other. As with the selection described above, Arg15 was the most conserved residue in final pools. The selected peptides all bound their targets with nanomolar KD values and were able to distinguish between different RNA hairpins.146 Subsequent studies further examined the function of these selected peptides as well as their affinity and specificity for different RNA targets.148–150
6.2.2. G-Protein Signaling
Peptides can also be used to modulate intracellular signaling. Many of the most important intracellular signaling networks are controlled by G-Protein Coupled Receptors (GPCRs).151, 152 Binding of cognate ligands to GPCR extracellular domains results in activation of G-proteins bound to the cytosolic portion of the receptor.153–155 There are three classes of G-protein subunits (α, β, γ), that associate in a combinatorial fashion.156 Furthermore, G-proteins α subunits have different states (GDP-bound, GTP-bound, or apo) which dictate their biochemical and biological function.157
To target proteins in the Gα family, Ja et al. designed a library based on the G-protein regulatory motif (L19 GPR) and performed a selection using Giα1 as the target. The library fixed 10 amino acids of the L19 GPR motif to act as an “anchor” for binding to Giα1, followed by six random amino acids. The selection resulted in a peptide called R6A (KD of 60 nM) which was selective for the GDP-bound form of Giα1. R6A was truncated to a nine amino acid version (called R6A-1) that retained a KD of 200 nM for Giα1. Surprisingly, R6A-1 only includes two amino acids from the original GPR “anchor,” suggesting that the majority of the fixed GPR residues were dispensable for binding in the context of the newly selected residues. However, R6A and R6A-1 still compete with L19 GPR for binding, suggesting that they bind the same site, though the authors could not rule out allosteric competition. In addition, both peptides inhibit the guanine nucleotide dissociation of Giα1 and compete with Gβγ for binding to Giα1.153, 158
Further studies examined the specificity profile of the selected peptides. While L19 GPR has selectivity for the Gαi class, R6A was able to bind to three Gα classes, and R6A-1 was able to bind to all four Gα classes (Gα, Go, Gq, and Gs).159 Since truncation of R6A to R6A-1 resulted in a decrease of specificity, the authors hypothesized that performing the inverse experiment – i.e., addition of amino acids to R6A-1 – might confer binding specificity to different Gα classes. To test this hypothesis, Austin and co-workers designed a library based on R6A-1 of the form MXXXXXXDQLYWWEYLXXXXXX, where the central R6A-1 sequence (bold) was doped at ~50% wild type amino acids and six random amino acids (X positions) were added to the N- and C-terminus.160 This library was used in a positive selection for Gαs(s) binding, which resulted in a peptide called GSP that was able to bind with some specificity to Gαs(s) but still retained some binding to Giα1. A second library based on the GSP sequence was used in a second selection that included a negative selection for binding to Giα1. Here, a molar excess of soluble Giα1 was added to the binding buffer to act as a decoy in the presence of immobilized Gαs(s). This dual selection strategy resulted in highly specific Gαs(s) peptides with KDs in the 130-300 nM range, representing an over 8,000-fold change in specificity from Giα1 to Gαs(s).160
This library was also used in a second selection to generate state-specific peptides. In this selection, the library described above was targeted to Giα1-AlF (AlF3 or AlF4−), which mimics the transition state of GTP hydrolysis. This selection also included a negative selection against Giα1-GDP to remove Giα1-GDP binding sequences. Two peptides with the highest ratio of binding Giα1-AlF versus Giα1-GDP (AR6–04 and AR6–05) were chosen for further characterization. Even with the negative selection, AR6–05 had a KD of 10 nM for Giα1-GDP, blocked Gβγ binding, and enhanced the basal current of a downstream G-protein-activated inwardly rectifying potassium (GIRK) channel. AR6-04 was the only peptide that bound more strongly to Giα1-AlF versus Giα1-GDP. It also enhanced Gβγ binding to Giα1, in contrast with the AR6-05 and the parental R6A-1 peptides. One reason why Giα1-AlF-specific peptides might have not been readily found is that AR6-04 also has a much weaker KD of 10 μM, yet was still able to decrease downstream basal GIRK current.161
Ja and co-workers attempted to modulate G-Protein/GPCR signaling by engineering peptides that directly target a GPCR.162 Using mRNA display, a 27-mer random library was selected against the ectodomain of Methuselah (Mth), a Drosophila melanogaster GPCR associated with longevity. Several peptides in the final pool contained a [R/P]XXWXXR motif and showed dissociation constants ranging from 18 to 31 nM. These peptides antagonized Mth signaling with IC50 values ranging from 45 to 115 nM. Consistent with previous results showing that Mth antagonism increases fly lifespan, transgenic flies expressing one of the selected peptides showed a 38% increase in mean life span and a 26% increase in maximal life span.
6.2.3. Cell Penetrating Peptides
mRNA display has also been used to discover peptides that penetrate cell membranes and enhance the delivery of otherwise non-penetrant molecules. In many of these selections, an mRNA display library is first incubated with cells, the molecules on the outside of the cells are removed, and the cells lysed to recover mRNA-peptide fusions that successfully crossed the cell membrane. Kamide and co-workers performed such a selection using a 15-mer random library fused to a C-terminal cargo protein (a POU domain).163 This library was selected for entry into Jurkat cells and yielded several putatively alpha helical peptides that contained a cationic helical face. One of these peptides was labeled with fluorescein and showed higher intracellular fluorescence in CHO, HeLa, and Jurkat cells relative to the TAT peptide, a previously identified cell penetrating peptide.
In a similar study, the same library was used to select for cell penetrating peptides in the glioblastoma multiforme (GBM) cell line U87MG with the goal of using these peptides for cell-specific delivery.164 After selection and screening of fluorescein-labeled peptides, peptide1 resulted in the highest internal fluorescence in U87MG cells. Peptide1 was specific for the U87MG cell line and could deliver a small peptidic inhibitor of p16, triggering apoptotic cell death.
Lee et al. used a 29-mer random library to select for peptides that would increase peptide uptake in HEK 293 and HeLa cells.165 After selection and sequencing of the library, two peptide sequences were identified and fused to the red fluorescent protein, DsRed. Incubation of these constructs with HeLa and HEK293 cells resulted in higher intracellular fluorescence compared to a DsRed fusion tagged with Arg11, a previously identified cell penetrating peptide.
6.2.4. Epitope Mapping
mRNA display has been used as a tool to identify peptides that interact with antibodies for epitope mapping and identification of epitope mimics, or “mimotopes”, to act as vaccine antigens.166–170 One of the first examples was the selection of epitope-like motifs that bound to the 9E10 anti-c-myc antibody. 9E10 recognizes the linear epitope EQKLISEEDL and 61% of selected clones contained the “LISE” sequence, and another 25% of clones showed single homologous substitutions in this motif. A similar selection was performed against the M2 anti-FLAG antibody that binds the sequence DYKDDDDK.29 Four different libraries of varying levels of randomization were tested using both mRNA display and cDNA display. Both techniques showed that the antibody preferred an eDYKxxDp motif (where upper case indicate strong conservation and lower case indicate weaker conservation).
mRNA display was also used to identify peptides that bind to an anti-polyhistidine mAb. A linear X27 library was panned against immobilized antibody yielding several peptides containing stretches of 2 to 5 histidine residues. Surprisingly, the selected sequences also revealed a high bias for arginine.168 In order to minimize the length of the dominant peptide sequence (Peptide C), the authors constructed a deletion library and reselected against the anti-polyhistidine mAb. By aligning the fragments resulting from this second selection Peptide C (KD = 18.5 nM) was minimized to 15 amino acids (KD = 38 nM). Interestingly, most sequences recovered in the second selection were not homologous to Peptide C and a new consensus motif (ARRXA) was observed.
Traditional vaccine development for pathogens such as Hepatitis C Virus (HCV) has been challenging. To address this, Guo et al. performed an mRNA display selection to identify mimotopes, or short peptides that mimic the structure of native epitopes. These mimotopes could then be used to elicit the production of neutralizing antibodies.167 Using mRNA display, a random 27 amino acid library was sieved against the HCV-neutralizing antibody mAb41, which resulted in peptides with a common W(L/I)XX(L/I) motif. The conserved amino acids in this motif align with W437, L438, and L441 in the wild type sequence of the HCV Genotype 1a E2 protein, indicating which amino acids are important for mAb41 binding. Interestingly, a previous PD selection against mAb41 only identified peptides with W437 and L438 but not L441.171 When administered to mice, the selected peptides elicited an immune response and the production of neutralizing antibodies similar to the original mAb41 antibody.
Mimotopes were also generated against cetuximab, an anti-human EGFR therapeutic mAb.170 After six rounds of mRNA display, one dominant peptide, M077-A02 showed the highest level of selective binding to cetuximab. Rabbits immunized with M077-A02 conjugated to keyhole limpet hemocyanin (KLH) developed antibodies that were reactive with human EGFR. These studies indicate that mRNA display can be used to reliably generate effective peptides mimotopes against antibodies of interest.
6.2.5. Enzymatic Substrate Mapping
mRNA display also provides a powerful tool to identify the optimal sequence for enzyme substrates.172, 173 This is typically accomplished by incubating an mRNA display library with the enzyme of interest and purifying the modified sequences. This approach was first used to determine residues critical for the interaction between Ecballium elaterium trypsin inhibitor two (EETI-II) and trypsin. In this study, positions 3-8 of EETI-II, which interact with trypsin, were randomized. A consensus sequence, PRXLXX, was identified, with 20% of the clones having the wild type PRILMR sequence. This study confirmed that mRNA display could be used to optimize enzyme substrates as well as target-binding ligands. mRNA display was also used to determine the consensus sequence for the substrates of the v-abl tyrosine kinase.172 A random peptide library containing a central fixed tyrosine (GCGGX5-Y-X5GCG) was incubated with v-abl, followed by immunoprecipitation with a phosphotyrosine-specific antibody. The selection resulted in peptides closely resembling the previously known consensus sequence (I/L/V-Y-X1-5-P/F). In a second selection, the authors used a cDNA library to identify cellular proteins that were substrates for v-abl.172 In addition to identifying previously known substrate proteins of v-abl, novel v-abl substrates were also identified and may indicate a more extensive interactome for this kinase.
An mRNA display selection was also used to identify substrates of transglutaminase, an enzyme that catalyzes the crosslinking of glutamine and lysine side chains. A random 10 amino acid peptide library was first incubated with Streptomyces mobaraensis transglutaminase (STG) in the presence of beads with immobilized hexalysine (K6). Functional STG peptide substrates thus could be immobilized on the beads and amplified.173 Two peptides were minimized to pentamers which showed selective reactivity to Streptomyces mobaraensis transglutaminase relative to guinea pig transglutaminase, demonstrating some specificity for the enzyme used for selection.
Fleming et al. used mRNA display to investigate the interaction of PaaA enzyme with its substrates and to evaluate its promiscuity, recognition mode, and processing.174 PaaA is involved in the biosynthesis of Pantocin A, a ribosomally synthesized and post-translationally modified peptide natural product (RiPP). PaaA catalyzes a bicyclization reaction through dehydration and decarboxylation of two glutamic acids within the precursor peptide, making the bicyclic peptide products resistant to proteolytic cleavage by GluC. This property was used in a selection to probe PaaA activity where a peptide library was first incubated with PaaA, treated with GluC to remove the non-PaaA modifiable sequences, and the uncleaved sequences that were modified by PaaA were recovered. Using a 6-position library, the authors obtained a consensus sequence of XBXXRB (where B = V, L, or I), which matched a previously determined FXXBXXRB consensus sequence (underlined amino acids correspond to the randomized positions). Surprisingly, the enzyme accepts a wide range of substrates outside of residues involved in peptide binding. An analogous study by Vinogradov and co-workers was recently published and described the use of reactivity-profiling mRNA display to identify the substrate preference of the lactazole biosynthetic pathway.175 Post-translational modification by these and other similar enzymes could be used to expand the number of unnatural structures that could be used in mRNA display selections (see the Unnatural Peptide section below).
6.2.6. Peptides Targeting Cancer Relevant Proteins
Immune surveillance provides a critical role in preventing the formation and expansion of malignant tumors. However, one of the hallmarks of cancer is the development of molecular mechanisms to evade immune surveillance.176 The interaction between the PD-L1 receptor on tumor cell surfaces with the PD-1 receptor on T cell surfaces dampens T-cell activity and dramatically attenuates their anti-tumor activity. A dual-stage mRNA display strategy was used to identify SPAM (Signal Peptide-based Affinity Maturated ligand), an 18 amino acid linear peptide that binds to glycosylated and non-glycosylated human PD-L1 with dissociation constants of 67 and 119 nM, respectively.177 SPAM was highly selective for human PD-L1 relative to murine PD-L1 and human PD-L2. SPAM was found to disrupt binding of PD-1 as well as the mAbs Atezolizumab and Avelumab, suggesting that the SPAM binding site overlaps with the native PD-1 binding interface. SPAM is also resistant to modification in human serum, showing no proteolytic degradation after five hours in serum ex vivo. While additional affinity maturation of this peptide is needed for in vivo applications, its relative ease of synthesis and biological stability make it an attractive lead compound.
MDM2 (Mouse double minute 2 homolog) is an E3 ubiquitin ligase that ubiquitinates the p53 tumor suppressor protein, thereby tagging p53 for destruction by the proteasome. Upregulation of MDM2 is found in many tumors and represents a potential target for anticancer therapeutic development. An mRNA display selection targeting MDM2 resulted in a peptide called MIP (MDM2 Inhibitory Peptide) that inhibited the interaction of MDM2 or MDMX (an MDM2 homologue) with p53.154 MIP was shown to interact with MDM2 inside cells, resulting in stabilization of p53 and reactivation of the p53 pathway. Further characterization of the peptide by NMR showed that the peptide formed an α-helical structure. Key hydrophobic residues that participate in the peptide:MDM2 interface were also identified.178
Several mRNA display selections have targeted the anti-apoptotic protein Bcl-xL (B-cell lymphoma-extra large). Using a random 16-mer library and a partially randomized library based on the Bak peptide (an endogenous peptide that binds Bcl-xL), Matsumura and co-workers selected for peptides that bound to Bcl-xL. Two peptides called A01 and B01 showed IC50 values of 0.9 μM and 6.4 μM, respectively, in a competition binding assay with Bak. These peptides bound Bcl-xL when transfected into cells as GFP fusions and could block the anti-apoptotic function of Bcl-xL to induce cell death. Additionally, using mRNA display, 21-amino acid peptides with picomolar affinities to Bcl-xL were also isolated from a primary selection followed by a second selection using a doped library.179
6.2.7. Non-biological Target Selections
While the majority of mRNA display selections have been carried out against macromolecular targets, selections have also been performed against non-biological targets. In several cases, the goal of these selections is to discover peptides that react with small molecules, enabling the selective modification of proteins and peptides without enzymatic assistance. Kawakami et al. developed the Directed In Vitro Evolution of Reactive peptide tags via Sequential Enrichment (DIVERSE) system to discover peptides that react with small molecules.180 This group designed a selection where peptides that react with a p-(chloromethyl)benzamide (p-ClBz) moiety would lead to immobilization of the peptide on the solid phase resin. The selective pressure was increased throughout the selection by decreasing the reaction time between the library and the immobilized p-ClBz. After 19 rounds of selection, two cysteine-containing peptides were isolated. A second-generation peptide (WFWCPYWCCWVP) was isolated after a maturation selection. This peptide covalently reacted with p-ClBz-containing molecule within 30 min and could be used to add fluorescent dyes the peptide. Fusion of the tag with GFP demonstrated that the conjugation reaction could be catalyzed inside living cells.
An alternate strategy was used to select for peptides that could undergo cysteine arylation with perfluoroarenes. Evans and co-workers designed an mRNA display selection where a peptide library was incubated with a biotinylated water soluble perfluoroarene, enabling peptides that successfully reacted with the perfluoroarene target to be selectively purified.181 Previous studies had identified a FCPF sequence that performed the cysteine arylation and this sequence was doped into the library. Interestingly, the FCPF sequence was not enriched after selection, and the winning MP01 sequence instead used a cysteine encoded in the random region. The MP01 sequence was quantitatively arylated in 6-8 hours. The reaction was found to require some peptide structure, as addition of urea into the reaction significantly inhibited MP01 arylation (but did not affect the arylation of a control sequence). Addition of the MP01 sequence to Sortase A showed that the reaction was regioselective where the MP01 cysteine was arylated specifically, even in the presence of a cysteine in the active site of the enzyme. Further investigation showed many other perfluoroarene-reactive peptides, some which were predicted to have β-sheet and α-helical conformations.182
A similar selection was performed to isolate peptides that would react with dibenzocyclooctyne (DBCO), a strained cyclooctyne that is commonly used for copper-free click chemistry.183 Using cDNA display, a 23 amino acid peptide was discovered that contained a single cysteine that reacted with DBCO. The peptide was shown to react with DBCO by MALDI-TOF mass spectrometry and was minimized to a 16-mer peptide called sDRP. However, the reaction yield was relatively poor as only about 20% of sDRP could be modified by DBCO. sDRP was also fused to Dihydrofolate reductase (DHFR), which could be specifically labeled with DBCO-tetramethyl rhodamine even the presence of other cysteine-containing proteins.
Peptides that bind to small molecules have also been isolated using cDNA display. Phenolphthalein was immobilized on a solid support and used as a target for a cDNA display selection.184 In this study, the authors used a library with a higher fraction of aromatic residues at the randomized positions and selected a hydrophobic peptide called LV59 (LVFLIWWM). LV59 binds phenolphthalein at neutral pH but is eluted at alkaline pH. Control experiments where aromatic residues were mutated to alanine abrogated binding. However, LV59 showed some weak cross-reactivity with phenol, suggesting that additional selection experiments are required to develop highly selective dye-binding peptides.
Surfaces and membranes may also serve as the target in mRNA display selection.185, 186 Artificial linear peptides that can bind to the c-face of hydroxyapatite were selected by Ono and co-workers through mRNA display. An alanine-asparagine-threonine (ANT) motif was recognized to be essential for the adsorption of the peptides on the hydroxyapatite surface.185 Kobayashi and co-workers used liposomes as an artificial lipid bilayer target and selected for peptides that can bind to such surfaces. The selected peptide contained both cationic and amphiphilic regions and bound to a wide variety of liposomes with different compositions. One selected peptide was successfully used as an anchor to immobilize macromolecules on the liposome surface.186
7. Toward the Selection Of “Drug-Like” Peptides By mRNA Display
The development of peptide drugs has been limited in large part by poor stability in biological systems and poor cell permeability. Looking to nature, there are numerous examples of drug-like, bioactive peptides that are stable in biological systems, modulate protein-protein interactions, and are cell permeable (Figure 10). Many of these peptides are classified as NRSPs, and are assembled by large, multi-protein complexes found in prokaryotic and eukaryotic organisms.190, 191 These peptides have several features in common. First, they contain a mixture of both natural and non-natural amino acids191 (Figure 10), especially N-methylated amino acids. N-methyl amino acids are known to enhance cell permeability192, 193 and impart resistance to proteolytic degradation.50, 194 Secondly, a significant number of bioactive natural peptides are covalently cyclized (as opposed to cyclized by redox-labile disulfide bonds). Cyclization can enhance binding affinity by increasing conformational restraint and reducing the unfavorable loss of entropy upon target binding.195 Cyclization has also been shown to enhance stability to proteolytic degradation and may enhance cell uptake.196, 197
Figure 10. Examples of bioactive macrocyclic peptides.

Non-natural amino acids are shown in red.
In theory, introduction of unnatural amino acids or cyclization into a natural peptide might be achieved through systematic medicinal chemistry optimization without in vitro selection. However, introducing these modifications into a sequence is rarely straightforward. For example, starting with a Gαi1-binding peptide, Fiacco and co-workers systematically substituted each position with its corresponding N-methyl amino acid analog followed by measuring binding for Gαi1.50 Many N-methyl substitutions resulted in a significant increase in stability toward degradation by trypsin even when placed within two amino acids (either N- or C-terminal) of the scissile amide bond. However, while protease resistance was improved, almost all substitutions resulted in a reduction of binding affinity, apart from one position that showed a 2.5-fold increase in binding. More concerning, a single N-methyl amino acid substitution at one position in the peptide changed the binding specificity from Gαi1 towards a different family member, Gα12. While introduction of N-methylated amino acids can successfully increase the protease resistance of selected peptides, these modifications may also be accompanied by a reduction in affinity and/or selectivity.50 This observation argues that a multi-parametric selection strategy that balances affinity, selectivity, stability, and potentially cell permeability is needed to generate drug-like peptides by mRNA display.
Directed evolution of mRNA display libraries containing unnatural amino acids and/or cyclization offers a potential solution. In vitro selection can simultaneously optimize multiple biochemical and physiochemical properties provided that appropriate selection pressures can be incorporated into the selection workflow. While the natural set of amino acids is sufficient for high affinity target binding, incorporation of non-natural modifications may facilitate the selection of more metabolically stable, cell-permeable peptides for in vivo applications. The challenge, however, is how best to efficiently incorporate these non-natural modifications into mRNA display libraries with high fidelity and minimal impact on the labile nucleic acid component. In the past twenty years, there have been significant efforts by multiple groups to 1) incorporate non-natural amino acids and, 2) incorporate covalent macrocyclization into mRNA display libraries for the purpose of selecting intrinsically “drug-like” peptides.
7.1. Addition of Non-Natural Amino Acids to mRNA Display Libraries
Incorporation of non-natural (non-proteogenic) amino acids in biological display systems via translation requires that the non-natural amino acid be coupled, or “charged” onto the 3’ end of a tRNA molecule. This tRNA can recognize either a sense or nonsense codon and its aminoacylation can be accomplished in a variety of ways. In the chemoenzymatic approach (Figure 11A),198, 199 an unnatural amino acid is chemically coupled to a dinucleotide, which is then ligated to tRNA. In the most widely adopted approach, the carboxylic acid is activated as a cyanomethyl ester following protection of the α-amino group. The activated amino acid is then coupled to phospho-deoxyC-riboA (pdCpA), a 5’ phosphorylated dinucleotide. This “charged” dinucleotide is then enzymatically ligated to a tRNA lacking the last two nucleotides via T4 RNA Ligase to form an aminoacyl tRNA. Deprotection of the amino group is carried out immediately prior to translation.
Figure 11: Methods for charging tRNA with non-natural amino acids for mRNA display selections.

(A) In the chemoenzymatic method, the amino acid (R1 = H, CH3; R2 = various side chains) is first protected at the α-amino group, typically with the photolabile NVOC group, and activated at the α-carboxylic acid as a cyanomethyl ester (CME). The resulting product is reacted with the 5’ phosphorylated di-nucleotide pdCpA to form the aminoacylated product which is ligated to tRNA using T4 RNA ligase. The NVOC group is removed by photolysis (λ = 350 nm) immediately prior to translation. (B) In the ribozymatic method, the α-carboxylic acid is activated as an ester (CME = cyanomethyl ester, DBE = 3,5-dinitrobenzyl ester, CBT = 4-chlorobenzyl thioester, ABT = amino-modified benzyl thioester) which is then ligated to the 3’ end of a tRNA by a small ribozyme (“flexizyme”). The aminoacylated tRNA is then added to the translation mixture. (C) Using a reconstituted translation system, non-natural amino acids are ligated to cognate tRNAs by aminoacyl tRNA synthetases (AARS) during the translation reaction. In this approach, a single tRNA molecule can undergo multiple rounds of charging and amide bond formation. ACL designates the anticodon loop of the tRNA.
This strategy was first applied in the context of mRNA display libraries in 2002 by Li and co-workers.200 In this work, the non-natural amino acid biocytin (biotinyl-lysine) was protected with the photolabile NVOC protecting group and coupled to an amber suppressor tRNA derived from Tetrahymena thermophilia (THG73).201 After removal of the NVOC group by UV photolysis, the charged tRNA was then added to an in vitro translation mixture containing an mRNA display library with a single randomized codon. After purification and selection for streptavidin binding, the selection resulted in near-total convergence to the UAG stop codon at the randomized position, indicating that the library underwent selection for the non-natural biotin functionality. This work demonstrated that encoding of non-natural amino acids in mRNA display was efficient and that the addition of non-proteogenic chemical complexity provided a selective advantage in directed evolution experiments. A similar experiment was carried out by Sisido and co-workers which demonstrated that biocytin could be incorporated into mRNA display libraries by both nonsense suppression and four-base suppression and that selections against streptavidin resulted in enrichment of four-base or nonsense codons.202
While nonsense suppression of stop codons provides a facile way to incorporate non-natural chemical functionalities, it can only be used to incorporate a single non-natural amino acid (Figure 12A). While the use of the UAA and UGA stop codons is theoretically possible, release factors compete with these nonsense suppressors resulting in suboptimal suppression at these codons.203 An alternative strategy of suppressing sense codons was first described by Frankel and co-workers in 2003,204 where low-usage sense codons, particularly the valine codon GUA in the rabbit reticulocyte extract, were efficiently suppressed with chemoenzymatically acylated THG73 tRNA bearing a complementary UAC anticodon (Figure 12B). This approach enabled the production of mRNA-peptide fusions with up to 10 tandem N-methyl phenylalanine residues. These poly-N-methyl amino acid fusions were remarkably resistant to proteolytic degradation by Proteinase K, and this proteolytic resistance could be attenuated by replacement of even a single N-methyl phenylalanine with phenylalanine.
Figure 12. Strategies for non-natural amino acid incorporation into mRNA display libraries.

(A) A single non-natural amino acid can be incorporated into mRNA-peptide fusions at UAG (amber) stop codons through the use of pre-charged nonsense suppressor tRNA. This strategy is typically used in non-reconstituted translation systems. (B) Multiple non-natural amino acids can be incorporated at sense codons by pre-charged aminoacyl sense tRNA. Although this strategy can be carried out in non-reconstituted translation systems such as RRL (rabbit reticulocyte lysate),204 it is typically used in reconstituted systems where native AARS (aminoacyl tRNA synthetase) enzymes, tRNA, and amino acids are omitted. (C) In a fully reconstituted translation system, non-natural amino acids, tRNA, and AARS enzymes (native or re-engineered) can be selectively added to incorporate multiple non-natural amino acids at sense codons. In this strategy, AARS enzymes continuously recharge tRNAs with non-natural amino acids in the absence of their natural counterparts.
The chemoenzymatic approach for introducing non-natural amino acids is highly versatile virtually any non-proteogenic amino acid that is accepted by the translation system can be introduced into a peptide library. However, despite its flexibility, the chemoenzymatic approach requires multiple synthetic steps and a final deprotection step, which must be highly efficient and compatible with tRNA stability. To address this, Suga and co-workers developed a ribozyme-based (“ribozymatic”) approach where amino acids are conjugated to tRNAs by engineered ribozymes (“Flexizymes”)205–207 prior to addition to a cell-free translation system (Figure 11B). This technology is based on in vitro selected ribozymes that recognize and bind activated amino acids without recognition of specific R-groups. These flexizymes must also bind to the 3’ end of tRNA, where the ribozyme catalyzes the aminoacylation of the terminal 3’ hydroxyl group of the tRNA with concomitant release of the activating group. Although the first prototype ribozyme (Fx3)206 only recognized and acetylated tRNAs with aromatic side chains, subsequent work resulted in the development of three additional flexizymes (dFx, eFx, and aFx)205, 208, 209 that efficiently acylated tRNAs with a wide range of functionalities including amino acids with different R-groups, α-hydroxy acids, D-amino acids, N-methyl amino acids, and N-acyl amino acids.208 By judiciously choosing the combination of activating group (CME (cyanomethyl ester); DBE (3, 5-dinitrobenzyl ester); CBT (4-chlorobenzyl thioester); or ABT (amino-modified benzyl thioester)), amino acid side chain, and flexizyme, individual tRNAs can be charged with a wide range of non-proteogenic amino and hydroxy acids. Introduction of these ribozymatically charged tRNAs into a reconstituted cell-free expression system where corresponding aminoacyl tRNA synthetases (AARS) and sense tRNAs are omitted (the Flexible In vitro Translation (FIT) system) allows for the reassignment of multiple sense codons and the expression of mRNA-peptide fusions with multiple non-natural amino acids.210 While the ribozymatic approach supports robust peptide and protein translation in vitro, reassigned aminoacyl-tRNA species are still consumed stoichiometrically and cannot be regenerated during translation (Figure 11C and Figure 12C).
A third approach for incorporating non-natural amino acids involves using the natural substrate promiscuity of aminoacyl tRNA synthetases to recognize unnatural amino acid analogs and charge tRNAs with these amino acids. In 2001, Shimizu and co-workers developed a wholly reconstituted translation system based on purified recombinant components from E. coli.211 This PURE system was later adapted for incorporation of multiple non-natural amino acids into a single peptide chain.212–215 In 2012, Szostak and co-workers demonstrated the translation of mRNA display libraries in the PURE system with 12 codons reassigned to non-proteogenic amino acids.216 Previously, the Szostak group had developed a method for screening non-natural amino acids that were able to be both activated and then charged onto tRNA by endogenous AARS,217–219 including some N-methyl amino acids.220 Using this knowledge, the PURE system could be simply supplied with non-natural amino acids, which would be faithfully incorporated via the natural translation machinery. One advantage of this co-translational charging strategy is that it is simpler to charge tRNAs with unnatural amino acids compared to the ribozyme or chemoenzymatic methods discussed above. Secondly, the system is more robust since aminoacyl tRNAs are constantly being replenished, resulting in high translational efficiency. Conversely, the disadvantage of this strategy is that the choice of non-natural amino acid is generally limited to those amino acids close in chemical space to their natural counterparts since they must be an efficient substrate for natural AARS enzymes. In the future, addition of engineered AARS enzymes221 into the PURE system will likely further expand the non-natural amino acids that can be incorporated by this strategy.
7.2. Incorporation of Small Molecule “Warheads” And Branched Oligosaccharides In mRNA Display Libraries
The affinity and target selectivity of small molecule lead compounds are often suboptimal for in vivo applications. The affinity and/or specificity of a ligand can be dramatically enhanced by conjugating the ligand to a peptide selected by mRNA display. Several strategies for conjugating a ligand to an mRNA display library include post-translational ligand conjugation or incorporation of the ligand into the library as a non-proteogenic amino acid.
In the first strategy, an mRNA display library is post-translationally conjugated with a small molecule through reactive moieties on amino acid side chains. Li and Roberts first demonstrated this strategy by conjugating 6-bromoacetyl penicillanate to a fixed cysteine residue within a randomized decapeptide library.222 This library was used to target the S. Aureus penicillin-binding protein 2A (PBP2A), a major source of resistance against β-lactam antibiotics in Methicillin-Resistant S. Aureus (MRSA). After completion of the selection, the resulting penicillin-peptide conjugates were found to have a 100-fold greater inhibitory potency than the unconjugated penicillin alone.
A second strategy for addition of small molecule ligands uses the co-translational incorporation of a non-natural amino acid. One example of this strategy was described by Suga and co-workers where ε-N-trifluoroacetyl lysine (KTfa) was incorporated via a reassigned AUG codon/elongator tRNA-AA within the context of a randomized macrocyclic dodecapeptide library.223 Selection against the human deacetylase SIRT2 resulted in the identification of a 3 nM inhibitor of SIRT2, approximately 3 orders of magnitude better than a control peptide where the KTfa residue was incorporated into an α-tubulin substrate motif.
A more recent study by Krauss and co-workers incorporated small molecules co-translationally by employing a reassigned AUG codon to direct the incorporation of homopropargylglycine (HPG) at either fixed or random positions within a randomized linear 27-mer mRNA display library.224 Following translation in the presence of HPG, an azide-modified branched mannose oligosaccharide (Man9-azide) was conjugated to HPG side chains via the copper-catalyzed azide-alkyne cycloaddition (“click chemistry”). This was one of the first studies to demonstrate the compatibility of click chemistry with mRNA display libraries and showed that the reaction does not affect mRNA integrity. The oligosaccharide libraries were then selected against the antibody 2G12, which recognizes a glycosylated epitope on the HIV envelope protein gp120. The resulting sequences showed a similar pattern of HPG incorporation with similar spacing between each site in both the fixed and random libraries. A consensus peptide sequence was observed in a fraction of the round 10 clones, although subsequent analysis showed very little correlation between binding affinity and the presence of the consensus sequence. The most promising variant, which incorporated HPG at five positions, was found to have a KD of 1 nM against the 2G12 antibody, representing a >300,000-fold enhancement over a single Man9 oligosaccharide. These results suggest that the mRNA display selection optimized the number and location of glycosylations within the peptide scaffold rather than the peptide sequence itself. Lastly, although different strategies for addition of the ligand were used, all studies thus far incorporating small molecule ligands indicate that the selected peptide sequence is critical for increasing the affinity of the “warhead” for the target.
7.3. Macrocyclization
Several strategies have been used for macrocyclization in the context of mRNA display (Figure 13). Millward and co-workers were the first to demonstrate the cyclization of mRNA display libraries by post-translationally cross-linking the N-terminal amine and the ε-amino group of a fixed C-terminal lysine within a peptide using a bi-functional N-hydroxysuccinimide (NHS) ester, disuccinimidyl glutarate (DSG)225 (Figure 13A). While disulfide bond-mediated cyclization has been described in PD methods for years,226 disulfide bonds are not stable in reducing environments, thus this NHS crosslinking strategy is better suited for generating redox-insensitive binding peptides. While the efficiency of disulfide bond and other cyclization strategies in display techniques is rarely quantified, (i.e., the fraction of the library that is cyclized), in this study the efficiency of cyclization was determined by incorporation of an α-hydroxy amino acid that allowed specific and quantitative cleavage of the library. The authors reasoned that cyclization would result in retention of a radioactive label in the library after the α-hydroxy position was cleaved, whereas a linear peptide would lose the radioactive label, allowing a facile method to quantify cyclization efficiency. The cyclization efficiency was found to be dependent on the number of intervening residues between the amino groups, with the shortest libraries having the highest efficiency. Although this efficiency decreased as the chain length increased, 30% cyclization efficiency was still observed in libraries of ten random positions.
Figure 13. Strategies used for library macrocyclization in mRNA display.

(A) Disuccinimidyl glutarate (DSG) mediated cyclization (B) Crosslinking cysteine residues with dibromo-m-xylene (DBX) (C) Oxidative conversion of 4-selenalysine (K’) to dehydroalanine (Dha) using oxidized glutathione (GSHox) and hydrogen peroxide and subsequent intramolecular formation of a thioether linkage in the presence of tris(2-carboxyethyl)-phosphine (TCEP) (D) N-terminal chloroacetyl group crosslinking with cysteine residues (E) Spontaneous thioether exchange, formation of a thioether bond, deprotection and ring opening of an N-terminal thiazolidine, and finally native chemical ligation allow display of head-to-tail cyclized peptides as mRNA-peptide fusions. (F) Bicyclic peptide formation by a combination of cysteine residues crosslinking and copper mediated azide alkyne cycloaddition.
This strategy was later improved to allow DSG-mediated cyclization of mRNA-peptide libraries directly on oligo-deoxythymidine (dT)-cellulose. This resin is commonly used for purification of mRNA display libraries, although its commercial discontinuation has led to the development of alternate dT-modified resins227, 228. On-resin reaction with DSG increased the efficiency of cyclization and reduced the number of library manipulations required for each selection step.229 In this study, the DSG-cyclized pentapeptide library was used to select cyclic peptides against the Fc region of human IgG for use in chromatographic purification of mAbs. The selection resulted in several peptides, one with a 7.6 μM KD for human IgG at room temperature. This peptide was linked to a solid support and used for purification of mAbs from cell culture, facilitating antibody elution with less harsh conditions (pH 4) as compared with Protein A (pH 2.5). This on-resin DSG cyclization approach has recently been employed in the selection of peptide macrocycles targeting the WW domains of Yes-associated protein and the TOM22 mitochondrial import receptor230 and to select for cyclic peptides that cross cell membranes.231
An alternative strategy for cyclizing mRNA-peptide fusions to generate redox-insensitive cycles was achieved by cross-linking cysteine residues with dibromo-m-xylene (DBX). This strategy was described by Timmerman et al.232 and first used in mRNA display by Guillen Schlippe and co-workers to select macrocyclic inhibitors of thrombin216 (Figure 13B). In a recent study by Iqbal and Hartman, DBX crosslinking of i and i+4 α-methyl cysteine residues in an E2-binding peptide was found to result in a cyclized peptide with comparable affinity to a hydrocarbon stapled variant where crosslinking was carried out by olefin metathesis between similarly spaced (S)-2-(4-pentenyl)alanine residues.233 The authors then demonstrated efficient incorporation of two α-methyl cysteine residues into an mRNA-peptide fusion through a combination of pre-charged tRNA and an editing-deficient AARS (ValRS T222P). These results suggest that DBX-mediated crosslinking of mRNA display libraries could be used as a nucleic acid-compatible alternative to hydrocarbon stapling by olefin metathesis.
Seebeck and co-workers described an alternative strategy where oxidative decomposition converts the non-natural amino acid 4-selenalysine into dehydroalanine (Dha),234 which then undergoes a Michael addition with an intramolecular cysteine to form a thioether-linked macrocycle235 (Figure 13C). While the latter method is highly selective, the chemistry of lanthionine bridge formation may limit the size of the macrocycle and therefore the chemical complexity of the constrained ring.
Suga and co-workers frequently employ the reaction between an N-terminal chloroacetyl group and a C-terminal cysteine to generate redox-stable thioether macrocycles (Figure 13D). The N-terminal chloroacetyl residue is charged onto an initiator tRNA using the flexizyme system and can have either L or D stereochemistry. Indeed, the choice of stereochemistry at this position can have significant effects on the sequence and affinity of the selected peptides.236 mRNA display libraries cyclized in this manner have been employed in selections against a wide range of targets including SIRT2,223 VEGFR2,237 Akt2,238 the Multidrug and Toxic Compound Extrusion (MATE) transporter239, K-Ras,240 and PlexinB1.241 This cyclization strategy is highly selective and spontaneous allowing rapid and efficient macrocyclization of mRNA display libraries of various chain lengths. A recent study has expanded the repertoire of the N-terminal chloroacetyl-bearing amino acids to include cyclopropane-containing γ-amino acids which are proposed to provide additional constraint to selected macrocycles.242
The chloroacetyl-cyclization strategy was also used to discover cyclic peptides against the RBD of the SARS-CoV2 Spike protein.243 Norman and co-workers used three libraries cyclized with an N-terminal N-chloroacetyl-Tyr, an N-chloroacetyl-D-Tyr, or with a Cys-Cys disulfide bond to develop cyclic peptides against the Spike RBD. All three libraries encoded 4-15 random positions and three selected sequences from each library were studied in more detail. Dissociation constants were obtained by SPR and were found to range from 15-550 nM. Interestingly, all three peptides from the disulfide cyclized library showed non-specific binding. Surprisingly, the highest affinity sequence did not compete with the ACE2/RBD interaction but could be used in an ELISA to detect Spike protein as low as 30 ng/mL. A crystal structure of the peptide in complex with RBD showed that the cyclic peptide displaces a β-strand in the C-terminal region of RBD and shows some sequence homology to the primary sequence in this region of the spike protein.
Recently, chloroacetyl cyclization chemistry has been combined with native chemical ligation to generate head-to-tail peptide macrocycles in the context of mRNA-peptide fusions.244 Indeed, all previously described methods for cyclizing mRNA display libraries employ N-terminal to side-chain crosslinking (Figure 13A and Figure 13D) or side-chain to side-chain crosslinking (Figure 13B and Figure 13C). While these approaches have resulted in the selection of high affinity macrocycles, most bioactive peptides found in nature are cyclized by an N-terminus-to-C-terminus main chain linkage. Indeed, the fundamental mechanism of mRNA-peptide fusion formation (the covalent linkage between puromycin and the C-terminus of the peptide chain) precludes this macrocyclic structure. Suga and co-workers have described an intricate and elegant approach where mRNA-peptide fusions containing a backbone thioester linkage undergo thioester exchange with a C-terminal cysteine side chain (Figure 13E). The resulting thiol group reacts with an N-terminal chloroacetyl side-chain to form the first cyclic intermediate. Reaction of an N-terminal thiazolidine capping group with NaBH3CN results in the formation of an N-terminal N-methyl cysteine which undergoes an intramolecular native chemical ligation reaction with the newly formed backbone thioether to afford a backbone cyclized peptide that retains the puromycin linkage with the encoding mRNA. In this manner, head-to-tail macrocycles can be selected for function by mRNA display.
7.4. Bicyclic Peptides – If One Cycle Is Good, Why Not Two?
Extending the concept that macrocyclization can confer favorable properties on peptides, several groups have worked to create even more highly constrained peptides with more than one intramolecular bridge. Bicyclic peptides have been discovered with PD245 using a trifunctional version of the bromoxylene chemistry described above, which in principle could be compatible with mRNA display. Choice of cyclization chemistry is important as it must not react with the mRNA portion of the fusions. The multiple crosslinking steps to form a multicyclic peptide also requires the cyclization chemistry to be efficient, as lower efficiency will reduce the fraction of the library that is correctly cyclized. Finally, the cyclization chemistry must be highly selective to avoid generation of multiple, topologically distinct molecules from the same sequence.
Toward this end, Hacker et. al. designed a library encoding bicyclic peptides using mRNA display technology to select for highly constrained peptide sequences formed through two orthogonal cyclization steps. This cyclization strategy results in the formation of theta (θ)-bridged bicyclic peptides consisting of three loops with different sizes (Figure 13F). Two chemistries were utilized for cyclization; the copper-mediated azide–alkyne cycloaddition (CuAAC; click chemistry) and DBX-mediated cysteine bisalkylation. To enable the CuAAC reaction, non-natural α-amino acids carrying azide and alkyne groups were incorporated into the sequence. The authors performed a model selection using a bicyclic peptide library and a monocyclic peptide library produced through cysteine bisalkylation to generate high-affinity streptavidin binders. Mid-nanomolar peptides were isolated using both libraries but, interestingly, addition of the second cyclization seemed to only result in a relatively modest 2-fold increase in affinity relative to clones isolated from the monocyclic library. Both monocyclized and bicyclized peptides showed an exceptional resistance to protease digestion with 57% of the monocyclic and 96% of the bicyclic peptide retaining binding to streptavidin after a 24 hour incubation with chymotrypsin.246
7.5. Integrating Non-Natural Amino Acids and Macrocyclization in mRNA Display Libraries
Millward and co-workers took the first step in integrating non-natural amino acid incorporation and macrocyclization by integrating nonsense suppression of the UAG stop codon with N-methyl phenylalanine and covalent macrocyclization to select cyclic peptides with antibody-like affinity for the Giα1 G-protein.247 In this study, a randomized 10-mer peptide library was translated in the presence of THG73 amber suppressor tRNA charged with N-methyl phenylalanine, cyclized with DSG, and selected for binding to immobilized Giα1. After seven rounds of selection, the library converged on a (T/S)WYEF(V/L) consensus sequence, which was found to be highly conserved in a specific registry within the context of the macrocycle. The most promising sequence, cycGIBP, was found to have low single-digit nanomolar affinity for Gαi (KD ≈ 2.1 nM) and binding was highly dependent on macrocyclization. Surprisingly, no amber stop codons were present in any of the clones in the final pool suggesting that N-methyl Phe was lost either due to poor translation efficiency or the absence of a survival advantage in sequences that contained this residue.
Roberts and co-workers expanded on this work and attempted to answer the question of how much protease resistance could be achieved with only macrocyclization. They demonstrated that functional, protease-resistant Gαi1 binding cyclic peptides consisting of only natural amino acids could be obtained using a novel, dual challenge selection strategy.52 In these experiments, a cyclic mRNA display library was first exposed to chymotrypsin digestion, followed by selection for binding to Gαi1. Thus, a sequence would need to both survive protease digestion with and bind to the target to survive and be amplified. Starting with Pool 7 of the previous Gαi1 selection above, three additional rounds of protease/binding selection were performed. Three cyclic protease resistant peptides (cycPRPs) were isolated with significant binding to Gαi1 along with a 100- to 400-fold increase in protease resistance relative to cycGIBP. The increased resistance was found to arise from a combination of reduced cleavage rate (kcat) and a lower enzyme affinity (increased KM). Although the cycPRPs were only exposed to chymotrypsin in this dual selection, the cycPRPs showed enhanced stability in human serum ex vivo, with one cycPRP showing a half-life in serum of up to 28 hours. Part of this stability may be attributable to cyclization which protects the peptide termini from aminopeptidases and carboxyesterases.52 Although significant improvements in stability were obtained from selections with wholly natural peptide libraries, it was still unclear whether the inclusion of non-natural N-methyl amino acids could further enhance cyclic peptide stability in the context of selection.
7.5.1. SUPR Peptide mRNA Display
In 2016, a study by Roberts and co-workers shed additional light on this question.51 A >106-member library based on cycGiBP was constructed so that each position in the sequence was randomized to code for a high fraction of wild type or UAG codons. This library was then translated in the presence of N-methyl alanine-charged amber suppressor tRNA to facilitate incorporation of N-methyl Ala at UAG stop codons. Using the strategy described above, the library was subjected to a cocktail of proteases prior to selection for target binding to enrich protease-resistant sequences. After five rounds of selection, the library converged to a macrocyclic peptide with the sequence MFYAYEYAQWSK where A represents the incorporation of N-methyl Ala (1, Figure 14). This sequence, which differs significantly from the Gαi1 consensus sequence in the previous study, maintained high affinity and isoform selectivity for Gαi1 and showed dramatically enhanced stability towards in vitro proteolysis by chymotrypsin (~400-fold) and proteinase K (>5,000-fold) relative to cycGiBP. The selected peptide also showed reduced susceptibility to modification in human liver microsomes and reduced degradation in human serum ex vivo, even though the library had not been exposed to these metabolic processes during selection. Surprisingly, stability was profoundly cooperative as the protease resistance was dependent on the presence of both N-methyl Ala residues. Substitution of either N-methyl Ala with alanine resulted in complete abrogation of protease resistance. These highly stable non-natural macrocycles were named SUPR peptides (Scanning Unnatural Protease Resistant).
Figure 14: Cyclic Unnatural Peptides Identified by mRNA Display.

Unnatural amino acids are shown in red. For clarity, stereochemistry is not shown.
Although SUPR peptides showed dramatically enhanced hydrolytic stability relative to their linear and non-natural counterparts, it was unclear if this molecular architecture could support the selection of cell permeable peptide macrocycles. Although cell permeability is challenging to predict, even in the case of positively charged and/or highly N-methylated macrocyclic peptides,197 the presence of an aliphatic crosslinker (DSG) and multiple N-methylated amino acids suggested that reasonable cell permeability was possible. Indeed, the hydrophobic DSG crosslinker is analogous to the hydrocarbon crosslinker employed in stapled peptides, many of which show excellent membrane permeability and cytosolic access.248 A recent study by Gray and co-workers provided empirical evidence of SUPR peptide cell permeability and intracellular target engagement.249 In this work, a SUPR peptide mRNA display library consisting of eight randomized positions within the macrocycle was translated in the presence of amber suppressor tRNA aminoacylated with N-methyl alanine, pre-selected for proteinase K resistance, and panned against the autophagy protein LC3. After seven rounds of selection, three peptide sequence families were found to be present. One of these families contained a F/Y-XXX-V motif in constant registration within the macrocycle which was analogous to the known LC3 interacting motif (F/Y/W-XX-V/L/I) albeit with an additional amino acid residue between the aromatic and aliphatic positions. Almost all selected sequences contained an N-methyl alanine residue immediately adjacent to the invariant C-terminal lysine used for DSG cyclization. One of the most promising sequences in the round 7 pool (SUPR4B) showed mid-nanomolar affinity for LC3 which was enhanced by a factor of three by a single amino acid substitution (SUPR4B1W 2, Figure 14). Cyclization was found to enhance the binding affinity by a factor of 3 while substitution of the N-methyl alanine for alanine showed little effect on affinity. Conversely, cyclization only improved protease stability by a factor of two while the N-methyl alanine residue enhanced protease resistance by a factor of 18. Assessment of cell permeability and cytosolic access by the chloroalkane penetration assay250 revealed a low micromolar CP50 for SUPR4B1W (1.3 μM) which was increased by a factor of 7 following linearization and a factor of nearly 3 when the N-methyl alanine residue was replaced with alanine. These results indicated that cyclization and N-methylation played key roles in driving affinity, stability, and cell permeability of the selected SUPR peptide. SUPR4B1W inhibited autophagy in cell culture models and, in combination with carboplatin, almost completely eliminated intraperitoneal tumor burden in an orthotopic mouse model of ovarian cancer. These results indicate that the SUPR peptide architecture supports the selection of cell permeable cyclic peptides for the disruption of intracellular protein-protein interactions in vitro and in vivo.
7.5.2. Integration of unnatural amino acids and cyclization in mRNA Display Libraries
In 2012 Szostak and co-workers reported a selection against thrombin using an mRNA display library where 12 sense codons were reprogrammed with non-natural amino acids in a reconstituted translation system (co-translational enzymatic charging).216 Cyclization of the resulting libraries using dibromo-m-xylene and selection for thrombin binding resulted in 13-membered macrocycles with 0–80% of the randomized positions within the macrocycle occupied by non-natural amino acids. Functional analysis of two non-natural variants and one selected from a wholly natural library showed that all three sequences bound to thrombin with low nanomolar affinities. Replacement of natural amino acids in the non-natural variants or linearization (all variants) resulted in significant attenuation of affinity. Interestingly, a natural variant showed the highest affinity and inhibitory potency and was the least dependent on cyclization. The structure of the most potent non-natural variant (3) is shown in Figure 14. This suggests that selection of non-natural amino acids is dependent on conformation and that merely increasing the content of non-natural amino acids within the macrocycle does not necessarily result in increased function.
In 2011, Suga and co-workers combined ribozymatic tRNA charging, thiol-chloroacetyl cyclization, and a reconstituted translation system to select numerous drug-like peptides (the Random non-standard Peptide Integrated Discovery (RaPID) system). This methodology was employed to select macrocyclic peptides that bound with sub-nanomolar affinity to the E6AP HECT domain.194 The most promising peptides from this selection contained up to four N-methyl amino acids within the context of the macrocycle (>33% of the randomized positions). Similar selections against VEGFR2 have also been carried out using a combination of N-methyl, D-, and α,α-dimethyl amino acids.237
Recently, mRNA display selections using the RaPID system have yielded several high affinity macrocyclic peptides where unnatural amino acids have been selected within the randomized region. Nitsche and co-workers have described the identification of a potent inhibitor of the Zika virus protease from a library of libraries with 8-10 randomized positions within the macrocyclic scaffold (4, Figure 14).251 In this selection, six N-methyl amino acids were included in the genetic code and the most promising selected peptide contained three unique N-methyl amino acids in the randomized region (N-methyl serine, N-methyl phenylalanine, and N-methyl-4-O-methyl-tyrosine). This peptide bound the Zika virus protease with a KD of 9 nM and showed sub-micromolar inhibitory potency in in vitro assays of protease activity. The effect of cyclization and N-methyl amino acid incorporation on the affinity, selectivity, and stability of the selected compounds was not addressed in this study.
Johansen-Leete and co-workers recently described the selection of macrocyclic inhibitors of the chemokine CCL11. In this selection, a library of libraries with 4-15 randomized positions within the macrocycle was translated in the presence of tRNA charged with the unnatural amino acids sulfotyrosine (sY) and the sulfonate mimic of sY, Phe(p-CH2SO3−).252 sY is frequently found on chemokine receptors and chemokine-binding proteins and therefore represents a potentially useful chemical functionality to enhance the affinity of selected CCL11-binding macrocyclic peptides. Indeed, the selection resulted in the identification of a 17-mer peptide containing an internal four residue macrocycle. Three Phe(p-CH2SO3−) residues were present in the peptide with two located in the macrocyclic portion. A variant of the selected peptide, where all three Phe(p-CH22SO3−) residues were replaced with sY bound to CCL11 with a KD of 10.4 nM and inhibited CCL11 signaling in HEK cells with an IC50 of 160 nM. Replacing the sY residues with tyrosine residues resulted in almost complete abrogation of binding while replacement with Phe(p-CH2SO3−) resulted in a more modest (2- to 4-fold) reduction in affinity. This study illustrates the critical role of unnatural amino acids in selections where these residues are specifically chosen for their favorable interaction with the biological target.
In a recent elegant study by Katoh and co-workers, thiol-chloroacetyl cyclized mRNA display libraries were translated in the presence of three cyclic β-amino acids and selected for binding to the human factor XIIa (hXIIa).253 The library was sequenced after four rounds of selection and each of most abundant clones contained at least one β-amino acid and each bound to hXIIa with low nanomolar affinity. Replacement of the two selected β-amino acids with alanine in the highest affinity sequence resulted in undetectable target binding by SPR. Similarly, substitution of selected β-amino acids with alanine in the highest affinity sequence (5, Figure 14) resulted in >20-fold reduction in serum stability. These results indicate 1) β-amino acids with five- and six-membered rings can be efficiently incorporated into mRNA display libraries through the translation machinery and 2) these residues can make substantial contributions to both target affinity and serum stability.
The first clinical-stage peptide resulting from mRNA display Zilucoplan (RA101495) is currently in Phase III trials for treatment of Myasthenia Gravis. This cyclic peptide prevents the cleavage of the complement component, C5, by binding to the C5b domain and blocking binding to complement component C6.254 While literature is scarce on the exact development path of Zilucoplan, mRNA display using the co-translational enzymatic charging method resulted in several peptides binding C5, containing a mixture of natural, non-natural, and N-methyl amino acids.255 Many peptides were tested for their ability to inhibit red blood cell hemolysis, with some peptides showing IC50 constants in the single-digit nM range. One sequence was also shown to have >1,440 min half-life in human plasma. A synthesis of Zilucoplan has been published.256
A comprehensive list of mRNA display selections for drug-like peptides is shown in Table 3.
Table 3. mRNA Display Selections for Macrocyclic Peptides with And Without the Incorporation of Non-Natural Amino Acids.
Residues shown in bold indicate points of macrocyclization.
| Target | # random positions | Library Diversity1 | # non-natural Monomers2 | Selected non-natural amino acids3 | KD (nM) | Affinity Measurement Method | Affinity Fold-increase from cyclization | t1/2 Fold-increase (protease) from cyclization | Peptide Sequence | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| Gαi1 | 10 | 1.7X1012 | 1(1) | 0 | 2.1 | Radioligand Displacement | 15 | 2.6 (chymotrypsin) | MITWYEFVAGTK | 247 |
| E6AP | 8-15 | >1012 | 5(4) | mS, mF, mG | 0.6 | SPR | 300 | N/S | ClAc-wCDV-mS-GR-mF-mG-Y-mF-PCG | 194 |
| Akt2 | 4-12 | >1012 | 1(0) | - | 1104 | Enzyme Inhibition | N/S | N/S | ClAc-YILVRNRLLRVDCG | 238 |
| SIRT2 | 7-11 | >1012 | 2(0) | - | 3.7 | SPR | 2.7 | N/S | ClAc-yHDYRI-KTfa-RYHTYPC | 223 |
| 3.8 | 3.4 | ClAc-YSNFRI-KTfa-RYSNSSC | ||||||||
| Thrombin | 10 | 2.3X1013 | 12(5) | Ma, La, Ya, Ta, Ka | 4.5 | Equilibrium Ultrafiltration | 78 | N/S | Ma-C-La-QNS-Ya-IA-Ta-Ka-GC | 216 |
| 1.9X1013 | 0(0) | - | 1.5 | 11 | MCIIKKSRDPGRC | |||||
| Sortase A | 9 | 5X1011 | 1(0) | - | 3000 | FP | >133-fold | N/S | MRGLWY-KSe-LS-C-WGRIGRR | 235 |
| Fc | 5 | N/S | 0(0) | - | 7600 | Solid-phase adsorption | N/S | N/S | MWFRHYK | 229 |
| Human Serum Albumin | 8-12 | 1012 – 1013 | 1(0) | - | 41 | FP | N/S | N/S | ClAB-FTYNERLFWC | 15 |
| VEGFR2 | 8-15 | ≤1013 | 1(0) | - | 94 | BLI | N/S | N/S | ClAc-FVVVSTDPWVNGLYIDC | 237 |
| 1(0) | - | 8 | ClAB-FIGHYRVKVHPISLERC | |||||||
| 1(0) | - | 2 | ClAB-fKPDWWTYYDLRHPC | |||||||
| 5(4) | D-Tyr | 33 | ClAB-fDCGLWRRNIPSNC | |||||||
| MATE transporter | 7-15 | ≥1012 | 1(0) | - | >100004 | Enzyme Inhibition in cells | N/S | N/S | ClAc-ClAcfSVACSAFVRIAHHASC | 239, 257 |
| Gαi1 | 10 | N/S | 0(0) | - | 9 | Radioligand Displacement | N/S | 133 (serum) 0.72 (Chymotrypsin) | MTWFEFLSSTSK | 52 |
| c-Met | 4-15 | >1X1012 | 1(0) | - | 2.3 | SPR | 83 | N/S | ClAc-yWYYAWDQTYKAFPC | 236,258 |
| EpCAM | 4-12 | 1014 | 1(0) | - | 1.7 | SPR | N/S (strongly dependent) | N/S | Ac-wRPTRYRLLPWWIC | 259 |
| Gαi1 | 10 | 4X106 | 1(1) | mA | 60 | Radioligand Displacement | N/S | 19 (chymotrypsin) 1.9 (proteinase K) 570 (serum) | MFY-mA-YEY-mA-QWSK | 51 |
| Her2 | 8 | 1.6X107 | 1(1) | mnV | 76 | ELISA | N/S | N/S | MVC-mnV-mnV-LYDDK | |
| Plexin B1 | 10-15 | >1012 | 1(0) | - | 3.5 | SPR | N/S | N/S | ClAc-wRPRVARWTGQIIYC | 260 |
| Streptavidin | 8 | 2.6X1010 | 2(0) | - | 361 (monocyclic) 593 (bicyclic) | Radioligand binding | N/S | ~ 200 (monocyclic) ~350 (bicyclic) |
AzHA-TDPNC-yneF-GNPC (monocyclic) AzHA-HPQNC-yneF-HVFSC (bicyclic) | 246 |
| α-amylase | 6-15 | >1012 | 2(1) | - | 15 | Enzyme Inhibition | N/S | N/S | ClAc-yPYSCWARHVRIREN | 261 |
| KDM4A | 8-10 | 6.8X1012 | 1(0) | - | 64 | Enzyme Inhibition | N/S | N/S | ClAc-yTRFRRSGIVFYYC | 262 |
| NS2B-NS3 protease | 8-10 | >5X1013 | 7(6) | mYm, mF, mS | 9 | SPR | N/S | N/S | ClAc-Y-mYm-K-mF-K-mS-mYm-K-mYm-mYm-KC | 251 |
| CCL11 | 4-15 | >1012 | 2(1) | sY | 10.4 | FP | N/S | N/S | ClAc-y-sY-sY-CVDWGL-sY-RPIET-sY-S | 252 |
| K-Ras (G12D) | 8-12 | >1012 | 1(0) | - | 9000 | ITC | N/S | N/S | ClAc-y-FVNFRNFRTFRC | 240 |
| WW-YAP | 5, 7, or 10 | 1012 | 0(0) | - | 36,000 | Solid phase adsorption | 0.14 | N/S | M-AFRLC-K | 230 |
| CeiPGM | 6-12 | >1012 | 1(0) | - | 0.3 | SPR | N/S | N/S | ClAc-Cp-F-FSISGIEWNGPEI-C | 242 |
| hFXIIa | 6-15 | NS | 5(3) | 2ACHC | 0.98 | SPR | N/S | N/S | ClAc-y-FAYDRR-2ACHC-LSNN-2ACHC-RNY-cG | 253 |
| IFNGR1 | 6-15 | NS | 5(3) | RR2ACPC, SS2ACPC | 1.87 | SPR | N/S | N/S | ClAc-y-FGV-SS2ACPC-RRACPC-FYNRT-cG | 253 |
| Ubiquitin tetramer | 6-12 | >1012 | 7(6) | Aoc, mA, mG | 9 | SPR | N/S | N/S | ClAc-f-QYW-Aoc-Y-mA-T-mG-VCG | 263 |
| Plexin B1 | 13 | ~1011 | 1(0) | - | 0.28 | SPR | N/S | N/S | ClAc-w-RPYIERWTGRLIV-CG | 241 |
| LC3A | 8 | NS | 1(1) | mA | 120 | FP | 3.1 | 1.9 (proteinase K) | M-WPHRVTA-mA-K | 249 |
Refers to the stated complexity of the library in the original reference. N/S is used if this value is not provided.
Refers to the total number of non-natural amino acids present during translation including those that were held constant in the library. The number in parentheses indicates the number of non-natural amino acids that were not held constant during the selection.
Refers to the unnatural amino acids obtained in the selected peptide which were not held constant in the library.
IC50 value.
Ki value.
7.6. Cyclic Unnatural mRNA Display Libraries – What Have We Learned?
The past 15 years have shown considerable advances in the selection of drug-like peptides from mRNA display libraries. However, given the technical challenges and time cost of unnatural amino acid incorporation, post-translational chemical modification, and macrocyclization, it is vital to consider their actual contribution to the drug-like properties of selected lead compounds. Put another way, how do these design elements enhance binding affinity, specificity, resistance to protease, and cell permeability?
In the case of macrocyclization, in almost all selections where cyclization was enforced, it was found to be essential for affinity as evidenced by the loss of activity versus the linear versions of the peptides. In some cases the effect was modest (3-fold)223 and in some cases it was quite dramatic (1,000-fold) (Table 3).194 Furthermore, although consensus motifs in linear peptides can be found in various registers within the peptide sequence, they are typically found to be conserved in a single register within the macrocycle. This suggests that the position within the macrocyclic architecture plays a role in the optimal configuration of the molecule and that binding motifs may only be compatible with specific 3D conformations.52, 194, 216, 247
These observations suggest that the benefits of macrocyclization will be highly context-dependent. Indeed, a recent study provided direct experimental evidence to address the contribution of cyclization to peptide fitness.264 In this work, mixed libraries containing linear, cyclic, and bicyclic peptides were panned against streptavidin to identify the relative representation of each topology in the final pool. Surprisingly, sequencing of the late round pools revealed that all the top 100 sequences were linear as evidenced by the absence of residues required for cyclization. When the selection was repeated with a protease pre-selection step, eight of the 10 most highly enriched peptides contained the residues required for cyclization. This indicates that cyclization is not absolutely required for high affinity, but when other selection pressures are present (e.g., protease challenge), cyclic peptides may emerge as a preferred topology.
The case for non-natural amino acids is more complex. In cases where the non-natural functionality is explicitly200, 202, 252 or implicitly51, 249 selected for, non-natural amino acids are typically well-represented in the final pool and are essential for the function of the resulting peptides. However, when there is no explicit selection pressure, the results are somewhat mixed. In selections against thrombin216 and Akt2,238 libraries containing only natural amino acids produced the most highly functional sequences as measured by affinity and inhibitory potency. In the aforementioned selection against the E6AP domain,194 replacement of the selected N-methyl amino acids with their natural counterparts resulted in complete abrogation of target binding, although some individual N-methyl amino acids could be replaced with natural residues with only minimal effect on peptide function. In a recent report describing the optimization of a macrocyclic peptide obtained from a selection against proprotein convertase subtilisin/kexin type 9 (PCSK9), two fluorinated tryptophan residues and a fluorinated N-methyl phenylalanine residue were found in one of the most promising selected peptides.265 Elimination of the fluorinated N-methyl phenylalanine decreased the IC50 of the selected peptide by more than an order or magnitude while substitution of the fluorinated tryptophan at position 3 with tryptophan only increased the IC50 by a factor of 2. In contrast, replacement of the fluorinated tryptophan at position 4 with tryptophan increased the IC50 by >20-fold. In initial cyclic selections against Gαi1, no non-natural N-methyl Phe was found in the final pool despite being available throughout the selection. In all five cases, no explicit or implicit selection pressure was placed on the library to enforce non-natural amino acid incorporation. While unnatural amino acids are often found to drive peptide affinity and/or stability249, 253, their presence in selected sequences is not guaranteed nor are they certain to enhance peptide function.
In selections that include a protease challenge prior to target binding, N-methyl amino acid incorporation was strongly enforced and was essential for peptide function.51, 249 N-methyl amino acids increase protease resistance of peptides,204, 266, 267 and the presence of the non-natural amino acid has a significant positive effect on the overall fitness of a selected peptide. From this, albeit limited data set, we could draw the provisional conclusion that in the absence of explicit selection pressure for non-natural function there is no guarantee that including unnatural amino acids in the library will confer additional fitness to selected sequences relative to the set of 20 natural amino acids. Indeed, protease resistance and serum stability can be obtained in mRNA display selections with only the natural set of amino acids provided that a protease pre-selection is included.52
7.7. Evolving Drug-like peptides for molecular imaging
Real-time imaging of cancer biomarkers by molecular imaging can inform diagnosis and monitor the progress of treatment. Increasingly, macrocyclic peptide scaffolds are being exploited as targeting agents for molecular imaging applications. mRNA display is well-positioned to create metabolically stable peptides that combine rapid peptide pharmacokinetics with the high target affinities characteristic of mAbs. While antibodies generally have excellent target affinities (nanomolar or below), they suffer from slow tumor uptake and blood pool clearance, requiring the use of long half-life radionuclides (e.g., Zr-89).268 In contrast, metabolically stable macrocyclic peptides can persist in circulation long enough to be taken up into the tumor, but are cleared rapidly enough for same-day imaging. Antibodies often suffer from high liver uptake making them poor contrast agents for the detection of primary tumors and lesions in the abdominal cavity.269
mRNA display targeting of the immune checkpoint programmed death ligand-1 (PD-L1) resulted in the identification of highly potent macrocyclic peptides for non-invasive imaging of this critical immune checkpoint receptor.270–272 Recently, Bristol Myers Squibb (BMS) disclosed patents describing mRNA display-generated macrocyclic peptides that bind PD-L1, which are described in a patent review by Shaabani et al.273 Independent characterization confirmed that a subset of these peptides bound to a site on PD-L1 that abrogated the engagement with PD-1.274 One of the macrocyclic peptides, termed WL12 (6, Figure 14), was investigated as a PET imaging probe using murine subcutaneous xenograft tumor models.272 DOTA was conjugated to WL-12 via an ornithine side chain for stable chelation of 64Cu. This study found that PD-L1 expressing CHO xenograft tumors showed significantly higher uptake of [64Cu]-WL12 relative to control tumors (14.9% ID/g versus 4.0% ID/g).
To help mitigate trans-chelation of 64Cu in the liver,272 68Ga was investigated as an alternative radiometal.270 The 67.6 min half-life of 68Ga is also predicted to result in improved dosimetry (lower total dose to patients) and is well-matched to the biological half-life of WL-12. [68Ga]-WL12 showed significantly decreased liver uptake relative to [64Cu]-WL12, but maintained its affinity for PD-L1 in the same xenograft subcutaneous models (11.56 ± 3.18 %ID/g for PD-L1 tumors versus 1.33 ± 0.21 %ID/g for negative controls).270 [64Cu]-WL12 uptake was also shown to be useful in informing anti-PD-L1 antibody dosing and efficacy271 as reduction of WL12 uptake was correlated to increased target engagement by anti-PD-L1 antibodies. By employing mathematical models of WL12 biodistribution, the authors could predict the optimal antibody dose required to saturate systemic PD-L1271 which may allow real-time optimization of immune checkpoint blockade therapy.
The Her2 binding peptide, SUPR4,51 was used as a model system for developing 18F-based peptide radiolabeling strategies and SUPR peptide-based PET imaging. In a recent study, SUPR4 was radiolabeled via click chemistry-mediated conjugation of 2-[18F]-fluoroethylazide (FEA) to a C-terminal alkyne on an automated radiochemical synthetic platform.275 This methodology also introduced a novel azide-linked resin to scavenge unreacted alkyne in the precursor peptide which eliminated the need for high performance liquid chromatography (HPLC) purification and resulted in tracers with high specific activity (>200 GBq/μmol). [18F]-SUPR4 showed Her2-selective tumor uptake and rapid renal clearance.275 As expected, the probe was not degraded or modified in vivo prior to imaging and showed no appreciable liver uptake suggesting that SUPR peptides are attractive scaffolds for the development of molecular imaging probes.
8. mRNA Display Technology Development
As library design becomes more robust, it is worth turning our attention to technologies that increase the rate and throughput of selection rounds. As the number of validated targets for drug development increases, these technologies will increasingly become part of the mRNA design toolbox as they enable a more rapid progression from library to lead. Indeed, given the technical challenges inherent in the mRNA display platform, reducing the number of steps in a selection, or increasing the rate of each step vastly simplifies the directed evolution process and makes this technology accessible to non-expert users.
Several detailed protocols for performing mRNA display experiments have been published.276–280 Improvements in mRNA display methods have been made across all steps in the mRNA display cycle. In some cases, improvements have been out of necessity as key reagents are not commercially available. For example, two groups describe the production of dT beads used for mRNA-protein purification since dT-cellulose typically used for purification after translation is no longer commercially available.227, 228 Other improvements have focused on the production of mRNA display libraries, the production of target, increasing the selection speed, or integrating high-throughput sequencing into selection analysis.
8.1. mRNA display Library Production
Construction of long random protein libraries (e.g., > 100 amino acids) introduces a number of challenges including the presence of stop codons, frameshifts introduced by insertions or deletions during DNA synthesis, and the inherently low synthetic yields associated with long DNA oligos. Cho et al., described strategies to construct long randomized reading frames by systematically addressing these challenges. First, to reduce stop codons and frameshifts, a pre-selection for full-length translation products was performed, where random libraries were translated with N-terminal and C-terminal tags (e.g., FLAG or His) and affinity purified to select for full-length open reading frames. Next, these pre-selected fragments were assembled into longer open reading frames using Type IIS restriction enzymes followed by ligation of the restriction fragments. This strategy resulted in an increase in full-length ORFs in the library by up to 84-fold.
Open reading frame assembly was also achieved by incorporating deoxyinosine (I) into DNA primers followed by endonuclease V digestion.281 Endonuclease V specifically cleaves at I bases and proper placement of I into PCR primers allows for generation of sticky ends, allowing multiple DNA fragments to be ligated. Using this strategy, a single domain (nanobody) library was constructed.
8.2. Target Production
Target production is one of the most important steps for an mRNA display selection but is often overlooked. Often, proteins are produced with a biotin tag153 (e.g., the Avitag) that allows immobilization of the target on Neutravidin or Streptavidin beads. For bacterial systems, expression and biotinylation is straightforward,282 however many human proteins are unsuitable for bacterial expression. While mammalian expression systems can often produce these proteins, biotinylation of these targets must be done in vitro using purified enzyme. To address this, Grindel and co-workers developed a mammalian co-expression system that resulted in production of both target protein and BirA protein (responsible for biotinylating the Avitag) in a transient transfection system.283 Using this in situ biotinylation system, milligram quantities of >90% pure biotinylated proteins could be produced and used as targets for mRNA display selections.
While purified proteins are often used as targets for mRNA display selections, some proteins – such as membrane proteins – are challenging or even impossible to purify. To address this, selections have been performed on cells expressing the protein target on the cell surface. However, one disadvantage with this strategy is that a random selection may not result in ligands against the target of interest but instead against other cell surface proteins. Fiacco et al. designed a doped library based on a known Her2-binding peptide to direct the library to bind to Her2 on a Her2-overexpressing cell line.51 In another study, the Yes-Associated Protein 1 (YAP) was expressed on the surface of yeast cells and a counter selection against cells expressing an unrelated protein were used as a negative selection to remove non-YAP binding sequences.230
8.3. Increasing mRNA Display Selection Speed
Decreasing the time required to perform mRNA display selections is an attractive goal since it narrows the gap between selection and lead and allows more selections to be attempted in a given time. These speed improvements can either be focused on decreasing the time to perform a selection round or decreasing the number of selection cycles that are needed to find a functional sequence.
A microfluidics selection strategy called Continuous Flow Magnetic Separation (CFMS) was implemented to increase the speed of mRNA display selections. In CFMS, biotinylated antigens are first immobilized on neutravidin-coated superparamagnetic beads and incubated with an mRNA display library. The mixture is then flowed through a microfluidic system where the magnetic beads are trapped by a series of rare-earth NdFeB magnets. Using the CFMS system, enrichment efficiency is improved by >1,000-fold versus traditional mRNA display methods, resulting in a selection that converges in 3-4 rounds versus 6-10 rounds for typical agarose-based mRNA display selections.115 The CFMS system also allows off-rate selections to be performed easily.284 The magnetic beads containing the target-bound fusions are eluted directly into PCR buffer for amplification and sequencing. Using a fibronectin library, the CFMS system was used to successfully identify a fibronectin ligand against Interleukin-6 that bound with a 21 nM KD and an off-rate of 8.8 x 10−4 s−1 after only four rounds of selection.115
In a follow-up study, the CFMS system was used in the first mRNA display selection to combine high-throughput selection with high-throughput sequencing (HTS).116 By pairing the high levels of enrichment achieved via the CFMS system with the millions of sequence reads achieved via high-throughput sequencing, the authors were able to engineer functional fibronectin ligands with only a single round of mRNA display. A two loop 10Fn3 library (Figure 9C) was used to target maltose binding protein (MBP) and human Fc (from IgG) and the CFMS system was used to carry out one round of selection. The target-bound fusions were eluted, PCR amplified, and sequenced on an Illumina GAIIx. Clones were ranked by copy number and clones with high copy numbers were selected for further study. Sequences present in both libraries were omitted from further analysis, as they were hypothesized to be artifacts or non-target binding sequences (e.g., matrix-binding sequences). High copy number, target-unique clones were expressed and analyzed for target binding. Interestingly, almost all target-unique clones tested showed binding to their respective target, with the most promising ligands showing a KD value of ~28 nM vs. IgG and 129-282 nM towards MBP.116 With current HTS instruments capable of providing billions of sequence reads, novel selection platforms such as CFMS/HTS have extraordinary potential to improve the current selections by reducing the time needed to generate lead clones for further characterization and affinity maturation.
An alternative approach to microfluidic selection was developed using a 3D printed microfluidic device.285 A MicroFluidic Enrichment Device (MFED) was assembled using 3D printed parts and a frit and was designed to capture the beads from an mRNA display selection, allow highly controlled washing. Similar to the CFMS system above, the MFED allows off-rate selections to be performed easily, without the need to add competitor target. Using this device, a >2-fold increase in the enrichment of functional sequences was observed compared with manual washing methods.
These technologies enable parallel selections, selections against multiple targets, selections under different binding conditions, and selections with different libraries. The ability to perform multiple selections in parallel allows different experimental conditions to be tested and provides a greater chance of success by reducing the risks of selection failure present in with manual, single target selections. As microfluidic devices and 3D printing become cheaper and more accessible to non-expert users, we expect these technologies to make rapid, high-throughput selection experiments routine.
8.4. Post Selection Analysis of mRNA Display Libraries
After an mRNA display selection, peptide sequences in the final pool are determined using DNA sequencing. In the age of Sanger Sequencing, practical and economic considerations typically limited this analysis to several hundred DNA sequences.146 With the advent of next-generation sequencing technologies, millions to billions of DNA sequences in the final library can now be analyzed. It is clear from these experiments that, typically, there are tens of thousands of putative functional sequences in these final, “converged” libraries. An emerging question is thus how to best use these data to determine which sequences should be selected for further study.
To a first approximation, a sequence’s functionality correlates with the number of times it appears in the final pool.116 However, more recent experiments have shown that abundance does not always correlate with affinity.179 This could be due to various non-binding selective pressures that are exerted on the pool during the selection cycle (i.e., PCR, transcription, translation, efficiency of fusion formation, etc.). Thus, rank order might not always correlate with affinity if these other steps affect overall fitness of a sequence in the context of the entire selection cycle. As more sequence information is obtained from mRNA display selections by high throughput sequencing, the cost and time of traditional lead validation precludes the synthesis of all high-frequency sequences that appear in the final pool. These constraints provide the impetus to develop new methods that enable rapid, high throughput, high-quality synthesis and purification of proteins and peptides. Similarly, there is also a need to be able to test these sequences quickly, giving quantitative data about binding affinity, binding specificity, protease resistance, cell permeability, and serum stability.
One strategy for high throughput synthesis and purification of peptides is to use mRNA display technology to test individual sequence clones. This strategy is attractive since DNA sequences can easily and cheaply be ordered, synthesized, and quickly converted into purified mRNA-peptide fusions. Using the ribosome to translate a peptide allows for high quality peptide synthesis (as opposed to chemical peptide synthesis that often requires HPLC purification) and easy purification by oligo-dT chromatography. Translating putative sequences as mRNA-peptide fusions is a relatively straightforward method to making high-quality peptides for further testing.
Jalali-Yazdi et al. used this strategy to quantitatively characterize the binding of several in vitro translated peptides using an acoustic sensing device.286 In this method, a peptide ligand of interest is translated in vitro with a C-terminal HA-tag. After purification by oligo-dT chromatography, the mRNA-peptide fusions are incubated with magnetic beads that contain the target of interest, in this case, Bcl-xL. The sample is then added to an acoustic sensor surface that contains an anti-HA antibody, which binds to the HA-tag on the mRNA-peptide fusion. If the peptide binds the target, a sandwich is created between the magnetic bead and acoustic sensor, generating a binding signal. The authors tested a number of in vitro translated HA-tagged peptides that have affinity toward Bcl-xL and showed that they could measure dose-dependent binding.
However, to rank order the affinities of different sequences, this assay would need to also determine relative binding affinities. Different sequences are translated with different efficiencies, resulting in unequal peptide concentrations and the need to correct for peptide expression in order to determine relative binding affinities. The authors used a second competition assay where a synthetic peptide doubly labeled with an HA-tag and fluorescein was used to create a sandwich between magnetic beads and an anti-HA antibody on an acoustic sensor, similar to the assay described above. This signal was then decreased by addition of the same in vitro translated HA-tagged peptides or proteins, which compete with the synthetic peptide for binding to anti-HA beads. However, since these translated peptides lack fluorescein, their binding to the anti-HA antibody results in a decreased signal, allowing the concentration of the translated peptide to be determined. With both concentration and binding of each peptide, the relative affinity of the peptides could be determined, and the sequences ranked by affinity for the target. Compared to ELISA, this method provides the same quantitation range but is more automated, sensitive, and requires less antibody and target protein.286
In an alternate method to rank in vitro translated peptides based on their binding affinity, Larsen et al. designed a membrane-based 96-well dot blot apparatus that allows the partitioning of peptide-protein complexes and unbound peptides.287 In this method, the peptides are translated in vitro and are incubated with their cognate protein target until equilibrium is reached. The reaction is then passed through a double filter system consisting of a cellulose top membrane, that can bind the protein-peptide complexes, and a nylon bottom membrane that retains unbound peptides that pass through the first filter. The KD can be determined by quantifying the complexed and unbound populations over a range of concentrations using phosphor imaging. Using this method, the binding affinity was investigated for peptides ranging from 22 to 74 residues and with KD values ranging from 1 to 500 nM. The authors were able to analyze 22 new peptides from a previous mRNA display selection targeting human α-thrombin, where two peptides had been previously tested and characterized for binding.187 The double-filter method identified five new peptides that bound α-thrombin with equal or superior binding affinity compared to previously characterized peptides. The KD values of these peptides were then determined using the same assay and were found to be consistent with KD values measured by microscale thermophoresis.287 Overall, the double filter method shows promise for screening different clones as well as generating accurate equilibrium binding constants.
Arrays of proteins or peptides offer an even higher-throughput method of testing thousands or more compounds simultaneously to determine binding affinity, selectivity, and epitope mapping. The ability to test many interactions in parallel on a protein chip would be an immensely powerful technique for screening potential ligands or for understanding off-target binding of a lead compound However, the challenges of generating a protein chip can easily hamper such experiments. For example, in typical protein arrays, each protein must be expressed, purified, spotted, and immobilized on a surface. Conjugation of the protein to the surface must not lead to protein unfolding.288
mRNA-protein fusions offer an elegant solution to the construction of peptide and protein chips. In a pioneering study, Weng et al. used mRNA display to make a self-assembling protein microarray.289 In this study, the authors first synthesized a chip with DNA capture probes that were specific for mRNA coding for a unique epitope (Myc, FLAG, or HA11). When mRNA-protein fusions were incubated with this chip, the DNA capture probes specifically hybridized to their corresponding mRNA-protein fusions, resulting in self-assembly of the protein chip. These immobilized mRNA-protein fusions were then specifically recognized by their corresponding antibody, demonstrating that they retained functionality when immobilized on the chip surface. If this approach can be extended to cover a significant fraction of the proteome, it may provide a rapid and inexpensive route to a comprehensive protein microarray.
8.5. Integration of mRNA display with Next Generation Sequencing
The impact of Next Generation Sequencing technologies on mRNA display has been discussed above. Next Generation sequencing has enabled unparalleled analysis of the sequences obtained during mRNA display selections and has facilitated a dramatic acceleration of the mRNA display process. As described in Section 8.4, the emerging challenge in post-mRNA display analysis is choosing, synthesizing, and characterizing lead sequences in a rapid and cost-effective manner. Combining next generation sequencing with chip-based analysis to characterize millions of peptides or proteins represents another potential solution to this problem. In such a system, an mRNA displayed peptide or protein can either be hybridized to the DNA on a next generation sequencing chip or generated in situ by performing translation and mRNA display on the surface of the chip. In theory, this would allow a DNA sequence to be obtained and binding characterization to be performed on the peptide or protein coded by that DNA sequence.
A next generation sequencing analysis platform was developed by Swenson et al. to perform high-throughput characterization of the binding affinities of peptides displayed on the surface of an Illumina flow-cell. In a typical Illumina sequencing experiment, millions of monoclonal clusters of DNA strands are generated in the form of bound oligos on the solid surface of the flow-cell chip through a bridge amplification process.290 These DNA clusters, called “polonies,” are then sequenced to determine the DNA sequence of that cluster. In order to adapt this process to an mRNA display format, the DNA clusters not only needed to be transcribed into mRNA that could be translated on the chip, but also requires the mRNA remains covalently attached to the surface near its corresponding cluster. A covalent linkage in this system is important to ensure the fidelity of information so that a DNA sequence corresponds to its encoded peptide and the binding information for that peptide. To solve this problem, the authors identified a primer-dependent RNA polymerase, poliovirus polymerase 3DPol, which can utilize DNA or RNA templates in vitro, but also transcribes the mRNA strand so that it remains covalently attached to the initiating primer. A second challenge was that 3DPol requires an RNA primer for initiation, but a typical Illumina workflow uses DNA primers. To solve this issue, the authors used terminal transferase (TdT) to add several guanosine ribonucleotides to the 3’ end of the flow-cell bound DNA primers, generating hybrid DNA-RNA primers. The authors further confirmed that the mRNA strands synthesized by 3DPol were full-length and the synthesis using this approach was very efficient. mRNA protein fusions were then formed by hybridizing a puromycin-containing oligo to the 3’ end of the mRNA, in a manner analogous to TRAP display.15 To demonstrate functionality in this system, the authors used templates encoding Myc and FLAG peptides. Anti-FLAG and anti-Myc antibodies were then incubated with the immobilized mRNA-peptide fusions and showed specific binding to their cognate sequences. While only two peptides were displayed in the system and no sequencing of the DNA was performed, the core elements of this system show potential for a high throughput sequencing and characterization platform.291
A study using High-Throughput Sequencing Kinetics (HTSK) of mRNA display libraries was used to obtain the association and dissociation rate constants of thousands of peptides simultaneously.179 To do this, the authors collected fractions of an mRNA display pool of Bcl-xL binding peptides at different time points during the association or the dissociation phase of target binding. The cDNA from the mRNA-peptide fusions at each time point was amplified and subjected to high-throughput sequencing, which provided the normalized frequency of each peptide sequence at that time point. By fitting the rate of increase of each peptide sequence, a kinetic association rate constant could be determined for each peptide sequence that appeared in the pool. Likewise, fitting the rate of decrease of each peptide sequence during the dissociation phase yielded kinetic dissociation rate constants. Using both rate constants together, the KD for each sequence could then be determined. The authors demonstrated strong correlation between rate constants determined using this method and those measured using radioactive assays. Further, using this method, rare sequences with lower frequency rank but higher binding affinities were found, demonstrating that the abundance of a sequence in the final mRNA display pool does not always correlate with binding affinity. While this method might be best suited for very strong affinity pools (i.e., it could not be used on pools with too fast on- or off-rates), it is a powerful method for analysis and characterization of multiple individual sequence in mRNA display pools.
Lastly, Illumina sequencing of mRNA display libraries is challenging because processing of the sequencing data does not fit well with standard genomic workflows. Although some specialized tools exist for processing of selection sequencing data, Blanco et al. developed a set of user-friendly scripts called EasyDIVER for specifically processing high throughput sequence data for mRNA display.292 Using EasyDIVER, the DNA sequence data is extracted, trimmed to remove primers and Illumina adapters, translated into peptide sequences if necessary, and the counts of individual sequences are determined. Such scripts will help to make the use of high-throughput sequencing data for mRNA display selections more accessible to a wider array of researchers.
9. Conclusions and Outlook
mRNA display is a powerful technique for performing directed evolution of proteins and peptides. The linkage of protein to mRNA via puromycin allows the amplification of minute quantities of functional proteins and the facile identification of these proteins via DNA sequencing. As a wholly in vitro technique, the minimalist architecture of mRNA display libraries to provides significant advantages over other display platforms which include 1) reduction in library bias and access to a wide range of selection conditions, 2) the ability to search large libraries (over a quadrillion sequences) for function and 3) access to unnatural amino acids and non-proteogenic chemical functionalities.
Over the past 15 years, mRNA display selections have yielded many novel functional proteins and peptides. mRNA display libraries of antibody and antibody-mimetic proteins have resulted in new molecules that have found use as tools for chemical biology, in vivo imaging, and therapeutic development. mRNA display selections have also yielded natural, linear peptides as well as unnatural, cyclic, NRSP-like peptides with high target affinity, superior metabolic stability, and in some cases, high cell permeability. These selections have set the stage for the use of mRNA display to evolve potent lead compounds for drug development, and potentially, for the direct selection of translation-ready therapeutics. As the list of targets generated from genomic and proteomic analyses increases in size, mRNA display is well-positioned to play a critical role in identifying new ligands to modulate these targets in vivo.
Despite its potential as a directed evolution platform, mRNA display is often seen as technically challenging and time-intensive which may have prevented its adoption by non-experts. However, we believe that this is likely to change in the next twenty years for a variety of reasons. First, the synthesis of high-quality DNA libraries has been simplified by the dramatically lower costs of synthetic DNA in conjunction with the commensurate increase in the quality of commercial oligos. Wholly reconstituted in vitro translation systems are now commercially available which provides a facile path for the incorporation of non-natural amino acids into mRNA display libraries. Protein expression has been greatly simplified and accelerated by the advent of synthetic DNA technologies (e.g., the rapid and inexpensive synthesis of entire genes) and can be readily integrated with in situ biotinylation to generate multi-milligram quantities of target protein for directional immobilization on solid phase.283 The rapidly diminishing cost of next generation sequencing, one of the few techniques in biology to roughly follow Moore’s Law, allows deep analysis of selection results and may facilitate hit identification in early rounds. Finally, automated selection technologies offer the prospect of high throughput, massively parallel selections with minimal consumption of library and target. We believe that these enabling technologies, in conjunction with advances in library design and selection workflow, will lead to the wide adoption of mRNA display as a design tool to meet the challenges of drug discovery in the post-genomic world.
Table 2: Linear Peptides Consisting of Canonical Amino Acids Selected By mRNA Display.
Affinities are obtained directly from the corresponding reference and are not normalized for temperature. The randomized residues are underlined.
| Target | # of random residues | Library Diversity (#of unique sequences) | Affinity measurement method | Top Peptide Sequences | Affinity/IC50/Kinetic Properties | Notes | Ref | |
|---|---|---|---|---|---|---|---|---|
| boxBR RNA hairpin | 2 | 150 | FT | MDAQTRRRERRAEKQAQWKAANDYKDDDDKNSCA | KD= 1.0±0.2 nM | GNRA: KD= 9.0±0.7 nM P22: KD= 41±3 nM |
146 | |
| boxBR RNA hairpin | 2 | 1600 | FT | MDAQTRRRERRAEERVAQWKAANDYKDDDDKNSCA | KD= 8.5±2 nM | GNRA: KD= 800±150 nM P22: KD= 580±20 nM |
||
| Equimolar mixture of three hairpins: (i) boxBR RNA (ii) GNRA tetraloop and (iii) P22 | 10 | ~9 X 1012 | FT | MDAQTRRRERRALERTKLEKALQLRNNSCA | KD= 3.4±0.1 nM | GNRA: KD= 81±2 nM P22: KD= 60±3 nM |
||
| MDAQTRRRERRAMERATLPQVLQLRNNSCA | KD= 3.0±0.2 nM | GNRA: KD= 288±7 nM P22: KD= 246±7 nM |
||||||
| MDAQTRRRERRALQRSRARHALQLRNNSCA | KD= 0.5±0.1 nM | GNRA: KD= 6.5±0.7 nM P22: KD= 8.4±0.8 nM |
||||||
| MDAQTRRRERRAALRNEKFWVVQLRNNSCA | KD= 1.9±0.5 nM | GNRA: KD= 93±9 nM P22: KD= 140±10 nM |
||||||
| MDAQTRRRERRANMRMYRSLVIQLRNNSCA | KD= 0.4±0.1 nM | GNRA: KD= 9±2 nM P22: KD= 14±1 nM |
||||||
| Human PD-L1 | 17 | >1012 | FP | MPIFLDHILNKFWILHYA | KD= 67–119 nM | Selective binding for human PD-L1 | ||
| v-abl kinase | tyrosine | 10 | ~1012-1013 | Consensus motif with fixed tyrosine: GCGGXXXXXYXXXXXGCG Consensus motif with upstream tyrosine: GCGGY1-5YXXXXXGCG Consensus motif with downstream tyrosine: GCGGXXXXXYY1-5GCG |
172 | |||
| Bovine trypsin | 6 | 6.4 X 107 | RA | PRILMR | KD= 16±3.2 nM | 166 | ||
| PRVLAL | KD= 52±21.0 nM | |||||||
| PRLVQR | KD= 53±9.0 nM | |||||||
| PRLLAP | KD= 81±21.0 nM | |||||||
| PKLLAP | KD= 21±2.4 nM | |||||||
| Anti-c-myc antibody 9E10 | 27 | 2 X1013 | - | CASVISEREC EEYLVSEYVM RQYLISEYEH LQRLISEQMF IVRLLSEYHM EEYLLSEYVM MQNLISEHEL TMDLIPEHYM EQKLISEEDL DMMLISEKEL FQALIAEEEL QRVLISEFWL |
||||
| Human α-thrombin | 15 | 1.2 X 1011 | SPR | ERNYNDFCDPGRVGL | KD= 520±6 nM | 187 | ||
| FFDRYDSARDPGRLL | KD= 166±18 nM | |||||||
| Giα1-GDP | 6 | 6.4 X 107 | SPR | MSQTKRLDDQLYWWEYL | KD= 60 nM; IC50~0.5uM | 153 | ||
| Anti-polyhistidine mAb | 27 | ~1012 | SPR | DGHPERHDAGDHHHHHGVRQWRLISTG | KD= 18 nM | 168 | ||
| RHDAGDHHHHHGVRQ | KD= 38 nM | |||||||
| ITNSPGRFRHHHVLARRHALYR | KD= 15.5 nM | |||||||
| MKVRRDVMRWHHHHRMARRKANR | KD= 7.6 nM | |||||||
| Botulinum neurotoxin serotype A (light chain) | 4 | FT | MWMTSCRATKML | IC50= 56.3 ± 2.9 uM Ki= 7.3 uM |
188 | |||
| Giα1 GDP-AlF | 21 | ~2 X 1013 | SPR | DESDPEELMYWWEFLSEDPSS | KD=10 nM | 161 | ||
| Mth ectodomain | 27 | ~1.5 X 1013 | SPR | MRLVWIVRSRHFGPRLRMALLGSDRKMW | KD= 4.1 nM IC50=45±10 nM |
N-stunted induced Mth signaling antagonist | 162 | |
| MAPRAVWIQRAIQAMFRLASRQESKAFN | KD= 7.0 nM IC50=115±25 nM |
N-stunted induced Mth signaling antagonist | ||||||
| MNVSWGSFPSSWLQRYYLAKRREADVTL | KD= 6.3 nM | |||||||
| MLKYPDTWLARSLSVFYLRKSARQGKSV | KD= 9.5 nM | |||||||
| MVRIGYTSKPGGMNPGNSYTMSIIRMLI | KD= 6.1 nM | |||||||
| 5’ upstream binding site of Thymidylate Synthase mRNA | 21 | >1013 | HCYPLDESILRLVRGELGAVH YWFAWAKSWQLAKREGHMGFD NWSGFYNLKLNTLKSVRQVHY NWHHALLSDIVSALNKSSLWN NCGTEMFCMDKINEWREVYVD NWSLIHKKYLQQAKMKYARCH DWTLMKVHVKQCVRKLESFTN HCRFVRRVAGLMFRERLEDDH NWRLSHNAVLDDARVQYGVHH |
The selected pool of peptides suppressed thymidylate synthase mRNA translation in a dose dependent manner | 147 | |||
| Gαs(s) | 21 | 1.6 X 1013 | SPR | MAMSDRNKRLTVWEFLALPSST | KD=100±15nM | 160 | ||
| 21 | ~2.5 X 1012 | SPR | MAMSDQNKRMTVREFLALPSSL | KD=300 nM | ||||
| MYTSDHNKLLTVREFLALPSST | KD=130 nM | |||||||
| Anti-human TP53 | 10 | ~1013 | SPR | TFSDLWKLLP | KD= 6 nM | 169 | ||
| LFSDLNKLKP | KD= 11 nM | |||||||
| TDSDLHKLKS | KD= 59 nM | |||||||
| U87MG glioblastoma cell line | 10 | >1012 | NTCTWLKYHS | 163, 164 | ||||
| Bcl-XL | 16 | FP | FPRWKLLAHWADRWWF | IC50= 0.9 uM | 189 | |||
| 11 | FP | YQVARMLRRVADQMAS | IC50=6.4 uM | |||||
| Bcl-XL | 20 | MIETITIYNYKKAADHFSMSM | KD= 23 pM | 179 | ||||
| 20 | MIEAISIYNYKKAADHYAMTK | KD= 15 pM | ||||||
| 20 | MIDTNVILNYKKAADHFSITM | KD= 2.4 pM | ||||||
| Interleukin-6 | 11 | INTLLSEINSILLDIISLL | IC50= 2.8 uM | 155 | ||||
| cetuximab | 10 | CLFSLSTRRLDC | The peptides did not show significant binding in vitro but showed binding after conjugation to keyhole limpet hemocyanin (KLH) | 170 | ||||
| 20 | MEGCLRTWSLFDRRLKCMVG | |||||||
| 20 | CYDVNGVWIRLRNPTHAGIRFC | |||||||
| MDM2 | 12 | 5.1 X 1011 | SPR | PRFWEYWLRLME | KD= 18.4 nM IC50 = 0.01 uM |
154, 178 | ||
| HEK293 cells | 20 | >1012 | MAMPGEPRRANVMAHKLEPASLQLRNSCA | 165 | ||||
| HeLa cells | 20 | >1012 | MAPQRDTVGGRTTPPSWGPAKAQLRNSCA | 165 | ||||
| Streptomyces mobaraensis Transglutaminase | 10 | RLQQP RTQPA | 173 | |||||
| Hydroxyapatite (C face) | 16 | FT | NPPTRQTKPKRVANTN | Adsorption constant= 114 nm2 mg−1 | Peptides selected were highly specific to the c face of hydroxyapatite rather than the unoriented surfaces | 185 | ||
| SAANTTQLNTPTEDNEP | Adsorption constant= 70 nm2 mg−1 | |||||||
| TTDPHRTDNNRTKYQT | Adsorption constant= 34 nm2 mg−1 | |||||||
| TDPPSPKHHCLPTTAN | Adsorption constant= 23 nm2 mg−1 | |||||||
| TKTSPTPENPTQQHRT | Adsorption constant= 14 nm2 mg−1 | |||||||
| HCV neutralizing mAb41 | 15 or 27 | BLI | MMECEKGKYVQGWLCHLFWEDCQDRGWLEQLRNSCA | KD= 16.7 nM | 167 | |||
| MTMPTMVEHSEVAWLDRLVTPYEGWIECIQLRNSCA | KD= 32.9 nM | |||||||
| MYMTSPMSQPCGWIEYLAHEDQPVP-WLDQLRNSCA | KD= 69.7 nM | |||||||
| MQHWTQEYTTMWNEDTGWLDWLIEQDHCQLRNSCA | KD= 11.0 nM | |||||||
| DOPC liposomes | 30 | 6 X 1012 | BLI | RHSKSLPSRVIPRADPRTKTRRRRRRKRTL | The binding was not specific for this liposome | 186 | ||
| PEGylated pentafluoroarene | 30 | ~5 X 1013 | MHQKYKMTKDCFFSFLAHHKKRKLYPMSGSGSLGHHHHHHRL | Rate constant: 0.28 M−1s−1 | 181, 182 |
(Abbreviations: SPR (Surface Plasmon Resonance); FT (Fluorescence Titration); FP (Fluorescence polarization); BLI (Bio-Layer Interferometry); AlF (Aluminum fluoride), Gαs(s) (the short isoform of Gαs), RA (Radioactive Assay); Mth (Methuselah); DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine); HCV (Hepatitis C Virus); KLH (Keyhole Limpet Hemocyanin)
Acknowledgments
This work was supported by UT MDACC Startup Funds (SWM), a Cancer Prevention and Research Institute of Texas (CPRIT) Individual Investigator Award (RP200166-IIRA, SWM), a University of Texas MD Anderson Cancer Center Institutional Research Grant, the National Institutes of Health R44CA206771 (SWM), R01CA170820 (R.W.R., T.T.T.), and R01CA231509 (SWM). We also acknowledge support from the Glioblastoma Research Foundation (The Lee Project), a G.E. In-kind Multi-Investigator Imaging (MI2) Research Award (SWM, RWR, TTT), the Department of Defense/CDMRP CA140291 (TTT), the Ming Hsieh Institute for Research on Engineering-Medicine for Cancer (R.W.R., TTT) as well as T32A196561 (BJG).
List of Acronyms and Abbreviations
- 2ACHC
(1S, 2S)-2-aminocyclohexanecarboxyllic acid
- 10Fn3
10th subunit of Fibronectin type III
- AARS
Aminoacyl tRNA synthetase
- ABT
amino-modified benzyl thioester
- ACL
Anti Codon Loop
- ADCC
Antibody-dependent cell-mediated toxicity
- AlF
Aluminum fluoride
- Aoc
L-2-aminooctanoic acid
- AzHA
β-azidohomoalanine
- Bcl-xL
B-cell lymphoma-extra large
- BLI
Bio-Layer Interferometry
- CaMKIIα
Ca2+/calmodulin-dependent protein kinase II α
- CAR
Chimeric antigen receptor
- CBT
4-chlorobenzyl thioester
- CCR1
CC Chemokine Receptor-1
- CDRs
Complementarity determining regions
- CFMS
Continuous flow magnetic separation
- CH
Constant heavy chain
- CL
Constant light chain
- ClAB-f
N-[3-2-chloroacetamido)benzoyl]-D-phenylalanine
- ClAB-F
N-[3-2-chloroacetamido)benzoyl]-L-phenylalanine
- ClAB-Y
N-[3-2-chloroacetamido)benzoyl]-L-tyrosine
- ClAc-F
a-N-(2-chloroacetyl)-L-phenylalanine
- ClAc-w
a-N-(2-chloroacetyl)-D-tryptophan
- ClAc-y
a-N-(2-chloroacetyl)-D-tyrosine
- ClAc-Y
a-N-(2-chloroacetyl)-L-tyrosine
- CME
Cyanomethyl ester
- CPG
Controlled pore glass
- CNVK
3-cyanovinylcarbazole nucleoside
- CuAAC
Copper-mediated azide–alkyne cycloaddition
- cycPRPs
Cyclic protease resistant peptides
- DBCO
dibenzocyclooctyne
- DBE
3,5-dinitrobenzyl ester
- DBX
dibromo-m-xylene
- DC-SIGN
Dendritic cell-specific ICAM3-grabbing non-integrin receptor
- DHFR
Dihydrofolate reductase
- Dha
dehydroalanine
- DIVERSE
Directed In Vitro Evolution of Reactive peptide tags via Sequential Enrichment
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DSG
disuccinimidyl glutarate
- ED
E. coli display
- EETI-II
Ecballium elaterium trypsin inhibitor two
- EGFR
Epidermal growth factor receptor
- ELISA
enzyme-linked immunosorbent assay
- Fab
Fragment antigen binding
- FANGs
Fibronectin Antibody-mimetic Nicotinic acetylcholine receptor Generated ligands
- Fc
Fragment crystallizable
- FEA
2-[18F]-fluoroethylazide
- FingRs
Fibronectin intrabodies generated with mRNA display
- FIT
Flexible in vitro Translation
- Fn3
Fibronectin type III
- FP
Fluorescence polarization
- FRET
Fluorescence resonance energy transfer
- FT
Fluorescence Titration
- Fv
Immunoglobulin variable domains
- GFP
Green fluorescent protein
- GIRK
G-protein activated inwardly rectifying potassium channels
- GPCRs
G-Protein Coupled Receptors
- GPR
G-protein regulatory motif
- GSHox
Oxidized glutathione
- Gαs(s)
The short isoform of Gαs
- HCV
Hepatitis C Virus
- HA-tag
Hemagglutinin tag
- HIV
human immunodeficiency virus
- HPLC
high performance liquid chromatography
- HPG
homopropargylglycine
- HTS
High throughput sequencing
- HTSK
High-Throughput Sequencing Kinetics
- ID
injected dose
- IGF-1R
Insulin-like growth factor-I receptor
- IL
interleukin
- IVV
in vitro virus
- Ka
Trans-4,5-dehydro-DL-lysine
- KSe
4-selenalysine
- KD
Dissociation constant
- KDR
Kinase insert domain receptor
- KLH
Keyhole Limpet Hemocyanin
- KTfa
ε-N-trifluoroacetyl lysine
- La
β-t-butyl-DL-alanine
- Ma
DL-2-amino-hex-5-ynoic acid
- mA
N-methyl-L-alanine
- mAbs
Monoclonal antibodies
- MATE
Multidrug and Toxic Compound Extrusion
- MBP
Maltose binding protein
- MDM2
Mouse Double Minute 2 homolog
- MFED
MicroFluidic Enrichment Device
- mF
N-methyl-L-phenylalanine
- mG
N-methyl glycine
- MIP
MDM2 Inhibitory Peptide
- mnV
N-methyl-L-norvaline
- MRSA
Methicillin-Resistant S. Aureus
- mS
N-methyl-L-serine
- Mth
Methuselah
- mYm
N-methyl-4-O-methyl-tyrosine
- NHS
N-hydroxysuccinimide
- NRPS
Non-ribosomal peptide synthetase
- OBOC
One-bead-one-compound
- ORF
Open reading frame
- PBP2A
penicillin-binding protein 2A
- p-CIBz
p-(chloromethyl)benzamide
- PCR
Polymerase chain reaction
- PCSK9
subtilisin/kexin type 9
- PD
Phage display
- pdCpA
phospho-deoxyC-riboA
- PD-L1
Programmed death ligand 1
- PET
Positron emission tomography
- pI
isoelectric point
- PEG
polyethylene glycol
- PKS
Polyketide synthetase
- PSD-95
Post Synaptic Density-95
- PSD
Post Synaptic Density
- PURE
Protein Synthesis from Recombinant Elements
- PXR
human pregnane x receptor
- RA
Radioactive Assay
- RANK
Receptor Activator of Nuclear Factor κB
- RaPID
Random non-standard Peptide Integrated Discovery
- RasIn
Ras Intrabody
- Cp-puromycin
Ribocytidyl-puromycin
- RBD
Receptor binding domain
- RiPP
Ribosomally synthesized and post-translationally modified peptide
- RR2ACPC
(1R, 2R)-2-aminocyclopentanecarboxyllic acid
- RRL
Rabbit reticulocyte lysate
- SARS-CoV-2
severe acute respiratory syndrome (SARS) Coronavirus-2
- SARS-N
Severe acute respiratory syndrome nucleocapsid protein
- scFv
Single chain fragment variable
- SPAM
Signal Peptide-based Affinity Maturated ligand
- SPR
Surface Plasmon Resonance
- SRC-1
steroid receptor co-activator-1
- SS2ACPC
(1S, 2S)-2-aminopentanecarboxyllic acid
- STG
Streptomyces mobaraensis transglutaminase
- SUPR
Scanning Unnatural Protease Resistant
- sY
sulfotyrosine
- t1/2
Half-life
- Ta
DL-hydroxy-norvaline
- TCEP
Tris(2-carboxyethyl)-phosphine
- TdT
Terminal transferase
- TNFR
Tumor Necrosis Factor α Receptor
- TNFα
Tumor Necrosis Factor α
- TRAP
Transcription–Translation coupled with Association of Puromycin linker
- UTR
Untranslated region
- VEGFR-2
Vascular endothelial growth factor receptor-2
- VH
Variable heavy chain
- VL
Variable light chains
- y
D-tyrosine
- Ya
3-fluoro-L-tyrosine
- YAP
Yes-Associated Protein 1
- YD
Yeast display
- yneF
p-ethynyl phenylalanine
- ZF
zinc-finger
Footnotes
Conflicts of Interest
There are no conflicts of interest to declare
References
- 1.Huang PS; Boyken SE; Baker D, The coming of age of de novo protein design. Nature 2016, 537 (7620), 320–7. [DOI] [PubMed] [Google Scholar]
- 2.Smith GP, Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228 (4705), 1315–1317. [DOI] [PubMed] [Google Scholar]
- 3.Smith GP; Petrenko VA, Phage display. Chemical reviews 1997, 97 (2), 391–410. [DOI] [PubMed] [Google Scholar]
- 4.Roberts RW; Szostak JW, RNA-peptide fusions for the in vitro selection of peptides and proteins. Proceedings of the National Academy of Sciences of the United States of America 1997, 94 (23), 12297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanes J; Pluckthun A, In vitro selection and evolution of functional proteins by using ribosome display. Proceedings of the National Academy of Sciences of the United States of America 1997, 94 (10), 4937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cull MG; Miller JF; Schatz PJ, Screening for receptor ligands using large libraries of peptides linked to the C terminus of the lac repressor. Proceedings of the National Academy of Sciences of the United States of America 1992, 89 (5), 1865–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Odegrip R; Coomber D; Eldridge B; Hederer R; Kuhlman PA; Ullman C; FitzGerald K; McGregor D, CIS display: In vitro selection of peptides from libraries of protein-DNA complexes. Proceedings of the National Academy of Sciences of the United States of America 2004, 101 (9), 2806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winter G; Griffiths AD; Hawkins RE; Hoogenboom HR, Making antibodies by phage display technology. Annual review of immunology 1994, 12 (1), 433–455. [DOI] [PubMed] [Google Scholar]
- 9.Sergeeva A; Kolonin MG; Molldrem JJ; Pasqualini R; Arap W, Display technologies: application for the discovery of drug and gene delivery agents. Advanced drug delivery reviews 2006, 58 (15), 1622–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galán A; Comor L; Horvatić A; Kuleš J; Guillemin N; Mrljak V; Bhide M, Library-based display technologies: where do we stand? Molecular BioSystems 2016, 12 (8), 2342–2358. [DOI] [PubMed] [Google Scholar]
- 11.Tizei PA; Csibra E; Torres L; Pinheiro VB, Selection platforms for directed evolution in synthetic biology. Biochemical Society Transactions 2016, 44 (4), 1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colas P, Combinatorial protein reagents to manipulate protein function. Current opinion in chemical biology 2000, 4 (1), 54–59. [DOI] [PubMed] [Google Scholar]
- 13.Liu R; Barrick JE; Szostak JW; Roberts RW, Optimized synthesis of RNA-protein fusions for in vitro protein selection. Methods Enzymol 2000, 318, 268–93. [DOI] [PubMed] [Google Scholar]
- 14.Starck SR; Roberts RW, Puromycin oligonucleotides reveal steric restrictions for ribosome entry and multiple modes of translation inhibition. RNA 2002, 8 (7), 890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishizawa T; Kawakami T; Reid PC; Murakami H, TRAP display: a high-speed selection method for the generation of functional polypeptides. J Am Chem Soc 2013, 135 (14), 5433–40. [DOI] [PubMed] [Google Scholar]
- 16.Kreider BL, PROfusion: genetically tagged proteins for functional proteomics and beyond. Med Res Rev 2000, 20 (3), 212–5. [DOI] [PubMed] [Google Scholar]
- 17.Nemoto N; Miyamoto-Sato E; Husimi Y; Yanagawa H, In vitro virus: bonding of mRNA bearing puromycin at the 3’-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett 1997, 414 (2), 405–8. [DOI] [PubMed] [Google Scholar]
- 18.Yamaguchi J; Naimuddin M; Biyani M; Sasaki T; Machida M; Kubo T; Funatsu T; Husimi Y; Nemoto N, cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions. Nucleic acids research 2009, 37 (16), e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore MJ; Sharp PA, Site-specific modification of pre-mRNA: the 2’-hydroxyl groups at the splice sites. Science 1992, 256 (5059), 992–7. [DOI] [PubMed] [Google Scholar]
- 20.Kurz M; Gu K; Al-Gawari A; Lohse PA, cDNA–protein fusions: covalent protein–gene conjugates for the in vitro selection of peptides and proteins. Chembiochem 2001, 2 (9), 666–672. [DOI] [PubMed] [Google Scholar]
- 21.Kurz M; Gu K; Lohse PA, Psoralen photo-crosslinked mRNA-puromycin conjugates: a novel template for the rapid and facile preparation of mRNA-protein fusions. Nucleic acids research 2000, 28 (18), E83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mochizuki Y; Suzuki T; Fujimoto K; Nemoto N, A versatile puromycin-linker using cnvK for high-throughput in vitro selection by cDNA display. J Biotechnol 2015, 212, 174–80. [DOI] [PubMed] [Google Scholar]
- 23.Barrick JE; Takahashi TT; Balakin A; Roberts RW, Selection of RNA-binding peptides using mRNA-peptide fusions. Methods 2001, 23 (3), 287–293. [DOI] [PubMed] [Google Scholar]
- 24.Tabuchi I; Soramoto S; Nemoto N; Husimi Y, An in vitro DNA virus for in vitro protein evolution. FEBS Lett 2001, 508 (3), 309–12. [DOI] [PubMed] [Google Scholar]
- 25.Nishigaki K; Taguchi K; Kinoshita Y; Aita T; Husimi Y, Y-ligation: an efficient method for ligating single-stranded DNAs and RNAs with T4 RNA ligase. Mol Divers 1998, 4 (3), 187–90. [DOI] [PubMed] [Google Scholar]
- 26.Tabuchi I; Soramoto S; Suzuki M; Nishigaki K; Nemoto N; Husimi Y, An Efficient Ligation Method in the Making of an in vitro Virus for in vitro Protein Evolution. Biol Proced Online 2002, 4, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biyani M; Husimi Y; Nemoto N, Solid-phase translation and RNA-protein fusion: a novel approach for folding quality control and direct immobilization of proteins using anchored mRNA. Nucleic acids research 2006, 34 (20), e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naimuddin M; Kubo T, A High Performance Platform Based on cDNA Display for Efficient Synthesis of Protein Fusions and Accelerated Directed Evolution. ACS Comb Sci 2016, 18 (2), 117–29. [DOI] [PubMed] [Google Scholar]
- 29.Reyes SG; Kuruma Y; Fujimi M; Yamazaki M; Eto S; Nishikawa S; Tamaki S; Kobayashi A; Mizuuchi R; Rothschild L; Ditzler M; Fujishima K, PURE mRNA display and cDNA display provide rapid detection of core epitope motif via high-throughput sequencing. Biotechnol Bioeng 2021, 118 (4), 1736–1749. [DOI] [PubMed] [Google Scholar]
- 30.Hanes J; Plückthun A, In vitro selection and evolution of functional proteins by using ribosome display. Proceedings of the National Academy of Sciences 1997, 94 (10), 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lam KS; Lebl M; Krchnak V, The “One-Bead-One-Compound” Combinatorial Library Method. Chem Rev 1997, 97 (2), 411–448. [DOI] [PubMed] [Google Scholar]
- 32.Wong FT; Khosla C, Combinatorial biosynthesis of polyketides—a perspective. Current opinion in chemical biology 2012, 16 (1-2), 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walsh CT, Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science 2004, 303 (5665), 1805–1810. [DOI] [PubMed] [Google Scholar]
- 34.Lancet D; Sadovsky E; Seidemann E, Probability model for molecular recognition in biological receptor repertoires: significance to the olfactory system. Proceedings of the National Academy of Sciences 1993, 90 (8), 3715–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaughan TJ; Williams AJ; Pritchard K; Osbourn JK; Pope AR; Earnshaw JC; McCafferty J; Hodits RA; Wilton J; Johnson KS, Human Antibodies with Sub-nanomolar Affinities Isolated from a Large Non-immunized Phage Display Library. Nat Biotech 1996, 14 (3), 309–314. [DOI] [PubMed] [Google Scholar]
- 36.Rosenwald S; Kafri RAN; Lancet D, Test of a Statistical Model for Molecular Recognition in Biological Repertoires. Journal of Theoretical Biology 2002, 216 (3), 327–336. [DOI] [PubMed] [Google Scholar]
- 37.Wilson DS; Keefe AD; Szostak JW, The use of mRNA display to select high-affinity protein-binding peptides. Proceedings of the National Academy of Sciences 2001, 98 (7), 3750–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voss S; Skerra A, Mutagenesis of a flexible loop in streptavidin leads to higher affinity for the Strep-tag II peptide and improved performance in recombinant protein purification. Protein engineering 1997, 10 (8), 975–982. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt TG; Skerra A, The random peptide library-assisted engineering of a C-terminal affinity peptide, useful for the detection and purification of a functional Ig Fv fragment. Protein Engineering, Design and Selection 1993, 6 (1), 109–122. [DOI] [PubMed] [Google Scholar]
- 40.Knight R; Yarus M, Analyzing partially randomized nucleic acid pools: straight dope on doping. Nucleic acids research 2003, 31 (6), e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brule CE; Grayhack EJ, Synonymous codons: choose wisely for expression. Trends in Genetics 2017, 33 (4), 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall MN; Gabay J; Débarbouillé M; Schwartz M, A role for mRNA secondary structure in the control of translation initiation. Nature 1982, 295 (5850), 616–618. [DOI] [PubMed] [Google Scholar]
- 43.Gold L, mRNA display: diversity matters during in vitro selection. Proceedings of the National Academy of Sciences of the United States of America 2001, 98 (9), 4825–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sha F; Salzman G; Gupta A; Koide S, Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci 2017, 26 (5), 910–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi L; Wheeler JC; Sweet RW; Lu J; Luo J; Tornetta M; Whitaker B; Reddy R; Brittingham R; Borozdina L, De novo selection of high-affinity antibodies from synthetic fab libraries displayed on phage as pIX fusion proteins. Journal of molecular biology 2010, 397 (2), 385–396. [DOI] [PubMed] [Google Scholar]
- 46.Cwirla SE; Peters EA; Barrett RW; Dower WJ, Peptides on phage: a vast library of peptides for identifying ligands. Proceedings of the National Academy of Sciences of the United States of America 1990, 87 (16), 6378–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zade HM; Keshavarz R; Shekarabi HSZ; Bakhshinejad B, Biased selection of propagation-related TUPs from phage display peptide libraries. Amino acids 2017, 49 (8), 1293–1308. [DOI] [PubMed] [Google Scholar]
- 48.Krumpe LR; Atkinson AJ; Smythers GW; Kandel A; Schumacher KM; McMahon JB; Makowski L; Mori T, T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics 2006, 6 (15), 4210–4222. [DOI] [PubMed] [Google Scholar]
- 49.Yamane K; Mizushima S, Introduction of basic amino acid residues after the signal peptide inhibits protein translocation across the cytoplasmic membrane of Escherichia coli. Relation to the orientation of membrane proteins. Journal of Biological Chemistry 1988, 263 (36), 19690–19696. [PubMed] [Google Scholar]
- 50.Fiacco SV; Roberts RW, N-Methyl Scanning Mutagenesis Generates Protease-Resistant G Protein Ligands with Improved Affinity and Selectivity. ChemBioChem 2008, 9 (14), 2200–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiacco SV; Kelderhouse LE; Hardy A; Peleg Y; Hu B; Ornelas A; Yang P; Gammon ST; Howell SM; Wang P; Takahashi TT; Millward SW; Roberts RW, Directed Evolution of Scanning Unnatural-Protease-Resistant (SUPR) Peptides for in Vivo Applications. Chembiochem 2016, 17 (17), 1643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Howell SM; Fiacco SV; Takahashi TT; Jalali-Yazdi F; Millward SW; Hu B; Wang P; Roberts RW, Serum stable natural peptides designed by mRNA display. Scientific reports 2014, 4, 6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nichols AL; Noridomi K; Hughes CR; Jalali-Yazdi F; Eaton JB; Lai LH; Advani G; Lukas RJ; Lester HA; Chen L; Roberts RW, alpha1-FANGs: Protein Ligands Selective for the alpha-Bungarotoxin Site of the alpha1-Nicotinic Acetylcholine Receptor. ACS Chem Biol 2018, 13 (9), 2568–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cotten SW; Zou J; Valencia CA; Liu R, Selection of proteins with desired properties from natural proteome libraries using mRNA display. Nat Protoc 2011, 6 (8), 1163–82. [DOI] [PubMed] [Google Scholar]
- 55.Wang H; Liu R, Advantages of mRNA display selections over other selection techniques for investigation of protein-protein interactions. Expert Rev Proteomics 2011, 8 (3), 335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kintzing JR; Filsinger Interrante MV; Cochran JR, Emerging Strategies for Developing Next-Generation Protein Therapeutics for Cancer Treatment. Trends Pharmacol Sci 2016, 37 (12), 993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freise AC; Wu AM, In vivo imaging with antibodies and engineered fragments. Mol Immunol 2015, 67 (2 Pt A), 142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gebauer M; Skerra A, Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol 2009, 13 (3), 245–55. [DOI] [PubMed] [Google Scholar]
- 59.Rossmann M; S JG; Moschetti T.; Dinan M; Hyvonen M, Development of a multipurpose scaffold for the display of peptide loops. Protein Eng Des Sel 2017, 30 (6), 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hosse RJ; Rothe A; Power BE, A new generation of protein display scaffolds for molecular recognition. Protein Sci 2006, 15 (1), 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zoller F; Haberkorn U; Mier W, Miniproteins as phage display-scaffolds for clinical applications. Molecules 2011, 16 (3), 2467–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koide A; Bailey CW; Huang X; Koide S, The fibronectin type III domain as a scaffold for novel binding proteins1. Journal of molecular biology 1998, 284 (4), 1141–1151. [DOI] [PubMed] [Google Scholar]
- 63.Forrer P; Jung S; Pluckthun A, Beyond binding: using phage display to select for structure, folding and enzymatic activity in proteins. Curr Opin Struct Biol 1999, 9 (4), 514–20. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi TT; Austin RJ; Roberts RW, mRNA display: ligand discovery, interaction analysis and beyond. Trends in biochemical sciences 2003, 28 (3), 159–165. [DOI] [PubMed] [Google Scholar]
- 65.Keefe AD; Szostak JW, Functional proteins from a random-sequence library. Nature 2001, 410 (6829), 715–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho G; Keefe AD; Liu R; Wilson DS; Szostak JW, Constructing high complexity synthetic libraries of long ORFs using in vitro selection. J Mol Biol 2000, 297 (2), 309–19. [DOI] [PubMed] [Google Scholar]
- 67.Schmidt MM; Wittrup KD, A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Molecular cancer therapeutics 2009, 8 (10), 2861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Doshi R; Chen BR; Vibat CR; Huang N; Lee CW; Chang G, In vitro nanobody discovery for integral membrane protein targets. Scientific reports 2014, 4, 6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanaka J; Yanagawa H; Doi N, Comparison of the frequency of functional SH3 domains with different limited sets of amino acids using mRNA display. PLoS One 2011, 6 (3), e18034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sumida T; Yanagawa H; Doi N, In vitro selection of fab fragments by mRNA display and gene-linking emulsion PCR. J Nucleic Acids 2012, 2012, 371379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu L; Aha P; Gu K; Kuimelis RG; Kurz M; Lam T; Lim AC; Liu H; Lohse PA; Sun L, Directed evolution of high-affinity antibody mimics using mRNA display. Chemistry & biology 2002, 9 (8), 933–942. [DOI] [PubMed] [Google Scholar]
- 72.Emanuel SL; Engle LJ; Chao G; Zhu R-R; Cao C; Lin Z; Yamniuk AP; Hosbach J; Brown J; Fitzpatrick E In A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin-like growth factor-I receptor, MAbs, Taylor & Francis: 2011; pp 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Getmanova EV; Chen Y; Bloom L; Gokemeijer J; Shamah S; Warikoo V; Wang J; Ling V; Sun L, Antagonists to human and mouse vascular endothelial growth factor receptor 2 generated by directed protein evolution in vitro. Chemistry & biology 2006, 13 (5), 549–556. [DOI] [PubMed] [Google Scholar]
- 74.Grilo AL; Mantalaris A, The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol 2019, 37 (1), 9–16. [DOI] [PubMed] [Google Scholar]
- 75.Kohler G; Milstein C, Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256 (5517), 495–7. [DOI] [PubMed] [Google Scholar]
- 76.Geyer CR; McCafferty J; Dubel S; Bradbury AR; Sidhu SS, Recombinant antibodies and in vitro selection technologies. Methods Mol Biol 2012, 901, 11–32. [DOI] [PubMed] [Google Scholar]
- 77.Kipriyanov SM; Le Gall F, Generation and production of engineered antibodies. Mol Biotechnol 2004, 26 (1), 39–60. [DOI] [PubMed] [Google Scholar]
- 78.Liu H; May K, Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs 2012, 4 (1), 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hsieh CM; Kutskova YA; Memmott JE Methods of RNA Display. US8916504, December 23, 2014.
- 80.Nagumo Y; Fujiwara K; Horisawa K; Yanagawa H; Doi N, PURE mRNA display for in vitro selection of single-chain antibodies. J Biochem 2016, 159 (5), 519–26. [DOI] [PubMed] [Google Scholar]
- 81.Chen L; Kutskova YA; Hong F; Memmott JE; Zhong S; Jenkinson MD; Hsieh CM, Preferential germline usage and VH/VL pairing observed in human antibodies selected by mRNA display. Protein Eng Des Sel 2015, 28 (10), 427–35. [DOI] [PubMed] [Google Scholar]
- 82.Tabata N; Sakuma Y; Honda Y; Doi N; Takashima H; Miyamoto-Sato E; Yanagawa H, Rapid antibody selection by mRNA display on a microfluidic chip. Nucleic acids research 2009, 37 (8), e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shibui T; Kobayashi T; Kanatani K, A completely in vitro system for obtaining scFv using mRNA display, PCR, direct sequencing, and wheat embryo cell-free translation. Biotechnol Lett 2009, 31 (7), 1103–10. [DOI] [PubMed] [Google Scholar]
- 84.Fukuda I; Kojoh K; Tabata N; Doi N; Takashima H; Miyamoto-Sato E; Yanagawa H, In vitro evolution of single-chain antibodies using mRNA display. Nucleic acids research 2006, 34 (19), e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shibui T; Kobayashi T; Kanatani K; Koga H; Misawa S; Isomura T; Sasaki T, In vitro selection of scFv and its production: an application of mRNA display and wheat embryo cell-free and E. coli cell production system. Appl Microbiol Biotechnol 2009, 84 (4), 725–32. [DOI] [PubMed] [Google Scholar]
- 86.Ducancel F; Muller BH, Molecular engineering of antibodies for therapeutic and diagnostic purposes. MAbs 2012, 4 (4), 445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chan CE; Chan AH; Lim AP; Hanson BJ, Comparison of the efficiency of antibody selection from semi-synthetic scFv and non-immune Fab phage display libraries against protein targets for rapid development of diagnostic immunoassays. Journal of immunological methods 2011, 373 (1–2), 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Steinwand M; Droste P; Frenzel A; Hust M; Dübel S; Schirrmann T In The influence of antibody fragment format on phage display based affinity maturation of IgG, MAbs, Taylor & Francis: 2014; pp 204–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gebauer M; Skerra A, Engineered Protein Scaffolds as Next-Generation Therapeutics. Annu Rev Pharmacol Toxicol 2020, 60, 391–415. [DOI] [PubMed] [Google Scholar]
- 90.16 - Development issues: antibody stability, developability, immunogenicity, and comparability. In Therapeutic Antibody Engineering, Strohl WR; Strohl LM, Eds. Woodhead Publishing: 2012; pp 377–595. [Google Scholar]
- 91.Chung WK; Russell B; Yang Y; Handlogten M; Hudak S; Cao M; Wang J; Robbins D; Ahuja S; Zhu M, Effects of antibody disulfide bond reduction on purification process performance and final drug substance stability. Biotechnol Bioeng 2017, 114 (6), 1264–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zollinger AJ; Smith ML, Fibronectin, the extracellular glue. Matrix Biol 2017, 60-61, 27–37. [DOI] [PubMed] [Google Scholar]
- 93.Koide S; Koide A; Lipovšek D, Target-binding proteins based on the 10th human fibronectin type III domain (10Fn3). In Methods in enzymology, Elsevier: 2012; Vol. 503, pp 135–156. [DOI] [PubMed] [Google Scholar]
- 94.Olson CA; Roberts RW, Design, expression, and stability of a diverse protein library based on the human fibronectin type III domain. Protein science 2007, 16 (3), 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bannas P; Hambach J; Koch-Nolte F, Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Frontiers in immunology 2017, 8, 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ramamurthy V; Krystek SR Jr.; Bush A; Wei A; Emanuel SL; Das Gupta R; Janjua A; Cheng L; Murdock M; Abramczyk B; Cohen D; Lin Z; Morin P; Davis JH; Dabritz M; McLaughlin DC; Russo KA; Chao G; Wright MC; Jenny VA; Engle LJ; Furfine E; Sheriff S, Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure 2012, 20 (2), 259–69. [DOI] [PubMed] [Google Scholar]
- 97.Khan JA; Camac DM; Low S; Tebben AJ; Wensel DL; Wright MC; Su J; Jenny V; Gupta RD; Ruzanov M; Russo KA; Bell A; An Y; Bryson JW; Gao M; Gambhire P; Baldwin ET; Gardner D; Cavallaro CL; Duncia JV; Hynes J Jr., Developing Adnectins that target SRC co-activator binding to PXR: a structural approach toward understanding promiscuity of PXR. J Mol Biol 2015, 427 (4), 924–42. [DOI] [PubMed] [Google Scholar]
- 98.Han X; Cinay GE; Zhao Y; Guo Y; Zhang X; Wang P, Adnectin-Based Design of Chimeric Antigen Receptor for T Cell Engineering. Mol Ther 2017, 25 (11), 2466–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Donnelly DJ; Smith RA; Morin P; Lipovsek D; Gokemeijer J; Cohen D; Lafont V; Tran T; Cole EL; Wright M; Kim J; Pena A; Kukral D; Dischino DD; Chow P; Gan J; Adelakun O; Wang XT; Cao K; Leung D; Bonacorsi SJ Jr.; Hayes W, Synthesis and Biologic Evaluation of a Novel (18)F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J Nucl Med 2018, 59 (3), 529–535. [DOI] [PubMed] [Google Scholar]
- 100.Waldo GS; Standish BM; Berendzen J; Terwilliger TC, Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol 1999, 17 (7), 691–5. [DOI] [PubMed] [Google Scholar]
- 101.Lipovsek D, Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel 2011, 24 (1-2), 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xiao L; Hung KC; Takahashi TT; Joo KI; Lim M; Roberts RW; Wang P, Antibody-mimetic ligand selected by mRNA display targets DC-SIGN for dendritic cell-directed antigen delivery. ACS Chem Biol 2013, 8 (5), 967–77. [DOI] [PubMed] [Google Scholar]
- 103.Mangan PR; Su LJ; Jenny V; Tatum AL; Picarillo C; Skala S; Ditto N; Lin Z; Yang X; Cotter PZ; Shuster DJ; Song Y; Borowski V; Thomas RL; Heimrich EM; Devaux B; Das Gupta R; Carvajal I; McIntyre KW; Xie J; Zhao Q; Struthers M; Salter-Cid LM, Dual Inhibition of Interleukin-23 and Interleukin-17 Offers Superior Efficacy in Mouse Models of Autoimmunity. J Pharmacol Exp Ther 2015, 354 (2), 152–65. [DOI] [PubMed] [Google Scholar]
- 104.Stutvoet TS; van der Veen EL; Kol A; Antunes IF; de Vries EF; Hospers GA; de Vries EG; de Jong S; Lub-de Hooge MN, Molecular imaging of PD-L1 expression and dynamics with the adnectin-based PET tracer 18F-BMS-986192. Journal of Nuclear Medicine 2020, 61 (12), 1839–1844. [DOI] [PubMed] [Google Scholar]
- 105.Huisman MC; Niemeijer A-LN; Windhorst AD; Schuit RC; Leung D; Hayes W; Poot A; Bahce I; Radonic T; Oprea-Lager DE, Quantification of PD-L1 Expression with 18F-BMS-986192 PET/CT in Patients with Advanced-Stage Non–Small Cell Lung Cancer. Journal of Nuclear Medicine 2020, 61 (10), 1455–1460. [DOI] [PubMed] [Google Scholar]
- 106.Robu S; Richter A; Gosmann D; Seidl C; Leung D; Hayes W; Cohen D; Morin P; Donnelly DJ; Lipovšek D, Synthesis and Preclinical Evaluation of 68Ga-labeled Adnectin, 68Ga-BMS-986192 as a PET Agent for Imaging PD-L1 Expression. Journal of Nuclear Medicine 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wensel D; Sun Y; Li Z; Zhang S; Picarillo C; McDonagh T; Fabrizio D; Cockett M; Krystal M; Davis J, Discovery and Characterization of a Novel CD4-Binding Adnectin with Potent Anti-HIV Activity. Antimicrob Agents Chemother 2017, 61 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wensel D; Sun Y; Davis J; Li Z; Zhang S; McDonagh T; Fabrizio D; Cockett M; Krystal M, A Novel gp41-Binding Adnectin with Potent Anti-HIV Activity Is Highly Synergistic when Linked to a CD4-Binding Adnectin. J Virol 2018, 92 (14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wensel D; Sun Y; Davis J; Li Z; Zhang S; McDonagh T; Langley D; Mitchell T; Tabruyn S; Nef P, GSK3732394: a Multi-specific Inhibitor of HIV Entry. Journal of virology 2019, 93 (20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lipovsek D; Carvajal I; Allentoff AJ; Barros A Jr.; Brailsford J; Cong Q; Cotter P; Gangwar S; Hollander C; Lafont V; Lau WL; Li W; Moreta M; O’Neil S; Pinckney J; Smith MJ; Su J; Terragni C; Wallace MA; Wang L; Wright M; Marsh HN; Bryson JW, Adnectin-drug conjugates for Glypican-3-specific delivery of a cytotoxic payload to tumors. Protein Eng Des Sel 2018, 31 (5), 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kondo T; Iwatani Y; Matsuoka K; Fujino T; Umemoto S; Yokomaku Y; Ishizaki K; Kito S; Sezaki T; Hayashi G, Antibody-like proteins that capture and neutralize SARS-CoV-2. Science advances 2020, 6 (42), eabd3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parker M; Chen Y; Danehy F; Dufu K; Ekstrom J; Getmanova E; Gokemeijer J; Xu L; Lipovsek D, Antibody mimics based on human fibronectin type three domain engineered for thermostability and high-affinity binding to vascular endothelial growth factor receptor two. Protein Engineering Design and Selection 2005, 18 (9), 435–444. [DOI] [PubMed] [Google Scholar]
- 113.Olson CA; Liao H-I; Sun R; Roberts RW, mRNA display selection of a high-affinity, modification-specific phospho-IκBα-binding fibronectin. ACS chemical biology 2008, 3 (8), 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liao HI; Olson CA; Hwang S; Deng H; Wong E; Baric RS; Roberts RW; Sun R, mRNA display design of fibronectin-based intrabodies that detect and inhibit severe acute respiratory syndrome coronavirus nucleocapsid protein. The Journal of biological chemistry 2009, 284 (26), 17512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Olson CA; Adams JD; Takahashi TT; Qi H; Howell SM; Wu TT; Roberts RW; Sun R; Soh HT, Rapid mRNA-Display Selection of an IL-6 Inhibitor Using Continuous-Flow Magnetic Separation. Angewandte Chemie International Edition 2011, 50 (36), 8295–8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Olson CA; Nie J; Diep J; Al-Shyoukh I; Takahashi TT; Al-Mawsawi LQ; Bolin JM; Elwell AL; Swanson S; Stewart R; Thomson JA; Soh HT; Roberts RW; Sun R, Single-round, multiplexed antibody mimetic design through mRNA display. Angew Chem Int Ed Engl 2012, 51 (50), 12449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gross GG; Junge JA; Mora RJ; Kwon H-B; Olson CA; Takahashi TT; Liman ER; Ellis-Davies GC; McGee AW; Sabatini BL, Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 2013, 78 (6), 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mora RJ; Roberts RW; Arnold DB, Recombinant probes reveal dynamic localization of CaMKIIalpha within somata of cortical neurons. J Neurosci 2013, 33 (36), 14579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cetin M; Evenson WE; Gross GG; Jalali-Yazdi F; Krieger D; Arnold D; Takahashi TT; Roberts RW, RasIns: genetically encoded intrabodies of activated Ras proteins. Journal of molecular biology 2017, 429 (4), 562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dineen SP; Sullivan LA; Beck AW; Miller AF; Carbon JG; Mamluk R; Wong H; Brekken RA, The Adnectin CT-322 is a novel VEGF receptor 2 inhibitor that decreases tumor burden in an orthotopic mouse model of pancreatic cancer. BMC cancer 2008, 8 (1), 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mamluk R; Carvajal IM; Morse BA; Wong HK; Abramowitz J; Aslanian S; Lim A-C; Gokemeijer J; Storek MJ; Lee J In Anti-tumor effect of CT-322 as an adnectin inhibitor of vascular endothelial growth factor receptor-2, MAbs, Taylor & Francis: 2010; pp 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bloom L; Calabro V, FN3: a new protein scaffold reaches the clinic. Drug discovery today 2009, 14 (19-20), 949–955. [DOI] [PubMed] [Google Scholar]
- 123.Tolcher AW; Sweeney CJ; Papadopoulos K; Patnaik A; Chiorean EG; Mita AC; Sankhala K; Furfine E; Gokemeijer J; Iacono L, Phase I and pharmacokinetic study of CT-322 (BMS-844203), a targeted Adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clinical Cancer Research 2011. [DOI] [PubMed] [Google Scholar]
- 124.Evans JB; Syed BA, Next-generation antibodies. Nature Publishing Group: 2014. [DOI] [PubMed] [Google Scholar]
- 125.Schiff D; Kesari S; de Groot J; Mikkelsen T; Drappatz J; Coyle T; Fichtel L; Silver B; Walters I; Reardon D, Phase 2 study of CT-322, a targeted biologic inhibitor of VEGFR-2 based on a domain of human fibronectin, in recurrent glioblastoma. Invest New Drugs 2015, 33 (1), 247–53. [DOI] [PubMed] [Google Scholar]
- 126.El-Husseini AE; Schnell E; Chetkovich DM; Nicoll RA; Bredt DS, PSD-95 involvement in maturation of excitatory synapses. Science 2000, 290 (5495), 1364–8. [PubMed] [Google Scholar]
- 127.Proba K; Worn A; Honegger A; Pluckthun A, Antibody scFv fragments without disulfide bonds made by molecular evolution. J Mol Biol 1998, 275 (2), 245–53. [DOI] [PubMed] [Google Scholar]
- 128.Ishikawa FN; Chang HK; Curreli M; Liao HI; Olson CA; Chen PC; Zhang R; Roberts RW; Sun R; Cote RJ; Thompson ME; Zhou C, Label-free, electrical detection of the SARS virus N-protein with nanowire biosensors utilizing antibody mimics as capture probes. ACS Nano 2009, 3 (5), 1219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yushen D; Tian-hao Z; Xiangzhi M; Yuan S; Menglong H; Shuofeng Y; Chit Ying L; Shu-xing L; Siwei L; Jiayan L; Haigen H; Hong J; Dongdong C; Li S; Mengying H; Anders O; Hsiang IL; Xiaojiang C; Xinmin L; Gexin Z; Richard WR; Jasper FWC; Dong-Yan J; Irvin SYC; Honglin C; Kwok-Yung Y; Quan H; Ren S, Development of high affinity monobodies recognizing SARS-CoV-2 antigen. Research Square 2021. [Google Scholar]
- 130.Koide S, Engineering of recombinant crystallization chaperones. Curr Opin Struct Biol 2009, 19 (4), 449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Son JH; Keefe MD; Stevenson TJ; Barrios JP; Anjewierden S; Newton JB; Douglass AD; Bonkowsky JL, Transgenic FingRs for Live Mapping of Synaptic Dynamics in Genetically-Defined Neurons. Scientific reports 2016, 6, 18734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bensussen S; Shankar S; Ching KH; Zemel D; Ta TL; Mount RA; Shroff SN; Gritton HJ; Fabris P; Vanbenschoten H; Beck C; Man HY; Han X, A Viral Toolbox of Genetically Encoded Fluorescent Synaptic Tags. iScience 2020, 23 (7), 101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li Y; Junge JA; Arnesano C; Gross GG; Miner JH; Moats R; Roberts RW; Arnold DB; Fraser SE, Discs large 1 controls daughter-cell polarity after cytokinesis in vertebrate morphogenesis. Proceedings of the National Academy of Sciences of the United States of America 2018, 115 (46), E10859–E10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fosgerau K; Hoffmann T, Peptide therapeutics: current status and future directions. Drug discovery today 2015, 20 (1), 122–128. [DOI] [PubMed] [Google Scholar]
- 135.Arkin MR; Randal M; DeLano WL; Hyde J; Luong TN; Oslob JD; Raphael DR; Taylor L; Wang J; McDowell RS, Binding of small molecules to an adaptive protein–protein interface. Proceedings of the National Academy of Sciences 2003, 100 (4), 1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Conte LL; Chothia C; Janin J, The atomic structure of protein-protein recognition sites. Journal of molecular biology 1999, 285 (5), 2177–2198. [DOI] [PubMed] [Google Scholar]
- 137.Stites WE, Protein–protein interactions: interface structure, binding thermodynamics, and mutational analysis. Chemical reviews 1997, 97 (5), 1233–1250. [DOI] [PubMed] [Google Scholar]
- 138.Manning MC; Chou DK; Murphy BM; Payne RW; Katayama DS, Stability of protein pharmaceuticals: an update. Pharmaceutical research 2010, 27 (4), 544–575. [DOI] [PubMed] [Google Scholar]
- 139.Larsen BD, Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis. Google Patents: 2008. [Google Scholar]
- 140.Timmerman P; Puijk W; Boshuizen R; Dijken PV; Slootstra J; Beurskens F; Parren P; Huber A; Bachmann M; Meloen R, Functional reconstruction of structurally complex epitopes using CLIPS™ Technology. The Open Vaccine Journal 2009, 2 (1). [Google Scholar]
- 141.Sim S; Kim Y; Kim T; Lim S; Lee M, Directional assembly of α-helical peptides induced by cyclization. Journal of the American Chemical Society 2012, 134 (50), 20270–20272. [DOI] [PubMed] [Google Scholar]
- 142.Thurber GM; Schmidt MM; Wittrup KD, Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Advanced drug delivery reviews 2008, 60 (12), 1421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ladner RC; Sato AK; Gorzelany J; de Souza M, Phage display-derived peptides as therapeutic alternatives to antibodies. Drug discovery today 2004, 9 (12), 525–529. [DOI] [PubMed] [Google Scholar]
- 144.Li ZJ; Cho CH In Peptides as targeting probes against tumor vasculature for diagnosis and drug delivery, Journal of translational medicine, BioMed Central: 2012; p S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ruoslahti E, Peptides as targeting elements and tissue penetration devices for nanoparticles. Advanced Materials 2012, 24 (28), 3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Barrick JE; Takahashi TT; Ren J; Xia T; Roberts RW, Large libraries reveal diverse solutions to an RNA recognition problem. Proceedings of the National Academy of Sciences 2001, 98 (22), 12374–12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yan S; Niu R; Wang Z; Lin X, In vitro selected peptides bind with thymidylate synthase mRNA and inhibit its translation. Science in China Series C: Life Sciences 2007, 50 (5), 630–636. [DOI] [PubMed] [Google Scholar]
- 148.Barrick JE; Roberts RW, Sequence analysis of an artificial family of RNA-binding peptides. Protein Science 2002, 11 (11), 2688–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Austin RJ; Xia T; Ren J; Takahashi TT; Roberts RW, Differential modes of recognition in N peptide–BoxB complexes. Biochemistry 2003, 42 (50), 14957–14967. [DOI] [PubMed] [Google Scholar]
- 150.Austin RJ; Xia T; Ren J; Takahashi TT; Roberts RW, Designed arginine-rich RNA-binding peptides with picomolar affinity. Journal of the American Chemical Society 2002, 124 (37), 10966–10967. [DOI] [PubMed] [Google Scholar]
- 151.Kinch MS; Hoyer D; Patridge E; Plummer M, Target selection for FDA-approved medicines. Drug discovery today 2015, 20 (7), 784–789. [DOI] [PubMed] [Google Scholar]
- 152.Hauser AS; Attwood MM; Rask-Andersen M; Schiöth HB; Gloriam DE, Trends in GPCR drug discovery: new agents, targets and indications. Nature reviews Drug discovery 2017, 16 (12), 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ja WW; Roberts RW, In vitro selection of state-specific peptide modulators of G protein signaling using mRNA display. Biochemistry 2004, 43 (28), 9265–9275. [DOI] [PubMed] [Google Scholar]
- 154.Shiheido H; Takashima H; Doi N; Yanagawa H, mRNA display selection of an optimized MDM2-binding peptide that potently inhibits MDM2-p53 interaction. PLoS One 2011, 6 (3), e17898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kobayashi T; Kakui M; Shibui T; Kitano Y, In vitro selection of a peptide inhibitor of human IL-6 using mRNA display. Molecular biotechnology 2011, 48 (2), 147–155. [DOI] [PubMed] [Google Scholar]
- 156.Flock T; Hauser AS; Lund N; Gloriam DE; Balaji S; Babu MM, Selectivity determinants of GPCR–G-protein binding. Nature 2017, 545 (7654), 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Goricanec D; Stehle R; Egloff P; Grigoriu S; Plückthun A; Wagner G; Hagn F, Conformational dynamics of a G-protein α subunit is tightly regulated by nucleotide binding. Proceedings of the National Academy of Sciences 2016, 113 (26), E3629–E3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.William WJ; Roberts RW, G-protein-directed ligand discovery with peptide combinatorial libraries. Trends in biochemical sciences 2005, 30 (6), 318–324. [DOI] [PubMed] [Google Scholar]
- 159.William WJ; Adhikari A; Austin RJ; Sprang SR; Roberts RW, A peptide core motif for binding to heterotrimeric G protein α subunits. Journal of Biological Chemistry 2005, 280 (37), 32057–32060. [DOI] [PubMed] [Google Scholar]
- 160.Austin RJ; William WJ; Roberts RW, Evolution of class-specific peptides targeting a hot spot of the Gαs subunit. Journal of molecular biology 2008, 377 (5), 1406–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ja WW; Wiser O; Austin RJ; Jan LY; Roberts RW, Turning G proteins on and off using peptide ligands. ACS chemical biology 2006, 1 (9), 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.William WJ; West AP Jr; Delker SL; Bjorkman PJ; Benzer S; Roberts RW, Extension of Drosophila melanogaster life span with a GPCR peptide inhibitor. Nature chemical biology 2007, 3 (7), 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kamide K; Nakakubo H; Uno S; Fukamizu A, Isolation of novel cell-penetrating peptides from a random peptide library using in vitro virus and their modifications. International journal of molecular medicine 2010, 25 (1), 41–51. [PubMed] [Google Scholar]
- 164.Higa M; Katagiri C; Shimizu-Okabe C; Tsumuraya T; Sunagawa M; Nakamura M; Ishiuchi S; Takayama C; Kondo E; Matsushita M, Identification of a novel cell-penetrating peptide targeting human glioblastoma cell lines as a cancer-homing transporter. Biochemical and biophysical research communications 2015, 457 (2), 206–212. [DOI] [PubMed] [Google Scholar]
- 165.Lee JH; Song HS; Lee SG; Park TH; Kim BG, Screening of cell-penetrating peptides using mRNA display. Biotechnology journal 2012, 7 (3), 387–396. [DOI] [PubMed] [Google Scholar]
- 166.Baggio R; Burgstaller P; Hale SP; Putney AR; Lane M; Lipovsek D; Wright MC; Roberts RW; Liu R; Szostak JW, Identification of epitope-like consensus motifs using mRNA display. Journal of Molecular Recognition 2002, 15 (3), 126–134. [DOI] [PubMed] [Google Scholar]
- 167.Guo N; Duan H; Kachko A; Krause BW; Major ME; Krause PR, Reverse Engineering of Vaccine Antigens Using High Throughput Sequencing-enhanced mRNA Display. EBioMedicine 2015, 2 (8), 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ja WW; Olsen BN; Roberts RW, Epitope mapping using mRNA display and a unidirectional nested deletion library. Protein Engineering Design and Selection 2005, 18 (7), 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shiratori M; Kobayashi T; Shibui T, Identification of amino acids essential for antibody binding by mRNA-display using a random peptide library: an anti-human tumor protein p53 antibody as a model. Molecular biotechnology 2009, 41 (2), 99–105. [DOI] [PubMed] [Google Scholar]
- 170.Shibui T; Kobayashi T, Mimotopes to Cetuximab Isolated by mRNA-Display using Random Peptide Libraries and their Characteristics. International Journal of Peptide Research and Therapeutics 2011, 17 (1), 69–74. [Google Scholar]
- 171.Duan H; Kachko A; Zhong L; Struble E; Pandey S; Yan H; Harman C; Virata-Theimer ML; Deng L; Zhao Z; Major M; Feinstone S; Zhang P, Amino acid residue-specific neutralization and nonneutralization of hepatitis C virus by monoclonal antibodies to the E2 protein. J Virol 2012, 86 (23), 12686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cujec TP; Medeiros PF; Hammond P; Rise C; Kreider BL, Selection of v-abl tyrosine kinase substrate sequences from randomized peptide and cellular proteomic libraries using mRNA display. Chemistry & biology 2002, 9 (2), 253–264. [DOI] [PubMed] [Google Scholar]
- 173.Lee JH; Song C; Kim DH; Park IH; Lee SG; Lee YS; Kim BG, Glutamine (Q)-peptide screening for transglutaminase reaction using mRNA display. Biotechnology and bioengineering 2013, 110 (2), 353–362. [DOI] [PubMed] [Google Scholar]
- 174.Fleming SR; Himes PM; Ghodge SV; Goto Y; Suga H; Bowers AA, Exploring the Post-translational Enzymology of PaaA by mRNA Display. J Am Chem Soc 2020, 142 (11), 5024–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vinogradov AA; Nagai E; Chang JS; Narumi K; Onaka H; Goto Y; Suga H, Accurate Broadcasting of Substrate Fitness for Lactazole Biosynthetic Pathway from Reactivity-Profiling mRNA Display. J Am Chem Soc 2020. [DOI] [PubMed] [Google Scholar]
- 176.Hanahan D; Weinberg RA, Hallmarks of cancer: the next generation. Cell 2011, 144 (5), 646–74. [DOI] [PubMed] [Google Scholar]
- 177.Kamalinia G; Engel BJ; Srinivasamani A; Grindel BJ; Ong JN; Curran MA; Takahashi TT; Millward SW; Roberts RW, mRNA Display Discovery of a Novel Programmed Death Ligand 1 (PD-L1) Binding Peptide (a Peptide Ligand for PD-L1). ACS Chem Biol 2020, 15 (6), 1630–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nagata T; Shirakawa K; Kobayashi N; Shiheido H; Tabata N; Sakuma-Yonemura Y; Horisawa K; Katahira M; Doi N; Yanagawa H, Structural basis for inhibition of the MDM2: p53 interaction by an optimized MDM2-binding peptide selected with mRNA display. PloS one 2014, 9 (10), e109163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jalali-Yazdi F; Lai LH; Takahashi TT; Roberts RW, High-Throughput Measurement of Binding Kinetics by mRNA Display and Next-Generation Sequencing. Angew Chem Int Ed Engl 2016, 55 (12), 4007–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kawakami T; Ogawa K; Goshima N; Natsume T, DIVERSE System: De Novo Creation of Peptide Tags for Non-enzymatic Covalent Labeling by In Vitro Evolution for Protein Imaging Inside Living Cells. Chem Biol 2015, 22 (12), 1671–9. [DOI] [PubMed] [Google Scholar]
- 181.Evans ED; Pentelute BL, Discovery of a 29-Amino-Acid Reactive Abiotic Peptide for Selective Cysteine Arylation. ACS Chem Biol 2018, 13 (3), 527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Evans ED; Pentelute BL, Studies on a landscape of perfluoroaromatic-reactive peptides. Organic & biomolecular chemistry 2019, 17 (7), 1862–1868. [DOI] [PubMed] [Google Scholar]
- 183.Ando T; Takamori Y; Yokoyama T; Yamamoto M; Kawakami T, Directed evolution of dibenzocyclooctyne-reactive peptide tags for protein labeling. Biochem Biophys Res Commun 2021, 534, 27–33. [DOI] [PubMed] [Google Scholar]
- 184.Terai T; Anzai H; Nemoto N, Selection of Peptides that Associate with Dye-Conjugated Solid Surfaces in a pH-Dependent Manner Using cDNA Display. ACS Omega 2019, 4 (4), 7378–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ono S; Tsuji T; Oaki Y; Imai H, Artificial peptides binding to the c face of hydroxyapatite obtained by molecular display technology. RSC Advances 2013, 3 (6), 1885–1889. [Google Scholar]
- 186.Kobayashi S; Terai T; Yoshikawa Y; Ohkawa R; Ebihara M; Hayashi M; Takiguchi K; Nemoto N, In vitro selection of random peptides against artificial lipid bilayers: A potential tool to immobilize molecules on membranes. Chemical Communications 2017, 53 (24), 3458–3461. [DOI] [PubMed] [Google Scholar]
- 187.Raffler NA; Schneider-Mergener J; Famulok M, A novel class of small functional peptides that bind and inhibit human α-thrombin isolated by mRNA display. Chemistry & biology 2003, 10 (1), 69–79. [DOI] [PubMed] [Google Scholar]
- 188.Yiadom KP; Muhie S; Yang DC, Peptide inhibitors of botulinum neurotoxin by mRNA display. Biochemical and biophysical research communications 2005, 335 (4), 1247–1253. [DOI] [PubMed] [Google Scholar]
- 189.Matsumura N; Tsuji T; Sumida T; Kokubo M; Onimaru M; Doi N; Takashima H; Miyamoto-Sato E; Yanagawa H, mRNA display selection of a high-affinity, Bcl-XL-specific binding peptide. The FASEB Journal 2010, 24 (7), 2201–2210. [DOI] [PubMed] [Google Scholar]
- 190.Thell K; Hellinger R; Schabbauer G; Gruber CW, Immunosuppressive peptides and their therapeutic applications. Drug discovery today 2014, 19 (5), 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hancock RE; Chapple DS, Peptide antibiotics. Antimicrobial agents and chemotherapy 1999, 43 (6), 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chatterjee J; Laufer B; Kessler H, Synthesis of N-methylated cyclic peptides. Nature protocols 2012, 7 (3), 432. [DOI] [PubMed] [Google Scholar]
- 193.Ovadia O; Greenberg S; Chatterjee J; Laufer B; Opperer F; Kessler H; Gilon C; Hoffman A, The effect of multiple N-methylation on intestinal permeability of cyclic hexapeptides. Molecular pharmaceutics 2011, 8 (2), 479–487. [DOI] [PubMed] [Google Scholar]
- 194.Yamagishi Y; Shoji I; Miyagawa S; Kawakami T; Katoh T; Goto Y; Suga H, Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chemistry & biology 2011, 18 (12), 1562–1570. [DOI] [PubMed] [Google Scholar]
- 195.Khan AR; Parrish JC; Fraser ME; Smith WW; Bartlett PA; James MNG, Lowering the Entropic Barrier for Binding Conformationally Flexible Inhibitors to Enzymes. Biochemistry 1998, 37 (48), 16839–16845. [DOI] [PubMed] [Google Scholar]
- 196.Josephson K; Ricardo A; Szostak JW, mRNA display: from basic principles to macrocycle drug discovery. Drug Discovery Today 2014, 19 (4), 388–399. [DOI] [PubMed] [Google Scholar]
- 197.Dougherty PG; Sahni A; Pei D, Understanding Cell Penetration of Cyclic Peptides. Chem Rev 2019, 119 (17), 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Heckler TG; Chang LH; Zama Y; Naka T; Chorghade MS; Hecht SM, T4 RNA ligase mediated preparation of novel “chemically misacylated” tRNAPheS. Biochemistry 1984, 23 (7), 1468–73. [DOI] [PubMed] [Google Scholar]
- 199.Noren C; Anthony-Cahill S; Griffith M; Schultz P, A general method for site-specific incorporation of unnatural amino acids into proteins. Science 1989, 244 (4901), 182–188. [DOI] [PubMed] [Google Scholar]
- 200.Li S; Millward S; Roberts R, In vitro selection of mRNA display libraries containing an unnatural amino acid. J Am Chem Soc 2002, 124 (34), 9972–3. [DOI] [PubMed] [Google Scholar]
- 201.Saks ME; Sampson JR; Nowak MW; Kearney PC; Du F; Abelson JN; Lester HA; Dougherty DA, An engineered Tetrahymena tRNAGln for in vivo incorporation of unnatural amino acids into proteins by nonsense suppression. The Journal of biological chemistry 1996, 271 (38), 23169–75. [DOI] [PubMed] [Google Scholar]
- 202.Muranaka N; Hohsaka T; Sisido M, Four-base codon mediated mRNA display to construct peptide libraries that contain multiple nonnatural amino acids. Nucleic acids research 2006, 34 (1), e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Johnson DB; Xu J; Shen Z; Takimoto JK; Schultz MD; Schmitz RJ; Xiang Z; Ecker JR; Briggs SP; Wang L, RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nature chemical biology 2011, 7 (11), 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Frankel A; Millward SW; Roberts RW, Encodamers: unnatural peptide oligomers encoded in RNA. Chem Biol 2003, 10 (11), 1043–50. [DOI] [PubMed] [Google Scholar]
- 205.Murakami H; Ohta A; Ashigai H; Suga H, A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat Methods 2006, 3 (5), 357–9. [DOI] [PubMed] [Google Scholar]
- 206.Murakami H; Saito H; Suga H, A versatile tRNA aminoacylation catalyst based on RNA. Chem Biol 2003, 10 (7), 655–62. [DOI] [PubMed] [Google Scholar]
- 207.Ohuchi M; Murakami H; Suga H, The flexizyme system: a highly flexible tRNA aminoacylation tool for the translation apparatus. Curr Opin Chem Biol 2007, 11 (5), 537–42. [DOI] [PubMed] [Google Scholar]
- 208.Goto Y; Katoh T; Suga H, Flexizymes for genetic code reprogramming. Nat Protoc 2011, 6 (6), 779–90. [DOI] [PubMed] [Google Scholar]
- 209.Niwa N; Yamagishi Y; Murakami H; Suga H, A flexizyme that selectively charges amino acids activated by a water-friendly leaving group. Bioorg Med Chem Lett 2009, 19 (14), 3892–4. [DOI] [PubMed] [Google Scholar]
- 210.Hipolito CJ; Suga H, Ribosomal production and in vitro selection of natural product-like peptidomimetics: the FIT and RaPID systems. Curr Opin Chem Biol 2012, 16 (1–2), 196–203. [DOI] [PubMed] [Google Scholar]
- 211.Shimizu Y; Inoue A; Tomari Y; Suzuki T; Yokogawa T; Nishikawa K; Ueda T, Cell-free translation reconstituted with purified components. Nat Biotech 2001, 19 (8), 751–755. [DOI] [PubMed] [Google Scholar]
- 212.Forster AC; Cornish VW; Blacklow SC, Pure translation display. Analytical biochemistry 2004, 333 (2), 358–364. [DOI] [PubMed] [Google Scholar]
- 213.Tan Z; Blacklow SC; Cornish VW; Forster AC, De novo genetic codes and pure translation display. Methods 2005, 36 (3), 279–290. [DOI] [PubMed] [Google Scholar]
- 214.Forster AC; Tan Z; Nalam MN; Lin H; Qu H; Cornish VW; Blacklow SC, Programming peptidomimetic syntheses by translating genetic codes designed de novo. Proceedings of the National Academy of Sciences 2003, 100 (11), 6353–6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zhang B; Tan Z; Dickson LG; Nalam MN; Cornish VW; Forster AC, Specificity of translation for N-alkyl amino acids. Journal of the American Chemical Society 2007, 129 (37), 11316–11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Guillen Schlippe YV; Hartman MCT; Josephson K; Szostak JW, In Vitro Selection of Highly Modified Cyclic Peptides That Act as Tight Binding Inhibitors. Journal of the American Chemical Society 2012, 134 (25), 10469–10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hartman MC; Josephson K; Szostak JW, Enzymatic aminoacylation of tRNA with unnatural amino acids. Proceedings of the National Academy of Sciences of the United States of America 2006, 103 (12), 4356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Josephson K; Hartman MC; Szostak JW, Ribosomal synthesis of unnatural peptides. J Am Chem Soc 2005, 127 (33), 11727–35. [DOI] [PubMed] [Google Scholar]
- 219.Hartman MC; Josephson K; Lin CW; Szostak JW, An expanded set of amino acid analogs for the ribosomal translation of unnatural peptides. PLoS One 2007, 2 (10), e972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Subtelny AO; Hartman MC; Szostak JW, Ribosomal synthesis of N-methyl peptides. J Am Chem Soc 2008, 130 (19), 6131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Liu CC; Schultz PG, Adding new chemistries to the genetic code. Annu Rev Biochem 2010, 79, 413–44. [DOI] [PubMed] [Google Scholar]
- 222.Li S; Roberts RW, A Novel Strategy for In Vitro Selection of Peptide-Drug Conjugates. Chemistry & biology 2003, 10 (3), 233–239. [DOI] [PubMed] [Google Scholar]
- 223.Morimoto J; Hayashi Y; Suga H, Discovery of Macrocyclic Peptides Armed with a Mechanism-Based Warhead: Isoform-Selective Inhibition of Human Deacetylase SIRT2. Angewandte Chemie International Edition 2012, 51 (14), 3423–3427. [DOI] [PubMed] [Google Scholar]
- 224.Horiya S; Bailey JK; Temme JS; Guillen Schlippe YV; Krauss IJ, Directed evolution of multivalent glycopeptides tightly recognized by HIV antibody 2G12. J Am Chem Soc 2014, 136 (14), 5407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Millward SW; Takahashi TT; Roberts RW, A general route for post-translational cyclization of mRNA display libraries. J Am Chem Soc 2005, 127 (41), 14142–3. [DOI] [PubMed] [Google Scholar]
- 226.O’Neil KT; Hoess RH; Jackson SA; Ramachandran NS; Mousa SA; DeGrado WF, Identification of novel peptide antagonists for GPIIb/IIIa from a conformationally constrained phage peptide library. Proteins 1992, 14 (4), 509–15. [DOI] [PubMed] [Google Scholar]
- 227.Engel BJ; Grindel BJ; Gray JP; Millward SW, Purification of poly-dA oligonucleotides and mRNA-protein fusions with dT25-OAS resin. Bioorg Med Chem Lett 2020, 30 (4), 126934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Sau SP; Larsen AC; Chaput JC, Automated solid-phase synthesis of high capacity oligo-dT cellulose for affinity purification of poly-A tagged biomolecules. Bioorg Med Chem Lett 2014, 24 (24), 5692–5694. [DOI] [PubMed] [Google Scholar]
- 229.Menegatti S; Hussain M; Naik AD; Carbonell RG; Rao BM, mRNA display selection and solid-phase synthesis of Fc-binding cyclic peptide affinity ligands. Biotechnol Bioeng 2013, 110 (3), 857–70. [DOI] [PubMed] [Google Scholar]
- 230.Bacon K; Bowen J; Reese H; Rao BM; Menegatti S, Use of Target-Displaying Magnetized Yeast in Screening mRNA-Display Peptide Libraries to Identify Ligands. ACS Comb Sci 2020, 22 (12), 738–744. [DOI] [PubMed] [Google Scholar]
- 231.Bowen J; Schloop AE; Reeves GT; Menegatti S; Rao BM, Discovery of Membrane-Permeating Cyclic Peptides via mRNA Display. Bioconjugate chemistry 2020, 31 (10), 2325–2338. [DOI] [PubMed] [Google Scholar]
- 232.Timmerman P; Beld J; Puijk WC; Meloen RH, Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem 2005, 6 (5), 821–4. [DOI] [PubMed] [Google Scholar]
- 233.Iqbal ES; Richardson SL; Abrigo NA; Dods KK; Osorio Franco HE; Gerrish HS; Kotapati HK; Morgan IM; Masterson DS; Hartman MCT, A new strategy for the in vitro selection of stapled peptide inhibitors by mRNA display. Chem Commun (Camb) 2019, 55 (61), 8959–8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Seebeck FP; Szostak JW, Ribosomal synthesis of dehydroalanine-containing peptides. J Am Chem Soc 2006, 128 (22), 7150–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hofmann FT; Szostak JW; Seebeck FP, In vitro selection of functional lantipeptides. J Am Chem Soc 2012, 134 (19), 8038–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ito K; Sakai K; Suzuki Y; Ozawa N; Hatta T; Natsume T; Matsumoto K; Suga H, Artificial human Met agonists based on macrocycle scaffolds. Nature communications 2015, 6 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kawakami T; Ishizawa T; Fujino T; Reid PC; Suga H; Murakami H, In Vitro Selection of Multiple Libraries Created by Genetic Code Reprogramming To Discover Macrocyclic Peptides That Antagonize VEGFR2 Activity in Living Cells. ACS Chemical Biology 2013, 8 (6), 1205–1214. [DOI] [PubMed] [Google Scholar]
- 238.Hayashi Y; Morimoto J; Suga H, In Vitro Selection of Anti-Akt2 Thioether-Macrocyclic Peptides Leading to Isoform-Selective Inhibitors. ACS Chemical Biology 2012, 7 (3), 607–613. [DOI] [PubMed] [Google Scholar]
- 239.Hipolito C; Tanaka Y; Katoh T; Nureki O; Suga H, A Macrocyclic Peptide that Serves as a Cocrystallization Ligand and Inhibits the Function of a MATE Family Transporter. Molecules 2013, 18 (9), 10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Zhang Z; Gao R; Hu Q; Peacock H; Peacock DM; Dai S; Shokat KM; Suga H, GTP-State-Selective Cyclic Peptide Ligands of K-Ras(G12D) Block Its Interaction with Raf. ACS Cent Sci 2020, 6 (10), 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bashiruddin NK; Hayashi M; Nagano M; Wu Y; Matsunaga Y; Takagi J; Nakashima T; Suga H, Development of cyclic peptides with potent in vivo osteogenic activity through RaPID-based affinity maturation. Proceedings of the National Academy of Sciences of the United States of America 2020, 117 (49), 31070–31077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Okuma R; Kuwahara T; Yoshikane T; Watanabe M; Dranchak P; Inglese J; Shuto S; Goto Y; Suga H, A Macrocyclic Peptide Library with a Structurally Constrained Cyclopropane-containing Building Block Leads to Thiol-independent Inhibitors of Phosphoglycerate Mutase. Chem Asian J 2020, 15 (17), 2631–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Norman A; Franck C; Christie M; Hawkins PME; Patel K; Ashhurst AS; Aggarwal A; Low JKK; Siddiquee R; Ashley CL; Steain M; Triccas JA; Turville S; Mackay JP; Passioura T; Payne RJ, Discovery of Cyclic Peptide Ligands to the SARS-CoV-2 Spike Protein using mRNA Display. bioRxiv 2020, 2020.12.22.424069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Takatsuji R; Shinbara K; Katoh T; Goto Y; Passioura T; Yajima R; Komatsu Y; Suga H, Ribosomal Synthesis of Backbone-Cyclic Peptides Compatible with In Vitro Display. J Am Chem Soc 2019, 141 (6), 2279–2287. [DOI] [PubMed] [Google Scholar]
- 245.Heinis C; Rutherford T; Freund S; Winter G, Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol 2009, 5 (7), 502–7. [DOI] [PubMed] [Google Scholar]
- 246.Hacker DE; Hoinka J; Iqbal ES; Przytycka TM; Hartman MC, Highly constrained bicyclic scaffolds for the discovery of protease-stable peptides via mRNA display. ACS chemical biology 2017, 12 (3), 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Millward SW; Fiacco S; Austin RJ; Roberts RW, Design of cyclic peptides that bind protein surfaces with antibody-like affinity. ACS Chem Biol 2007, 2 (9), 625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Chu Q; Moellering RE; Hilinski GJ; Kim Y-W; Grossmann TN; Yeh JTH; Verdine GL, Towards understanding cell penetration by stapled peptides. MedChemComm 2015, 6 (1), 111–119. [Google Scholar]
- 249.Gray JP; Uddin MN; Chaudhari R; Sutton MN; Yang H; Rask P; Locke H; Engel BJ; Batistatou N; Wang J; Grindel BJ; Bhattacharya P; Gammon ST; Zhang S; Piwnica-Worms D; Kritzer JA; Lu Z; Bast RC; Millward SW, Directed evolution of cyclic peptides for inhibition of autophagy. Chemical Science 2021, 12 (10), 3526–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Peraro L; Deprey KL; Moser MK; Zou Z; Ball HL; Levine B; Kritzer JA, Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J Am Chem Soc 2018, 140 (36), 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Nitsche C; Passioura T; Varava P; Mahawaththa MC; Leuthold MM; Klein CD; Suga H; Otting G, De Novo Discovery of Nonstandard Macrocyclic Peptides as Noncompetitive Inhibitors of the Zika Virus NS2B-NS3 Protease. ACS Med Chem Lett 2019, 10 (2), 168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Johansen-Leete J; Passioura T; Foster SR; Bhusal RP; Ford DJ; Liu M; Jongkees SAK; Suga H; Stone MJ; Payne RJ, Discovery of Potent Cyclic Sulfopeptide Chemokine Inhibitors via Reprogrammed Genetic Code mRNA Display. J Am Chem Soc 2020, 142 (20), 9141–9146. [DOI] [PubMed] [Google Scholar]
- 253.Katoh T; Sengoku T; Hirata K; Ogata K; Suga H, Ribosomal synthesis and de novo discovery of bioactive foldamer peptides containing cyclic beta-amino acids. Nat Chem 2020, 12 (11), 1081–1088. [DOI] [PubMed] [Google Scholar]
- 254.Howard JF Jr.; Nowak RJ; Wolfe GI; Freimer ML; Vu TH; Hinton JL; Benatar M; Duda PW; MacDougall JE; Farzaneh-Far R; Kaminski HJ; Zilucoplan MGSG; Barohn R; Dimachkie M; Pasnoor M; Farmakidis C; Liu T; Colgan S; Benatar MG; Bertorini T; Pillai R; Henegar R; Bromberg M; Gibson S; Janecki T; Freimer M; Elsheikh B; Matisak P; Genge A; Guidon A; David W; Habib AA; Mathew V; Mozaffar T; Hinton JL; Hewitt W; Barnett D; Sullivan P; Ho D; Howard JF Jr.; Traub RE; Chopra M; Kaminski HJ; Aly R; Bayat E; Abu-Rub M; Khan S; Lange D; Holzberg S; Khatri B; Lindman E; Olapo T; Sershon LM; Lisak RP; Bernitsas E; Jia K; Malik R; Lewis-Collins TD; Nicolle M; Nowak RJ; Sharma A; Roy B; Nye J; Pulley M; Berger A; Shabbir Y; Sachdev A; Patterson K; Siddiqi Z; Sivak M; Bratton J; Small G; Kohli A; Fetter M; Vu T; Lam L; Harvey B; Wolfe GI; Silvestri N; Patrick K; Zakalik K; Duda PW; MacDougall J; Farzaneh-Far R; Pontius A; Hoarty M, Clinical Effects of the Self-administered Subcutaneous Complement Inhibitor Zilucoplan in Patients With Moderate to Severe Generalized Myasthenia Gravis: Results of a Phase 2 Randomized, Double-Blind, Placebo-Controlled, Multicenter Clinical Trial. JAMA Neurol 2020, 77 (5), 582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Modulation of complement activity.
- 256.Gorman DM; Lee J; Payne CD; Woodruff TM; Clark RJ, Chemical synthesis and characterisation of the complement C5 inhibitory peptide zilucoplan. Amino Acids 2021, 53 (1), 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Tanaka Y; Hipolito CJ; Maturana AD; Ito K; Kuroda T; Higuchi T; Katoh T; Kato HE; Hattori M; Kumazaki K, Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 2013, 496 (7444), 247–251. [DOI] [PubMed] [Google Scholar]
- 258.Bahng N; Ito K; Ha S; Kim MY; Lee E; Suga H; Lee DS, In vivo targeting of c-Met using a non-standard macrocyclic peptide in gastric carcinoma. Cancer letters 2017, 385, 144–149. [DOI] [PubMed] [Google Scholar]
- 259.Iwasaki K; Goto Y; Katoh T; Yamashita T; Kaneko S; Suga H, A fluorescent imaging probe based on a macrocyclic scaffold that binds to cellular EpCAM. Journal of molecular evolution 2015, 81 (5-6), 210–217. [DOI] [PubMed] [Google Scholar]
- 260.Matsunaga Y; Bashiruddin NK; Kitago Y; Takagi J; Suga H, Allosteric inhibition of a semaphorin 4D receptor plexin B1 by a high-affinity macrocyclic peptide. Cell chemical biology 2016, 23 (11), 1341–1350. [DOI] [PubMed] [Google Scholar]
- 261.Jongkees SA; Caner S; Tysoe C; Brayer GD; Withers SG; Suga H, Rapid discovery of potent and selective glycosidase-inhibiting de novo peptides. Cell chemical biology 2017, 24 (3), 381–390. [DOI] [PubMed] [Google Scholar]
- 262.Passioura T; Bhushan B; Tumber A; Kawamura A; Suga H, Structure-activity studies of a macrocyclic peptide inhibitor of histone lysine demethylase 4A. Bioorganic & medicinal chemistry 2018, 26 (6), 1225–1231. [DOI] [PubMed] [Google Scholar]
- 263.Rogers JM; Nawatha M; Lemma B; Vamisetti GB; Livneh I; Barash U; Vlodavsky I; Ciechanover A; Fushman D; Suga H; Brik A, In vivo modulation of ubiquitin chains by N-methylated non-proteinogenic cyclic peptides. RSC Chemical Biology 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Hacker DE; Abrigo NA; Hoinka J; Richardson SL; Przytycka TM; Hartman MCT, Direct, Competitive Comparison of Linear, Monocyclic, and Bicyclic Libraries Using mRNA Display. ACS Comb Sci 2020, 22 (6), 306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Alleyne C; Amin RP; Bhatt B; Bianchi E; Blain JC; Boyer N; Branca D; Embrey MW; Ha SN; Jette K; Johns DG; Kerekes AD; Koeplinger KA; LaPlaca D; Li N; Murphy B; Orth P; Ricardo A; Salowe S; Seyb K; Shahripour A; Stringer JR; Sun Y; Tracy R; Wu C; Xiong Y; Youm H; Zokian HJ; Tucker TJ, Series of Novel and Highly Potent Cyclic Peptide PCSK9 Inhibitors Derived from an mRNA Display Screen and Optimized via Structure-Based Design. J Med Chem 2020, 63 (22), 13796–13824. [DOI] [PubMed] [Google Scholar]
- 266.Haviv F; Fitzpatrick TD; Swenson RE; Nichols CJ; Mort NA; Bush EN; Diaz G; Bammert G; Nguyen A; Rhutasel NS; et al. , Effect of N-methyl substitution of the peptide bonds in luteinizing hormone-releasing hormone agonists. J Med Chem 1993, 36 (3), 363–9. [DOI] [PubMed] [Google Scholar]
- 267.Ostresh JM; Husar GM; Blondelle SE; Dorner B; Weber PA; Houghten RA, “Libraries from libraries”: chemical transformation of combinatorial libraries to extend the range and repertoire of chemical diversity. Proceedings of the National Academy of Sciences of the United States of America 1994, 91 (23), 11138–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.van de Watering FC; Rijpkema M; Perk L; Brinkmann U; Oyen WJ; Boerman OC, Zirconium-89 labeled antibodies: a new tool for molecular imaging in cancer patients. Biomed Res Int 2014, 2014, 203601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Vaickus L; Foon KA, Biotechnology Update: Overview of Monoclonal Antibodies in the Diagnosis and Therapy of Cancer. Cancer investigation 1991, 9 (2), 195–209. [DOI] [PubMed] [Google Scholar]
- 270.De Silva RA; Kumar D; Lisok A; Chatterjee S; Wharram B; Venkateswara Rao K; Mease R; Dannals RF; Pomper MG; Nimmagadda S, Peptide-Based (68)Ga-PET Radiotracer for Imaging PD-L1 Expression in Cancer. Mol Pharm 2018, 15 (9), 3946–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Kumar D; Lisok A; Dahmane E; McCoy M; Shelake S; Chatterjee S; Allaj V; Sysa-Shah P; Wharram B; Lesniak WG; Tully E; Gabrielson E; Jaffee EM; Poirier JT; Rudin CM; Gobburu JV; Pomper MG; Nimmagadda S, Peptide-based PET quantifies target engagement of PD-L1 therapeutics. J Clin Invest 2019, 129 (2), 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Chatterjee S; Lesniak WG; Miller MS; Lisok A; Sikorska E; Wharram B; Kumar D; Gabrielson M; Pomper MG; Gabelli SB; Nimmagadda S, Rapid PD-L1 detection in tumors with PET using a highly specific peptide. Biochem Biophys Res Commun 2017, 483 (1), 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Shaabani S; Huizinga HPS; Butera R; Kouchi A; Guzik K; Magiera-Mularz K; Holak TA; Domling A, A patent review on PD-1/PD-L1 antagonists: small molecules, peptides, and macrocycles (2015-2018). Expert Opin Ther Pat 2018, 28 (9), 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Magiera-Mularz K; Skalniak L; Zak KM; Musielak B; Rudzinska-Szostak E; Berlicki L; Kocik J; Grudnik P; Sala D; Zarganes-Tzitzikas T; Shaabani S; Domling A; Dubin G; Holak TA, Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint. Angew Chem Int Ed Engl 2017, 56 (44), 13732–13735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Pisaneschi F; Kelderhouse LE; Hardy A; Engel BJ; Mukhopadhyay U; Gonzalez-Lepera C; Gray JP; Ornelas A; Takahashi TT; Roberts RW; Fiacco SV; Piwnica-Worms D; Millward SW, Automated, Resin-Based Method to Enhance the Specific Activity of Fluorine-18 Clicked PET Radiotracers. Bioconjugate chemistry 2017, 28 (2), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Takahashi TT; Roberts RW, In vitro selection of protein and peptide libraries using mRNA display. In Nucleic Acid and Peptide Aptamers, Springer: 2009; pp 293–314. [DOI] [PubMed] [Google Scholar]
- 277.Seelig B, mRNA display for the selection and evolution of enzymes from in vitro-translated protein libraries. Nat Protoc 2011, 6 (4), 540–52. [DOI] [PubMed] [Google Scholar]
- 278.Barendt PA; Ng DT; McQuade CN; Sarkar CA, Streamlined protocol for mRNA display. ACS Comb Sci 2013, 15 (2), 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Arai H; Kumachi S; Nemoto N, cDNA Display: A Stable and Simple Genotype-Phenotype Coupling Using a Cell-Free Translation System. Methods Mol Biol 2020, 2070, 43–56. [DOI] [PubMed] [Google Scholar]
- 280.Nemoto N; Kumachi S; Arai H, In Vitro Selection of Single-Domain Antibody (VHH) Using cDNA Display. Methods Mol Biol 2018, 1827, 269–285. [DOI] [PubMed] [Google Scholar]
- 281.Yamamoto Y; Terai T; Kumachi S; Nemoto N, In Vitro Construction of Large-scale DNA Libraries from Fragments Containing Random Regions using Deoxyinosine-containing Oligonucleotides and Endonuclease V. ACS Comb Sci 2020, 22 (4), 165–171. [DOI] [PubMed] [Google Scholar]
- 282.Tsao KL; DeBarbieri B; Michel H; Waugh DS, A versatile plasmid expression vector for the production of biotinylated proteins by site-specific, enzymatic modification in Escherichia coli. Gene 1996, 169 (1), 59–64. [DOI] [PubMed] [Google Scholar]
- 283.Grindel BJ; Engel BJ; Hall CG; Kelderhouse LE; Lucci A; Zacharias NM; Takahashi TT; Millward SW, Mammalian Expression and In Situ Biotinylation of Extracellular Protein Targets for Directed Evolution. ACS Omega 2020, 5 (39), 25440–25455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Boder ET; Midelfort KS; Wittrup KD, Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proceedings of the National Academy of Sciences of the United States of America 2000, 97 (20), 10701–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Evenson WE; Lin WS; Pang K; Czaja AT; Jalali-Yazdi F; Takahashi TT; Malmstadt N; Roberts RW, Enabling Flow-Based Kinetic Off-Rate Selections Using a Microfluidic Enrichment Device. Anal Chem 2020, 92 (15), 10218–10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Jalali-Yazdi F; Corbin JM; Takahashi TT; Roberts RW, Robust, quantitative analysis of proteins using peptide immunoreagents, in vitro translation, and an ultrasensitive acoustic resonant sensor. Anal Chem 2014, 86 (10), 4715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Larsen AC; Gillig A; Shah P; Sau SP; Fenton KE; Chaput JC, General approach for characterizing in vitro selected peptides with protein binding affinity. Analytical chemistry 2014, 86 (15), 7219–7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Duarte JG; Blackburn JM, Advances in the development of human protein microarrays. Expert Rev Proteomics 2017, 14 (7), 627–641. [DOI] [PubMed] [Google Scholar]
- 289.Weng S; Gu K; Hammond PW; Lohse P; Rise C; Wagner RW; Wright MC; Kuimelis RG, Generating addressable protein microarrays with PROfusion covalent mRNA-protein fusion technology. Proteomics 2002, 2 (1), 48–57. [PubMed] [Google Scholar]
- 290.Bentley DR; Balasubramanian S; Swerdlow HP; Smith GP; Milton J; Brown CG; Hall KP; Evers DJ; Barnes CL; Bignell HR; Boutell JM; Bryant J; Carter RJ; Keira Cheetham R; Cox AJ; Ellis DJ; Flatbush MR; Gormley NA; Humphray SJ; Irving LJ; Karbelashvili MS; Kirk SM; Li H; Liu X; Maisinger KS; Murray LJ; Obradovic B; Ost T; Parkinson ML; Pratt MR; Rasolonjatovo IM; Reed MT; Rigatti R; Rodighiero C; Ross MT; Sabot A; Sankar SV; Scally A; Schroth GP; Smith ME; Smith VP; Spiridou A; Torrance PE; Tzonev SS; Vermaas EH; Walter K; Wu X; Zhang L; Alam MD; Anastasi C; Aniebo IC; Bailey DM; Bancarz IR; Banerjee S; Barbour SG; Baybayan PA; Benoit VA; Benson KF; Bevis C; Black PJ; Boodhun A; Brennan JS; Bridgham JA; Brown RC; Brown AA; Buermann DH; Bundu AA; Burrows JC; Carter NP; Castillo N; Chiara ECM; Chang S; Neil Cooley R; Crake NR; Dada OO; Diakoumakos KD; Dominguez-Fernandez B; Earnshaw DJ; Egbujor UC; Elmore DW; Etchin SS; Ewan MR; Fedurco M; Fraser LJ; Fuentes Fajardo KV; Scott Furey W; George D; Gietzen KJ; Goddard CP; Golda GS; Granieri PA; Green DE; Gustafson DL; Hansen NF; Harnish K; Haudenschild CD; Heyer NI; Hims MM; Ho JT; Horgan AM; Hoschler K; Hurwitz S; Ivanov DV; Johnson MQ; James T; Huw Jones TA; Kang GD; Kerelska TH; Kersey AD; Khrebtukova I; Kindwall AP; Kingsbury Z; Kokko-Gonzales PI; Kumar A; Laurent MA; Lawley CT; Lee SE; Lee X; Liao AK; Loch JA; Lok M; Luo S; Mammen RM; Martin JW; McCauley PG; McNitt P; Mehta P; Moon KW; Mullens JW; Newington T; Ning Z; Ling Ng B; Novo SM; O’Neill MJ; Osborne MA; Osnowski A; Ostadan O; Paraschos LL; Pickering L; Pike AC; Pike AC; Chris Pinkard D; Pliskin DP; Podhasky J; Quijano VJ; Raczy C; Rae VH; Rawlings SR; Chiva Rodriguez A; Roe PM; Rogers J; Rogert Bacigalupo MC; Romanov N; Romieu A; Roth RK; Rourke NJ; Ruediger ST; Rusman E; Sanches-Kuiper RM; Schenker MR; Seoane JM; Shaw RJ; Shiver MK; Short SW; Sizto NL; Sluis JP; Smith MA; Ernest Sohna Sohna J; Spence EJ; Stevens K; Sutton N; Szajkowski L; Tregidgo CL; Turcatti G; Vandevondele S; Verhovsky Y; Virk SM; Wakelin S; Walcott GC; Wang J; Worsley GJ; Yan J; Yau L; Zuerlein M; Rogers J; Mullikin JC; Hurles ME; McCooke NJ; West JS; Oaks FL; Lundberg PL; Klenerman D; Durbin R; Smith AJ, Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456 (7218), 53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Svensen N; Peersen OB; Jaffrey SR, Peptide Synthesis on a Next-Generation DNA Sequencing Platform. ChemBioChem 2016, 17 (17), 1628–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Blanco C; Verbanic S; Seelig B; Chen IA, EasyDIVER: A Pipeline for Assembling and Counting High-Throughput Sequencing Data from In Vitro Evolution of Nucleic Acids or Peptides. J Mol Evol 2020, 88 (6), 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]