

Abstract
Cyclin D2 (CCND2) is a critical cell cycle regulator and key member of the cyclin D2-CDK4 (DC) complex. De novo variants of CCND2 clustering in the distal part of the protein have been identified as pathogenic causes of brain overgrowth (megalencephaly, MEG) and severe cortical malformations in children including the megalencephaly-polymicrogyria-polydactyly-hydrocephalus (MPPH) syndrome. Megalencephaly-associated CCND2 variants are localized to the terminal exon and result in accumulation of degradation-resistant protein. We identified five individuals from three unrelated families with novel variants in the proximal region of CCND2 associated with microcephaly, mildly simplified cortical gyral pattern, symmetric short stature, and mild developmental delay. Identified variants include de novo frameshift variants and a dominantly inherited stop-gain variant segregating with the phenotype. This is the first reported association between proximal CCND2 variants and microcephaly, to our knowledge. This series expands the phenotypic spectrum of CCND2-related disorders and suggests that distinct classes of CCND2 variants are associated with reciprocal effects on human brain growth (microcephaly and megalencephaly due to possible loss or gain of protein function, respectively), adding to the growing paradigm of inverse phenotypes due to dysregulation of key brain growth genes.
Keywords: cyclin D2, inverse phenotypes, megalencephaly, megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome (MPPH syndrome), microcephaly
1 |. INTRODUCTION
Overgrowth syndromes are disorders characterized by cellular hyperplasia and/or hypertrophy, which may involve one or more of the three embryonic tissue layers (endoderm, ectoderm, or mesoderm) and present clinically with generalized or segmental overgrowth of the brain or the body (Burkardt et al., 2019; Tatton-Brown & Weksberg, 2013). Undergrowth syndromes or syndromes of growth restriction, on the other hand, are groups of disorders characterized by tissue hypoplasia and growth restriction. Collectively, these syndromes may manifest with localized or generalized growth abnormalities and display considerable heterogeneity in onset, tissue involvement and distribution among other features (Grissom & Reyes, 2013).
The Cyclin D2 Cyclin-dependent kinases 4 (CDK4) family of genes regulates the cell cycle by modulating the G1-to-S phase transition, critical for cell proliferation and development (Jeong et al., 2016). Posttranslational modification by phosphorylation of CCND2, downstream of the phosphatidylinositol 3-kinase (PI3K)/ AK murine Thymoma (AKT)/mammalian target of the rapamycin (MTOR) signaling pathway inhibits its binding to the transcription factor E2F, allowing the cell to exit the G1 phase of the cell cycle (Becker et al., 2010; Bouchard et al., 1999; Sicinski et al., 1996). A schematic representation of the PI3K-AKT-MTOR pathway and CCND2 interaction is provided in Figure 1a. Cyclin D2 has well-studied roles in brain development and is expressed in dividing cells derived from neuronal precursors in the adult hippocampus (Ross et al., 1996). Hyperactivation of CCND2 has been linked to hematologic malignancies and increased proliferation in human cancer cell lines (Hung et al., 2018; Murai et al., 2001). Further, genetic variants of CCND2 (MIM #123833), as well as two other key PI3K-AKT-MTOR pathway genes, namely AKT3 and PIK3R2, have been identified as pathogenic causes of the megalencephaly-polymicrogyria-polydactyly-hydrocephalus (MPPH) syndrome (Hiraiwa et al., 2021; G. Mirzaa et al., 2014). MPPH is a rare megalencephaly (MEG) syndrome characterized by congenital or early postnatal brain overgrowth and cortical malformations, most commonly polymicrogyria (PMG). MPPH is also associated with variable degree of ventriculomegaly, with 40–50% of affected individuals progressing to hydrocephalus and necessitating surgical intervention (G. Mirzaa et al., 1993). Most affected children have mild to severe intellectual disability and oromotor dysfunction, including expressive language or speech delays, difficulties handling oral secretions, and dysphagia; with seizures reported in at least 50% of individuals. CCND2-related MPPH is characterized by severe and widespread polymicrogyria, typically bilateral perisylvian PMG (BPP) extending to the frontal and/or occipital lobes and correlating with increased severity of epilepsy and intellectual disability (G. Mirzaa et al., 2014). An additional report of a child with the MPPH phenotype in association with the CCND2 variant, p.Thr280Ile, further expanded the phenotypic spectrum as this individual had additional brain abnormalities including hippocampal hypoplasia with malrotation, diffuse hypomyelination, and abnormalities of the midbrain and cerebellum (Cappuccio et al., 2019).
FIGURE 1.

CCND2 gene diagram and role in cell signaling. (a) A schematic representation of the PI3K-AKT-MTOR pathway and its’ interaction with the CCND2 protein. Proteins outlined in magenta are the ones with known gain of function and loss of function mutations leading to opposite phenotypes (PTEN, AKT3, MYCN, CCND2). Created with BioRender.com. (b) Scheme of the human CCND2 gene (Homo sapiens chromosome 12, GRCh38.p12 CCND2: NC_000012.12 mRNA NM_001759.4 protein NP_001750.1). Blue rectangles represent exons (1–5), while magenta lines represent introns. Exon, coding and intron base pairs length (bp) is shown in the relative colors. Above the gene scheme, putative CCND2 loss of function (LOF, in black) reported here, and previously published gain of function (GOF, in blue) mutations are reported
While overexpression and hyperactivation of CCND2 have been identified in humans and mice, the effects of CCND2 deficiency or loss of function have not been studied in humans. In mice, Ccnd2 knock-out leads to lack of cerebellar stellate interneurons (Garthe et al., 2014; Glickstein et al., 2007) and striking microcephaly (Huard et al., 1999). CCND2 haploinsufficiency due to 12p13.32 contiguous microdeletion syndromes has been reported in individuals with microcephaly, although the critical region for this microdeletion also encompasses other key genes (Firth et al., 2009; Leyser et al., 2016). In this study, we describe three novel proximal variants in CCND2 in five individuals from three unrelated families presenting with microcephaly, symmetric short stature, hypotonia and developmental delays. This series suggest that frameshift and stop-gain variants in the proximal portion of CCND2 are linked to human microcephaly and adds CCND2 to the growing list of genes associated with reciprocal brain growth dysregulation. All microcephaly- and megalencephaly (including MPPH)-associated variants in CCND2 identified to date are schematically shown in Figure 1b. Examples of reciprocal gain and loss of protein function resulting in inverse phenotypes are seen in a multitude of abnormal growth syndromes, including mutations in other genes involved in cell growth and cell cycle regulation such as AKT3, PTEN, NSD1, and MYCN. These inverse brain growth phenotypes due to reciprocal gene defects are summarized in Table 1 and discussed in depth in the literature review we provide here.
TABLE 1.
A summary of clinical and molecular features of inverse brain growth phenotype due to variants in CCND2, AKT3, PTEN, NSD1, MYCN, EZH2, and MIR17HG
| Gene | Megalencephaly/macrocephaly Disease (OMIM) | Protein function | Clinical features | Inheritance | Type of variants | Microcephaly Disease (OMIM) | Protein function | Clinical features | Inheritance | Type of variants |
|---|---|---|---|---|---|---|---|---|---|---|
| CCND2 | MPPH3 (#615938) | Gain | Megalencephaly, postaxial polydactyly, hypotonia, epilepsy, intellectual disability, oromotor dysfunction, polymicrogyria | De novo/AD | Missense, nonsense | Microcephaly | Lossa | Microcephaly, symmetric short stature, hypotonia, mild intellectual disability, mild motor delay | De novo/AD | Missense, small indels |
| AKT3 | MPPH2 (#615937) | Gain | Megalencephaly, polymicrogyria/cortical dysplasia, postaxial polydactyly, hypotonia, epilepsy, intellectual disability, oromotor dysfunction, connective tissue laxity, vascular malformations | De novo/AD | Missense, duplication | Postnatal microcephaly | Haploin sufficiency/loss | Microcephaly, callosal abnormalities, seizures, hypotonia, dysmorphic features | De novo/AD | Microdeletion 1q43-q44 |
| PTEN | Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome (BRRS); Lhermitte-Duclos disease (LDD); (#158350) | Loss | Megalencephaly, facial trichilemmomas, acral keratoses, papillomatous papules, hamartoma, increased risk for breast, thyroid, and endometrial carcinoma; hamartomatous polyps of the gastrointestinal tract, mucocutaneous lesions, developmental delay, lipomas, hemangiomas | De novo/AD | Missense, nonsense, small/large deletions | Microcephaly | Gain | Primary microcephaly, autism spectrum disorder, intellectual disability | AD | Microduplication 10q23.31 |
| Autism spectrum disorder with macrocephaly (#605309) | Loss | Megalencephaly, abnormal facial features, and delayed psychomotor development resulting in autistic behavior or mental retardation | De novo/AD | Missense, nonsense, small/large deletions | ||||||
| NSD1 | Sotos syndrome (#117550) | Loss | Megalencephaly, somatic overgrowth, dysmorhpic facial features, cardiac issues, skeletal issues, renal anomalies, developmental delay/intellectual disability | De novo/AD | Missenese, nonsense, splicing defects, frameshift, deletions | Microcephaly | Gain | Microcephaly, short stature, seizures, developmental delay/intellectual disability, dysmorphic facial features | De novo/AD | Microduplication 5q35 |
| MYCN | MYCN related megalencephaly syndrome | Gain | Brain abnormalities (megalencephaly, ventriculomegaly, hypoplastic corpus callosum); Intellectual disability, polydactyly; neuroblastoma | De novo | Missense (c.173C>T) | Feingold syndrome type 1 (#164280) | Loss | Microcephaly; Limb abnormalities (clinodactyly, syndactyly, thumb hypoplasia); Esophageal and or duodenal atresia; Learning disabilities; Characteristic facial features (micrognathia, short palpebral fissures) | AD/unknown de novo | Nonsense, missense, large deletions, partial and entire gene deletions |
| EZH2 | Weaver syndrome (#277590) | Loss | Megalencephaly, overgrowth, distinctive facial gestalt, accelerated bone maturation, developmental delays | De novo | Missense | Microcephaly and developmental delays | Gain | Growth failure, hypotonia, “clover-leaf” shaped skull, large anterior fontanelle, sparse eyebrows, upslanting palpebral fissures, and small ears, developmental delays | De novo | Missense |
| MIR17HG | MIR17HG related overgrowth syndrome | Gain | Megalencephaly, developmental delay, skeletal and digital abnormalities, tall stature | De novo | 13q31.3 microduplications | Feingold syndrome type 2 (#614326) | Loss | Microcephaly, brachymesophalangy, toe syndactyly, short stature, cardiac anomalies, growth hormone deficiency, aortic dilation, phalangeal joint contractures, memory, and sleep problems | De novo | 13q31.3 microdeletions |
Notes: When available, the OMIM number is listed, otherwise the disorder has not yet been described in OMIM.
Abbreviations: AD, autosomal dominant; IUGR, intrauterine growth restriction; MPPH, megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome; UK, unknown.
Predicted loss of function due to premature stop codon based on the five cases reported here.
2 |. METHODS
2.1 |. Human subjects and cell lines
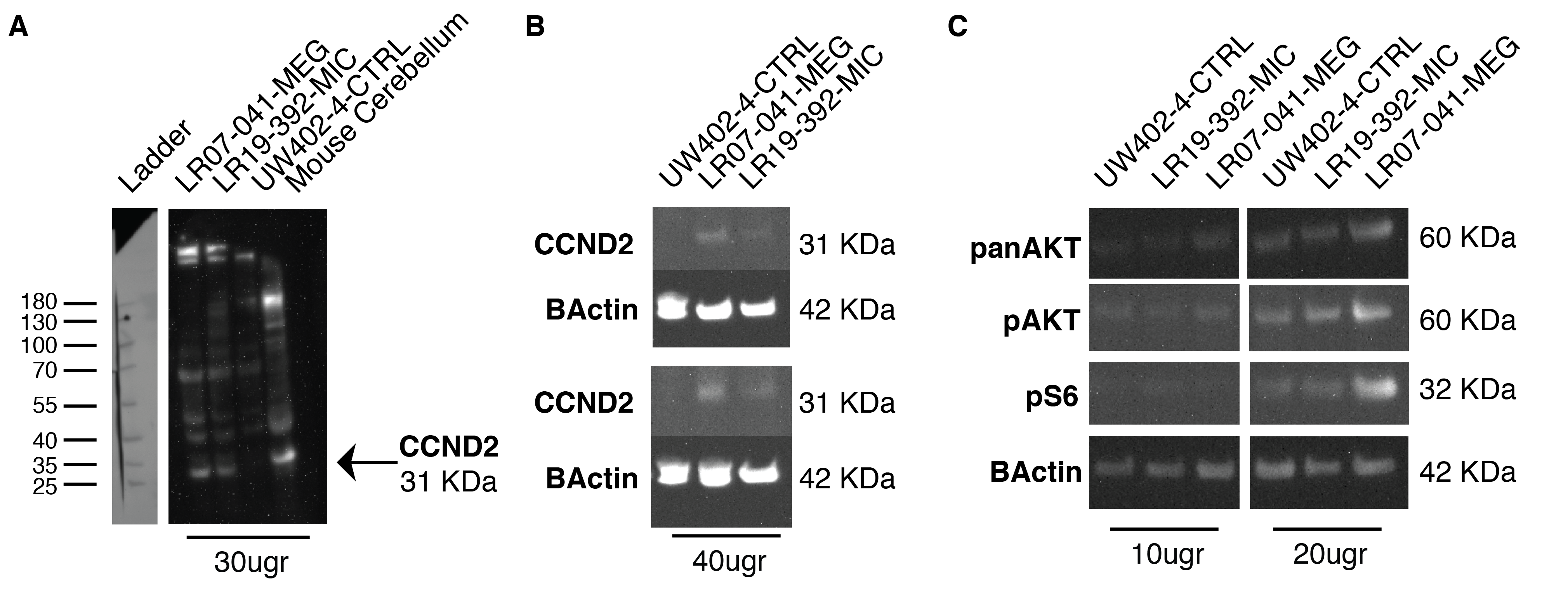
Families were identified through our clinical and research programs, personal communication, as well as the MatchMaker Exchange (MME) including GeneMatcher (http://www.genematcher.org) (Sobreira et al., 2015). Informed consent for publication and analysis of photos, imaging and clinical data was obtained from the patients’ legal guardians under an IRB approved protocol at Seattle Children’s Hospital (IRB#13291). Brain magnetic resonance imaging (MRI) studies were performed on three individuals (LR19–002, LR20–198a1, and LR20–198a2) and reviewed by the investigators. Fibroblast cell lines were established from one of the patients (LR19–392) with the c.305delG, p.G102Vfs*17 variant (referred to as LR19–392-MIC throughout the manuscript), and compared to a MEG/MPPH fibroblast cell line from a previously published patient (LR07–041) with the CCND2, c.839C>A, p.T280N variant (referred to as LR07–041-MEG hereon) (G. Mirzaa et al., 2014), as well as fibroblasts obtained from a healthy unrelated control (UW402–4-CTRL).
2.2 |. Western blot analysis
Fibroblast cell lines (LR19–392-MIC, LR07–041-MEG, and UW402–4-CTRL) were expanded to six confluent T-75 flasks each before protein extraction. Cell culture flasks were placed on ice and washed once with 10 mL ice cold TBS. Flasks were then incubated on ice for 5 min with 500 μl cold RIPA buffer (Thermo Scientific, 89900) containing phosphatase and protease inhibitors (Thermo Scientific, 78,430, P5726, and P2850), cells were scraped and collected in 15 mL conical tubes and agitated for 30 minutes at 4C before being centrifuged at 14,000g for 15 min. The resulting supernatant was aliquoted into 0.2 mL tubes, snap frozen with liquid N2, and stored at −80°C. Protein concentrations were assessed via the standard Bicinchoninic Acid (BCA) Assay (Thermofisher, 23227). Equal amounts of protein extracts (10 and 20 μg) were heated to 95°C for 5 mins with NuPAGE® LDS Sample Buffer (4×) (Thermofisher, NP0007) and run on NuPAGE® Novex® 4–12% Bis-Tris gels (Thermofisher, NP0326BOX), together with a protein ladder (Pageruler prestained protein ladder, Thermofisher, 26617). Proteins were transferred to nitrocellulose membranes (Thermofisher, 77010). Due to low expression of CCND2 in human fibroblasts, we used 40 μg of protein extracts for its detection via Western Blot analysis. Membranes were blocked at room temperature for 1 h in blocking solution (3% BSA and 10% Donkey Serum in TBS with 0.1% Tween-20). For the CCND2 antibody, the blocking buffer contained 5% nonfat milk in addition to 3% BSA and 10% Donkey Serum to improve specificity. Membranes were incubated with primary antibody diluted in blocking solution overnight at 4C on an orbital shaker. Primary antibodies were diluted as follows: 1:200 for CCND2 (D52F9, Cell Signaling), 1:2500 for pAKT (Cell signaling, C31E5E), 1:2500 for pan-AKT (Cell Signaling, C67E7), 1:1000 for pS6 (Cell Signaling, 2215), and 1:5000 for β-actin (Abcam, ab8226). Membranes were washed three times for 15 min at room temperature in TBS-Tween 0.1% then incubated with secondary antibody in blocking solution for 1 h at RT. The secondary antibodies Donkey Anti-Mouse IgG H&L HRP (Abcam, ab205724) or Donkey Anti-Rabbit IgG H&L HRP (Abcam, ab97064) were used at 1:10,000 in blocking solution. After three washes at room temperature with TBS-Tween 0.1%, protein signal was detected with Pierce™ ECL Western Blotting Substrate (Thermo Scientific, 32109) and visualized using a ChemiDoc-It®2810 Imager (Ultra-Violet Products Ltd., Cambridge, UK). If necessary, membranes were stripped and re-probed, probing the antibody for the protein with the weakest signal first, following standard protocols. Six independent experiments were performed for each antibody and images were analyzed, quantified, and normalized against B-Actin using ImageJ24. Statistical analysis and graphs were generated using GraphPad Prism version 9.0.2.
2.3 |. High-content imaging
Fibroblast cell lines LR19–392 MIC, LR07–041 MEG, and UW402-CTRL were passaged in an optical 96 well plate coated with 0.1% porcine gelatin and cultured for 48 hours before fixing with cold 4% PFA in TBS (20 min at room temperature). Four wells per cell line were used for immunostaining. Cells were washed twice with cold TBS and permeabilized with TBS-Tween 0.1% for 5 min at room temperature. Immunostaining was performed using standard protocols. Briefly, cells were incubated for 1 hour at room temperature with blocking buffer (TBS-Tween 0.1% with 10% Donkey serum) and then incubated overnight with primary antibodies at 4C. Primary antibodies were diluted as follows: mouse anti-Ki-67 1:300 (Cell Signaling, 9449S) and rabbit anti-pS6 1:100 (Cell Signaling, 2215S). The following day, cells were washed three times with TBS-Tween 0.1% for 15 min and incubated with secondary antibodies diluted in blocking buffer for 1 h at room temperature. Secondary antibodies Donkey Anti-mouse Alexa 594 (Abcam, ab150112) and Donkey Anti-Rabbit Alexa 488 (Abcam, ab150065) and were used at 1:1000 dilution. Cells were washed three times in TBS-Tween 0.1%, followed by one wash in TBS, and stored in Glycerol-TBS (70:30) for acquisition and long-term storage. Image acquisition was performed using the CellInsight CX7 LZR system (Thermofisher). Fifteen fields for each of the 4 replicate wells per cell lines were acquired at 10× and 20× magnification. Images (10× magnification) were analyzed and quantified with a custom algorithm using the HCS Studio software. Statistical analysis was performed using GraphPad Prism version 9.0.2. and specific tests used are described in figure legends.
3 |. RESULTS
3.1 |. Clinical reports
Clinical and genetics features for all patients are summarized in Table 2.
TABLE 2.
Summary of clinical and molecular features for individuals with microcephaly and novel CCND2 variants
| LR19-002 | LR19-392 | LR20-198 m | LR20-198a1 | LR20-198a2 | ||
|---|---|---|---|---|---|---|
| Genetic testing | Identified variant | CCND2: NM_001759.4: c.416_419dupGGGA, p.L141GfsX19 | CCND2: NM_001759.4:c.305delG, p.G102VfsX17 | CCND2: NM_001759.4:c.544C>T, p.Q182X(het) | ||
| Molecular methods and other molecular findings [inheritance] | Exome sequencing; GBA (c.882T>G, p.H294Q), het, VUS [pat]; TENM1 (c.679 C>A, p.P227T), VUS, [mat] | NGS panel; no | Array CGH, targeted sequencing, arr 18q12.3 (37.877.518–37.961.503)x3 (hg18) | Exome sequencing, Array CGH, arr 18q12.3 (37.877.518–37.961.503)x3mat (hgl8) | Array CGH, Targeted sequencing, arr 8q23.1 (106.455.702–107.566.961) x3pat,18q12.3 (37.877.518–37.961.503)x3mat (hgl8) | |
| Demographics | Sex | M | F | F | M | F |
| Age at last assessment | 7m | 12y2m | 30y | 12y11m | 6y8m | |
| Ethnicity | ND | ND | Caucasian (Germany) | Caucasian (Germany) | Caucasian (Germany) | |
| Notable family history | Non-consanguineous, conceived via intrauterine insemination, family history unremarkable | Non-consanguineous, family history unremarkable | Father has boderline ID, short stature and microcephaly without genetic diagnosis | See LR20-198 m. One sibling and two maternal half siblings show normal OFC and normal development and do not carry CCND2 variant | See LR20-198a1 | |
| Pregnancy and birth | Pregnancy | G1P1 mother, suspected preeclampsia, vaginal bleeding, IUGR at 32-33gw | G2P2, IUGR at end of second trimester | G1P1 | G1P1 | G4P3 |
| Gestational age (weeks/days) | 35 + 6 | 39 + 0 | ND | 38 + 5 | 37 + 5 | |
| Birth weight (%le/z-score) | 1870 g (2.13%, −2.05 SD) | 2320 g (3%, −1.8 SD) | ND | 2900 g (10–25%, −1.2 SD) | 2480 g (3%, −1.8 SD) | |
| Birth length (%le/z-score) | 43.5 cm (3%, −1.8 SD) | 46 cm (3–10%, −1.5 SD) | ND | 49 cm (25–50%, −1 SD) | 47 cm (10–25%, −1.3 SD) | |
| Birth head circumference (%le/z-score) | 30.5 cm (3–10%, −1–2 SD) | 32 cm (2%, −2 SD) | ND | 29.5 cm (<1%, −4.3 SD) | 32 cm (2%, −2 SD) | |
| Growth | Weight at last assessment (%le/z-score) | 6.1 kg (0.48%, −2.6 SD) | 25.50 kg (<0.1%, −2.8 SD) | 85 kg (95%, +1.68 SD) | 34.2 kg (5%, −1.7 SD) | 15.2 kg (<1%, −2.8 SD) |
| Height at last assessment (%le/z-score) | 64.5 cm (5%, −1.6 SD) | 140.5 cm (7%, −1.4 SD) | 150 cm (2%, −2 SD) | 144.5 cm (7%, −1.5 SD) | 111 cm (2%, −2 SD) | |
| Head circumference at last assessment (%1e/z-score) | 40.2 cm (<1%, −3.1 SD) | 48 cm (<1%, −4.3SD) | 51 cm (<1%, −3.1 SD) | 48 cm (<1%, −4.2 SD) | 45.2 cm (<1%, −5 SD) | |
| Growth failure | Yes | Yes | Yes | No | Yes | |
| Neurologic findings | Degree of developmental delay or ID | N.E. | Mild | Mild | Mild | |
| Age–1st walked | N.E. | 13 m | 2 years | 12 month | 11 month | |
| Age–1st words | N.E. | Unknown | Unknown | Unknown | ||
| Speech delays | N.E. | Yes | Unknown | Yes (with 3 years less than 100 words and two-word-sentences) | Yes | |
| Seizures | No | No | No | No | No | |
| EEG findings | N.E. | N.E. | N.E. | normal | yes, among others occipital epileptic potentials | |
| Tone abnormalities | Hypotonia | No | N.E. | No | No | |
| Brain MRI findings | Microcephaly with a mildly simplified gyral pattern | Microcephaly with a mildly simplified gyral pattern | N.E. | Normal | Normal | |
| Autism | N.E. | No | N.E. | No | No | |
| ADHD | N.E. | Yes, mild | N.E. | Suspected | N.E. | |
| Other behavioral issues | N.E. | No | aggressive, impulsive and antisocial behavior | recurring (breath-holding spells), impusive behavior, reduced interaction with peers | ||
| Other neurologic findings | No | No | No | No | No | |
| Formal developmental or autism assessments | N.E. | Special school | Attended a special school | Attended a special school | Bayley II (23 month): in the lower standard range | |
| Other findings | Dysmorphic facial features | Mild right occipital flattening | Large ears, short philtrum, incomplete single transverse right palmar crease | Sloping forehead, broad nasal tip, upslanting palpebral fissures, long philtrum, thin upper lip vermilion | Sloping forehead, broad nasal base, broad nasal tip, long philtrum, thin upper lip vermilion | Sloping forehead, broad nasal tip |
| Heart | N.E. | N.E. | N.E. | N.E. | N.E. | |
| Eyes | N.E. | Mild myopia | N.E. | N.E. | Normal | |
| Urogenital system | N.E. | N.E. | N.E. | N.E. | N.E. | |
| Skeletal | N.E. | Bone age delayed at 2y10m, normalized later | N.E. | Pes planus | Talus obliqus and pes planus unilateral | |
| GI | N.E. | N.E. | N.E. | N.E. | Nutritional difficulties | |
| Other medical issues | Mild motor delays at birth, responded well to physical therapy | No | Bilateral inguinal hernia after birth | Asphyxia, anchyloglossia | Respiratory adaptation disorder with CPAP-therapy, failure to thrive | |
Notes: The table summarizes the clinical features, growth data, and family history for the five individuals with proximal CCND2 frameshift or stop-gain variants.
Abbreviation: N.E., not evaluated.
3.1.1 |. Patient LR19–002
This 7-month-old boy was referred to the genetics clinic for microcephaly, intrauterine growth restriction (IUGR), hypotonia, and mild motor delays. He was the first child of non-consanguineous parents of Armenian descent, a 30-year-old G1P1 mother and 30-year-old father and conceived by intrauterine insemination. Pregnancy was complicated by suspected preeclampsia and vaginal bleeding. No teratogenic exposures were reported. Family anamnesis was unremarkable with no history of microcephaly, delayed growth, or development. At 32–33 weeks of gestation, IUGR and placental insufficiency were reported. The boy was delivered at 35 + 5 weeks of gestation by a scheduled caesarian section. Birth weight was 1870 grams (2nd centile, −2.05 SD), length 42.5 cm (3rd centile, −1.8 SD) and occipitofrontal circumference (OFC) 30.5 cm (3–10th centile, − 1–2 SD). OFC measured again at 36 weeks was 29.9 cm (1–2nd centile, −2.25 SD). The child was also diagnosed with gastroesophageal reflux and allergic colitis. At age 7 months, he was sitting up without assistance, had begun rolling in both directions, grabbed objects with both hands and was beginning to vocalize sounds. OFC was 40.2 cm (<1st centile, − 3.1 SD), weight was 6.1 kg (<1st centile, −2.6 SD), and length was 64.5 cm (5th centile, −1.6 SD). His body habitus was proportionately small with symmetric limbs, clinically apparent microcephaly with mild right occipital flattening but otherwise normal skull shape and no other notable dysmorphic features.
Brain magnetic resonance imaging (MRI) without contrast performed at age 6 months revealed apparent microcephaly with a diffusely simplified gyral pattern (Figure 2a–c). An echocardiogram performed at age 4 months revealed a small patent foramen ovale versus secundum atrial septal defect with left to right shunting.
FIGURE 2.

Clinical photographs and brain MR images for three individuals with CCND2-associated microcephaly. (a–c) T2-weighted brain MR images of patient LR19–002 at age 6 months. (a) Midsagittal image showing a mildly thin corpus callosum, with a relatively preserved cerebellar vermis; (b) axial image showing an overall simplified gyral pattern with foreshortened frontal lobes, normal ventricles and (c) coronal image showing also an overall simplified gyral pattern. (d–f) MRI of patient LR20–198a1 at age 4 years showing a mildly foreshortened frontal lobe and subtle simplified gyrification with no cortical malformations and no other major anomalies. (g and h) frontal and lateral pictures of individual LR20–198a1. (i–k) MRI of patient LR20–198a2 at age 2 years 5 months showing a normal appearance with no cortical malformations and no other major anomalies. (l and m) frontal and lateral pictures of patient LR10–198a2
Chromosomal microarray analysis was normal. Trio exome sequencing on buccal samples identified a heterozygous de novo variant in CCND2 (NM_001759.4:c.416_419dupGGGA, p.L141GfsX19) (Figure 1b). This frameshift variant causes a premature stop codon at position 19 of the new reading frame and is predicted to cause a loss of normal protein function either through protein truncation or nonsense-mediated mRNA decay. This novel variant has not been observed in over 60,000 apparently normal individuals (Lek et al., 2016) (Table 3). Other reported findings unrelated to the patient’s phenotype include a heterozygous paternally-inherited variant of uncertain significance (VUS) in GBA (NM_000157.4: c.882T>G, p.H294Q), and a maternally inherited hemizygous VUS in TENM1 (NM_001163278.1:c.679 C>A, p.P227T) a gene not currently known to be associated with microcephaly or other human Mendelian disorders, but has been reported as possibly associated with cerebral palsy (McMichael et al., 2015), autism spectrum disorder (Yuen et al., 2015), schizophrenia (Gulsuner et al., 2013), and congenital general anosmia (Alkelai et al., 2016).
TABLE 3.
Analysis of variants pathogenicity and frequency in the population
| Variant |
Variant frequency |
|||||
|---|---|---|---|---|---|---|
| Gene | CDS | Protein | Mutation Taster | dbSNP | gnomAD (Mut/refa) | 1000G |
| CCND2 | c.305delG | p.G102Vfsx17 | Disease causing | Not present | 0/251466 | Not present |
| CCND2 | c.416_419dupGGGA | p.L141GfsX19 | Disease causing | Not present | 0/249026 | Not present |
| CCND2 | c.544C>T | p.Q182X | Disease causing | Not present | 0/248742 | Not present |
| GBA | c.882T>G | p.H294Q | Disease causing | Not present | 0/279868 | Not present |
| TENM1 | c.679C>A | p.P227T | Disease causing | Not present | 0/156640 | Not present |
Notes: Genetic variants found in individuals LR19-002, LR19-392, LR20-198 m, LR20-198a1, and LR20-198a2 are listed with their respective prediction of pathogenicity (Mutation Taster) and frequency in the population based on data collected in dbSNP, gnomAD, and 1000 Genome project (1000G) (results as of April 5, 2021).
For gnomAD reference allele frequency, data for the closest variant in the database were used to estimate the coverage of the region.
3.1.2 |. Patient LR19–392
This 12-year-old girl presented with a history of IUGR, microcephaly, delayed language skills, mild attention deficit hyperactivity disorder (ADHD), and short stature. She was the second child born to non-consanguineous parents. Family history was unremarkable with no microcephaly, growth, or developmental delay. Pregnancy was uneventful until the end of the second trimester when IUGR was identified. Amniocentesis revealed a normal female karyotype (46,XX). She was delivered at 39 weeks of gestation by scheduled cesarean section due to IUGR. Her birth weight was 2320 grams (3rd centile, − 1.8 SD), length was 46 cm (3–10th centile, −1.5 SD), and OFC was 32 cm (2nd centile, −2 SD). At age 2 years and 10 months, her weight was 9.8 kg (<1st centile, −2.8 SD), length was 89 cm (18th centile, −0.93 SD), and OFC was 43.6 cm (<1st centile, −3.1 SD). Bone age was delayed by 6 months but normalized at later follow-up. Evaluation of growth hormone response to GHRH plus arginine at 8 years of age was normal.
Her motor developmental milestones were within the normal range. She sat without assistance at age 6–7 months and walked unsupported at age13 months. Her language skills were delayed. She had mild attention deficit hyperactivity disorder (ADHD) and attended a special education program at school. Vision assessment revealed mild myopia. At her last evaluation at 12 years and 2 months, weight was 25.50 kg (<1st centile, −2.8 SD), height 140.50 cm (8th centile, −1.42 SD) and OFC was 48 cm (<1st centile, −4.27 SD). Physical examination revealed marked microcephaly with relatively large ears, short philtrum, and an incomplete right single transverse palmar crease.
Head ultrasound performed at birth did not show any structural abnormalities. A brain MRI has not been performed due to the presence of dental braces. An electroencephalogram (EEG) was performed at 12 years and 5 months and was normal.
Targeted multigene NGS panel testing for microcephaly using the NimbleGen SeqCap Target Enrichment kit (Roche) by NextSeq550 Platform (Illumina) revealed a heterozygous de novo variant in CCND2 (NM_001759.4:c.305delG). This single base deletion is expected to cause a premature stop codon, p.G102Vfsx17 (Figure 1b), with the de novo status confirmed by segregation analysis performed by Sanger sequencing of both parents. This novel variant was not observed in large population cohorts (Table 3). Other microcephaly genes in the panel include MCPH1, WDR62, CDK5RAP2, CASC5, ASPM, CENPJ, STIL, CEP135, CEP152, CDK6, ATR, RBBP8, CEP63, NIN, and ATRIP.
3.1.3 |. Family LR20–198
This family includes two siblings: a 12-year-old boy (LR20–198a1) and 6-year-old girl (LR20–198a2) born to their 30-year-old mother (LR20–198 m). LR20–198a1 presented prenatally with IUGR and microcephaly. He was born at 38 + 5 weeks of gestation by emergency cesarean section and had severe perinatal asphyxia (Apgar scores of 1, 6, and 9 at 1, 5, and 10 min, respectively). Developmental delay was documented at the age of 3 years. The child attends special education classes at school and has abnormal behaviors with frequent temper tantrums and aggression. A brain MRI at the age of 4 years shows mildly foreshortened frontal lobes and very subtle simplification of the cortical gyral pattern. Brain MR images and clinical photographs are shown in Figure 2d–h. The younger sister, LR20–198a2, similarly presented with IUGR. Microcephaly was documented at the age of 12 months. She also had short stature from the 2nd year of life, with failure to thrive, feeding difficulties and speech delays. At the age of 4 years, she had several seizure-like episodes and an abnormal EEG pattern, but no anti-epileptic treatments were initiated. A brain MRI performed at age 2 years and 5 months was normal. Brain MR images and clinical photographs are shown in Figure 2i–m. The mother of these two siblings, LR20–198 m, also had microcephaly (OFC 51 cm <1st centile, − 3.1 SD) and had short stature (height 150 cm, 2nd centile, −2 SD). She was also identified to have mild intellectual and learning disabilities. She attended special education classes at school and did not graduate. Her health history was otherwise unremarkable.
Exome sequencing performed on LR20–198a1 showed a heterozygous CCND2 variant (NM_001759.4:c.544C>T, p.Q182X) which was not observed in gnomADv2.1.1 or in-house databases. This nonsense variant is predicted to be deleterious by in silico prediction programs including Combined Annotation Dependent Depletion (CADD) and Mutation Taster (Kircher et al., 2014; Schwarz et al., 2014). Through targeted sequencing, this variant was confirmed in the heterozygous state in both LR20–198a2 and LR20–198 m but was absent in three unaffected siblings. A chromosomal microarray on LR20–198a1 demonstrated an 85 kb intragenic microduplication affecting exons 20–25 of PIK3C3. This microduplication was present in LR20–198a2 and LR20–198 m but also in several unaffected relatives and was therefore considered to be benign.
Table 2 summarizes the clinical features and molecular features of the five individuals in this series. We investigated the potential pathogenicity of the three CCND2 variants, as well as the TNEM1 variant identified in individual LR19–002 using several in silico prediction programs. All CCND2 variants were classified as disease causing and were not present in public databases such as dbSNP, 1000Genomes and gnomADv2.1.1 (Auton et al., 2015; Karczewski et al., 2020; Sherry et al., 2001). These results are summarized in Table 3. In gnomAD, coverage of the regions surrounding the three novel CCND2 variants reported here was good (gnomADv2.1.1, over 240,000 reference alleles), and seven putative loss of function (pLoF) variants are listed in the gene. However, none of them overlap with our variants, six of these are classified as low confidence (variant annotation or quality dubious) pLoF calls, with the remaining pLoF variant, p.Glu272Ter, listed as pathogenic.
3.2 |. Western blot analysis of CCND2 mutant fibroblasts
To identify differences in CCND2 expression levels and compare them to expression levels of proteins in the PI3K-AKT-MTOR pathway, including AKT, phosphorylated AKT (pAKT), and phosphorylated S6 (pS6), we performed western blot experiments using protein extracts from control fibroblasts (UW402–4-CTRL), one CCND2 MEG patient (LR07–041-MEG, p.T280N) and one CCND2 MIC patient (LR19–392-MIC, p.G102VfsX17). Due to low expression of CCND2 in primary human fibroblasts, we confirmed the specificity of the CCND2 antibody band at 31KDa using a mouse cerebellar protein extract (Supplementary Figure 1A). When compared to the control human fibroblast cell line, the LR07–041-MEG line had significantly increased levels of CCND2 (average 1.6-fold change) as well as increased levels of panAKT (average 1.71-fold change), pAKT (1.64-fold change) and pS6 (2.41-fold change), as expected (Figure 3a–c, Supplementary Table 1, Supplementary Figure 1C). Interestingly, the LR19–392-MIC line also had significantly increased levels of CCND2 compared to controls (average 1.17-fold change). The increase in CCND2 correlated with a small increase in pS6 levels (average 1.16-fold change), but not of panAKT or pAKT (Figure 3a–c, Supplementary Table 1, Supplementary Figure 1C).
FIGURE 3.

Western blot analysis of MIC- and MEG-associated CCND2 cell lines. Six independent experiments were performed using 10, 20, or 40 μg of protein extracts from fibroblasts cell lines obtained from a wild-type (UW402–4-CTRL), a CCND2 MIC patient (LR19–392-MIC) and a CCND2 MEG patient (LR07–041-MEG). (a) Representative blots for CCND2 and beta-actin for the three cell lines, with relative molecular weight shown on the left; due to low expression of CCND2 in fibroblasts, 40 μg of protein extracts was used. (b) Representative blots for PanAKT, pAKT, pS6, and beta-actin for the three cell lines. Two different protein extract concentrations were loaded (10 and 20 μg), the relative molecular weight of each detected protein is shown on the left. (c) Quantification of the analyzed proteins as average of the six independent experiments. The box plot represents the 5–95 centile interval, with the line in the box representing the median value, and the error bars indicating the minimum and maximum value. A one-way ANOVA (mixed effect analysis) was performed to identify significant differences among the cell lines. LR07–041-MEG presented increased expression of CCND2, panAKT, pAKT, and pS6 compared to control. LR19–392-MIC showed increased levels of CCND2 compared to control, but lower than LR07–041. While PanAKT and pAKT were not affected by the increase in CCND2, LR19–392-MIC presented increased levels of pS6 compared to control, albeit at lower levels than LR07–041-MEG. Ns, nonsignificant; *p-value < 0.05; **p-value < 0.01. The full range of quantifications is listed in Supplementary Table 1, additional representative western blots are shown in Supplementary Figure 1
3.3 |. High-content imaging of CCND2 mutant fibroblasts
To assess the morphological and functional differences between LR19–392-MIC and LR07–041-MEG cell lines, we performed immunostaining of key markers for proliferation (Ki-67) and MTOR hyperactivation (pS6) using high-content imaging performed with the CellInsight CX7 LZR (Thermofisher). We analyzed 33,218 cells in total for UW402–4-CTRL line; 36,104 cells for the LR19–392-MIC line; and 12,508 cells for the LR07–041-MEG line, as shown in Figure 4. We identified a significant increase in the percentage of pS6 positively staining cells in LR07–041-MEG, but not in LR19–392-MIC cells (Figure 4). The increase in pS6+ cells was accompanied by a significant increase in the percentage of double positive cells (pS6+/Ki-67 +) in LR07–041-MEG. In addition, the intensity (and thus the levels of expression) of both pS6 and Ki-67 were significantly higher in LR07–041-MEG but not in LR19–392-MIC (Figure 4). More interestingly, cytoplasmic area assessments via pS6 staining revealed marked hypertrophy in LR07–041-MEG cells (average area 590.93 pixel2) compared to controls and to the LR19–392-MIC line (376.18 pixel2 in UW402–4, and 329.13 pixel2 in LR19–392) (Figure 4b).
FIGURE 4.

High-content imaging on fibroblast cell lines with CCND2 MIC and MEG mutations. Fibroblast cell lines from one control (UW402–4-CTRL), one CCND2 MIC (LR07–041-MIC), and one CCND2 MEG (LR19–392-MEG) patients were plated in an optical 96 well plate (4 replicates each) and immunostained for proliferation marker Ki-67 and MTOR hyperactivation marker pS6; nuclei were counterstained with DAPI. Image acquisition and quantification was performed using the high-content imaging platform CellInsight CX7 LZR and the HCS studio software. (a) Single channel and overlay immunofluorescence images of one representative field per well per cell line are shown (20× magnification, scale bar 100 μm). For LR07–041-MEG, a hypertrophic fibroblast is indicated by the white arrow. (b) Quantification of the cells analyzed (4 wells per cell line, 15 fields per well) using the HCS software. Each value represents the average per well per cell line. In each graph and for each cell line, the thick bar represents the median, and the error bars represent the 95% confidence interval. One-way ANOVA analysis with multiple comparison was performed using GraphPad Prism software. Ns, nonsignificant; *p-value < 0.05; **p-value < 0.01; ***p-value < 0.0001; ****p-value < 0.00001. LR07–041-MEG shows increase in the percentage of pS6+ and ps6/Ki-67+ cells, with a reduction of Ki-67+ cells. The same line also presented increased intensity (level of expression) for pS6 and Ki-67, as well as increased cytoplasmic area. LR19–392-MIC did not show any significant difference when compared to the control. The table below panel B lists the number of cells analyzed per well per cell line, and the relative total number of cells per cell line
3.4 |. Literature review of reciprocal brain growth phenotypes
In light of our findings, we reviewed the literature of inverse brain phenotypes similar to CCND2 associated with reciprocal functional effects of genes. A brief overview of these genes, namely AKT3, PTEN, NSD1, MYCN, EZH2, and MIR17HG, is provided below, and their associated phenotypes are summarized in Table 1.
AKT3
AKT3, also known as protein kinase B (PKB), is located on chromosome 1q43–44 and encodes a serine/threonine protein kinase expressed primarily in the brain. AKT3 is one of three closely related AKT isoforms and is the primary downstream effector of PI3K signaling. PI3K-AKT-mTOR signaling regulates many important cell functions, including protein synthesis, cell proliferation and survival (Diez et al., 2012; Ersahin et al., 2015; Ling Wang et al., 2017). AKT3 exerts its regulatory effects through serine and/or threonine phosphorylation of various downstream substrates, one of which is linked to influencing cyclin-D2 levels by S9 residue phosphorylation and subsequent inactivation of glycogen synthase kinase-3 β (GSK-3β) (P. Cohen & Frame, 2001; Hermida et al., 2017; Miao et al., 2016; Ouimet et al., 2019). GSK-3β is a negative regulator of cyclin D1 and D2 by ubiquitin dependent degradation. The AKT family of proteins are implicated in a wide range of human diseases including cancer, diabetes, cardiovascular and neurological disease (Alcantara et al., 2017; Chung et al., 2014; Conti et al., 2015; Dobyns & Mirzaa, 2019; Gai et al., 2015; Grabinski et al., 2011; Hers et al., 2011; Lee et al., 2012; Lopes et al., 2019; Mure et al., 2010; Poduri et al., 2012; Riviere et al., 2012; D. Wang et al., 2013; Yang et al., 2005). Gain-of-function variants in AKT3 lead to inactivation of GSK-3β which in turn results in stabilization and increased levels of cyclin D2 which has been proposed as the unifying mechanism for MPPH syndrome (G. Mirzaa et al., 2014). Data from humans and animal models suggest a gene dosage effect of AKT3 resulting in inverse phenotypes. Akt3 knockout mice display microcephaly suggesting that it plays a critical role in determining brain size (Easton et al., 2005; Yang et al., 2005). Mosaic and constitutional gain-of-function variants of AKT3 are associated with MPPH syndrome (Lee et al., 2012; Lopes et al., 2019; Poduri et al., 2012; Riviere et al., 2012), with more than 20 patients reported to date with molecularly confirmed AKT3 related megalencephaly (Alcantara et al., 2017; Harada et al., 2015; Jamuar et al., 2014; Nakamura et al., 2014; Negishi et al., 2017; Nellist et al., 2015; Riviere et al., 2012). A subset of affected individuals is mosaic for the common p.E17K mutation causing hemimegalencephaly (Jansen et al., 2015; Lee et al., 2012; Poduri et al., 2012; Riviere et al., 2012). Duplications of 1q43-q44 encompassing AKT3 have also been reported to cause macrocephaly (Chung et al., 2014; Hemming et al., 2016; Luo et al., 2018; D. Wang et al., 2013). Heterozygous deletions of 1q43-q44 involving AKT3, on the other hand, result in postnatal microcephaly and agenesis of the corpus callosum (Gai et al., 2015; Hemming et al., 2016; Lopes et al., 2019).
PTEN
Loss-of-function variants in PTEN, which encodes for phosphatase and tensin homolog and tumor suppressor, cause a spectrum of clinical megalencephaly disorders including Cowden syndrome (CS), Bannayan–Riley–Ruvalcaba syndrome (BRRS), and autism spectrum disorder with macrocephaly (Gorlin et al., 1992; Herman et al., 2007; Marsh et al., 1998; Orrico et al., 2009; Varga et al., 2009; Zhou et al., 2003). These syndromes share characteristic features including overgrowth and predisposition to various tumors. PTEN suppresses phosphoinositide 3-kinase (PI3K) signaling that activates cell growth and tumorigenesis. Mice deficient in nuclear PTEN have microcephaly persisting into adulthood (Igarashi et al., 2018; Kwon et al., 2001; Kwon et al., 2003; Kwon et al., 2006). Further, deletions of the 10q23 locus encompassing PTEN are associated with macrocephaly, hypotonia, and increased predisposition to various malignancies, including juvenile polyposis syndrome (Babovic et al., 2010). In contrast, microduplications of this locus have recently been identified in association with autosomal dominant primary microcephaly and decreased mTOR signaling (Oliveira et al., 2019).
NSD1
NSD1, located on chromosome 5q35.3, encodes for nuclear receptor binding (SET) domain histone methyltransferase that methylates lysine 36 of histone 3 (H3K36) (Graham et al., 2016). This histone modifier regulates the activity of several genes involved in growth and development. Haploinsufficiency of NSD1 causes Sotos syndrome characterized by brain and somatic overgrowth, distinctive facial features (broad, prominent forehead, dolichocephaly, spares frontotemporal hair, long chin, long narrow face, malar flushing, down-slanting palpebral fissures), and intellectual disability (Kurotaki et al., 2002). Other major features include behavioral problems, advanced bone age, cardiac defects, cranial abnormalities, skeletal abnormalities including joint hyperlaxity and scoliosis, renal defects, and seizures. A minority of affected individuals (<15%) have other features such as hemihypertrophy, conductive hearing loss, pectus excavatum, and strabismus. Loss-of-function variants and deletions of 5q35 encompassing NSD1 have been identified in approximately 90% of individuals with Sotos syndrome (Tatton-Brown et al., 2005). Conversely, NSD1 duplications result in growth retardation and microcephaly (Dikow et al., 2013; Franco et al., 2010; Lucio-Eterovic et al., 2010; Sachwitz et al., 2017; Sellars et al., 2011; Tracy et al., 2011; J. C. Wang et al., 2007; Zhang et al., 2011). Further, murine models carrying a heterozygous 1.5-Mb deletion of 36 genes including Nsd1 syntenic with 5q35.2-q35.3 in humans had a phenotype characterized by small gestational age and decreased postnatal growth (Migdalska et al., 2012).
MYCN
MYCN, located on chromosome 2p24.3, encodes for N-Myc protein, a transcription factor critical to normal embryonic development and postnatal brain size. The MYCN gene is part of the Myc family of proto-oncogenes critical for regulating cell differentiation and growth (Henriksson & Luscher, 1996). Much of the function of the Myc family of proteins is through downstream activation and repression of specific target genes (Eisenman, 2001). These targets include the cyclin-dependent kinase CDK4, a Cdc25A phosphatase that activates CDKs, cyclin D2 and the E2F family (Bouchard et al., 1999; Galaktionov et al., 1996; Leone et al., 2001). Amplification of MYCN is associated with various cancers including prostate cancer (Dardenne et al., 2016), retinoblastoma (Rushlow et al., 2013), and aggressive forms of neuroblastoma (Higashi et al., 2019; Otto et al., 2009; Tavana et al., 2016; Xue et al., 2016). The carcinogenic potential of N-Myc is likely due to its ability to drive cells into the cell cycle. A mouse model showed that N-myc plays a vital role in neurogenesis and that homozygous absence of N-myc results in a twofold decrease in brain mass that disproportionately affects the cerebellum and cerebral cortex (Knoepfler et al., 2002). This same study suggested that the decrease in brain size is due to downregulation of a subset of cyclin-dependent kinase inhibitors and disruption of CCND2 expression. Germline loss of function variants of MYCN cause Feingold syndrome type 1 characterized by microcephaly, limb abnormalities, atresia of the gastrointestinal tract, learning disabilities, and dysmorphic facial features. Deletions involving the MYCN locus have also been reported as a cause of Feingold Syndrome (Chen et al., 2012; Mercedes Bloch et al., 2014). Although there is variable expressivity, there are no significant genotype–phenotype differences between individuals with Feingold Syndrome (Marcelis et al., 2008). Further, an MYCN gain-of-function variant resulting in increased CCND1 and CCND2 expression has recently been reported to cause a novel megalencephaly syndrome that is similar but distinct from MPPH syndrome (Kato et al., 2018).
EZH2
Enhancer of Zeste, Drosophila, homolog 2 (EZH2, MIM: 601573) encodes for part of the polycomb repressive complex 2 (PRC2), which mediates suppression of transcription via chromatin condensation through methylation of histone H3 (lysine 27) (Cao et al., 2002). Variants in this gene are the primary cause of Weaver Syndrome (WS), characterized by overgrowth, facial gestalt, accelerated bone maturation and variable degree of intellectual disability (A. S. Cohen et al., 2016; Gibson et al., 2012; Lui et al., 2018; Tatton-Brown et al., 2011; Tatton-Brown et al., 2013). Studies in humans and mice have demonstrated that EZH2 variants associated with WS are mainly due to reduced methyltransferase activity, and thus are classified as loss of function (A. S. Cohen & Gibson, 2016; Lui et al., 2018). Recently, an elegant study utilized DNA methylation signature to classify missense variants in EZH2 as pathogenic or benign, and provided insights into gain of function mutations and mosaicism (Choufani et al., 2020). Notably, gain of methylation at the same CpG sites associated with WS, lead to transcriptional changes during development resulting in growth restriction in one patient (Choufani et al., 2020). This evidence adds EZH2 to the growing list of genes with epigenetic functions that can affect cell growth through changes in their transcriptional profiles, similarly to NSD1 (Aref-Eshghi et al., 2018; Butcher et al., 2017; Choufani et al., 2015).
MIR17HG and 13q31.3
MIR17HG gene encodes the miR-17 ~92 polycistronic microRNA (miRNA) cluster, a family of six highly conserved miRNA (Han et al., 2015). Focal amplifications of this region are present in many types of cancer, with ectopic expression of this locus associated with acceleration of tumor formation in murine models (Han et al., 2015). Microdeletions of chromosome 13q that include the MIR17HG gene are associated with Feingold 2 syndrome, with features of microcephaly, brachymesophalangy, toe syndactyly, short stature, cardiac anomalies, growth hormone deficiency, aortic dilation, phalangeal joint contractures, memory, and sleep problems (de Pontual et al., 2011; Muriello et al., 2019). Recently, a de novo 13q31.3 microduplication encompassing MIR17HG was described in a female patient with developmental delay, skeletal and digital abnormalities, and tall stature and macrocephaly (Siavriene et al., 2020).
4 |. DISCUSSION
The CCND2 gene, located on 12p13.32, encodes the protein Cyclin D2 and is a member of the highly conserved cyclin family, regulating the G1 to S phase transition in the cell cycle (Z. Wang et al., 2008; Xiong et al., 1992). Gain-of-function (GoF) variants in CCND2, AKT3 and PIK3R2 have been implicated in MPPH syndrome. Megalencephaly-associated variants in components of the PI3K-AKT-MTOR pathway, including variants within the terminal exon of CCND2, share the same functional endpoint, namely inhibition of proteasomal degradation of cyclin D2 (G. Mirzaa et al., 2014). This influences many important cell functions including cell growth and division, protein synthesis and cell survival. Although not directly in the PI3K-AKT-MTOR pathway, MCYN indirectly participates through upstream modulation of CCND1 and CCND2 expression (Kato et al., 2018). Further, copy number abnormalities, namely 12p13.32 deletions encompassing CCND2, have been identified in individuals with microcephaly and a wide spectrum of clinical features including intellectual disability, short stature, developmental delays, and dysmorphic features. However, the highly variable presentation and high gene content of this microdeletion syndrome provided limited evidence regarding the role of CCND2. (Firth et al., 2009). This is the first series reporting on individuals with heterozygous frameshift and stop gain CCND2 variants resulting in microcephaly, to our knowledge. All affected individuals demonstrated poor growth, in association with neurodevelopmental issues, among other features.
CCND2 is highly expressed in pluripotent human embryonic stem cells (Becker et al., 2010), and is postnatally expressed at high levels in the human brain, adrenal gland, heart, and colon (Fagerberg et al., 2014). CCND2 is a regulatory subunit of the Cyclin D2-CDK4 complex, which phosphorylates and inhibits members of the retinoblastoma (RB) family of proteins including RB1. Phosphorylation of RB1 allows dissociation of the transcription factor E2F from the RB/E2F complex and the subsequent transcription of E2F target genes responsible for progression through the G1 phase of the cell cycle. Cyclin D2 is also a substrate for SMAD3, phosphorylating SMAD3 in a cell-cycle-dependent manner and repressing its transcriptional activity. Cyclin D2 has been found to be highly expressed in the adult mouse brain suggesting that cyclin D2 carries out a unique role in terminally differentiated neurons (Ross et al., 1996). Murine knockout models of Ccnd2 lack adult neurogenesis almost entirely (Kondratiuk et al., 2015). A deficit in parvalbumin positive (PV+) GABAergic interneurons in Ccnd2 null mice have been observed in both the cortex and the hippocampus leading to a decrease in cortical inhibition (Glickstein et al., 2007). Evidence of this cortical inhibition was demonstrated on EEGs of awake cD2-null mice showing increased frequency and amplitude of paroxysmal cortical discharges (Glickstein et al., 2007). Furthermore, gene dose dependent differences in brain cortical surface area have been observed in mouse models with heterozygous knockouts of Ccnd2 resulting in an intermediate phenotype (Glickstein et al., 2007). Ccnd2 null mice had microcephaly with either a relatively preserved or mildly decreased body size with catch up growth and an increase in seizure activity as they age (M. Elizabeth Ross, personal communication, April 25, 2019).
Our series of five individuals with microcephaly and three novel variants in CCND2 expands the genotype–phenotype spectrum for this gene. Our functional analysis revealed increased levels of CCND2 and other key PI3K-AKT-MTOR markers in CCND2 mutant fibroblasts derived from a child with MPPH, confirming previous findings by our group (G. Mirzaa et al., 2014). Immunostaining also revealed marked increase in Ki-67 expression, suggesting that increased proliferation is a mechanism for CCND2-related MEG, as demonstrated by our prior work on CCND2 (G. Mirzaa et al., 2014). Our data further show that an increase in cell size, or cellular hypertrophy, is likely another feature of CCND2-related MEG, as demonstrated by the significant enlargement of cytoplasmic area, and similar to the MTOR-dependent hypertrophy identified in Pten-deficient mice (Kwon et al., 2001; Kwon et al., 2003). Despite the increase in pS6+ and Ki-67+/pS6+ cells, the LR07–041 MEG cell line showed a reduced overall percentage of Ki67+ cells, due to the overall lower number of cells present in the wells, as cells occupy a larger volume (12,508 cells for LR07–041 MEG vs. 33,218 cells in the control). Similar assays on the CCND2-associated microcephaly cell line revealed lower levels of CCND2 compared to the MEG cell line. However, levels were surprisingly slightly higher than the control line. We were able to detect the CCND2 protein in the LR19–392 MIC cell line despite the premature stop codon as the antibody recognized an epitope in the N-terminus, suggesting expression of a truncated isoform of the protein in this patient. We therefore hypothesize that the increase in CCND2 levels in the MIC cell line could be due to accumulation of unphosphorylated degradation-resistant cyclin D2, similarly to what demonstrated for CCND2 -related MPPH (G. Mirzaa et al., 2014), albeit with distinct functional consequences in the developing brain. We also hypothesize that this mild increase in protein level is likely insufficient to cause hyperplasia or hypertrophy, as the LR19–392-MIC cells were not significantly different in size or number from controls. We previously identified a nonsense variant in the terminal exon of CCND2 in association with MPPH (p.Lys270X) (G. Mirzaa et al., 2014), thus we postulate that variants in distinct regions of the gene (proximal vs. distal) are directly relevant to the functional consequences and associated phenotypes of CCND2-related disorders.
We considered whether the TENM1 hemizygous variant in individual LR19–002 could contribute to the phenotype. However, we believe this is less likely as TENM1 is not known to be associated with microcephaly, to date. Notably, individual LR19–392 in our study underwent only targeted multigene sequencing. Therefore, we cannot rule out the possibility that this individual may carry other variants that might contribute to the phenotype. Finally, additional functional studies in more relevant models, such as human induced pluripotent stem cell-derived neurons, are needed to confirm some of the above hypotheses regarding the role of CCND2 in regulating brain size and the exact molecular mechanisms of CCND2-related microcephaly in humans.
In summary, our study expands the phenotypic spectrum of CCND2-related disorders in humans and suggests that distinct classes of genetic variants within different domains of the gene—namely proximal versus distal variants—have different functional consequences on the developing human brain and are likely associated with reciprocal effects on early brain growth—with more proximal and potentially loss of function variants causing protein truncation associated with microcephaly, and distal variants abolishing phosphorylation-dependent ubiquitination sites resulting in megalencephaly. This study further highlights the diversity of the molecular mechanisms that cause brain growth syndromes and the paradigm of reciprocal genetic variants leading to inverse brain phenotypes.
Supplementary Material
{kind=link}
ACKNOWLEDGMENTS
We thank the patients, their families and care providers for their contribution to this study. We thank Dr. M. Elizabeth Ross, Professor of Neurology and Neuroscience, and the Ross Laboratory at Weill Cornell Medicine for their expert input regarding the function and role of CCND2 in brain development. We also thank Dr. Daniel Doherty, Professor of Pediatrics and Developmental Medicine, for providing the control fibroblast cell line used in this manuscript (UW402-4). Research reported in this publication was supported by Jordan’s Guardian Angels and the Sunderland Foundation (to Filomena Pirozzi and Ghayda M. Mirzaa), and the Brotman-Baty Institute (to Ghayda M. Mirzaa); Nataliya Di Donato work was supported by German Research Foundation (DI 2170/3-1). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding sources had no role in the design and conduct of the study, collection, management, analysis and interpretation of the data, preparation, review or approval of the manuscript, or decision to submit the manuscript for publication.
Funding information
Brotman-Baty Institute; German Research Foundation, Grant/Award Number: DI 2170/3-1; Jordan’s Guardian Angels; Sunderland Foundation
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material. The raw data that support the findings of this study, including the genetic analyses and the high content imaging results are available on request to the corresponding authors.
REFERENCES
- Alcantara D, Timms AE, Gripp K, Baker L, Park K, Collins S, Cheng C, Stewart F, Mehta SG, Saggar A, Sztriha L, Zombor M, Caluseriu O, Mesterman R, Van Allen MI, Jacquinet A, Ygberg S, Bernstein JA, Wenger AM, … Mirzaa GM (2017). Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly. Brain, 140(10), 2610–2622. 10.1093/brain/awx203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkelai A, Olender T, Haffner-Krausz R, Tsoory MM, Boyko V, Tatarskyy P, Gross-Isseroff R, Milgrom R, Shushan S, Blau I, Cohn E, Beeri R, Levy-Lahad E, Pras E, & Lancet D (2016). A role for TENM1 mutations in congenital general anosmia. Clinical Genetics, 90(3), 211–219. 10.1111/cge.12782 [DOI] [PubMed] [Google Scholar]
- Aref-Eshghi E, Rodenhiser DI, Schenkel LC, Lin H, Skinner C, Ainsworth P, Paré G, Hood RL, Bulman DE, Kernohan KD, Care4Rare Canada Consortium, Boycott KM, Campeau PM, Schwartz C, & Sadikovic B (2018). Genomic DNA methylation signatures enable concurrent diagnosis and clinical genetic variant classification in neurodevelopmental syndromes. American Journal of Human Genetics, 102(1), 156–174. 10.1016/j.ajhg.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, & Genomes Project C (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babovic N, Simmons PS, Moir C, Thorland EC, Scheithauer B, Gliem TJ, & Babovic-Vuksanovic D (2010). Mucinous cystadenoma of ovary in a patient with juvenile polyposis due to 10q23 microdeletion: Expansion of phenotype. American Journal of Medical Genetics. Part A, 152A(10), 2623–2627. 10.1002/ajmg.a.33637 [DOI] [PubMed] [Google Scholar]
- Becker KA, Ghule PN, Lian JB, Stein JL, van Wijnen AJ, & Stein GS (2010). Cyclin D2 and the CDK substrate p220(NPAT) are required for self-renewal of human embryonic stem cells. Journal of Cellular Physiology, 222(2), 456–464. 10.1002/jcp.21967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, & Eilers M (1999). Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. The EMBO Journal, 18(19), 5321–5333. 10.1093/emboj/18.19.5321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkardt DD, Tatton-Brown K, Dobyns W, & Graham JM Jr. (2019). Approach to overgrowth syndromes in the genome era. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 181(4), 483–490. 10.1002/ajmg.c.31757 [DOI] [PubMed] [Google Scholar]
- Butcher DT, Cytrynbaum C, Turinsky AL, Siu MT, Inbar-Feigenberg M, Mendoza-Londono R, Chitayat D, Walker S, Machado J, Caluseriu O, Dupuis L, Grafodatskaya D, Reardon W, Gilbert-Dussardier B, Verloes A, Bilan F, Milunsky JM, Basran R, Papsin B, … Weksberg R (2017). CHARGE and kabuki syndromes: Gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. American Journal of Human Genetics, 100(5), 773–788. 10.1016/j.ajhg.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, & Zhang Y (2002). Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science, 298(5595), 1039–1043. 10.1126/science.1076997 [DOI] [PubMed] [Google Scholar]
- Cappuccio G, Ugga L, Parrini E, D’Amico A, & Brunetti-Pierri N (2019). Severe presentation and complex brain malformations in an individual carrying a CCND2 variant. Molecular Genetics & Genomic Medicine, 7(6), e708. 10.1002/mgg3.708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Lin SP, Chern SR, Wu PS, Chang SD, Ng SH, Liu YP, Su JW, & Wang W (2012). A de novo 4.4-Mb microdeletion in 2p24.3→p24.2 in a girl with bilateral hearing impairment, microcephaly, digit abnormalities and Feingold syndrome. European Journal of Medical Genetics, 55(11), 666–669. 10.1016/j.ejmg.2012.07.003 [DOI] [PubMed] [Google Scholar]
- Choufani S, Cytrynbaum C, Chung BH, Turinsky AL, Grafodatskaya D, Chen YA, Cohen AS, Dupuis L, Butcher DT, Siu MT, Luk HM, Lo IF, Lam ST, Caluseriu O, Stavropoulos DJ, Reardon W, Mendoza-Londono R, Brudno M, Gibson WT, … Weksberg R (2015). NSD1 mutations generate a genome-wide DNA methylation signature. Nature Communications, 6, 10207. 10.1038/ncomms10207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choufani S, Gibson WT, Turinsky AL, Chung BHY, Wang T, Garg K, Vitriolo A, Cohen ASA, Cyrus S, Goodman S, Chater-Diehl E, Brzezinski J, Brudno M, Ming LH, White SM, Lynch SA, Clericuzio C, Temple IK, Flinter F, … Weksberg R (2020). DNA methylation signature for EZH2 functionally classifies sequence variants in three PRC2 complex genes. American Journal of Human Genetics, 106(5), 596–610. 10.1016/j.ajhg.2020.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung BK, Eydoux P, Van Karnebeek CD, & Gibson WT (2014). Duplication of AKT3 is associated with macrocephaly and speech delay. American Journal of Medical Genetics. Part A, 164A(7), 1868–1869. 10.1002/ajmg.a.36521 [DOI] [PubMed] [Google Scholar]
- Cohen AS, & Gibson WT (2016). EED-associated overgrowth in a second male patient. Journal of Human Genetics, 61(9), 831–834. 10.1038/jhg.2016.51 [DOI] [PubMed] [Google Scholar]
- Cohen AS, Yap DB, Lewis ME, Chijiwa C, Ramos-Arroyo MA, Tkachenko N, Milano V, Fradin M, McKinnon ML, Townsend KN, Xu J, Van Allen MI, Ross CJD, Dobyns WB, Weaver DD, & Gibson WT (2016). Weaver syndrome-associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Human Mutation, 37(3), 301–307. 10.1002/humu.22946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, & Frame S (2001). The renaissance of GSK3. Nature Reviews. Molecular Cell Biology, 2(10), 769–776. 10.1038/35096075 [DOI] [PubMed] [Google Scholar]
- Conti V, Pantaleo M, Barba C, Baroni G, Mei D, Buccoliero AM, Giglio S, Giordano F, Baek ST, Gleeson JG, & Guerrini R (2015). Focal dysplasia of the cerebral cortex and infantile spasms associated with somatic 1q21.1-q44 duplication including the AKT3 gene. Clinical Genetics, 88(3), 241–247. 10.1111/cge.12476 [DOI] [PubMed] [Google Scholar]
- Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T, Cheung C, Yuen KS, Gao D, Chen Y, Eilers M, Mosquera JM, Robinson BD, Elemento O, Rubin MA, … Rickman DS (2016). N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell, 30(4), 563–577. 10.1016/j.ccell.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pontual L, Yao E, Callier P, Faivre L, Drouin V, Cariou S, Van Haeringen A, Geneviève D, Goldenberg A, Oufadem M, Manouvrier S, Munnich A, Vidigal JA, Vekemans M, Lyonnet S, Henrion-Caude A, Ventura A, & Amiel J (2011). Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nature Genetics, 43(10), 1026–1030. 10.1038/ng.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez H, Garrido JJ, & Wandosell F (2012). Specific roles of Akt iso forms in apoptosis and axon growth regulation in neurons. PLoS One, 7 (4), e32715. 10.1371/journal.pone.0032715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikow N, Maas B, Gaspar H, Kreiss-Nachtsheim M, Engels H, Kuechler A, Garbes L, Netzer C, Neuhann TM, Koehler U, Casteels K, Devriendt K, Janssen JWG, Jauch A, Hinderhofer K, & Moog U (2013). The phenotypic spectrum of duplication 5q35.2-q35.3 encompassing NSD1: Is it really a reversed Sotos syndrome? American Journal of Medical Genetics. Part A, 161A(9), 2158–2166. 10.1002/ajmg.a.36046 [DOI] [PubMed] [Google Scholar]
- Dobyns WB, & Mirzaa GM (2019). Megalencephaly syndromes associated with mutations of core components of the PI3K-AKT-MTOR pathway: PIK3CA, PIK3R2, AKT3, and MTOR. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 181, 582–590. 10.1002/ajmg.c.31736 [DOI] [PubMed] [Google Scholar]
- Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VMY, Szabolcs M, De Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, & Birnbaum MJ (2005). Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Molecular and Cellular Biology, 25(5), 1869–1878. 10.1128/MCB.25.5.1869-1878.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman RN (2001). Deconstructing myc. Genes & Development, 15(16), 2023–2030. https://doi.org/10.1101/gad928101 [DOI] [PubMed] [Google Scholar]
- Ersahin T, Tuncbag N, & Cetin-Atalay R (2015). The PI3K/AKT/mTOR interactive pathway. Molecular BioSystems, 11(7), 1946–1954. 10.1039/c5mb00101c [DOI] [PubMed] [Google Scholar]
- Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjöstedt E, Lundberg E, Szigyarto CA, Skogs M, Takanen JO, Berling H, Tegel H, Mulder J, … Uhlén M (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & Cellular Proteomics, 13(2), 397–406. 10.1074/mcp.M113.035600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, van Vooren S, Moreau Y, Pettett RM, & Carter NP (2009). DECI-PHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources. https://doi.org/10/1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed]
- Franco LM, de Ravel T, Graham BH, Frenkel SM, Van Driessche J, Stankiewicz P, Lupski JR, Vermeesch JR, & Cheung SW (2010). A syndrome of short stature, microcephaly and speech delay is associated with duplications reciprocal to the common Sotos syndrome deletion. European Journal of Human Genetics, 18(2), 258–261. 10.1038/ejhg.2009.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai D, Haan E, Scholar M, Nicholl J, & Yu S (2015). Phenotypes of AKT3 deletion: A case report and literature review. American Journal of Medical Genetics. Part A, 167A(1), 174–179. 10.1002/ajmg.a.36710 [DOI] [PubMed] [Google Scholar]
- Galaktionov K, Chen X, & Beach D (1996). Cdc25 cell-cycle phosphatase as a target of c-myc. Nature, 382(6591), 511–517. 10.1038/382511a0 [DOI] [PubMed] [Google Scholar]
- Garthe A, Huang Z, Kaczmarek L, Filipkowski RK, & Kempermann G (2014). Not all water mazes are created equal: Cyclin D2 knockout mice with constitutively suppressed adult hippocampal neurogenesis do show specific spatial learning deficits. Genes, Brain, and Behavior, 13 (4), 357–364. 10.1111/gbb.12130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, Marra MA, FORGE Canada Consortium, Chitayat D, Boycott KM, Weaver DD, & Jones SJ (2012). Mutations in EZH2 cause Weaver syndrome. American Journal of Human Genetics, 90(1), 110–118. 10.1016/j.ajhg.2011.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickstein SB, Moore H, Slowinska B, Racchumi J, Suh M, Chuhma N, & Ross ME (2007). Selective cortical interneuron and GABA deficits in cyclin D2-null mice. Development, 134(22), 4083–4093. 10.1242/dev.008524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Cohen MM Jr., Condon LM, & Burke BA (1992). Bannayan–Riley–Ruvalcaba syndrome. American Journal of Medical Genetics, 44(3), 307–314. 10.1002/ajmg.1320440309 [DOI] [PubMed] [Google Scholar]
- Grabinski N, Bartkowiak K, Grupp K, Brandt B, Pantel K, & Jucker M (2011). Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cellular Signalling, 23(12), 1952–1960. 10.1016/j.cellsig.2011.07.003 [DOI] [PubMed] [Google Scholar]
- Graham SE, Tweedy SE, & Carlson HA (2016). Dynamic behavior of the post-SET loop region of NSD1: Implications for histone binding and drug development. Protein Science, 25(5), 1021–1029. 10.1002/pro.2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissom NM, & Reyes TM (2013). Gestational overgrowth and undergrowth affect neurodevelopment: Similarities and differences from behavior to epigenetics. International Journal of Developmental Neuroscience, 31(6), 406–414. 10.1016/j.ijdevneu.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Consortium on the Genetics of Schizophrenia (COGS), PAARTNERS Study Group, Nimgaonkar VL, Go RC, Savage RM, Swerdlow NR, Gur RE, Braff DL, King MC, & McClellan JM (2013). Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell, 154(3), 518–529. 10.1016/j.cell.2013.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YC, Vidigal JA, Mu P, Yao E, Singh I, Gonzalez AJ, Concepcion CP, Bonetti C, Ogrodowski P, Carver B, Selleri L, Betel D, Leslie C, & Ventura A (2015). An allelic series of miR-17 approximately 92-mutant mice uncovers functional specialization and cooperation among members of a microRNA polycistron. Nature Genetics, 47(7), 766–775. 10.1038/ng.3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Miya F, Utsunomiya H, Kato M, Yamanaka T, Tsunoda T, Kosaki K, Kanemura Y, & Yamasaki M (2015). Sudden death in a case of megalencephaly capillary malformation associated with a de novo mutation in AKT3. Child’s Nervous System, 31(3), 465–471. 10.1007/s00381-014-2589-y [DOI] [PubMed] [Google Scholar]
- Hemming IA, Forrest AR, Shipman P, Woodward KJ, Walsh P, Ravine DG, & Heng JI (2016). Reinforcing the association between distal 1q CNVs and structural brain disorder: A case of a complex 1q43-q44 CNV and a review of the literature. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics, 171B(3), 458–467. 10.1002/ajmg.b.32427 [DOI] [PubMed] [Google Scholar]
- Henriksson M, & Luscher B (1996). Proteins of the Myc network: Essential regulators of cell growth and differentiation. Advances in Cancer Research, 68, 109–182. [DOI] [PubMed] [Google Scholar]
- Herman GE, Butter E, Enrile B, Pastore M, Prior TW, & Sommer A (2007). Increasing knowledge of PTEN germline mutations: Two additional patients with autism and macrocephaly. American Journal of Medical Genetics. Part A, 143A(6), 589–593. 10.1002/ajmg.a.31619 [DOI] [PubMed] [Google Scholar]
- Hermida MA, Dinesh Kumar J, & Leslie NR (2017). GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Advances in Biological Regulation, 65, 5–15. 10.1016/j.jbior.2017.06.003 [DOI] [PubMed] [Google Scholar]
- Hers I, Vincent EE, & Tavare JM (2011). Akt signalling in health and disease. Cellular Signalling, 23(10), 1515–1527. 10.1016/j.cellsig.2011.05.004 [DOI] [PubMed] [Google Scholar]
- Higashi M, Sakai K, Fumino S, Aoi S, Furukawa T, & Tajiri T (2019). The roles played by the MYCN, Trk, and ALK genes in neuroblastoma and neural development. Surgery Today, 49(9), 721–727. 10.1007/s00595-019-01790-0 [DOI] [PubMed] [Google Scholar]
- Hiraiwa A, Matsui K, Nakayama Y, Komatsubara T, Magara S, Kobayashi Y, Hojo M, Kato M, Yamamoto T, & Tohyama J (2021). Polymicrogyria with calcification in Pallister-Killian syndrome detected by microarray analysis. Brain Dev, 43(3), 448–453. 10.1016/j.braindev.2020.11.003 [DOI] [PubMed] [Google Scholar]
- Huard JM, Forster CC, Carter ML, Sicinski P, & Ross ME (1999). Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development, 126(9), 1927–1935. [DOI] [PubMed] [Google Scholar]
- Hung CS, Wang S, Yen YT, Lee TH, Wen WC, & Lin RK (2018). Hypermethylation of CCND2 in lung and breast cancer is a potential biomarker and drug target. International Journal of Molecular Sciences, 19(10), 3096. 10.3390/ijms19103096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi A, Itoh K, Yamada T, Adachi Y, Kato T, Murata D, Sesaki H, & Iijima M (2018). Nuclear PTEN deficiency causes microcephaly with decreased neuronal soma size and increased seizure susceptibility. The Journal of Biological Chemistry, 293(24), 9292–9300. 10.1074/jbc.RA118.002356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamuar SS, Lam AT, Kircher M, D’Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, Servattalab S, Mehta BK, Topcu M, Amrom D, Andermann E, Dan B, Parrini E, Guerrini R, Scheffer IE, … Walsh CA (2014). Somatic mutations in cerebral cortical malformations. The New England Journal of Medicine, 371(8), 733–743. 10.1056/NEJMoa1314432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen LA, Mirzaa GM, Ishak GE, O’Roak BJ, Hiatt JB, Roden WH, Gunter SA, Christian SL, Collins S, Adams C, Riviére JB, St-Onge J, Ojemann JG, Shendure J, Hevner RF, & Dobyns WB (2015). PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain, 138(Pt 6), 1613–1628. 10.1093/brain/awv045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong OS, Chae YC, Jung H, Park SC, Cho SJ, Kook H, & Seo S (2016). Long noncoding RNA linc00598 regulates CCND2 transcription and modulates the G1 checkpoint. Scientific Reports, 6, 32172. 10.1038/srep32172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581 (7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Miya F, Hamada N, Negishi Y, Narumi-Kishimoto Y, Ozawa H, Ito H, Hori I, Hattori A, Okamoto N, Kato M, Tsunoda T, Kanemura Y, Kosaki K, Takahashi Y, Nagata KI, & Saitoh S (2018). MYCN de novo gain-of-function mutation in a patient with a novel megalencephaly syndrome. Journal of Medical Genetics, 56, 388–395. 10.1136/jmedgenet-2018-105487 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, & Eisenman RN (2002). N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes & Development, 16(20), 2699–2712. 10.1101/gad.1021202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratiuk I, Plucinska G, Miszczuk D, Wozniak G, Szydlowska K, Kaczmarek L, Filipkowski RK, & Lukasiuk K (2015). Epileptogenesis following kainic acid-induced status epilepticus in cyclin D2 knock-out mice with diminished adult neurogenesis. PLoS One, 10(5), e0128285. 10.1371/journal.pone.0128285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, Sugimoto T, Sonoda T, Hasegawa T, Chinen Y, Tomita HA, Kinoshita A, Mizuguchi T, Yoshiura KI, Ohta T, … Matsumoto N (2002). Haploinsufficiency of NSD1 causes Sotos syndrome. Nature Genetics, 30(4), 365–366. 10.1038/ng863 [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, & Parada LF (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron, 50(3), 377–388. 10.1016/j.neuron.2006.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, & Baker SJ (2003). mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proceedings of the National Academy of Sciences of the United States of America, 100 (22), 12923–12928. 10.1073/pnas.2132711100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, & Baker SJ (2001). Pten regulates neuronal soma size: A mouse model of Lhermitte–Duclos disease. Nature Genetics, 29(4), 404–411. 10.1038/ng781 [DOI] [PubMed] [Google Scholar]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, & Gleeson JG (2012). De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nature Genetics, 44(8), 941–945. 10.1038/ng.2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, … Exome Aggregation Consortium (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone G, Sears R, Huang E, Rempel R, Nuckolls F, Park CH, Giangrande P, Wu L, Saavedra HI, Field SJ, Thompson MA, Yang H, Fujiwara Y, Greenberg ME, Orkin S, Smith C, & Nevins JR (2001). Myc requires distinct E2F activities to induce S phase and apoptosis. Molecular Cell, 8(1), 105–113. [DOI] [PubMed] [Google Scholar]
- Leyser M, Dias BL, Coelho AL, Vasconcelos M, & Nascimento OJ (2016). 12p deletion spectrum syndrome: A new case report reinforces the evidence regarding the potential relationship to autism spectrum disorder and related developmental impairments. Molecular Cytogenetics, 9, 75. 10.1186/s13039-016-0278-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Wang DH, Jiang Z, Luo Y, Norris C, Zhang M, Tian X, & Tang Y (2017). Akt3 is responsible for the survival and proliferation of embryonic stem cells. Biol Open, 6(6), 850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes F, Torres F, Soares G, van Karnebeek CD, Martins C, Antunes D, Silva J, Muttucomaroe L, Botelho LF, Sousa S, Rendeiro P, Tavares P, Van Esch H, Rajcan-Separovic E, & Maciel P (2019). The role of AKT3 copy number changes in brain abnormalities and neurodevelopmental disorders: Four new cases and literature review. Frontiers in Genetics, 10, 58. 10.3389/fgene.2019.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucio-Eterovic AK, Singh MM, Gardner JE, Veerappan CS, Rice JC, & Carpenter PB (2010). Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proceedings of the National Academy of Sciences of the United States of America, 107(39), 16952–16957. 10.1073/pnas.1002653107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JC, Barnes KM, Dong L, Yue S, Graber E, Rapaport R, Dauber A, Nilsson O, & Baron J (2018). Ezh2 mutations found in the weaver overgrowth syndrome cause a partial loss of H3K27 histone methyltransferase activity. The Journal of Clinical Endocrinology and Metabolism, 103(4), 1470–1478. 10.1210/jc.2017-01948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo A, Cheng D, Yuan S, Li H, Du J, Zhang Y, Yang C, Lin G, Zhang W, & Tan YQ (2018). Maternal interchromosomal insertional translocation leading to 1q43-q44 deletion and duplication in two siblings. Molecular Cytogenetics, 11, 24. 10.1186/s13039-018-0371-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelis CL, Hol FA, Graham GE, Rieu PN, Kellermayer R, Meijer RP, Lugtenberg D, Scheffer H, van Bokhoven H, Brunner HG, & de Brouwer AP (2008). Genotype–phenotype correlations in MYCN-related Feingold syndrome. Human Mutation, 29(9), 1125–1132. 10.1002/humu.20750 [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, Liaw D, Caron S, Duboué B, Lin AY, Richardson AL, Bonnetblanc JM, Bressieux JM, Cabarrot-Moreau A, Chompret A, Demange L, Eeles RA, Yahanda AM, Fearon ER, … Eng C (1998). Mutation spectrum and genotype–phenotype analyses in Cowden disease and Bannayan–Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Human Molecular Genetics, 7(3), 507–515. [DOI] [PubMed] [Google Scholar]
- McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, Van Bon BWM, Van Eyk CL, Broadbent J, Reynolds C, O’Callaghan ME, Nguyen LS, Adelson DL, Russo R, Jhangiani S, Doddapaneni H, Muzny DM, Gibbs RA, Gecz J, & MacLennan AH (2015). Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Molecular Psychiatry, 20(2), 176–182. 10.1038/mp.2014.189 [DOI] [PubMed] [Google Scholar]
- Mercedes Bloch AL, Diplas AA, Pepermans X, Emanuel BS, Rocca MS, Revencu N, & Sznajer Y (2014). Further phenotype description, genotype characterization in patients with de novo interstitial deletion on 2p23.2–24.1. American Journal of Medical Genetics. Part A, 164A(7), 1789–1794. 10.1002/ajmg.a.36516 [DOI] [PubMed] [Google Scholar]
- Miao L, Yang L, Huang H, Liang F, Ling C, & Hu Y (2016). mTORC1 is necessary but mTORC2 and GSK3beta are inhibitory for AKT3-induced axon regeneration in the central nervous system. eLife, 5, e14908. 10.7554/eLife.14908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migdalska AM, van der Weyden L, Ismail O, Sanger Mouse Genetics P, Rust AG, Rashid M, White JK, Sánchez-Andrade G, Lupski JR, Logan DW, Arends MJ, & Adams DJ (2012). Generation of the Sotos syndrome deletion in mice. Mammalian Genome, 23(11–12), 749–757. 10.1007/s00335-012-9416-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa GM (1993). MPPH syndrome. In Adams MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa GM, & Anemiya A (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. [Google Scholar]
- Mirzaa G, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, Logan CV, Roberts N, Johnson CA, Singh S, Kholmanskikh SS, Adams C, Hodge RD, Hevner RF, Bonthron DT, Braun K, Faivre L, Riviére JB, St-Onge J, Gripp KW, … Sheridan EG (2014). De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nature Genetics, 46(5), 510–515. 10.1038/ng.2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai Y, Dobashi Y, Okada E, Ishizawa S, Shiota M, Mori S, & Takano Y (2001). Study on the role of G1 cyclins in Epstein-Barr virus-associated human lymphomas maintained in severe combined immune deficiency (SCID) mice. International Journal of Cancer, 92(2), 232–239. [DOI] [PubMed] [Google Scholar]
- Mure H, Matsuzaki K, Kitazato KT, Mizobuchi Y, Kuwayama K, Kageji T, & Nagahiro S (2010). Akt2 and Akt3 play a pivotal role in malignant gliomas. Neuro-Oncology, 12(3), 221–232. 10.1093/neuonc/nop026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriello M, Kim AY, Sondergaard Schatz K, Beck N, Gunay-Aygun M, & Hoover-Fong JE (2019). Growth hormone deficiency, aortic dilation, and neurocognitive issues in Feingold syndrome 2. American Journal of Medical Genetics. Part A, 179(3), 410–416. 10.1002/ajmg.a.61037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kato M, Tohyama J, Shiohama T, Hayasaka K, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Matsumoto N, & Saitsu H (2014). AKT3 and PIK3R2 mutations in two patients with megalencephaly-related syndromes: MCAP and MPPH. Clinical Genetics, 85(4), 396–398. 10.1111/cge.12188 [DOI] [PubMed] [Google Scholar]
- Negishi Y, Miya F, Hattori A, Johmura Y, Nakagawa M, Ando N, Hori I, Togawa T, Aoyama K, Ohashi K, Fukumura S, Mizuno S, Umemura A, Kishimoto Y, Okamoto N, Kato M, Tsunoda T, Yamasaki M, Kanemura Y, … Saitoh S (2017). A combination of genetic and biochemical analyses for the diagnosis of PI3K-AKT-mTOR pathway-associated megalencephaly. BMC Medical Genetics, 18 (1), 4. 10.1186/s12881-016-0363-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellist M, Schot R, Hoogeveen-Westerveld M, Neuteboom RF, van der Louw EJ, Lequin MH, Bindels-de Heus K, Sibbles BJ, de Coo R, Brooks A, & Mancini GM (2015). Germline activating AKT3 mutation associated with megalencephaly, polymicrogyria, epilepsy and hypoglycemia. Molecular Genetics and Metabolism, 114(3), 467–473. 10.1016/j.ymgme.2014.11.018 [DOI] [PubMed] [Google Scholar]
- Oliveira D, Leal GF, Sertie AL, Caires LC Jr., Goulart E, Musso CM, Oliveira J, Krepischi A, Vianna-Morgante AM, & Zatz M (2019). 10q23.31 microduplication encompassing PTEN decreases mTOR signalling activity and is associated with autosomal dominant primary microcephaly. Journal of Medical Genetics, 56(8), 543–547. 10.1136/jmedgenet-2018-105471 [DOI] [PubMed] [Google Scholar]
- Orrico A, Galli L, Buoni S, Orsi A, Vonella G, & Sorrentino V (2009). Novel PTEN mutations in neurodevelopmental disorders and macrocephaly. Clinical Genetics, 75(2), 195–198. 10.1111/j.1399-0004.2008.01074.x [DOI] [PubMed] [Google Scholar]
- Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, Berwanger B, & Eilers M (2009). Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell, 15(1), 67–78. 10.1016/j.ccr.2008.12.005 [DOI] [PubMed] [Google Scholar]
- Ouimet B, Pepin E, Bergeron Y, Chagniel L, Beaulieu JM, Massicotte G, & Cyr M (2019). Motor learning deficits and striatal GSK-3 hyperactivity in Akt3 knockout mice. Behavioral Neuroscience, 133(1), 135–143. 10.1037/bne0000292 [DOI] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BFD, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, & Walsh CA (2012). Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron, 74(1), 41–48. 10.1016/j.neuron.2012.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, … Dobyns WB (2012). De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nature Genetics, 44(8), 934–940. 10.1038/ng.2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross ME, Carter ML, & Lee JH (1996). MN20, a D2 cyclin, is transiently expressed in selected neural populations during embryogenesis. The Journal of Neuroscience, 16(1), 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Theriault BL, Prigoda-Lee NL, Spencer C, Dimaras H, Corson TW, Pang R, Massey C, Godbout R, Jiang Z, Zacksenhaus E, Paton K, Moll AC, Houdayer C, Raizis A, … Gallie BL (2013). Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. The Lancet Oncology, 14(4), 327–334. 10.1016/S1470-2045(13)70045-7 [DOI] [PubMed] [Google Scholar]
- Sachwitz J, Meyer R, Fekete G, Spranger S, Matuleviciene A, Kucinskas V, Bach A, Luczay A, Brüchle NO, Eggermann K, Zerres K, Elbracht M, & Eggermann T (2017). NSD1 duplication in Silver–Russell syndrome (SRS): Molecular karyotyping in patients with SRS features. Clinical Genetics, 91(1), 73–78. 10.1111/cge.12803 [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: Mutation prediction for the deep-sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Sellars EA, Zimmerman SL, Smolarek T, & Hopkin RJ (2011). Ventricular noncompaction and absent thumbs in a newborn with tetrasomy 5q35.2–5q35.3: An association with hunter-McAlpine syndrome? American Journal of Medical Genetics. Part A, 155A(6), 1409–1413. 10.1002/ajmg.a.33997 [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, & Sirotkin K (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29(1), 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siavriene E, Preikšaitiene E, Maldžiene Ž, Mikstiene V, Rančelis T, Ambrozaityte L, Gueneau L, Reymond A, & Kučinskas V (2020). A de novo 13q31.3 microduplication encompassing the miR-17 ~92 cluster results in features mirroring those associated with Feingold syndrome 2. Gene, 753, 144816. 10.1016/j.gene.2020.144816 [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JAS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, & Weinberg RA (1996). Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature, 384(6608), 470–474. 10.1038/384470a0 [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, & Hamosh A (2015). GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, Waggoner D, Turkmen S, Cormier-Daire V, Irrthum A, Rahman N, & Childhood Overgrowth Collaboration (2005). Genotype–phenotype associations in Sotos syndrome: An analysis of 266 individuals with NSD1 aberrations. American Journal of Human Genetics, 77(2), 193–204. 10.1086/432082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte S, Ramsay E, Snape K, Murray A, Perdeaux ER, Seal S, Loveday C, Banka S, Clericuzio C, Flinter F, Magee A, McConnell V, Patton M, Raith W, Rankin J, … Rahman N (2011). Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget, 2(12), 1127–1133. 10.18632/oncotarget.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, Bird LM, Clericuzio CL, Cormier-Daire V, Cushing T, Flinter F, Jacquemont ML, Joss S, Kinning E, Lynch SA, Magee A, McConnell V, Medeira A, Ozono K, … Rahman N (2013). Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. American Journal of Medical Genetics. Part A, 161A(12), 2972–2980. 10.1002/ajmg.a.36229 [DOI] [PubMed] [Google Scholar]
- Tatton-Brown K, & Weksberg R (2013). Molecular mechanisms of childhood overgrowth. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 163C(2), 71–75. 10.1002/ajmg.c.31362 [DOI] [PubMed] [Google Scholar]
- Tavana O, Li D, Dai C, Lopez G, Banerjee D, Kon N, Chen C, Califano A, Yamashiro DJ, Sun H, & Gu W (2016). HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nature Medicine, 22(10), 1180–1186. 10.1038/nm.4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy B, Graham JM Jr., Feldman G, Perin J, Catherwood A, Knowlton R, Rappaport EF, Emanuel B, Driscoll DA, & Saitta SC (2011). High-resolution genomic arrays identify CNVs that phenocopy the chromosome 22q11.2 deletion syndrome. Human Mutation, 32(1), 91–97. 10.1002/humu.21395 [DOI] [PubMed] [Google Scholar]
- Varga EA, Pastore M, Prior T, Herman GE, & McBride KL (2009). The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genetics in Medicine, 11(2), 111–117. 10.1097/GIM.0b013e31818fd762 [DOI] [PubMed] [Google Scholar]
- Wang D, Zeesman S, Tarnopolsky MA, & Nowaczyk MJ (2013). Duplication of AKT3 as a cause of macrocephaly in duplication 1q43q44. American Journal of Medical Genetics. Part A, 161A(8), 2016–2019. 10.1002/ajmg.a.35999 [DOI] [PubMed] [Google Scholar]
- Wang JC, Steinraths M, Dang L, Lomax B, Eydoux P, Stockley T, Yong SL, & Van Allen MI (2007). Craniosynostosis associated with distal 5q-trisomy: Further evidence that extra copy of MSX2 gene leads to craniosynostosis. American Journal of Medical Genetics. Part A, 143A(24), 2931–2936. 10.1002/ajmg.a.31946 [DOI] [PubMed] [Google Scholar]
- Wang Z, Xie Y, Zhang L, Zhang H, An X, Wang T, & Meng A (2008). Migratory localization of cyclin D2-Cdk4 complex suggests a spatial regulation of the G1-S transition. Cell Structure and Function, 33(2), 171–183. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Menninger J, Beach D, & Ward DC (1992). Molecular cloning and chromosomal mapping of CCND genes encoding human D-type cyclins. Genomics, 13(3), 575–584. [DOI] [PubMed] [Google Scholar]
- Xue C, Yu DM, Gherardi S, Koach J, Milazzo G, Gamble L, Liu B, Valli E, Russell AJ, London WB, Liu T, Cheung BB, Marshall GM, Perini G, Haber M, & Norris MD (2016). MYCN promotes neuroblastoma malignancy by establishing a regulatory circuit with transcription factor AP4. Oncotarget, 7(34), 54937–54951. 10.18632/oncotarget.10709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZZ, Tschopp O, Di-Poi N, Bruder E, Baudry A, Dummler B, Wahli W, & Hemmings BA (2005). Dosage-dependent effects of Akt1/protein kinase Balpha (PKBalpha) and Akt3/PKBgamma on thymus, skin, and cardiovascular and nervous system development in mice. Molecular and Cellular Biology, 25(23), 10407–10418. 10.1128/MCB.25.23.10407-10418.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, Chrysler C, Nalpathamkalam T, Pellecchia G, Liu Y, Gazzellone MJ, D’Abate L, Deneault E, Howe JL, Liu RS, Thompson A, Zarrei M, Uddin M, Marshall CR, Ring RH, … Scherer SW (2015). Whole-genome sequencing of quartet families with autism spectrum disorder. Nature Medicine, 21(2), 185–191. 10.1038/nm.3792 [DOI] [PubMed] [Google Scholar]
- Zhang H, Lu X, Beasley J, Mulvihill JJ, Liu R, Li S, & Lee JY (2011). Reversed clinical phenotype due to a microduplication of Sotos syndrome region detected by array CGH: Microcephaly, developmental delay and delayed bone age. American Journal of Medical Genetics. Part A, 155A(6), 1374–1378. 10.1002/ajmg.a.33769 [DOI] [PubMed] [Google Scholar]
- Zhou XP, Waite KA, Pilarski R, Hampel H, Fernandez MJ, Bos C, Dasouki M, Feldman GL, Greenberg LA, Ivanovich J, Matloff E, Patterson A, Pierpont ME, Russo D, Nassif NT, & Eng C (2003). Germline PTEN promoter mutations and deletions in Cowden/Bannayan–Riley–Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. American Journal of Human Genetics, 73(2), 404–411. 10.1086/377109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material. The raw data that support the findings of this study, including the genetic analyses and the high content imaging results are available on request to the corresponding authors.